
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn imidazole-based covalent-organic framework enabling a super-efficiency in sunlight-driven uranium extraction from seawater†
Lizhen
Zhong
a,
Xuefeng
Feng
*a,
Qingyun
Zhang
a,
Xianqing
Xie
b and
Feng
Luo
 *a
*a
aSchool of Chemistry and Materials Science, East China University of Technology, Nanchang 330013, China. E-mail: xffeng@ecut.edu.cn; ecitluofeng@163.com
bNational Engineering Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang, 330027, China
First published on 7th June 2024
Abstract
Uranium extraction from seawater represents an effective way to solve the difficulty of the insufficient uranium supply chain. However, this route is still restricted by the low extraction efficiency of reported adsorbents. Here, we find that reversing the donor–acceptor in imidazole-based COFs (covalent-organic frameworks) would be effective for enhancing the extraction efficiency of uranium. As a result, the TI-COF is found to enable a uranium extraction efficiency up to 8.8 mg g−1 day−1 from seawater under visible light irradiation, exceeding all established adsorbents for such use, and an unprecedented uranium extraction efficiency up to 6.9 mg g−1 day−1 from seawater under natural sunlight.
1. Introduction
Nuclear energy has become an effective way to solve the energy and dual carbon problems due to its high energy density and low pollution.1,2 However, the scarcity of uranium mines still seriously restricts the sustainable development of nuclear energy.3,4 As we know, the uranium reserves on land are estimated to be 1.45 million tons, which can only supply for nuclear energy up to 80 years, according to the current development rate.5–7 By contrast, seawater contains approximately 4.5 billion tons of uranium, which is thousands of times of the uranium reserves on land. Accordingly, generation of uranium from seawater should be the best way to solve the problem of insufficient reserves of uranium. But, the trace uranium concentration (∼3.3 ppb) along with the presence of a large number of other competing ions in a higher abundance in seawater will inherently prevent us from separating uranium from seawater.8–13 Thereby, there emerges an urgent and long-term demand in designing new materials and methods for acquiring uranium from seawater.Previous findings have revealed the advantage of adsorption-based separation strategies for such use. And several effective adsorbents have been developed including, but not limited to, porous polymers, metal–organic frameworks (MOFs), covalent-organic frameworks (COFs), and hydrogen-bonded organic frameworks (HOFs).14–29 It was also found that anchoring special strong adsorption sites to fix uranyl ions on the porous scaffold can largely boost its uranium acquisition capability from seawater (Scheme 1a). In this content, Kushwaha et al. achieved a remarkable value up to 17.9 mg g−1 by means of a HOF adsorbent after 30 days,29a while a benchmark value up of 28.2 mg g−1 after 25 days was reported by Ghosh et al. through using an ionic MOF.29b It is worth emphasizing that although the adsorption method can obtain a high uranium adsorption capacity from seawater, it generally requires a particularly long working time (often more than 30 days, Fig. 1a), which will largely increase the overall cost of extracting uranium from seawater and consequently restrict its practical application in uranium manufacture from seawater. Moreover, current adsorption methods also face some challenges such as the recycling of adsorbents and the precise recognition of uranium.
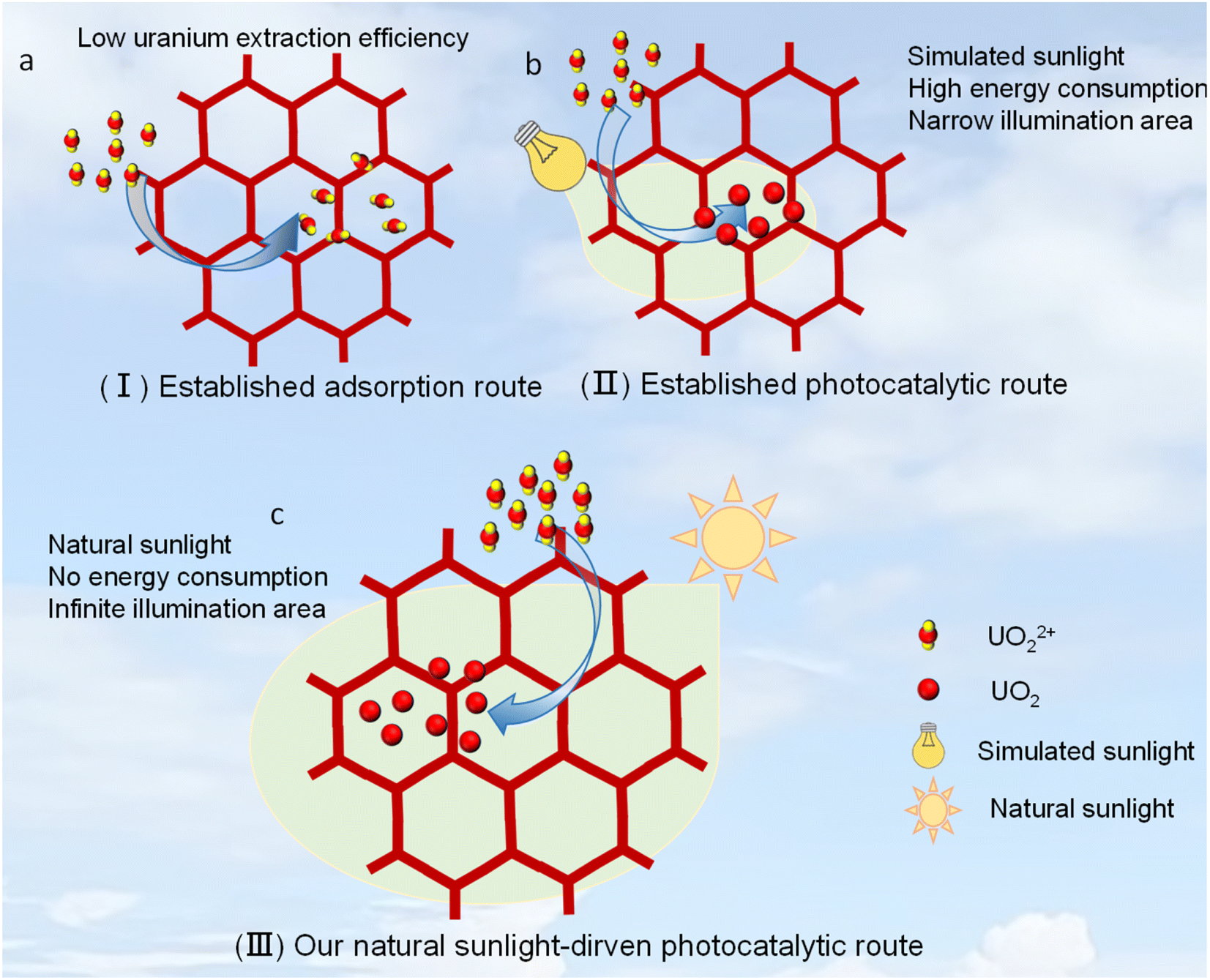 | ||
| Scheme 1 (a) View of the established adsorbent route for uranium extraction from seawater. (b) View of the established photocatalytic route for uranium extraction from seawater under simulated sunlight. (c) View of our photocatalytic route for uranium extraction from seawater under natural sunlight. | ||
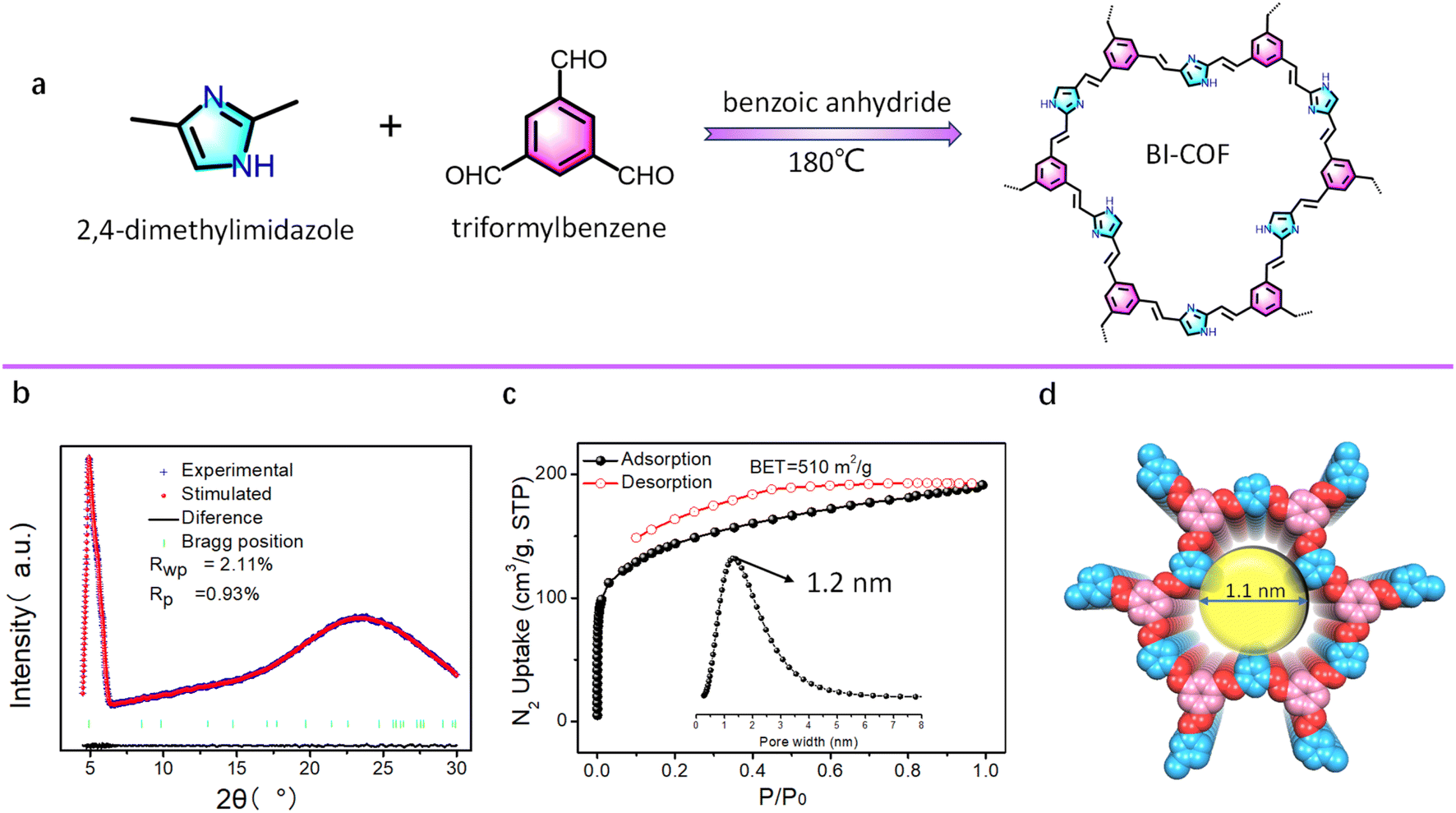 | ||
| Fig. 1 (a) The synthesis route of the BI-COF based on benzene-1,3,5-tricarboxaldehyde and 2,4-dimethylimidazole building blocks. (b) Experimental PXRD patterns of the BI-COF with corresponding Pawley refinement (red) and Bragg positions (green), showing good fit to the experimental data (blue) with minimal differences (black). (c) N2 adsorption isotherm at 77 K with the inset of pore size distribution. (d) View of the structure of the BI-COF in the eclipsed (AA) stacking mode. | ||
Alternative to this adsorption method is the recently developed solar-driven uranium extraction method, which involves a conversion from solar energy to chemical energy and is viewed to be a green and powerful avenue.30–35 Such technology can theoretically solve all the problems faced by the adsorption method like that of long working time, unstable reusability and strong competing adsorption. For example, COF-4P through a photocatalytic route reported by Ma et al. exhibited a uranium extraction efficiency of 8.02 mg g−1 day−1,35 which is almost 7.3-fold that of the above-mentioned benchmark adsorbent reported by Ghosh et al (on average 1.1 mg g−1 day−1).29b In this context, COF photocatalysts are especially interesting,36,37 due to their outstanding advantages in porosity, diversity and conjugated structure. However, only a few photocatalysts have been explored for such use, and this field is still in its infancy. Thereby, it is highly desirable to design and prepare new photocatalysts with not only high chemical stability but also superior uranium extraction efficiency. Moreover, there remains a major issue that all established photocatalysts were just evaluated under simulated sunlight (usually using a high-intensity Xe lamp) rather than natural sunlight (Scheme 1b).30–35 In general, such simulated sunlight from a high-intensity Xe lamp often outputs a continuous high-intense visible light and a very small irradiation area (Scheme 1b). This is far from the energy-free sunlight driven mode we actually need, since natural sunlight has a low intensity and large irradiation area, as well as its intensity undergoes drastic changes with seasons and daily time periods. In this regard, developing real sunlight-driven photocatalysts for such use is the most desirable one (Scheme 1c), and in this work, we report a highly rare imidazole-based COF, which can exhibit an exciting uranium extraction efficiency up to 6.9 mg g−1 day−1 from seawater under natural sunlight.
2. Experimental section
2.1 Materials and methods
Uranium is radioactive, and thus we must perform experiments under special protection. Monomers of 2,4-dimethylimidazole (99%), benzene-1,3,5-tricarboxaldehyde (99%), and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (99%), and UO2(NO3)2·6H2O (99%), benzoic anhydride (99%) and organic solvents (99%) were purchased from Aladdin Biochemical Technology Co., Ltd. These were used as received without further purification. X-ray powder diffraction was performed using a Bruker AXSD8 Discover powder diffractometer at 40 kV, 40 mA for Cu Kλ (λ = 1.5406 Å). The simulated powder patterns were calculated using Mercury 1.4. Infrared Spectra (IR) were measured using a Bruker VERTEX70 spectrometer in the 800–3600 cm−1 region. The gas adsorption isotherms were collected on a Belsorp-max. Ultrahigh-purity-grade (>99.999%) N2 gases were used during the adsorption measurement. The analyses of concentrations of U ions in the solution were carried out using ThermoFisher iCap7600 ICP-OES or iCap RQplus ICP-MS instruments. X-ray photoelectron spectra (XPS) were collected using a Thermo Scientific ESCALAB 250 Xi spectrometer. Scanning electron microscopy (SEM) images were recorded on a Hitachi SU 8100 Scanning Electron Microscope. Solid-state NMR experiments were performed on a Varian Infinityplus 300 solid-state NMR spectrometer (300 MHz). Scanning electron microscopy (SEM) images were recorded on a Hitachi SU 8100 Scanning Electron Microscope. Photoelectrochemical measurements were performed on a CHI760 workstation using a three-electrode configuration. UV-vis spectroscopy results were recorded in diffuse reflectance (DR) mode at room temperature on a SHIMADZU UV-2700 spectrophotometer equipped with an integrating sphere attachment.2.2 Synthesis of BI-COF
A mixture of 2,4-dimethylimidazole (600 mg, 6 mmol), benzene-1,3,5-tricarboxaldehyde (724 mg, 4 mmol) and benzoic anhydride (2 g, 17.6 mmol) was added into a PTFE-lined reactor. The reactor was then transferred to a muffle furnace and heated at 180 °C for 5 days. The resulting reactants were collected, washed sequentially with DMF, water, methanol, and then dried under vacuum at 60 °C for 12 hours, giving the BI-COF in a yield of 80% (1.0 g).2.3 Synthesis of the TI-COF
A mixture of 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (800 mg, 2 mmol), 2,4-dimethylimidazole (300 mg, 3 mmol) and benzoic anhydride (5 g, 22 mmol) was charged into a PTFE-lined reactor. The reactor was then transferred to a muffle furnace and heated at 180 °C for 5 days. The resulting reactants were collected, washed sequentially with DMF, water, methanol, and then dried under vacuum at 60 °C for 12 hours. The TI-COF was obtained as a reddish-brown powder in a yield (1.0 g) of approximately 95%.2.4 Synthesis of the model compound
A mixture of 2,4-dimethylimidazole (30.0 mg, 0.3 mmol), benzaldehyde (21.2 mg, 0.2 mmol) and benzoic anhydride (500 mg, 2.2 mmol) was charged into a PTFE-lined reactor. The reactor was then transferred to a muffle furnace and heated at 180 °C for 5 days. The model compound was extracted with dichloromethane and used for MS analysis.2.5 Photocatalytic reduction of uranium
For visible light irradiation (simulated mode), a 300 W Xe lamp (Beijing NBET Technology Co., Ltd, SHX-F300) was used. The radiant output is 50 W, including UV output (6.6 W), IR output (26.8 W), and visible output (>390 nm). The window diameter is about 25.4 mm. The working spot diameter is about 32 mm. The distance between the window diameter and the samples is about 20 cm. To exclude the impact of heat generated by lighting, all uranium removal experiments were conducted in a quartz flask photoreactor under a constant temperature circulating water system. The illumination intensity of light on the sample surface was revealed by a xenon lamp irradiance meter of TS560 (Shenzhen Suderei Technology Co., Ltd), giving ∼5.8 W cm−2. No sacrificial agent was added in the photocatalytic reaction.A stock uranium solution of 1000 ppm by dissolving an appropriate amount of UO2 (NO3)2·6H2O in deionized water. pH = 5 was adjusted using 1 M HNO3 aqueous solution.
In kinetics experiments, U solutions with initial concentrations of 100, 10 and 1 ppm were used. The dose of adsorbent is 5 mg, while the U solution is 50 mL.
The adsorption amount, Qe (mg g−1), was calculated by the difference of U equilibrium concentration before and after adsorption (see eqn (1)):
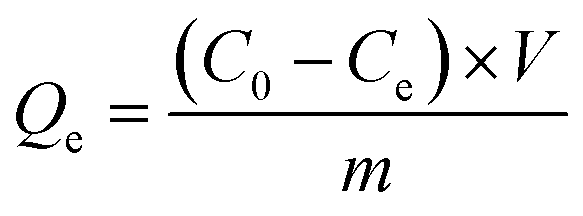 | (1) |
In the selective adsorption experiments, a 14-ion (Na+, K+, Cs+, Mg2+, Sr2+, Zn2+, Ni2+, Co2+, Cu2+, Cd2+, Cr3+, Th4+, VO3−, UO22+) mixed solution contains both U and other 13 metal ions with respective initial concentrations of 50 ppm. The dose of adsorbent is 5 mg, while the solution is 50 mL. Contacting time is 40 min.
2.6 Extraction of uranium from seawater
Seawater from Zhuhai, China, was used. The U concentration was 3.3 ppb. The dose of adsorbent was 5 mg, while the volume of natural seawater was 30 L. Contacting time was one day.For real sunlight irradiation, the experiment was carried out from December 23, 2023 to December 25, 2023 in Nanchang city in China. The weather was sunny. All weather temperature was 4–13 °C. The time period was 9 am to 5 pm, and the uranium extraction process under sunlight irradiation was performed for three days, which was equal to sunlight irradiation for about 24 h, since the TI-COF has little adsorption ability towards uranium.
2.7 DFT calculations
The first-principles calculations were performed within the framework of density functional theory (DFT) as implemented in the plane wave set Vienna Ab-initio Simulation Package (VASP) code, in which the Perdew–Burke–Ernzerhof (GGA-PBE) functionalization and the project-augmented wave generalized gradient approximation pseudopotentials (PAW-GGA) were employed to calculate the exchange-correlation energy and electron–ion interaction, respectively. Additionally, spin-polarization was considered in all calculations. Wave functions were expanded using a plane-wave basis set with a kinetic energy cutoff of 500 eV and the geometries were fully relaxed until the residual force convergence value on each atom was less than 0.02 eV Å−1. A gamma k-point mesh of 3 × 3 × 1 was used for Brillouin zone sampling for structural optimization and a gamma k-point mesh of 5 × 5 × 1 was used for Brillouin zone sampling for charge-related calculations.3. Results and discussion
3.1 COF design and synthesis
In general, the rational construction of a strong donor–acceptor system is the essential rule for accessing a good photocatalyst.30–35 In this regard, reversing the donor–acceptor in a material could be a wise choice to modulate the donor–acceptor system and then the photocatalytic performance, since such a reversing donor–acceptor approach would cause a big difference in the direction of electron transfer. In this regard, a new monomer of 2,4-dimethylimidazole was selected, since it holds a variable property as the electron donor or electron acceptor that is completely dependent on which type of monomer is connected to it, thus providing us a good platform to explore the effect from the reversing donor–acceptor approach on the photocatalytic performance. To ensure the tolerance and reusability of the adsorbent during the extraction of uranium from seawater, we herein adopt a synthesis of the inherently stable olefin-linked COF through the irreversible Knoevenagel condensation. This is different from the established COF photocatalyst, which was mainly constructed by the reversible imine linkage.25 The use of the 2,4-dimethylimidazole monomer to perform the irreversible Knoevenagel condensation is due to that the five-membered imidazole ring in 2,4-dimethylimidazole is comparable with the commonly used monomers involved in the six-membered pyridine ring for driving the irreversible Knoevenagel condensation.24,25 Moreover, before carrying out the synthesis of these 2,4-dimethylimidazole-derived COFs, we first performed a model reaction between 2,4-dimethylimidazole and benzaldehyde. And the model compound, 2,4-di((E)-styryl)-1H-imidazole was successfully produced, as shown in Fig. S1.†3.2 Characterization and structure of BI-COF
The new COF of the BI-COF was synthesized from 2,4-dimethylimidazole and benzene-1,3,5-tricarboxaldehyde through a [3 + 2] Knoevenagel condensation with benzoic anhydride as the melt solvent24 at a reaction temperature of 180 °C (Fig. 1a). To confirm the success in the synthesis of the BI-COF, we performed a series of characterization studies including infrared spectroscopy (IR), solid-state cross-polarization magic angle spinning (CP-MAS) 13C NMR, powder X-ray diffraction (PXRD), Brunauer–Emmett–Teller (BET), and scanning electron microscopy (SEM). The strong IR band at 1605 cm−1 suggests the formation of an olefin linkage in the BI-COF (Fig. S2†).24 In CP-MAS 13C NMR, two major peaks at 129 ppm and 135 ppm are observed, consistent with the COF skeleton (Fig. S3†). The PXRD test shows a sharp peak at 2θ = 4.8° (Fig. 1b), confirming its high crystallinity. Permanent porosity is confirmed by N2 adsorption at 77 K (Fig. 1c), giving a high BET specific surface area of 510 m2 g−1 and narrow pore size of 1.1 nm.The structure of the BI-COF was determined by a commonly used method through the first PXRD test and then corresponding Pawley refinement.24,25 As shown in Fig. 1a, we initially outline the model structure for such [3 + 2] assembly. After Pawley refinement, the resultant structure owns the unit cell parameters of a hexagonal system, P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) space group, a = b = 20.80 Å, c = 3.44 Å (Table S1†), while the simulated PXRD value is in good agreement with the experimental value (Fig. 1b), as evidenced by the small residual factor of Rp = 0.93% and Rwp = 2.11%. The 2D layered structure in the eclipsed (AA) stacking mode is shown in Fig. 2d, where such [3 + 2] assembly renders an uncommon hcb-derived topology.38 Along the c axis, we observed rare low-symmetry dodecagonal micropores (Fig. 1d) in this BI-COF, and its pore size is consistent with the experimental results.
space group, a = b = 20.80 Å, c = 3.44 Å (Table S1†), while the simulated PXRD value is in good agreement with the experimental value (Fig. 1b), as evidenced by the small residual factor of Rp = 0.93% and Rwp = 2.11%. The 2D layered structure in the eclipsed (AA) stacking mode is shown in Fig. 2d, where such [3 + 2] assembly renders an uncommon hcb-derived topology.38 Along the c axis, we observed rare low-symmetry dodecagonal micropores (Fig. 1d) in this BI-COF, and its pore size is consistent with the experimental results.
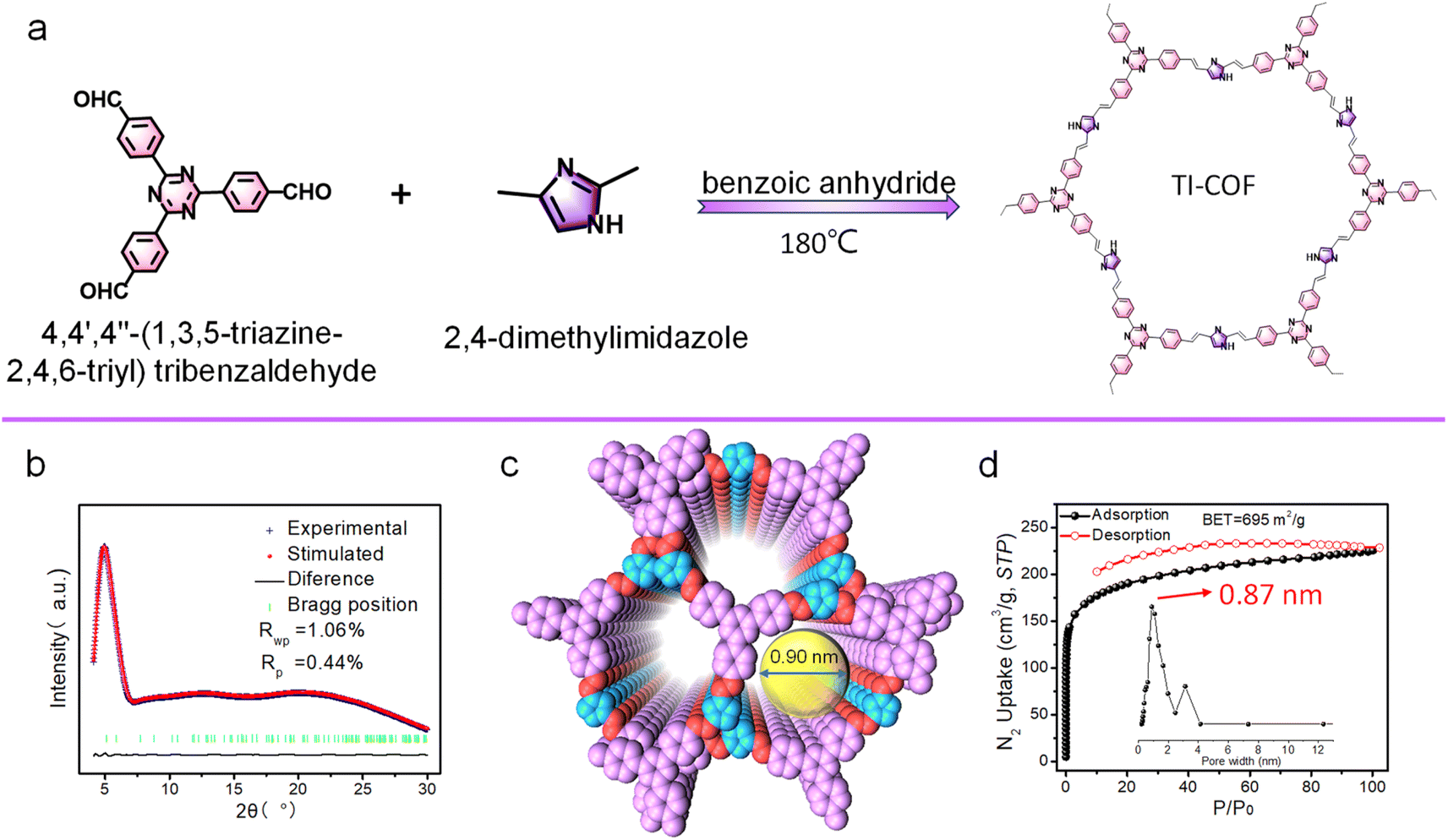 | ||
| Fig. 2 (a) Synthesis route of the TI-COF. (b) Experimental PXRD pattern and Pawley refinement. (c) View of the staggered (AB) stacking structure with a 1D channel. (d) N2 adsorption–desorption isotherms at 77 K with the inset of pore size distribution. | ||
3.3 Characterization and structure of the TI-COF
The synthesis of the TI-COF is similar to the synthesis of the BI-COF, except for the replacement of benzene-1,3,5-tricarboxaldehyde by 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde (Fig. 2a). Similarly, the peak at 1603 cm−1 in IR confirms the formation of the olefin linkage (Fig. S4†).24 All the carbon atoms in the TI-COF can be read out from CP-MAS 13C NMR spectra (Fig. S5†). A simulation method by the combination of PXRD plus Pawley refinement is also used to determine the structure of the TI-COF (Fig. 2b). The optimized solution gives a hexagonal crystalline system with the P space group, showing the unit cell parameter, a = b = 34.0869 Å, c = 6.6018 Å, Rp = 0.44% and Rwp = 1.06% (Table S2†), but a staggered (AB) stacking model is suggested. Such staggered (AB) stacking reduces the pores down to 0.9 nm (Fig. 2c), which is in good agreement with the experimental value from aperture analysis (0.87 nm). Moreover, a high Brunauer–Emmett–Teller (BET) surface area of 695 m2 g−1 is observed (Fig. 2d).
3.4 Uranium extraction experiments
We initially explored the uranium extraction performance of the BI-COF and TI-COF from a 50 mL 100 ppm UO22+ solution under visible light irradiation (simulated sunlight) without addition of any sacrificial agent. After photocatalysis of 40 min, the BI-COF afforded a 56.2% removal rate with an extraction capacity of 562 mg g−1, while the TI-COF gave a largely enhanced 80% removal rate with extraction capacity up to 902 mg g−1 (Fig. 3a). This confirms a positive effect on the uranium extraction from our reversing approach. The apparent quantum efficiency (400 nm) is about 3.4% for the BI-COF and 5.9% for the TI-COF.39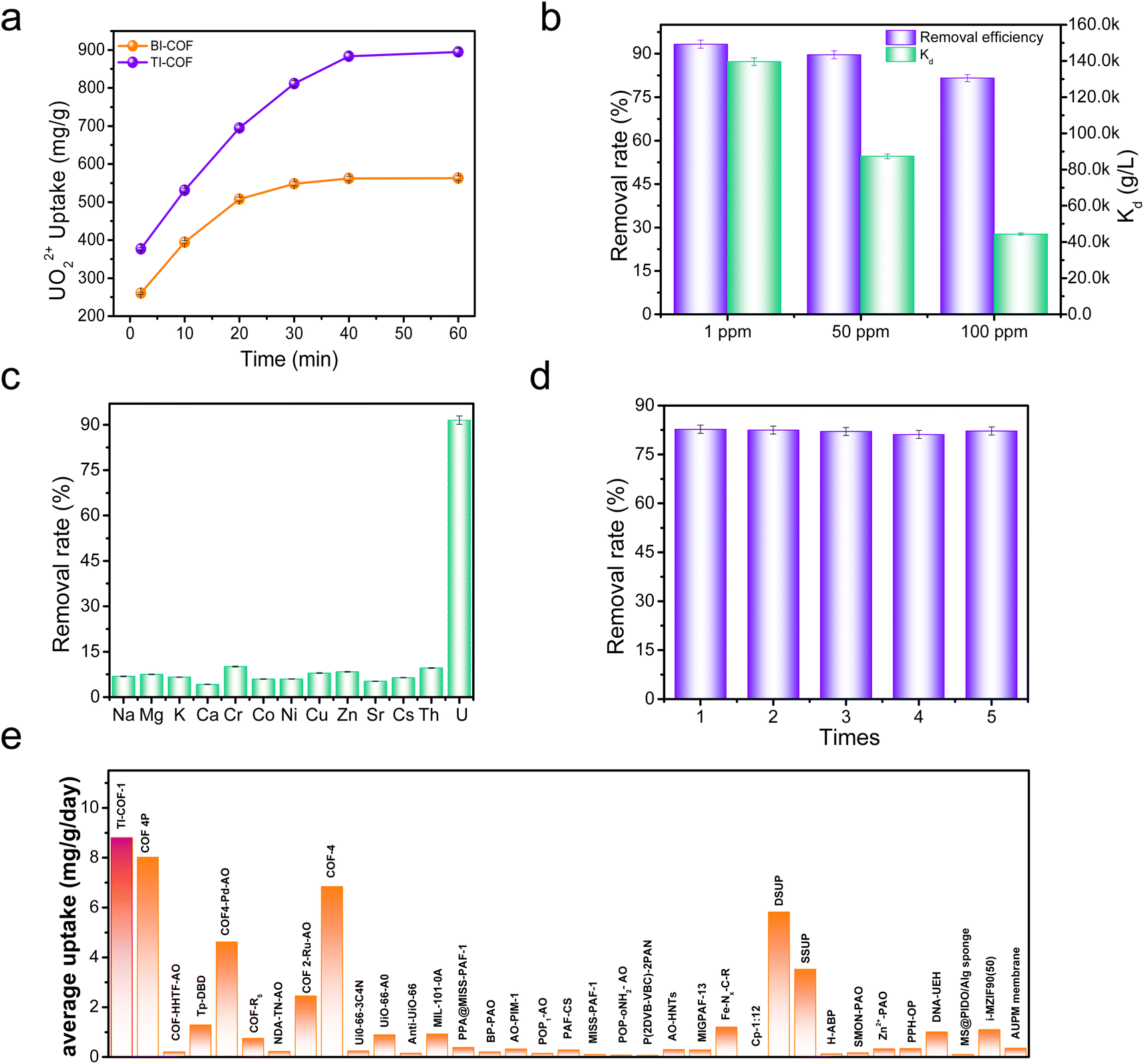 | ||
| Fig. 3 (a) Uranium extraction kinetics upon the BI-COF and TI-COF for a 50 mL 100 ppm U solution. (b) A comparison of the removal rate and Kd value for the TI-COF in 100, 50, and 1 ppm U solutions. (c) Selective extraction towards U over other ions. (d) Recycle of TI-COF. (e) A comparison of uranium extraction efficiency among reported adsorbents and our case. The error bars are based on the average value of three times. | ||
Note that such high extraction capacity and fast extraction kinetics observed in the TI-COF exceeds most reported adsorbents or photocatalysts for such use (Tables S3 and S4†). The outstanding performance in the TI-COF prompted us to make a further evaluation under low concentration such as 50 ppm and 1 ppm. It was found that the TI-COF afforded an impressive 90% and 94% removal rate, respectively (Fig. 3b), suggesting its potential for extraction of uranium from seawater. Based on these data, we can also deduce a preliminary judgment on the affinity between the COF and uranium through comparing their distribution coefficients (Kd). For the TI-COF, it affords a Kd value of 4.4 × 104 mL g−1 for a 100 ppm uranium solution (Fig. 3b), which is about 3.7-fold that of the BI-COF (1.2 × 104 mL g−1), which means higher affinity from the TI-COF skeleton for uranium; this is consistent with our molecule design. Meanwhile, the Kd value from 50 and 1 ppm uranium solutions is as high as 1.4 × 105 and 8.7 × 104 mL g−1 (Fig. 3b), respectively, further confirming the high affinity of the TI-COF towards uranium. It is worth emphasizing that such a high distribution coefficient is obtained in just 40 min, suggesting an ultralow time cost for the present TI-COF, which is better than most reported adsorbents and photocatalysts for such use and very beneficial for improving the uranium extraction efficiency from seawater.
Next, we investigated the selective extraction upon the TI-COF from a 14-ion mixed solution, containing monovalent Na+, K+, and Cs+ ions, divalent Mg2+, Ca2+, Zn2+, Co2+, Ni2+, Cu2+, and Sr2+, trivalent Cr3+, tetravalent Th4+, and VO3− ions (Fig. 3c), which is vital for the uranium extraction from seawater. Interestingly, we observed 91.5% removal rate for uranium and less than 10% removal rate for the other ions, implying selective adsorption of UO22+ over the other ions. The selectivity of SU/M is calculated to be as high as more than 100 for most other ions (Fig. S6†). Moreover, it was found that the loaded uranium on the TI-COF can be 100% desorbed through using 1 M Na2CO3 solution, and repeating such an extraction–desorption recycle for five times did not cause clear decrease in the uranium removal rate (Fig. 3d), indicative of the good reusability of our material.
3.5 Uranium extraction from seawater
The above results further prompted us to apply our TI-COF for uranium extraction from seawater and a 5 mg TI-COF sample with 30 L natural seawater (3.3 ppb uranium) was used for such research. For BI-COF, it affords a low uranium extraction capacity of 2.3 mg g−1 under simulated sunlight after one day contacting time, whereas the TI-COF could enable an ultrahigh uranium extraction capacity up to 8.8 mg g−1. Such a value for the TI-COF exceeds those of most reported adsorbents and photocatalysts for such use (Fig. 3e and Table S5†). More importantly, if taking time cost into account, the uranium extraction efficiency for the BI-COF and TI-COF is 2.3 and 8.8 mg g−1 day−1, respectively. It is clear that the uranium extraction efficiency of the TI-COF is far higher than that of the BI-COF, confirming the positive effect from our reversing approach on uranium extraction from seawater. More impressively, such high uranium extraction efficiency using the TI-COF exceeds those of all reported adsorbents and photocatalysts for such use (Fig. 3e and Table S5†), including i-MZIF90(50) (1.1 mg g−1 day−1),29b COF 4P (8.0 mg g−1 day−1),34 and COF-4 (6.84 mg g−1 day−1),35 creating a record in this field.Afterwards, the use of the TI-COF for uranium extraction from seawater under natural sunlight was explored. To the best of our knowledge, almost all reported photocatalysts are only evaluated under simulated sunlight for the uranium extraction from seawater, and few has been explored under natural sunlight. Notably, our TI-COF photocatalyst can be also efficient for uranium extraction from seawater under natural sunlight, resulting in 6.9 mg g−1 uranium uptake capacity and 6.9 mg g−1 day−1 uranium extraction efficiency. Such a value is even close to that under simulated conditions, and comparable with those of the reported best photocatalysts under simulated conditions such as COF 4P (8.0 mg g−1 day−1)34 and COF-4 (6.84 mg g−1 day−1).35
3.6 Uranium extraction mechanism of our COF under light
To disclose the uranium extraction mechanism in the TI-COF, we first carried out several control experiments (Fig. S7†). For comparison, the uranium extraction was performed in a 50 mL 100 ppm UO22+ solution with a 5 mg TI-COF sample under darkness or a 50 mL 100 ppm UO22+ solution without the photocatalyst under simulated sunlight. It is found that the former just captures 9.5% uranium within 40 min, equal to an extraction capacity of 90.0 mg g−1, while the later only captures less than 1% uranium. By contrast, the TI-COF under simulated sunlight performs a 80% uranium uptake. Thereby, a light-driven U(VI)-to-U(IV) reduction is deduced to be responsible for the current outstanding uranium extraction from the TI-COF. This conclusion can be further supported by SEM-EDS and XPS (X-ray photoelectron spectroscopic) analyses. In the SEM-EDS images, we can directly read out the extracted uranium in a rod-shaped shape morphology that is fixed on the surface of the TI-COF and the U element that shows a uniform distribution on the TI-COF (Fig. 4a and b). In XPS, two peaks at 380.0 eV and 390.9 eV for respective U4f7/2 and U4f5/2 are observed (Fig. 4c), confirming that the valence state of uranium of +4 resulted from photocatalytic U(VI)-to-U(IV) reduction.34,35 Moreover, the formation of UO2 after photocatalysis is further confirmed by both PXRD (Fig. S8†) and Raman spectra (Fig. S9†).23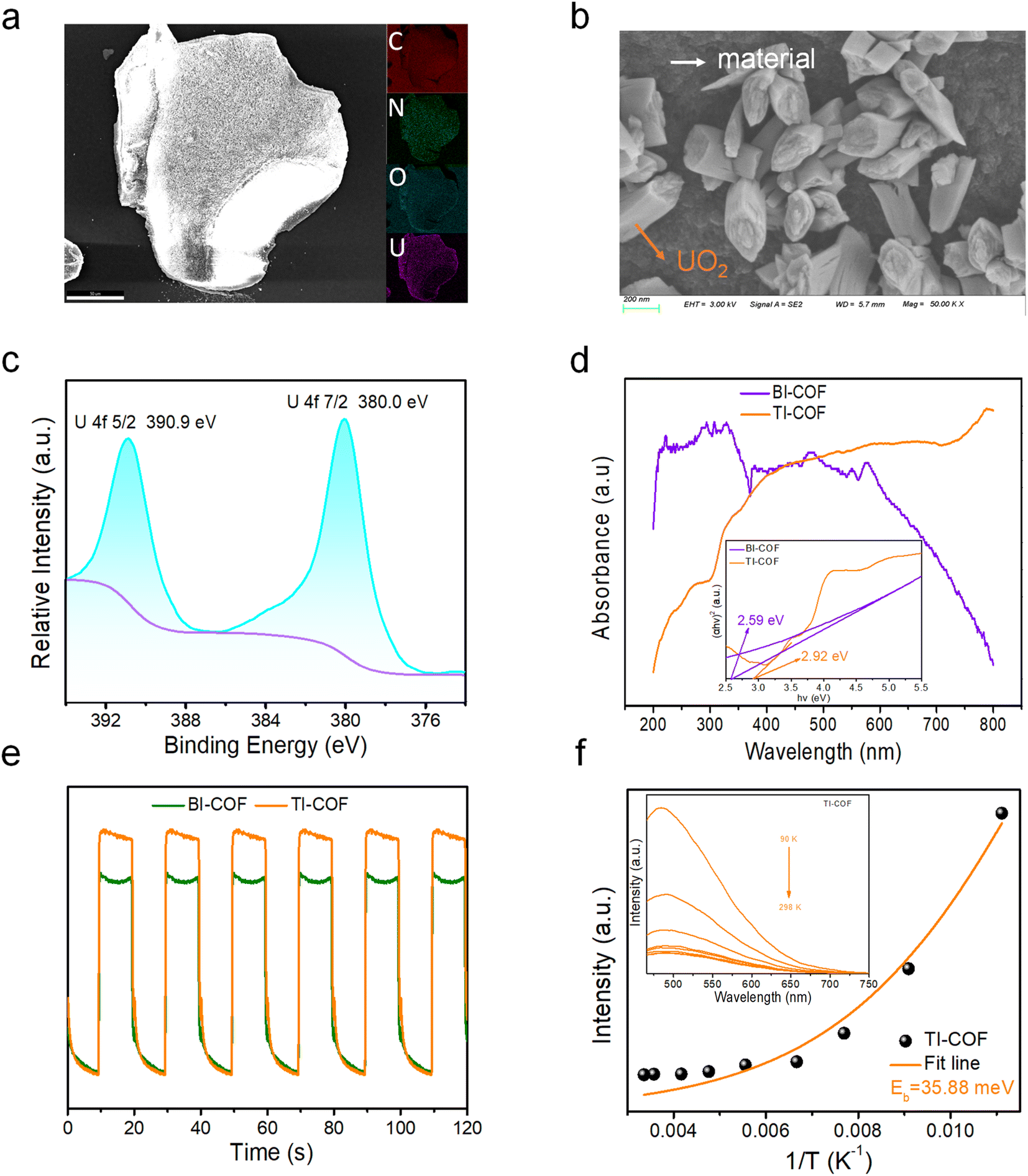 | ||
| Fig. 4 (a) SEM-EDS images of the U-loaded TI-COF with the inset of element mapping. (b) SEM image of the U-loaded TI-COF. (c) XPS U-element spectra of the U-loaded TI-COF. (d) UV-vis diffuse reflectance spectra of the BI-COF and TI-COF. (e) Photocurrent response of the BI-COF and TI-COF. (f) The temperature-dependent photoluminescence (TD-PL) spectra of the TI-COF with the inset of fitting results. | ||
Next, we carried out some characterization studies often used for photocatalysts. First, the optical band gaps of the BI-COF and TI-COF were deduced from UV-vis diffuse reflectance spectra by Tauc plots of the (αhν)1/2vs. hν curve, giving 2.59 eV for the BI-COF and 2.92 eV for the TI-COF (Fig. 4d). This means the semiconductor property of them and suggests their potential as photocatalysts. By means of Mott–Schottky tests, we estimated their lowest unoccupied molecular orbital (LUMO) level, giving −1.19 V for the BI-COF and −0.50 V for the TI-COF (Fig. S10 and S11†). And based on the formula, EHOMO = ELUMO + Eg, we obtained the highest occupied molecular orbital (HOMO) level with +1.4 V for the BI-COF and +2.42 V for the TI-COF. The LUMO level in the BI-COF and TI-COF is smaller than the reduction potential of UO22+ with E(UO22+/UO2) = +0.41 V (URR), suggesting that both the BI-COF and TI-COF are thermodynamically favorable for UO22+-to-UO2 reduction. This is in good agreement with the experimental results.
Moreover, for the TI-COF, we further verified the active intermediates during the photocatalytic process by adding different scavengers, i.e., benzoquinone (BQ, ˙O2− scavenger) and AgNO3 (e− scavenger). The presence of BQ can significantly reduce the uranium extraction ability, while AgNO3 afforded no detectable effect on the uranium extraction ability (Fig. S12†). The results disclosed that ˙O2− contributes to the photocatalytic U(VI)-to-U(IV) reduction in this work, and photogenerated electrons (e−) do not directly participate in such photocatalytic reaction. The LUMO level of the TI-COF (−0.50 V) is more negative than E(O2/˙O2−) = −0.33 V, suggesting an inherent driving force for the TI-COF to convert O2 to ˙O2− active intermediates for the photocatalytic U(VI)-to-U(IV) reduction.
In addition, we also carried out photocurrent and electrochemical impedance spectroscopy (EIS) to evaluate the charge dynamics of our photocatalyst. A stronger photocurrent response was observed in the TI-COF, relative to the BI-COF, indicative of higher efficiency in charge carrier transfer (Fig. 4e). At the same time, the TI-COF afforded smaller impedance, in contrast to the BI-COF (Fig. S13†), implying more excellent electron migration ability of the TI-COF. All these advantages strongly suggest more excellent photocatalytic performance of the TI-COF over the BI-COF; this is in agreement with the experimental results.
To further disclose the separation and transportation of photogenerated carriers in the TI-COF, we first measured its fluorescence lifetimes. Long fluorescence lifetimes up to 3.90 ns were observed (Fig. S14†), which is bigger than those of the reported photocatalysts for such use, e.g. PyN-DAB (3.4 ns),32 COF4P (0.96 ns)34 and COF-4 (1.42 ns),35 suggesting its advantage in inhibiting the recombination of photogenerated carriers and consequently photocatalysis. Second, the temperature-dependent photoluminescence (TD-PL) spectra were measured, and the exciton binding energy (Eb) of the TI-COF can be deduced through fitting the plot of temperature vs. PL intensity via the Arrhenius equation. Notably, the Eb value is as low as 35.88 meV (Fig. 4f), which is far lower than all reported organic semiconductors such as PyTTA-COF (113 meV)38 and TT-CTP (74.5 meV),39 implying a small energy loss for exciton dissociation into free charges and more effective charge separation. All these advantages strongly support its outstanding performance in uranium extraction from seawater under natural sunlight.
3.7 Theoretical calculation
To obtain a deep insight into the relationship between the structure and function, we then carried out DFT (density functional theory) calculation on the BI-COF and TI-COF. As shown in Fig. S15,† the HOMO level of the BI-COF is majorly located in the benzene ring unit from the benzene-1,3,5-tricarboxaldehyde section, while the LUMO level is majorly contributed from the imidazole unit from the 2,4-dimethylimidazole section, which means an electron transfer from benzene-1,3,5-tricarboxaldehyde to adjacent 2,4-dimethylimidazole. In this regard, in the BI-COF, the D–A system is composed of benzene-1,3,5-tricarboxaldehyde as the electron donor and 2,4-dimethylimidazole as the electron acceptor. By contrast, an opposite situation is observed in the TI-COF, whereas in the HOMO level the electron is mainly located in the imidazole unit from the 2,4-dimethylimidazole section (Fig. 5a), and correspondingly, in the LUMO level the electron is mainly located in the triazine unit from the 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde section (Fig. 5b). This corresponds to an electron transfer from 2,4-dimethylimidazole to adjacent 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde. And this leads to a reversed D–A system in the TI-COF composed of 2,4-dimethylimidazole as the electron donor and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl) tribenzaldehyde as the electron acceptor. And it is precisely because of such a reversed donor–acceptor system in the COF, the TI-COF can exhibit an exciting uranium extraction performance from seawater under natural sunlight. In addition, the adsorption site for UO22+ ions in the TI-COF was calculated and is shown in Fig. 5c and d, where two different adsorption sites are observed. Site I is around the triazine unit and fixed by three hydrogen bonds with a distance of 2.80–3.49 Å and a binding energy of −6.28 kJ mol−1, which is stronger than site II that is located around the imidazole unit and stabilized by two hydrogen bonds with a distance of 3.15–3.22 Å and binding energy of −4.55 kJ mol−1. Thereby, both triazine and imidazole units can be the active center for photocatalytic U(VI)-to-U(IV) reduction, and triazine units should play the major role.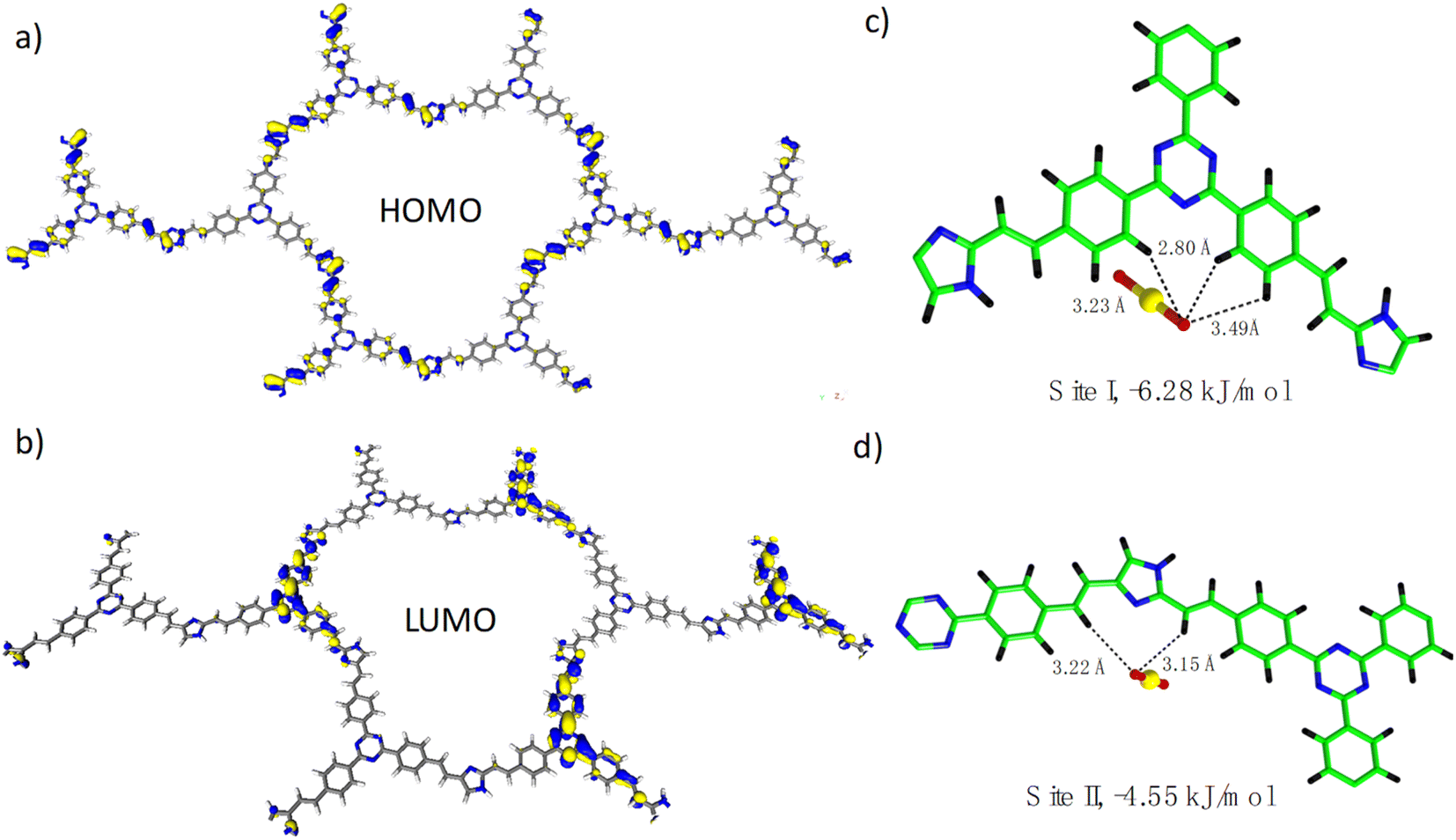 | ||
| Fig. 5 (a) View of the HOMO level of the TI-COF. (b) View of the LUMO level of the TI-COF. (c) View of the adsorption site I. (d) View of the adsorption site II. | ||
In light of these results, we can deduce the photocatalytic mechanism in the current cases, which is shown in Fig. S16.† From the analysis of their HOMO and LUMO levels, in conjunction with the experimental and DFT results, both the TI-COF and BI-COF adopt the uranium reduction reaction (URR, E(UO22+/UO2) = 0.41 V) to extract uranium from seawater with O2˙− as the active intermediate. In this URR process, the photogenerated e− is transferred from the triazine unit to UO22+ for the TI-COF, whereas it is transferred from the imidazole unit to UO22+ for the BI-COF. The higher electronic transfer in the triazine unit over the imidazole unit is responsible for the better photocatalytic performance in the TI-COF than BI-COF. On the other hand, the photogenerated h+ is captured by water oxidation reaction (WOR, E(H2O/O2) = 1.23 V) for both the BI-COF and TI-COF. In a word, the difference in the electronic structure and electron transfer ability between the BI-COF and TI-COF makes the major factor for the higher extraction efficiency of uranium for the TI-COF over the BI-COF.
4. Conclusions
In summary, we have presented the synthesis, structure, and photocatalytic properties of two new imidazole-based COFs, BI-COF and TI-COF. The two COFs exhibit an interesting contrary feature in the D–A system, which to some extent shows us a pathway to enhance the photocatalytic performance in uranium extraction from seawater. And the resultant TI-COF was found to show higher efficiency in both generation and transmission of photogenerated carriers, relative to BI-COF. Correspondingly, the TI-COF is found to show better performance in photocatalytic extraction of uranium from seawater, relative to BI-COF, resulting in a record value in the extraction efficiency of uranium from seawater. In addition, the synthetic cost of the TI-COF is as low as 0.6 $ per g, estimated from the used raw materials of 2,4-dimethylimidazole/0.04 $ per g, benzoic anhydride/0.01$ per g, and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)tribenzaldehyde/0.55 $ per g. These results, in conjunction with the solid-state large-scale synthesis, make our photocatalyst a promising candidate for the ultrahigh, rapid, and large-scale extraction of uranium from seawater.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
L. Z. Z.: methodology, formal analysis, investigation, writing. X. F. F., Q. Y. Z., X. Q. X.: methodology, formal analysis, validation. F. L.: conceptualisation, writing – review & editing, supervision, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (22376024), the Youth Leading Talent Project of Fuzhou (no. 2020ED64), and the Jiangxi Project (DHSQT22021007). The authors would also like to thank Wenqian Liu from Shiyanjia Lab (http://www.Shiyanjia.com) for the XRD and SEM analyses.References
- J. Deutch, Joule, 2020, 4, 2237–2240 CrossRef PubMed.
- A. Q. Gilbert and M. D. Bazilian, Joule, 2020, 4, 1839–1843 CrossRef PubMed.
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- W. Peng, F. Wagner, M. V. Ramana, H. Zhai, M. J. Small, C. Dalin, X. Zhang and D. L. Mauzerall, Nature Sustainability, 2018, 1, 693–701 CrossRef.
- C. R. DeRolph, R. A. McManamay, A. M. Morton and S. S. Nair, Nature Sustainability, 2019, 2, 412–420 CrossRef.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed.
- Y. Xie, Z. Liu, Y. Geng, H. Li, N. Wang, Y. Song, X. Wang, J. Chen, J. Wang, S. Ma and G. Ye, Chem. Soc. Rev., 2023, 52, 97–162 RSC.
- T. Chen, K. Yu, C. Dong, X. Yuan, X. Gong, J. Lian, X. Cao, M. Li, L. Zhou, B. Hu, R. He, W. Zhu and X. Wang, Coord. Chem. Rev., 2022, 467, 214615 CrossRef CAS.
- D. Mei, L. Liu and B. Yan, Coord. Chem. Rev., 2023, 475, 214917 CrossRef CAS.
- S. Dai, Chem, 2021, 7, 537–539 CAS.
- Q. Sun, B. Aguila and S. Ma, Trends Chem., 2019, 1, 292–303 CrossRef CAS.
- R. V. Davies, J. Kennedy, R. W. McIlroy, R. Spence and K. M. Hill, Nature, 1964, 203, 1110–1115 CrossRef.
- X. Xu, H. Zhang, J. Ao, L. Xu, X. Liu, X. Guo, J. Li, L. Zhang, Q. Li, X. Zhao, B. Ye, D. Wang, F. Shen and H. Ma, Energy Environ. Sci., 2019, 12, 1979–1988 RSC.
- Y. Yuan, Y. Yang, X. Ma, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef PubMed.
- W. R. Cui, C. R. Zhang, W. Jian, F. F. Li, R. P. Liang, J. Liu and J. D. Qiu, Nat. Commun., 2020, 11, 436 CrossRef CAS PubMed.
- L. Zhou, M. Bosscher, C. Zhang, S. Ozçubukçu, L. Zhang, W. Zhang, C. J. Li, J. Liu, M. P. Jensen, L. Lai and C. He, Nat. Chem., 2014, 6, 236–241 CrossRef CAS PubMed.
- Y. Yuan, Q. Yu, J. Wen, C. Li, Z. Guo, X. Wang and N. Wang, Angew. Chem., Int. Ed., 2019, 58, 11785–11790 CrossRef CAS PubMed.
- Y. Yuan, T. Liu, J. Xiao, Q. Yu, L. Feng, B. Niu, S. Feng, J. Zhang and N. Wang, Nat. Commun., 2020, 11, 5708 CrossRef CAS PubMed.
- Z. Wang, R. Ma, Q. Meng, Y. Yang, X. Ma, X. Ruan, Y. Yuan and G. Zhu, J. Am. Chem. Soc., 2021, 143, 14523–14529 CrossRef CAS PubMed.
- Y. Song, C. Zhu, Q. Sun, B. Aguila, C. W. Abney, L. Wojtas and S. Ma, ACS Cent. Sci., 2021, 7, 1650–1656 CrossRef CAS PubMed.
- Y. Xu, Z. Yu, Q. Zhang and F. Luo, Adv. Sci., 2023, 10, 2300408 CrossRef CAS PubMed.
- C. Liu, P. C. Hsu, J. Xie, J. Zhao, T. Wu, H. Wang, W. Liu, J. Zhang, S. Chu and Y. Cui, Nat. Energy, 2017, 2, 17007 CrossRef CAS.
- X. H. Xiong, Z. W. Yu, L. L. Gong, Y. Tao, Z. Gao, L. Wang, W. H. Yin, L. X. Yang and F. Luo, Adv. Sci., 2019, 6, 1900547 CrossRef PubMed.
- Y. Yuan, Q. Yu, M. Cao, L. Feng, S. Feng, T. Liu, T. Feng, B. Yan, Z. Guo and N. Wang, Nature Sustainability, 2021, 4, 708–714 CrossRef.
- B. Yan, C. Ma, J. Gao, Y. Yuan and N. Wang, Adv. Mater., 2020, 32, 1906615 CrossRef CAS PubMed.
- L. Yang, H. Xiao, X. Zhao, X. Y. Kong, P. Liu, W. Xin, L. Fu, L. Jiang, L. Wen and Y. Qian, Nature Sustainability, 2022, 5, 71–80 CrossRef.
- Q. Zhang, L. Zhang, J. Zhu, L. Gong, Z. Huang, F. Gao, J. Wang, X. Xie and F. Luo, Nat. Commun., 2024, 15, 453 CrossRef CAS PubMed.
- (a) A. Kaushik, K. Marvaniya, Y. Kulkarni, D. Bhatt, J. Bhatt, M. Mane, E. Suresh, S. Tothadi, K. Patel and S. Kushwaha, Chem, 2022, 10, 2749–2765 CrossRef; (b) S. Mollick, S. Saurabh, Y. D. More, S. Fajal, M. M. Shirolkar, W. Mandal and S. K. Ghosh, Energy Environ. Sci., 2022, 15, 3462–3469 RSC.
- H. Li, F. Zhai, D. Gui, X. Wang, C. Wu, D. Zhang, X. Dai, H. Deng, X. Su, D. Juan, Z. Lin, Z. Chai and S. Wang, Appl. Catal., B, 2019, 254, 47–54 CrossRef CAS.
- L. Feng, Y. Yuan, B. Yan, T. Feng, Y. Jian, J. Zhang, W. Sun, K. Lin, G. Luo and N. Wang, Nat. Commun., 2022, 13, 1389 CrossRef CAS PubMed.
- C. P. Niu, C. R. Zhang, X. Liu, R. P. Liang and J. D. Qiu, Nat. Commun., 2023, 14, 4420 CrossRef CAS PubMed.
- H. Zhang, W. Liu, A. Li, D. Zhang, X. Li, F. Zhai, L. Chen, L. Chen, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 16110–16114 CrossRef CAS PubMed.
- H. Yang, M. Hao, Y. Xie, X. Liu, Y. Liu, Z. Chen, X. Wang, G. I. N. Waterhouse and S. Ma, Angew. Chem., Int. Ed., 2023, 62, e202303129 CrossRef CAS PubMed.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS PubMed.
- J. Chang, Q. Li, Y. Yan, J. W. Shi, J. Zhou, M. Lu, M. Zhang, H. M. Ding, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202209289 CrossRef CAS PubMed.
- J. M. Buriak, P. V. Kamat and K. S. Schanze, ACS Appl. Mater. Interfaces, 2014, 6, 11815–11816 CrossRef CAS PubMed.
- L. Zou, Z. A. Chen, D. H. Si, S. L. Yang, W. Q. Gao, K. Wang, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2023, 62, e202309820 CrossRef CAS PubMed.
- S. Wang, Z. Xie, D. Zhu, S. Fu, Y. Wu, H. Yu, C. Lu, P. Zhou, M. Bonn, H. I. Wang, Q. Liao, H. Xu, X. Chen and C. Gu, Nat. Commun., 2023, 14, 6891 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis in detail, additional figures, and tables. See DOI: https://doi.org/10.1039/d4sc02554g |
| This journal is © The Royal Society of Chemistry 2024 |