
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of polyurethanes through the oxidative decarboxylation of oxamic acids: a new gateway toward self-blown foams†
Quentin
Jaussaud
a,
Ikechukwu Martin
Ogbu
b,
Govind Goroba
Pawar
b,
Etienne
Grau
 a,
Frédéric
Robert
b,
Thomas
Vidil
a,
Yannick
Landais
*b and
Henri
Cramail
*a
a,
Frédéric
Robert
b,
Thomas
Vidil
a,
Yannick
Landais
*b and
Henri
Cramail
*a
aUniversity of Bordeaux, CNRS, Bordeaux INP, LCPO, UMR 5629, 16 Avenue Pey-Berland, F-33600, Pessac, France. E-mail: cramail@enscbp.fr
bUniversity of Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, 351, Cours de la Libération, F-33400, Talence, France. E-mail: yannick.landais@u-bordeaux.fr
First published on 25th July 2024
Abstract
Polyurethane (PU) thermoplastics and thermosets were prepared through the step-growth polymerization of in situ generated polyisocyanates through the decarboxylation of polyoxamic acids, in the presence of phenyliodine diacetate (PIDA), and polyols. The CO2 produced during the reaction allowed the access to self-blown polyurethane foams through an endogenous chemical blowing. The acetic acid released from ligand exchange at the iodine center was also shown to accelerate the polymerization reaction, avoiding the recourse to an additional catalyst. Changing simple parameters during the production process allowed us to access flexible PU foams with a wide range of properties.
Introduction
Polyurethanes (PUs) were developed during World War II as an alternative to rubber and have become the 6th most produced polymer in the world.1,2 They represent a significant portion of the polymer market, estimated to be 3.81 Mt in the European Union in 2020, i.e. 7.8% of the EU total polymer demand.3,4 PU main products include rigid and flexible foams, coatings, adhesives, sealants and elastomers for applications in furniture and interiors, the automotive industry, electronics and appliances, packaging and footwear industries.2,5 PUs are commonly accessed through the straightforward addition of polyols to polyisocyanates (Bayer reaction).6,7 Conveniently, the partial hydrolysis of the isocyanates can be used to liberate gaseous CO2 during the polymerization, leading to the formation of self-blown PU foams.8,9 The importance of PU foams has for instance become central in building insulation and renovation for energy conservation.10,11However, the use of isocyanates in industrial applications is increasingly subjected to international regulations,12,13 as they are known to be powerful irritants to mucous membranes, leading to occupational asthma sensitization, eyes and skin irritation.14,15 In addition, they are classified as carcinogenic to animals and potentially to humans. Finally, isocyanates are produced from amines and phosgene, a very hazardous gas.16,17 In this context, substantial efforts have been dedicated to the development of isocyanate-free PUs syntheses.18 Among the various available options, polyhydroxyurethanes (PHUs) obtained from the reaction of poly(cyclic carbonate)s and polyamines are foreseen as one of the most promising alternatives to traditional PUs.19,20 For instance, tremendous efforts were recently made to develop self-foaming PHUs through the partial decarboxylation of the cyclic carbonates.21–24 However, PHUs are structurally different from conventional PUs due to the numerous hydroxyl functions they contain, and they cannot supplant them in all applications.
Another way around to circumvent the toxicity of isocyanates is to generate them in situ from stable and innocuous precursors, i.e. blocked or latent isocyanates, that can be premixed with polyols.25–27 This way, polyisocyanates can be safely created, on demand, in closed reactors, when all safety measures are thoroughly ensured for the operators. Additionally, this strategy increases the shelf life of single component formulations and offers a good control over the initiation of the polymerization in space and time.
One simple approach to obtain blocked isocyanate consists in reacting a pre-existing isocyanate with a compound containing an active hydrogen.25 The most representative examples are phenol,28 oxime29 and pyrazole.30 The isocyanate is then regenerated at high temperature (typically 100 °C ≤ T ≤ 200 °C) through the reverse reaction (Fig. 1a). Despite the high industrial applicability of this approach, notably for the design of one-pot formulation used in coating applications, it still relies on the use of free isocyanates for the synthesis of the blocked precursors. Moreover, the blocking compounds are not volatile enough to be valorized as potential blowing agents during the polymerization reaction.
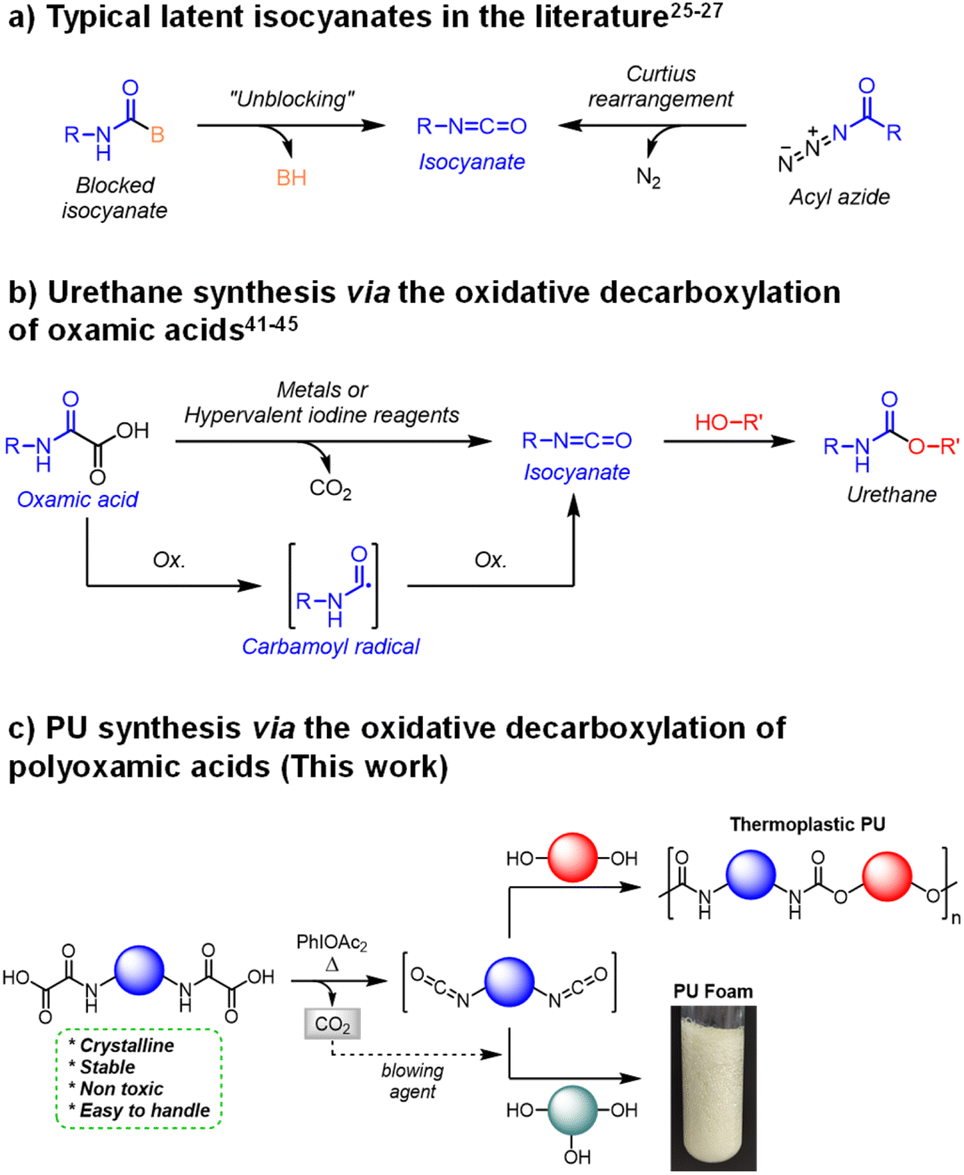 | ||
| Fig. 1 Urethane and Polyurethane (PU) synthesis from oxamic and polyoxamic acids under oxidative conditions. | ||
Alternatively, well-known chemical compounds can undergo intramolecular rearrangements to provide isocyanates.31 For instance acyl azides can be converted into isocyanates through the Curtius32,33 rearrangement (Fig. 1a). The latter has been successfully employed in single-component formulations to synthesize thermoplastic PUs, notably through the self-condensation of AB-type hydroxyl–acyl azide monomers.34–37 Additionally, the gaseous nitrogen liberated during the rearrangement reaction was valorized to fabricate self-blown thermosetting PU foams.38 Nevertheless, acyl azides are considered to be highly unstable and potentially explosive intermediates.39,40
In 1995, Minisci et al. introduced a new latent precursor of isocyanate: oxalic acid monoamides, also called oxamic acids (OA, Fig. 1b).41 They can undergo metal-catalyzed (e.g. Ag(I), Cu(II)) oxidative decarboxylation to deliver isocyanate, possibly in the presence of alcohols for the direct in situ formation of the corresponding urethane. The decarboxylation was shown to proceed through a free-radical pathway involving a carbamoyl radical intermediate. Conveniently, oxamic acids are very stable and easy to handle precursors, which can be prepared from amine and oxalic acid derivatives using a simple and efficient procedure.42 Furthermore, several oxamic acid analogs are considered as safe prodrugs in pharmaceutical applications.43,44 It is thus anticipated that they pose minimal risk to both human health and the environment.
Very recently, we developed metal-free approaches to access isocyanates from oxamic acids.45–47 The methodology is based on the use of hypervalent iodine reagents as lipophilic oxidants (e.g. (acetoxy)benziodoxolone (Bi-OAc) or the commercially available phenyliodine diacetate (PIDA)). Conveniently, we demonstrated that the decarboxylation reaction can be either photo-46,47 or thermo-initiated45 under very mild conditions. In the first case, the oxidation is mediated at room temperature by a photocatalyst under visible or near infrared light (NIR). In the second case, the whole oxidative process is mediated by the hypervalent iodine itself at moderated temperatures (≥60 °C). The reactions were conducted in organic solvent (e.g. dichloroethane) in the presence of mono-alcohols (resp. diol) to deliver mono-urethanes (resp. diurethanes) in high yield (up to 95%). We anticipated that the use of these highly efficient and liposoluble oxidants could unlock the use of oxamic acids for the one-pot synthesis of PU in bulk.
Reported here is the first example of the thermal decarboxylation of bis-oxamic acids in the presence of polyols to produce both thermoplastic and thermosetting PUs (Fig. 1c). Advantageously, we demonstrate that the isocyanate-alcohol polyaddition reaction is strongly catalyzed by the acetic acid released from the hypervalent iodine reagent, PIDA. The resulting polymerization kinetics are faster than those of conventional isocyanate-alcohol systems. Furthermore, in the case of thermosetting PUs, the CO2 gas generated upon oxamic acid oxidation was valorized as an endogenous foaming agent. A series of flexible foams, with well-controlled porous structures, was successfully synthesized according to this procedure. Eventually, the oxidative decarboxylation of oxamic acid represents a new versatile methodology for the on-demand synthesis of PUs, while circumventing the direct handling of isocyanates.
Results and discussion
Synthesis of thermoplastic PUs
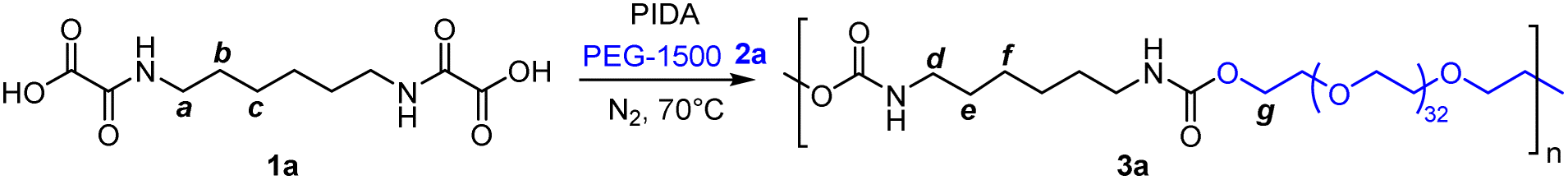
In a first trial, hexamethylene bis-oxamic acid 1a (HBOA, 1 eq., Table 1), obtained according to a previously reported procedure,45 was used as a latent-isocyanate precursor. Di-hydroxy telechelic PEG-1500 2a (1 eq.) was chosen as the diol and phenyliododiacetate (PIDA, 2.5 eq.) 3 as the oxidant. At 70 °C under nitrogen flux and magnetic stirring, HBOA and PIDA, both crystalline solids, were homogeneously dispersed in the liquid diol (melting point ∼50 °C). The mixture quickly evolves from a white paste to a transparent liquid with the formation of bubbles, attesting the liberation of gaseous CO2. After 12 h of reaction, the mixture was diluted in dichloromethane (DCM) and precipitated in diethyl ether (Et2O). The resulting product, 3a (Table 1, entry 1), was first analyzed by size exclusion chromatography in HFIP (SEC), confirming the presence of a polymer with Mn = 19 kg mol−1 and Đ = 1.8 (ESI, Fig. S1†). Moreover, the signature of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond vibration typical of PUs at about 1710 cm−1 was observed by FT-IR (ESI, Fig. S2†). 1H NMR in DMSO-d6 further confirms the formation of urethane linkages with the presence of a triplet typical of the N–H proton of urethane around 7.16 ppm (ESI, Fig. S3†).
O bond vibration typical of PUs at about 1710 cm−1 was observed by FT-IR (ESI, Fig. S2†). 1H NMR in DMSO-d6 further confirms the formation of urethane linkages with the presence of a triplet typical of the N–H proton of urethane around 7.16 ppm (ESI, Fig. S3†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Time (h) | M n (g mol−1) | M w (g mol−1) | Đ | DP |
| a All reactions were performed under solvent free conditions, using a Schlenk tube equipped with a magnetic stirrer. b Reaction performed at 100 °C. c Obtained from an instrument calibrated with poly(methyl methacrylate) standards. | ||||||
| 1 | 1:1 |
12 | 19300 |
34900 |
1.8 | 23 |
| 2 | 1.1:1 |
12 | 25200 |
44400 |
1.8 | 30 |
| 3 | 1.1:1 |
48 | 34600 |
64100 |
1.9 | 41 |
| 4 | 1.25:1 |
48 | 25300 |
46400 |
1.8 | 30 |
| 5b | 1.1:1 |
6 | 36400 |
91100 |
2.5 | 43 |
For comparison purposes, a PU analog was synthesized by reacting commercial hexamethylene diisocyanate (HDI) with the same diol 2a ([HDI]:[2a] = 1:1). After 24 h of reaction at 70 °C, the resulting polyurethane, PUref, was analyzed by FTIR, 1H NMR and SEC in the same conditions than 3a. The FTIR spectra of 3a and PUref are nearly identical (Fig. S2†), thus supporting the idea that 3a corresponds to the expected PU. When comparing the NMR analysis of the two polymers (Fig. S3†), the spectra are also superimposable. However, the spectrum of 3a contains additional peaks of small integrals suggesting the presence of residual side products. The latter could not be removed despite further precipitations in Et2O but were fully resolved and quantified later in this study. The SEC traces of 3a and PUref (Mn = 28 kg mol−1 and Đ = 2.0, DP = 33) are roughly the same (Fig. S4†), implying that both polymers exhibit similar macromolecular features. Overall, these preliminary results demonstrate that 3a is a polyurethane that compares well with those obtained from the conventional isocyanate-alcohol route.
Before going any further in the understanding of the polymerization mechanism, the synthesis of 3a was optimized by playing with the different parameters of the reaction. A decrease of the proportion of diol 2a from 1 eq. to 0.9 eq. (Table 1, entry 2) resulted in a substantial increase of the PU molecular weight from Mn = 19 kg mol−1 to 25 kg mol−1. Under these latter conditions, an increase of the reaction time from 12 h to 48 h fostered the molecular weight to 35 kg mol−1 (Table 1, entry 3). Further decrease in the polyol amount to 0.8 eq. resulted in a decrease of Mn (Table 1, entry 4). When the reaction was performed at 100 °C, a slightly larger molecular weight was observed (Table 1, entry 5).
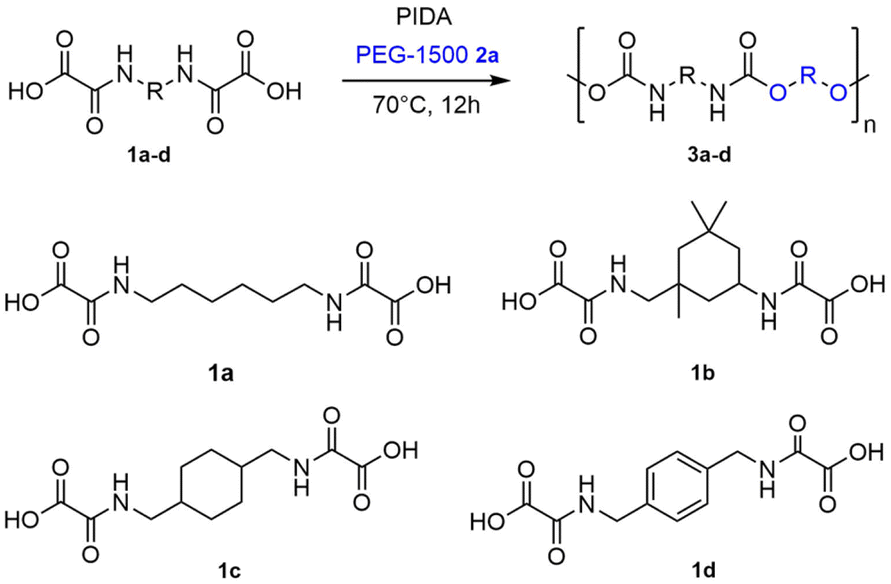
With these optimized conditions in hand (T = 70 °C, [oxamic acid]:[OH] = 1.1:1, t = 48 h, entry 3), a series of thermoplastic PUs were then prepared starting from bis-oxamic acids 1b–d and PEG-1500 2. In all cases (Table 2), 1H NMR (Fig. S5†) and FT-IR (Fig. S6†) are consistent with the formation of the expected PU, while SEC analyses (Fig. S7†) confirm that polymers with molecular weight ranging from 25 to 45 kg mol−1 and dispersity between 1.7 and 3.2 are formed. Thus, 1b–d are all competent latent-diisocyanate monomers delivering the corresponding PUs 3b–d. The thermal properties of 3a–d, as well as PUref, were studied using TGA and DSC (Fig. S8 and S9†). Data are collected in (Table 2). TGA indicates that the PU 3d containing aromatic ring exhibits the highest thermal stability with Td5% = 309 °C.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| 1a–d | Diol | PU | M n (g mol−1) | M w (g mol−1) | Đ | T d5% (°C) | T g (°C) | T m (°C) |
| a All reactions were carried out in a Schlenk tube equipped with magnetic stirrer under solvent free conditions at 70 °C. The corresponding bis-oxamic acid (1.0 mmol), diol (0.9 mmol), PIDA (2.5 mmol) were used unless indicated otherwise. | ||||||||
| 1a | 2a | 3a | 45400 |
79200 |
1.7 | 298 | −50.8 | 42.9 |
| 1b | 2a | 3b | 39500 |
71700 |
1.8 | 296 | N/A | 32.2 |
| 1c | 2a | 3c | 40000 |
128600 |
3.2 | 268 | −47.4 | 35.5 |
| 1d | 2a | 3d | 26000 |
45100 |
1.7 | 305 | −45.8 | 37 |
| 2a | PUref | 28000 |
56300 |
2.0 | 308 | N/A | 48.2 | |
DSC traces showed that all PUs are semicrystalline with glass transition temperatures, Tg, comprised between −51 °C and −45 °C, and melting points, Tm, varying from 35 °C to 43 °C.
To further test the generality of this new process, PEG diols of different molecular weights as well as a PTMO diol were polymerized with HBOA 1a as the model bis-oxamic acid (ESI, Table S1†). The SEC traces as well as FTIR and 1H NMR spectra of the resulting polymers, 3e–g, are represented in Fig. S10–S12.† They are all consistent with the formation of the expected PU.
Using the optimized monomer proportions, the kinetic of the bulk polymerization was studied by 1H NMR. The reaction of 1a with PEG-1500 was carried out in bulk, at 70 °C, under nitrogen flow, using mechanical stirring in a sealed Schlenk tube. Fig. 2 represents the 1H NMR spectra in DMSO-d6. On the blue spectrum recorded after 30 min of reaction, the multiplet a (3.08 ppm) corresponding to the proton located in α-position of the oxamic functions drops dramatically, indicating that most of the oxamic functions are converted during the first hour of reaction. It is replaced by the multiplet d (2.94 ppm) which is diagnostic of the α-protons of urethane functions, indicating that the polymerization reaction has already started during this period. In parallel, the signal of the β-protons b (1.37 ppm) splits in two major multiplets: e (1.36 ppm) and e′ (1.53 ppm). Peak e is consistent with the β-protons of urethane functions while e′ fits well with the β-protons of isocyanates. This is confirmed by superimposition with the NMR spectrum of commercial hexamethylene diisocyanate and with the NMR spectrum of the reactive medium during the synthesis of PUref at the same reaction time (ESI, Fig. S13†). The NMR spectra recorded for longer periods indicate the total disappearance of the signals distinctive of the oxamic (a and b) and the isocyanate (e′) functions while the integrals of the α and β-protons of the urethane functions (d and e) increase. Thus, the NMR monitoring of the polymerization reaction is consistent with the rapid conversion of the oxamic acids into isocyanates and the concomitant reaction of the later with hydroxyl groups to afford PU. The percentage of oxamic acid that is effectively converted in urethane, i.e. the urethane yield, was measured over time by integrating the signal of protons g at 4.03 ppm, using the multiplets of the eight protons of the hexamethylene backbone located in between 1.1 and 1.7 ppm as an internal reference. Fig. 2b represents the evolution of the urethane yield as a function of time. After approximately 3 hours of reaction, it tends to an asymptote around 75%. This value is consistent with the formation of side products as suggested earlier by the presence of unattributed peaks in the NMR spectrum of 3a (Fig. S4†). In fact, the formation of these peaks is well distinguished in the spectra recorded during the NMR monitoring of the polymerization (Fig. 2a, singlets at 2.10 ppm and 1.77 ppm, multiplets at 3.02 ppm and 2.99 ppm). Considering the formation of acetic acid (AcOH) as side product of the oxamic acid decarboxylation using PIDA, it was anticipated that AcOH might react with isocyanate after their in situ formation. Indeed, isocyanates can react with carboxylic acids to form mixed carboxylic-carbamic anhydrides, also referred as linear N-carboxyanhydrides I (NCA) (Fig. 2).48–50 They are considered unstable and several authors observed their decarboxylation in the form of the corresponding amides II.
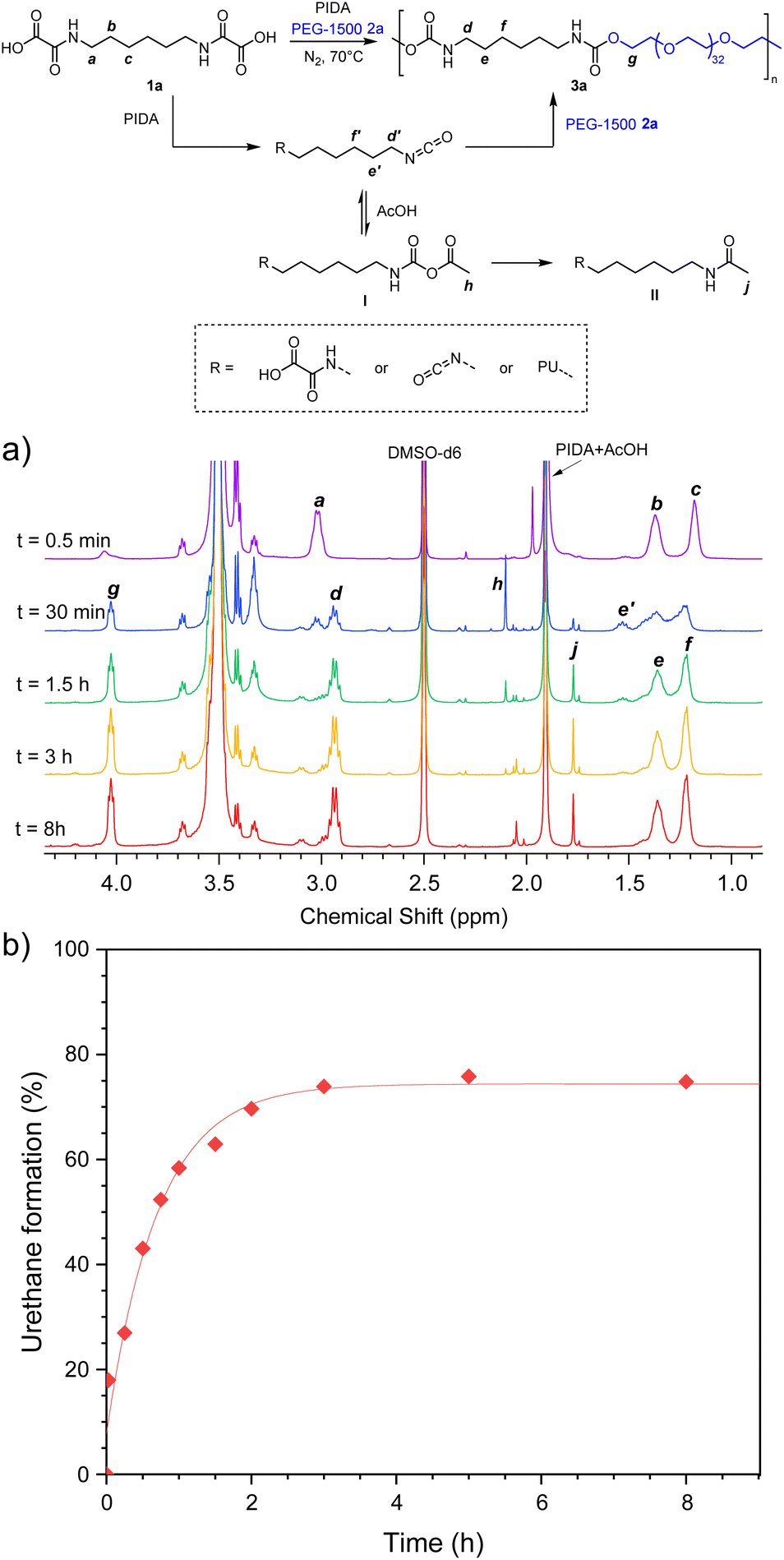 | ||
| Fig. 2 (a) 1H NMR spectra (in DMSO-d6) of the kinetics of PU 3a synthesis through the decarboxylation of oxamic acid HBOA 1a, and (b) corresponding urethane bond formation kinetics. | ||
In order to gain a better insight on the formation of the side products, a series of experiments using model molecules was conducted.
Model reactions
First of all, n-butylisocyanate 4 was reacted with acetic acid 5 ([4]:[5] = 1:1) at 70 °C in bulk. This model reaction is noted M0 and the NMR spectra recorded over time (in CDCl3) are represented in Fig. 3 (full scale spectra in Fig. S14†). Based on the reported mechanism for the reaction between isocyanates and carboxylic acids,48 the singlet o observed at 2.19 ppm after a few seconds of reaction, then disappearing, can be attributed to the formation of the unstable NCA 6. Moreover, the singlet p at 1.98 ppm as well as the quadruplet q at 3.24 ppm are distinctive of the amide 7. The attribution of the peaks relative to 7 is well confirmed by comparing the spectra with that of the pure amide (Fig. S15†) synthesized according to a procedure described in the ESI.† Thus, M0 confirms the known reactivity of isocyanates with carboxylic acids.48–50 In a second model reaction, M1 (Fig. 3 and S16†), 4 was reacted with n-hexanol 9 in conditions mimicking those of the conventional isocyanate-alcohol polymerization (70 °C, bulk, [4]:[9] = 1:1). The NMR spectra indicate that the reaction yields the expected urethane 10, which is identified by its characteristic broad triplet t and broad quadruplet s around 4.03 ppm and 3.16 ppm, respectively. The model reaction M2 (Fig. 3 and S17†) was conducted on the same basis than M1 with the addition of 1 eq. of AcOH 5. In this case, after 5.5 min of reaction, NCA 6, amide 7 and urethane 10 are all three present in the reactive medium. After 30 min of reaction, NCA 6 is no longer detected. 10 represent 86 mol% of the products against 13 mol% for 7 after 2 h. These results indicate that, when isocyanates are in the presence of both alcohols and carboxylic acids, the urethane is the main product of the reaction while the amide is formed as a side product.
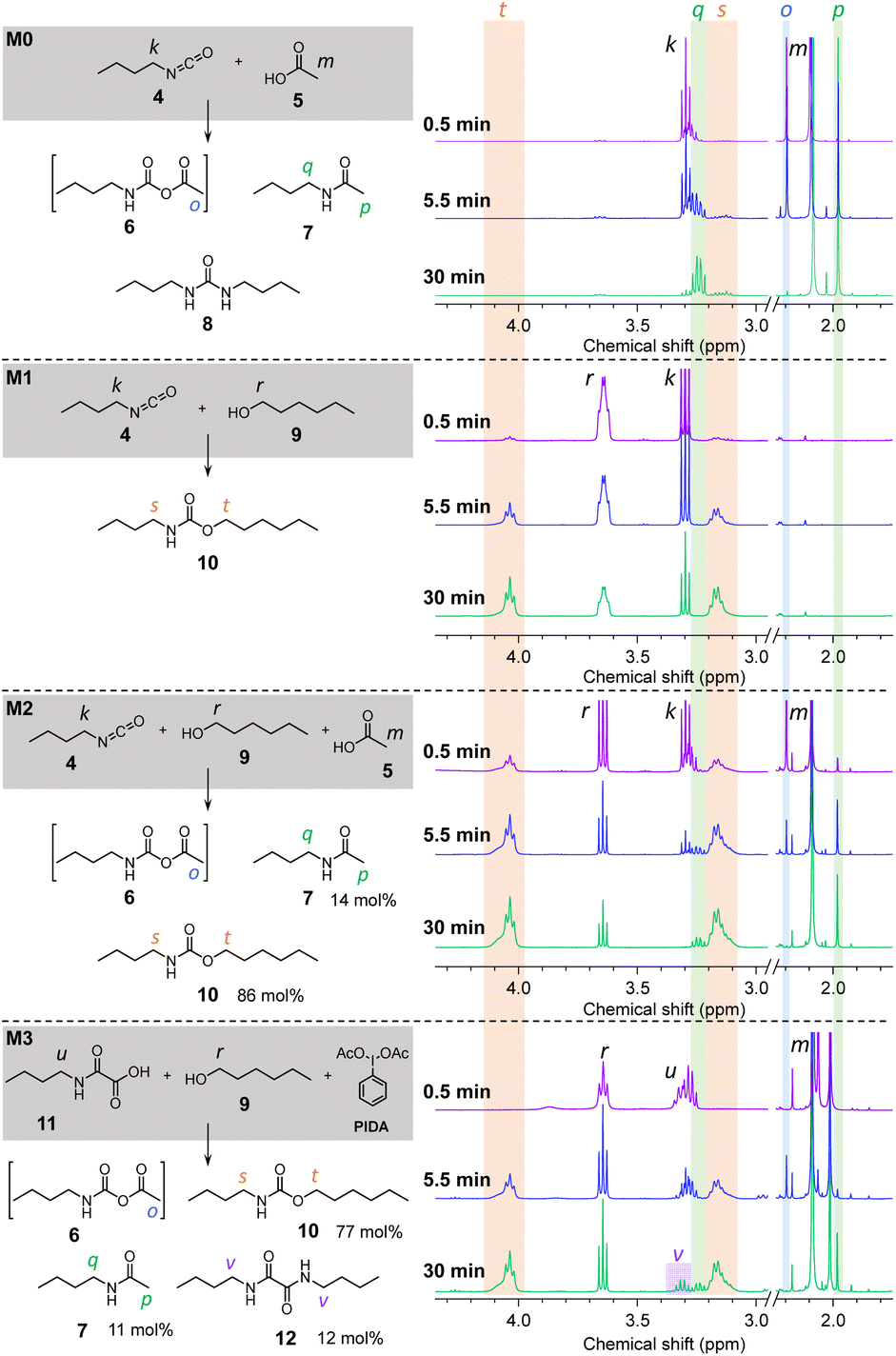 | ||
| Fig. 3 Model reactions at 70 °C and their corresponding 1H NMR traces (in CDCl3) evolution in time. | ||
Finally, the model reaction M3 (Fig. 3 and S18†) was conducted by reacting a mono-oxamic acid, n-butyloxamic acid 11, with n-hexanol 9 and PIDA in the conditions used for the other model reactions (70 °C, bulk, [11]:[9]:[PIDA] = 1:1:1.2). In a similar fashion than M2, the NMR spectrum after 5.5 min of reaction indicates the simultaneous presence of NCA 6, amide 7 and urethane 10. After 30 min, NCA 6 is undetectable while 7 and 10 are both present in significant proportions. However, the multiplet v at 3.31 ppm indicates the presence of an additional impurity. Based on our previous works,45 it was anticipated that this species might be the oxalamide 12 resulting from the recombination of two carbamoyl radicals, the reactive intermediates during the transformation of oxamic acid into isocyanates (see Scheme 1). In order to confirm this hypothesis, 12 was synthesized by reacting n-butylamine with oxalyl chloride (procedure described in the ESI†). The comparison of the NMR spectrum of the resulting molecule with that of M3 (ESI, Fig. S19†) corroborates that oxalamide is formed as a side product. In the end, the mixture of products is composed of 82 mol% of urethane 10, 12 mol% of amide 7 and 6 mol% of oxalamide 12.
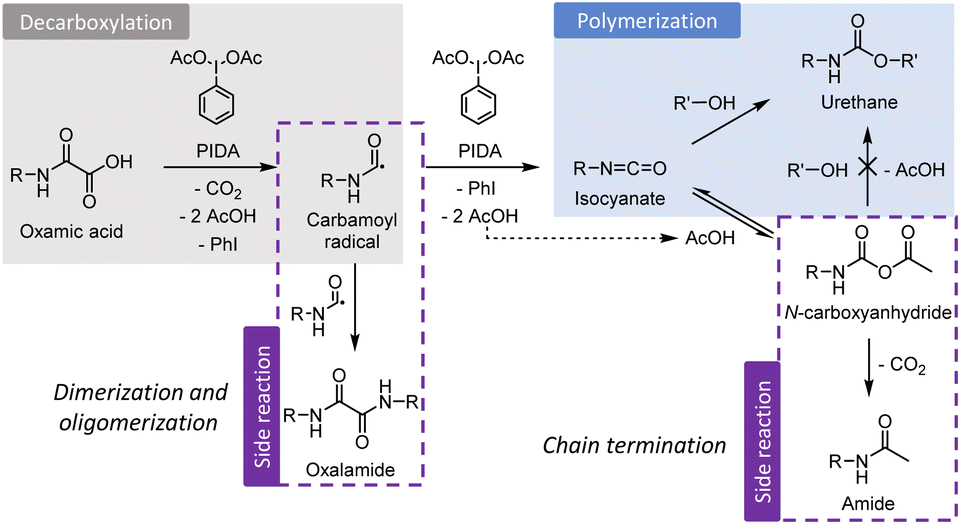 | ||
| Scheme 1 Reaction steps leading to polyurethane from poly(oxamic acids) and encountered side reactions. | ||
It is worth noting that the urethane yield of the model reaction M3 (77%)51 is roughly the same than that of the polymerization reaction of the bis-oxamic acid 1a with PEG-1500 2a (75%, Fig. 2b). In fact, by comparing the NMR spectra of the two reactions (ESI, Fig. S20†), we can confirm that the amide and the oxalamide are also formed during the polymerization reaction. Approximately 18% of the initial oxamic acid functions are transformed into the amide, and 9% into the oxalamide. For both the model reaction and the polymerization, we did not observe any product suggesting the addition of oxamic acids onto isocyanates. DFT calculations (ESI, Fig. S21†) indicate that this reaction would be slightly endothermic, in contrast with the addition of acetic acid which is thus much more favorable. Thus, it is possible to establish the global mechanism during the bulk polymerization of poly(oxamic acid) with polyols activated by PIDA as represented in Scheme 1. After the oxidative decarboxylation step, during which oxalamides can be formed, the isocyanates react either directly with an alcohol to yield a urethane or with acetic acid to provide a NCA. The latter can restitute the isocyanate through the reverse reaction or decompose into an acetamide. Theoretically, NCA could also react with an alcohol to provide the desired urethane. However, DFT calculations presented below suggest that it is unlikely. Noteworthily, the formation of oxalamides can result in dimers and short oligomers that can participate in the growing of the main polyurethane chains, while the formation of methyl amides results in a chain termination.
Despite the additional oxidative step to deliver the isocyanate, the kinetic of the urethane formation during the oxamic route is competitive with that of the conventional isocyanate-alcohol route. This is well confirmed by comparing the profiles of the urethane yields over time for the different model reactions (Fig. 4). Surprisingly, the kinetics of the oxamic route M3 is even faster than that of the conventional isocyanate route M1. The final urethane yield is however lower in the case of M3 due to the above-mentioned side reactions. Interestingly, the kinetics of M2, i.e. the isocyanate route in the presence of AcOH, is faster than M1 as well, thus suggesting that AcOH is responsible for the acceleration of the kinetics.
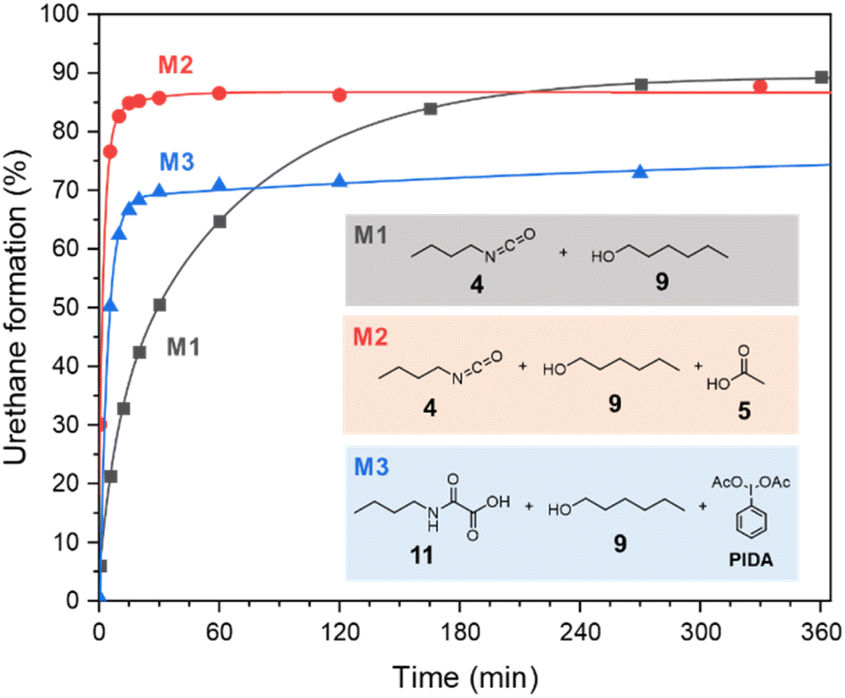 | ||
| Fig. 4 Urethane bond formation kinetics for model reactions M1, M2 and M3. | ||
Computational modeling
Catalysis of the isocyanate-alcohol reaction by strong Brønsted acids, such as sulfonic or triflic acids, has been reported by Sardon et al.52 Interestingly, under their experimental conditions, i.e. diluted solution and room temperature, AcOH exhibited no catalytic activity. Using computational modeling, the authors showed that the efficiency of the catalytic process relied on the formation of a very favorable ternary transition state involving the isocyanate, the alcohol and the acid. Surprisingly, the transition state calculated with AcOH was found to be only slightly higher than those calculated for triflic or sulfonic acids, in contrast with experimental observations. Considering that Sardon et al. did not take into account the possible formation of NCA adducts, further computational studies were carried out to rationalize the rate increase of the isocyanate-alcohol reaction with AcOH under our conditions, i.e. bulk and T > 70 °C. Preliminary calculations showed that NCA adduct I is formed through a low barrier of activation TS1 and is more stable than the isocyanate-acid mixture, in good agreement with NMR observations (Scheme 2). However, the lowest energy pathway leading to urethane from I through TS3 is quite high (see ESI, Fig. S22† for a more detailed study). A more realistic pathway would involve the regeneration of the isocyanate from I and the formation of the urethane through the more favorable 3-component transition state TS2 closely related to that of Sardon et al.52 NCA adduct I would thus behave as an isocyanate reservoir, able to liberate at higher temperature (at least 70 °C) the isocyanate, ready for the carboxylic acid-catalyzed addition of the alcohol through the low-energy TS2. This would rationalize the pronounced catalytic effect of AcOH under our bulk conditions, contrasting with Sardon's investigation where catalysis was notably absent at room temperature in solution.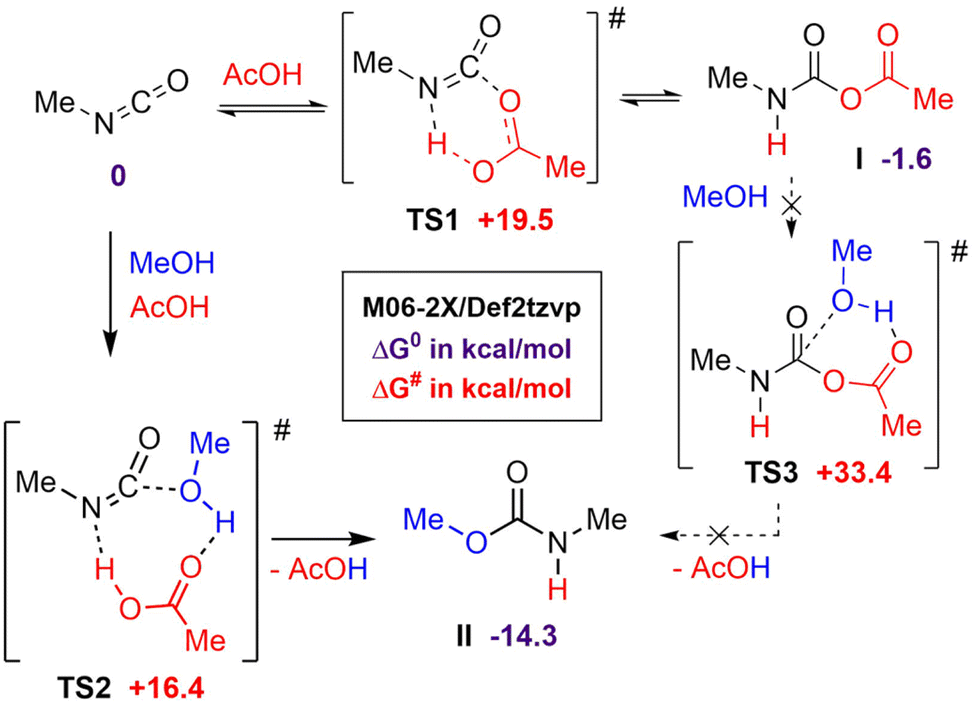 | ||
| Scheme 2 DFT calculations for a pathway toward urethane formation. | ||
The possible mechanisms leading to the formation of amide were also simulated (see ESI, Fig. S23†). Contrarily to what was suggested in some references of the literature,48–50 the spontaneous decarboxylation of NCA I is unlikely due to the very high energy of the transition state associated to this mechanism. Our calculations indicate that the reaction becomes much more favorable in the presence of a basic catalyst such as an amine. Catalytic amounts of amines can easily be obtained in our system via the hydrolysis of isocyanates with traces of water, or through the attack of an alcohol on NCA I (Fig. S23†).
Overall, the combination of model reactions and computational modeling provide a clear picture of the polymerization mechanism in bulk, and a rationalization of both the formation of by-products and the remarkable acceleration of the kinetics as compared to the conventional isocyanate-alcohol reaction.
Synthesis of thermosetting PU foams
By extending the methodology to the synthesis of thermosetting PU, we envisioned to valorize CO2 as an endogenous blowing agent to deliver PU foams. CO2 can be trapped within the growing crosslinked network when using monomers with 3 or more reactive functions, providing that the kinetics of the polymerization and that of the gas production are in the same time frame (Fig. 5a). Advantageously, this requirement should be attainable here because both kinetics are related to the same reaction, e.g. the decarboxylation of the oxamic acid into isocyanate.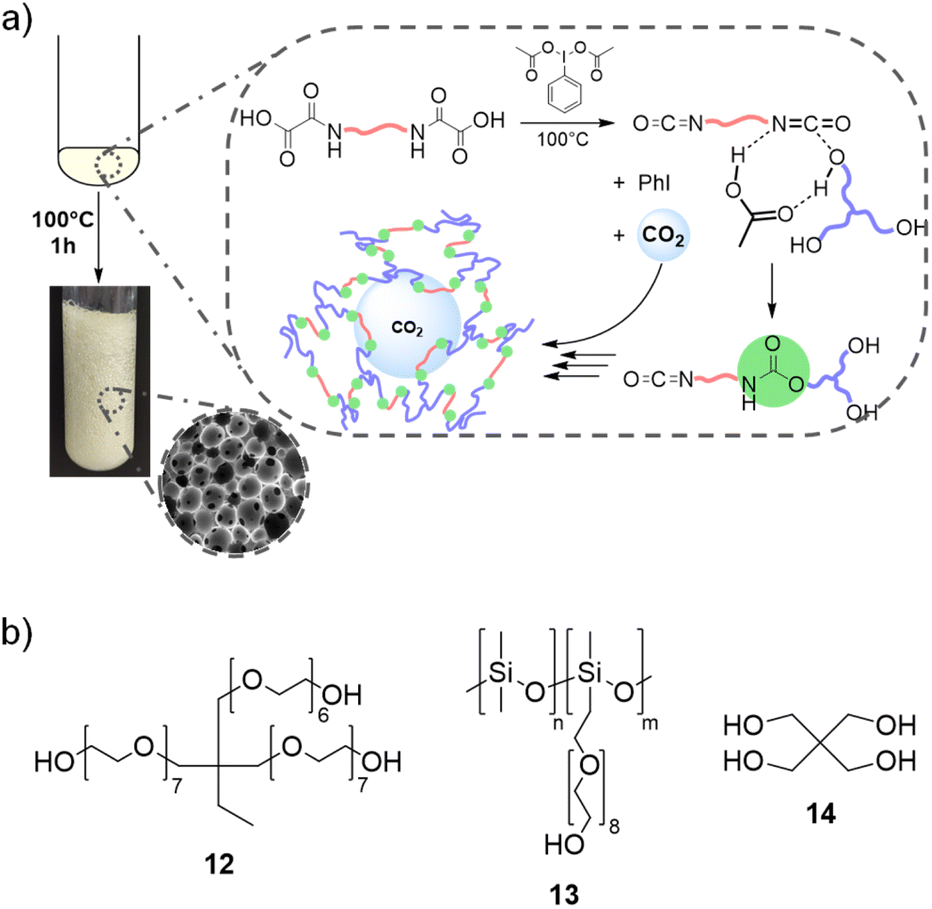 | ||
| Fig. 5 (a) PU foam synthesis strategy. (b) Components used in formulations. | ||
To exemplify this strategy, a tri-arm polyethylene glycol, trimethylolpropane ethoxylate (Mn = 1000 g mol−1, 12, 0.6 eq.), was used as a triol (Fig. 5b), in combination with a bis-oxamic acid (HBOA, 1a, 1 eq.) and PIDA (2.2 eq.) to produce a thermosetting polyurethane. The molar ratios are chosen so that [OA]:[OH]:[PIDA] = 1:0.9:1.1. The solid reagents, 1a and PIDA, were dispersed in the liquid triol to provide a white paste that was further introduced in an open reactor. The mixture was heated at 70 °C, the temperature used for the synthesis of thermoplastic PU and for the model reactions. Upon temperature increase and with a very slow magnetic stirring (∼50 rpm), it quickly homogenized in the form of a translucid liquid. The initiation of the decarboxylation was attested by the bubbling of CO2. However, none of the bubbles remained entrapped in the PU matrix and the final material was a gel swollen with excess of AcOH and iodobenzene (ESI, Fig. S24†).
Foaming was then attempted at higher temperature, e.g. 100 °C. Moreover, a surfactant was added to the formulation to stabilize the gas–polymer interfaces. We selected a polydimethylsiloxane grafted with polyethylene glycol arms (Fig. 5, PDMS-g-PEG, 13, 0.4 wt% vs. the total mass of 3.17 g), a commercial surfactant that is routinely used for the synthesis of conventional PU foams. Under these conditions, after a bubbling period of about 6 min, the rapid increase of the viscosity enables the rise of the foam. The rise time, trise, defined as the period between the beginning of the reaction and the stabilization of the height of the foam, is approximately of 480 s. Screenshots of the video of the foaming process are available in Fig. S25.† In parallel, the foaming reaction was monitored through FT-IR analyses. The vibration of the isocyanate bond (–NCO) is clearly visible around 2270 cm−1, next to the vibration band of CO2 (Fig. 6a, full scale spectra in Fig. S26†). The evolution of the relative intensity of the NCO band as a function of time (Fig. 6b) indicates that the full conversion of the isocyanate is observed after 60 min of reaction. Thus, the foam was post-cured at 100 °C for 1 hour and it was further dried in a vacuum oven at 60 °C for 4 hours to eliminate the remaining traces of AcOH and iodobenzene.
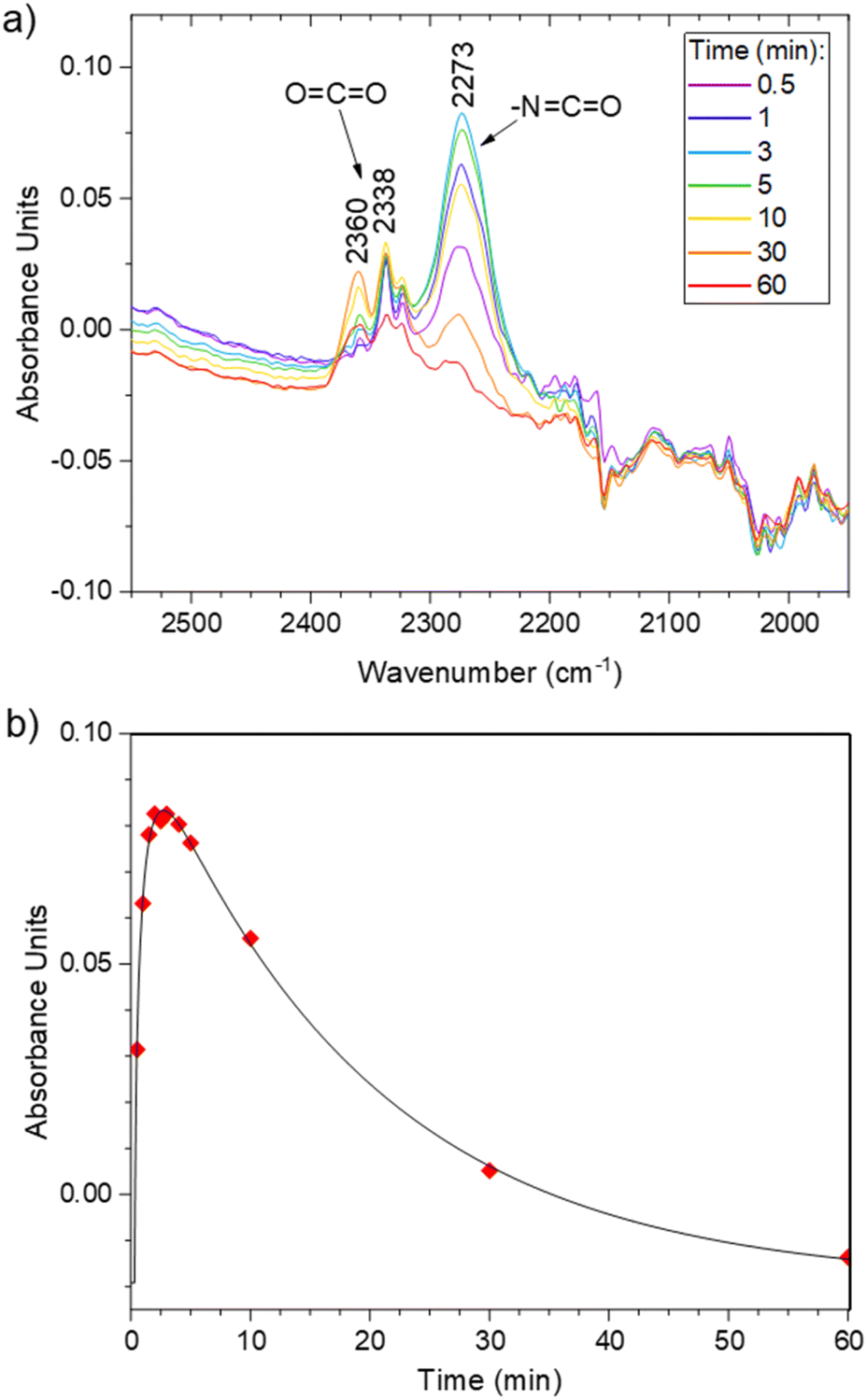 | ||
| Fig. 6 (a) FTIR kinetics of the F1 synthesis. (b) Evolution of the isocyanate peak during the reaction. | ||
The resulting foam, noted F1, is a well crosslinked material with a gel fraction of about 95% (measured in DCM). A picture of the foam is available in the ESI, Fig. S27.† It is made of a homogeneous open-cell structure as assessed by scanning electron microscopy (SEM, Fig. 7) with an average diameter of 0.62 ± 22 mm and a density of 228 kg m−3. Thermogravimetric analysis (TGA) indicates that the material is thermally stable with a 5 wt% loss at Td5% = 269 °C, thus confirming that AcOHand iodobenzene are fully eliminated from the material (ESI, Fig. S28†). From a thermo-mechanical standpoint, F1 is a flexible foam with a glass transition temperature (Tg) of −46.5 °C as measured by dynamic scanning calorimetry (DSC, ESI, Fig. S29†). The mechanical properties were evaluated by compression testing. Fig. 8a represents the compressive stress–strain diagram of F1. We performed a cyclic loading–unloading up to 70% strain (deformation rate of 100% per min). The hysteresis loop between the loading and the unloading cycle is consistent with the foam's ability to dissipate energy. Moreover, the residual strain after the unloading cycle is small, ∼3%, and indicates the good relaxation ability of the foam.
 | ||
| Fig. 7 SEM images of foams F1–8. | ||
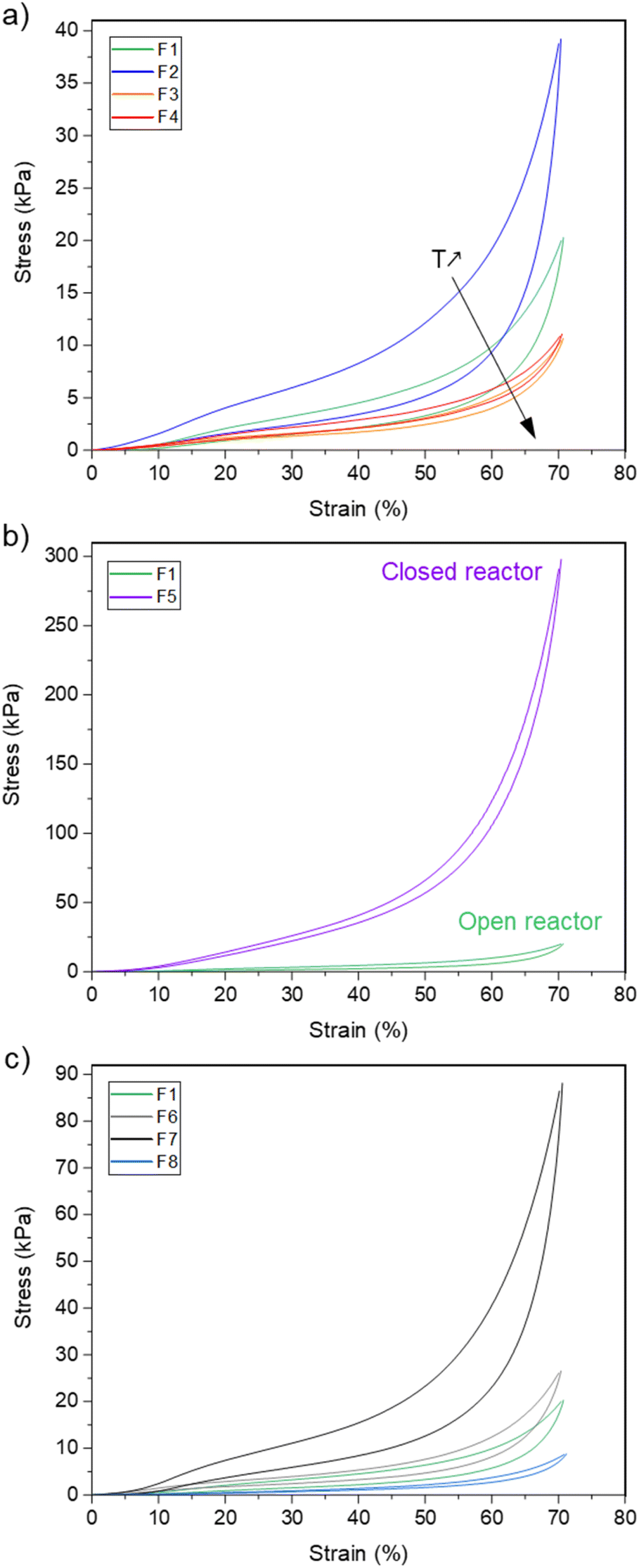 | ||
| Fig. 8 Compression testing of the foams showing (a) the impact of the foaming temperature, (b) open vs. closed reactor process, (c) the impact of the chemical composition. | ||
With this efficient system in hand, we further investigated the impact of the foaming temperature, T. Stable foams were obtained for T = 80 °C, 115 °C and 130 °C. They are noted F2, F3 and F4 respectively (pictures provided in the ESI, Fig. S27†). For all temperatures, FTIR (ESI, Fig. S30†) confirmed the full conversion of the isocyanates. The SEM images of the foams are represented in Fig. 7 and their characteristics are collected in Table 3. As expected, trise and the density decrease with T, while the cells size increases. The dispersity of the apparent cells size of F4 is probably indicative of the initiation of Ostwald ripening at higher foaming temperature.53Td5% and Tg are not impacted by the foaming temperature. The comparison of the stress–strain curves of the foams (Fig. 8a) reveals a significant increase of the compressive stress when T decreases. This is consistent with the concurrent increase of the density of the foams.
| Entry | Oxamic acid | Polyols | T (°C) | t rise (s) | Density (kg m−3) | Average cell size (mm ± SD) | T g (°C) | T d5% (°C) | |
|---|---|---|---|---|---|---|---|---|---|
| Polyol 1 (OH%) | Polyol 2 (OH%) | ||||||||
| a Closed reactor process. | |||||||||
| F1 | 1a | 12 | — | 100 | 480 | 228 | 0.62 ± 0.22 | −46.5 | 269 |
| F2 | 1a | 12 | — | 80 | 1080 | 272 | 0.26 ± 0.13 | −45.4 | 274 |
| F3 | 1a | 12 | — | 115 | 240 | 165 | 0.75 ± 0,25 | −45.4 | 292 |
| F4 | 1a | 12 | — | 130 | 180 | 156 | 0.75 ± 0.47 | −44.8 | 292 |
| F5 | 1a | 12 | — | 100 | 295 | 318 | 0.33 ± 0.10 | −44.9 | 288 |
| F6 | 1a | 12 (60) | 14 (40) | 100 | 360 | 214 | 0.37 ± 0.10 | −35.9 | 247 |
| F7 | 1a | 12 (20) | 14 (80) | 100 | 550 | 223 | 0.39 ± 0.09 | −14.8 | 209 |
| F8 | 1b | 12 | — | 100 | 720 | 213 | 0.85 ± 0.30 | −33.2 | 277 |
The same formulation was also foamed in conditions simulating a closed reactor. The tri-arm polyethylene glycol 12 (0.6 eq.), the bis-oxamic acid 1a (1 eq.), PIDA (2.2 eq.) and the surfactant (0.4 wt%) were charged in a vial hermetically closed with a septum cap. The mixture was heated at 100 °C, while maintaining a slow magnetic stirring (∼50 rpm). As the viscosity increased – attested by the reduction of the magnetic stirrer's speed – it was necessary to slowly depressurize the vial to trigger the foaming process. After a short rise time, trise ∼5 min, the vial was vacuum pumped for 5 min and the resulting foam, F5, was post-cured at 100 °C for 1 hour (picture provided in the ESI, Fig. S27†). F5 exhibits a homogeneous open-cell structure (SEM, Fig. 7) with an average cell size of 0.33 ± 0.10 mm and a density of 318 kg m−3. The thermal properties, Tg and T5wt%, appear unchanged as compared to the open reactor strategy (Fig. S28, S29† and Table 3). However, the mechanical behavior of the foam in compression testing is dramatically impacted. Fig. 8b compares the cyclic compression of F1 and F5. Clearly, the compressive stress of F5 is much higher than that of F1 during the whole cycle. This is consistent with the increase of the density and the reduction of the cell size in the case of F5.
Eventually, other polyol and oxamic acid precursors were considered for foam synthesis. The tetraol, pentaerythritol 14 (Fig. 5b), was used in combination with the triol 12. Two foams, noted F6 and F7, were synthesized using 12:14 functional ratios of 60:40 and 20:80, respectively. In both cases, the mixture of polyols was reacted at 100 °C with the bis-oxamic acid 1a and PIDA in the presence of the PDMS-g-PEG surfactant (0.4 wt%), while respecting the same functional ratio than that used for F1, i.e [OA]:[OH]:[PIDA] = 1:0.9:1.1. F6 and F7 are homogeneous foams with an open cell structure (pictures in the ESI, Fig. S27† and SEM images in Fig. 7). Their thermomechanical properties are reported in Table 3. The large increase of their compressive strength at 70% strain (Fig. 8c) as compared to F1 is consistent with their higher crosslinking density. An additional foam, F8, was successfully synthesized by replacing the bis-oxamic acid 1a with the isophorone bis-oxamic acid 1b (Table 2) in the same formulation as F1. F8 density is roughly the same than F1, but its cells are larger with an evident increase of the open to close cell ratio (Fig. 7 and Table 3). This correlate well with the decrease of the compressive stress during the loading–unloading cycle (Fig. 8c).
These experiments demonstrate that bis-oxamic acids are promising precursors of polyurethane foams. By playing with simple parameters such as the chemical nature of the backbone, the functionality of the reactants or the reaction procedure (open versus closed reactor), flexible foams with well-defined porous structures and a broad range of mechanical behavior are accessible. Moreover, it is worth mentioning that it is possible to obtain larger pieces of foam by using ∼20 g of the precursors (tri-arm PEG + HBOA). Pictures of the resulting foam are represented in the ESI, Fig. S31.†
Conclusions
In summary, we reported a new method for the production of polyurethane through the oxidative decarboxylation of polyoxamic acids in the presence of polyols and a hypervalent iodine reagent as an oxidant. Polyoxamic acids are prepared from benign polyamines and oxalyl acid chlorides in a high yield two-step process. They are used to generate polyisocyanates in situ, which react with polyols at moderate temperature (≤100 °C). Conveniently, the acetic acid released during the oxidation step is able to accelerate the polyaddition reaction, thus avoiding the recourse to conventional catalysts of the isocyanate-alcohol reaction such as toxic tin compounds. Moreover, the CO2 generated during the reaction may be exploited as an endogenous chemical blowing agent for the production of flexible polyurethane foams. Eventually, this strategy opens new avenues for the synthesis of commodity polyurethane materials, while circumventing the isolation of carcinogenic isocyanates. It introduces oxamic acids as promising latent precursors of isocyanates and will surely stimulate new research to improve the polymerization procedure, notably the atom economy of the oxidative step.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H. C. and Y. L. directed the project. H. C., T. V. and Y. L. wrote the project, and supervised the research. E. G. and F. R. co-supervised the research. G. G. P. and I. M. O. carried out the syntheses and the characterizations of the thermoplastic PUs. I. M. O. and Q. J. carried out the syntheses and the characterizations of the PUs foams. Q. J. carried out the model reactions. F. R. performed the DFT calculations. All co-authors contributed to the formal analyses of the results. Q. J. provided the first draft of the paper that was first corrected by H. C., T. V. and Y. L. All co-authors then corrected the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Agence Nationale de la Recherche (NCO-INNOV, No. 20-CE07-0015-01) is gratefully acknowledged for a PhD grant to Q. J. and for financial support. We are grateful to the University of Bordeaux (UBx) for financial support through the 2017 IdEx Bordeaux Post-Doctoral Program (G. G. P.). I. M. O. thanks the Alex-Ekwueme Federal University Ndufu-Alike Ikwo (AE-FUNAI) for PhD funding. We acknowledge (Cesamo, UBx) and the CNRS for financial help. Cedric Le Coz is acknowledged for his help related to the thermo-mechanical characterizations of the foams.Notes and references
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- PlasticsEurope, Plastics – the Facts 2021 Plastics Europe, https://plasticseurope.org/knowledge-hub/plastics-the-facts-2021/, accessed 25 September 2023.
- PrecedenceResearch, Polyurethane Market Size, Trends, Share, Growth, Report 2032, https://www.precedenceresearch.com/polyurethane-market, accessed 25 September 2023.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- O. Bayer, Angew. Chem., 1947, 59, 257–272 CrossRef.
- S. Ozaki, Chem. Rev., 1972, 72, 457–496 CrossRef.
- F. Monie, T. Vidil, B. Grignard, H. Cramail and C. Detrembleur, Mater. Sci. Eng., 2021, 145, 100628 CrossRef.
- W. J. Seo, H. C. Jung, J. C. Hyun, W. N. Kim, Y.-B. Lee, K. H. Choe and S.-B. Kim, J. Appl. Polym. Sci., 2003, 90, 12–21 CrossRef CAS.
- A. Serrano-Jiménez, C. Díaz-López, K. Verichev and Á. Barrios-Padura, Energy Build., 2023, 278, 112595 CrossRef.
- V. Masatin, A. Volkova, A. Hlebnikov and E. Latosov, Energy Procedia, 2017, 113, 265–269 CrossRef.
- The European Commission, Commission Regulation (EU) 2020/1149 of 3 August 2020 amending Annex XVII to Regulation (EC) No 1907/2006 of the European Parliament and of the Council concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) as regards diisocyanates, http://data.europa.eu/eli/reg/2020/1149/oj, accessed 18 September 2023.
- C. Six and F. Richter, in Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, Ltd, 2003 Search PubMed.
- D. Bello, S. R. Woskie, R. P. Streicher, Y. Liu, M. H. Stowe, E. A. Eisen, M. J. Ellenbecker, J. Sparer, F. Youngs, M. R. Cullen and C. A. Redlich, Am. J. Ind. Med., 2004, 46, 480–491 CrossRef CAS PubMed.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Environ. Health Perspect., 2007, 115, 328–335 CrossRef CAS PubMed.
- J. Borak and W. F. Diller, J. Occup. Environ. Med., 2001, 43, 110–119 CrossRef CAS PubMed.
- W. F. Diller, Toxicol. Ind. Health, 1985, 1, 7–15 CrossRef CAS PubMed.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- C. Carré, Y. Ecochard, S. Caillol and L. Avérous, ChemSusChem, 2019, 12, 3410–3430 CrossRef.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- F. Monie, B. Grignard and C. Detrembleur, ACS Macro Lett., 2022, 236–242 CrossRef CAS.
- F. Monie, B. Grignard, J.-M. Thomassin, R. Mereau, T. Tassaing, C. Jerome and C. Detrembleur, Angew. Chem., 2020, 132, 17181–17189 CrossRef.
- M. Bourguignon, B. Grignard and C. Detrembleur, Angew. Chem., Int. Ed., 2022, 61, e202213422 CrossRef CAS PubMed.
- G. Coste, C. Negrell and S. Caillol, Macromol. Rapid Commun., 2022, 43, 2100833 CrossRef CAS PubMed.
- M. S. Rolph, A. L. J. Markowska, C. N. Warriner and R. K. O'Reilly, Polym. Chem., 2016, 7, 7351–7364 RSC.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 2001, 41, 1–83 CrossRef CAS.
- S. Kalaimani and A. S. Nasar, Eur. Polym. J., 2017, 91, 221–231 CrossRef CAS.
- S. J. Phillips, N. C. Ginder and B. J. Lear, Macromolecules, 2022, 55, 7232–7239 CrossRef CAS.
- S. Jana, D. Samanta, M. M. Fahad, S. N. Jaisankar and H. Kim, Polymers, 2021, 13, 2875 CrossRef CAS PubMed.
- O. Kreye, H. Mutlu and M. A. R. Meier, Green Chem., 2013, 15, 1431–1455 RSC.
- A. K. Ghosh, A. Sarkar and M. Brindisi, Org. Biomol. Chem., 2018, 16, 2006–2027 RSC.
- J. Lee, S. Baek, H. H. Moon, S. U. Son and C. Song, ACS Appl. Polym. Mater., 2021, 3, 5767–5777 CrossRef CAS.
- D. V. Palaskar, A. Boyer, E. Cloutet, C. Alfos and H. Cramail, Biomacromolecules, 2010, 11, 1202–1211 CrossRef CAS PubMed.
- A. S. More, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2012, 3, 1594–1605 RSC.
- A. S. More, L. Maisonneuve, T. Lebarbé, B. Gadenne, C. Alfos and H. Cramail, Eur. J. Lipid Sci. Technol., 2013, 115, 61–75 CrossRef CAS.
- S. S. Kuhire, C. V. Avadhani and P. P. Wadgaonkar, Eur. Polym. J., 2015, 71, 547–557 CrossRef CAS.
- C. N. D. Neumann, W. D. Bulach, M. Rehahn and R. Klein, Macromol. Rapid Commun., 2011, 32, 1373–1378 CrossRef CAS PubMed.
- J.-P. Hagenbuch, Chimia, 2003, 57, 773 CrossRef CAS.
- M. M. Sebeika and G. B. Jones, Curr. Org. Synth., 2014, 11, 732–750 CrossRef CAS.
- F. Minisci, F. Fontana, F. Coppa and Y. M. Yan, J. Org. Chem., 1995, 60, 5430–5433 CrossRef CAS.
- I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 7593–7607 RSC.
- J. Cordero-Martínez, C. Aguirre-Alvarado, C. Wong and L. Rodríguez-Páez, Syst. Biol. Reprod. Med., 2014, 60, 189–198 CrossRef.
- S. Choi, A. B. Beeler, A. Pradhan, E. B. Watkins, J. M. Rimoldi, B. Tekwani and M. A. Avery, J. Comb. Chem., 2007, 9, 292–300 CrossRef CAS PubMed.
- I. M. Ogbu, G. Kurtay, M. Badufle, F. Robert, C. S. Lopez and Y. Landais, Chem.–Eur. J., 2023, 29, e202202963 CrossRef CAS PubMed.
- G. G. Pawar, F. Robert, E. Grau, H. Cramail and Y. Landais, Chem. Commun., 2018, 54, 9337–9340 RSC.
- I. M. Ogbu, D. M. Bassani, F. Robert and Y. Landais, Chem. Commun., 2022, 58, 8802–8805 RSC.
- W. R. Sorenson, J. Org. Chem., 1959, 24, 978–980 CrossRef CAS.
- P. Babusiaux, R. Longeray and J. Dreux, Justus Liebigs Ann. Chem., 1976, 1976, 487–495 CrossRef.
- K. Sasaki and D. Crich, Org. Lett., 2011, 13, 2256–2259 CrossRef CAS PubMed.
- The urethane yield of M3 (77%), i.e. the percentage of oxamic acid converted into urethane, differs from the molar percentage of urethane in the final mixture of product, 82 mol%, because one molecule of oxalamide is obtained from two molecules of oxamic acid.
- H. Sardon, A. C. Engler, J. M. W. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice, H. W. Horn and J. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS PubMed.
- J. Peyrton and L. Avérous, Mater. Sci. Eng., 2021, 145, 100608 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and synthesis methods, additional 1H NMR and FTIR spectra, SEC traces, TGA and DSC thermograms, and DFT calculations. See DOI: https://doi.org/10.1039/d4sc02562h |
| This journal is © The Royal Society of Chemistry 2024 |