
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe merger of electro-reduction and hydrogen bonding activation for a radical Smiles rearrangement†
Liyuan
Lan
,
Kun
Xu
 * and
Chengchu
Zeng
* and
Chengchu
Zeng
College of Chemistry and Life Science, Beijing University of Technology, Beijing 100124, China. E-mail: kunxu@bjut.edu.cn
First published on 19th July 2024
Abstract
The reductive activation of chemical bonds at less negative potentials provides a foundation for high functional group tolerance and selectivity, and it is one of the central topics in organic electrosynthesis. Along this line, we report the design of a dual-activation mode by merging electro-reduction with hydrogen bonding activation. As a proof of principle, the reduction potential of N-phenylpropiolamide was shifted positively by 218 mV. Enabled by this strategy, the radical Smiles rearrangement of N-arylpropiolamides without external radical precursors and prefunctionalization steps was accomplished. [DBU][HOAc], a readily accessible ionic liquid, was exploited for the first time both as a hydrogen bonding donor and as a supporting electrolyte.
Introduction
Organic electrosynthesis provides a simple and sustainable way to alter the redox-states of organic molecules, and it has merged as an increasingly viable platform in modern organic synthesis.1 While selectivity and functional group tolerance are deciding factors in how useful a methodology may ultimately be, the electro-activation of chemical bonds under low operating potentials (absolute value) to fulfill the criteria of good selectivity and broad functional group tolerance is a sought-after goal in organic electrosynthesis.2 In the context of electro-reduction, the reduction of functionalities at less negative potentials is much appreciated (Scheme 1a). To meet this requirement, the concept of mediated cathodic reduction has shown great promise for reducing functionalities at lower operating potentials.3 However, this strategy loses its utility when functionalities possessing high reduction potentials are used, considering the limited availability of strongly reducing mediators. Against this background, the merger of electro-reduction and photoredox catalysis to generate strongly reducing species at less negative potentials has been developed in recent years.4 Another conceptually appealing activation mode is the strategic exploitation of the collaboration between electro-reduction and hydrogen bonding (Scheme 1b). Hydrogen bonding, a classical noncovalent interaction, has wide applications in organic synthesis due to its high directionality.5 In this sense, the synergism of electro-reduction with hydrogen bonding activation would, in principle, decrease the activation potentials of hydrogen bonding acceptors, thus providing valuable advantages in terms of functional group tolerance, as well as selectivity. Although voltammetric studies have shown that electro-reduction could be enhanced by hydrogen bonding,6 the application of this noncovalent interaction in reductive bulk electrolysis is still in its infancy, mainly due to the fact that hydrogen bonding donors are susceptible to H2 evolution under highly reductive conditions. As such, overcoming this synthetic challenge requires the identification of an electro-reductively stable hydrogen bonding donor. | ||
| Scheme 1 The collaboration between electro-reduction and hydrogen bonding and the reported strategies to radical Smiles rearrangement. | ||
To implement this dual activation strategy, the electrochemical radical Smiles rearrangement of N-arylpropiolamides possessing high reduction potentials was investigated. Radical Smiles rearrangement offers a powerful approach to arylation via ipso substitution, thus leading to an increased interest from the synthetic community in recent years.7 Taking the widely studied desulfonylative variant for example (Scheme 1c), the radical Smiles rearrangement was often triggered by the generation of a key carbon-centered radical (intermediate I) either by an intermolecular radical addition8 or by an intramolecularly reductive C–X bond cleavage.9 In contrast, the generation of intermediate I that relied on Mn(III)-catalyzed hydrogen atom transfer or Fe(III)-mediated alkene oxidation has been developed by Shenvi and Studer groups, respectively.10 Encouraged by the recent achievements in electro-reductive synthesis,11,12 we envisioned that the electro-reduction of N-arylpropiolamides would generate the corresponding vinyl radicals to trigger the following radical Smiles rearrangement. Considering that N-arylpropiolamides are difficult to be reduced, the introduction of hydrogen bonding activation would decrease their reduction potentials to provide broader functional group tolerance and improved selectivity. Herein, we report a dual-activation strategy integrating electro-reduction and hydrogen bonding activation to shift the reduction potential of N-phenylpropiolamide positively by 218 mV (Scheme 1d). [DBU][HOAc], a typical ionic liquid,13 was exploited for the first time both as a hydrogen bonding donor and as a supporting electrolyte. Taking advantage of this strategy, the radical Smiles rearrangement of N-arylpropiolamides was realized, providing a rapid access to diarylpropanamides that widely exist in biologically relevant molecules.14 Distinct from prior-art on radical Smiles rearrangements, this electro-reductive variant obviates the use of external radical precursors, prefunctionalization steps to install C–X bonds, and transition metal catalysts.
Results and discussion
We began our study with the electrochemical rearrangement of 1a as the model reaction for reaction condition optimization (Table 1). When the Smiles rearrangement was carried out in an undivided cell with [DBU][HOAc] (2) as the additive under constant current conditions, the desired product 3 was obtained in 61% yield (entry 1). Replacing [DBU][HOAc] with [DBU][TFA] or [DBU][MSA] afforded 3 in 52% and 50% yields, respectively (entries 2 and 3), while [DBU][PhCO2H] as the additive gave 3 in the highest yield of 68% (entry 4). However, [DBU][HOAc] was selected as the optimal additive for further optimization due to its low cost and high atom economy. When the Pt plate or graphite was employed as the cathode instead of nickel foam, the product 3 was obtained in 44% and 47% yields, respectively (entry 5). The solvent screening showed that dimethyl sulfoxide (DMSO) was the optimal solvent, while other commonly used solvents such as CH3CN, N,N-dimethylacetamide (DMA) and N,N-dimethylformamide (DMF) gave 3 in trace to low yields (entries 6–8). Next, the addition of external supporting electrolytes such as n-Bu4NPF6, n-Bu4NClO4, LiClO4, and n-Bu4NBF4 failed to improve the yield of 3 (entries 9–12). Further optimization showed that 10 mA cm−2 was the optimal current density, while decreased current densities led to lower yields (entries 13). The yield of 3 decreased to 54% when the electrolysis was carried out in the absence of N2 protection (entry 14). The control experiments revealed that a complex mixture involving a trace amount of 3 was generated in the absence of [DBU][HOAc] with n-Bu4NBF4 as the supporting electrolyte (entry 15), while no reaction occurred without electrolysis (entry 16).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| a Reaction conditions: 1a (0.3 mmol), 2 (0.3 mmol), DMSO (4 mL), graphite anode (1 cm × 1 cm × 0.2 cm), foamed nickel cathode (1 cm × 1 cm × 0.2 cm), J = 10 mA cm−2, N2, rt, 8 h; isolated yield; TFA = trifluoroacetic acid, MSA = methanesulfonic acid. | ||
| 1 | None | 61 |
| 2 | [DBU][TFA] instead of 2 | 52 |
| 3 | [DBU][MSA] instead of 2 | 50 |
| 4 | [DBU][PhCO2H] instead of 2 | 68 |
| 5 | Pt plate or graphite as the cathode | 44 or 47 |
| 6 | CH3CN instead of DMSO | 32 |
| 7 | DMA instead of DMSO | Trace |
| 8 | DMF instead of DMSO | Trace |
| 9 | n-Bu4NPF6 (0.05 M) as the supporting electrolyte | 44 |
| 10 | n-Bu4NClO4 (0.05 M) as the supporting electrolyte | 46 |
| 11 | LiClO4 (0.05 M) as the supporting electrolyte | Trace |
| 12 | n-Bu4NBF4 (0.05 M) as the supporting electrolyte | 58 |
| 13 | 6 mA cm−2 or 8 mA cm−2 | 41 or 57 |
| 14 | No N2 | 54 |
| 15 | No 2, n-Bu4NBF4 (0.05 M) as the supporting electrolyte | Trace |
| 16 | No electrolysis | 0 |
Having established the optimal conditions for this radical Smiles rearrangement, the substrate scope was then investigated (Scheme 2). For the mono-substituted Ar1 moiety, both electron-donating and electron-withdrawing substituents were well tolerated to give the corresponding products 3–16 with up to 72% yield. When the substituent was shifted from the para-position to the ortho- or meta-position, the Smiles rearrangement also proceeded smoothly to afford the corresponding products 17–18 with up to 69% yield. For the di-substituted Ar1 moiety, substituents can be located relative to each other in the ortho, meta, or para position, yielding diarylpropanamides 19–25 with up to 68% yield. In addition, both 1-naphthyl and 2-naphthyl substituted substrates proved to be efficient to generate diarylpropanamides 26–27 in 62–63% yields. Noteworthy, N-heteroaryl substituted propiolamides were also suitable substrates to afford products 28–30 in 30–45% yields. Altering the R group from the methyl group to the ethyl or isopropyl group led to a slightly decreased efficiency, delivering products 31–32 in 59–66% yields. Finally, the scope of the Ar2 moiety was examined. It was found that both electron-rich and electron-deficient substituents attached on the Ar2 moiety were well tolerated to give 3, 9, 11, 12, and 13 in 61–74% yields. The moderate yields result from the incomplete conversion of starting materials and the side reaction of alkyne hydrogenation. This electrochemical Smiles rearrangement features excellent functional group compatibility, as evidenced by the tolerance of various reducible functionalities such as carbonyl (11), cyan (12, 18, 24), ester (13, 25, 28), sulfonyl (14), alkynyl (15, 17), and amide (29). As a limitation, the reaction did not occur when the R group was replaced with hydrogen (33). In addition, the alkyl-substituted N-arylpropiolamides did not work under the optimal conditions.
 | ||
| Scheme 2 The substrate scope. aReaction conditions: 1a (0.3 mmol), 2 (0.3 mmol), DMSO (4 mL), graphite anode (1 cm × 1 cm × 0.2 cm), foamed nickel cathode (1 cm × 1 cm × 0.2 cm), J = 10 mA cm−2, N2, rt, 8 h; isolated yield. aJ = 8 mA cm−2. bJ = 5 mA cm−2. | ||
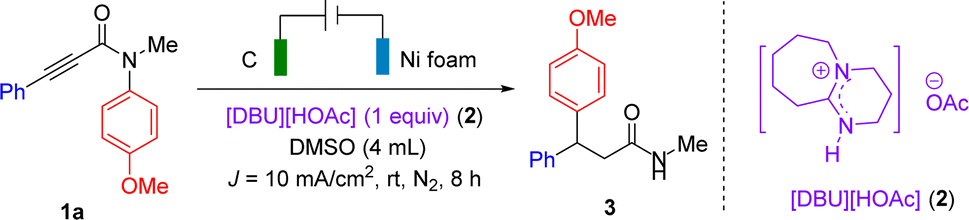
First, cyclic voltammetry (CV) experiments were carried out to reveal the hydrogen bonding between 1a and [DBU][HOAc] (2). As shown in Fig. 1, left, the CV of substrate 1a showed an obvious reduction peak at −2.5 V vs. Ag/AgNO3 (curve a), while 2 exhibited no obvious reduction behaviour within the range of 0 to −2.7 V (curve b). These results suggest that the electro-reduction of 1a is much preferred to that of 2. As shown in Fig. 1, middle, the CV of the mixture of 2 (0.5 equiv.) and 1a showed the evolution of a new reduction peak at −2.3 V vs. Ag/AgNO3 (curve c). Compared with curve b, the reduction potential of 1a was shifted positively by 218 mV. Notably, the corresponding reduction current was enhanced upon gradually increasing the molar ratio of 2/1a (curves d and e). However, DBU cannot shift the reduction potential of 1a positively upon addition of DBU to the mixture of 1a/DMSO (Fig. 1, right). Collectively, the CV analyses suggest the hydrogen bonding between 1a and 2.
 | ||
| Fig. 1 CV experiments. | ||
To further demonstrate the proposed hydrogen bonding, 1H NMR experiments were carried out (Fig. 2). However, considering that 1a doesn't have alkyne protons and the signal of the NH proton in 2 can't be observed in d6-DMSO (see the ESI† for details), an analog compound (34) was employed for 1H NMR analysis. As shown in Fig. 2, the signal of the alkyne proton in 34 appeared at 4.18 ppm. Upon addition of 2 (1 equiv.) into the mixture of 34/d6-DMSO, the chemical shift value of the alkyne proton shifted to down-field. This value shifted to down-field again when another portion of 2 was added. The 1H NMR analyses in combination with CV experiments support the hydrogen bonding between 1a and [DBU][HOAc] (2).
 | ||
| Fig. 2 1H NMR experiments. | ||
To gain more mechanistic insight into this electrochemical Smiles rearrangement, some control experiments were carried out (Scheme 3). When the electrochemical rearrangement was performed in the presence of D2O, 3-d3 was obtained in 23% yield in which 85% and 80% deuterations were found at the two benzylic positions, respectively (Scheme 3a). In contrast, the deuteration ratios were very low when d6-DMSO or [DBU][CD3COOD] was used as the deuterium source (Scheme 3a). These deuterium-labeling studies demonstrate that carbanions were generated at these two benzylic positions during the electrolysis. When 1-(4-methylpiperidin-1-yl)-3-phenylprop-2-yn-1-one (35) was electrolyzed under the standard conditions, the complete hydrogenation product 36 was obtained in 50% yield (Scheme 3b). However, the semi-reduction of alkyne (37), as well as the completely hydrogenated product (36), was observed when shortening the reaction time to 2 h (Scheme 3b). These results suggest that the electro-reduction of 35 proceeds via a stepwise electron transfer-proton transfer process. To demonstrate that the radical Smiles rearrangement was initiated by a transient vinyl radical instead of an alkyl radical, the electrolysis of 38 was performed (Scheme 3c). As expected, no Smiles rearrangement product 3 was observed. This result excludes the possibility that the Smiles rearrangement was triggered by an alkyl radical resulting from the SET reduction of 38. The selectivity experiment suggests that the vinyl radical would more likely attack the electron-rich site of substrate 39 to yield 40 in 32% yield (Scheme 3d).
 | ||
| Scheme 3 The control experiments. | ||
Based on the control experiments, CV and 1H NMR analyses, the generation of 3 through the relay of electro-reduction of alkyne via a proton-coupled electron-transfer (PCET) process, radical Smiles rearrangement, and alkene hydrogenation was proposed (Scheme 4). Initially, the mixture of 1a and [DBU][HOAc] (2) forms a complex of 42 through a hydrogen bonding interaction, which was supported by the CV analysis and 1H NMR analyses. The hydrogen bonding enables the SET reduction of 42 at decreased potential to afford a radical anion 43, which undergoes protonation to yield a transient vinyl radical 44. Subsequently, radical 44 triggers a Smiles rearrangement via an unusual C–N cleavage to afford a postulated amidyl radical 46. Upon hydrogen atom abstraction or SET reduction of 46 followed by protonation, intermediate 47 is generated. The cathodic reduction of 47 followed by protonation generates 48, which proceeds via a radical-polar crossover (RPC)15 pathway to deliver product 3. Simultaneously, the anodic oxidation of DMSO maintains the charge balance.
 | ||
| Scheme 4 A plausible mechanism accounting for the generation of 3. | ||
Conclusions
In conclusion, this work details the strategic integration of hydrogen bonding into electro-reduction to activate N-arylpropiolamides at less negative potentials, which in turn provides a basis for high selectivity and functional group compatibility. This dual activation strategy led to a radial Smiles rearrangement of N-arylpropiolamides to deliver diarylpropanamides that are otherwise difficult to access. Distinct from prior-art on radical Smiles rearrangement, this electro-reductive variant obviates the use of external radical precursors or prefunctionalization steps, and features 100% atom efficiency. Uniquely, [DBU][HOAc] serves both as a hydrogen bonding donor and as a supporting electrolyte. The stable nature of [DBU][HOAc] under highly reductive conditions is the key to the success of this work. The proposed hydrogen bonding was supported by CV and 1H NMR studies. We hope this dual activation strategy would offer new possibilities for reductive electrosynthesis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. Y. L. performed the experiment and data analyses. K. X. conceived the idea and designed the research. K. X. drafted the manuscript; C. C. Z. helped supervise the project and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22171015 and 22271009), and Beijing Natural Science Foundation (2222003). We also extend our thanks to the Large-scale Instruments and Equipment Sharing Platform of Beijing University of Technology for NMR experiments.Notes and references
- Recent reviews on organic electrosynthesis, see: (a) L. F. T. Novaes, J.-J. Liu, Y.-F. Shen, L.-X. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941 RSC; (b) K. Lam and K. M. P. Wheelhouse, Org. Process Res. Dev., 2021, 25, 2579 CrossRef CAS; (c) N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487 CrossRef CAS PubMed; (d) M. Klein and S. R. Waldvogel, Angew. Chem., Int. Ed., 2022, 61, e202204140 CrossRef CAS PubMed; (e) C. A. Malapit, M. B. Prater, J. R. Cabrera-Pardo, M. Li, T. D. Pham, T. P. McFadden, S. Blank and S. D. Minteer, Chem. Rev., 2022, 122, 3180 CrossRef CAS PubMed; (f) Y.-W. Liu, P.-F. Li, Y.-W. Wang and Y.-A. Qiu, Angew. Chem., Int. Ed., 2023, 62, e202306679 CrossRef CAS PubMed; (g) Y.-L. Wang, S. Dana, H. Long, Y. Xu, Y.-J. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269 CrossRef CAS PubMed; (h) Y. Kawamata and P. Baran, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-glvvh; (i) R. Francke and R. D. Little, Curr. Opin. Electrochem., 2023, 40, 101315 CrossRef CAS; (j) L. Zeng, J.-X. Wang, D.-X. Wang, H. Yi and A.-W. Lei, Angew. Chem., Int. Ed., 2023, 62, e202309620 CrossRef CAS PubMed; (k) G. Hilt, Curr. Opin. Electrochem., 2024, 43, 101425 CrossRef CAS; (l) N. Chen, Z.-J. Wu and H.-C. Xu, Isr. J. Chem., 2024, 64, e202300097 CrossRef and reviews cited therein..
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561 CrossRef CAS PubMed.
- (a) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492 RSC; (b) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (c) S. Inagi and T. Fuchigami, Curr. Opin. Electrochem., 2017, 2, 32 CrossRef CAS; (d) R. Krueger and K. D. Moeller, J. Org. Chem., 2021, 86, 15847 CrossRef CAS PubMed.
- (a) T. Hardwick and N. Ahmed, ACS Sustain. Chem. Eng., 2021, 9, 4324 CrossRef CAS; (b) H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567 CrossRef CAS PubMed; (c) S.-Z. Wu, J. Kaur, T. A. Karl, X.-H. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed; (d) L. Grimaud, S. Lakhdar and M. R. Vitale, Curr. Opin. Electrochem., 2023, 40, 101307 CrossRef CAS; (e) M. Lepori, S. Schmid and J. P. Barham, Beilstein J. Org. Chem., 2023, 19, 1055 CrossRef CAS PubMed; (f) J. M. Edgecomb, S. N. Alektiar, N. G. W. Cowper, J. A. Sowin and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 20169 CrossRef CAS PubMed.
- A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS PubMed.
- (a) M. E. Tessensohn and R. D. Webster, Electrochem. Commun., 2016, 62, 38 CrossRef CAS; (b) A. M. Costero, G. M. Rodríguez-Muňiz, P. Gaviňa, S. Gil and A. Domenech, Tetrahedron Lett., 2007, 48, 6992 CrossRef CAS; (c) A. N. Hellman, R. Haiges and S. C. Marinescu, ChemElectroChem, 2021, 8, 1864 CrossRef CAS.
- (a) T. J. Snape, Chem. Soc. Rev., 2008, 37, 2452 RSC; (b) C. M. Holden and M. F. Greaney, Chem.–Eur. J., 2017, 23, 8992 CrossRef CAS PubMed; (c) A. R. Allen, E. A. Noten and C. R. J. Stephenson, Chem. Rev., 2021, 122, 2695 CrossRef PubMed; (d) D. M. Whalley and M. F. Greaney, Synthesis, 2022, 54, 1908 CrossRef CAS; (e) S. M. Wales, R. K. Saunthwal and J. Clayden, Acc. Chem. Res., 2022, 55, 1731 CrossRef CAS PubMed; (f) X.-H. Chang, Q.-L. Zhang and C. Guo, Org. Lett., 2019, 21, 10 CrossRef CAS PubMed; (g) C.-K. Liu, Q. Jiang, Y. Lin, Z. Fang and K. Guo, Org. Lett., 2020, 22, 795 CrossRef CAS PubMed; (h) E. Derat, G. Masson and A. Claraz, Angew. Chem., Int. Ed., 2024, e202406017 CAS.
- (a) M. Wang, H.-H. Zhang, J.-G. Liu, X.-X. Wu and C. Zhu, Angew. Chem., Int. Ed., 2019, 58, 17646 CrossRef CAS PubMed; (b) Z.-S. Wang, Y.-B. Chen, H.-W. Zhang, Z. Sun, C.-Y. Zhu and L.-W. Ye, J. Am. Chem. Soc., 2020, 142, 3636 CrossRef CAS PubMed; (c) J.-M. Yan, H.-W. Cheo, W.-K. Teo, X.-C. Shi, H. Wu, S. B. Idres, L.-W. Deng and J. Wu, J. Am. Chem. Soc., 2020, 142, 11357 CrossRef CAS PubMed; (d) C. Hervieu, M. S. Kirillova, T. Suárez, M. Müller, E. Merino and C. Nevado, Nat. Chem., 2021, 13, 327 CrossRef CAS PubMed; (e) J.-Y. Lan, K.-J. Lin, X. Zhang and T.-S. Zhu, Green Chem., 2022, 24, 6138 RSC; (f) J.-P. Huang, F. Liu, L.-H. Zeng, S.-Y. Li, Z.-Y. Chen and J. Wu, Nat. Commun., 2022, 13, 7081 CrossRef CAS PubMed; (g) E. A. Noten, C. H. Ng, R. M. Wolesensky and C. R. J. Stephenson, Nat. Chem., 2024, 16, 599 CrossRef CAS PubMed; (h) C. Hervieu, M. S. Kirillova, Y.-W. Hu, S. Cuesta-Galisteo, E. Merino and C. Nevado, Nat. Chem., 2024, 16, 607 CrossRef CAS PubMed.
- (a) W. B. Motherwell and A. M. K. Pennell, J. Chem. Soc., Chem. Commun., 1991, 13, 877 RSC; (b) A. Gheorghe, B. Quiclet-Sire, X. Vila and S. Z. Zard, Tetrahedron, 2007, 63, 7187 CrossRef CAS; (c) J. J. Douglas, H. Albright, M. J. Sevrin, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 14898 CrossRef CAS PubMed; (d) D. Alpers, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2018, 57, 12167 CrossRef CAS PubMed; (e) N. Radhoff and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 3561 CrossRef CAS PubMed.
- (a) S. W. M. Crossley, R. M. Martinez, S. Guevara-Zuluaga and R. A. Shenvi, Org. Lett., 2016, 18, 2620 CrossRef CAS PubMed; (b) N. Radhoff and A. Studer, Nat. Commun., 2022, 13, 3083 CrossRef CAS PubMed.
- Recent reviews and examples on reductive electrosynthesis, see: (a) P.-F. Hu, B. K. Peters, C. A. Malapit, J. C. Vantourout, P. Wang, J.-J. Li, L. Mele, P.-G. Echeverria, S. D. Minteer and P. S. Baran, J. Am. Chem. Soc., 2020, 142, 20979 CrossRef CAS PubMed; (b) S. Manabe, C. M. Wong and C. S. Sevov, J. Am. Chem. Soc., 2020, 142, 3024 CrossRef CAS PubMed; (c) X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W.-J. Xia, Org. Lett., 2021, 23, 3472 CrossRef CAS PubMed; (d) C. Zhu, S.-C. Lee, H.-F. Chen, H.-F. Yue and M. Rueping, Angew. Chem., Int. Ed., 2022, 61, e202204212 CrossRef CAS PubMed; (e) A. Claraz and G. Masson, ACS Org. Inorg. Au, 2022, 2, 126 CrossRef CAS PubMed; (f) W. Zhang, W.-Y. Guan, J. I. M. Alvarado, L. F. T. Novaes and S. Lin, ACS Catal., 2023, 13, 8038 CrossRef CAS PubMed; (g) P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed; (h) M.-R. Wang, C.-Q. Zhang, C.-G. Ci, H.-F. Jiang, P. H. Dixneuf and M. Zhang, J. Am. Chem. Soc., 2023, 145, 10967 CrossRef CAS PubMed; (i) Y.-Z. Wang, Z.-H. Wang, I. L. Eshel, B. Sun, D. Liu, Y.-C. Gu, A. Milo and T.-S. Mei, Nat. Commun., 2023, 14, 2322 CrossRef CAS PubMed; (j) J. Luo, M. T. Davenport, C. Callister, S. D. Minteer, D. H. Ess and L. T. Liu, J. Am. Chem. Soc., 2023, 145, 16130 CrossRef CAS PubMed; (k) P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed; (l) G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z.-P. Lu, D.-G. Yu and S. Lin, Nature, 2023, 615, 67 CrossRef CAS PubMed and references cited therein..
- (a) S. Zhang, L.-J. Li, J.-J. Li, J.-X. Shi, K. Xu, W.-C. Gao, L.-Y. Zong, G.-G. Li and M. Findlater, Angew. Chem., Int. Ed., 2021, 60, 7275 CrossRef CAS PubMed; (b) F. Lian, K. Xu and C.-C. Zeng, CCS Chem., 2023, 5, 1973 CrossRef CAS; (c) F. Lian, K. Xu and C.-C. Zeng, Sci. China: Chem., 2023, 66, 540 CrossRef CAS; (d) F. Xiang, D.-H. Wang, K. Xu and C.-C. Zeng, Org. Lett., 2024, 26, 411 CrossRef CAS PubMed.
- [DBU][HOAc] was typically used to increase the nucleophilicity of amines, see: (a) R. Singh, D. S. Raghuvanshi and K. N. Singh, Org. Lett., 2013, 15, 4202 CrossRef CAS PubMed; (b) N.-L. Luo, M.-S. Li, T. Wang, Y.-X. Li and C.-D. Wang, ChemistrySelect, 2019, 4, 11121 CrossRef CAS; (c) S. K. Boddu, N. U. Rehman, T. K. Mohanta, A. Majhi, S. K. Avula and A. Al-Harrasi, Green Chem. Lett. Rev., 2022, 15, 765 CrossRef CAS.
- (a) J. R. Huh, E. E. Englund, H. Wang, R.-L. Huang, P.-X. Huang, F. Rastinejad, J. Inglese, C. P. Austin, R. L. Johnson, W.-W. Huang and D. R. Littman, ACS Med. Chem. Lett., 2013, 4, 79 CrossRef CAS PubMed; (b) K. Sugimoto, K. Toyoshima, S. Nonaka, K. Kotaki, H. Ueda and H. Tokuyama, Angew. Chem., Int. Ed., 2013, 52, 7168 CrossRef CAS PubMed.
- Recent reviews on electrochemical RPC reactions, see: (a) Z.-M. Tan, H.-N. Zhang, K. Xu and C.-C. Zeng, Sci. China: Chem., 2024, 67, 450 CrossRef CAS; (b) A. Choi and H. Kim, Curr. Opin. Electrochem., 2024, 44, 101449 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02821j |
| This journal is © The Royal Society of Chemistry 2024 |