
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective recognition and enrichment of C70 over C60 using an anthracene-based nanotube†
Hao
Nian‡
ab,
Song-Meng
Wang‡
a,
Yan-Fang
Wang
a,
Yu-Tao
Zheng
a,
Li-Shuo
Zheng
a,
Xiaoping
Wang
a,
Liu-Pan
Yang
 *c,
Wei
Jiang§
*a and
Liping
Cao
*b
*c,
Wei
Jiang§
*a and
Liping
Cao
*b
aDepartment of Chemistry, Southern University of Science and Technology, Xueyuan Blvd 1088, Shenzhen, 518055, China. E-mail: jiangw@sustech.edu.cn
bCollege of Chemistry and Materials Science, Northwest University, Xi'an, 710069, P. R. China. E-mail: chcaoliping@nwu.edu.cn
cSchool of Pharmaceutical Science, University of South China, Hengyang, Hunan 421001, China. E-mail: yanglp@usc.edu.cn
First published on 30th May 2024
Abstract
Selective recognition and enrichment of fullerenes (e.g., C60 and C70) remains challenging due to the same diameter and geometrical similarity. Herein, we report a hexagonal anthracene-based nanotube (1) through a one-pot Suzuki–Miyaura cross-coupling reaction. With anthracene-based side walls and pyridine linkers, 1 features a nano-scale tubular cavity measuring 1.2 nm in diameter and 0.9 nm in depth, along with pH-responsive properties. Interestingly, the electron-rich 1 shows high binding affinity (Ka ≈ 106 M−1) and selectivity (Ks ≈ 140) to C70 over C60 in toluene, resulting from their different contribution of π–π interactions with the host. The protonation of 1 simultaneously alters the electronic properties within the nanotube, resulting in the release of the fullerene guests. Lastly, the selective recognition and pH stimuli-responsive properties of the nanotube have been utilized to enrich C70 from its low-content mixtures of fullerenes in chloroform.
Introduction
Since Krätschmer and coworkers reported the extraction of C60 and C70 from carbon soot by using aromatic solvents in 1990,1 a number of artificial receptors have been extensively developed for the recognition and separation of fullerenes.2,3 Most traditional macrocyclic hosts and their derivatives, such as γ-cyclodextrin (γ-CD),4–7 calix[n]arenes,8–13 pillar[10]arene,14 and theoretical cucurbit[9]uril (CB[9]),15 have shown similar binding affinities to fullerene compounds including C60 and C70. There are still many challenges in efficiently distinguishing C60 and C70 through molecular recognition. For example, C60 and C70 have the same diameter but differ in length, leading to challenges in their selective recognition. This requirement implies that there is variation in the geometric-shape matching between the receptors' cavities and different fullerenes. On the other hand, fullerenes can be regarded as electron-deficient guests in the host–guest interaction. The electron-rich aromatic units, such as porphyrins,16–18 anthracene,19,20 and nanographene21 can be used as building blocks to achieve the complementary charge interactions with fullerenes. As a result, several fullerene-selective receptors with various geometric-shape cavities have been reported (Scheme 1a), including molecular cages,22 coordination capsules,23 deep cavitands,24 and so on. These three-dimensional (3D) structures with both restricted cavities and buried π–π interactions have achieved selective binding for fullerenes based on the shape matching and/or the charge complementation.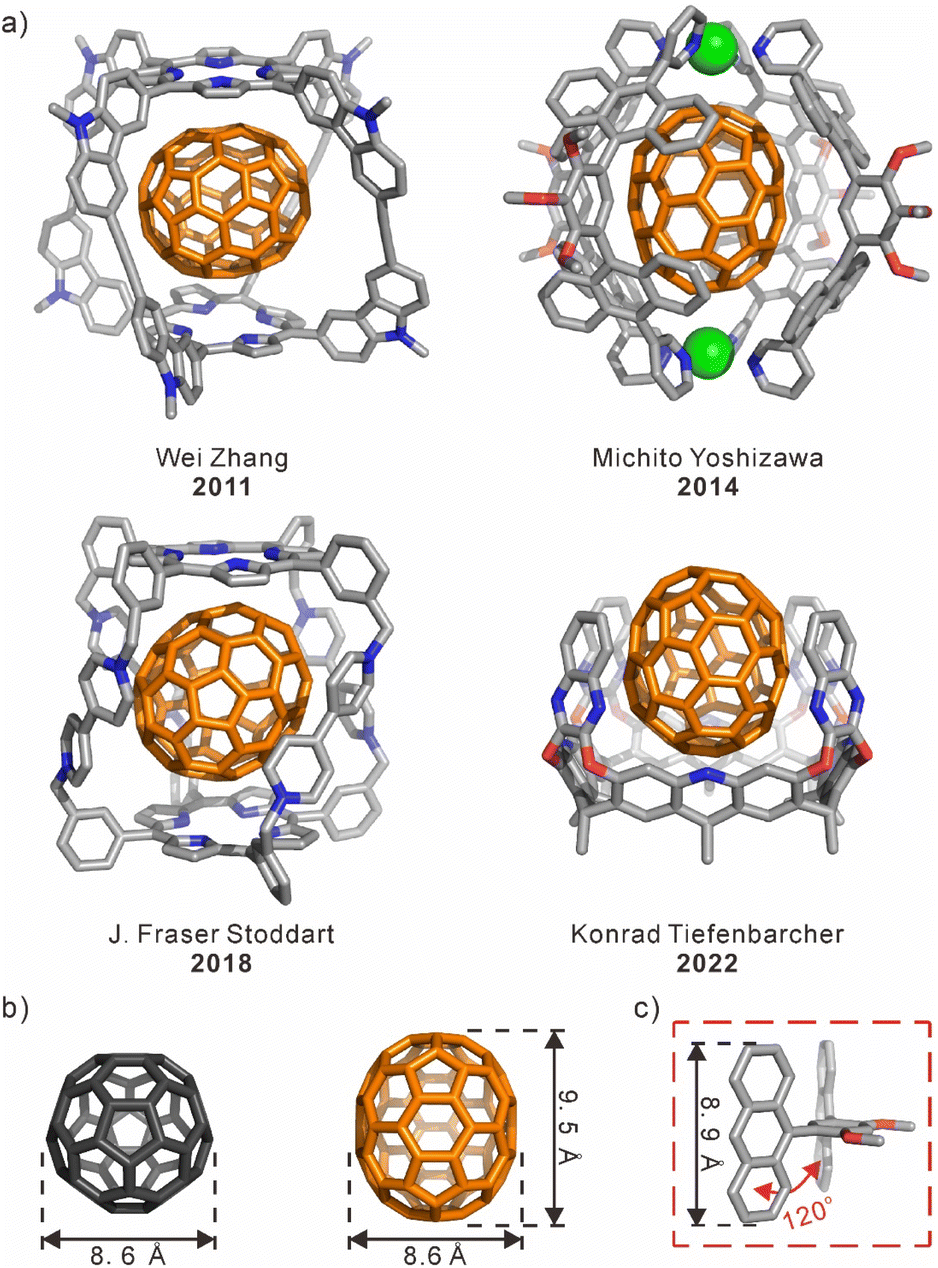 | ||
| Scheme 1 (a) Representative samples of the host⊃C70 complexes. (b) Comparison of the sizes of C60 and C70. (c) The bent structure of the anthracene-dimer building block. | ||
The design and synthesis of macrocyclic compounds has been one of the interesting areas for supramolecular chemists.25,26 Especially, a class of aromatic macrocycles with nano-scale cavities, such as cycloparaphenylenes (CPPs)27–29 and cyclophanes,30–32 can bind to the outer convex surface of a fullerene within their inner concave surfaces via the charge- and shape-complementary π–π interactions. However, the selective recognition of fullerenes by aromatic macrocycles or nanotubes has been rarely achieved. Although there are some examples, the selectivity of fullerenes is still not satisfactory for their separation.33,34 In fact, there are enormous challenges associated with selective recognition of macrocyclic hosts for fullerenes, including issues related to synthesis procedures, molecular structure design, and control over flexibility for size expansion. In general, large macrocycles that are tailored to the size of fullerenes can be synthesized by increasing both the number14 and size35,36 of building blocks.37,38 However, the flexibility may increase with the enlargement of the ring size, which may cause the collapse of the macrocyclic compounds.39 Additionally, a more intricate molecular structure is required to differentiate the small size disparity between C60 and C70 (Scheme 1b). Therefore, an effective solution is to achieve selective recognition by matching the size of a macrocyclic cavity with different fullerenes.
The anthracene-dimer building block19,40 with a bent structure of 120° angle was used to construct a series of coordination capsules/tubes,23 covalent nanotubes,41 and polyaromatic capsules (Scheme 1c).42,43 And, anthracene, as a large aromatic electron-rich panel with a length comparable to that of C70, could be a suitable building block for constructing receptors to selectively recognize C70 over C60.42,44 Herein, we have designed and synthesized a hexagonal anthracene-based nanotube (1) formed from three anthracene-dimer building blocks and three 2,6-dibromo-4-methoxypyridine rings via a one-pot Suzuki–Miyaura cross-coupling reaction. In the macrocyclic structure of 1, six anthracene panels serve as side walls and are covalently linked through alternating benzene and pyridine rings to form a hexagonal cavity. Interestingly, the size of the inner cavity with the diameter of ∼1.2 nm and the depth of ∼0.9 nm perfectly matches with the size of C70. As a result, the nanotube can selectively encapsulate C70 along the long axis via enhanced π–π interactions over C60, showing a high binding selectivity (Ks) of ∼140 times. On the other hand, the pH stimuli-responsive properties of the nanotube were verified through NMR, UV-vis, and fluorescence spectroscopy, which are attributed to the protonation and deprotonation of the pyridine rings in the nanotube. As a result, this anthracene-based nanotube was successfully employed in the enrichment of C70 from a fullerene mixture enriched with C60, owing to its selectivity towards C70 and pH stimuli-responsive properties.
Results and discussion
The synthetic route of the nanotube is shown in Fig. 1a. First, 2Bpin can be directly obtained by 2Br through the Miyaura borylation reaction (Fig. S1–S4†). The anthracene-based nanotube 1·was synthesized in ∼5% yield from a one-pot Suzuki–Miyaura cross-coupling of 2Bpin and 2,6-dibromo-4-methoxypyridine (3) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio (Fig. S5†). 1 was fully characterized by 1H, 13C, and 2D NMR spectroscopy as well as high-resolution mass spectrometry (HR-MS) measurements (Fig. S6–S10†). The nanotube displayed good solubility in toluene, chloroform, and dichloromethane, but was insoluble in methanol. A highly symmetric structure was indicated by 1H NMR spectra, and no significant aggregation or other conformational isomers were observed at NMR concentrations (Fig. S6†). Notably, the inward-directed nitrogen atoms of pyridine rings can be protonated by adding excess trifluoroacetic acid (TFA) (Fig. 1b). The simplicity of the NMR signals suggested that all pyridine rings were protonated, and significant splitting of anthracene rings was observed (Hf–i), which indicated that anthracene rings were situated in a more asymmetric chemical environment. The electron cloud density of pyridine rings decreased after full protonation, resulting in the proton resonances of anthracene (Hi) and pyridine (Ha) groups displayed obvious upfield shifts because these protons are located at the deshielding area of pyridine rings. However, the other proton resonances (Hb, Hd, and Hg–h) were significantly shifted downfield. There are little shifts of protons Hc and He on the benzene rings, due to a more positive electronic environment of the inner cavity. The broad peaks implied that the solubility of the protonated nanotube in toluene-d8 was slightly reduced under the acidic conditions.
:1 molar ratio (Fig. S5†). 1 was fully characterized by 1H, 13C, and 2D NMR spectroscopy as well as high-resolution mass spectrometry (HR-MS) measurements (Fig. S6–S10†). The nanotube displayed good solubility in toluene, chloroform, and dichloromethane, but was insoluble in methanol. A highly symmetric structure was indicated by 1H NMR spectra, and no significant aggregation or other conformational isomers were observed at NMR concentrations (Fig. S6†). Notably, the inward-directed nitrogen atoms of pyridine rings can be protonated by adding excess trifluoroacetic acid (TFA) (Fig. 1b). The simplicity of the NMR signals suggested that all pyridine rings were protonated, and significant splitting of anthracene rings was observed (Hf–i), which indicated that anthracene rings were situated in a more asymmetric chemical environment. The electron cloud density of pyridine rings decreased after full protonation, resulting in the proton resonances of anthracene (Hi) and pyridine (Ha) groups displayed obvious upfield shifts because these protons are located at the deshielding area of pyridine rings. However, the other proton resonances (Hb, Hd, and Hg–h) were significantly shifted downfield. There are little shifts of protons Hc and He on the benzene rings, due to a more positive electronic environment of the inner cavity. The broad peaks implied that the solubility of the protonated nanotube in toluene-d8 was slightly reduced under the acidic conditions.
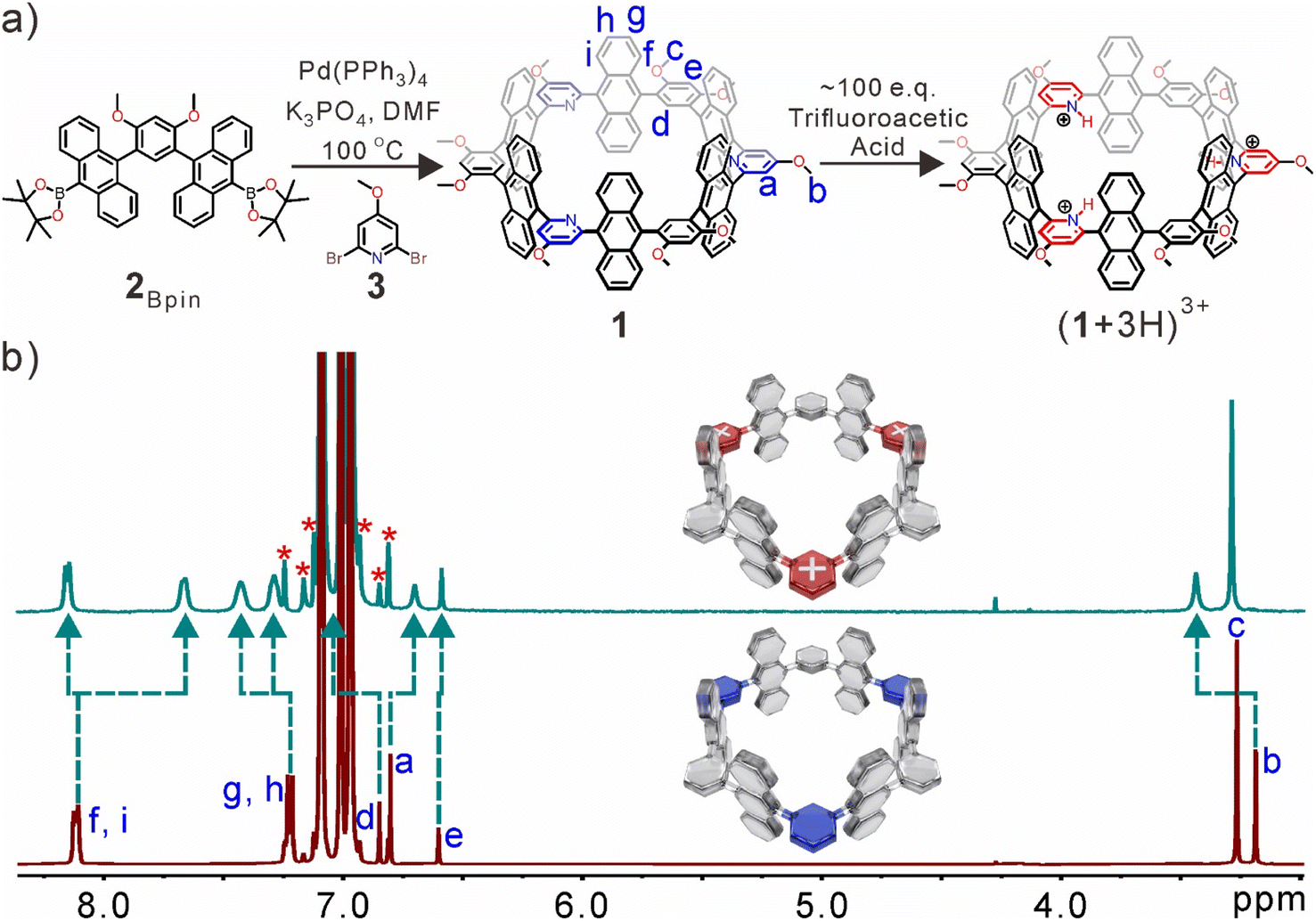 | ||
| Fig. 1 (a) Synthesis of 1 and its protonation. (b) Partial 1H NMR spectra of 1 (bottom) and (1 + 3H)3+ (top) in toluene-d8 at 298 K. The satellite peaks of solvents are indicated by the asterisk symbol. | ||
Fluorescence experiments were conducted to investigate the pH stimuli-responsive properties of 1. The unprotonated 1 exhibited a distinct anthracene monomer emission at 436 nm (Fig. S11a†). Upon gradual addition of TFA, the monomer emission decreased while the anthracene excimer emission at ∼518 nm increased progressively, indicating the formation of the protonated nanotube. As anticipated, the protonated nanotube could be neutralized by the addition of triethylamine (TEA), resulting in a blueshift from excimer to monomer emission after excessive TEA addition (Fig. S11b†). The reversible response of the nanotube to pH was supported by the UV-vis absorption spectra (Fig. S11c†). Furthermore, dilution of the protonated nanotube led to an increase in excimer emission (Fig. S11d†), followed by a gradual blueshift. These findings highlight the pH stimuli-responsive nature and intriguing photophysical properties of the nanotube, which will be utilized for the release of guests. Fortunately, single crystals of the protonated nanotube suitable for X-ray measurement were obtained by adding an appropriate amount of trifluoroacetic acid (TFA) to a solution of 1 in toluene because the protonated nanotube has lower solubility (Fig. 2, S12 and Table S2†). In the X-ray structure of (1 + 3H)3+·(TFA−)3, six anthracene rings, three benzene rings, and three protonated pyridine rings mainly formed the hexagonal nanotube with a diameter above 1.2 nm (Fig. 2a). Among them, six anthracene rings are perpendicular to the equatorial plane of the nanotube, while the benzene rings and protonated pyridine rings are parallel to the equatorial plane of the nanotube, forming a tubular cavity with a depth of ∼0.9 nm (Fig. 2b). Furthermore, adjacent nanotubes were inserted into each other, where the cavities of the nanotubes or the gaps between the nanotubes are filled with TFA− and toluene molecules (Fig. 2c). This insertion was accompanied by a slight curvature of the equatorial plane, leading to the blockage of the pore structure of the nanotubes (Fig. 2d).
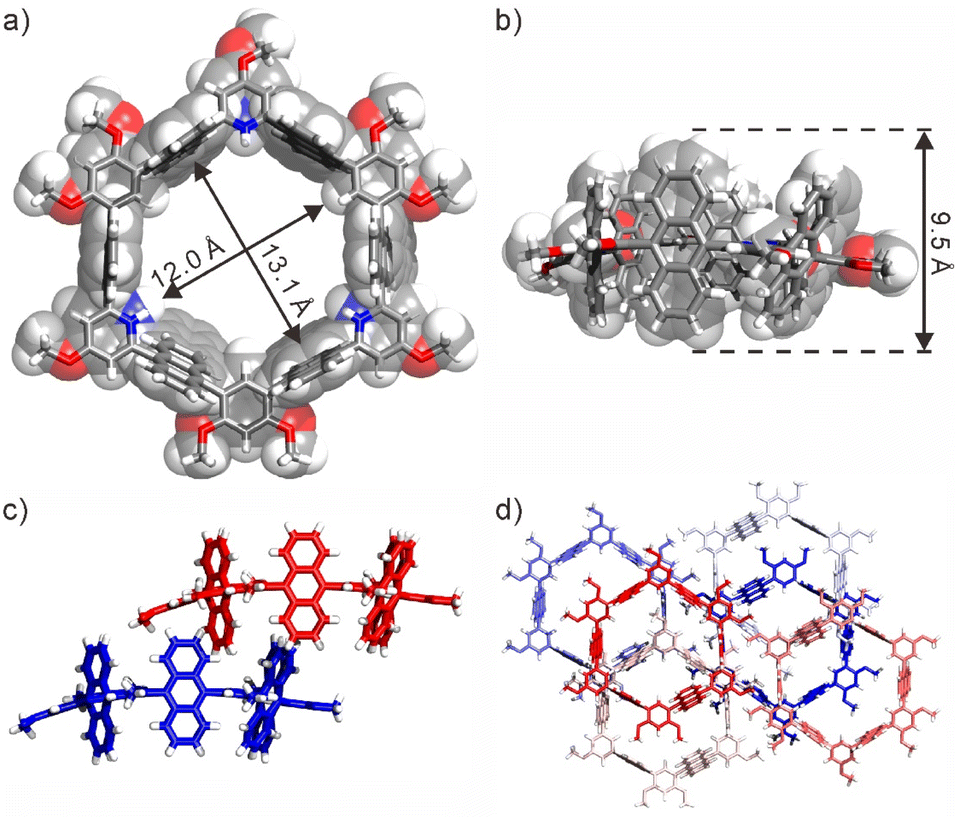 | ||
| Fig. 2 X-ray structures of (1 + 3H)3+(TFA−)3: (a) top view and (b) side view of a single molecule; (c) side view of neighboring nanotubes inserted into each other; (d) top view of the structure of the packing nanotubes. For clarity, all counterions and solvent molecules are omitted. CCDC No.: 2323307. | ||
Based on the X-ray structure, the diameter size of the inner cavity of the nanotube is well matched with the size of the fullerenes. Initially, the 1:1 host–guest complexes formed from 1 and fullerenes (including C60 and C70) were investigated using 1H NMR spectra (Fig. 3a). Fullerenes can be considered as electron acceptors. Consequently, the addition of fullerenes to nanotubes results in a significant chemical shift of the proton Hd, which is directed towards the inner part of the cavity. The π–π interactions between fullerenes and nanotubes greatly diminish the shielding effects of the anthracene rings. As a result, the proton Hd, which is located within the shielding area in the complex, experienced a significant downfield shift. Additionally, the CH⋯π interactions between Hd and fullerenes also play a role in the observed downfield shift. Similarly, the resonances of the methoxy proton (Hc) and the proton (He) on the benzene rings exhibit slight downfield shifts. The protons of the anthracene rings exhibit distinct splitting patterns. Specifically, Hi and Hf split into two separate sets of peaks, while Hg and Hh, which are located at the port of the nanotube, experience only a slight split. These findings suggest that the fullerenes are completely encapsulated within the cavity and primarily bind to the equatorial plane of the nanotube. The electron-deficient nature of the pyridine ring leads to a significant upfield shift of the protons Ha,b in the presence of the fullerene ring current in complexes. The chemical shifts of 1 ⊃ C70 were observed to be more significant compared to those of 1 ⊃ C60, suggesting a stronger interaction between 1 and C70 than with C60. To determine the affinities of nanotubes to fullerenes, 1H NMR titration experiments were used to calculate the association constants (Ka) of the host–guest complexes. The Ka of 1 ⊃ C60 could be measured to be 2.3 × 104 M−1 by using the 1:1 binding mode in toluene-d8 (Fig. S13†). The competitive 1H NMR experiment confirmed a higher binding affinity for 1 ⊃ C70 (Ka = 3.3 × 106 M−1), in which C60 was used as a competitor (Fig. 3b and S14†). Fluorescence titration experiments (Fig. S15 and S16†) were employed to further confirm the association constants (Ka = 3.3 × 104 M−1 and 3.1 × 106 M−1 for 1 ⊃ C60 and 1 ⊃ C70, respectively), which were consistent with the results from 1H NMR titration experiments. Job plots and HR-MS strongly supported the 1:1 binding stoichiometry between 1 and fullerenes (Fig. 3c, and S17–S19†). The results of all host–guest experiments consistently indicated that the nanotube cavity is sufficiently spacious to fully encapsulate the entire fullerene molecules.
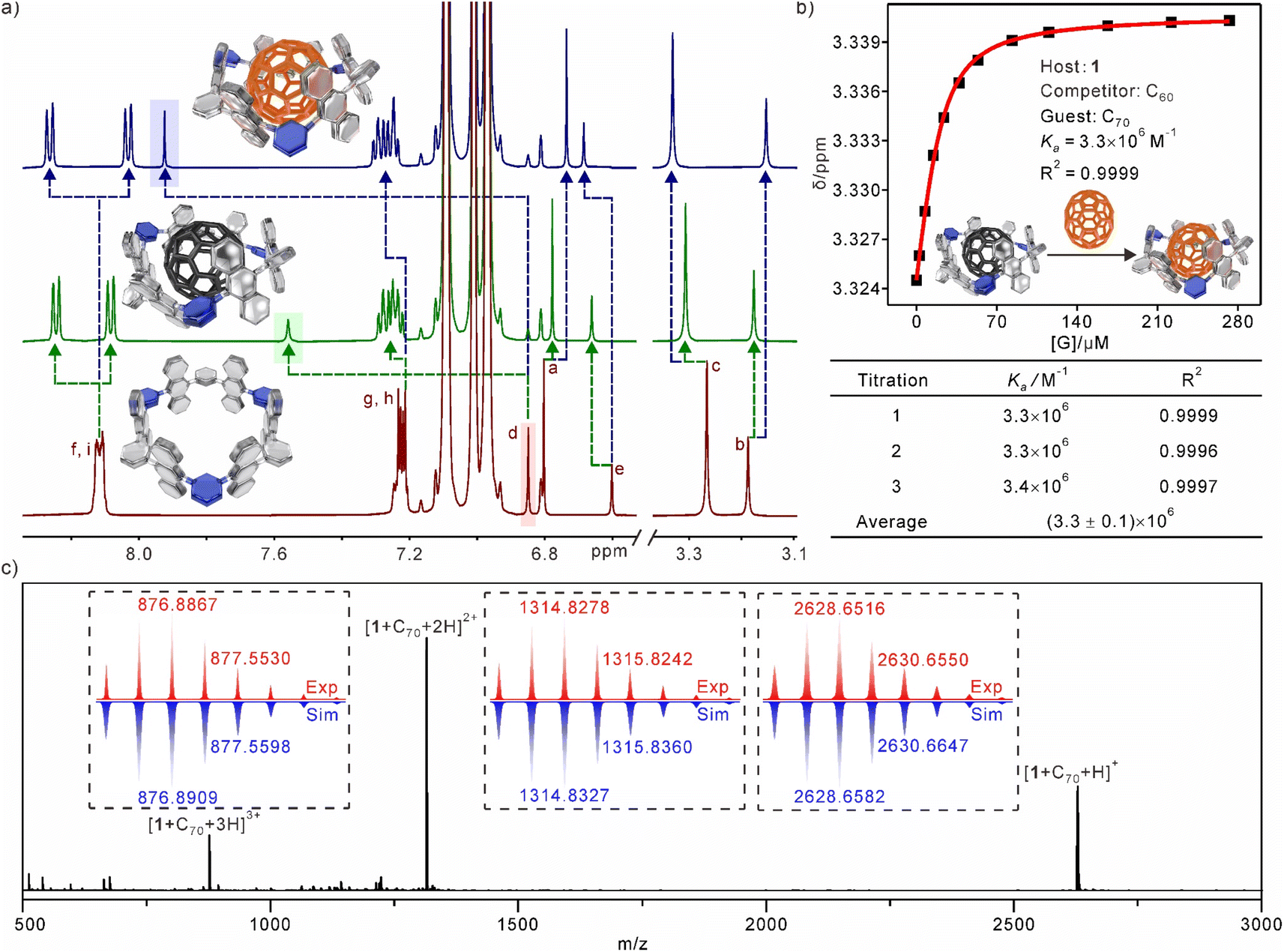 | ||
| Fig. 3 (a) Partial 1H NMR spectra (500 MHz, toluene-d8, 298 K) recorded for (bottom) 1, (medium) 1 ⊃ C60, and (top) 1 ⊃ C70 ([1] = 2.0 mM; [C60 and C70] = 2.0 mM); (b) competitive 1H NMR experiment and association constant of 1 ⊃ C70; (c) experimental and simulated ESI mass spectra of 1 ⊃ C70. | ||
Despite the similarities in structure and properties among fullerenes, the nanotube demonstrated a remarkable binding selectivity (Ks ≈ 140) between C70 and C60. To gain deep insights into the selectivity of 1 for fullerenes, theoretical calculations were performed using Density Functional Theory (DFT) and with the Multiwfn program to process wavefunction files.48 As shown in Fig. 4a, the concave cavity of 1 is electron-rich and complements the electron-deficient convex surface of fullerenes. In both 1 ⊃ C60 and 1 ⊃ C70, the shortest atom-to-atom distances between the aromatic ring and fullerenes were ≈3.1–3.3 Å. The CH⋯π interactions were also observed arising from benzene rings and guests, which were around 2.5 Å and 2.4 Å for 1 ⊃ C60 and 1 ⊃ C70, respectively. In the host–guest binding structures, the anthracene planes undergo rotation along the equatorial plane of the nanotube. This conformational adaptation optimizes the π–π interactions between the anthracene fragments and fullerenes (Fig. 4b, c and S20†).
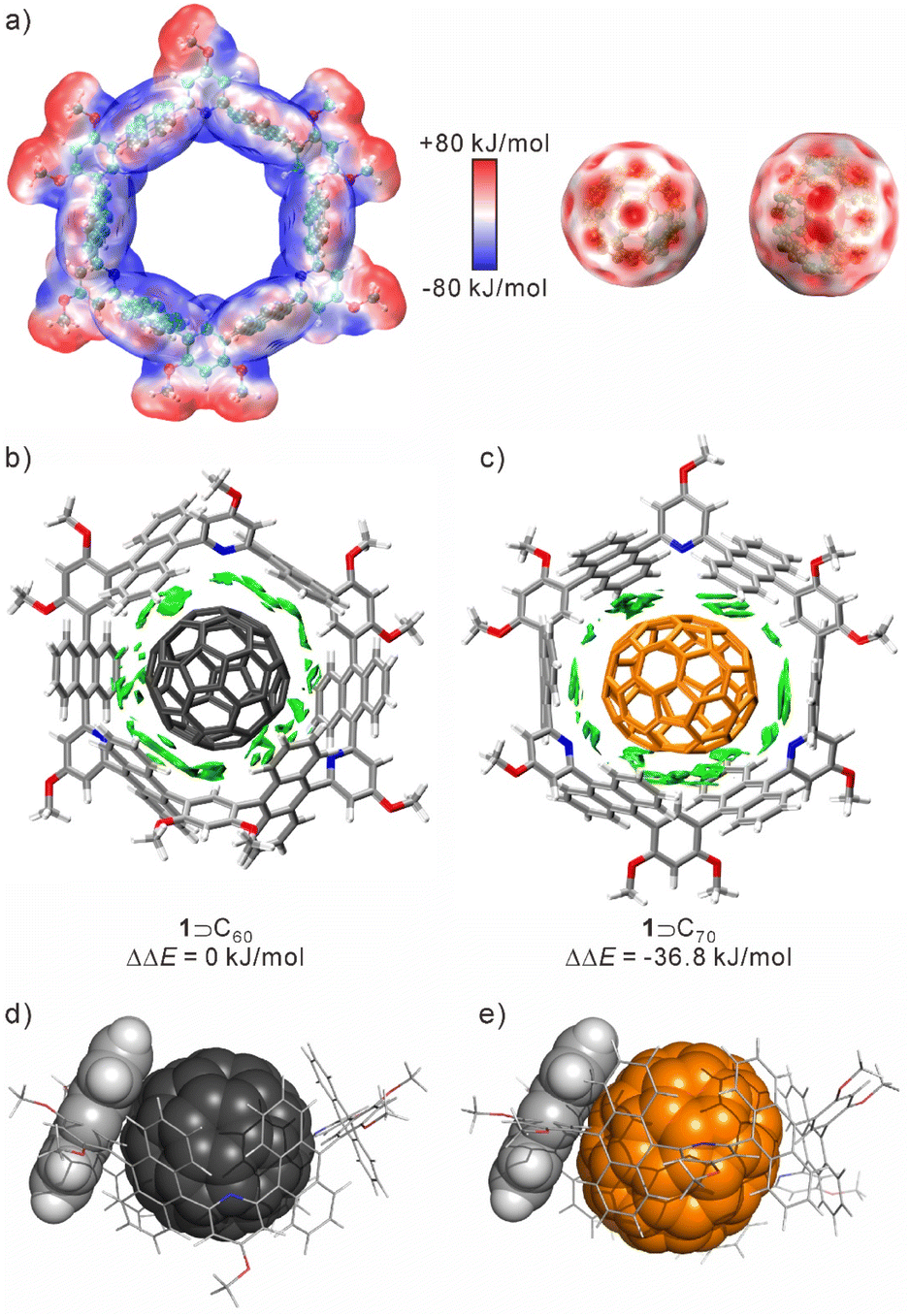 | ||
| Fig. 4 (a) Electrostatic potential surface of 1, C60, and C70; non-covalent interaction analysis (using an independent gradient model based on Hirshfeld partition, IGMH)45 for (b) 1 ⊃ C70 and (c) 1 ⊃ C60, using the ωB97XD functional46 together with the Pople basis set;47 side view of the anthracene fragment and (d) C60 and (e) C70. | ||
Moreover, the lengths of the anthracene planes and C70 are more matched, resulting in an increased contact area between the host and guest. This matching could potentially contribute to the selectivity of 1 towards C70 over C60 (Fig. 4d and e). Meanwhile, the HOMO and LUMO of the complexes were mainly located on the anthracene rings and fullerenes, and the energy gaps were 5.38 eV for 1 ⊃ C60 and 5.32 eV for 1 ⊃ C70, respectively (Fig. S21 and S22†). By comparing the binding energy of 1 ⊃ C60 and 1 ⊃ C70, the binding affinity of 1 ⊃ C70 is stronger than that of 1 ⊃ C60, with a lower free energy of −36.8 kJ mol−1. The selectivity is primarily driven by enthalpic differences, with a slight entropic contribution, as revealed by the analysis of thermodynamic parameters (Table S2†). This suggests that the nanotube may undergo more shape changes to achieve maximum contact with the guest during the binding process of C60. Additionally, due to its longer size and larger volume, C70 is better suited to the volume and shape of the host cavity compared to C60.
The protonation of the inward-directed pyridine rings can alter the electronic environment of the inner cavity, leading to a decrease in electron cloud density within the cavity. As a result, the binding between the nanotube and fullerenes could be dissociated under the low pH conditions. Based on the binding selectivity for fullerenes and pH stimuli-responsive properties of 1, the separation of C70 from C60-enriched fullerene mixtures by using 1 as extractant was explored.17 First, the release of the fullerenes was observed by adding excess TFA through UV-vis and 1H NMR experiments (Fig. S23 and S24†). In the NMR spectra, signals corresponding to complexes disappeared, and a new set of signals appeared, which corresponded to the protonated nanotube. The protonated nanotube was also subjected to theoretical calculations (Fig. S25†). The electrostatic potential surface (EPS) of the (1 + 3H)3+ species exhibited a significantly higher positive charge compared to the unprotonated 1. As a result, the EPS of the nanotube and fullerenes, which was previously in equilibrium, became repulsive. This repulsion, along with the change in the internal volume of the cavity before and after protonation, contributes to the release of fullerenes.
Next, the enrichment of C70 from the C60/C70 (n:n = 9:1) mixture was investigated using UV-vis absorption spectra (Fig. 5). A mixture of C60/C70 with 1 in chloroform was set as the test group, while the mixture of C60/C70 was set without the nanotube as the control experiment. The two mixed groups were sonicated for 1 min, and the solvent was separated by centrifugation. The solution color of the test group was darker compared with the control group, indicating that 1 may increase the solubility of the fullerene by forming 1 ⊃ fullerene complexes in chloroform. After the addition of acid and sonication, a black solid precipitated in the test group, while no precipitation occurred in the control group. The solid was separated, and 1 could be regenerated by neutralization with TEA. The UV-vis absorption revealed an increase in C70 content from ∼10% to ∼50% after just one cycle.17 This result demonstrates the selective binding and enrichment capability of 1 for C70 in a mixture of fullerenes.
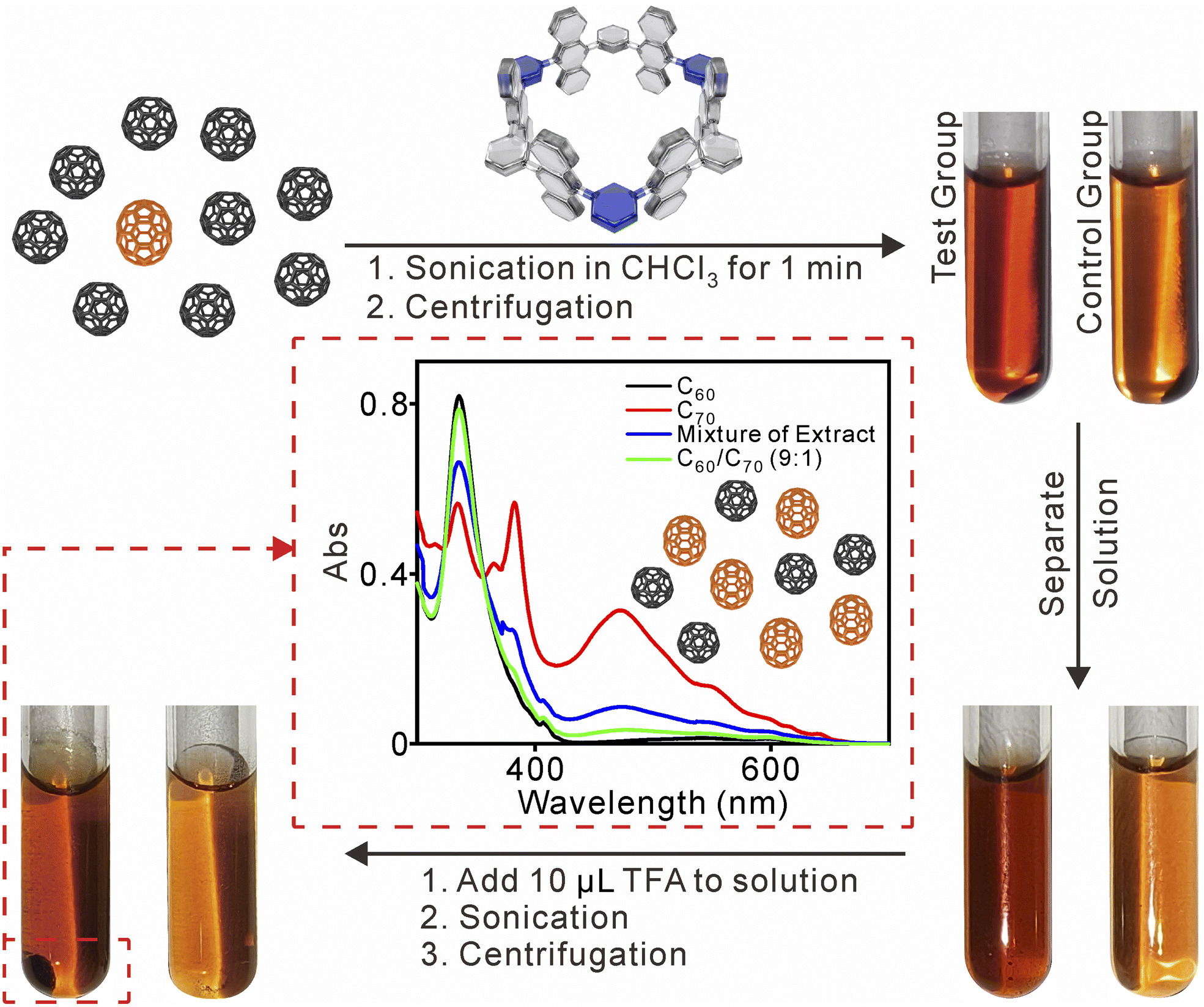 | ||
| Fig. 5 Schematic presentation and UV-vis spectra of the C70 enrichment process. | ||
Conclusions
In conclusion, we have developed the synthesis of an anthracene-based nanotube with a hexagonal cavity of ∼1.2 nm × 0.9 nm. The selective binding and pH stimuli-responsive behaviors of the nanotube with C60 and C70 were investigated by NMR, fluorescence, UV-vis, and HR-MS experiments. The electron-rich cavity formed by anthracene-based side walls exhibited high affinity and selectivity to C70 over C60. Theoretical calculations showed that the selectivity is primarily driven by enhanced π–π interactions between anthracene fragments and fullerenes. Moreover, this study successfully achieved the enrichment of C70 from its low-content mixtures of fullerenes. As a result, these findings provide significant insights into the selective recognition of fullerene molecules and contribute to the expansion of the family of supramolecular hosts with nano-scale recognition cavities.Data availability
All data, including experimental details, computational details, characterization data, 1H NMR spectra of the complexes, 1H NMR titration data, UV-vis spectra, fluorescent titration data, are available in Supplementary Information.Author contributions
W. J. conceived and designed the experiments. L. C., and L.-P. Y. supervised the research. H. N. and S.-M. W. carried out the experiments and performed the DFT calculations. Y.-F. W. solved crystal structures. L. C., L.-P. Y., H. N., S.-M. W., Y.-F. W., Y.-T. Z., L.-S. Z., and X. W. analyzed the data. H. N., S.-M. W. and L. C. wrote, reviewed and edited the manuscript with contributions from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the technical support from SUSTech-CRF and the Center for Computational Science and Engineering of SUSTech. The calculations were also performed by using the CHEM high-performance supercomputer cluster (CHEM-HPC) located at the Department of chemistry, SUSTech. L. C. acknowledges the funding support from the National Natural Science Foundation of China (22122108 and 22371229).Notes and references
- W. Krätschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- F. Diederich and M. Gómez-López, Chem. Soc. Rev., 1999, 28, 263–277 RSC.
- T. Kawase, in Supramolecular Chemistry of Fullerenes and Carbon Nanotubes, ed. N. Martín and J.-F. Nierengarten, Wiley-VCH, Weinheim, Germany, 2012, ch. 3, pp. 55–78 Search PubMed.
- T. Andersson, K. Nilsson, M. Sundahl, G. Westman and O. Wennerström, J. Chem. Soc., Chem. Commun., 1992, 604–606 RSC.
- Y. Takeda, T. Nagamachi, K. Nishikori and S. Minakata, Asian J. Org. Chem., 2012, 2, 69–73 CrossRef.
- A. Ikeda, M. Ishikawa, R. Aono, J. Kikuchi, M. Akiyama and W. Shinoda, J. Org. Chem., 2013, 78, 2534–2541 CrossRef CAS PubMed.
- K. Miki, Z. Dan Zhang, K. Kaneko, Y. Kakiuchi, K. Kojima, A. Enomoto, M. Oe, K. Nogita, Y. Murata, H. Harada and K. Ohe, Mater. Adv., 2022, 3, 312–317 RSC.
- M. Yanase, T. Haino and Y. Fukazawa, Tetrahedron Lett., 1999, 40, 2781–2784 CrossRef CAS.
- T. Haino, Y. Matsumoto and Y. Fukazawa, J. Am. Chem. Soc., 2005, 127, 8936–8937 CrossRef CAS PubMed.
- S.-Q. Liu, D.-X. Wang, Q.-Y. Zheng and M.-X. Wang, Chem. Commun., 2007, 3856–3858 RSC.
- T. Haino, M. Yanase and Y. Fukazawa, Angew. Chem., Int. Ed., 1998, 37, 997–998 CrossRef CAS PubMed.
- T. Haino, C. Fukunaga and Y. Fukazawa, Org. Lett., 2006, 8, 3545–3548 CrossRef CAS PubMed.
- T. Haino, C. Fukunaga and Y. Fukazawa, J. Nanosci. Nanotechnol., 2007, 7, 1386–1388 CrossRef CAS PubMed.
- T. Ogoshi, N. Ueshima, F. Sakakibara, T. A. Yamagishi and T. Haino, Org. Lett., 2014, 16, 2896–2899 CrossRef CAS PubMed.
- G. Colherinhas, E. E. Fileti and T. Malaspina, J. Mol. Model., 2018, 24, 181 CrossRef PubMed.
- P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487–10495 CrossRef CAS.
- C. Zhang, Q. Wang, H. Long and W. Zhang, J. Am. Chem. Soc., 2011, 133, 20995–21001 CrossRef CAS PubMed.
- H. Liu, C. Guo, L. Li, Z. Zhang, Y. Hou, C. Mu, G.-L. Hou, Z. Zhang, H. Wang, X. Li and M. Zhang, J. Am. Chem. Soc., 2024 DOI:10.1021/jacs.4c01873.
- N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed.
- H. Nian, L. Cheng, L. Wang, H. Zhang, P. Wang, Y. Li and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 15354–15358 CrossRef CAS PubMed.
- H. He, Y. J. Lee, Z. Zong, N. Liu, V. M. Lynch, J. Kim, J. Oh, D. Kim, J. L. Sessler and X.-S. Ke, J. Am. Chem. Soc., 2023, 146, 543–551 CrossRef PubMed.
- Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q.-H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS PubMed.
- N. Kishi, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2014, 53, 3604–3607 CrossRef CAS PubMed.
- J. Pfeuffer-Rooschüz, S. Heim, A. Prescimone and K. Tiefenbacher, Angew. Chem., Int. Ed., 2022, 61, e202209885 CrossRef PubMed.
- S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- I. Roy, A. H. G. David, P. J. Das, D. J. Pe and J. F. Stoddart, Chem. Soc. Rev., 2022, 51, 5557–5605 RSC.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572–576 CrossRef CAS PubMed.
- P. Seitz, M. Bhosale, L. Rzesny, A. Uhlmann, J. S. Wössner, R. Wessling and B. Esser, Angew. Chem., Int. Ed., 2023, 62, e202306184 CrossRef CAS PubMed.
- J. C. Barnes, E. J. Dale, A. Prokofjevs, A. Narayanan, I. C. Gibbs-Hall, M. Juríček, C. L. Stern, A. A. Sarjeant, Y. Y. Botros, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 2392–2399 CrossRef CAS PubMed.
- V. G. Jiménez, A. H. G. David, J. M. Cuerva, V. Blanco and A. G. Campaña, Angew. Chem., Int. Ed., 2020, 59, 15124–15128 CrossRef PubMed.
- P. Kumar, V. B. R. Mani Kandan, P. Balakrishnan, P. K. S. Antharjanam and V. Parthasarathy, Angew. Chem., Int. Ed., 2023, 62, e202305005 CrossRef CAS PubMed.
- S.-X. Fa, L.-X. Wang, D.-X. Wang, L. Zhao and M.-X. Wang, J. Org. Chem., 2014, 79, 3559–3571 CrossRef CAS PubMed.
- I. González-Veloso, J. Rodríguez-Otero and E. M. Cabaleiro-Lago, Phys. Chem. Chem. Phys., 2016, 18, 31670–31679 RSC.
- B. Li, B. Wang, X. Huang, L. Dai, L. Cui, J. Li, X. Jia and C. Li, Angew. Chem., Int. Ed., 2019, 58, 3885–3889 CrossRef CAS PubMed.
- Z.-Y. Zhang and C. Li, Acc. Chem. Res., 2022, 55, 916–929 CrossRef CAS PubMed.
- H. Han, R. Fu, R. Wang, C. Tang, M.-M. He, J.-Y. Deng, D.-S. Guo, J. F. Stoddart and K. Cai, J. Am. Chem. Soc., 2022, 144, 20351–20362 CrossRef CAS PubMed.
- Y. Tian, Y. Guo, X. Dong, X. Wan, K.-H. Cheng, R. Chang, S. Li, X. Cao, Y.-T. Chan and A. C. H. Sue, Nat. Synth., 2023, 2, 395–402 CrossRef.
- Y. F. Wang, H. Yao, L. P. Yang, M. Quan and W. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202211853 CrossRef CAS PubMed.
- M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392–2404 CrossRef CAS PubMed.
- K. Hagiwara, Y. Sei, M. Akita and M. Yoshizawa, Chem. Commun., 2012, 48, 7678–7680 RSC.
- K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- K. Matsumoto, S. Kusaba, Y. Tanaka, Y. Sei, M. Akita, K. Aritani, M.-a. Haga and M. Yoshizawa, Angew. Chem., Int. Ed., 2019, 58, 8463–8467 CrossRef CAS PubMed.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem.–Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2323307. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02814g |
| ‡ These authors contributed equally to this work. |
| § Deceased (2022.12). |
| This journal is © The Royal Society of Chemistry 2024 |