
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEthanol synthesis via catalytic CO2 hydrogenation over multi-elemental KFeCuZn/ZrO2 catalyst†
Pengfei
Du
a,
Abdellah
Ait El Fakir
*a,
Shirun
Zhao
a,
Nazmul Hasan M. D.
Dostagir
 a,
HongLi
Pan
a,
Kah Wei
Ting
a,
Shinya
Mine
b,
Yucheng
Qian
a,
Ken-ichi
Shimizu
*a and
Takashi
Toyao
*a
a,
HongLi
Pan
a,
Kah Wei
Ting
a,
Shinya
Mine
b,
Yucheng
Qian
a,
Ken-ichi
Shimizu
*a and
Takashi
Toyao
*a
aInstitute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan. E-mail: toyao@cat.hokudai.ac.jp; Abdellah@cat.hokudai.ac.jp; kshimizu@cat.hokudai.ac.jp
bNational Institute of Advanced Industrial Science and Technology (AIST), Research Institute for Chemical Process Technology, 4-2-1 Nigatake, Miyagino, Sendai 983-8551, Japan
First published on 22nd August 2024
Abstract
Technological enablers that use CO2 as a feedstock to create value-added chemicals, including ethanol, have gained widespread appeal. They offer a potential solution to climate change and promote the development of a circular economy. However, the conversion of CO2 to ethanol poses significant challenges, not only because CO2 is a thermodynamically stable and chemically inert molecule but also because of the complexity of the reaction routes and uncontrollability of C–C coupling. In this study, we developed an efficient catalyst, K–Fe–Cu–Zn/ZrO2 (KFeCuZn/ZrO2), which enhances the EtOH space time yield (STYEtOH) to 5.4 mmol gcat−1 h−1, under optimized conditions (360 °C, 4 MPa, and 12 L gcat−1 h−1). Furthermore, we investigated the roles of each constituent element using in situ/operando spectroscopy such as X-ray absorption spectroscopy (XAS) and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). These results demonstrate that all components are necessary for efficient ethanol synthesis.
Introduction
CO2 hydrogenation into chemicals and fuels is recognized as a pivotal process for achieving a sustainable carbon cycle.1–4 The CO2 hydrogenation into C2+ alcohols has great industrial significance but is scientifically challenging, primarily because of the intricate reaction mechanisms involved and necessity of forming carbon–carbon (C–C) bonds, resulting in the low selectivity of C2+ alcohols.5–8 Among the various C2+ alcohol products,6,7 ethanol (EtOH) has received widespread attention in recent years as an alternative fuel,9–11 promising hydrogen carrier,9,12 and versatile building block chemical for producing high-value products.9,13–15 EtOH generation not only involves competition from several parallel reactions, including the formation of C2+ hydrocarbons and methanol, but also requires precise control of the dissociative and non-dissociative activation of C–O bonds to obtain surface alkyl and CO (H) species, respectively.5,6,12,16 Therefore, achieving high selectivity and yield toward EtOH is challenging but urgently desired for future CO2 utilization.5,9,13,14Various catalytic systems have been reported for the hydrogenation of CO2 into EtOH. Among these catalytic materials, noble-metal-based catalysts (Pd17–20 and Rh21–25) are usually applied to promote C–O activation and the subsequent C–C coupling for EtOH synthesis.7,13 The high price of noble metal catalysts limits their further application; thus, researchers have shifted their attention to 3d transition metal catalysts, such as Cu-,19,26–29 Fe-,30–34 and Co4,35–38-based catalysts, coupled with promoter elements such as alkali metal oxides. Among the 3d transition metal catalysts, Fe–Cu catalysts stand out as a cost-effective option with exceptional catalytic activity to produce C2+ alcohols from CO2.30,32,39 However, they also generate a substantial amount of hydrocarbons as byproducts.32,39,40 Although some recent work has suggested that the introduction of Zn into Cs-promoted Fe–Cu based catalysts would be more efficient in producing EtOH,30 the catalytic performance does not meet industrial requirements. In addition, little is known about the role of each element. Further improvements in the catalyst systems are essential for developing industrially valuable reaction processes.
Herein, we present a highly efficient catalytic system, namely K-promoted FeCuZn/ZrO2 (KFeCuZn/ZrO2), which significantly enhances the rate of EtOH production through CO2 hydrogenation. Under optimized conditions (360 °C, 4 MPa, and 12 L gcat−1 h−1), our catalyst (KFeCuZn/ZrO2) exhibits high activity (STYEtOH: 5.4 mmol gcat−1 h−1) in the EtOH synthesis, compared to different supports. We thoroughly investigated the role of each constituent element using in situ/operando spectroscopic techniques and various characterizations.
Experimental details
Chemicals and catalyst preparation
Chemicals and materials were purchased from commercial suppliers and used without further purification. ZrO2 (RC-100), equivalent to JRC-ZRO-3, was supplied by Daiichi Kigenso Kagaku Kogyo. TiO2 (P25) was obtained from Evonik. γ-Al2O3 (Puralox) was obtained from Sasol. SiO2 (CariACT Q-10) was purchased from Fuji Silysia Chemical Company, Ltd.KFeCuZn/support catalysts were prepared using a simple impregnation method. In this process, the support material was impregnated with an aqueous solution of KNO3 (>98%; Wako Pure Chemical Industries), Fe (NO3)3·9H2O (>98%; Wako Pure Chemical Industries), Zn (NO3)2·6H2O (>98%; Kanto Chemical), and Cu (NO3)2·3H2O (>99%; Aldrich). For example, KFeCuZn/ZrO2 (3, 15, 10, and 5% wt of K, Fe, Zn, and Cu, respectively) was prepared by adding a certain amount of K, Fe, Cu, and Zn precursors and ZrO2 to a glass vessel (100 mL) containing 20 mL of deionized water. The mixture was stirred at 200 rpm for 60 min at room temperature. Subsequently, water was removed from the mixture by evaporation in vacuo, followed by drying at 120 °C under ambient pressure, for ∼12 h. The resulting material was calcined for 3 h at 500 °C in air.
Characterization
X-ray diffraction (XRD) analysis was conducted on a Miniflex (Rigaku) diffractometer using Cu Kα radiation. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) were performed using an FEI Titan G2 microscope. The samples were prepared by dropping an ethanolic solution containing a catalyst onto carbon-supported Mo grids. N2 adsorption measurements were conducted using an AUTOSORB 6AG (Yuasa Ionics) instrument at 77 K. Before the measurements, the calcined pieces of samples were ground and outgassed under vacuum at 200 °C for 3 h. The specific surface areas and pore size distribution of our samples were acquired by the Brunauer–Emmett–Teller (BET) N2 adsorption measurements.H2 temperature-programmed reduction (H2-TPR) was performed on a BELCat II instrument with a TCD detector. Briefly, ∼100 mg of the catalyst was placed into a quartz tube and purged with Ar at 200 °C (2 h) to remove physically adsorbed water and surface carbonates. Then, the sample was cooled to 50 °C, followed by subsequent heating to 800 °C in 10 vol% H2 balanced with Ar, at a ramping rate of 10 °C min−1.
CO2 temperature programmed surface reaction (CO2-TPSR) was conducted on a BELCat II instrument with a mass spectrometer (BELMass; MicrotracBEL Corp). ∼100 mg of catalyst was placed in a quartz tube and reduced by 10% H2/Ar at 400 °C for 0.5 hours. The carrier gas was then changed to He and the sample cooled to 50 °C. Subsequently, 10% CO2/He was introduced to allow CO2 adsorption on the catalyst surface for 1 hour. The sample was then heated to 700 °C in a mixture of 10% H2/Ar at a ramp rate of 10 °C min−1. Ion fragmentation was monitored by BELMass at m/z = 40 for Ar, m/z = 15 for CH4, m/z = 28 for CO and m/z = 44 for CO2.
Pulse hydrogenation measurements were performed in a fixed-bed reactor. In detail, 100 mg of the used KFeCuZn/ZrO2 catalyst was reactivated under the reaction gas (40 mL min−1) at 320 °C for 1 h. After that, a pulse of CH3CHO (500 μL for every injection) was introduced every 10 min for four cycles under a carrier gas (H2/CO2/Ar in a ratio of 74.4/24.8/0.8 and the total flow rate is 40 mL min−1). The signals of CH3CHO and CH3CH2OH were monitored using an online mass spectrometer.
In situ/operando diffuse reflectance infrared Fourier transform (DRIFT) spectra were recorded on a JASCO FT/IR-4600 instrument equipped with a mercury–cadmium–telluride (MCT) detector. The sample was pressed into a DRIFT cell (DR-650 Ci) using a CaF2 window. The spectra were measured by accumulating 20 scans at a resolution of 8 cm−1, 0.5 MPa and temperature range of 200–320 °C. The reference spectrum in He flow (20 scans), taken at the measurement temperature, was subtracted from each spectrum. A high-sampling-rate GC-TCD (490 Micro GC; Agilent Technologies Inc.) was installed at the outlet for the analysis of methanol and ethanol.
Fe K-edge, Cu K-edge, and Zn K-edge X-ray absorption spectroscopy (XAS) were performed in transmission mode at BL01B1 of SPring-8 at the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No.: 2023A1931). A Si (111) double crystal monochromator was used. Boron nitride (BN) was used to make a pellet sample when the required amount was less than 20 mg. The spectra of reference compounds were recorded at room temperature, in air. The obtained XAS spectra were analyzed using the Athena and Artemis software ver. 0.9.26, included in the Demeter package.41
For in situ XAS measurements, samples in pellet forms (φ: 7 mm) were introduced into a cell equipped with Kapton film windows and gas lines connected to the micro-gas chromatograph. Pretreatment of the samples involved heating under a flow of H2 (300 mL min−1) at 300 °C for 30 min. Subsequently, 25% CO2/He (400 mL min−1), 75% H2/He (400 mL min−1), and CO2 (100 mL min−1) + H2 (300 mL min−1) were introduced into the cell with He purge intervals between gas introductions.
Catalytic reaction
CO2 hydrogenation reactions were conducted in a fixed-bed continuous-flow reactor operating at a total pressure of 4 MPa (40 bar). We used 1% Ar/99% H2 (purity; H2: 99.99999%, Ar: 99.9999%) and CO2 (purity; 99.995%) gas cylinders to supply the reaction gases. The reactor was supplied with a gas mixture of H2/CO2/Ar in a ratio of 74.4/24.8/0.8, with Ar serving as an internal standard gas. Prior to the catalytic measurements, 200 mg of the catalyst was placed between quartz wool inside a 1/4 inch fixed-bed reactor (inside diameter: 3.79 mm). The catalyst was pretreated under ambient pressure at a H2/Ar (99%) flow rate of 20 mL min−1 and temperature of 400 °C for 0.5 h. Following the pretreatment, a H2/CO2/Ar (74.4/24.8/0.8) flow at a total flow rate of 40 mL min−1 was passed through the catalyst bed at 4 MPa. The weight hourly space velocity (WHSV) was 12 L gcat−1 h−1. We conducted an aging treatment for 10 h at 400 °C under the reaction gas atmosphere as an accelerated aging test before recording the results of the catalytic reaction (Fig. S1†). The products were analyzed using an online gas chromatograph (Shimadzu GC-2014) equipped with TCD (Shincarbon-ST column) and FID (Porapak Q column) detectors, within a reaction temperature range of 240–400 °C.The CO2 conversion (XCO2) was calculated as follows.
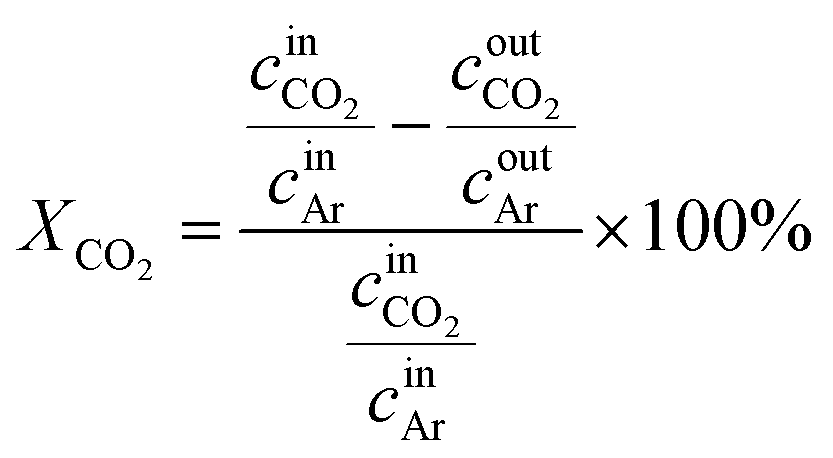
The selectivity (Si) for individual products was calculated by the following equation:

The EtOH decomposition experiments were conducted in a batch reactor using the spent catalyst. The spent catalyst (50 mg) was placed in a quartz tube and reduced using H2 at 300 °C for 30 min. Subsequently, ethanol (0.3 mL) and octane (0.7 mL) were introduced into the quartz tube and fixed in a batch reactor. The above mixture was magnetically stirred at 260 °C under N2 (0.5 MPa) for 3 h. Gas-phase products were collected and analyzed using a GC-FID (Shimadzu GC-2014; Porapak Q column) with a methanizer (Shimadzu MTN-1), whereas the liquid-phase products were analyzed using a gas chromatograph with a GC-FID (Shimadzu GC-14B; Ultra ALLOY capillary column UA+-1; Frontier Laboratories, Ltd).
CO hydrogenation and additional CO2 hydrogenation over the KFeCuZn/ZrO2 catalyst reactions were conducted in the same fixed-bed continuous-flow reactor with the same WHSV (18.45 L gcat−1 h−1) and operated at the same pressure of 3 MPa for comparison. The reactor was supplied with a gas mixture of H2/CO (CO2)/Ar in a ratio of 74.4/24.8/0.8 (CO or CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2 = 1:3), with Ar serving as an internal standard gas. We conducted an aging treatment for 10 h at 400 °C under the reaction gas atmosphere as an accelerated aging test before recording the results of the catalytic reaction (as described in Fig. S1†). The products were analyzed using an online gas chromatograph (Shimadzu GC-2014) equipped with TCD (Shincarbon-ST column) and FID (Porapak Q column) detectors, within a reaction temperature range of 240–400 °C.
:H2 = 1:3), with Ar serving as an internal standard gas. We conducted an aging treatment for 10 h at 400 °C under the reaction gas atmosphere as an accelerated aging test before recording the results of the catalytic reaction (as described in Fig. S1†). The products were analyzed using an online gas chromatograph (Shimadzu GC-2014) equipped with TCD (Shincarbon-ST column) and FID (Porapak Q column) detectors, within a reaction temperature range of 240–400 °C.
Results and discussion
Catalytic CO2 hydrogenation to EtOH
CO2 hydrogenation reactions were conducted under specific operating conditions of P = 4 MPa, T = 280–400 °C, and WHSV = 12 L gcat−1 h−1. As shown in Fig. 1a, the KFeCuZn/ZrO2 catalyst exhibits the highest selectivity towards C2+ alcohol products, with ∼40 and 16.5% selectivity towards C2+ alcohols and EtOH, respectively, while achieving a CO2 conversion of 52.4%. In particular, the KFeCuZn/ZrO2 catalyst also displays an excellent space time yield (STY) of EtOH, with a STYEtOH of 5.4 mmol gcat−1 h−1. In comparison, the CO2 conversion fell within a comparable range of 49.6–56.7% for the other studied catalysts (KFe/ZrO2, KFeCu/ZrO2 and KFeZn/ZrO2). Additionally, for the Cu-added catalyst (KFeCu/ZrO2), the EtOH selectivity was approximately 14%, accompanied by a high ethanol space-time yield (STYEtOH) reaching up to 4.0 mmol gcat−1 h−1, which surpasses those of catalysts without Cu (KFe/ZrO2 and KFeZn/ZrO2). This suggests that Cu plays a significant role in facilitating the activation and coupling of CO2 molecules for ethanol production.40 Although the addition of Zn (KFeZn/ZrO2) did not directly result in an obvious enhancement in EtOH production, the catalyst incorporating both Cu and Zn (KFeCuZn/ZrO2) demonstrated the highest selectivity and STYEtOH. Literature data on EtOH synthesis from CO2/H2 (Table S1†) are compared with our best catalytic system. Our catalyst displayed the best STYEtOH for EtOH synthesis even after the accelerated aging test at 400 °C for 10 h. | ||
| Fig. 1 Catalytic performance. (a) CO2 conversion and product selectivity at 360 °C and STYEtOH over KFe/ZrO2 based catalysts; (b) CO2 conversion, product selectivity, and STYEtOH over different supporting; and (c) stability test for KFeCuZn/ZrO2 at 360 °C. Pretreatment condition: 400 °C, 0.1 MPa, H2 (99%), and 0.5 h. Reaction condition: 12 L gcat−1 h−1, 4.0 MPa, and H2/CO2/Ar (74.4/24.8/0.8%). Ar was used as an internal standard gas. The data was collected after 3 h, when the temperature and reaction were stable. Accelerated aging treatment was performed before reaction at 400 °C for 10 h under the reaction environment. Others include acetaldehyde and ethyl formate. In (a) and (b), the gray dot is CO2 conversion; red dot is EtOH STY. | ||
To emphasize the pivotal roles of potassium and iron, we intentionally omitted these elements from the compared catalysts, as illustrated in Fig. 1a. Notably, the FeCuZn/ZrO2 catalyst, while maintaining a CO2 conversion of 47% comparable to that of the KFeZnCu/ZrO2 catalyst, exhibited a significant increase in CH4 selectivity, reaching 37%. Simultaneously, the selectivity for total alcohols and EtOH decreased sharply to 9 and 3.4%, respectively. Furthermore, the STYEtOH for the FeCuZn/ZrO2 catalyst was measured at 0.6 mmol gcat−1 h−1. This significantly reduced alcohol activity in the absence of K underscores the essential role of K in C2+ alcohol synthesis and the inhibition of over-hydrogenation.34,39,40 The EtOH production significantly decreased over the KCuZn/ZrO2 catalyst under the same conditions because of the absence of Fe, which resulted in the loss of the ability of the catalyst for C–C coupling.42,43 Furthermore, Fig. S2† displayed a volcano-shaped curve for STYEtOH with increasing Fe loading, and the most suitable loading is 15%wt. Fig. 1b illustrates CO2 hydrogenation experiments using different supports, such as Al2O3, TiO2, and SiO2. All three catalysts showed an obvious decrease in the EtOH selectivity to less than 2%. In terms of STYEtOH, the KFeZnCu/ZrO2 catalyst significantly outperformed the others, indicating that ZrO2 is an excellent carrier for EtOH synthesis.
In addition, the effects of the reaction conditions, including reaction temperature, pressure, and WHSV, on EtOH synthesis were investigated in detail. On varying the reaction temperature within the range of 280–400 °C (Fig. S3†), the STY of EtOH and C2+ alcohol distribution exhibited a characteristic volcano-shaped curve. The appropriate temperature for acquiring EtOH is 360 °C. Additionally, the reaction pressure was varied from 3 to 5 MPa (Fig. S4a†), and the highest STYEtOH (5.4 mmol gcat−1 h−1) were achieved at 4 MPa, making it a more favorable pressure regime for C2+ alcohol synthesis. Furthermore, we explored the influence of the WHSV (Fig. S4b†) by varying it between 9–15 L gcat−1 h−1 and observed the most suitable WHSV was 12 L gcat−1 h−1. Moreover, the stability test was performed for the KFe-based catalysts (Fig. 1c and S5†). Among them, KFeCu/ZrO2, KFeZn/ZrO2 and KFeCuZn/ZrO2 showed robust stability, maintaining their performance for at least 60 h at 360 °C. Only KFe/ZrO2 displayed slight decrease in STYEtOH after 35 h. Additionally, a simplified version of Fig. 1 was provided as Fig. S6,† by replacing the selectivity of C=2–C=4, C2−–C4−, and C5+ to C2+ hydrocarbons.
Structural characterization
Multiple characterization methods were used to reveal the physical characterizations and structural evolution. The BET specific surface areas and pore volumes (Table S2†), obtained from the N2 adsorption measurements at 77 K, decreased upon increase in loading amount of the supported species. Pore size distributions were not significantly different among samples measured (Fig. S7†). XRD patterns (Fig. 2) show that the ZrO2 support is composed of monoclinic and minor tetragonal phases based on the diffraction angles and intensities. Phase transformations of the loading elements were also observed at different reaction stages. For example, for the KFeCuZn/ZrO2 catalyst, metallic Fe was obtained after H2 reduction pretreatment (Fig. 2); subsequently, metallic Fe was oxidized to Fe3O4 and carburized to Fe5C2 after the reaction.42,44 Iron carbide (Fe5C2) is an essential phase for C–C coupling in the Fischer–Tropsch (F–T) process over Fe-based catalysts.42 Cu-derived peaks were not observed by XRD after calcination and reduction, probably due to the highly dispersed nature of Cu. Metallic copper was observed after the reaction owing to the sintering of Cu under a reductive atmosphere for a longer time. For the Zn species, only ZnO diffraction peaks were observed, indicating that no phase changes occurred during the entire reaction process. In comparison with the other control catalysts (Fig. S8†), the same evolution processes (reduction and carburization of Fe, reduction and aggregation of Cu, and stability of ZnO on Zn) were observed for the catalysts containing the relative elements. | ||
| Fig. 2 XRD patterns of KFeCuZn/ZrO2 before and after H2 reduction at 400 °C as a pretreatment and after reaction. “Spent catalyst” refers to a sample that has undergone a 20 hours accelerated aging treatment at 400 °C under the reaction conditions. | ||
To further clarify the phase transformations of Fe, Cu, and Zn species, X-ray adsorption spectroscopy (XAS) was performed on KFeCuZn/ZrO2. The ex situ X-ray absorption near edge structure (XANES) spectra of the Fe K-edge (Fig. 3a) show that after H2 reduction as the pretreatment, the absorption edge shifted to almost the same energy as that of the Fe foil, suggesting that the Fe species in KFeZnCu/ZrO2 were completely reduced. Similarly, after the reaction, the absorption edge moved to a higher energy compared to that of the Fe foil, suggesting the partial oxidation of Fe, in line with the XRD results. Extended X-ray absorption fine structure (EXAFS) results further helped to understand the local environmental changes (Fig. 3b and Table S3†). After the H2 reduction pretreatment, the Fe–Fe bonds at 2.45 and 2.83 Å appeared, whereas the Fe–O bond at 1.99 Å disappeared.45,46 After the reaction, Fe–C and Fe–O appeared, and the Fe–Fe bond weakened, indicating the generation of FeOx and FeCx, consistent with the XRD results. In the case of Cu (Fig. 3c), as exhibited in ex situ XANES spectra, CuO was reduced to metallic Cu after reduction, which was maintained until the end of the reaction, as supported by the curve fitting of the EXAFS spectra (Fig. 3d and Table S4†). Additionally, as indicated by the Zn K-edge XANES (Fig. 3e), after reduction, a weak shift to a lower energy was observed, implying the presence of a minor amount of metallic Zn, as confirmed by the EXAFS curve fitting results (Fig. 3f and Table S5†). In contrast, after the reaction, the energy increased to that of the closed ZnO reference and only one component of ZnO was captured (Table S5†), indicating that the zinc species prefers oxygen over hydrogen.47
 | ||
| Fig. 3 Ex situ XAS spectra of KFeCuZn/ZrO2 before and after H2 reduction at 400 °C as a pretreatment and after reaction (spent). (a) Normalized Fe K-edge XANES spectra; (b) FT-EXAFS spectra for Fe; (c) normalized Cu K-edge XANES spectra; (d) FT-EXAFS spectra for Cu; (e) normalized Zn K-edge XANES spectra; and (f) FT-EXAFS spectra for Zn. “Spent catalyst” refers to a sample that has undergone a 20 hours accelerated aging treatment at 400 °C under the reaction condition. | ||
H2-TPR experiments were carried out to clarify the reduction ability of different additive elements and H2 activation capacity over different catalysts. As depicted in Fig. S9,† considering mono-component supported catalysts such as Fe/ZrO2, Cu/ZrO2, and Zn/ZrO2, Cu displayed the lowest reduction temperature and was the most readily reduced element, followed by Fe. In agreement with the XRD and XAS results, no reduction was observed in the ZnO content. In contrast, the introduction of K could promoted the generation of reduction peak at 600 °C, unlike with pure ZrO2.40 Contrastingly, after loading the promoter, the reduction temperatures of Fe and Cu increased for KFe/ZrO2 and KCu/ZrO2, inhibiting the reduction of the active elements.39,40 Furthermore, in comparison with KFe/ZrO2 and KFeCu/ZrO2, the temperature for the reduction of Fe2O3 to Fe3O4 on the KFeCu/ZrO2 catalyst decreased to 390 °C.48 In contrast, the reduction temperature of FeOx remained virtually unchanged over the KFeZn/ZrO2 catalyst, suggesting that the introduction of Zn was ineffective in promoting the reduction of the Fe species. However, after loading Zn into KFeCu/ZrO2, the reduction temperature increased slightly, indicating that the introduction of Zn led to a decrease in the reduction ability.47,49
HAADF-STEM and EDS were performed for fresh and spent KFeCuZn/ZrO2, as shown in Fig. 4 and 5, respectively. Because of the weak Z-contrast, identifying the crystal phase and interplanar spacing of the Fe, Cu, and Zn species was challenging. Therefore, we only observed the elemental distributions. The HAADF-STEM and EDS elemental mapping images revealed that K, Fe, Cu, and Zn over the KFeCuZn/ZrO2 catalyst were homogeneously distributed on ZrO2 for fresh KFeCuZn/ZrO2 (Fig. 4). After the reaction, K, Fe, and Zn remained highly dispersed in ZrO2 (Fig. S10† and 5). The particle size of Cu increased a little, indicating the aggregation of Cu over KFeCuZn/ZrO2. In comparison, larger Cu nanoparticles were observed over the spent KFeCu/ZrO2, elucidating that the introduction of Zn limited Cu sintering (Fig. 5 and S11–S13†). Inhibition of sintering of Cu nanoparticles was also reported for Cs–CuZnFe30 and CuZnO–Al2O3 (ref. 50) systems.
 | ||
| Fig. 4 HAADF-STEM image and corresponding EDS mapping images of the fresh KFeCuZn/ZrO2 that has undergone calcination at 500 °C under air. | ||
 | ||
| Fig. 5 HAADF-STEM image and corresponding EDS mapping images of the spent KFeCuZn/ZrO2 that has undergone a 20 hours accelerated aging treatment at 400 °C under the reaction condition. | ||
Mechanistic studies
In situ XAS was employed to clarify the chemical states under He, CO2, and CO2 + H2 atmospheres at 400 °C and ambient pressure after H2 pretreatment. The Fe K-edge XANES spectra (Fig. 6a) of KFeCuZn/ZrO2 clearly showed that the absorption edge shifted to higher energies after the introduction of CO2 (Fig. 6b), indicating that the Fe species were oxidized by CO2. These results clearly proved that CO2 functioned as an oxidizing agent, facilitating the oxidation of Fe species.51 | ||
| Fig. 6 In situ XAFS spectra of KFeCuZn/ZrO2. (a) Normalized Fe K-edge XANES spectra; (b) X-ray energy at normalized absorption (μ = 0.4) under different atmospheres; (c) Cu K-edge XANES spectra; and (d) Zn K-edge XANES spectra. Conditions: 400 °C and 1 bar. | ||
For the Cu K-edge and Zn K-edge, although there were minor shifts upon the introduction of CO2, the edge positions remained almost unchanged upon the introduction of CO2 + H2. These results suggested that the redox reactions of Cu and Zn were not involved in the CO2 hydrogenation reaction.
The in situ/operando DRIFTS was performed to elucidate the mechanism underlying the hydrogenation of CO2 to EtOH and role of each element. These in situ/operando DRIFTS experiments were conducted under specific conditions, including a pressure of 0.5 MPa, temperature of 320 °C (with an exception for KFeCuZn/ZrO2, where the temperature range was 200–320 °C), and H2:CO2 ratio of 3:1. The critical assignments of the surface species and adsorbed methanol and ethanol species are provided in Table S6 and Fig. S14.† Observations at 200 °C for KFeCuZn/ZrO2 indicate the presence of adsorbed CO2 at 1269 and 1514 cm−1,30,52 which can be attributed to the carbonate species (bicarbonate species at 1620 cm−1)53 and surface formate located at 2775/1593/1393 cm−1.22,40,54 No CH3O* or CH3CH2O* were detected (Fig. S15a† and 7a). After the reaction temperature increased to 240 °C (Fig. S15b†), a weak peak, which can be attributed to an important intermediate of CH3CHO* in EtOH synthesis, was observed at 1410 cm−1.40 When the temperature reached 280 °C (Fig. S16a†), with an increase in the exposure time, the intensity of the CH3CHO* assignment gradually increased at 1410 cm−1. After ∼25 min, it was consumed, and subsequently, peaks assignable to ethoxy (2852/2922/2956 cm−1) were observed.27,30,39 In addition, EtOH in gas phase was tracked and quantified using online gas chromatography (Fig. S16b†). A CH3CHO pulse experiment was also performed in a CO2 + H2 environment using the spent KFeZnCu/ZrO2 catalyst (Fig. S17†). The introduction of CH3CHO into the reaction environment resulted in a rapid increase in the intensity of the CH3CH2OH signal. This clearly indicates that the hydrogenation of CH3CHO to CH3CH2OH is highly facile under the given conditions. Furthermore, the DRIFTS spectra also showed a minor peak at 2820 cm−1, which can be assigned to CH3O*,30,54 along with a decrease in the intensity of the peak at 2770 cm−1, assignable to formate. Upon reaching a temperature of 320 °C, the intensity of CH3CH2O* became more pronounced (Fig. 7b). In addition, another assignment appeared, with CH3CH2O* characterized by δ(CH2) vibrations at 1462 cm−1.40 The methane appeared at 3011 cm−1,39,40 which was not seen at lower temperatures. Notably, some papers reported that the peaks for CH3CH2O* and CH3O* overlapped in the 2940–2980 cm−1 region.27,30 According to the quantification of the micro-GC results the formation rate of EtOH was much higher than that of methanol (Fig. 7c). Thus, we assigned the peaks at 2852, 2922, and 2956 cm−1 to CH3CH2O*, which is regarded as an important intermediate for EtOH formation.27,30
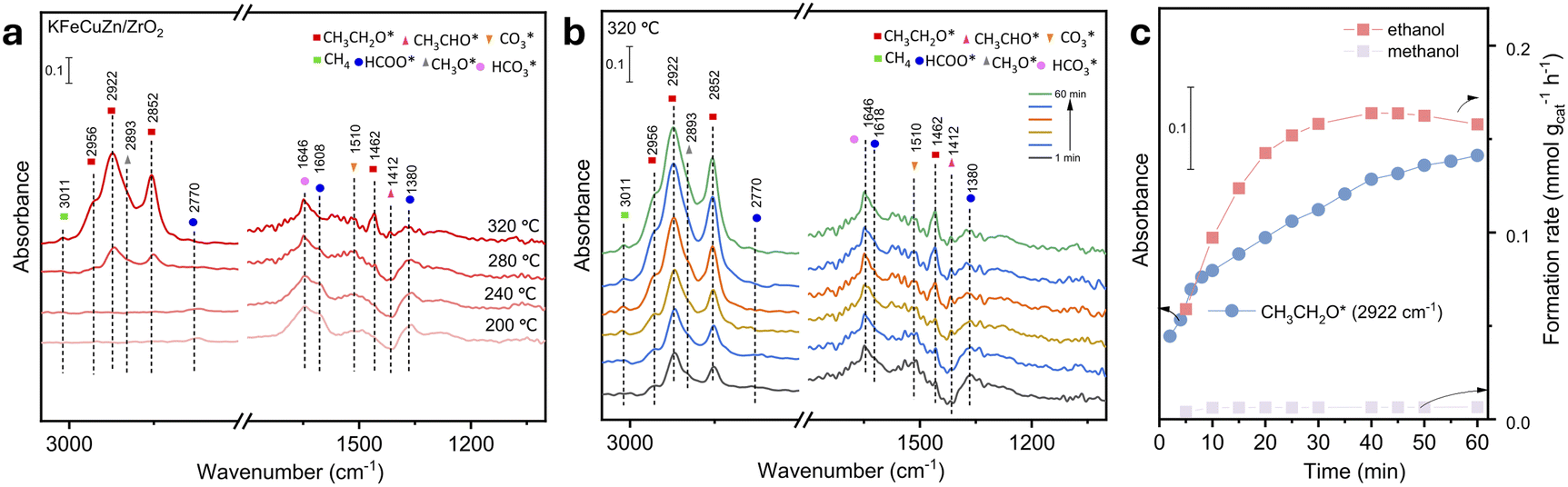 | ||
| Fig. 7 In situ/operando DRIFTS spectra. (a) DRIFTS spectra of the CO2 + H2 reaction over KFeCuZn/ZrO2 after 60 min at 0.5 MPa and different temperatures; (b) time resolution spectra of the CO2 + H2 reaction over KFeCuZn/ZrO2 at 0.5 MPa and 320 °C; (c) dynamic IR peak intensities of CH3CH2O* (2922 cm−1) and formation rate of ethanol and methanol, quantified by micro-GC at 320 °C. | ||
DRIFTS experiments were also conducted on various catalysts to identify the roles of the different elements. KFeCuZn/ZrO2 catalyst displayed the highest CH3CH2O* intensity after 60 min (Fig. 8a). In comparison, the FeCuZn/ZrO2 catalyst exhibited strong CH4 peaks at 1302 and 3011 cm−1 during DRIFTS analysis (Fig. 8a and S18†),55 which indicated K enabled suppression of CH4 formation.40,56 Simultaneously, there was a significant decrease in produced EtOH, the amount of adsorbed carbonate30,52 at 1268 and 1510 cm−1 and formate species22,40,54 at 1388 and 1594 cm−1. These results indicate that the addition of potassium has a positive impact on enhancing CO2 adsorption/activation and facilitates alcohol synthesis. In contrast, KFe/ZrO2, KFeZn/ZrO2, and KFeCu/ZrO2 showed similar surface species but with variations in intensity compared to the KFeCuZn/ZrO2 catalyst, indicating the same mechanism with different efficiencies (Fig. 8a and S19†). The evolution of CH3CH2O* is shown in Fig. 8b. The peak intensities of CH3CH2O* over KFeCuZn/ZrO2 and KFeCu/ZrO2 are similar and higher than those of the catalysts without Cu. This indicates that the addition of Cu is helpful for the evolution of CH3CH2O*, leading to EtOH formation. Concretely, introduction of Cu significantly promotes the formation of CO (CO2-TPSR, Fig. S20†). It was reported30,39,40,46 that increased CO provides a great chance of coupling with CHx on iron carbide surface to form CH3CHO, which in turn is hydrogenated to CH3CH2O* and subsequently CH3CH2OH, over Fe–Cu based catalysts. Because of the similar catalyst components, it is thought that EtOH was produced via the same pathway over our catalyst. Simultaneously, as previously reported, Cu helps non-dissociative activation of CO and its coupling with alkyl (CHx) species to form ethanol.30,39,40,46 Therefore, for KFe/ZrO2 and KFeZn/ZrO2 catalysts, the lack of Cu would result in the slight increase of CO and CH4 selectivity. Although the Zn-assisted KFeCu/ZrO2 catalyst showed a higher selectivity for EtOH and STYEtOH, the difference between KFeCuZn/ZrO2 and KFeCu/ZrO2 remains unclear, according to DRIFT results (Fig. 1a).
 | ||
| Fig. 8 In situ DRIFTS spectra. (a) Different catalysts under the CO2 + H2 reaction after 60 min at 320 °C and 0.5 MPa; (b) dynamic IR peak intensities of CH3CH2O* (2922 cm−1) at 320 °C for different catalysts. | ||
CO2 adsorption and EtOH decomposition experiments were conducted to elucidate the role and impact of Zn in EtOH synthesis. The DRIFTS results for CO2 adsorption (Fig. S21†) revealed that the addition of Zn to the KFeZn/ZrO2 and KFeCuZn/ZrO2 catalysts enhanced CO2 adsorption, leading to more formate and carbonates. The results of the ethanol decomposition experiments over KFeCu/ZrO2 and KFeCuZn/ZrO2 using a batch reactor are shown in Fig. 9. The carbonaceous products by the ethanol decomposition are COx, CH4 and acetaldehyde over KFeCuZn/ZrO2 and KFeCu/ZrO2 catalysts, with the CH3CH2OH decomposition rate of 0.13 and 0.28 mg gcat−1 h−1, respectively. This result indicates that the presence of Zn restricts EtOH decomposition (backward reaction), leading to higher selectivity toward EtOH.
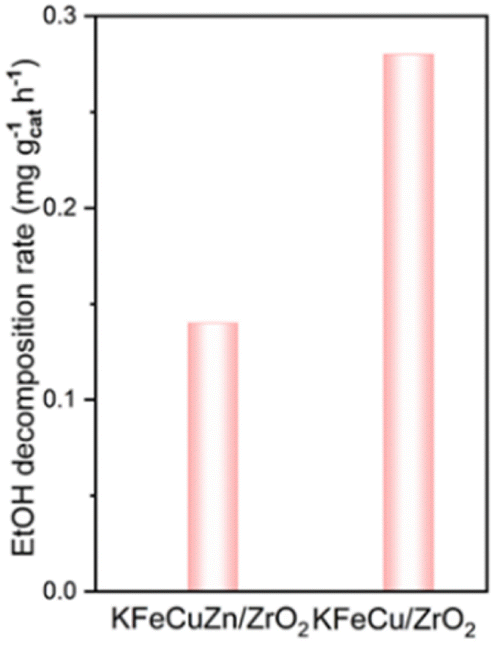 | ||
| Fig. 9 EtOH decomposition over the KFeCuZn/ZrO2 and KFeCu/ZrO2 catalysts (conditions: batch reactor, spent catalysts: 50 mg, N2: 0.5 MPa, at 260 °C, 3 h, ethanol (0.3 mL) + octane (0.7 mL)). | ||
CO hydrogenation over KFeCuZn/ZrO2
Although the one-step conversion of CO2 to value-added chemicals such as EtOH is attractive, the kinetically unreactive and thermodynamically stable nature of CO2 pose significant challenges.8,28,57,58 CO hydrogenation to EtOH is another promising method for producing EtOH;59–63 thus, we briefly explored the application of our catalysts in CO hydrogenation.64–69 CO2 and CO hydrogenation experiments were conducted on the KFeCuZn/ZrO2 catalyst under the same experimental conditions of 280 °C, 3 MPa, and CO or CO2:H2 = 1:3. The performance results revealed a higher STY of EtOH (1.8 mmol gcat−1 h−1) in CO hydrogenation (Fig. 10a). Furthermore, in a comparison of the CO2 + H2 DRIFTS experiments, a higher amount of CH3CH2O* (2852, 2922, 2956 cm−1)27,30 was observed in the CO2 (20% CO) + H2 DRIFTS experiments on KFeCuZn/ZrO2 (Fig. S22†). These two experiments imply that CO hydrogenation to EtOH is more facile than CO2 hydrogenation. In addition, CO + H2in situ DRIFTS was performed to clarify the potential roles of each element. According to the acquired IR spectra (Fig. 10b), the intermediates were similar to those observed in the CO2 + H2 DRIFTS results. The intensity of the peak at 1712 cm−1, attributed to CH3CHO* over KFeCuZn/ZrO2, was found to be higher compared to the other catalysts.30,70 Additionally, KFeCuZn/ZrO2 displayed stronger peaks at 2852, 2922, and 2956 cm−1, corresponding to CH3CH2O* (Fig. 10b, c and S23†), in comparison to the other catalysts.27,30,39,40 The CH3CH2O* intensities of KFe/ZrO2 and KFeZn/ZrO2 were similar and much weaker than those of KFeCuZn/ZrO2 (Fig. 10c).
 | ||
| Fig. 10 (a) Catalytic performance over the KFeCuZn/ZrO2 catalyst under a CO/H2 or CO2/H2 flow at 280 °C and 3 MPa; (b) in situ DRIFTS spectra of the CO + H2 reaction over the different catalysts under a CO/H2 flow at 280 °C and 0.5 MPa; and (c) dynamic IR peak intensities of CH3CH2O* (2922 cm−1) at 280 °C. | ||
Conclusion
In summary, we reported an efficient catalyst for ethanol synthesis, namely KFeCuZn/ZrO2, which achieves the STYEtOH of 5.4 mmol gcat−1 h−1 under the optimized conditions (360 °C, 4 MPa, and 12 L gcat−1 h−1) and elucidated the essential roles of K, Fe, Cu, and Zn in EtOH synthesis. In situ/operando spectroscopic techniques and various characterizations revealed the essential roles of K and Fe in EtOH synthesis. The introduction of Cu accelerated the generation of CH3CH2O*, which is an important intermediate in EtOH production. The addition of Zn inhibited EtOH decomposition, thereby improving the efficiency of EtOH synthesis. The importance of all the components was proven and emphasized in CO2 hydrogenation to EtOH. Finally, this catalyst demonstrated high performance in EtOH synthesis from syngas (CO + H2).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Takashi Toyao and Ken-ichi Shimizu supervised this research. Pengfei Du and Abdellah Ait El Fakir co-conducted the experiments and co-wrote the manuscript. Shirun Zhao, Yucheng Qian and HongLi Pan helped to perform the experiments and analyze the data. Nazmul Hasan MD Dostagir, Kah Wei Ting, and Shinya Mine discussed the data and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was financially supported by KAKENHI (21K18185, 21H04626, 22K14538, 23KF0129, and 23H01997) from the Japan Society for the Promotion of Science (JSPS), JST-FOREST Program JPMJFR211U, the JST-SPRING project JPMJSP2119, “Nanotechnology Platform Program” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) project (JPMXP09A21HK0027), and the Joint Usage/Research Center for Catalysis. A. F. acknowledges a JSPS Postdoctoral Fellowship (P23351). The XAS measurements were conducted at the BL01B1 beamline of SPring-8 at JASRI (Proposal No.: 2023A1931).References
- S. S. Ali, S. S. Ali and N. Tabassum, J. Environ. Chem. Eng., 2022, 10, 106962 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- T. Tatsumi, A. Muramatsu and H.-O. Tominaga, Chem. Lett., 1985, 593–594 CrossRef CAS.
- M. Irshad, H. J. Chun, M. K. Khan, H. Jo, S. K. Kim and J. Kim, Appl. Catal., B, 2024, 340, 123201 CrossRef CAS.
- D. Xu, Y. Wang, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, Chem, 2021, 7, 849–881 CAS.
- A. I. Latsiou, N. D. Charisiou, Z. Frontistis, A. Bansode and M. A. Goula, Catal. Today, 2023, 420, 114179 CrossRef CAS.
- S. Hafeez, E. Harkou, S. M. Al-Salem, M. A. Goula, N. Dimitratos, N. D. Charisiou, A. Villa, A. Bansode, G. Leeke, G. Manos and A. Constantinou, React. Chem. Eng., 2022, 7, 795–812 RSC.
- Y. Wang, D. Xu, X. Zhang, X. Hong and G. Liu, Catal. Sci. Technol., 2022, 12, 1539–1550 RSC.
- P. Iodice and M. Cardone, Energies, 2021, 14, 4034 CrossRef CAS.
- M. A. S. Al-Baghdadi, Renewable Energy, 2003, 28, 1471–1478 CrossRef CAS.
- D. Xu, H. Yang, X. Hong, G. Liu and S. C. Edman Tsang, ACS Catal., 2021, 11, 8978–8984 CrossRef CAS.
- S. Zhang, Z. Wu, X. Liu, K. Hua, Z. Shao, B. Wei, C. Huang, H. Wang and Y. Sun, Top. Catal., 2021, 64, 371–394 CrossRef CAS.
- X. Li, J. Ke, R. Li, P. Li, Q. Ma and T. S. Zhao, Chem. Eng. Sci., 2023, 282, 119226 CrossRef CAS.
- A. Z. Mendiburu, C. H. Lauermann, T. C. Hayashi, D. J. Mariños, R. B. Rodrigues da Costa, C. J. R. Coronado, J. J. Roberts and J. A. de Carvalho, Energy, 2022, 257, 124688 CrossRef CAS.
- M. Al-Faliti, B. Dvorak and A. Aly Hassan, J. Air Waste Manage. Assoc., 2022, 72, 602–616 CrossRef CAS PubMed.
- T. Inui, T. Yamamoto, M. Inoue, H. Hara, T. Takeguchi and J.-B. Kim, Appl. Catal., A, 1999, 186, 395–406 CrossRef CAS.
- Y. Lou, F. jiang, W. Zhu, L. Wang, T. Yao, S. Wang, B. Yang, B. Yang, Y. Zhu and X. Liu, Appl. Catal., B, 2021, 291, 120122 CrossRef CAS.
- F. J. Caparrós, L. Soler, M. D. Rossell, I. Angurell, L. Piccolo, O. Rossell and J. Llorca, ChemCatChem, 2018, 10, 2365–2369 CrossRef.
- S. Bai, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139, 6827–6830 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, X. Zhang, M. Wang, T. Liu, J. Wei, G. Zhang, X. Hong and G. Liu, Appl. Catal., B, 2024, 345, 123691 CrossRef CAS.
- F. Zhang, W. Zhou, X. Xiong, Y. Wang, K. Cheng, J. Kang, Q. Zhang and Y. Wang, J. Phys. Chem. C, 2021, 125, 24429–24439 CrossRef CAS.
- K. Zheng, Y. Li, B. Liu, F. Jiang, Y. Xu and X. Liu, Angew. Chem., Int. Ed., 2022, 61, e2022109 Search PubMed.
- A. Goryachev, A. Pustovarenko, G. Shterk, N. S. Alhajri, A. Jamal, M. Albuali, L. van Koppen, I. S. Khan, A. Russkikh, A. Ramirez, T. Shoinkhorova, E. J. M. Hensen and J. Gascon, ChemCatChem, 2021, 13, 3324–3332 CrossRef CAS.
- X. Ye, J. Ma, W. Yu, X. Pan, C. Yang, C. Wang, Q. Liu and Y. Huang, J. Energy Chem., 2022, 67, 184–192 CrossRef CAS.
- H. Kusama, K. Okabe, K. Sayama and H. Arakawa, Energy, 1997, 22, 343–348 CrossRef CAS.
- S. Liu, C. Yang, S. Zha, D. Sharapa, F. Studt, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2022, 61, e202109027 CrossRef CAS PubMed.
- L. Ding, T. Shi, J. Gu, Y. Cui, Z. Zhang, C. Yang, T. Chen, M. Lin, P. Wang, N. Xue, L. Peng, X. Guo, Y. Zhu, Z. Chen and W. Ding, Chem, 2020, 6, 2673–2689 CAS.
- S. Li, H. Guo, C. Luo, H. Zhang, L. Xiong, X. Chen and L. Ma, Catal. Lett., 2013, 143, 345–355 CrossRef CAS.
- A. H. M. da Silva, L. H. Vieira, C. S. Santanta, M. T. M. Koper, E. M. Assaf, J. M. Assaf and J. F. Gomes, Appl. Catal., B, 2023, 324, 122221 CrossRef CAS.
- D. Xu, M. Ding, X. Hong, G. Liu and S. C. E. Tsang, ACS Catal., 2020, 10, 5250–5260 CrossRef CAS.
- Y. Wang, W. Wang, R. He, M. Li, J. Zhang, F. Cao, J. Liu, S. Lin, X. Gao, G. Yang, M. Wang, T. Xing, T. Liu, Q. Liu, H. Hu, N. Tsubaki and M. Wu, Angew. Chem., Int. Ed., 2023, 62, e20231178 Search PubMed.
- W. Guo, W. G. Gao, H. Wang and J. J. Tian, Adv. Mater. Res., 2014, 827, 20–24 Search PubMed.
- Z. Si, L. Wang, Y. Han, J. Yu, Q. Ge, C. Zeng and J. Sun, ACS Sustain. Chem. Eng., 2022, 10, 14972–14979 CrossRef CAS.
- X. Xi, F. Zeng, H. Zhang, X. Wu, J. Ren, T. Bisswanger, C. Stampfer, J. P. Hofmann, R. Palkovits and H. J. Heeres, ACS Sustain. Chem. Eng., 2021, 9, 6235–6249 CrossRef CAS.
- H. Zhang, H. Han, L. Xiao and W. Wu, ChemCatChem, 2021, 13, 3333–3339 CrossRef CAS.
- K. An, S. Zhang, J. Wang, Q. Liu, Z. Zhang and Y. Liu, J. Energy Chem., 2021, 56, 486–495 CrossRef CAS.
- P. Riani, G. Garbarino, T. Cavattoni and G. Busca, Catal. Today, 2021, 365, 122–131 CrossRef CAS.
- S. Zhang, Z. Wu, X. Liu, Z. Shao, L. Xia, L. Zhong, H. Wang and Y. Sun, Appl. Catal., B, 2021, 293, 120207 CrossRef CAS.
- D. Xu, M. Ding, X. Hong and G. Liu, ACS Catal., 2020, 10, 14516–14526 CrossRef CAS.
- T. Liu, D. Xu, M. Song, X. Hong and G. Liu, ACS Catal., 2023, 13, 4667–4674 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- C. Wang, W. Fang, Z. Liu, L. Wang, Z. Liao, Y. Yang, H. Li, L. Liu, H. Zhou, X. Qin, S. Xu, X. Chu, Y. Wang, A. Zheng and F. S. Xiao, Nat. Nanotechnol., 2022, 17, 714–720 CrossRef CAS PubMed.
- N. Lohitharn, J. G. Goodwin and E. Lotero, J. Catal., 2008, 255, 104–113 CrossRef CAS.
- N. Boreriboon, X. Jiang, C. Song and P. Prasassarakich, Top. Catal., 2018, 61, 1551–1562 CrossRef CAS.
- K. Asakura, M. Nomura and H. Kuroda, Bull. Chem. Soc. Jpn., 1985, 58, 1543–1550 CrossRef CAS.
- Y. Wang, K. Wang, B. Zhang, X. Peng, X. Gao, G. Yang, H. Hu, M. Wu and N. Tsubaki, ACS Catal., 2021, 11, 11742–11753 CrossRef CAS.
- L. R. Merte, G. Peng, R. Bechstein, F. Rieboldt, C. A. Farberow, L. C. Grabow, W. Kudernatsch, S. Wendt, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, Science, 2012, 336, 889–893 CrossRef CAS PubMed.
- D. Peña, L. Jensen, A. Cognigni, R. Myrstad, T. Neumayer, W. van Beek, M. Rønning, D. Peña, L. Jensen, T. Neumayer, A. Cognigni, M. Rønning and W. van Beek, ChemCatChem, 2018, 10, 1300–1312 CrossRef.
- F. Amet, C. T. Ke, I. V. Borzenets, J. Wang, K. Watanabe, T. Taniguchi, R. S. Deacon, M. Yamamoto, Y. Bomze, S. Tarucha and G. Finkelstein, Science, 2016, 352, 966–969 CrossRef CAS PubMed.
- Š. Hajduk, V. D. B. C. Dasireddy, B. Likozar, G. Dražić and Z. C. Orel, Appl. Catal., B, 2017, 211, 57–67 CrossRef.
- P. Du, R. Qi, Y. Zhang, Q. Gu, X. Xu, Y. Tan, X. Liu, A. Wang, B. Zhu, B. Yang and T. Zhang, Chem, 2022, 8, 3252–3262 CAS.
- M. Mohtar and R. Omar, Solid State Sci. Technol., 2005, 13, 24–31 Search PubMed.
- N. H. M. Dostagir, R. Rattanawan, M. Gao, J. Ota, J. Y. Hasegawa, K. Asakura, A. Fukouka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef CAS.
- X. Chang, X. Han, Y. Pan, Z. Hao, J. Chen, M. Li, J. Lv and X. Ma, Ind. Eng. Chem. Res., 2022, 61, 6872–6883 CrossRef CAS.
- J. Wu, S. Li, G. Li, C. Li Bv and Q. Xin, Appl. Surf. Sci., 1994, 81, 37–41 CrossRef CAS.
- M. Wang, P. Wang, G. Zhang, Z. Cheng, M. Zhang, Y. Liu, R. Li, J. Zhu, J. Wang, K. Bian, Y. Liu, F. Ding, T. P. Senftle, X. Nie, Q. Fu, C. Song and X. Guo, Sci. Adv., 2023, 9, eadg0167 CrossRef CAS PubMed.
- D. Goud, S. R. Churipard, D. Bagchi, A. K. Singh, M. Riyaz, C. P. Vinod and S. C. Peter, ACS Catal., 2022, 12, 11118–11128 CrossRef CAS.
- T. Inoue, T. Iizuka and K. Tanabe, Appl. Catal., 1989, 46, 1–9 CrossRef CAS.
- Y. Ge, T. Zou, A. J. Martín and J. Pérez-Ramírez, ACS Catal., 2023, 13, 9946–9959 CrossRef CAS PubMed.
- P. Preikschas, M. Plodinec, J. Bauer, R. Kraehnert, R. Naumann D'Alnoncourt, R. Schlögl, M. Driess and F. Rosowski, ACS Catal., 2021, 11, 4047–4060 CrossRef CAS.
- X. Wang, P. J. Ramírez, W. Liao, J. A. Rodriguez and P. Liu, J. Am. Chem. Soc., 2021, 143, 13103–13112 CrossRef CAS PubMed.
- J. Hu, Z. Wei, Y. Zhang, R. Huang, M. Zhang, K. Cheng, Q. Zhang, Y. Qi, Y. Li, J. Mao, J. Zhu, L. Wu, W. Wen, S. Yu, Y. Pan, J. Yang, X. Wei, L. Jiang, R. Si, L. Yu, Y. Wang and D. Deng, Nat. Commun., 2023, 14, 6808 CrossRef CAS PubMed.
- Y. Lu, F. Yu, J. Hu and J. Liu, Appl. Catal., A, 2012, 429–430, 48–58 CrossRef CAS.
- M. Schumann, M. R. Nielsen, T. E. L. Smitshuysen, T. W. Hansen, C. D. Damsgaard, A. C. A. Yang, M. Cargnello, J. D. Grunwaldt, A. D. Jensen and J. M. Christensen, ACS Catal., 2021, 11, 5189–5201 CrossRef CAS.
- B. J. Kip, E. G. F. Hermans, J. H. M. C. Van Wolput, N. M. A. Hermans, J. Van Grondelle and R. Prins, Appl. Catal., 1987, 35, 109–139 CrossRef CAS.
- M. Gupta, M. L. Smith and J. J. Spivey, ACS Catal., 2011, 1, 641–656 CrossRef CAS.
- Y. Liu, K. Murata, M. Inaba, I. Takahara and K. Okabe, Catal. Today, 2011, 164, 308–314 CrossRef CAS.
- T. Ishida, T. Yanagihara, X. Liu, H. Ohashi, A. Hamasaki, T. Honma, H. Oji, T. Yokoyama and M. Tokunaga, Appl. Catal., A, 2013, 458, 145–154 CrossRef CAS.
- F. G. A. Van Den Berg, J. H. E. Glezer and W. M. H. Sachtler', J. Catal., 1985, 93, 340–352 CrossRef CAS.
- A. K. P. Mann, Z. Wu, F. C. Calaza and S. H. Overbury, ACS Catal., 2014, 4, 2437–2448 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02588a |
| This journal is © The Royal Society of Chemistry 2024 |