
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-heteroatom doped nanographenes: enhancing photosensitization capacity by forming an electron donor–acceptor architecture†
Ranran
Li‡
a,
Bin
Ma‡
a,
Meng
Li
a,
Dan
Wang
a,
Peng
Liu
a and
Peng
An
 *ab
*ab
aSchool of Chemical Science and Technology Yunnan University, Kunming 650091, P. R. China
bKey Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan University, Kunming 650091, P. R. China. E-mail: anp@ynu.edu.cn
First published on 17th June 2024
Abstract
Systematically tuning and optimizing the properties of synthetic nanographenes (NGs) is particularly important for NG applications in diverse areas. Herein, by devising novel electron donor–acceptor (D–A) type structures, we reported a series of multi-heteroatom-doped NGs possessing an electron-rich chalcogen and electron-deficient pyrimidine or pyrimidinium rings. Comprehensive experimental and theoretical investigations revealed significantly different physical, optical, and energetic properties compared to the non-doped HBC or chalcogen-doped, non-D–A analogues. Some intriguing properties of the new NGs such as unique electrostatically oriented molecular stacking, red-shifted optical spectra, solvatochromism, and enhanced triplet excitons were observed due to the formation of the D–A electron pattern. More importantly, these D–A type structures can serve as photosensitizers to generate efficiently reactive-oxygen species (ROS), and the structure-related photosensitization capacity that strengthens the electron transfer (ET) process leads to significantly enhanced ROS which was revealed by experimental and calculated studies. As a result, the cell-based photodynamic therapy (PDT) indicated that the cationic NG 1-Me+ is a robust photosensitizer with excellent water-solubility and biocompatibility.
Introduction
Synthetic nanographenes (NGs), as large polycyclic aromatic hydrocarbons (PAHs) or polycyclic conjugated hydrocarbons (PCHs) with a nanoscale size, have fascinated synthetic chemists in recent decades.1 Due to their aesthetic structures and intriguing physicochemical properties, so far, a variety of planar and non-planar NGs have been developed by bottom-up strategies and widely applied in various branches of science, such as optoelectronic devices,2 biological probes,3 supramolecular systems,4 and so on. As a benchmark for NGs, the hexa-peri-hexabenzocoronene (HBC), a D6h−symmetric planar π scaffold with 42 sp2 carbon atoms, has garnered significant attention in terms of novel nanocarbon materials,2a,5 as well as a building block for large-size NGs with different topological structures.4c,6 However, HBC generally exhibits relatively monotonous photophysical properties and aggregation-caused low solubility in water and other solvents, which sometimes hampers its application, particularly in the biological area. Accordingly, the conventional method to increase the solubility is to add hydrophilic substituents at the peripheral positions.7 Alternatively, the insertion of heteroatoms into NG frameworks, so-called heteroatom-doping, is a powerful tool to modulate the properties of NGs such as three-dimensional structures, chemical reactivity, electronic energy gap, photophysical behaviours, and water solubility.8 Therefore, many efforts have been devoted to developing heteroatom-doped NGs, for example, heteroatom-doped, HBC-like structures.9Electron donor–acceptor (D–A) type organic molecules possessing both electron-donating and electron-withdrawing groups separated by a conjugated π system represent commonly adopted scaffolds for versatile materials like organic photoredox catalysts, organic fluorophores, and organic solar cells.10 Such D–A type molecules provide tremendous intriguing physicochemical properties such as charge or energy transfer in the excited state, redistribution of frontier orbitals, and intensive low-energy behaviours which generally provide red-shifted spectra, solvatochromism, enhanced populations of the triplet excited state, and so on.10,11 Meanwhile, the increased dipole moment and potential charge separation of the D–A skeleton can significantly decrease the intermolecular π–π stacking and increase the solvent–solute interactions, leading to significantly higher solubility in polar solvents, like water. Some typical synthetic nanocarbon structures employing a D–A architecture are the cycloparaphenylenes (CPPs), which are also known as carbon nanohoops.12 As shown in Fig. 1a, by insertion of different electron acceptors like pyridinium, benzothiadiazole, anthraquinone, and phthalimide groups or an electron donor, a variety of D–A type CPPs were constructed, which bring about new features in electronic and optical properties.13 However, this D–A pattern seldom appeared in synthetic nanographenes, which are usually designed as symmetrical, non-polar structures.
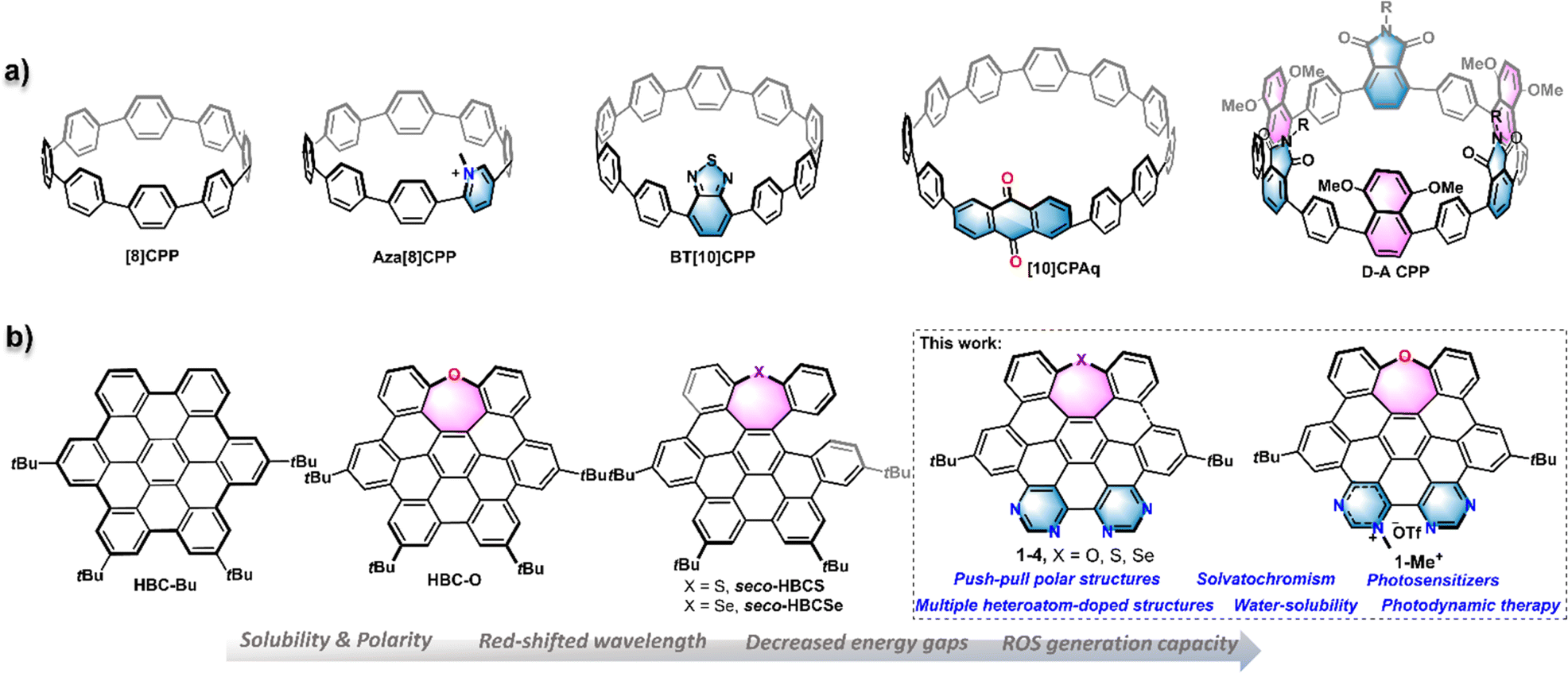 | ||
| Fig. 1 (a) Structure of [8]-cycloparaphenylene (CPP) and representative structures of D–A type CPPs. (b) Structure of tert-butyl-substituted hexa-peri-hexabenzocoronene (HBC)–HBC-Bu, chalcogen-doped HBC or seco-HBCs (HBC-O, seco-HBCS, and seco-HBCSe), and D–A type, and chalcogen/nitrogen-doped nanographenes (1–4, and 1-Me+) described in the current study. | ||
We recently reported a series of chalcogen-doped NGs HBC-O, seco-HBCS, and seco-HBCSe by incorporation of one chalcogen as an electron donor into the HBC backbone (Fig. 1b).14 Compared to HBC, such heteroatom-doped, nonplanar structures exhibit improved solubility and tunable electrooptical properties. To incorporate the well-known D–A pattern into the HBC-like nanographenes, we herein disclose a series of multi-heteroatom doped nanographenes 1–4 and 1-Me+ that exhibit the electron D–A architecture by the introduction of pyrimidine or pyrimidinium electron acceptors into the above-mentioned chalcogen-doped, electron-rich NGs (Fig. 1b). Their overall experimental and theoretical studies about structural geometry, photophysical properties, photosensitization capacity, and biocompatibility were presented and compared with those of some reference structures like HBC-Bu, and HBC-O (Fig. 1b). Specifically, the resulting D–A structures exhibited significant water solubility, thereby enabling the intracellular study. In contrast to HBC or mono-chalcogen-doped NGs, these new molecules exhibit red-shifted spectra and pronounced solvatochromism, which would facilitate long-wavelength light excitation. Particularly, they displayed remarkable photosensitization capacity to generate reactive oxygen species (ROS), especially for the cationic NG 1-Me+ arranged with an electron-withdrawing pyrimidinium unit. Possessing appropriate water solubility and extraordinary light absorption capability, NG 1-Me+ exhibited excellent cell compatibility and can efficiently kill cancer cells by generating ROS in vivo, which makes it a suitable candidate as a photosensitizer for photodynamic therapy (PDT).
Results and discussion
Synthesis
The synthesis of nanographenes 1–4 and 1-Me+ was carried out as displayed in Scheme 1. The dialdehydes 5–7 embedded with different chalcogens were prepared from SNAr-substitution or chalcogen insertion processes (Scheme S1 and 2†). Intramolecular Benzoin condensation reactions of 5–7 catalyzed by N-heterocyclic carbene with further spontaneous oxidation provided diones 8–10 in 65–75% yields. Subsequently, condensation reactions of 8–10 with acetone 11 were performed in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to furnish chalcogen embedded tetraphenylcyclopentadienones 12–14 in over 60% yield, respectively. Cyclopentadienones 12–14 were then subjected to a Diels–Alder reaction with dipyrimidylacetylene 15 at 230 °C to obtain the precursors 16–18 in 71–85% yields, respectively. The cyclodehydrogenation proceeded by treatment of precursors 16–18 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in the presence of trific acid at room temperature, providing HBC or seco-HBC-like NGs 1–4 in 5–40% yields, which gave similar cyclodehydrogenation results to those previously observed for chalcogen-doped nanographenes.14 To further strengthen the electron-withdrawing ability and improve the water-solubility, a cationic NG was prepared by the methylation reaction of fully fused structure 1 with methyl trifluoromethanesulfonate (TfOMe) at 0 °C. As a result, one regioisomer 1-Me+, suggested by the 2D NOESY experiment (Fig. S2†), selectively formed in 67% yield. All the D–A NGs show reasonable solubility in organic solvents; therefore, they could be unequivocally confirmed utilizing 1H- and 13C-NMR spectroscopy and high-resolution mass spectrometry (NMR spectra see the ESI†).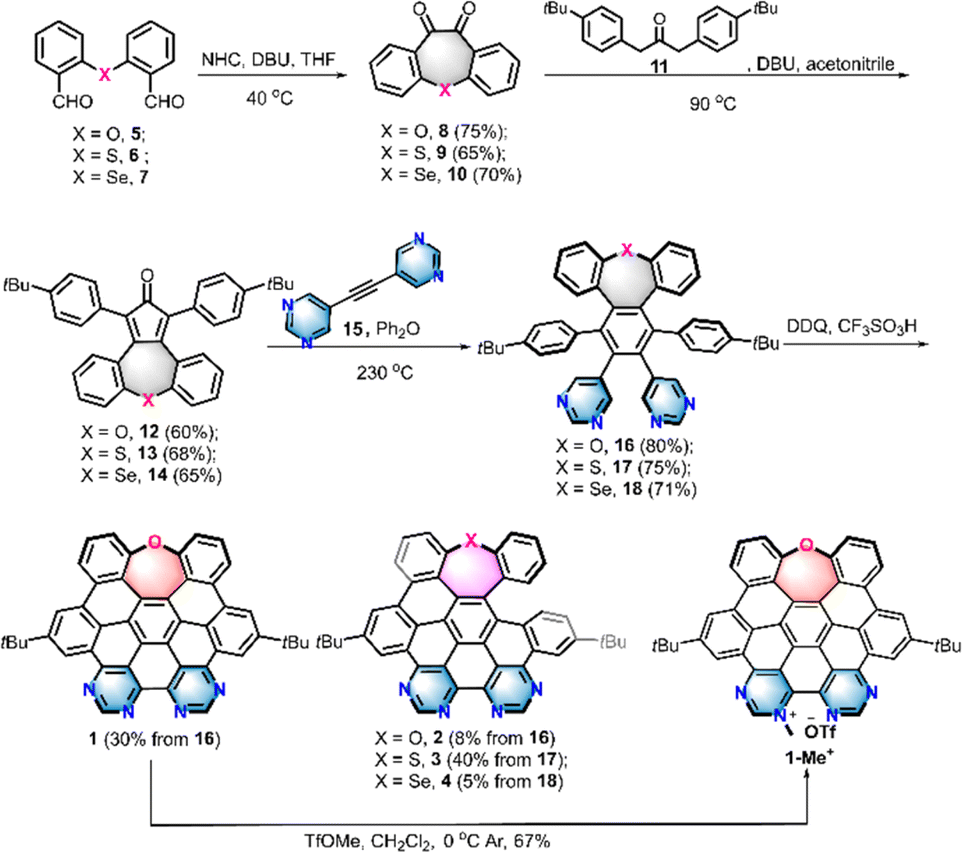 | ||
| Scheme 1 Synthesis of heteroatom-doped NGs 1, 2, 3, 4, and 1-Me+. | ||
Structural analysis
Single crystals of the fully cyclized structure 1 and seco-type NGs 2 and 4 suitable for X-ray diffraction analysis were obtained by slowly evaporating a chloroform–methanol solution of 1 and dichloromethane–methanol solutions of 2 and 4, respectively (Table S1†).15 As depicted in Fig. 2a, the X-ray crystallographic analysis of 1 reveals a twisted, negatively curved structure in which the oxygen atom protrudes from the π surface. Unlike the saddle-shaped solid structure of HBC-O,14 the twisted geometry of 1 revealed an asymmetrical torsional conformation, in which the torsion angles were 10.1 and 5.5° for C7–C8–C9–C10 and C11–C12–C13–C14, respectively. Moreover, the seven-membered oxepine ring exhibited an envelope conformation, in which the sum of seven angles is 852° (Fig. S3†), nearly 50° less than 900°, suggesting the nonplanarity of NG 1. The bond lengths of the oxepine ring adopted an uneven distribution: the two O–C bonds (O1–C1 and O1–C6) and two C–C bonds (C1–C2 and C5–C6) were around 1.40 Å, which were within the common range of carbon–carbon bonds in benzene rings, whereas the bond lengths of C2–C3, C3–C4, and C4–C5 were 1.47, 1.44, and 1.45 Å respectively, significantly longer than that of an aromatic carbon–carbon bond, which indicates a low aromaticity of the oxepine ring. Meanwhile, the valence electron density16 also suggested that the electron density of bonds C2–C3, C3–C4, and C4–C5 together with C8–C9, C12–C13 was remarkably lower than that of other carbon–carbon bonds (Fig. 2e). Therefore, the oxepine ring revealed local antiaromaticity with a calculated nucleus independent chemical shift (NICS)17 in the center of 6.7 as shown in the calculated 3D iso-chemical shielding surface (ICSS) map in Fig. S4.† The NICS(0) and NICS(1) of rings B/C were 0.4 and −3.4, suggesting a low aromaticity.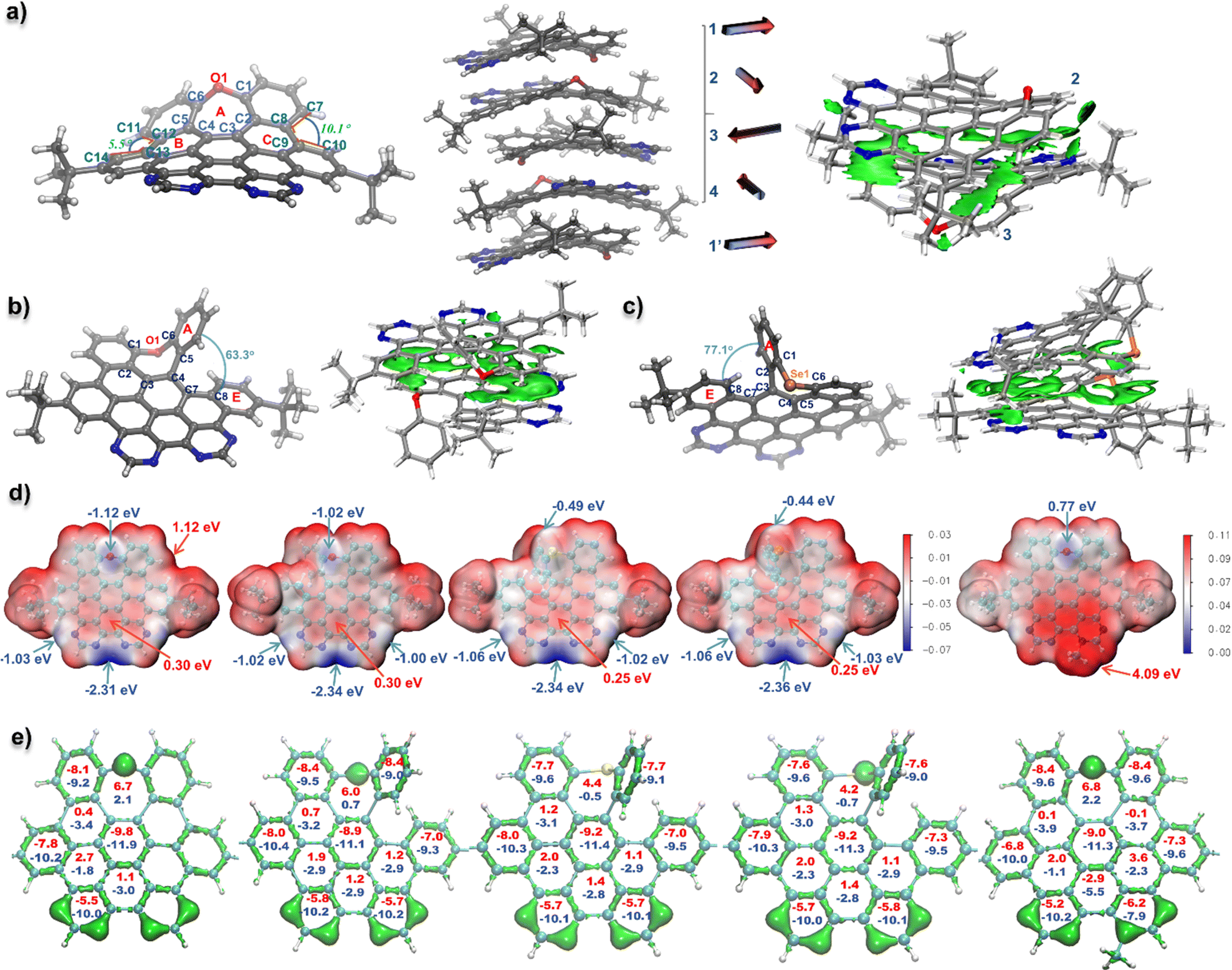 | ||
| Fig. 2 (a) (left): X-ray crystallographic structures of NG 1 with 50% probability of thermal ellipsoids; (middle): the packing mode of NG 1 with illustration of packing direction of each molecule in the image by arrows; (right): IGMH analysis of intermolecular interaction of 1 in the crystal. (b) X-ray crystallographic structures of NG 2 with 50% probability of thermal ellipsoids and its IGMH analysis of crystal packing. (c) X-ray crystallographic structures of NG 4 with 50% probability of thermal ellipsoids and its IGMH analysis of crystal packing. The wave functions for IGMH analysis obtained by the calculation of crystal structures at b3lyp-D3(BJ)/6-311+g(d,p) level (d) electrostatic potential (ESP) map and the energy color bar (au) of nanographenes 1–4, and 1-Me+, representative energy maximums and minimums were marked by red and blue arrows respectively. (energy color bar: −0.07–0.03 for 1–4; 0.00–0.11 for 1-Me+). (e) Isosurface map of valence electron density (isovalue = 0.28) and the NICS(0) and average NICS(1) values (NICS(0): red color; NICS(1): blue color) for each ring of nanographenes 1–4, and 1-Me+. The t-butyl groups were omitted for clarity. ESP, NICS, and valence electron density data were obtained based on the DFT-optimized structures at the cam-b3lyp/6-311+g(d,p) level. | ||
The solid structures of seco-type NGs 2 and 4 revealed a helical geometry containing a5 helicene motif (Fig. 2b and c). The end-to-end dihedral angles (dihedral angle between rings A and E) were 63.3 and 77.1° for 2 and 4, respectively, and a slightly larger dihedral angle was observed for 4 than that for NG seco-HBCSe (73.0°) due to the incorporation of pyrimidine rings instead of benzenes. Meanwhile, the sums of seven angles in the seven-membered rings in 2 and 4 are 839 and 827°, respectively, lower values than that in structure 1, suggesting more distorted geometry of helical nanographenes. Moreover, long C–Se bonds with over 1.9 Å (C1–Se1 and C6–Se1) were revealed in structure 4 due to the large atomic radius of Se. Similar aromaticity patterns to 1 were observed for NGs 2–4 and 1-Me+ based on the calculated valence electron density and NICS values (Fig. 2e), which suggested a low aromaticity of seven-membered ring in each structure. Specifically, even lower aromaticity of seven-membered rings was suggested for fully fused structures 1 and 1-Me+ than that in more distorted helical NGs 2–4 (Fig. S4†). In addition, the electrostatic potential (ESP) analysis was carried out to evaluate the electrostatic distribution of the D–A type nanographenes. As a result, anisotropic surfaces were elucidated due to the insertion of electronegative heteroatoms. As shown in Fig. 2d, for the neutral NGs 1–4, most regions surrounding the hydrocarbon backbone exhibited positive ESP. The maximum was around the bay area at the chalcogen side for each structure with 1.12 eV for 1 as a representative. Due to the electronegativity of heteroatoms, the regions around the chalcogen and nitrogen revealed negative values, indicating the electron-rich area. Moreover, the ESP minimums of NGs 1–4 were all located in the bay region of bipyrimidine, with less than −2.3 eV value for each structure, which is consistent with the site-specific alkylation of 1-Me+ at the most nucleophilic position (Scheme 1). Because of the anisotropic distribution of ESP, an interesting alternating molecular packing arrangement was observed in the crystal structures. As shown in Fig. 2a–c, a nearly 90° rotation relative to the adjacent molecule was observed in the molecular stacking of NGs 1, 2, and 4, due to the complementary intermolecular electrostatic interaction between positive/negative electrostatic regions. As a representative, a four-molecule repeating unit of NG 1 was observed in the molecular packing, reminiscent of the α-helical conformation of proteins (Fig. 2a). The corresponding independent gradient model based on Hirshfeld partition (IGMH) analysis18 of the crystal structure of 1 also suggested a significant non-covalent interaction between negatively charged bipyrimidine and adjacent aromatic rings with positive ESP (Fig. 2a). As for NGs 2 and 4, a similar torsional molecular stacking and intermolecular non-covalent interaction were observed based on the crystal packing and IGMH analysis (Fig. 2b and c). In comparison with neutral NGs 1–4, the ESP map of cationic NG 1-Me+ indicated a positive electrostatic surface with an ESP minimum of 0.77 eV above the oxygen and an ESP maximum of 4.09 eV around the pyrimidinium region, which would give strong electrostatic solvent–solute interactions in the polar solvents.
Photophysical properties and theoretical studies
UV-vis absorption and photoluminescence (PL) spectra for all the D–A type nanographenes, together with reference compounds HBC-Bu and HBC-O, were recorded in dichloromethane. As shown in Fig. 3a, when compared to HBC-Bu and HBC-O, the D–A type structures exhibited obvious red-shifted absorption and emission. The absorption maxima of 1–4 and 1-Me+ were located at 375–420 nm together with multiple shoulder peaks, which gave around 25–75 nm bathochromic shift compared to HBC-O (λabs = 350 nm). Meanwhile, the lower-energy bands were shifted to around 470 nm for neutral NGs 1–4 and 580 nm for 1-Me+. Notably, the absorption of cationic compound 1-Me+ can extend to 640 nm in dichloromethane and the near-infrared (NIR) region in an aqueous solution, like phosphate-buffered saline (Fig. S5†). As depicted in Fig. 3a, the emissive maxima of NGs 1–4 are located at 502, 517, 517, and 525 nm, respectively, whereas the cationic NG 1-Me+ displayed three emission peaks at 498, 545, and 660 nm, with the emission extending over 700 nm. In comparison with HBC-Bu (λem = 493 nm) and HBC-O (λem = 460 and 485 nm) under the same conditions, significant bathochromic emissions were observed due to the formation of an electron D–A structure. The fluorescence quantum yields (FQYs) of 1–4 ranged from 0.03 to 0.28 in the solvent of dichloromethane (Table S2,† the highest of 0.28 for compound 3 and lowest of 0.03 for compound 4), which provide the same chalcogen-dependent FQY as chalcogen-doped analogues.14 Meanwhile, NGs 1–4 were still emissive in the amorphous solid with different emissive colors (Fig. S6†). On the other hand, cationic compound 1-Me+ exhibited feeble luminescence both in the dichloromethane (Table S2,† FQY less than 0.01) and as the amorphous powder, suggesting possibly other decay pathways in the excited state.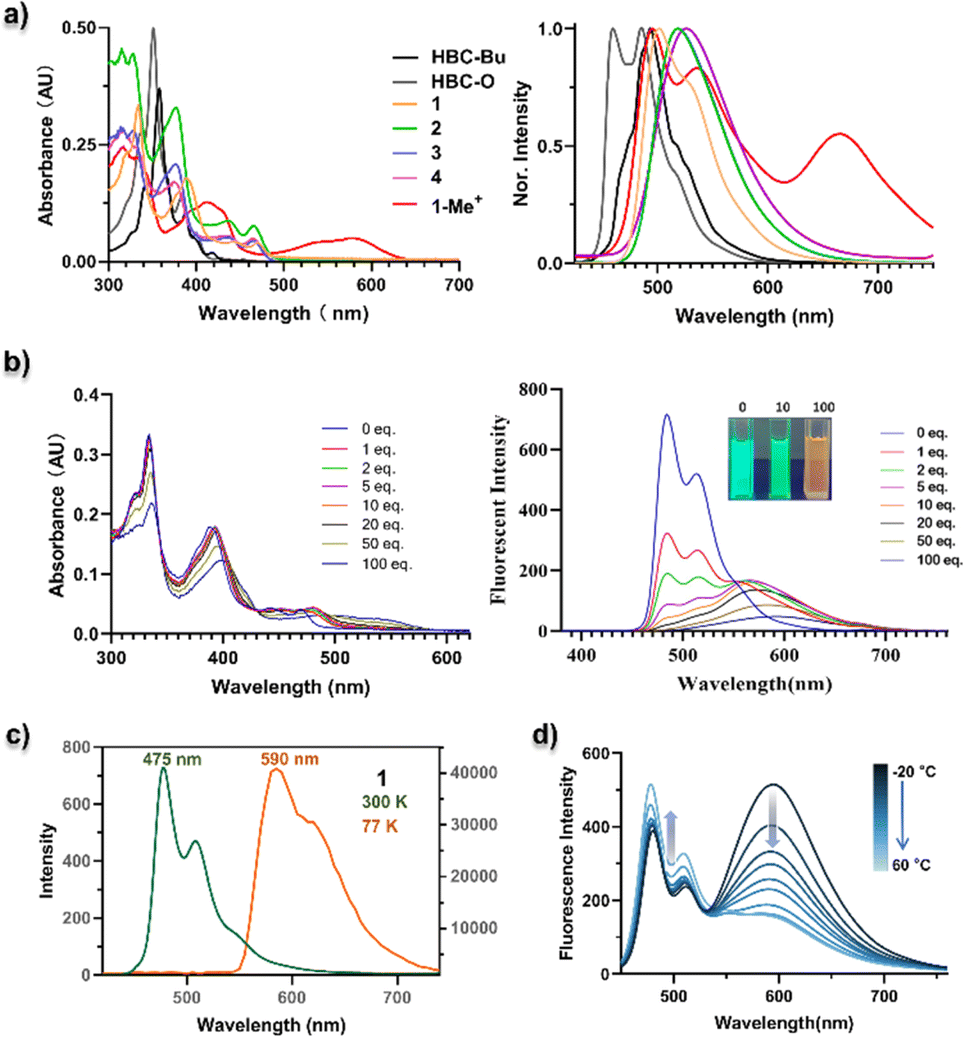 | ||
| Fig. 3 (a) UV/vis absorption (left) and normalized fluorescence (right) of NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ in CH2Cl2. (b) UV/vis absorption spectra (left) and fluorescence emission spectra (right) of addition of 0–100 equiv. of trifluoroacetic acid into a toluene solution of NG 1. 1 × 10−5 M concentration for each measurement. (c) Fluorescence (300 K) and phosphorescence (77 K) spectra of NG 1 (1 × 10−5 M in toluene). (d) Fluorescence spectra of 1-Me+ in toluene in response to temperature. For luminescence spectra, λex = 375 nm for HBC-Bu, HBC-O, and 1–4, λex = 400 nm for 1-Me+. | ||
To understand the frontier molecular orbitals (MOs) and obtain insights into the excited-state properties, we conducted time-dependent density-functional theory (TD-DFT) calculations. The DFT calculations indicated that the longest-lying absorption peaks (S0–S1) of all the D–A nanographenes were mainly contributed by the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) transition (Fig. S7 and Table S3–S7†), whose HOMO–LUMO energy gaps were also in line with the bathochromic shifted lower-energy bands. As shown in Fig. 4a, the NGs 1–4, with similar longest wavelength absorption at around 470 nm (Fig. 3a), adopted a similar HOMO–LUMO band gap of approximately 3.6 eV, which is smaller than that of HBC-Bu (3.85 eV) and HBC-O (3.94 eV), whereas the cationic structure 1-Me+ with the significantly smallest HOMO–LUMO energy gap of 2.58 eV displayed the lowest wavelength absorption peak of 580 nm. Additionally, the differences in MO distributions towards degrees of electronic D–A structures were observed. Compared to the local excitation (LE) character of HBC-Bu and HBC-O, whose HOMOs and LUMOs delocalized over the entire conjugated skeleton, a significant distinction of orbital distributions between HOMOs and LUMOs was revealed for the D–A type structures 1–4: the HOMOs were delocalized almost over the entire conjugated structures, but the majority of the LUMOs were located in the bipyrimidine regions. Furthermore, the cationic NG 1-Me+ exhibited nearly separated MOs of the HOMO and LUMO, with the HOMO located at the electron-rich oxygen-doped part and the LUMO delocalized over electron-deficient bipyrimidine. This MO redistribution indicated intramolecular charge transfer (ICT) from the ground state (S0) to the lowest singlet state (S1) due to the charge-separated D–A structure. Subsequently, the hole–electron analysis was performed to evaluate the charge transfer (CT) process in the lowest-lying excited state (S1).19 The wavefunction analysis of TD-DFT calculated molecules was performed using Multiwfn 3.7 (ref. 18a) to provide the electron/hole distribution and centroid positions. Then the isosurface of 0.0012 was used to quantify the surface of the centroids. As illustrated in Fig. 4b, for the specific S0–S1 transition, the hole and electron surfaces of NGs HBC-Bu and HBC-O were nearly overlapped, leading to a LE excited state, while partially separated hole and electron surfaces were observed for NGs 1–4 with the distance of the centroid between the hole and electron (D index) in the range of 1.1–1.2 Å, suggesting a significantly increased degree of CT in the S0–S1 transition. Moreover, installed with the strengthened electron-withdrawing unit, NG 1-Me+ displayed nearly separated hole–electron surfaces with a D index of 2.9 Å, indicating the strongest CT in the excited states. Collectively, the calculational results and experimental spectra indicated that the electron D–A pattern of nanographenes adopted obvious CT in the excited states. The CT in the excited was further confirmed by the solvatochromism experiment. As shown in Fig. 5, these D–A type nanographenes revealed a pronounced solvatochromic effect. All the NGs were dissolved in different solvents with gradually increased polarity namely hexane, toluene, chloroform, and methanol, and the fluorescence spectra were collected under identical conditions. Red-shifted emissions following solvent polarity were observed for NGs 1–4 and 1-Me+. Indeed, the PL has extended to approximately 700 nm in methanol with decreased intensity for all the D–A nanographenes. The red-shifted emissions and decreased intensity along with the increased solvent-polarity were likely due to the formation of the CT state and increased solvent–solute interactions in the polar environment in the excited states (Fig. S8–12†). In stark contrast, the HBC-Bu, together with HBC-O, seco-HBCS, and seco-HBCSe without electron-acceptor in their structures, showed no solvatochromism (Fig. 5 and S13†), which is consistent with overlapped holes and electrons. In addition, the cationic structure 1-Me+ with the strongest acceptor exhibited a more significant solvatochromic shift of 114 nm than those of NGs 1–4 of around 70 nm (Fig. S8–12†), suggesting a more CT character of 1-Me+ in the polar solvents. Meanwhile, the multiple emission peaks of cationic motif 1-Me+ in non-polar solvents like hexane, toluene, and chloroform (Fig. 5) likely belonged to the formation of excimers due to the strong intermolecular electrostatic interaction.20 To understand the monomer–excimer transformation, we performed temperature-dependent fluorescence measurements. As shown in Fig. 3d, the long-wavelength emission at around 600 nm was significantly high at −20 °C due to the excimer formation at low temperature. With the increase of the temperature, the long-wavelength emission peaks gradually decreased and the emission peaks at around 500 nm increased, indicating the excimer disassembly, while the excimers were not formed due to increased solvent–solute interactions in methanol. Consequently, the solvatochromism experiments revealed the CT excited-state properties of D–A nanographenes, which were highly consistent with the calculated molecular orbital (MO) energies and hole–electron analysis.
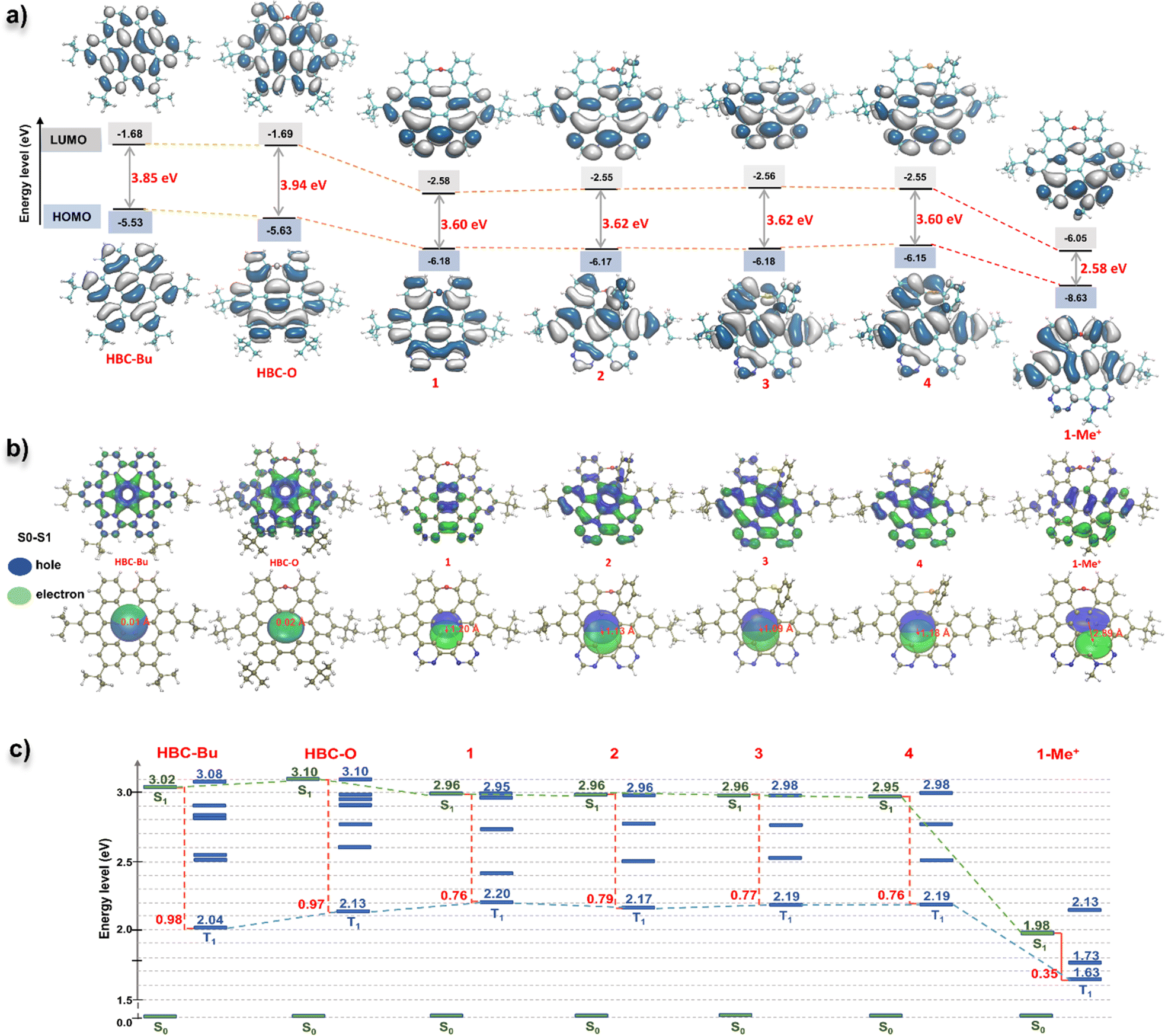 | ||
| Fig. 4 (a) TD-DFT calculated HOMO and LUMO orbitals and energies of NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ in the gas phase at the PBE0/def2-TZVP level. (b) Hole–electron and Chole–Celectron surface analysis for the S0–S1 transition states of NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ at the ground-state geometry based on TD-DFT calculations at the cam-b3lyp/def2-TZVP level. (c) Theoretical singlet and triplet vertical excitation energies in the gas phase at the PBE0/def2-TZVP level and the energy gaps (ΔEST) between the lowest singlet (S1) and lowest triplet (T1) excited states of each structure were shown in red and marked with a red line. | ||
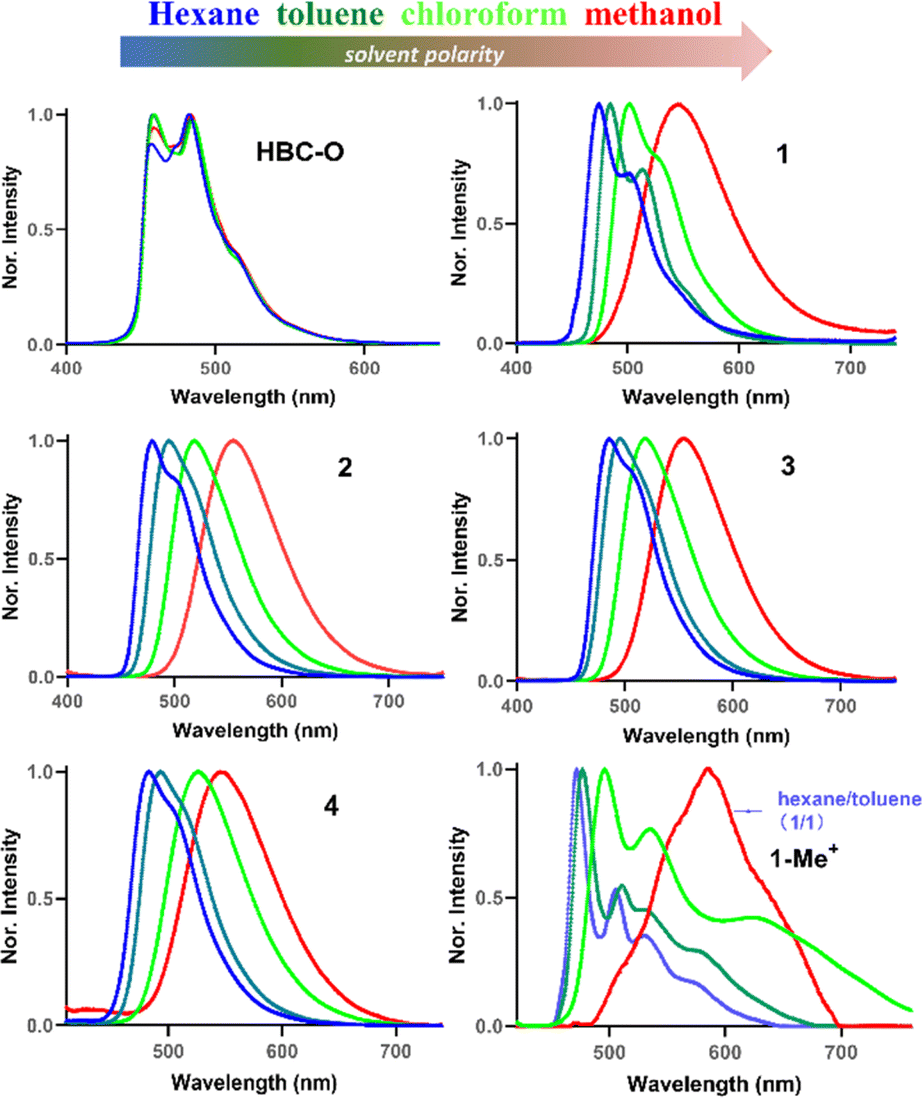 | ||
| Fig. 5 Solvatochromism experiment of NGs HBC-O, 1–4, and 1-Me+ (10 μM concentration, λex = 375 nm for HBC-O and 1–4, λex = 400 nm for 1-Me+). | ||
Considering the weak basicity of nitrogen-containing polyaromatic compounds and the potential applications in water with different pH, acid-induced changes of absorption and fluorescence spectra were observed. As illustrated in Fig. 3b and S14–17,† when mixing trifluoroacetic acid (0–100 equiv.) with NGs 1–4 in toluene, protonation of the nitrogen lone pairs caused a red shift of the whole absorption and emission spectra. Over 80 nm bathochromic shifts with obvious fluorescence color changes and intensity reductions were observed for these nitrogen-substituted NGs 1–4, which indicated that bathochromic absorption and fluorescence emission would be observed due to the formation of protonated nanographenes of 1–4 under physiological conditions.
Photosensitization capacity evaluation
Photodynamic therapy (PDT) is an emerging alternative treatment modality for malignant tumors, which is based on photosensitizers to transfer light energy into ROS to induce cell apoptosis and tissue damage. Due to the minimal invasiveness and spatiotemporal control by light, PDT has attracted more and more attention for tumor treatment. Seeking efficient photosensitizers is of paramount importance for PDT.21Synthetic nanographenes, possessing large conjugated structures and rigid ring systems, would have high light-absorption ability, low nonradiative decay, and reduced reorganization energy, which make them potential candidates for photosensitizers.3c–e Due to their unique D–A type structures, which can induce ISC through charge recombination,22 we then evaluated the photosensitization capacity of these newly synthesized NGs. Efficient and long-lived triplet excitons formed by singlet-to-triplet intersystem crossing (ISC) are extremely important for PDT, and can transfer the excitation energy to the surrounding oxygen molecules in their natural triplet state, leading to the generation of cytotoxic ROS or singlet oxygen (1O2). As a result, steady-state PL spectra of compounds 1–4, and 1-Me+ at 77 K exhibited intense phosphorescence emissions in air, which suggested a strong ISC process and inhibited non-radiative inactivation (Fig. 3c and S19†). Generally, a smaller energy gap (ΔEST) between the lowest singlet state (S1) and lowest triplet state (T1) is of paramount importance for ISC; in case the high energy-consuming and low-rate processes from S1 → Tn (n > 1) and Tn → T1 would be employed for cytotoxic ROS generation.23 Hence, we then evaluated the singlet and triplet excitation energies for NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ by time-dependent density functional theory (TD-DFT). As illustrated in Fig. 4c, the calculations revealed a high energy gap of around 1.00 eV between S1 and T1 of HBC-Bu, HBC-O, which is not accessible for ISC from S1 to T1. By introducing the pyrimidine units, smaller ΔEST of around 0.76 eV was observed for NGs 1–4, suggesting a higher transition possibility of ISC. Notably, the cationic NG 1-Me+ with a strong pyridinium acceptor provided significantly decreased singlet and triplet excitation energies with much smaller energy gap values of 0.35 and 0.25 eV for S1 → T1 and S1 → T2, indicating the highest ISC rate compared to other NG analogues.
We then experimentally assessed the photosensitization capacity of all the nanographenes by the detection of ROS and 1O2 generation in vitro in the phosphate-buffered saline (PBS) containing solvents (details see the ESI†). 2′,7′-Dichlorofluorescein (DCFH) was used as a fluorescent probe to identify ROS, which emits green fluorescence with 488 nm excitation after ROS oxidation. As shown in Fig. 6a, upon yellow-LED light (0.02 W cm−2) irradiation for over 100 min, slowly increasing fluorescence intensities at 525 nm were observed for the solutions containing NGs HBC-Bu and HBC-O (Fig. S20†). Similar fluorescence behaviours were observed for seco-HBCS and seco-HBCSe (Fig. S20†), while a faster ROS generation rate was observed for NGs 1–4, whose fluorescence reached saturation at the same time with the intensity gradually increasing to around two-fold higher compared to HBC-O. To our delight, the cationic NG 1-Me+ revealed an extremely faster ROS generation rate by reaching the fluorescence maximum within 2 min, more than 50 times faster than neutral NGs 1–4. On the other hand, the DCFH probe without a photosensitizer generates negligible emission (Fig. 6a and S20†). Together with the calculation results (Fig. 4), the ROS generation experiments indicated that the D–A type electron structure is beneficial to increase the photosensitization capacity. Notably, a similar ROS generation was observed for NG 1-Me+ by using a 630 nm laser (0.15 W cm−2) as the light source since it exhibited absorption in the red to near-infrared region (Fig. S21†). Meanwhile, the 1O2 generation ability of different nanographenes was further evidenced using the 1,3-diphenylisobenzofuran (DPBF) probe, which could be oxidized by 1O2, thus leading to the decrease of absorption intensity. A similar tendency to the ROS generation rate was observed. As indicated by the DPBF decay curves (Fig. 6b and S22†), HBC-Bu generated almost no 1O2 by giving the same decay profile as the control (with only the DPBF probe) upon irradiation with yellow-LED light, while slightly increased 1O2 generation was observed for chalcogen-doped analogues HBC-O, seco-HBCS, and seco-HBCSe. On the other hand, significantly enhanced 1O2 generation ability was suggested for D–A type structures: 25–40% declines for NGs 1–4, and a nearly complete decomposition of DPBF for 1-Me+ was observed within 10 min. In addition, the intracellular ROS generation of 1-Me+ in HeLa cells under 630 nm laser irradiation was further investigated by using confocal laser scanning microscopy (CLSM) with DCFH-DA as the fluorescence indicator. As shown in Fig. 6c, the cells were incubated with a medium containing 1-Me+ and DCFH-DA, respectively. After washing away the incubation medium, obvious green fluorescence was observed in HeLa cells upon 100 s laser irradiation due to the generation of ROS. However, there is negligible green fluorescence in the cells without the addition of 1-Me+ under the same irradiation conditions. These live-cell imaging studies indicated that NG 1-Me+ exhibited excellent photosensitization ability and cell permeability in live cells.
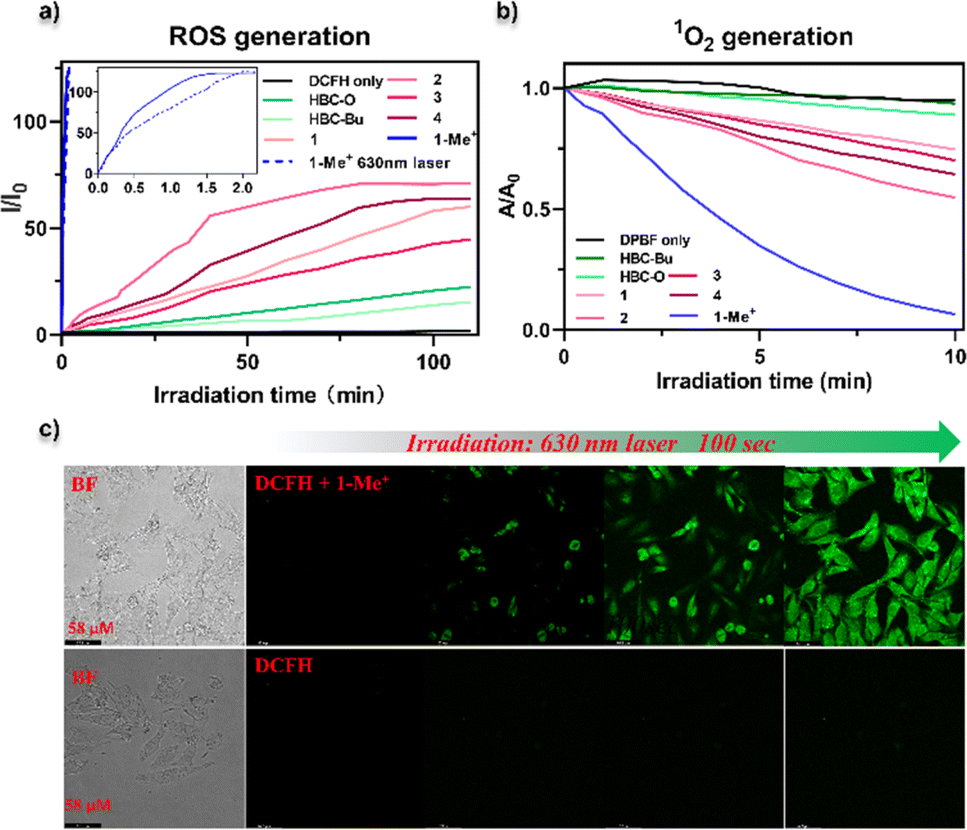 | ||
| Fig. 6 ROS or singlet oxygen detection by using different nanographenes as photosensitizers. (a) Time-dependent fluorescence changes of the fluorescence probe DCFH upon light irradiation coexisting with NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ in PBS. For all the nanographenes, yellow-LED light (0.02 W cm−2) was used as a light source, and a specific 630 nm laser (0.15 W cm−2) was used as a light source for NG 1-Me+. (b) Absorbance changes of DPBF at 411 nm in the presence of NGs HBC-Bu, HBC-O, 1–4, and 1-Me+ in PBS upon yellow-LED light irradiation as a function of irradiation time. (c) Confocal microscopy fluorescence imaging of HeLa cells with the DCFH-DA probe in the presence of (up) or in the absence of NG 1-Me+ photosensitizer by irradiation using a 630 nm femtosecond pulsed laser (0.15 W cm−2). The concentration of NG 1-Me+ was 10 μM. The excitation wavelength of DCF was 488 nm. | ||
We then investigated the cell-based PDT of NGs 1–4 and 1-Me+. The viabilities of HeLa after different treatments were studied via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays (Fig. S23†). The cells were treated with compounds 1–4 and 1-Me+ with various concentrations (2–25 μM), and irradiated with yellow-LED light for 30 min. Then, significantly decreased viabilities were observed with increasing concentrations of each compound with the morphology changes (Fig. S24†). In particular, 1-Me+ provided the highest PDT efficiency, which killed over 80% of cells at the concentration of 5 μM. In contrast, no obvious cytotoxicity was observed for cells without light illumination. Specifically, NG 1-Me+ with the lowest-energy absorption band displayed a similar PDT behaviour when using a low-power 630 nm laser. As illustrated in Fig. 7a, almost complete cell death was observed when the cell medium containing 10 μM 1-Me+ was irradiated with a 630 nm laser for 30 min. The morphology of most cells incubated with 1-Me+ changed to a round shape after only 5 min irradiation by 630 nm light (Fig. 7b), indicating that the cells were at the early apoptotic stage already. To visualize the photo-induced cell death, calcein-acetoxymethyl (Calcein-AM) and propidium iodide (PI) staining was employed to differentiate dead (red) and live (green) HeLa cells. The cells incubated with NGs 1–4 and 1-Me+ were irradiated with light (yellow-LED light for 1–4 and 1-Me+ and 630 nm for 1-Me+) for 30 min and then co-stained with Calcein-AM and PI fluorophores. The fluorescence imaging suggested the cells incubated with NGs 1–4 were partially killed (Fig. S25†), and nearly all the cells treated with cationic compound 1-Me+ were dead (Fig. 7c and S25†), which is consistent with the in vitro ROS generation ability.
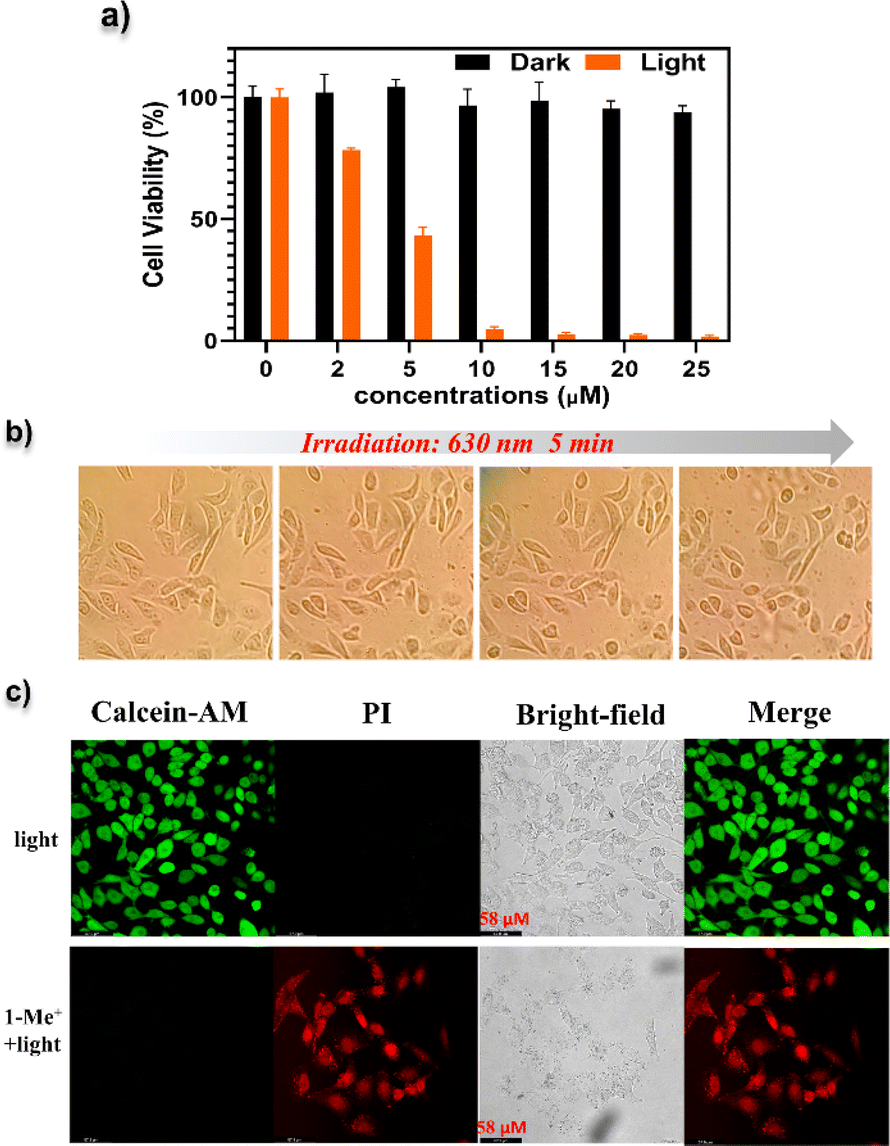 | ||
| Fig. 7 Phototherapy effect of NG 1-Me+. (a) Cell viability of HeLa cells incubated with 1-Me+ at different concentrations without or with 630 nm laser irradiation. Error bars were based on the standard error of mean (n = 4). (b) Cell morphology photographs of HeLa cells incubated with 1-Me+ (10 μM) upon irradiation with a 630 nm laser for 10 min. (c) Fluorescence images of Calcein AM (live cells, green) and PI (dead cells, red) co-stained HeLa cells after different treatments. (Up): intact HeLa cells irradiated with a 630 nm laser; (down): HeLa cells incubated with 1-Me+ (10 μM) and irradiated with a 630 nm laser. | ||
Conclusions
In summary, we have reported a series of multi-heteroatom-doped nanographenes, featuring electron donor–acceptor architecture. The solid-state structure, photophysical properties, and photosensitization capacity were experimentally and theoretically investigated. Compared to the non-doped analogues, the new D–A type structures exhibited bathochromic shifted absorption and emission, solvatochromic effect, and charge transfer in the excited states. Importantly, a pronounced ROS generation was observed by employing these NGs as photosensitizers. The structure–activity relationship was analyzed by comparison with reference compounds and DFT calculations. Notably, these D–A nanographenes displayed reasonable water solubility that the subcellular ROS generation and PDT studies were demonstrated. In particular, the cationic NG 1-Me+ with excellent water-solubility and cell permeability, exhibited the highest photosensitization capacity when irradiated with yellow-LED or 630 nm light, which makes it a valuable candidate metal-free photosensitizer for PDT. Overall, this work illustrates the process of property tunability for potential nanographene applications.Data availability
The full experimental details, methods, synthetic procedures, characterization data, UV-vis, fluorescence, NMR spectra, computational details, biological experiments, and supplementary discussions associated with this article are provided in the ESI.†Author contributions
R. Li did the synthetic work of cationic structure 1-Me+, reference structures, and 1–4 for property studies. R. Li performed the biological studies and parts of photophysical measurements. B. Ma initially explored the synthetic procedures of compounds 1–4 and performed parts of the photophysical studies. M. Li helped with biological studies. D. Wang helped with NMR and photophysical property measurements. P. Liu helped with the UV/vis and PL spectrum measurements. P. An conceived the concept and performed the DFT calculations. All the authors analyzed and interpreted the results. P. An prepared and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support provided by the National Natural Science Foundation of China (22061046 and 21901226), Yunnan Fundamental Research Projects (202401AT070475), and project of Innovative Research Team of Yunnan Province (202405AS350010). We appreciate the instrumentation support from the advanced analysis and measurement center of Yunnan University. We thank Professor Junqiao Ding and Jianhua Chen for the instrument assistance with the phosphorescence measurements.Notes and references
- (a) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC; (b) Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499–11524 CrossRef CAS PubMed; (c) Y. Zhang, S. H. Pun and Q. Miao, Chem. Rev., 2022, 122, 14554–14593 CrossRef CAS PubMed; (d) Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS; (e) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86–116 CrossRef CAS PubMed; (f) I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaňa, Chem. Commun., 2018, 54, 6705–6718 RSC; (g) M. Grzybowski, B. Sadowski, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 2998–3027 CrossRef CAS PubMed.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed; (b) Z. Liu, S. Fu, X. Liu, A. Narita, P. Samori, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, 2106055 CrossRef PubMed; (c) V. Bonal, R. Muñoz-Mármol, F. G. Gámez, M. Morales-Vidal, J. M. Villalvilla, P. G. Boj, J. A. Quintana, Y. Gu, J. Wu, J. Casado and M. A. Díaz-García, Nat. Commun., 2019, 10, 3327 CrossRef PubMed; (d) N. Bachar, L. Liberman, F. Muallem, X. Feng, K. Müllen and H. Gaick, ACS Appl. Mater. Interfaces, 2013, 5, 11641–11653 CrossRef CAS PubMed.
- (a) Q.-Q. Li, Y. Hamamoto, G. Kwek, B. Xing, Y. Li and S. Ito, Angew. Chem., Int. Ed., 2022, 61, e202112638 CrossRef CAS PubMed; (b) Y.-F. Wu, S.-W. Ying, L.-Y. Su, J.-J. Du, L. Zhang, B.-W. Chen, H.-R. Tian, H. Xu, M.-L. Zhang, X. Yan, Q. Zhang, S.-Y. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2022, 144, 10736–10742 CrossRef CAS PubMed; (c) H.-A. Lin, Y. Sato, Y. Segawa, T. Nishihara, N. Sugimoto, L. T. Scott, T. Higashiyama and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 2874–2878 CrossRef CAS PubMed; (d) Y.-Y. Ju, X.-X. Shi, S.-Y. Xu, X.-H. Ma, R.-J. Wei, H. Hou, C.-C. Chu, D. Sun, G. Liu and Y.-Z. Tan, Adv. Sci., 2022, 9, 2105034 CrossRef CAS PubMed; (e) X.-H. Ma, X. Gao, J.-Y. Chen, M. Gao, Q. Dai, Z.-K. Jia, Y.-B. Zhou, X.-J. Zhao, C. Chu, G. Liu and Y.-Z. Tan, J. Am. Chem. Soc., 2024, 146, 2411–2418 CrossRef CAS PubMed; (f) X. Zhu, Q. Chen, H. Zhao, Q. Yang, Goudappagouda, M. Gelléri, S. Ritz, D. Ng, K. Koynov, S. H. Parekh, V. K. Chetty, B. K. Thakur, C. Cremer, K. Landfester, K. Müllen, M. Terenzio, M. Bonn, A. Narita and X. Liu, J. Am. Chem. Soc., 2024, 146, 5195–5203 CrossRef CAS PubMed.
- (a) S. R. Peurifoy, T. J. Sisto, F. Ng, M. L. Steigerwald, R. Chen and C. Nuckolls, Chem. Rec., 2019, 19, 1050–1061 CrossRef CAS PubMed; (b) M. C. Stuparu, Acc. Chem. Res., 2021, 54, 2858–2870 CrossRef CAS PubMed; (c) J. Wang, X. Zhang, H. Jia, S. Wang and P. Du, Acc. Chem. Res., 2021, 54, 4178–4190 CrossRef CAS PubMed; (d) S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- (a) M. D. Watson, F. Jäckel, N. Severin, J. P. Rabe and K. Müllen, J. Am. Chem. Soc., 2004, 126, 1402–1407 CrossRef CAS PubMed; (b) K. Müllen and J. P. Rabe, Acc. Chem. Res., 2008, 41, 511–520 CrossRef PubMed; (c) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed; (d) H. Hölzel, P. Haines, R. Kaur, D. Lungerich, N. Jux and D. M. Guldi, J. Am. Chem. Soc., 2022, 144, 8977–8986 CrossRef PubMed.
- (a) J. Wu, M. D. Watson, N. Tchebotareva, Z. Wang and K. Müllen, J. Org. Chem., 2004, 69, 8194–8204 CrossRef CAS PubMed; (b) G. M. Beneventi, M. Krug, D. Reger, N. Jux and D. M. Guldi, J. Photochem. Photobiol. C Photochem. Rev., 2023, 56, 100602 CrossRef CAS; (c) R. Li, D. Wang, S. Li and P. An, Beilstein J. Org. Chem., 2023, 19, 736–751 CrossRef CAS PubMed; (d) J. Stanojkovic, R. William, Z. Zhang, I. Fernández, J. Zhou, R. D. Webster and M. C. Stuparu, Nat. Commun., 2023, 14, 803 CrossRef CAS PubMed; (e) J. N. Smith, J. M. Hook and N. T. Lucas, J. Am. Chem. Soc., 2018, 140, 1131–1141 CrossRef CAS PubMed; (f) H. J. Yen, H. Tsai, M. Zhou, E. F. Holby, S. Choudhury, A. Chen, L. Adamska, S. Tretiak, T. Sánchez, S. Iyer, H. Zhang, L. Zhu, H. Lin, L. Dai, G. Wu and H. L. Wang, Adv. Mater., 2016, 28, 10250–10256 CrossRef CAS PubMed; (g) A. Oró, F. Romeo-Gella, J. Perles, J. M. Fernández-García, I. Corral and N. Martín, Angew. Chem., Int. Ed., 2023, 62, e202312314 CrossRef PubMed; (h) J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernández, A. Molina-Ontoria, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2020, 142, 4162–4172 CrossRef CAS PubMed.
- (a) J. Wu, A. Fechtenkötter, J. Gauss, M. D. Watson, M. Kastler, C. Fechtenkötter, M. Wagner and K. Müllen, J. Am. Chem. Soc., 2004, 126, 11311–11321 CrossRef CAS PubMed; (b) J. Wu, J. Li, U. Kolb and K. Müllen, Chem. Commun., 2006, 48–50 RSC.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (c) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed; (d) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- (a) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed; (b) S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed; (c) M. Takase, V. Enkelmann, D. Sebastiani, M. Baumgarten and K. Müllen, Angew. Chem. Int. Ed., 2007, 46, 5524–5527 CrossRef CAS PubMed; (d) M. Takase, T. Narita, W. Fujita, M. S. Asano, T. Nashinaga, H. Benten, K. Yoza and K. Müllen, J. Am. Chem. Soc., 2013, 135, 8031–8040 CrossRef CAS PubMed; (e) S. M. Draper, D. J. Gregg and R. Madathil, J. Am. Chem. Soc., 2002, 124, 3486–3487 CrossRef CAS PubMed; (f) X.-Y. Wang, M. Richter, Y. He, J. Björk, A. Riss, R. Rajesh, M. Garnica, F. Hennersdorf, J. J. Weigand, A. Narita, R. Berger, X. Feng, W. Auwärter, J. V. Barth, C.-A. Palma and K. Müllen, Nat. Commun., 2017, 8, 1948 CrossRef PubMed; (g) C. J. Martin, B. Gil, S. D. Perera and S. M. Draper, Chem. Commun., 2011, 47, 3616–3618 RSC; (h) Y.-Z. Tan, B. Yang, K. Parvez, A. Narita, S. Osella, D. Beljonne, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2646 CrossRef PubMed; (i) P. An, R. Li, B. Ma, R.-Y. He, Y.-K. Zhang, M.-J. Xiao and B. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24478–24483 CrossRef CAS PubMed; (j) M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 60, 14998–15005 CrossRef CAS PubMed; (k) K. Nakamura, K. Ochiai, A. Yubuta, D. He, D. Miyajima and S. I. Precision, Chem, 2023, 1, 29–33 CAS; (l) B. Zhang, L. Ruan, Y.-K. Zhang, H. Zhang, R. Li and P. An, Org. Lett., 2023, 25, 732–737 CrossRef CAS PubMed.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) M. Dai, Y. J. Yang, S. Sarkar and K. H. Ahn, Chem. Soc. Rev., 2023, 52, 6344–6358 RSC; (c) G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H.-J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K.-Y. Jen and H.-L. Yip, Chem. Rev., 2022, 122, 14180–14274 CrossRef CAS PubMed.
- (a) W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS; (b) W.-C. Chen, Y. Yuan, Z.-L. Zhu, Z.-Q. Jiang, S.-J. Su, L.-S. Liao and C.-S. Lee, Chem. Sci., 2018, 9, 4062–4070 RSC; (c) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS PubMed.
- (a) E. R. Darzi, E. S. Hirst, C. D. Weber, L. N. Zakharov, M. C. Lonergan and R. Jasti, ACS Cent. Sci., 2015, 1, 335–342 CrossRef CAS PubMed; (b) H. Deng, Z. Guo, Y. Wang, K. Li, Q. Zhou, C. Ge, Z. Xu, S. Sato, X. Ma and Z. Sun, Chem. Sci., 2022, 13, 14080–14089 RSC.
- (a) T. Kuwabara, J. S. Orii, Y. Segawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 9646–9649 CrossRef CAS PubMed; (b) T. C. Lovell, Z. R. Garrison and R. Jasti, Angew. Chem., Int. Ed., 2020, 59, 14363–14367 CrossRef CAS PubMed; (c) S. Nishigaki, M. Fukui, H. Sugiyama, H. Uekusa, S. Kawauchi, Y. Shibata and K. Tanaka, Chem.–Eur. J., 2017, 23, 7227–7231 CrossRef CAS PubMed.
- R. Li, B. Ma, S. Li, C. Lu and P. An, Chem. Sci., 2023, 14, 8905–8913 RSC.
- Deposition numbers 2344508 (for 1), 2344509 (for 2), and 2344510 (for 4) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service..
- T. Lu and Q. Chen, Acta Phys.–Chim. Sin., 2018, 34, 0001–0009 Search PubMed.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed; (b) H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- X. Shan, W. Chi, H. Jiang, Z. Luo, C. Qian, H. Wu and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202215652 CrossRef CAS PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2344508, 2344509 and 2344510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02416h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |