
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpedient alkyne semi-hydrogenation by using a bimetallic AgCu–C3N4 single atom catalyst†
Jingting
Song‡
 a,
Xiangbin
Cai‡
b,
Zhongxin
Chen
c,
Tie
Wang
d,
Shibo
Xi
e,
Qikun
Hu
a,
Ning
Yan
d and
Kian Ping
Loh
*a
a,
Xiangbin
Cai‡
b,
Zhongxin
Chen
c,
Tie
Wang
d,
Shibo
Xi
e,
Qikun
Hu
a,
Ning
Yan
d and
Kian Ping
Loh
*a
aDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Singapore. E-mail: chmlohkp@nus.edu.sg
bDivision of Physics and Applied Physics, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore
cSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, Guangdong 518172, China
dDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 117585, Singapore
eInstitute of Chemical and Engineering Sciences, Agency for Science, Technology, and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
First published on 12th June 2024
Abstract
Metal-catalyzed semi-hydrogenation of alkynes is an important step in organic synthesis to produce diverse chemical compounds. However, conventional noble metal catalysts often suffer from poor selectivity owing to over-hydrogenation. Here, we demonstrate a high-loading bimetallic AgCu–C3N4 single-atom catalyst (SAC) for alkyne semi-hydrogenation. The AgCu–C3N4 SACs exhibit higher activity and selectivity (99%) than their low-loading variants due to the synergistic interaction of heteronuclear Ag–Cu sites at small inter-site distances. Using a combination of techniques such as phenylacetylene-DRIFTS, H2-temperature programmed desorption and DFT calculations, we showed that the cooperative bimetallic interaction during alkyne semi-hydrogenation was achieved by isolated Ag centers as hydrogen activation sites and isolated Cu centers as alkyne activation sites. Our work highlights the importance of achieving high catalyst loading to reduce the inter-site distance in bimetallic SACs for cooperative interactions, which can potentially open new catalytic pathways for synthesizing fine chemicals and pharmaceuticals.
Introduction
Metal-catalysed semi-hydrogenation of alkynes to alkenes is critical in producing a wide range of bulk and fine chemicals.1,2 Traditionally, heterogeneous noble metal-based catalysts such as Pd, Pt, and Ru are well-known hydrogen activation or hydride-transfer catalysts.3–8 However, these noble metals bind strongly to alkene and increase their residence time, which results in over-hydrogenation to alkane.9 Consequently, semi-poisoned catalysts such as Lindlar's catalyst10 are typically utilized to transform alkyne to alkene.11 However, this leads to sub-optimal utilization of noble metals and a trade-off between catalytic activity and selectivity.Single atom catalysts (SACs), where isolated individual metal atoms are dispersed on a solid support, represent a new paradigm in heterogeneous catalysis for maximizing atom utilization efficiency.12,13 Noble metal-based SACs have garnered considerable attention as potential replacements for semi-poisoned catalysts in alkyne semi-hydrogenation due to their ability to tune reactivity and selectivity via regulation of the coordination environment.14–17 It has also been demonstrated that ultrahigh loading SACs can trigger the multi-site mechanisms if two active metal neighbors enjoy proximity interactions via high metal loading (>10 wt%).18,19 Integrating bi- or multi-metallic active sites in such SACs allows the synergistic activation of reactants and drives a more efficient catalytic process than mono-metallic SACs.20
Silver (Ag), despite being the least expensive noble metal, is seldom used as SACs or in alkyne semi-hydrogenation owing to its low reactivity in activating alkyne molecules.21,22 Herein, we reported the incorporation of Ag–Cu bimetallic SACs on graphitic carbon nitride (g-C3N4) with high Ag and Cu loading (Ag: 9 wt%, Cu: 12 wt%). At high loading of the bimetallic SAC, the inter-site distance is <∼0.65 nm, and this characteristic feature is associated with superior activity and selectivity (99%) in the alkyne semi-hydrogenation. H2-TPD and DRIFTS investigation suggests that the Ag site activates the hydrogen, while the Cu site is responsible for alkyne adsorption. The high loading of SACs on C3N4 promotes the proximity of Ag and Cu SAC, facilitating the transfer of activated species and leading to a more efficient and selective semi-hydrogenation process.
Results and discussion
Preparation of bimetallic AgCu–C3N4 SAC
Ultrahigh-loading bimetallic AgCu–C3N4 SAC was prepared by an ion exchange method using K-intercalated g-C3N4 (C3N4-K) as a starting material (Fig. S1†). As shown in Fig. S2,† the C3N4-K synthesized by the molten salt method has a nanosheet morphology under transmission electron microscopy (TEM). X-ray diffraction (XRD) patterns show an intercalation peak emerging at 7.8°, and the most intense peak, corresponding to the (002) plane of g-C3N4, shifts to a higher angle in C3N4-K compared to pristine g-C3N4. These are evident in the successful intercalation of K+ ions, supported by the K 2p XPS signals (Fig. S3†).23,24 Incubation of C3N4-K with silver and copper ammonia solution initiates cationic exchange between Cu2+/Ag+ and K+ ions, where the larger-sized Ag+ and Cu2+ species are incorporated in the exfoliated C3N4 nanosheet.25,26 Subsequent thermal treatment of the SAC precursors under the N2 atmosphere enhances the coordination between Cu/Ag and substrate, giving a stable bimetallic AgCu–C3N4 SAC. By adjusting the concentration of Ag and Cu ammonia complex, bimetallic SACs with various Ag and Cu loadings up to 9 wt% Ag and 12 wt% Cu, as well as monometallic SACs with Ag (Ag–C3N4) up to 9 wt%, and Cu (Cu–C3N4) up to 21 wt% can be obtained for further exploration of their catalytic performance in alkyne semi-hydrogenation (Table S2†).Characterization of bimetallic AgCu–C3N4 SACs
Unless otherwise specified, the as-prepared bimetallic AgCu–C3N4 SAC (9 wt% Ag and 12 wt% Cu) exhibits good atomic dispersion and shows a similar morphology to C3N4-K. Fig. S4† demonstrates the retained nanosheet structure of the material as evidenced by TEM. Additionally, no visible particle can be identified within the nanosheet in AgCu–C3N4 SAC, consistent with the absence of diffraction peaks assignable to Ag or Cu nanoparticles in XRD patterns (Fig. S2†). Scanning transmission electron microscopy (STEM) in the annular dark field imaging mode (STEM-ADF) further verifies the atomic dispersion of Ag and Cu without detectable aggregation throughout the nanosheet in Fig. 1A. Energy-dispersive X-ray spectroscopy (EDS) analysis in Fig. 1B illustrates the co-existence of Ag and Cu species with high elemental uniformity on g-C3N4. Typical Ag and Cu sites can be identified in atomic-resolution STEM-ADF images and are marked with pink and green circles, respectively, in Fig. 1C. The statistical histogram of 196 atoms in Fig. 1D confirms a Z-contrast difference of 1.72 for Ag/Cu on the g-C3N4 support. This is reasonable, given their difference in atomic mass and the imaging condition used. Taking the atomic ratio of Cu/Ag (∼2.25) within the g-C3N4 support into consideration, the inter-distance of heteronuclear Ag–Cu is calculated from the average distance of one Ag atom to the nearest three Cu atoms, which is <0.65 ± 0.01 nm for AgCu–C3N4 SAC in Fig. 1E. We have also calculated the corresponding homonuclear Ag–Ag (1.36 ± 0.03 nm) and Cu–Cu distances (0.68 ± 0.01 nm) in the same sample. The Cu–Cu distance in AgCu–C3N4 is similar to our previous report for 21 wt% Cu–C3N4 SAC (0.74 ± 0.13 nm, no activity).18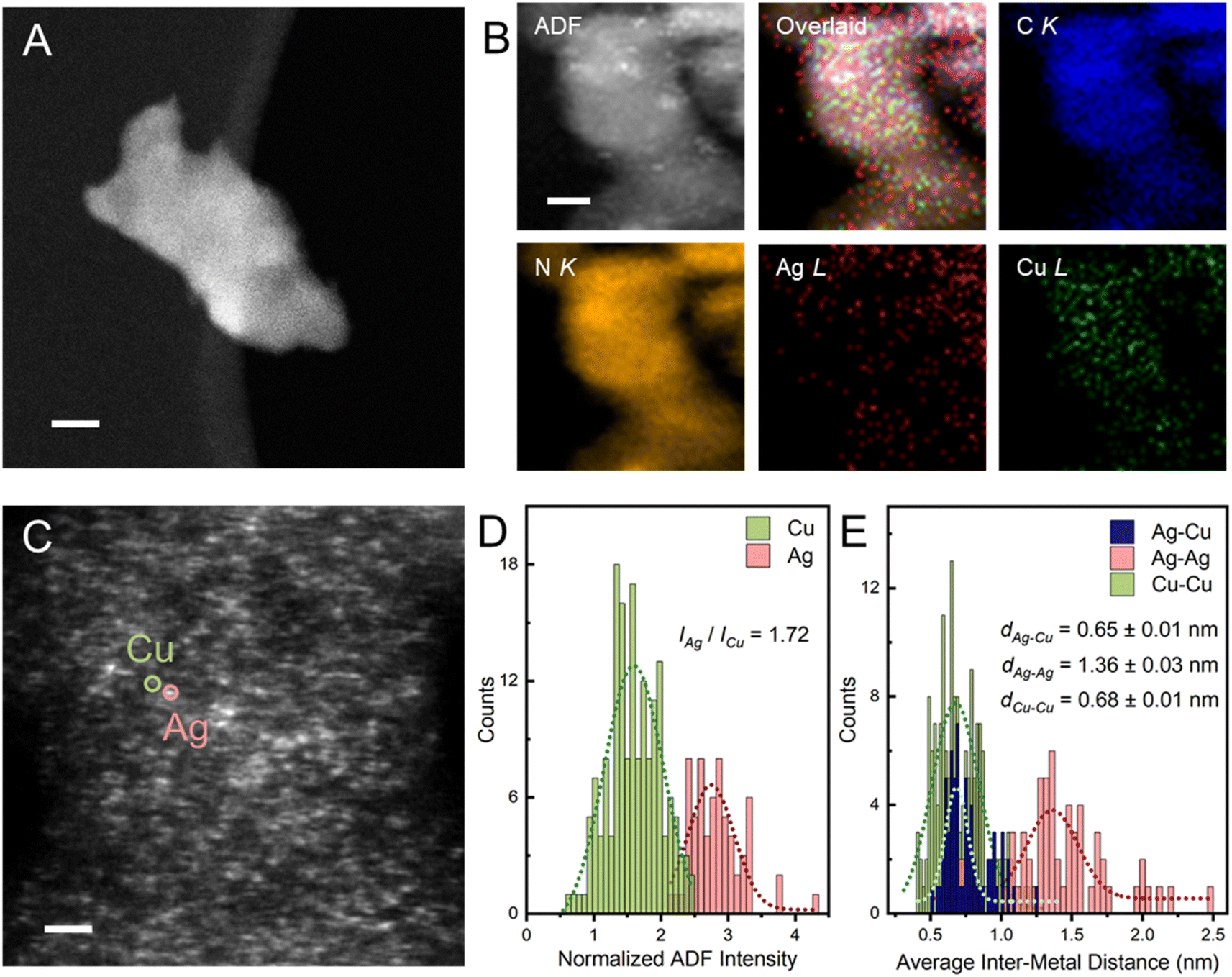 | ||
| Fig. 1 Microscopic characterizations of bimetallic AgCu–C3N4 SACs. (A) HAADF-STEM image of AgCu–C3N4 shows no particle aggregation. (B) EDS mapping of Ag, Cu, C, and N in AgCu–C3N4. (C) Atomic resolution HAADF-STEM image of AgCu–C3N4 SAC. (D) ADF intensity histogram of Ag and Cu atom sites of AgCu–C3N4. (E) Statistic distance distribution of the Ag–Cu, Ag–Ag, and Cu–Cu neighbors in the AgCu–C3N4 SACs from HAADF-STEM images, where 65 Ag atoms and 131 Cu atoms are considered. Samples with significant variation in thickness were pre-treated with ion-beam thinning (Ar+) for seconds to obtain very thin layers for high-resolution imaging. The Ag and Cu atomic coordinates are extracted by 2D-Gaussian fitting. The inter Ag–Ag, Cu–Cu, and Ag–Cu distances are calculated from the average distance from one metal to the nearest three homo/hetero metal atoms. The typical Ag and Cu single atoms are marked with pink and green circles. Scale bar: (A) 20 nm; (B and C) 2 nm. | ||
The electronic structure and coordination environment of mono and bimetallic SACs were first examined by X-ray photoelectron spectroscopy (XPS). Both AgCu–C3N4 and Ag–C3N4 SAC (Ag loading: 9 wt%) present two peaks at 368.3 and 374.3 eV in the Ag 3d XPS spectra (Fig. 2A), corresponding to the Ag 3d5/2 and Ag 3d3/2 of the AgI species, respectively. Auger spectra were further examined to distinguish between the Ag0 and AgI species. The Ag MNN Auger features two peaks at ∼350 and ∼355 eV in AgCu–C3N4 and Ag–C3N4 SAC, respectively.27 This agrees well with those in Ag2O, indicating the presence of AgI species in both SACs. In Cu 2p XPS spectra, the spin–orbit doublet at 932.7 eV for Cu 2p3/2 and 952.5 eV for Cu 2p1/2 (Fig. 2D) are assignable to CuI species in AgCu–C3N4 and Cu–C3N4 (Cu loading: 21 wt%) SACs.28 This result was further supported by X-ray absorption spectroscopy (XAS). In Ag K-edge X-ray absorption near-edge structure (XANES) spectra, the AgCu–C3N4 and Ag–C3N4 SACs show a white line intensity close to the Ag2O reference in Fig. 2B, suggesting an oxidation state of +1. Additionally, AgCu–C3N4 and Cu–C3N4 SACs exhibit similar absorption energy in the rising edge in the Cu K-edge XANES spectra in Fig. 2C, which agrees with the CuI species in Cu2O. The slight shift towards higher energy in absorption edge for SACs may arise from surface absorption of the O2 molecules in ambient conditions.18,29 The Fourier-transformed extended X-ray absorption fine structure (FT-XAFS) of AgCu–C3N4 and Ag–C3N4 SACs shows a dominant peak at ∼1.7 Å for the Ag–N scattering path (Fig. 2E), which is distinguished from the oxide (∼1.5 Å) and metallic species (∼2.7 Å). Meanwhile, AgCu–C3N4 and Cu–C3N4 SACs in Fig. 2F display a prominent peak at ∼1.5 Å from the scattering of the first coordination shell (Cu–N), in comparison with chloride (∼1.9 Å) and oxide (2nd shell coordination at ∼2.8 Å) and metallic species (∼2.5 Å). No metallic Ag–Ag, Cu–Cu, or Ag–Cu peak at >2.2 Å was observed for AgCu–C3N4 SAC, suggesting that Ag and Cu exist as isolated single atoms, consistent with the HAADF-STEM and XRD data.
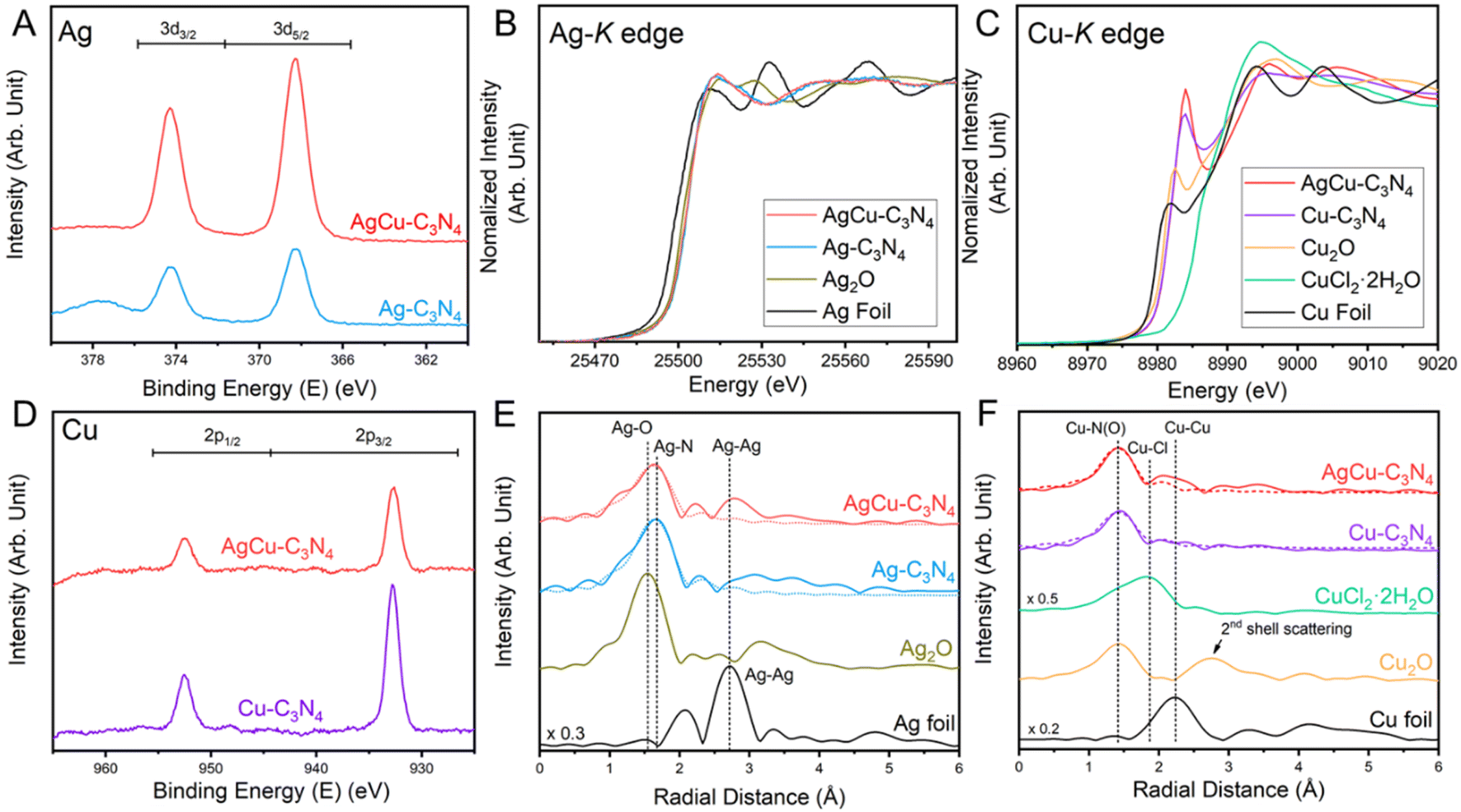 | ||
| Fig. 2 Electronic structure of AgCu–C3N4 SAC. (A) Ag3d XPS, (B) Ag K-edge XANES spectra, (C) Cu K-edge XANES spectra, (D) Cu2p XPS, (E) Ag and (F) Cu FT-EXAFS spectra of AgCu–C3N4 SACs. The dotted lines represent the FT-fitting curves. | ||
Alkyne semi-hydrogenation using AgCu–C3N4 SAC
We selected the liquid-phase hydrogenation of 4-ethynylanisole (1a) by ammonia borane complex (NH3·BH3) as a model reaction to evaluate the catalytic performance of AgCu–C3N4 SACs. As shown in Fig. 3A, pristine C3N4-K demonstrated no activity for alkyne semi-hydrogenation owing to the lack of metal sites for ammonia borane decomposition. Among all catalysts examined, AgCu–C3N4 (9 wt% Ag, 12 wt% Cu) yielded the highest selectivity and conversion (>99%) towards alkene 2a at room temperature for 20 hours. Monometallic SAC composites such as Ag–C3N4 (9 wt% Ag), Cu–C3N4 SACs (21 wt% Cu), and their physical mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, w/w) demonstrated poor or no performance (<5%). Particle catalysts such as commercial 10% Cu/C and Ag NP-C3N4 (23 wt%, a mixture of Ag nanoparticles and single atoms, Fig. S10†) also failed to catalyze the alkyne to alkene transformation. A homogeneous variant using AgNO3 and Cu(NO3)2 mixture gave poor activity (∼10%), even though it can provide free-moving Ag+ and Cu2+ ions. This probably arises from the deactivation of the catalyst due to the reduction of homogeneous Cu2+ and Ag+ ions in nanoparticles in a reducing environment. These results suggest that the coexistence of Ag and Cu single atoms on the C3N4 support is essential for the reaction. The stability test suggested the excellent catalytic performance of AgCu–C3N4 SAC without significant decay after 5 cycles. The structural stability of the spent catalyst was confirmed by TEM and XAS, where the atomic dispersion nature and identical coordination environment to fresh catalyst could be retained in view of their similar XANES features and FT-EXAFS profile in Fig. S11.†
:1, w/w) demonstrated poor or no performance (<5%). Particle catalysts such as commercial 10% Cu/C and Ag NP-C3N4 (23 wt%, a mixture of Ag nanoparticles and single atoms, Fig. S10†) also failed to catalyze the alkyne to alkene transformation. A homogeneous variant using AgNO3 and Cu(NO3)2 mixture gave poor activity (∼10%), even though it can provide free-moving Ag+ and Cu2+ ions. This probably arises from the deactivation of the catalyst due to the reduction of homogeneous Cu2+ and Ag+ ions in nanoparticles in a reducing environment. These results suggest that the coexistence of Ag and Cu single atoms on the C3N4 support is essential for the reaction. The stability test suggested the excellent catalytic performance of AgCu–C3N4 SAC without significant decay after 5 cycles. The structural stability of the spent catalyst was confirmed by TEM and XAS, where the atomic dispersion nature and identical coordination environment to fresh catalyst could be retained in view of their similar XANES features and FT-EXAFS profile in Fig. S11.†
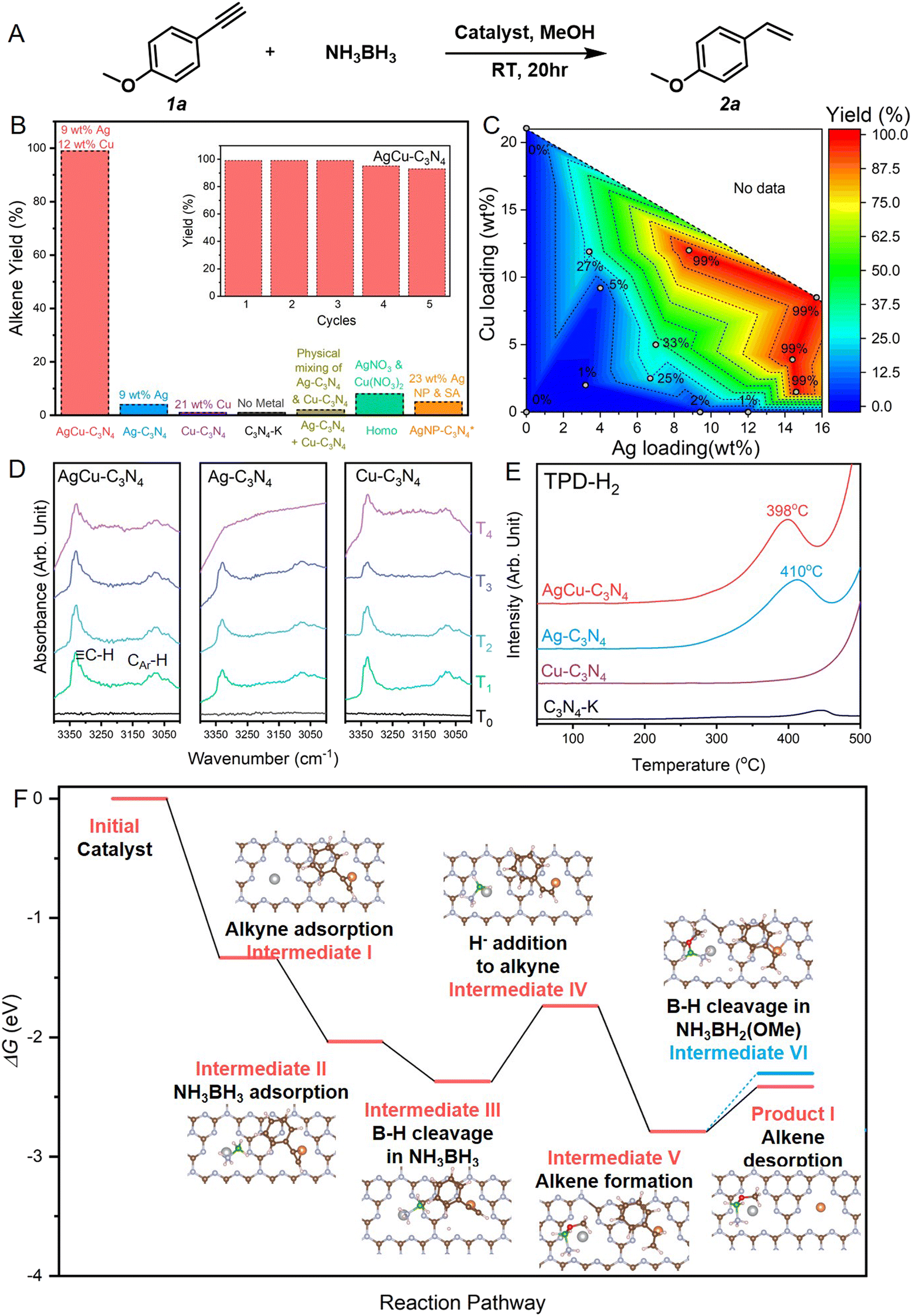 | ||
| Fig. 3 Alkyne semi-hydrogenation using bimetallic AgCu–C3N4 SACs. (A) Reaction scheme for the model alkyne semi-hydrogenation reaction. (B) Catalyst screening for the alkyne semi-hydrogenation. The inset shows the cycling stability of AgCu–C3N4 SAC. The 23 wt% Ag NP-C3N4 catalyst labelled with * contains Ag single atoms and clusters. (C) Dependence of catalytic performance on the Ag and Cu loading for mono- and bimetallic SACs. The metal amount charged to the reaction system was kept the same (8 mol%) for catalysts with different metal loadings. (D) DRIFTS spectra for phenylacetylene adsorption of AgCu–C3N4, Ag–C3N4 and Cu–C3N4 SACs, where black, green, sky blue, blue and purple lines refer to the FTIR spectra of fresh catalysts (T0), catalysts with 2 hours phenylacetylene adsorption (T1), catalysts with argon purging for 30 min at room temperature after phenylacetylene adsorption (T2), catalysts with subsequent argon purging for 30 min at 60 °C (T3), and catalysts with subsequent argon purging for 30 min at 110 °C (T4), respectively. (E) TPD-H2 profiles of AgCu–C3N4, Ag–C3N4, Cu–C3N4 SACs and C3N4-K. (F) DFT-calculated energy pathway for hydrogenation of 4-ethynylanisole by NH3BH3. Intermediates on the Ag1Cu1–C3N4 catalyst labelled in red are involved in the semi-hydrogenation to alkene, whereas the blue one refers to the full hydrogenation towards alkane. Inset shows the optimized configuration of each intermediates. The color scheme used: silver for Ag, orange for Cu, brown for C; grey for N, green for B, red for O, and pink for H. Energy diagram of further hydrogenation towards alkane product is illustrated in Fig. S14.† | ||
To investigate the role of the Ag–Cu bimetallic system in alkyne semi-hydrogenation, we have prepared a series of AgCu–C3N4 SACs at various Ag/Cu loadings for reaction screening. As shown in the contour in Fig. 3C, SACs with low metal loading (3 wt% Ag, 2 wt% Cu) suffered from poor activity. Increasing the Ag loading to 7 wt% (2 wt% Cu) gave a 25% yield of alkene, while further increasing the Cu loading to 5 wt% (7 wt% Ag) did not significantly improve the activity (33%). The increase in metal loading (>18 wt%) promoted higher catalytic efficiency (>99%). It was also noticed that SAC with high silver and low copper loading (15 wt% Ag, 1.5 wt% Cu) gave a 99% alkene yield. In comparison, the one with high copper and low silver loading (4 wt% Ag, 12 wt% Cu) only achieved 23% conversion towards alkene, indicating a direct correlation between the species activated at Ag sites and the rate-determining step (RDS) of the reaction.
We further utilized diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements and temperature-programmed desorption (TPD) to identify the intermediate species on Ag and Cu sites, respectively. In situ variable temperature DRIFTS experiments using phenylacetylene as a probe molecule were performed to detect the dynamic desorption behavior of alkyne on the samples. As shown in Fig. 3D, AgCu–C3N4, Ag–C3N4, and Cu–C3N4 SACs exhibit similar adsorption behavior upon introducing phenylacetylene at room temperature. The strong infrared bands at 3331 cm−1 and 3160–3025 cm−1 are associated with the C–H bond vibration modes from alkyne and aromatic ring originating from the surface adsorbed phenylacetylene. The intensity of these bands declines during argon purging. At 110 °C, the characteristic vibration bands of phenylacetylene are still visible on AgCu–C3N4 and Cu–C3N4 but gradually disappear on Ag–C3N4. This indicates that the strong binding of phenylacetylene on Cu-containing SACs promotes subsequent activation. In our experiments, NH3·BH3 serves as a reductant and provides the hydrogen source. Thus, we conducted H2-TPD measurement to investigate the relative strengths of hydrogen absorption on the surfaces of mono- and bimetallic SACs. As shown in Fig. 3E, the H2 desorption temperature of 398 °C on AgCu–C3N4 and 410 °C on Ag–C3N4 are higher than those reported on Ag particle catalysts, which can be attributed to strong binding of H with the atomic Ag+ sites. No prominent H2 desorption peak was detected for the Cu SACs, suggesting Ag sites bind more strongly to H than Cu sites. This is consistent with the adsorption energies calculated by DFT. Alkyne preferably binds on Cu while hydrogen binds more strongly on Ag (Fig. S12†).
It is noteworthy that the reaction will not proceed with H2 (1 atm) as the hydrogenation agent. This excludes the role of in situ generation of H2 (i.e., homo-cleavage) via Ag-catalysed dehydrogenation of NH3BH3 for subsequent hydrogenation. To better understand the hydrogenation process, we conducted the reaction using MeOD as the solvent. In the alkene product, the vinyl H near aryl can be detected in the 1H NMR spectrum (Fig. S13A†). Additionally, the 11B NMR spectrum of the reaction filtrate (Fig. S13B†) shows that the final product of the B species is B(OCD3)4−,30–32 indicating that the vinyl H close to aryl originates from the Hδ− attached to B. Furthermore, the deuteration of one terminal hydrogen in the alkene product suggests that the other hydrogen source for such semi-hydrogenation is the solvent methanol. This inference is supported by the absence of alkene product when using aprotic solvent instead of methanol (Table S4†).
Based on the experimental results mentioned above, we propose a mechanism for the semi-hydrogenation of alkyne with NH3BH3 catalyzed by AgCu–C3N4 through DFT calculations in Fig. 3F. Firstly, alkyne and NH3BH3 adsorb on the Cu and Ag sites, respectively (intermediate I & II). Then, the cleavage of the B–H bond at the Ag site generates Hδ− (intermediate III). Subsequently, this Hδ− undergoes nucleophilic attack on the alkyne, adding to the internal carbon of the alkyne (intermediate IV). Simultaneously, the O–H bond in MeOH breaks, with Hδ+ adding to the terminal carbon to afford the product alkene (intermediate V), while −OMe binds to the B center. Here, the Hδ− addition to alkyne is identified as the rate determining step (RDS) with an energy barrier of 0.634 eV. This underscores the significance of high loading of Ag, as more Ag sites can generate a higher concentration of Hδ− in the local area. Also, the short Ag–Cu distance further promotes efficient collision of Hδ− with alkyne to afford alkene. Next, the desorption of the alkene requires overcoming a smaller energy barrier (product I) compared to the release of the second Hδ− from the B–H bond (intermediate VI), which is the initial step of alkene hydrogenation. Therefore, semi-hydrogenation to alkene occurs rather than full hydrogenation to alkane.
The highly selective semi-hydrogenation of alkyne can be applied to remove trace amount of alkyne from alkene with an alkene/alkyne ratio as high as 99:1 (Fig. S15†). Several alkyne functionalities were also examined to extend the applicability of AgCu–C3N4 SAC in alkyne semi-hydrogenation. The reaction performs well with terminal aliphatic and aryl alkynes, tolerating boronate (2b), ester (2c), amine (2d), and hydroxyl (2e) functional groups in the starting material (Fig. 4A). It is worth noting that the successful semi-hydrogenation from pargyline to 2d in a clean and mild manner suggests excellent scope for synthesizing alkene-containing drugs via modification of alkynes. Semi-hydrogenation of internal alkynes can also be achieved. However, due to steric hindrance from the groups at both terminals,33 the activation of internal alkynes is more challenging. It requires a longer reaction time for complete transformation than terminal alkynes (Fig. 4B).
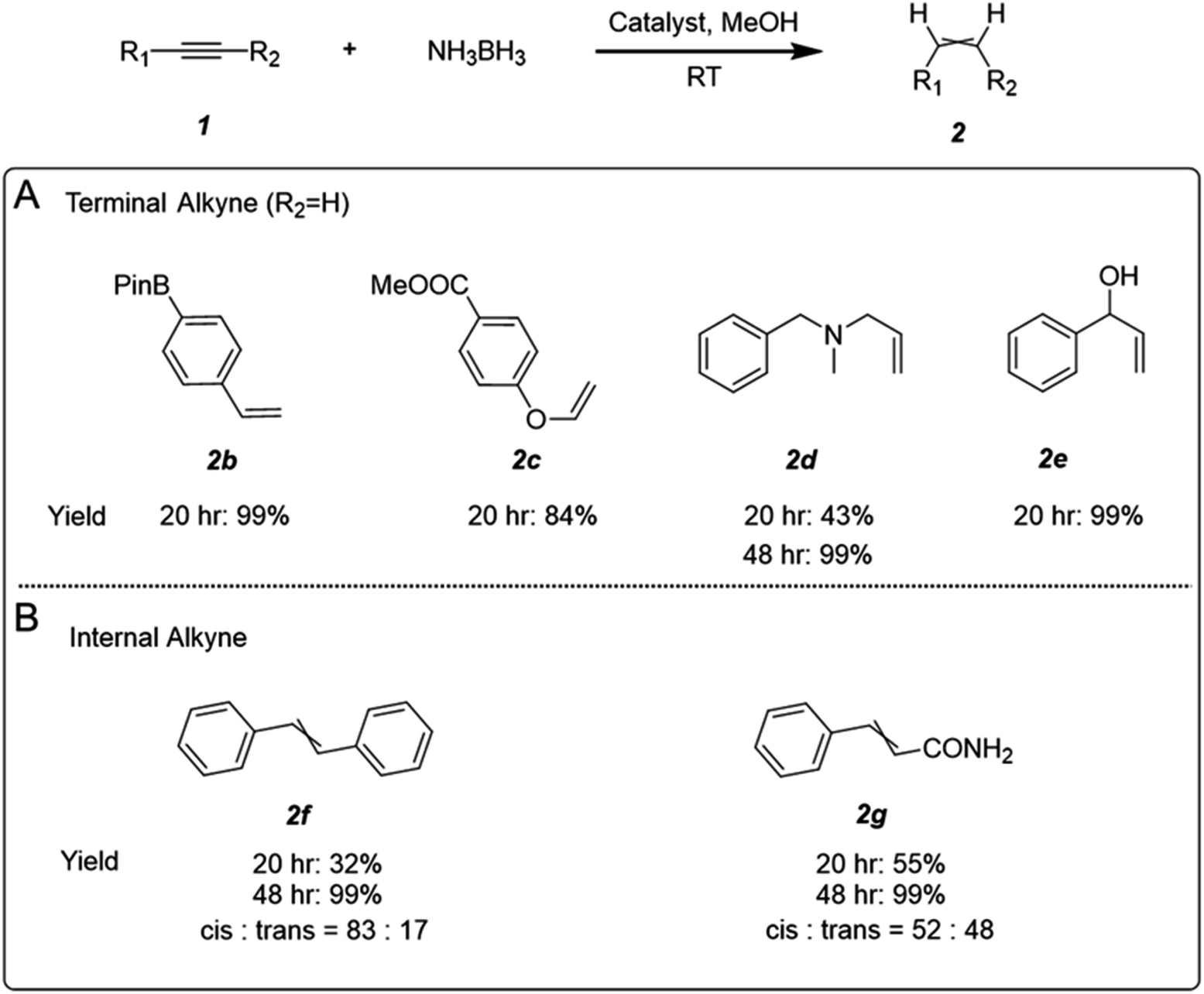 | ||
| Fig. 4 The substrate scope of the AgCu–C3N4 SAC catalysed alkyne semi-hydrogenation. (A) Multifunctional terminal aliphatic alkyne and aryl alkynes containing borate, amine, hydroxyl groups, and (B) internal alkynes can be processed with AgCu–C3N4 SAC. | ||
Conclusion
In summary, we have successfully synthesized bimetallic AgCu–C3N4 SAC, where the optimal composition for semi-hydrogenation of alkyne was 9 wt% Ag and 12 wt% Cu. Bimetallic SAC integrates the functionality of Ag SAC for hydrogen activation and Cu SAC for alkyne activation. By achieving high metal loadings, the inter-site distance is sufficiently small to facilitate the transfer of activated species, rendering excellent catalytic activities and selectivity in alkyne semi-hydrogenation towards alkene. Our work highlights the importance of high catalyst loading for reducing the inter-site distance between hetero-metal atoms in multi-metallic SACs. It provides a new strategy for designing efficient SACs to manufacture fine chemicals and pharmaceuticals.Methods
Synthesis of K-intercalated g-C3N4 (C3N4-K)
20 g of melamine was loaded into a covered crucible and heated in a muffle furnace at 550 °C for 4 h at 5 °C min−1 and then allowed to naturally cool down to room temperature to yield g-C3N4 as a light yellow solid. 1 g of the prepared g-C3N4 was ground into fine powder first and then further fine ground with a mixture of 1.10 g KCl and 0.90 g LiCl in an agate mortar. The mixture was then loaded into a covered quartz boat, transferred to a tube furnace under 50 sccm of Ar gas, and then heated at 550 °C for 4 h at 5 °C min−1. After cooling to room temperature, excessive salts were removed by repeated washing with deionized water and ethanol. Finally, the sample was dried at 60 °C in vacuum for 24 h to yield bright yellow powder.Synthesis of bimetallic AgCu–C3N4 SACs
For a typical loading of 9 wt% Ag and 12 wt% Cu, 200 mg of C3N4-K was first sonicated to disperse in deionized water and then slowly added with 3 mL of silver ammonia solution (72 mg of AgCl dissolved in 6.5 mL of 6.5% ammonia solution) and 3 mL of copper ammonia solution (15 mL of 0.1 M CuCl2 with 0.8 mL of 28% ammonia solution).34,35 The mixed suspension was stirred for 24 h at room temperature and then centrifuged. The centrifuged sediment was washed with deionized water and ethanol for a few times. After drying at 40 °C in vacuum for 24 h, the green powder was loaded into a covered quartz boat, transferred to a tube furnace under 50 sccm of Ar gas, heated at 550 °C for 4 h at 5 °C min−1, and allowed natural cooling down to room temperature to obtain the AgCu–C3N4 SAC. The Ag and Cu loading (including Ag–C3N4 and Cu–C3N4) can be tuned by the amount of silver and copper ammonia solution.Alkyne semi-hydrogenation using AgCu–C3N4 SACs
Typically, 8 mol% (regarding metal) of catalyst (or 5 mg of C3N4-K) was loaded in a pre-dried vial containing solvent (MeOH, 1 mL), alkyne (0.1 mmol), and ammonia borane complex (NH3·BH3, 0.5 mmol, 15 mg), and stirred at room temperature for 20 h. Then, the solid catalyst was separated, and the reaction suspension was treated with 1 M HCl (5 mL) and stirred vigorously to quench the excess NH3·BH3. The crude product was extracted with ethyl acetate (3 × 5 mL). The combined organic layer was washed with brine (8 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The crude product was analysed by GC-MS, and the pure product was obtained by flash column chromatography on silica gel or TLC plate.H2 temperature-programmed desorption (TPD)
H2-TPD was carried out on a Microtrac BELCat II instrument with a thermal conductivity detector (TCD). Typically, 40 mg of the sample was pre-dried at 80 °C for 1 h and cooled to 50 °C in He flow. An H2/Ar mixture (10 vol%) was introduced into the reactor at 50 mL min−1 and kept at 50 °C for 1 h. The sample was then swept with pure Ar for 30 min to remove physically adsorbed H2. Subsequently, the sample was heated in Ar up to 500 °C at 5 °C min−1, while the desorbed species was detected by TCD.Phenylacetylene DRIFTS measurement
DRIFTS measurements were performed on a Thermo Scientific Nicolet iS50 instrument with a Hg–Cd–Te (MCT) detector and a Praying Mantis high-temperature reaction chamber with ZnSe windows. The powder sample was placed in an in situ pool and pretreated in Ar at 20 mL min−1 at room temperature. Background spectrum was acquired until stable. After that, phenylacetylene was purged to the reaction cell by a mixture with Ar for 120 min at 50 mL min−1. Then, the gas was switched to pure Ar and purged at room temperature for 30 minutes before collecting DRIFTS spectra. DRIFTS spectra were also collected for the subsequent heating in Ar at 60 °C for 30 min and 100 °C for 30 min, respectively.Material characterization
The following equipment was used: STEM (JEOL ARM200F equipped with ASCOR probe corrector at 80 kV),36 TEM (JEM 2010F, 200 kV), XPS (AXIS UltraDLD, monochromatic Al Kα), XRD (Bruker D8), ICP-OES (PerkinElmer Avio 500, ppm level accuracy). XANES/EXAFS: 50 mg of sample was fine ground using a mortar and pestle before being pressed into an 8 mm pellet. Measurements were carried out at the RIKEN SPring-8 Center (RSC), the Australian Nuclear Science & Technology Organisation (ANSTO), and Singapore Synchrotron Light Sources (SSLS).37 Data analysis and simulation were conducted on Athena, Artemis, and Hephaestus (Version 0.9.23).38Data availability
All data are available from the authors upon reasonable request.Author contributions
J. S. conceived the research, synthesized the materials, conducted catalytic reactions, and wrote the draft under the supervision of Z. C. and K. P. L.; X. C. conducted TEM, STEM characterization, and data analysis; T. W. performed DRIFTS measurement and data analysis under the supervision of N. Y.; Q. H. and S. X. conducted XAS measurements and data processing; Q. H. performed DFT calculations; Z. C., X. C. and Q. H. assisted with materials characterization and data analysis. The draft was written by J. S. and X. C. and revised by Z. C. and K. P. L. All authors discussed and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
J. S. and X. C. contributed equally to this work. K. P. L. acknowledges funding support from NUS's Centre for Hydrogen Innovation program CHI-P2022-01. We appreciate Mr Haohan Li, Ms Jianhua Wu, and Mr Jinhui Pan's assistance in NMR and other experiments.References
- L. Zhang, Y. Ren, W. Liu, A. Wang and T. Zhang, Natl. Sci. Rev., 2018, 5(5), 653 CrossRef CAS.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137(33), 10484 CrossRef CAS PubMed.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. Diaz Fernandez, G. Nalin, D. Świtlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14(12), 1236 CrossRef CAS PubMed.
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6(1), 8550 CrossRef PubMed.
- A. Guthertz, M. Leutzsch, L. M. Wolf, P. Gupta, S. M. Rummelt, R. Goddard, C. Farès, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2018, 140(8), 3156 CrossRef CAS PubMed.
- Z. Wang, A. Garg, L. Wang, H. He, A. Dasgupta, D. Zanchet, M. J. Janik, R. M. Rioux and Y. Román-Leshkov, ACS Catal., 2020, 10(12), 6763 CrossRef CAS.
- T. Mitsudome, M. Yamamoto, Z. Maeno, T. Mizugaki, K. Jitsukawa and K. Kaneda, J. Am. Chem. Soc., 2015, 137(42), 13452 CrossRef CAS PubMed.
- Z. Chen, J. Song, R. Zhang, R. Li, Q. Hu, P. Wei, S. Xi, X. Zhou, P. T. T. Nguyen, H. M. Duong, P. S. Lee, X. Zhao, M. J. Koh, N. Yan and K. P. Loh, Nat. Commun., 2022, 13(1), 2807 CrossRef CAS PubMed.
- D. Teschner, Z. Révay, J. Borsodi, M. Hävecker, A. Knop-Gericke, R. Schlögl, D. Milroy, S. D. Jackson, D. Torres and P. Sautet, Angew. Chem., Int. Ed., 2008, 47(48), 9274 CrossRef CAS PubMed.
- H. Lindlar, Helv. Chim. Acta, 1952, 35(2), 446 CrossRef CAS.
- J. Prinz, C. A. Pignedoli, Q. S. Stöckl, M. Armbrüster, H. Brune, O. Gröning, R. Widmer and D. Passerone, J. Am. Chem. Soc., 2014, 136(33), 11792 CrossRef CAS PubMed.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144(40), 18155 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120(21), 11703 CrossRef CAS PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, P. Ren, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, J. Am. Chem. Soc., 2018, 140(41), 13142 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54(38), 11265 CrossRef PubMed.
- P. Wu, S. Tan, J. Moon, Z. Yan, V. Fung, N. Li, S.-Z. Yang, Y. Cheng, C. W. Abney, Z. Wu, A. Savara, A. M. Momen, D.-e. Jiang, D. Su, H. Li, W. Zhu, S. Dai and H. Zhu, Nat. Commun., 2020, 11(1), 3042 CrossRef CAS PubMed.
- Y. Liu, B. Wang, Q. Fu, W. Liu, Y. Wang, L. Gu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60(41), 22522 CrossRef CAS PubMed.
- J. Song, Z. Chen, X. Cai, X. Zhou, G. Zhan, R. Li, P. Wei, N. Yan, S. Xi and K. P. Loh, Adv. Mater., 2022, 34(33), 2204638 CrossRef CAS PubMed.
- B. Wang, C. Cheng, M. Jin, J. He, H. Zhang, W. Ren, J. Li, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61(33), e202207268 CrossRef CAS PubMed.
- L. Wang, J. Wu, S. Wang, H. Liu, Y. Wang and D. Wang, Nano Res., 2024, 17(4), 3261 CrossRef CAS.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60(11), 6170 CrossRef CAS PubMed.
- Z. Shen, Y. Yu, Z. Zhao, M. A. Mushtaq, Q. Ji, G. Yasin, L. N. U. Rehman, X. Liu, X. Cai, P. Tsiakaras and J. Zhao, Appl. Catal., B, 2023, 331, 122687 CrossRef CAS.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57(30), 9372 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, ACS Catal., 2016, 6(6), 3921 CrossRef CAS.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17(2), 174 CrossRef CAS PubMed.
- Z. Zeng, Y. Su, X. Quan, W. Choi, G. Zhang, N. Liu, B. Kim, S. Chen, H. Yu and S. Zhang, Nano Energy, 2020, 69, 104409 CrossRef CAS.
- K. Hakouk, P. Deniard, L. Lajaunie, C. Guillot-Deudon, S. Harel, Z. Wang, B. Huang, H.-J. Koo, M.-H. Whangbo, S. Jobic and R. Dessapt, Inorg. Chem., 2013, 52(11), 6440 CrossRef CAS PubMed.
- T. Yang, X. Mao, Y. Zhang, X. Wu, L. Wang, M. Chu, C.-W. Pao, S. Yang, Y. Xu and X. Huang, Nat. Commun., 2021, 12(1), 6022 CrossRef CAS PubMed.
- Z. Chen, J. Song, X. Peng, S. Xi, J. Liu, W. Zhou, R. Li, R. Ge, C. Liu, H. Xu, X. Zhao, H. Li, X. Zhou, L. Wang, X. Li, L. Zhong, A. I. Rykov, J. Wang, M. J. Koh and K. P. Loh, Adv. Mater., 2021, 33(34), 2101382 CrossRef CAS PubMed.
- E. Vasilikogiannaki, I. Titilas, G. Vassilikogiannakis and M. Stratakis, Chem. Commun., 2015, 51(12), 2384 RSC.
- Q. Zhao, J. Li, E. J. M. Hamilton and X. Chen, J. Organomet. Chem., 2015, 798, 24 CrossRef CAS.
- L. Li, W. Yang, Q. Yang, Q. Guan, J. Lu, S.-H. Yu and H.-L. Jiang, ACS Catal., 2020, 10(14), 7753 CrossRef CAS.
- G. J. Baker, A. J. P. White, I. J. Casely, D. Grainger and M. R. Crimmin, J. Am. Chem. Soc., 2023, 145(13), 7667 CrossRef CAS PubMed.
- T. Shoeib, R. K. Milburn, G. K. Koyanagi, V. V. Lavrov, D. K. Bohme, K. W. M. Siu and A. C. Hopkinson, Int. J. Mass Spectrom., 2000, 201(1), 87 CrossRef CAS.
- W. Burchard, N. Habermann, P. Klüfers, B. Seger and U. Wilhelm, Angew. Chem., Int. Ed. Engl., 1994, 33(8), 884 CrossRef.
- X. Cai, X. Chen, Z. Ying, S. Wang, Y. Chen, Y. Cai, G. Long, H. Liu and N. Wang, Mater. Des., 2021, 210, 110080 CrossRef CAS.
- Y. Du, Y. Zhu, S. Xi, P. Yang, H. O. Moser, M. B. H. Breese and A. Borgna, J. Synchrotron Radiat., 2015, 22(3), 839 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12(4), 537 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02469a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |