
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA strategy for the controllable generation of organic superbases from benchtop-stable salts†
Stephen J.
Sujansky‡
,
Garrett A.
Hoteling‡
and
Jeffrey S.
Bandar
 *
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: jeff.bandar@colostate.edu
First published on 29th May 2024
Abstract
Organic superbases are a distinct class of strong base that enable numerous modern reaction applications. Despite their great synthetic potential, widespread use and study of superbases are limited by their air sensitivity and difficult preparation. To address this, we report air-stable carboxylate salts of BTPP and P2-t-Bu phosphazene superbases that, when added to solution with an epoxide, spontaneously generate freebase. These systems function as effective precatalysts and stoichiometric prereagents for superbase-promoted addition, substitution and polymerization reactions. In addition to improving the synthesis, shelf stability, handling and recycling of phosphazenes, this approach enables precise regulation of the rate of base generation in situ. The activation strategy effectively mimics manual slow addition techniques, allowing for control over a reaction's rate or induction period and improvement of reactions that require strong base but are also sensitive to its presence, such as Pd-catalyzed coupling reactions.
Introduction
Brønsted bases are indispensable tools in synthetic chemistry. For a given application, the exact choice of base can be of critical importance as its properties often dictate reaction outcomes.1 Organic superbases are a valuable class of base that enable unique applications over more common metal-containing bases (Fig. 1, top).2 Defined as neutral organic compounds with basicity greater than Proton-sponge® (pKa′ = 18.6 in MeCN), superbases are distinguished by their high solubility in organic media, low nucleophilicity and the formation of conjugate acid ion pairs upon substrate deprotonation.3–5 These properties are frequently leveraged in the discovery of new base-promoted and -catalyzed reactions.2,6 Superbases also enable advances in other areas of methods development, such as cross-coupling and photoredox methodology and in high throughput experimentation technologies.7 However, as with all strong bases, organic superbases are high energy compounds that are unstable under ambient conditions, a significant limitation that hinders their wider use and study.8 To address this challenge, we herein exploit the unique ion pairing properties of organic superbases to establish a new and improved means for their use. We disclose air-stable superbase salts and additives that, once added to solution, controllably generate the freebase without the use of a separate strong base (Fig. 1, bottom). This process functions as a practical superbase precatalyst system and also provides a new way to regulate base introduction to a solution.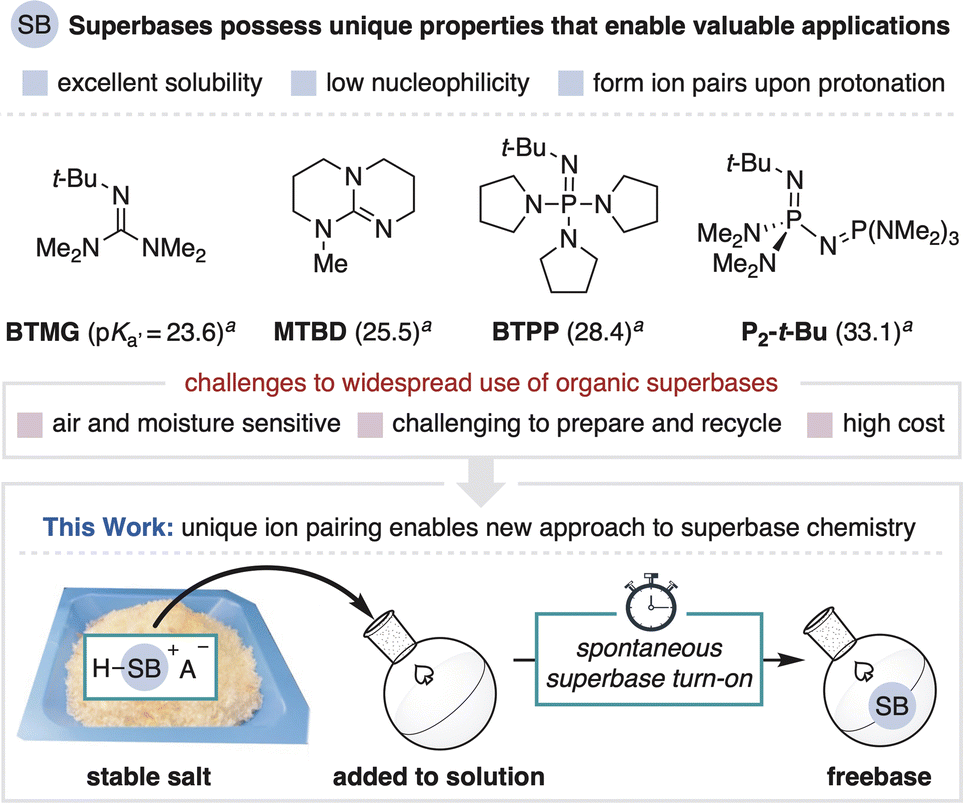 | ||
| Fig. 1 Organic superbases and a new approach to accessing superbases from precatalyst salts. a Reported pKa′ values in MeCN.13 | ||
Organic superbases form stable salts when combined with a strong acid, a fundamental contrast to commonly used anionic bases that form neutral conjugate acids (Fig. 2, top). Stable superbase salts are most commonly comprised of very weakly basic counteranions (e.g., BF4−) and their neutralization requires the use of a separate strong base.9 We reasoned, however, that the counteranion of a superbase salt could be a functional component of a design strategy for the controllable generation of freebase in solution. By definition, the counteranion of any stable salt is incapable of directly neutralizing the conjugate acid. Therefore, the key to spontaneous in situ activation of such a system is to implement a mechanism by which a weakly basic anion can facilitate superbase generation. This capability has been previously achieved within photocaged base technology wherein light-promoted counteranion decomposition leads to freebase formation.10 This approach, however, has thus far only been used for photocuring applications and has not been applied towards more traditional synthetic chemistry.11
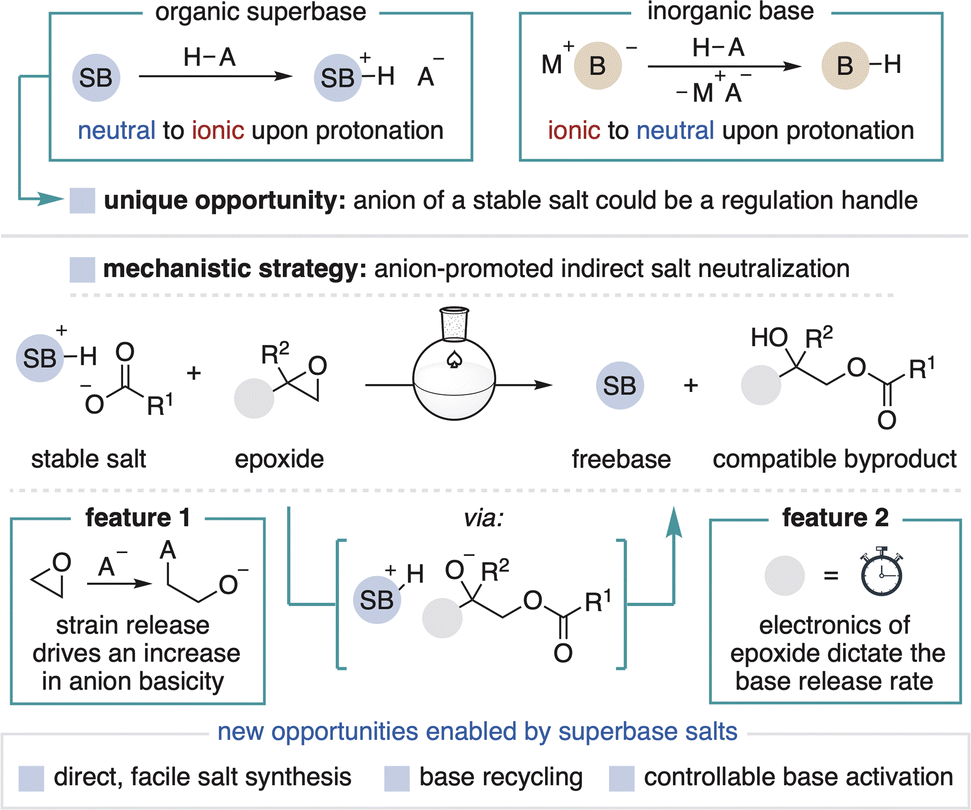 | ||
| Fig. 2 Mechanistic design for superbase precatalyst system. | ||
Herein, we describe a new strategy for the thermal generation of superbases from stable carboxylate salts in the presence of epoxides (Fig. 2, bottom). When in solution, the carboxylate (pKa′ ∼ 24 in MeCN) opens the epoxide, thereby harnessing potential energy (i.e., epoxide ring strain) to create a strongly basic alkoxide intermediate (pKa′ ∼ 43 in MeCN) that deprotonates the superbase conjugate acid.12,13 In principle, the epoxide can be stored independently for easy variation, premixed with the salt for convenience or incorporated into the counteranion structure, so long as activation takes place only in solution. This modular design allows for the carboxylate or epoxide to be adjusted to achieve desired physical properties and reactivity, including the rate of epoxide opening to control the speed of base generation. Importantly, the activation process generates tertiary alcohol byproducts that are compatible with most superbase applications.
The initial motivation for this work was to address key challenges associated with conducting superbase chemistry. Although superbase hydrochloride salts are readily prepared from commodity chemicals, their neutralization often involves nontrivial procedures such as distillation, air-free purification or the use of hazardous bases.14,15 The resulting freebases are air sensitive and typically stored and handled in an inert atmosphere glovebox, resulting in limited convenience and long-term fidelity, as well as high cost.16 In total, these concerns can stifle the translation of innovative breakthroughs made in discovery settings into more widely used chemistry.17 In this report, we focus on BTPP and P2-t-Bu phosphazenes as they are commercially available and among the most commonly used superbases for reaction discovery and methods development. The use of superbase carboxylate salts with this new activation method addresses the aforementioned challenges as the salts can be prepared without the need to access the freebase and are stored, handled and recycled under ambient conditions (Fig. 2, bottom).
Results and discussion
BTPP salt development and application
BTPP freebase irreversibly reacts with atmospheric CO2 to form a phosphoramide, thus necessitating storage and use under inert atmosphere.8 This deleterious process also rules out the use of a decarboxylative strategy for superbase salt activation. We therefore sought to identify a stable BTPP salt and effective activation system to overcome these challenges. Superbase carboxylate salts were targeted given that they can be stable salts while still possessing nucleophilic counteranions, as opposed to more commonly isolated salts comprised of nonnucleophilic counteranions (e.g., BF4−). After investigating various carboxylates, BTPP salt A was identified as a shelf-stable crystalline solid (Fig. 3a). BTPP salt A is accessible in scalable quantities via a one-pot process from PCl5, t-BuNH2 and pyrrolidine, followed by anion metathesis with potassium carboxylate salt 1 (Fig. 3a, >20 g prepared).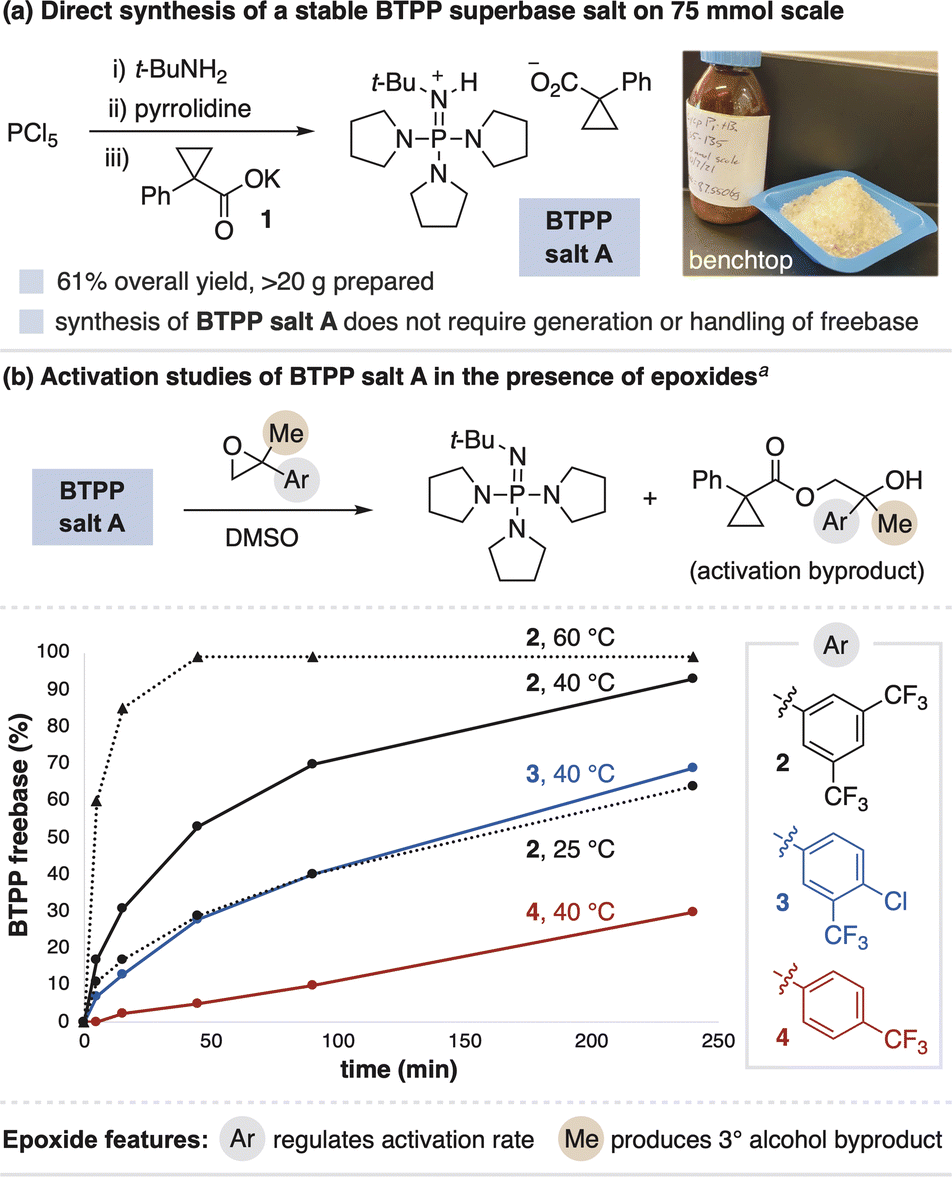 | ||
| Fig. 3 (a) Synthesis and (b) activation of BTPP salt A. a Activation studies run with 1 equiv BTPP salt A and 2 equiv epoxide; all conditions ultimately reach near-complete BTPP generation. See ESI† for details on data collection and analysis. | ||
We next studied the generation of freebase from BTPP salt A when mixed with epoxides in solution. We found that aryl-substituted epoxides readily facilitate BTPP generation, as tracked by 31P and 1H NMR spectroscopy to observe the freebase and activation byproduct, respectively. The activation curves for electronically-differentiated epoxides 2–4 show that the rate of BTPP formation directly correlates with epoxide electrophilicity (Fig. 3b). The activation rate is also dependent on the solvent identity, temperature and concentration, with full details in the ESI.† Thus, BTPP salt A activation occurs under a variety of conditions, which provides flexibility for its use in synthetic applications.
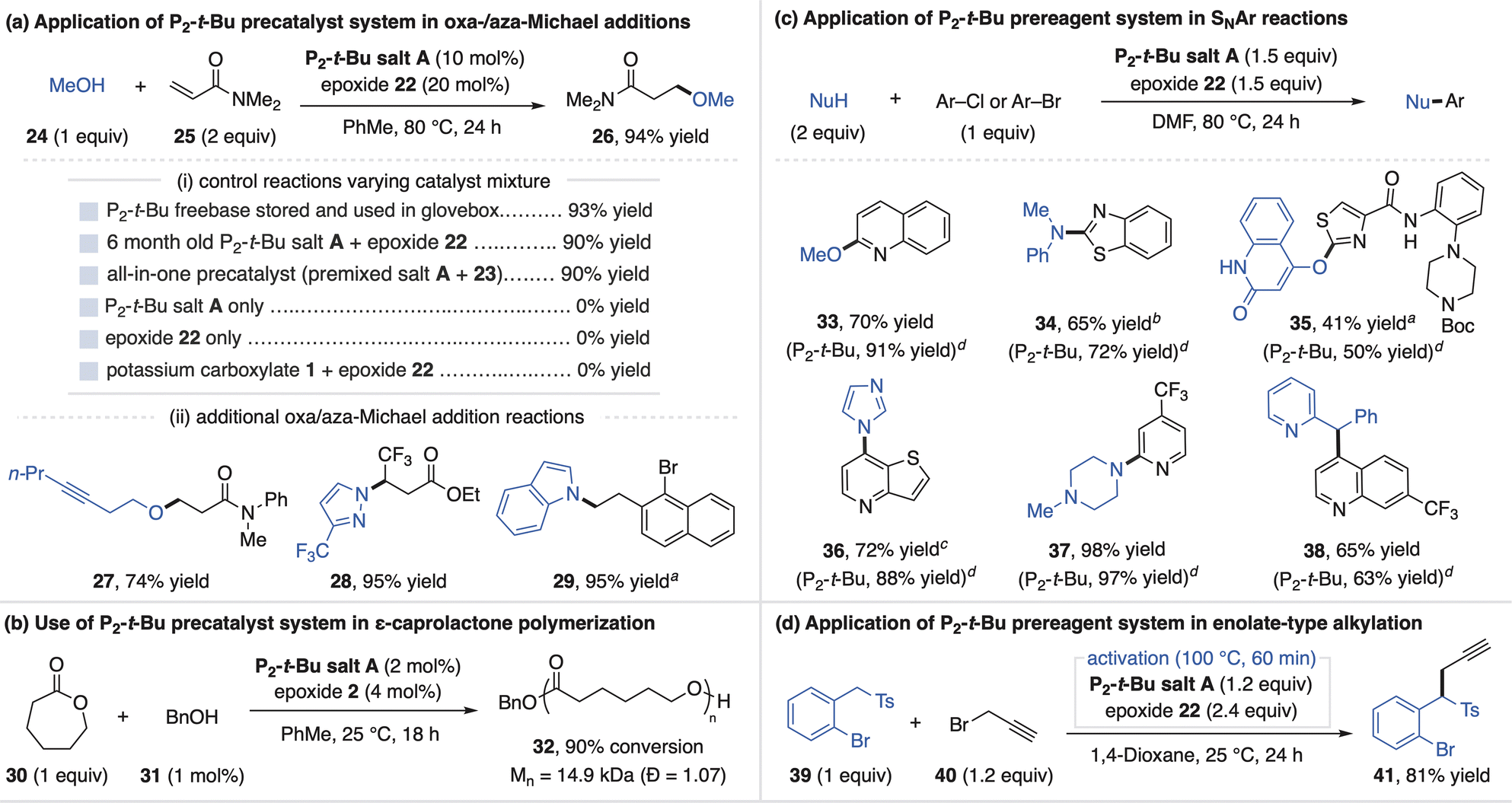
The successful activation of BTPP salt A with epoxides allows this system to function as a precatalyst for superbase-promoted reactions. All reaction applications of the superbase salts in this article were conducted outside of a glovebox with the use of a Schlenk line. To demonstrate this capability, we selected the Michael addition between deoxybenzoin (5) and tert-butyl acrylate (6) as a model reaction.18 The combination of BTPP salt A and epoxide 2 promotes this reaction in a similar high yield as commercial BTPP that is stored and handled in an inert atmosphere glovebox (Fig. 4ai). Consistent with a precatalyst activation process, reaction profiles for the Michael reaction show an induction period that reflects the electrophilicity of the epoxide activator used (Fig. 4aii). Further control experiments show that neither BTPP salt A nor epoxides on their own promote this reaction (Fig. 4ai). The fact that the combination of potassium carboxylate 1 with epoxide 2 also does not promote this reaction indicates BTPP is a necessary component of the precatalyst system.12 Additional Michael, aldol and Mannich products are shown in Fig. 4aiii to demonstrate the precatalyst system's generality (substrates 8–13). Finally, we note that BTPP salt A was regularly handled open-to-air for six months, after which it remained equally effective at promoting these reactions.
We found that direct use of the precatalyst system was not equally as effective as commercial BTPP freebase for certain applications. For example, in the ester amidation reaction in Fig. 4b, the aminoalcohol undergoes a competitive side reaction with the epoxide that inhibits BTPP activation.19 To address this, a five-minute preactivation process (stirring BTPP salt A and epoxide 2 at 80 °C in DMSO) generates a solution of freebase for use in catalytic ester amidation reactions (14–16).
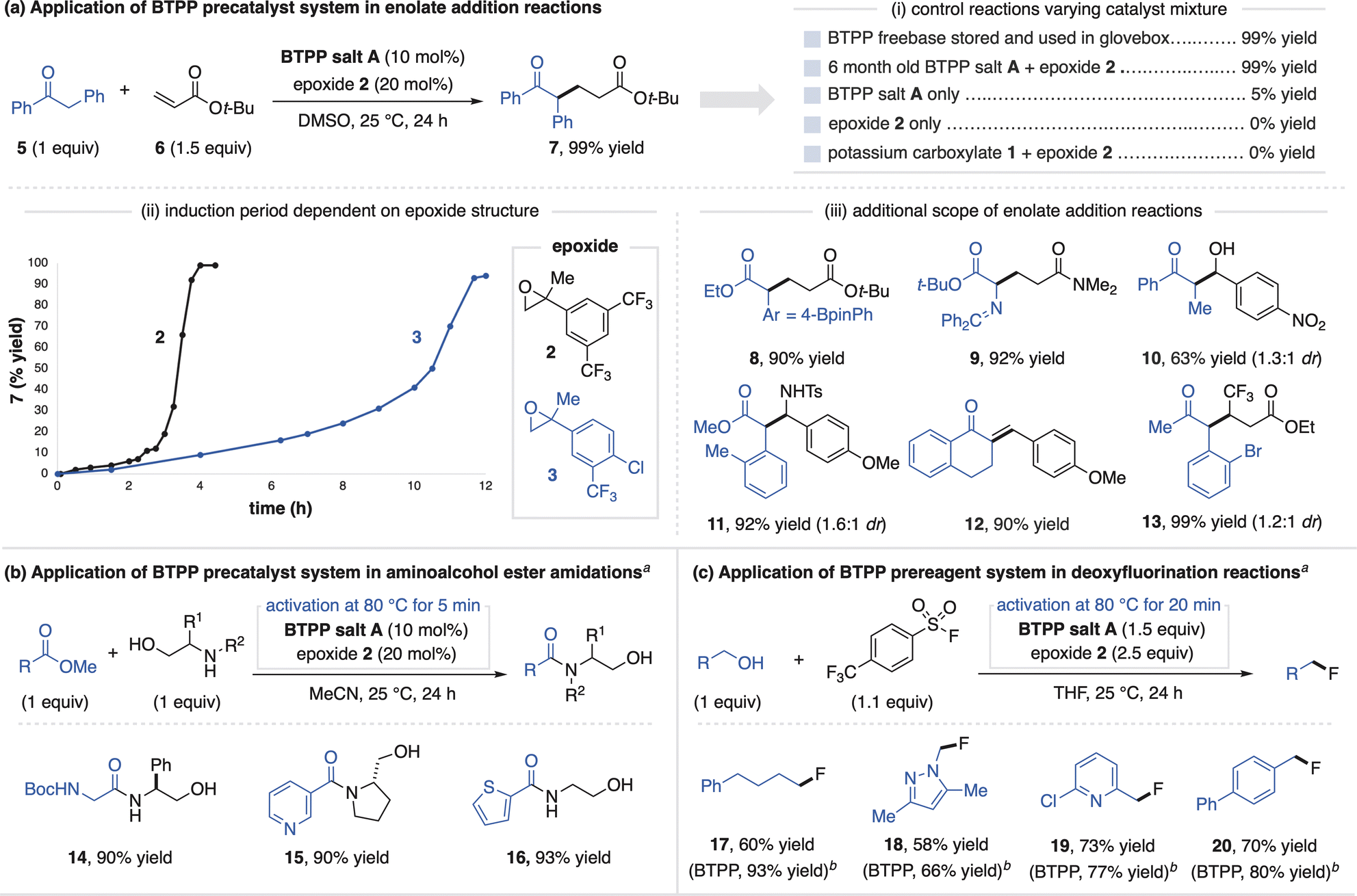 | ||
| Fig. 4 Use of BTPP salt A in catalytic (a and b) and stoichiometric (c) applications. Yields determined by 1H NMR spectroscopy. a Preactivation procedure conducted in DMSO for (b) and THF for (c). b Yields of reactions with commercial BTPP in a N2-filled glovebox for comparison. | ||
BTPP salt A and epoxide 2 can also be used in stoichiometric quantities as a prereagent system, a more demanding application that requires full generation of freebase. We studied this utility in the context of a BTPP-promoted deoxyfluorination method developed by the Doyle Group (Fig. 4c).20 This reaction was selected as a challenging test for the prereagent system, as the sulfonyl fluoride reagent can potentially react with the carboxylate of BTPP salt A or the activation byproduct. Using a preactivation procedure, the prereagent system provides alkyl fluorides (17–20) in good yields, albeit slightly lower than use of commercial BTPP. The tertiary alcohol activation byproduct does not undergo deoxyfluorination, demonstrating its compatibility in superbase-promoted alcohol functionalization reactions.
P2-t-Bu salt development and application
We next sought to develop salt systems for the stronger P2-t-Bu base, which is typically sold as a solution in THF under inert atmosphere as its pure, solid form rapidly absorbs ambient moisture.21 Slight modification of the carboxylate counteranion provided stable, crystalline P2-t-Bu salt A (Fig. 5a). However, activation of this salt with aryl-substituted epoxides (2 and 21) is reversible, with only 50% freebase generated at equilibrium (Fig. 5b), as confirmed by running the activation process in reverse. We reasoned this equilibrium effect is due to the greater thermodynamic challenge of generating P2-t-Bu as compared to BTPP. To address this, we found dialkyl-substituted epoxides provide a greater driving force for freebase generation, including spirocyclic epoxide 22 that produces >90% P2-t-Bu at equilibrium.22 We also identified epoxide 23 that, although only generates 75% freebase at equilibrium, can be stored together with P2-t-Bu salt A to serve as a premixed, all-in-one precatalyst system.23 Additional activation studies for P2-t-Bu salt A are described in the ESI.†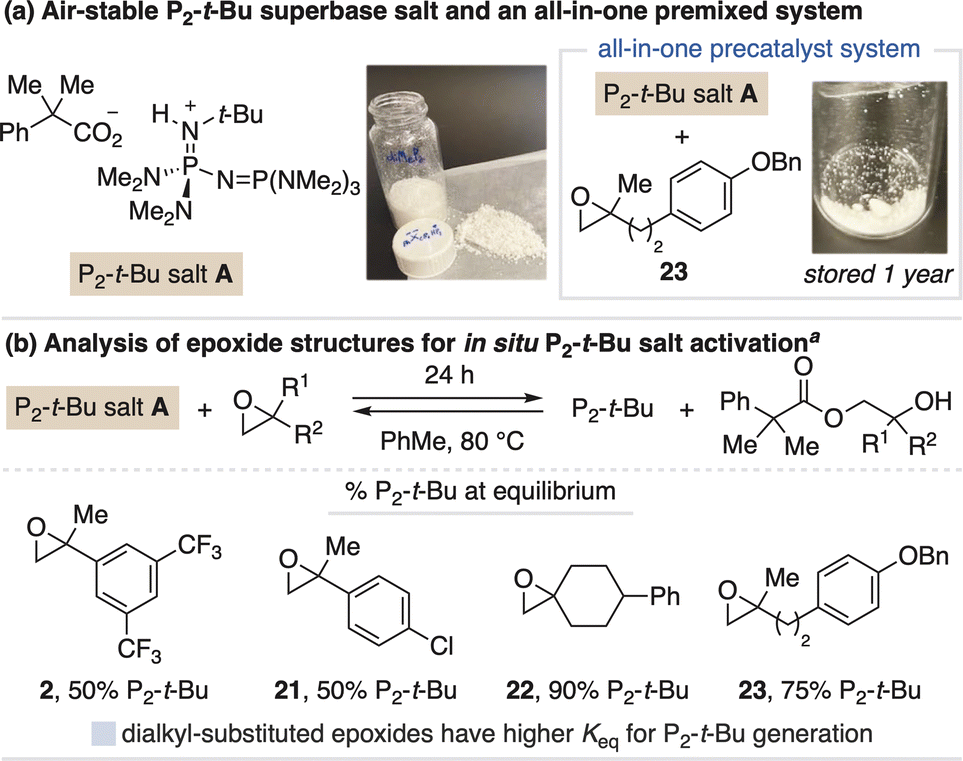 | ||
| Fig. 5 (a) Identification and (b) activation studies of P2-t-Bu salt A. Photos taken on the benchtop. a Activation with 1 equiv P2-t-Bu salt A and 2 equiv epoxide, assessed by 31P NMR spectroscopy. | ||
The P2-t-Bu precatalyst system was first investigated for the promotion of oxa-Michael addition reactions. This application was selected as such reactions are reversible and require strong, non-coordinating bases that operate in nonpolar media for high yields, making superbases ideal catalysts.24 The simple addition of P2-t-Bu salt A and epoxide 22 to a solution of methanol (24) and N,N-dimethylacrylamide (25) leads to a high-yielding oxa-Michael reaction, similar to the use of commercial P2-t-Bu in a glovebox (Fig. 6a). Notably, use of the all-in-one precatalyst (P2-t-Bu salt A and epoxide 23 stored together) or six-month-old P2-t-Bu salt A also provide high yield. Consistent with P2-t-Bu serving as the active catalyst, control studies show that neither P2-t-Bu salt A or epoxide 22 catalyze the reaction on their own (Fig. 6ai). The P2-t-Bu precatalyst system is general, as shown by an oxa-Michael reaction with an alkynol (27) and related hydroamination reactions between N-heterocycles and polarized alkenes (28 and 29, Fig. 6aii).25
 | ||
| Fig. 6 Use of P2-t-Bu salt A in catalytic (a and b) and stoichiometric (c and d) reaction applications. Yields determined by 1H NMR spectroscopy. a Reaction run in DMSO. b Reaction run in THF. c Preactivation at 100 °C for 1 h in PhMe. d Yields of reactions setup using commercial P2-t-Bu in a nitrogen-filled glovebox are provided for comparison. | ||
Beyond small molecule synthetic applications, phosphazene bases are also valued as organocatalysts for controlled anionic polymerization reactions.26 As these reactions are typically conducted at or near room temperature (rt), we used aryl-substituted epoxides that partially activate P2-t-Bu salt A at low temperature for polymerization.27 Thus, catalytic P2-t-Bu salt A and epoxide 2 promote the polymerization of ε-caprolactone (30) with benzyl alcohol (31) initiator to 90% conversion (32, Mn = 14.9 kDa, Đ = 1.07, Fig. 6b).28
We next used P2-t-Bu salt A and epoxide 22 as a prereagent system for nucleophilic aromatic substitution (SNAr) reactions. This represents an emerging application of P2-superbases as they have been shown in high throughput experimentation (HTE) to act as mild bases and uniquely enable challenging SNAr reactions.29 The prereagent system promotes SNAr for a broad range of O-, N-, and C-based pronucleophiles (33–38, Fig. 6c) with yields typically similar to direct use of commercial P2-t-Bu. We note that a 1 h preactivation procedure is required for substrate 36 as imidazole reacts with epoxide 22, precluding an effective single addition protocol. A separate stoichiometric application is shown in Fig. 6d, where preactivated P2-t-Bu salt A promotes the alkylation of benzyl sulfone 39 in 81% yield.30 A preactivation protocol was used as this reaction takes place at 25 °C whereas elevated temperature is required for full P2-t-Bu generation using epoxide 22.
Organic superbases also find utility in advanced applications of metal-catalyzed cross-coupling as they provide homogeneous reaction conditions, can enhance functional group tolerance and enable HTE for reaction screening.31 In this regard, scientists at Merck reported the advantages of P2-Et within Pd-catalyzed coupling reactions, which inspired us to apply the salt systems as prereagents for such methods.32 As shown in Fig. 7, superbase carboxylate salts and epoxide 2 added directly to standard Pd catalysis reaction conditions enable high-yielding amination of aryl halides and triflates (42–49). The P2-t-Bu prereagent system is optimal for alkyl amines, while the BTPP system can be used for more acidic aniline partners. Here, epoxide 2 is used for both bases as it activates the salts at 25 °C as compared to epoxide 22, a process driven to completion by consumption of the base in the coupling reaction. However, C–O (50 and 51) and Suzuki couplings (52, with water as an additive) require superbase preactivation with epoxide 22 as O-pronucleophiles can react with epoxide 2 and prevent superbase activation when all reagents are mixed simultaneously.
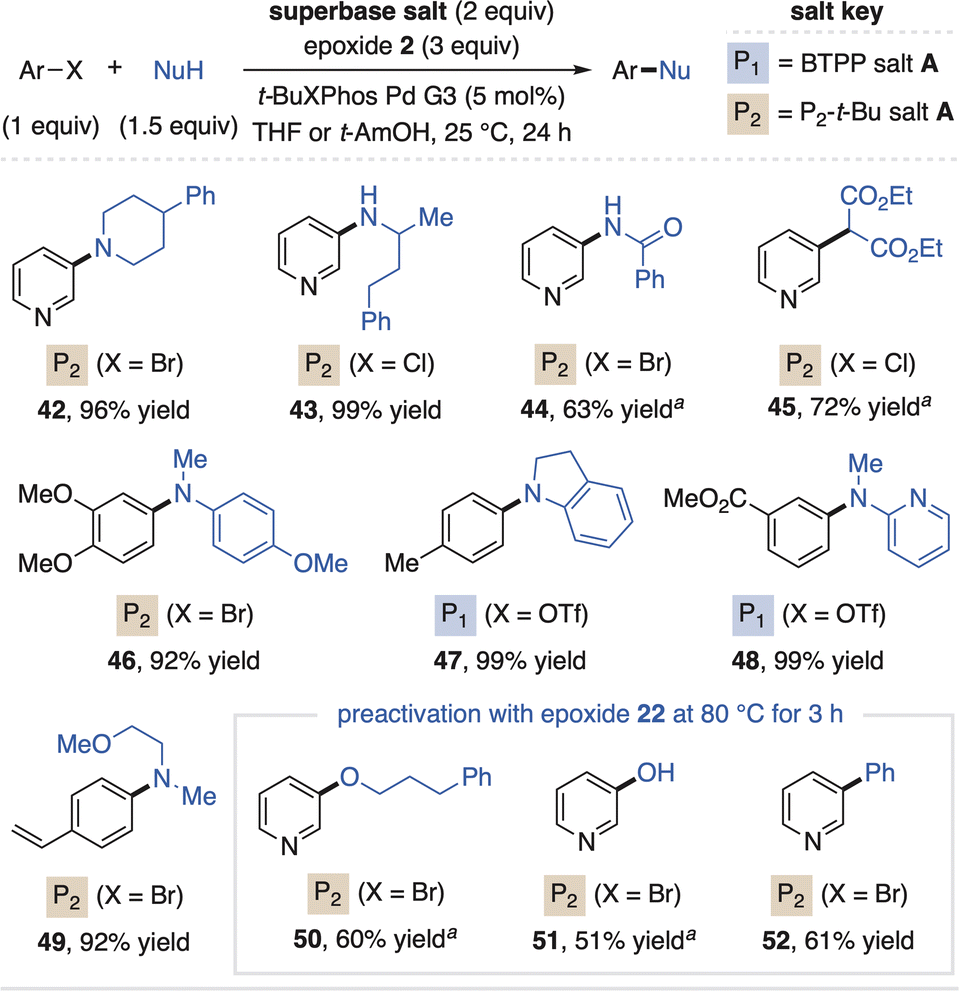 | ||
| Fig. 7 Use of superbase prereagents in Pd-catalyzed cross-coupling reactions. Yields determined by 1H NMR spectroscopy. at-BuBrettPhos Pd G3 (5 mol%) used as catalyst. | ||
Unique opportunities of superbase salt systems
The phosphazene salt activation systems provide several new opportunities compared to the manner in which strong base-promoted chemistry is typically conducted. First, salt synthesis and employment does not necessarily require a discrete neutralization step or handling of freebase. This was illustrated in the 75 mmol scale synthesis of BTPP salt A in Fig. 3a. This feature also enables recycling of the superbase salts, wherein the recovery of superbase hydrochloride salts from crude reaction mixtures, followed by anion metathesis, effectively regenerates the superbase carboxylate salts. Recovery is especially desirable in stoichiometric applications, as illustrated in Fig. 8a by a Pd-catalyzed amination reaction that used 1.1 g of P2-t-salt A that was regenerated in 71% yield.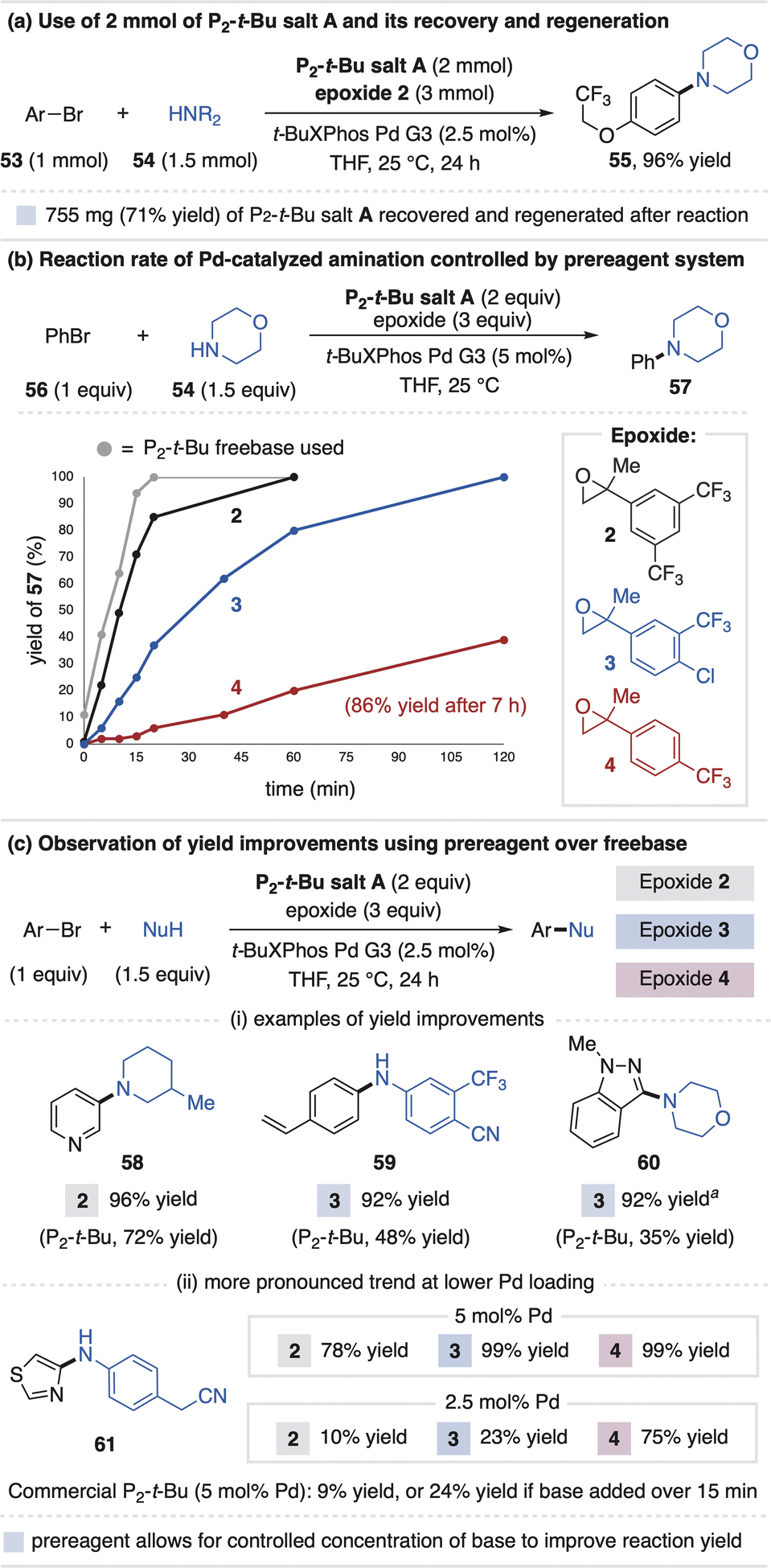 | ||
| Fig. 8 (a) Recovery of superbase salts and (b and c) use of rate-controlled base generation in Pd-catalyzed amination reactions. Yields determined by 1H NMR spectroscopy. a 5 mol% Pd used. | ||
A second feature of the salt system is that, once added to solution, the epoxide identity controls the rate of base generation. This effectively mimics the physical act of freebase addition to a reaction vessel that is traditionally achieved manually or by a syringe pump.33 Thus, one could view the epoxide component as a modular “timer” that can be adjusted through substituent modification, with more electrophilic epoxides generating base faster. We first observed this effect in the model Michael reaction for BTPP salt A, wherein the induction period onset is dictated by epoxide electrophilicity (Fig. 4aii). A similar correlation is observed for the rates of Pd-catalyzed coupling reactions of bromobenzene (56) and morpholine (54) that uses P2-t-Bu salt A with various epoxides (Fig. 8b).34 We anticipate this strategy could serve as a general way to regulate onset times, rates and potential exotherms of base-promoted reactions.35
While investigating Pd-catalyzed amination reactions, we noticed use of the P2-t-Bu prereagent system often provides higher yields than the commercial freebase, with examples in Fig. 8c (58–60). Despite their necessity for reaction promotion, strong bases can be detrimental to Pd catalysis due to competitive catalyst binding or undesired side reactions with catalytic intermediates and base-sensitive functional groups.36 Prior work has shown the advantage of slow base addition for Pd-catalyzed amination reactions33 and, in a similar sense, we speculate the yield improvements of the prereagent system are due to the epoxide opening process that governs base concentration in situ. This proposal was examined using substrate 61, where use of P2-t-Bu freebase provides only 9% yield using 5 mol% Pd catalyst. Control studies indicate the amine and aryl halide lose mass balance when mixed solely with stoichiometric P2-t-Bu, and that the Pd-catalyzed coupling yield increases to 24% when P2-t-Bu is added manually over 15 minutes. Use of P2-t-Bu salt A with epoxides 2, 3 and 4 provides increased yields of 78–99%. The yields are inversely correlated with epoxide electrophilicity, a trend that is amplified when the Pd catalyst loading is decreased to 2.5 mol%. Together, these results illustrate how the prereagent system provides a new approach to improving reactions that require strong base but are also sensitive to its presence.37
Improved superbase salt stability
Throughout our studies, BTPP salt A and P2-t-Bu salt A remained unchanged over six months during which time they were handled regularly open to air and stored in a benchtop desiccator while not in use.38 However, upon exposure to >60% humidity, the salts begin to absorb moisture and can become difficult to handle. This effect is exacerbated at higher humidity (80–90%) where the salts turn into oil hydrates. If this occurs, a toluene azeotrope restoration process via rotary evaporation can regenerate the anhydrous salts, as demonstrated in Fig. 9a for BTPP salt A. This restoration procedure exploits the fact that the hygroscopic nature of the carboxylate salts differs from freebase decomposition, as neutral phosphazenes react with CO2 or absorb water and are impossible or challenging to restore, respectively.8,14,15b A second approach we took to address salt hygroscopicity was to alter the carboxylate counteranion. To this end, we found BTPP salt B and P2-t-Bu salt B to be substantially less hygroscopic, with no change stored in glass vials open to 84% humidity for 24 h. Additionally, while spread out across weighing paper (a more moisture-sensitive state), the new BTPP and P2-t-Bu salts maintain crystallinity for 8 and 24 hours on weighing paper in 84% humidity, respectively. These more stable salts perform equally well in reaction applications compared to the original superbase carboxylate salts (details in the ESI†). Storage of the superbase salts in a desiccator or freezer while not in use is recommended.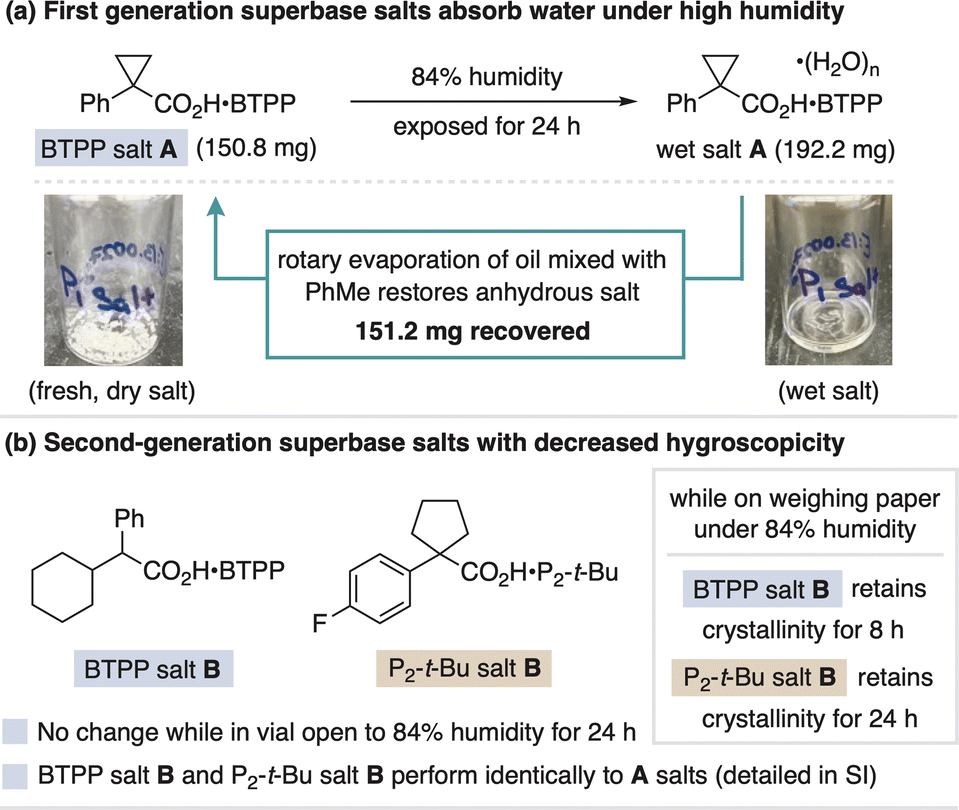 | ||
| Fig. 9 (a) Moisture sensitivity of superbase salts and (b) solutions to address this issue. | ||
Conclusions
In summary, the strategy presented here provides access to organic superbases from benchtop-stable carboxylate salts. These salts, with epoxide additives, are effective precatalysts and prereagents for BTPP and P2-t-Bu using standard Schlenk manifold protocols. The salts are easy to prepare, recyclable and can be regularly handled open to air. We described several scenarios where a preactivation procedure is required to illustrate potential challenges that may be encountered and how they can be addressed when using these salts. Improved activation systems are currently under development, including for the stronger P4-t-Bu superbase.4a,39 The precatalyst systems have the potential to accelerate the discovery of new superbases, superbase-promoted applications and their use by a wider community. More broadly, the mechanistic ability to regulate superbase introduction to solutions presents a myriad of possibilities for improving or manipulating base-promoted reactivity, a prospect that we are currently pursuing in numerous contexts.Data availability
Compound characterization data and experimental procedures are openly available in the ESI.†Author contributions
SJS and GAH conducted the experimental work and the manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Science Foundation under Grant No. CHE-1944478. We thank Professor Yiming Wang and Mr Austin Durham (University of Pittsburgh) for reproducing several reaction applications of the salts described in this report. We thank Mr Alexander Curtis for assistance with amination substrate 60 and Dr Ravikumar R. Gowda of the Chen Group at CSU for assistance with analysis of polymer 32. We also thank Colorado State University (CSU) for funding and the Analytical Resources Core (RRID: SCR_021758) at CSU for instrument access, training and assistance with sample analysis.Notes and references
- For selected reviews on the utility of strong bases and their structural effects on reactivity, see: (a) R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC; (b) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794–9824 CrossRef CAS; (c) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CrossRef CAS; (d) D. B. Collum, A. J. McNeil and A. Ramirez, Angew. Chem., Int. Ed., 2007, 46, 3002–3017 CrossRef CAS PubMed; (e) C. K. Patel, S. Banerjee, K. Kant, R. Sengupta, N. Aljaar and C. C. Malakar, Asian J. Org. Chem., 2023, 12, e2023003 Search PubMed; (f) T. L. Rathman and W. F. Bailey, Org. Process Res. Dev., 2009, 13, 144–151 CrossRef CAS.
- For reviews on organic superbases, see: (a) Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, ed. W. F. Ishikawa, Wiley, Chichester, UK, 2009 Search PubMed; (b) K. Vazdar, D. Margetić, B. Kovačević, J. Sundermeyer, I. Leito and U. Jahn, Acc. Chem. Res., 2021, 54, 3108–3123 CrossRef CAS PubMed; (c) R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Chem. Eur. J., 2021, 27, 10807–10825 CrossRef CAS; (d) Y.-H. Wang, Z.-Y. Cao, Q.-H. Li, G.-Q. Lin, J. Zhou and P. Tian, Angew. Chem., Int. Ed., 2020, 59, 8004–8014 CrossRef CAS.
- Phosphazene superbases are often sold as solutions in THF and hexanes, illustrating their high solubility in non-polar organic media.
- For discussions and measurements of the relative Brønsted basicity versus nucleophilicity of superbases, see: (a) R. Schwesinger and H. Schlemper, Angew. Chem., Int. Ed., 1987, 26, 1167–1169 CrossRef; (b) E. D. Nacsa and T. H. Lambert, J. Am. Chem. Soc., 2015, 137, 10246–10253 CrossRef CAS PubMed; (c) A. Gholamipour-Shirazi and C. Rolando, Org. Biomol. Chem., 2012, 10, 8059–8063 RSC.
- For example methods that rely on ion pairing of superbase conjugate acids, including in asymmetric catalysis, see: (a) H. Krawczyk, M. Dzięgielewski, D. Deredas, A. Albrecht and Ł. Albrecht, Chem.–Eur. J., 2015, 21, 10268–10277 CrossRef CAS; (b) H. J. A. Dale, G. R. Hodges and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 7181–7193 CrossRef CAS; (c) S. Martinez-Erro, A. Sanz-Marco, A. B. Gómez, A. Vázquez-Romero, M. S. G. Ahlquist and B. Martín-Matute, J. Am. Chem. Soc., 2016, 138, 13408–13414 CrossRef CAS PubMed; (d) H. Kawai, Z. Yuan, E. Tokunaga and N. Shibata, Org. Biomol. Chem., 2013, 11, 1446–1450 RSC; (e) D. L. Orsi, M. R. Yadav and R. A. Altman, Tetrahedron, 2019, 75, 4325–4336 CrossRef CAS.
- T. R. Puleo, S. J. Sujansky, S. E. Wright and J. S. Bandar, Chem. Eur. J., 2021, 27, 4216–4229 CrossRef CAS.
- For selected examples of phosphazene superbase use in photoredox catalysis, Pd-catalyzed cross-coupling reactions and HTE applications, see: (a) C. D. Matier, J. Schwaben, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 17707–17710 CrossRef CAS PubMed; (b) X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Nature, 2020, 580, 220–226 CrossRef CAS PubMed; (c) L. M. Baumgartner, J. M. Dennis, N. A. White, S. L. Buchwald and K. F. Jensen, Org. Process. Res. Dev., 2019, 23, 1594–1601 CrossRef CAS; (d) A. B. Santanilla, E. L. Regalado, T. Pereira, M. Shevlin, K. Bateman, L.-C. Campeau, J. Schneeweis, S. Berritt, Z.-C. Shi, P. Nantermet, Y. Liu, R. Helmy, C. J. Welch, P. Vachal, I. W. Davies, T. Cernak and S. D. Dreher, Science, 2015, 347, 49–53 CrossRef PubMed; (e) M. R. Uehling, R. P. King, S. W. Krska, T. Cernak and S. L. Buchwald, Science, 2019, 363, 405–408 CrossRef CAS PubMed; (f) D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS.
- For discussions of air sensitivity of strong organic and metal-containing Brønsted bases, see: (a) M.-A. Courtemanche, M.-A. Légaré, É. Rochette and F.-G. Fontaine, Chem. Commun., 2015, 51, 6858–6861 RSC; (b) C. Villiers, J.-P. Dognon, R. Pollet, P. Thuéry and M. Ephritikhine, Angew. Chem., Int. Ed., 2010, 49, 3465–3468 CrossRef CAS; (c) T. L. Rathman and J. A. Schwindeman, Org. Process. Res. Dev., 2014, 18, 1192–1210 CrossRef CAS; (d) L. Chen, J. Zhao, S.-F. Yin and C.-T. Au, RSC Advances, 2013, 3, 3799–3814 RSC; (e) R. Wethman, J. Derosa, V. T. Tran, T. Kang, O. Apolinar, A. Abraham, R. Kleinmans, S. R. Wisniewski, J. R. Coombs and K. M. Engle, ACS Catal., 2021, 11, 502–508 CrossRef CAS; (f) See also reviews on strong base chemistry covered in ref. 1 and 2, and the procedures for isolation of superbases in ref. 14..
- In situ generation of superbases from their conjugate acid salts can be enabled through the use of a separate strong base activator. For examples, see: (a) C. Luo, J. V. Alegre-Requena, S. J. Sujansky, S. P. Pajk, L. C. Gallegos, R. S. Paton and J. S. Bandar, J. Am. Chem. Soc., 2022, 144, 9586–9596 CrossRef CAS; (b) T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed; (c) D. Uraguchi, S. Sakaki and T. Ooi, J. Am. Chem. Soc., 2007, 129, 12392–12393 CrossRef CAS PubMed; (d) See also Ref. 4b..
- For reviews on photobase generators, see: (a) K. Suyama and M. Shirai, Prog. Polym. Sci., 2009, 34, 194–209 CrossRef CAS; (b) N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., Int. Ed., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- Several reports describe the synthesis of phosphazene photobase generators, although these reagents have not been extensively studied for reaction or polymerization applications; see: (a) X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130–8131 CrossRef CAS PubMed; (b) K. Arimitsu and R. Endo, Chem. Mater., 2013, 25, 4461–4463 CrossRef CAS; (c) D. Aoki, C. Gu, M. Suzuki, Y. Kawamura and K. Arimitsu, Chem. Lett., 2023, 52, 775–778 CrossRef CAS.
- For the use of epoxides as alkoxide-generating reagents, see: (a) J. Buddrus, Angew. Chem., Int. Ed., 1972, 11, 1041–1050 CrossRef CAS; (b) J. Buddrus and W. Kimpenhaus, Chem. Ber., 1974, 107, 2062–2078 CrossRef CAS; (c) S. Tanaka, T. Nakashima, N. Satou, H. Oono, Y. Kon, M. Tamura and K. Sato, Tetrahedron Lett., 2019, 60, 2009–2013 CrossRef CAS.
- For pKa values of carboxylic acids, alcohols and superbases shown in Fig. 1, see: (a) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS; (b) E. Rossini, A. D. Bochevarov and E. W. Knapp, ACS Omega, 2018, 3, 1653–1662 CrossRef CAS PubMed; (c) A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Eur. J. Org. Chem., 2021, 2021, 1407–1419 CrossRef; (d) S. Tshepelevitsh, A. Kütt, M. Lõkov, M. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 2019(40), 6735–6748 CrossRef CAS.
- For synthetic routes to P1-, P2- and P4-superbases and related strong bases, see: (a) R. Schwesinger, Chimia, 1985, 39, 269 CAS; (b) R. Schwesinger, H. Schlemper, C. Hasenfratz, J. Willaredt, T. Dambacher, T. Breuer, C. Ottaway, M. Fletschinger, J. Boele, H. Fritz, D. Putzas, H. W. Rotter, F. G. Bordwell, A. V. Satish, G.-Z. Ji, E.-M. Peters, K. Peters, H. G. von Schnering and L. Walz, Liebigs Ann. Chem., 1996, 1055–1081 CrossRef CAS; (c) R. Schwesinger and T. Dambacher, Z. Naturforsch. B., 2006, 61, 1229–1233 CrossRef CAS; (d) D. H. R. Barton, J. D. Elliot and S. D. Géro, J. Chem. Soc., Chem. Commun., 1981, 1136–1137 RSC; (e) K. Vazdar, R. Kunetskiy, J. Saame, K. Kaupmees, I. Leito and U. Jahn, Angew. Chem., Int. Ed., 2014, 53, 1435–1438 CrossRef CAS PubMed; (f) J. Tang and J. G. Verkade, Tetrahedron Lett., 1993, 34, 2903–2904 CrossRef CAS; (g) See also ref. 4a and 4b..
- Phosphazene freebases can be accessed by several other alternative routes. For a Staudinger reaction using organic azides and tris(dialkylamino)phosphines en route to P1 bases, see: (a) A. V. Alexandrova, T. Mašek, S. M. Polyakova, I. Císařová, J. Saame, I. Leito and I. M. Lyapkalo, Eur. J. Org. Chem., 2013, 2013, 1811–1823 CrossRef CAS . For high vacuum neutralization of hydrates of P4 phosphazenium hydroxide salts, see:; (b) R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2019, 58, 14633–14638 CrossRef CAS.
- The commercial cost of phosphazene superbases from Millipore Sigma are: BTPP (25 mL for $718, or $8780 per mol); P1-t-Bu (25 mL for $1600 or $15,954 per mol); P2-Et (5 mL for $740, or $49,247 per mol); P2-t-Bu (25 mL of 2 M solution for $1820, or $36,400 per mol); and P4-t-Bu (25 mL of 0.8 M solution for $1460, or $73,000 per mol).
- For examples and discussions of use of BTPP and cyclic guanidine superbases in production scale-up, see: (a) R. E. H. Jones, P. Aspin, S. H. Davies, I. Mann, C. Priestley, A. D. Roberts, N. Zotova-Eldridge and J. H. Leahy, Org. Process Res. Dev., 2022, 26, 2646–2655 CrossRef CAS; (b) A. M. Hyde, R. Calabria, R. Arvary, X. Wang and A. Klapars, Org. Process Res. Dev., 2019, 23, 1860–1871 CrossRef CAS; (c) C. Penverne, B. Hazard, C. Rolando and M. Penhoat, Org. Process Res. Dev., 2017, 21, 1864–1868 CrossRef CAS.
- D. Truong, B. L. Howard and P. E. Thompson, Tetrahedron, 2021, 85, 132034 CrossRef CAS.
- (a) N. Caldwell, C. Jamieson, I. Simpson and T. Tuttle, Org. Lett., 2013, 15, 2506–2509 CrossRef CAS PubMed; (b) N. Caldwell, P. S. Campbell, C. Jamieson, F. Potjewyd, I. Simpson and A. J. B. Watson, J. Org. Chem., 2014, 79, 9347–9354 CrossRef CAS PubMed.
- (a) M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed; (b) M. K. Nielsen, D. T. Ahneman, O. Riera and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 5004–5008 CrossRef CAS; (c) A. M. Żurański, S. S. Gandhi and A. G. Doyle, J. Am. Chem. Soc., 2023, 145, 7898–7909 CrossRef PubMed.
- Pure, solid P2-t-Bu freebase is very hygroscopic such that exposure to ambient conditions quickly leads to formation of an oily hydrated substance. For example, P2-t-Bu solid loses its solidity after 15 minutes on weigh paper under 84% humidity.
- Methylenecyclohexane oxides (e.g., 22) may provide a greater driving force due to increased stereoelectronic stabilization of the alcohol byproduct compared to the epoxide; for discussions, see: (a) R. G. Carlson and N. S. Behn, Chem. Commun., 1968, 339–340 RSC; (b) T. Hrenar, I. Primožič, D. Fijan and M. Majerić Elenkov, Phys. Chem. Chem. Phys., 2017, 19, 31706–31713 RSC; (c) I. V. Alabugin, L. Kuhn, N. V. Krivoshchapov, P. Mehaffy and M. G. Medvedev, Chem. Soc. Rev., 2021, 50, 10212–10252 RSC.
- Epoxide 23 is used for an all-in-on system in place of 22; we found that the epoxide 22 and P2-t-Bu salt A mixture is less stable over time compared to the mixture with 23.
- For reviews, see: (a) C. F. Nising and S. Bräse, Chem. Soc. Rev., 2012, 41, 988–999 RSC; (b) J. L. Kennemur, R. Maji, M. J. Scharf and B. List, Chem. Rev., 2021, 121, 14649–14681 CrossRef CAS PubMed . For mechanistic investigations of challenging alcohol addition reactions, see ref. 9a..
- For reviews on base-catalyzed hydroamination reactions, see: (a) J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795–813 CrossRef CAS; (b) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef PubMed; (c) B. N. Hemric, T. A. Garcia and A. E. Barni, Helv. Chim. Acta, 2024, 107, e202300160 CrossRef CAS.
- (a) S. Boileau and N. Illy, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS; (b) S. Liu, C. Ren, N. Zhao, Y. Shen and Z. Li, Macromol. Rapid Commun., 2018, 39, 1800485 CrossRef.
- For the use of P2-bases for ε-caprolactone polymerization, see: H. Alamri, J. Zhao, D. Pahovnik and N. Hadjichristidis, Polym. Chem., 2014, 5, 5471–5478 RSC.
- We note that when the precatalyst system is used, we observe approximately 10% of an oligomer side product that is not present with direct use of P2-t-Bu; see ESI†.
- For use of P2-bases in SNAr reactions, see ref. 7d and N. J. Gesmundo, B. Sauvagnat, P. J. Curran, M. P. Richards, C. L. Andrews, P. J. Dandliker and T. Cernak, Nature, 2018, 557, 228–232 CrossRef CAS PubMed.
- (a) A. Costa, C. Nájera and J. M. Sansano, Synlett, 2001, 12, 1881–1884 CrossRef; (b) A. Costa, C. Nájera and J. M. Sansano, J. Org. Chem., 2002, 67, 5216–5225 CrossRef CAS PubMed.
- For selected examples of organic bases in metal-catalyzed coupling reactions, see ref. 7 and: (a) J. M. Dennis, N. A. White, R. Y. Liu and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 4721–4725 CrossRef CAS; (b) S. H. Lau, P. Yu, L. Chen, C. B. Madsen-Duggan, M. J. Williams and B. P. Carrow, J. Am. Chem. Soc., 2020, 142, 20030–20039 CrossRef CAS; (c) G. L. Beutner, J. R. Coombs, R. A. Green, B. Inankur, D. Lin, J. Qiu, F. Roberts, E. M. Simmons and S. R. Wisniewski, Org. Process Res. Dev., 2019, 23, 1529–1537 CrossRef CAS; (d) S. K. Kashani, J. E. Jessiman and S. G. Newman, Org. Process Res. Dev., 2020, 24, 1948–1954 CrossRef CAS; (e) J. W. M. MacMillan, R. T. McGuire and M. Stradiotto, Chem.–Eur. J., 2022, e202200764 CrossRef CAS.
- For early work using phosphazenes in Pd-catalyzed coupling reactions, see ref. 7d and: (a) A. B. Santanilla, M. Christensen, L.-C. Campeau, I. W. Davies and S. D. Dreher, Org. Lett., 2015, 17, 3370–3373 CrossRef PubMed; (b) See also: C. Palomo, M. Oiarbide, R. López and E. Gómez-Bengoa, Tetrahedron Lett., 2000, 41, 1283–1286 CrossRef CAS.
- For an example of a Pd-catalyzed amination protocol where slow addition of a superbase improves the reaction yield, see: (a) J. M. Dennis, N. A. White, R. Y. Liu and S. L. Buchwald, ACS Catal., 2019, 9, 3822–3830 CrossRef CAS PubMed . For other examples of slow addition improving base-promoted reactions, see:; (b) H.-G. Lombart and W. D. Lubell, J. Org. Chem., 1996, 61, 9437–9446 CrossRef CAS; (c) S. K. Bertilsson and P. G. Andersson, Tetrahedron, 2002, 58, 4665–4668 CrossRef CAS; (d) D. E. Knutson, M. S. Rashid Roni, M. Y. Mian, J. M. Cook, D. C. Stafford and L. A. Arnold, Org. Process Res. Dev., 2020, 24, 1467–1476 CrossRef CAS PubMed.
- The rates of product formation (57) in Fig. 8b also correlate with the rate of alcohol activation byproduct appearance, consistent with the epoxide opening process regulating the rate of amination.
- Numerous reports of safety hazards associated with strong base-promoted reactions are available; for examples, see: (a) Q. Yiang, N. R. Babij and S. Good, Org. Process Res. Dev., 2019, 23, 2608–2626 CrossRef; (b) Q. Yang, B. Canturk, K. Gray, E. McCusker, M. Sheng and F. Li, Org. Process Res. Dev., 2018, 22, 351–359 CrossRef CAS; (c) Q. Yang, M. Sheng, J. J. Henkelis, S. Tu, E. Wiensch, H. Zhang, Y. Zhang, C. Tucker and D. E. Ejeh, Org. Process Res. Dev., 2019, 23, 2210–2217 CrossRef CAS; (d) D. B. Brown, A. D. Allian and Y. Chen, Org. Process Res. Dev., 2021, 25, 1932–1936 CrossRef CAS; (e) M. Power, E. Alcock and G. P. McGlacken, Org. Process Res. Dev., 2020, 24, 1814–1838 CrossRef CAS; (f) See also ref. 1a and 8c..
- For discussions of base coordination to and inhibition of Pd catalytic intermediates in C–N coupling reactions, see ref. 31, 33 and: (a) Y. Sunesson, E. Limé, S. O. Nilsson Lill, R. E. Meadows and P.-O. Norrby, J. Org. Chem., 2014, 79, 11961–11969 CrossRef CAS PubMed; (b) S.-T. Kim, B. Pudasaini and M.-H. Baik, ACS Catal., 2019, 9, 6851–6856 CrossRef CAS.
- For discussions of strong base-promoted decomposition of functional groups seen in Fig. 8, such as azoles, indazoles, styrenes and nitriles, see: (a) E. C. Reichert, K. Feng, A. C. Sather and S. L. Buchwald, J. Am. Chem. Soc., 2023, 145, 3323–3329 CrossRef CAS PubMed; (b) A. Sather and T. A. Martinot, Org. Process Res. Dev., 2019, 23, 1725–1739 CrossRef CAS; (c) M. L. Casey, D. S. Kemp, K. G. Paul and D. D. Cox, J. Org. Chem., 1973, 38, 2294–2301 CrossRef CAS; (d) K. Inoue and K. Okano, Asian J. Org. Chem., 2020, 9, 1548–1561 CrossRef CAS; (e) T. R. Puleo, A. J. Strong and J. S. Bandar, J. Am. Chem. Soc., 2019, 141, 1467–1472 CrossRef CAS PubMed.
- The humidity in our laboratory was typically 10–30% during the course of this work, with occasional raises up to approximately 60%.
- R. Schwesinger and Y. Kondo, Encyclopedia of Reagents for Organic Synthesis, 2010, DOI:10.1002/047084289X.rp150.pub2.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02524e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |