
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ polymerization of 1,3-dioxolane and formation of fluorine/boron-rich interfaces enabled by film-forming additives for long-life lithium metal batteries†
Ting
Li
a,
Kai
Chen
a,
Borui
Yang
a,
Kun
Li
a,
Bin
Li
a,
Miao
He
b,
Liu
Yang
a,
Anjun
Hu
*a and
Jianping
Long
 *a
*a
aCollege of Materials and Chemistry & Chemical Engineering, Chengdu University of Technology, 1#, Dongsanlu, Erxianqiao, Chengdu 610059, Sichuan, P. R. China. E-mail: anjunhu@cdut.edu.cn; longjianping@cdut.cn
bState Key Laboratory of Electronic Thin Films and Integrated Devices, University of Electronic Science and Technology of China, Chengdu, 610054, Sichuan, P. R. China
First published on 3rd July 2024
Abstract
In situ polymerized 1,3-dioxolane (PDOL) is widely utilized to construct solid polymer electrolytes because of its high room-temperature ionic conductivity and good compatibility with lithium metal. However, the current polymerization additives used in PDOL do not effectively contribute to the formation of a robust solid electrolyte interphase (SEI), leading to decreased cycle life. Herein, a film-forming Lewis acid, tris(hexafluoroisopropyl) borate (THB), is demonstrated not only to be a catalyst for the ring-opening polymerization of DOL, but also an additive for the formation of a stable fluorine- and boron-rich SEI to improve the interfacial stability and suppress the Li dendrite growth. Moreover, molecular dynamics simulations and experimental results demonstrate that the introduction of THB can promote the dissociation of lithium salt and release more Li+ while the boron site can effectively restrict the free movement of TFSI− anion, thus increasing Li+ transference numbers (0.76) and ensuring the long-term cycling stability of cells. By using THB-PDOL, a stable cycling of Li‖Li symmetric cell for 600 h at a capacity of 0.5 mA h cm−2 can be achieved. Furthermore, employing THB-PDOL in Li‖LiFePO4 full cell enables a capacity retention of 98.64% after 300 cycles at 1C and a capacity retention of 95.39% after 200 cycles at a high temperature (60 °C). At the same time, this electrolyte is also suitable for the Li‖NCM523 full cell, which also achieves excellent stability of more than 180 cycles. This film-forming Lewis acid additive provides ideas for designing low-cost, high-performance PDOL-based lithium metal batteries.
1. Introduction
With the growing demand for energy and the pressing need for eco-friendly energy solutions, high-energy-density lithium metal batteries (LMBs) hold significant potential.1–6 Nevertheless, the conventional organic liquid electrolytes (LEs) are prone to volatilization, leading to continuous reactions with the lithium metal anode and high-voltage cathode, thereby depleting the electrolyte.7–9 Additionally, the uneven deposition of lithium ions in the LEs enhances the likelihood of lithium dendrites forming dead lithium, ultimately destabilizing the battery cycling process and impeding the advancement of LMBs.10–12 In response to these challenges associated with LEs, researchers have explored various strategies. Significant efforts have been dedicated to optimizing electrolyte formulations,13,14 developing solid-state electrolytes,15–18 and designing electrolyte/electrode interfaces.3,19 Solid electrolytes encompass inorganic solid electrolytes (ISEs) and gel polymer electrolytes (GPEs). While ISEs boast superior mechanical properties, they exhibit significant contact resistance with the electrode material, leading to heightened internal battery resistance, which can impact battery output power, as well as charging and discharging efficiency.20,21 On the other hand, GPEs combine the merits of both LEs and ISEs by featuring a flexible polymer matrix that offers a certain level of mechanical strength to withstand volume expansion and dendrite growth during battery operation. Notably, GPEs also address the leakage issue associated with LEs, positioning them as a promising replacement for conventional liquid electrolytes.22,23In order to enhance the electrode–electrolyte contact in LMBs, in situ polymerized GPEs have been developed, with polymerized 1,3-dioxolane (PDOL) being a notable example, showing superior cycling stability when compared to conventional LEs.24,25 PDOL-based GPEs exhibit exceptional lithium ion conductivity and impressive cycling stability when coupled with low-voltage cathode materials like LiFePO4.26,27 The ring-opening polymerization of DOL can be initiated by the catalytic activity of Lewis acids such as LiPF6, BF3 or Al(OTf)3.28,29 However, existing research on PDOL has revealed certain limitations, including the inability of some DOL initiators to form a robust SEI and their potential to compromise the performance of the polymer matrix. To address this, additional film-forming additives are incorporated to ensure SEI stability. For instance, Li et al. utilized SnF2 as an effective DOL polymerization catalyst, resulting in the formation of composite SEIs containing LiF/LixSn, which enhanced the wetting properties at the electrode–electrolyte interface and inhibited the growth of lithium dendrites.30 Despite these benefits, the presence of SnF2 has been found to be susceptible to chain breaking within these poly-ethers, which can undermine interface stabilization.31 In a different approach, Wang et al. employed Mg(OTf)2 as an initiator and introduced fluoroethylene carbonate as a film-forming additive to further enhance the interfacial stability of the battery cells.32 Ciucci et al. used LiPF6 to initiate the ring-opening polymerization of DOL and incorporated a nitrate-based additive to promote the formation of Li3N/LixNy-containing SEIs, thereby inhibiting the parasitic reaction and improving the coulombic efficiency (CE).33 Consequently, there is a need to develop and identify additives that can dual-function as in situ polymerization initiators and maintain stable SEI interfaces, which presents a significant challenge in current research efforts.
In this study, we present a method to in situ polymerize DOL and improve the stability of SEI by using a film-forming Lewis acid additive – tris(hexafluoroisopropyl) borate (THB). Due to the presence of sp2 hybridized boron atoms in THB, as electron-deficient atoms, they can accept the electrons provided by DOL, thus inducing the ring-opening polymerization of DOL. Moreover, the THB additive promotes the formation of a robust LiF- and LiBxOy-rich SEI to improve the interfacial stability and suppress the Li dendrite growth. At the same time, theoretical and experimental results show that the introduction of THB promotes the dissociation of LiTFSI, while the electron-deficient boron center effectively anchors the TFSI− anion, thereby increasing the Li+ transfer number (0.76). These unique structural features enabled stable cycling for 600 hours in Li‖Li symmetric cell at a current density of 0.5 mA cm−2. Additionally, the Li‖THB-PDOL‖LFP full cell achieved a capacity retention rate of 98.64% after 300 cycles at 1C. The cell also exhibits stable cycling performance at a high temperature of 60 °C. Moreover, when paired with an NCM523 cathode, the capacity retention of the cell remains at 80.19% over 180 cycles at 0.5C. This work emphasizes the importance of using a film-forming agents to improve the interfacial stability of in situ polymerized gel electrolytes (Fig. 1).
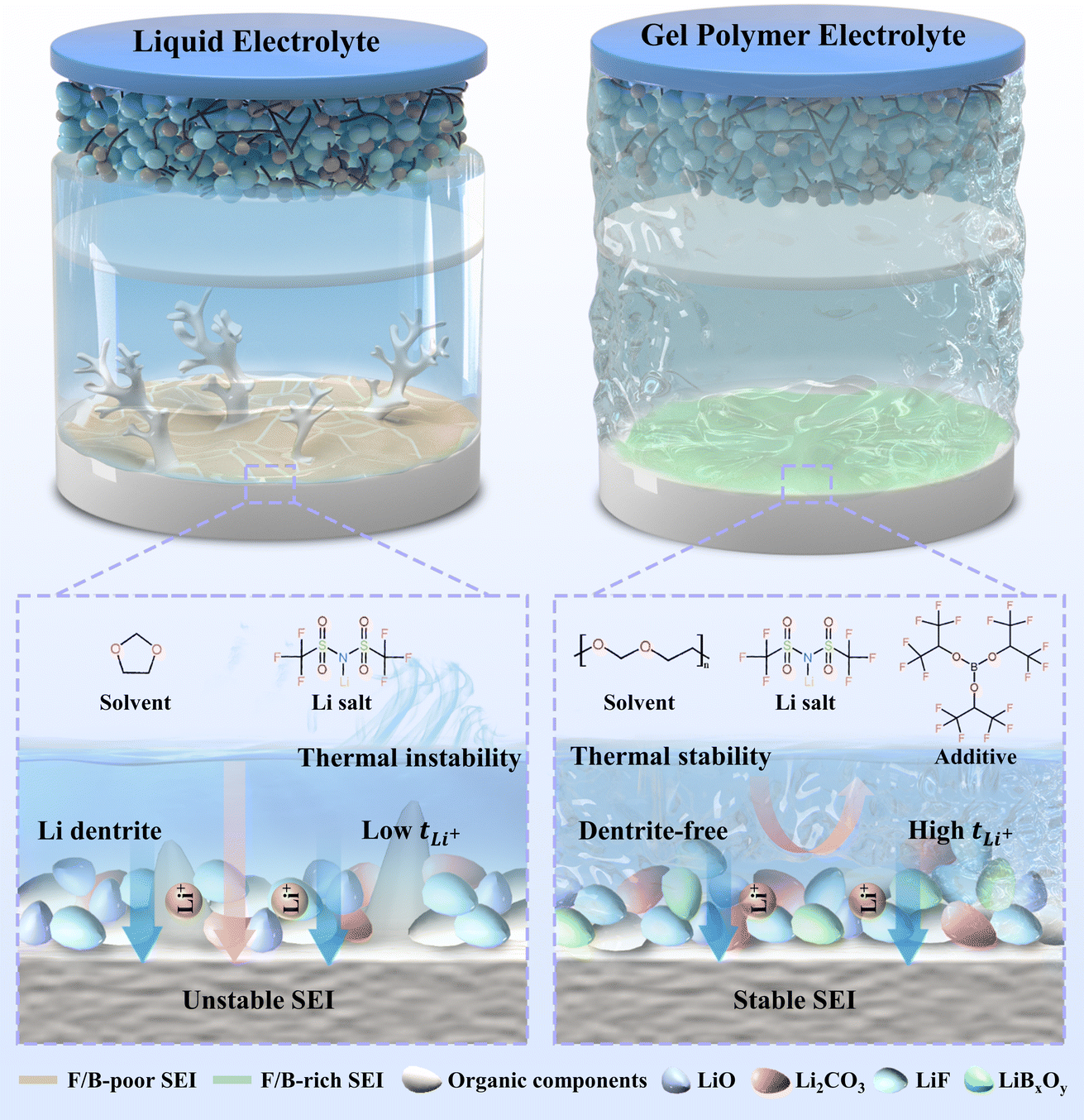 | ||
| Fig. 1 Schematic illustrations of the design principles of THB enhancing the performance of PDOL-based GPEs: The Lewis acid THB promotes the polymerization of DOL monomers to improve its electrochemical stability, and generates LiF LiBxOy rich ion conductive layer on the surface of lithium metal, which guides the uniform deposition of lithium ions and inhibits dendrite growth. | ||
2. Experimental section
2.1 Materials
Tris(hexafluoroisopropyl) borate (THB, >95.0% (GC)) was purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Lithium bis(trifluoromethanesulphonyl)imide (LiTFSI, 99.9%), 1,3-dioxolane (DOL, 99.9%), lithium foil (15.66 mm × 0.45 mm), and PP separator (25 μm thick) were acquired from DoDoChem Technology Co., Ltd. LiFePO4 (LFP), conductive graphite, N-methylpyrrolidone (NMP) and poly(vinylidene fluoride (PVDF)) were purchased from Shenzhen Battery New Material Group Co., Ltd. LiNi0.5Co0.2Mn0.3O2 (NCM523) was purchased from Guangdong Canrd New Energy Technology Co., Ltd. All reagents were not treated in any way prior to use and were used as received.2.2 Polymer electrolytes and cathode preparation
The liquid electrolyte (LE) was prepared by the dissolution of 2 M lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) in DOL. 1 wt% of THB was added to the LE, and under stirring, the THB is fully mixed and dissolved with the LE, 6 hours of resting time and the LE with THB is converted into a polymer electrolyte spontaneously. The entire preparation was carried out in an Ar-filled glove box (O2 < 0.01 ppm, H2O < 0.01 ppm). The LFP/NCM523 cathode consisted of LFP/NCM523, PVDF, and graphite in a ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 by mass, which was blended into NMP and stirred, and after the powder was fully dispersed, it was manually coated on the surface of the aluminum foil with an applicator, and then dried at 80 °C to remove the NMP solvent, and then cut into round electrode sheets. The loading of LFP cathode was approximately 1 mg cm−2, and the NCM523 was loaded with approximately 1.7 mg cm−2.
:1:1 by mass, which was blended into NMP and stirred, and after the powder was fully dispersed, it was manually coated on the surface of the aluminum foil with an applicator, and then dried at 80 °C to remove the NMP solvent, and then cut into round electrode sheets. The loading of LFP cathode was approximately 1 mg cm−2, and the NCM523 was loaded with approximately 1.7 mg cm−2.
2.3 Cell assembly and electrochemical measurements
Li‖Li and Li‖Cu symmetric cells were assembled for observing the stability of the electrolyte to lithium metal anode plating/stripping and the CE, respectively. The charge/discharge long cycle experiments were performed on a Neware electrochemical test system. The linear sweep voltammetry (LSV) measurements were performed on an electro chemical workstation (BioLogic SP-150) with a Li‖stain steel cell.Electrochemical impedance spectroscopy (EIS) measurements are performed on the as-prepared electrolyte with BioLogic SP-150 electrochemical workstation. The ionic conductivity of the electrolyte is evaluated over the frequency range at 0.1–106 Hz from 25–70 °C. The ionic conductivity was calculated using eqn (1):
 | (1) |
The activation energies of THB-PDOL could be obtained using the Arrhenius eqn (2):
 | (2) |
In order to calculate the lithium ion transference number (tLi+) of the cell, a Li‖Li symmetric cell was assembled, where a constant polarization voltage (ΔV = 10 mV) was applied to the cell by the chronoamperometry method, and the initial current (I0) and steady-state current (IS) before and after polarization were measured, and the lithium ion transference number of the electrolyte at room temperature was calculated through eqn (3).
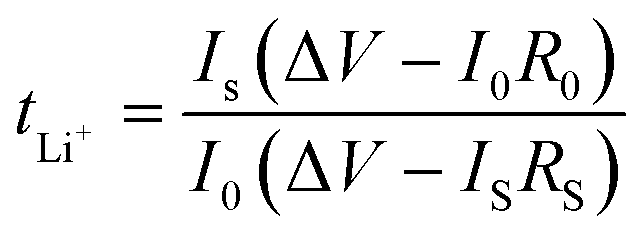 | (3) |
2.4 Material characterizations
The 1H NMR and 13C NMR spectra of the electrolytes were analyzed by Bruker 400 MHz NMR spectrometer after dissolution of the electrolytes with a deuterated solvent (DMSO-d6). The morphology of the lithium metal anode was observed on the Scanning Electron Microscope (SEM, ZEISS GeminiSEM 300). The FTIR spectra of the electrolytes were tested by Thermo Scientific Nicolet iS5 FTIR spectrometer. DSC measurements of the electrolyte were performed using a TA Q2000 with a temperature range of −80–60 °C under N2 atmosphere at a heating rate of 10 °C min−1. Raman spectra was analyzed using an i Horiba LabRAM HR Evolution at a laser wavelength of 633 nm/17.5 mW. SEI compositional information was analyzed by X-ray photoelectron spectroscopy (XPS; Thermo Scientific K-Alpha).2.5 Molecular dynamics simulations
The GRMOACS 2020.6 package was utilized for conducting molecular dynamics simulations. Visualizing structures was achieved through the VMD software. Use the PACKMOL package to blend molecules in a square box that has periodical boundary conditions.3. Results and discussion
3.1 Fabrication and properties of THB-PDOL
THB induces the ring-opening polymerization of DOL by accepting the electrons provided by DOL, thanks to the presence of electron-deficient sp2-hybridized boron atoms in THB, as illustrated in Fig. 2a. The LE composition involved dissolving 2 M LiTFSI in DOL. Upon adding 1 wt% THB to the LE and thorough mixing, a spontaneous transformation of the LE with THB into a polymer electrolyte occurs after a period of standing. This transition from a flowable liquid to a non-flowable polymer electrolyte is visually depicted in Fig. 2b. The hypothesis of THB-induced ring opening polymerization of DOL was corroborated through Fourier-transform infrared (FTIR). As shown in Fig. S1,† The FTIR spectrum of pure DOL shows an absorption region of C–H out-of-plane bending vibrations at 914 cm−1 and a band of C–O–C vibrations at 1030 cm−1. In comparison, THB-PDOL shows a characteristic peak of poly DOL, a characteristic band of long-chain vibration at 845 cm−1, a greatly weakened intensity of the C–H out-of-plane bending vibrational peak, and a shift of the C–O–C vibration.34,35 Moreover, nuclear magnetic resonance (NMR) was employed to identify the structure of polymer electrolytes. Divergent 1H and 13C NMR peaks were identified in THB-PDOL (Fig. 2c). The 1H NMR peaks at 3.61 and 4.64 ppm are attributed to H on the groups –O–CH2–CH2– and –O–CH2–O–, respectively, which corresponds to the structural unit (–CH2–O–CH2–CH2–O–) in poly DOL. The main chain carbons changed in chemical shifts from 64.27 ppm and 94.54 ppm to 66.92 ppm and 95.24 ppm, according to –O–CH2–CH2–O– and –O–CH2–O–, respectively.36,371H NMR and 13C NMR results confirmed the successful polymerization of DOL, with chemical shifts reflecting structural changes consistent with literature and theoretical expectations.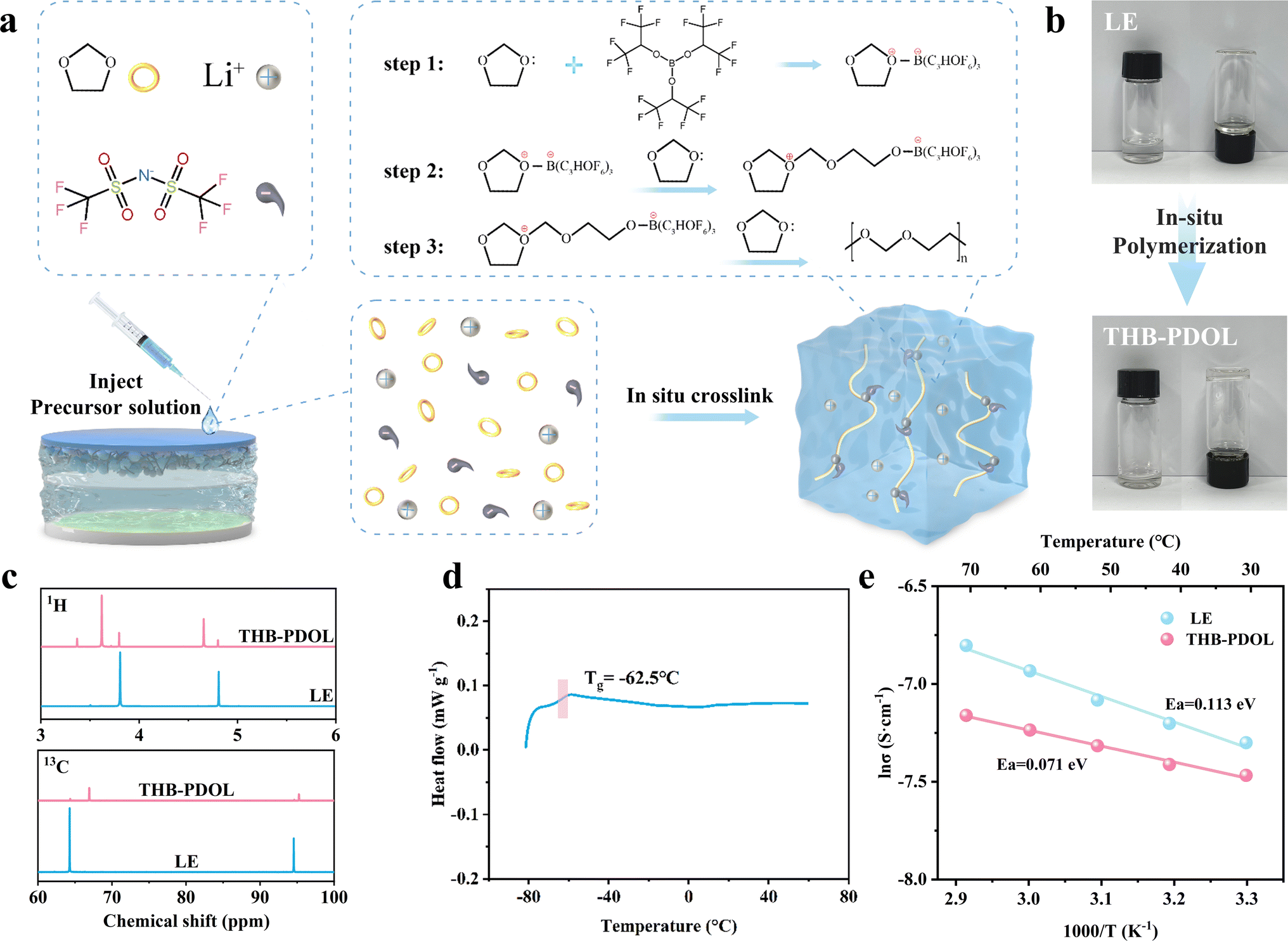 | ||
| Fig. 2 Fabrication and properties of THB-PDOL. (a) Schematic diagram of THB-initiated in situ polymerization of DOL. (b) Digital photos before and after in situ polymerization. (c) 1H and 13C NMR spectra of LE and THB-PDOL. (d) DSC curve of THB-PDOL. (e) Arrhenius plots of ionic conductivity of THB-PDOL. | ||
Ion transport in polymer electrolytes is influenced by the movement of the chain segments of the polymer, with research indicating that this transport primarily occurs in the amorphous region above the glass transition peak (Tg). Differential scanning calorimetry (DSC) was used to analyze the heat flow variations of THB-PDOL, revealing that no significant phase transition was observed below 60 °C. The Tg of THB-PDOL was determined to be −62.5 °C (Fig. 2d), indicating its amorphous structure at room temperature. Consequently, accelerated segmental motion is facilitated under ambient conditions, leading to enhanced ion transport performance. This behavior is further supported by the XRD spectra of THB-PDOL, which exhibited typical amorphous peak diffraction (Fig. S2†), conducive to improving ion conductivity and reducing the activation energy of ion transport through the promotion of segmental vibration of polymer chains.38 Additionally, the thermogravimetric analysis (TG) results displayed in Fig. S3† depicted a significant enhancement in the thermal stability of THB-PDOL compared to LE, with minimal mass loss occurring before 166.42 °C. This increase in thermal stability may be attributed to the long chain segment structure of PDOL, as a high molecular weight polymer, which hinders decomposition.39 Moreover, the presence of C–F bonds in THB contributes to its enhanced thermal stability, as these bonds are less prone to breakage, thereby reducing the risk of unexpected thermal hazards. Collectively, these characteristics indicate the potential of THB-PDOL to operate effectively in high-temperature environments.
As shown in Fig. 2e the activation energies of LE and THB-PDOL were calculated to be 0.113 eV and 0.071 eV, respectively, which suggests that the energy barrier for transferring Li+ in THB-PDOL is relatively low. The corresponding ionic conductivity results in Fig. S6 and S7† indicate that THB-PDOL achieves an ionic conductivity of 5.074 × 10−4 S cm−1 at room temperature, which is comparable to that of LE, thereby meeting the operational requirements of LMBs. An essential aspect for enhancing the energy density of polymer-based LMBs involves expanding the electrochemical window of the electrolyte to delay the high oxidation potential decomposition of the electrolyte when integrated with high-voltage cathode materials. To evaluate the electrochemical stability window of THB-PDOL, Linear Sweep Voltammetry (LSV) was performed. The in situ polymerized crosslinked electrolyte substantially broadened the primary oxidative stability of THB-PDOL to 4.2 V, as demonstrated in Fig. S8,† which is significantly higher than that of LE. The increase in oxidative stability can be attributed to the ring-opening polymerization of DOL and the formation of linear long chains.40,41
3.2 Structure-dependent ion transport properties
Chronoamperometry was used to measure the Li-ion transference numbers (tLi+) of LE and THB-PDOL. An important factor to consider is that low tLi+ can lead to the creation of concentrated polarization during the battery's charging and discharging processes, particularly at high rates. Initial analysis, depicted in Fig. 3a and b, showcases the variation in current values and impedance values before and after polarization of Li‖THB-PDOL‖Li and Li‖LE‖Li symmetrical cells under a constant bias voltage. Utilizing the Bruce–Vincent–Evans equation, it was determined that the tLi+ of THB-PDOL is 0.76, a significantly higher value compared to LE's tLi+ of 0.4. This disparity suggests that the transport of bulk anions in THB-PDOL was restricted due to the presence of a rapid Li+ transport channel, consequently reducing polarization during charging and discharging.42 Furthermore, Raman spectra analysis revealed the existence of Li+-TFSI− ion clusters and dissociated ions within the 730–760 cm−1 range (Fig. 3c).43,44 Specifically, the peak at 748 cm−1 signifies dissociated ions, while the one at 760 cm−1 indicates ion pairs. The differentiation in vibration modes by TFSI− in distinct molecular environments is believed to be responsible for the appearance of these two distinct peaks. Upon quantifying the ratio of ion pairs to dissociated ions, a noticeable increase in dissociated ions was observed in THB-PDOL, providing insight into the augmented tLi+ observed in this material.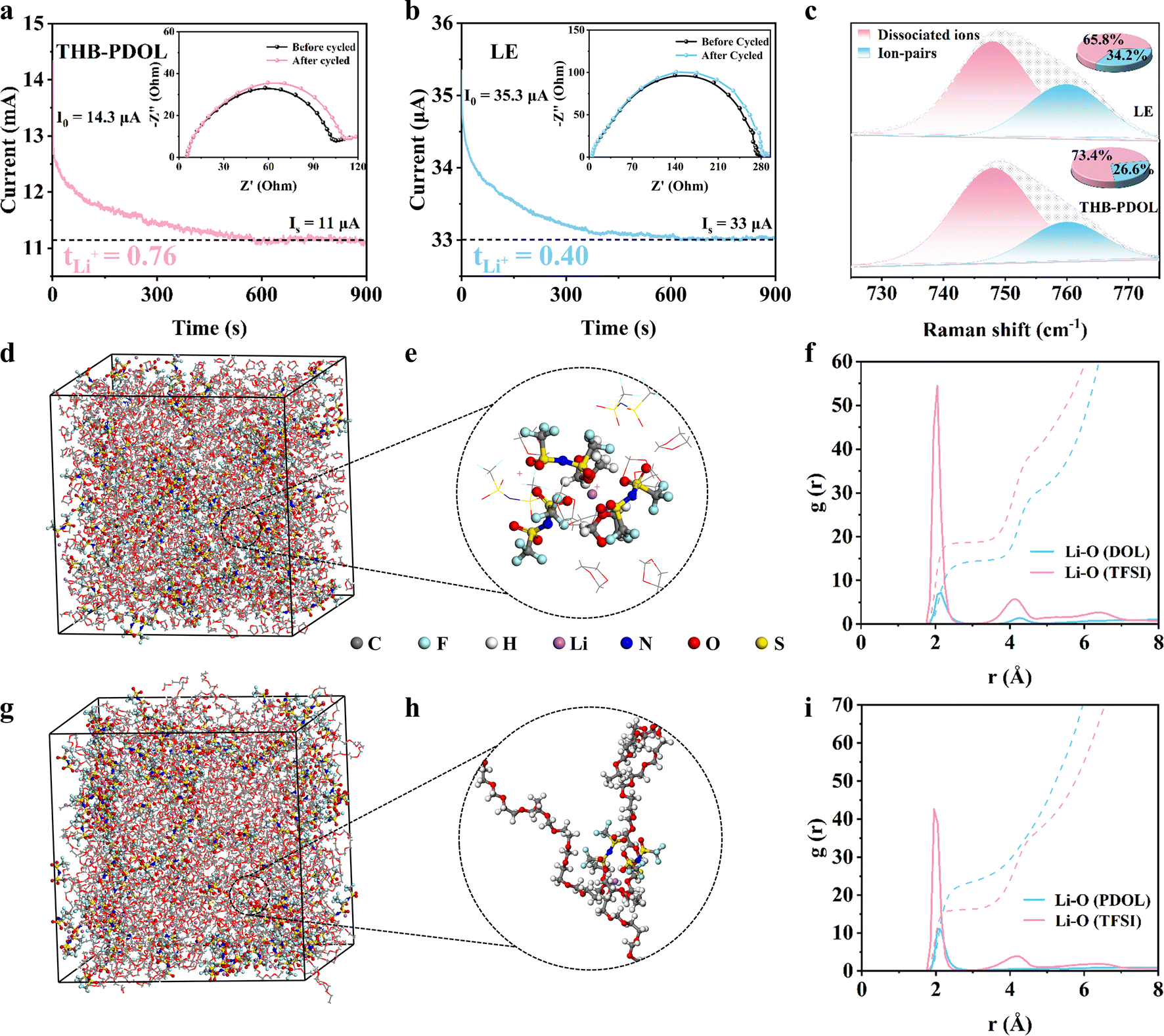 | ||
| Fig. 3 Structure-dependent ion transport properties. Polarization curves and initial and steady state impedance plots of (a) LE and (b) THB-PDOL at room temperature. (c) Raman spectrum of LE and THB-PDOL. Snapshots of (d) LE and (g) THB-PDOL obtained from MD simulations at 25 °C. Li+ coordination structures of (e) LE and (h) THB-PDOL. The radial distribution function (g(r), solid line) and the number of collocations (n(r), dashed line) for (f) LE and (i) THB-PDOL. | ||
Molecular dynamics (MD) simulations were conducted to analyze the solvation structure of LE and THB-PDOL systems. A snapshot of the simulated electrolytes can be observed in Fig. 3d–h. The radial distribution functions of Li+ with TFSI− and the electrolyte matrix were computed for both systems, as depicted in Fig. 3f and i. In the case of LE, the peaks at 2.05 Å for the Li–O (DOL) pair and 2.10 Å for the Li–O (TFSI) pair suggest that a significant portion of Li+ is directly coordinated to TFSI−. Contrasting these findings, observed shifts in the positions and intensities of peaks related to the Li–O (PDOL) pair and Li–O (TFSI) pair in THB-PDOL indicate a decrease in the coordination strength of Li+ with TFSI−. This shift leads to an increased number of Li+ in direct contact with the PDOL skeleton in the THB-PDOL system. This observation supports the conclusion that LiTFSI exhibits better dissociation in THB-PDOL, allowing for more Li+ to be released, consistent with Raman's findings. Furthermore, g(r) shown in Fig. S9† reveals that the dissociated TFSI− preferentially interacts with B+. This preferential interaction structure restricts the free movement of the anion, hence facilitating the transport of Li+ and ensuring the long-term stability of the cell.
3.3 Interfacial stability and electrochemical performances
As shown in Fig. S10,† Li‖Cu asymmetric cells were assembled to evaluate the reversibility of lithium plating/stripping efficiency at a current density of 0.5 mA cm−2. The initial CE of THB-PDOL (70.24%) is slightly higher than that of LE (69.09%). This improvement may be attributed to the interactions between B+ serving as anchors for the anions within the THB-PDOL system, thereby limiting their mobility. Additionally, the presence of high tLi+ effectively mitigates detrimental polarization effects, resulting in a more uniform lithium deposition and a superior CE performance.45,46 The CE of the LE cell exhibited fluctuations and gradual decline post 50 cycles, highlighting the inferior reversibility of its plating/stripping process. In contrast, THB-PDOL demonstrated consistent and stable CE with minimal decay over 120 cycles, showcasing its superior plating/stripping reversibility. This distinction is attributed to the formation of an unstable SEI in LE, leading to electrolyte side reactions with the lithium negative electrode that produce dead lithium. In contrast, the SEI of THB-PDOL offers greater resistance to lithium dendrite growth, facilitates ion transport, regulates lithium plating/stripping behavior, and enhances battery cycling stability through synergistic promotion of these functionalities.The maximum allowable current density of a cell is defined as critical current density (CCD), and it is associated with the capacity density of the battery.47 To avoid the impact of the initial interfacial evolution, the Li‖Li symmetric cell was initially tested at a low current density of 0.05 mA cm−2 for 20 h and then performed at a current density of 0.1–5 mA cm−2. The obtained test results depicted in Fig. 4a reveal that beyond a current density of 4 mA cm−2, spikes are observed in the polarization voltage plateau of the THB-PDOL, indicating its ability to function effectively up to 4 mA cm−2. Notably, this performance aligns with the industry standard for power battery operating current as stipulated in GB/T-31467.2-2015, thereby underscoring the robustness and functionality of the THB-PDOL in practical battery applications.
 | ||
| Fig. 4 Interfacial stability and electrochemical performances. (a) The critical current density of THB-PDOL was tested by constant-current charge/discharge tests at a current density of 0.1–5 mA cm−2, and each cycle consisted of half an hour of plating and half an hour of stripping. (b) Constant-current cycling and corresponding voltage distributions (inset) of Li‖Li symmetric cells at 0.5 mA cm−2 with a capacity of 0.5 mA h cm−2 for LE and THB-PDOL. SEM image of Li metal cross-section after 50 cycles at 0.5 mA cm−2 with a capacity of 0.5 mA h cm−2 in (c) LE and (g) THB-PDOL, Scale bar: 50 μm. XPS spectra corresponding to C 1s, F 1s, B 1s of Li metal after cycling of Li‖Li symmetrical cells at 0.5 mA cm−2 with a capacity of 0.5 mA h cm−2 in (d–f) LE and (h–j) THB-PDOL. | ||
In order to assess the influence of electrolyte on lithium plating/stripping on the anode side of Li metal, Li‖Li symmetrical cells were assembled and subjected to long cycling tests at a current density of 0.5 mA cm−2 with a specific capacity of 0.5 mA h cm−2. At the commencement of the cycling process, both LE and THB-PDOL exhibited smooth and low overpotentials. However, after 400 hours of cycling, the overpotential of LE experienced a gradual increase, in contrast to THB-PDOL, which maintained a stable overpotential (Fig. 4b). The escalation in the overpotential of LE can be attributed to the formation of lithium dendrites resulting from non-uniform lithium deposition during cycling, coupled with the accumulation of dead lithium at the electrolyte/electrode interface. This phenomenon destabilizes the interface and leads to an increase in overpotential. Conversely, THB-PDOL displayed significant stability in lithium plating/stripping, indicating that the SEI of THB-PDOL offers enhanced resistance to lithium dendrite growth. Upon reduction of the current density to 0.1 mA cm−2 (Fig. S11†), the THB-PDOL-based LMBs demonstrated an ability to cycle for over 4000 hours at an extremely low polarization potential of 20 mV. The characterization of the lithium metal anode post-cycling using scanning electron microscopy (SEM) revealed the presence of lithium dendrites on the surface of the lithium anode cycled in LE, while the surface cycled in THB-PDOL appeared relatively flat, and the lithium dendrites on the surface of the lithium anode deteriorated slightly with the cycling process (Fig. S12 and S13†). Specifically, the lithium anode interface in LE exhibited a porous lithium deposit measuring 93.99 μm in thickness, whereas THB-PDOL showcased a compact lithium deposit with a thickness of merely 21.95 μm (Fig. 4c and g).
X-ray photoelectron spectroscopy (XPS) was utilized to analyze the Li metal anode SEI after subjecting it to 20 and 50 cycles of electroplating/stripping at 0.5 mA h cm−2 in different electrolytes to investigate its chemical state. The composition of the SEI changed as the number of electrochemical cycles increased. As shown in Fig. 4d and h, the C 1s spectra of the lithium anode of LE exhibited organic components at 286.3 eV (C–O), 288.8 eV (O–C–O), and 289.7 eV (–CO32− and –COOR) arising from the DOL decomposition, which are deemed resistive. The SEI of THB-PDOL, on the other hand, primarily contained organic substances from the polymer chain at 285.9 eV. Analysis of the F 1s spectra (Fig. 4e and i) of LE and THB-PDOL revealed a higher LiF concentration in the SEI of THB-PDOL, attributing to the inclusion of THB that facilitates LiTFSI dissociation, leading to increased LiF formation. LiF, known as a vital component of stable SEIs, enhances Li deposition uniformity and cycling stability of LMBs.48,49 Additionally, the SEI of THB-PDOL displayed a peak of BxOy (Fig. 4f and j), indicating the involvement of THB in generating LiBxOy inorganic compounds that establish a cross-linked covalent backbone, ensuring the formation of stable and robust SEIs.43,50 Elemental analyses revealed that the THB was involved in the formation of SEIs enriched in B and F (Fig. S15 and Table S1†). The THB-PDOL forms polymer–inorganic composite SEIs, where the long-chain composition of polymer component enhanced flexibility to the SEI, enabling it to adapt to rapid changes in electrode volume during lithium plating/stripping.40,50 Additionally, the abundant inorganic component ensures the uniformity of lithium ion transport within the SEI, which is essential for preventing the formation of lithium dendrites on lithium metal anodes.
3.4 Electrochemical performances of THB-PDOL based batteries
To evaluate the feasibility of utilizing THB-PDOL in practical batteries, LiFePO4 (LFP) and LiNi0.5Co0.2Mn0.3O2 (NCM523) were selected as positive electrodes due to the exceptional electrode compatibility and electrochemical stability of in situ polymerized THB-PDOL. The electrochemical performance of full cells based on lithium electrode (LE) and THB-PDOL was assessed. Initially, the energy storage capabilities of THB-PDOL were examined by constructing Li‖LFP full cells with a gradual increase in current density from 0.1C to 3C. At a current density of 0.1C, the battery exhibited a discharge specific capacity of 163 mA h g−1 in the initial stages, which gradually decreased with rising current densities, maintaining a discharge specific capacity of 119 mA h g−1 at 3C. The reversible capacity of the battery was significantly recovered upon reducing the current density. Conversely, the LE-based full cell showcased comparatively poor rate performance, with a discharge specific capacity of 160 mA h g−1 at 0.1C and only 113 mA h g−1 at 3C. Subsequent reduction in the current density revealed a notably lower reversible capacity in the LE-based full cell compared to the THB-PDOL-based full cell. Evidently, the THB-PDOL-based full cell demonstrated superior rate performance as delineated by the negligible increase in overpotential with escalating current density, highlighted in Fig. 5b. This enhanced rate capability can be ascribed to the exceptional tLi+ behavior and uniform lithium deposition in the THB-PDOL-based full cell. | ||
| Fig. 5 Electrochemical performances of batteries based on THB-PDOL. (a) Rate performance of the Li‖LFP full cells using THB-PDOL and LE. The tests were conducted within a cutoff voltage of 2.5–3.8 V. (b) Corresponding charge–discharge voltage profiles at various rates. (c) EIS curve of Li‖THB-PDOL‖LFP at 1C. (d) Effect of LE and THB-PDOL on the cycling performance and CE of Li‖LFP (1 mg) full cell at 1C. (e) The charge–discharge voltage curve of Li‖THB-PDOL‖LFP full cell during 200 cycles. (f) Cycling performance of Li‖THB-PDOL‖LFP full cell at 60 °C. (g) Rate performance of the Li‖NCM523 full cell using THB-PDOL and LE. (h) Schematic of THB-PDOL in situ polymerization at the cathode structure. | ||
The electrochemical impedance spectra (EIS) of Li‖THB-PDOL‖Li full cell before and after cycling at 1C are shown in Fig. 5c. The ohmic resistance (Rs) and charge transfer resistance (Rct) of Li‖THB-PDOL‖Li full cell are both small, indicating stable and fast charge transfer during cycling. Moreover, the Rct after cycling is slightly smaller than that before cycling, suggesting improved compatibility between the electrolyte and electrodes in the first 10 cycles. As the number of cycles increases, there is a negligible change in total impedance, suggesting that the stabilized SEI formed during cycling effectively ensures the efficient operation of the cell. In contrast, the resistance of the Li‖LE‖LFP full cell gradually increases (Fig. S16†), indicating that the interfacial side reactions are continuously occurring during the cycling process. The cells underwent long cycling at a 1C current density, and their charge/discharge performances are depicted in Fig. 5d. All cells were electrochemically activated for two cycles at a low current of 0.1C, resulting in a stable and reinforced SEI. The LE based full cell exhibited an initial discharge capacity of 160.4 mA h g−1 at 1C current density, which decreased to 103.3 mA h g−1 after 300 cycles, retaining 65.14% of its capacity. This capacity decay is attributed to an uneven SEI causing increased overpotential, thereby impeding Li-ion transport kinetics and accelerating capacity loss. Conversely, the THB-PDOL-based full cell, post in situ polymerization, showed an initial discharge capacity of 160.7 mA h g−1, maintaining 158.24 mA h g−1 after 300 cycles, with an impressive capacity retention rate of 98.64% and stable CE of 99.98%. The charge/discharge platform curves of THB-PDOL under different cycles can be seen in Fig. 5e, comparable to the cells utilizing the LE (Fig. S17†). This similarity indicates that THB-PDOL exhibits minimal polarization, underscoring its superior ionic conductivity and low interfacial impedance. Furthermore, the charge–discharge curve of THB-PDOL remains stable, signifying its excellent oxidative stability. The thermal stability of THB-PDOL also allows the batteries to operate at temperatures as high as 60 °C. As shown in Fig. 5f and S18,† the capacity of the LE based full cell decayed rapidly due to the volatilization of the LE. In contrast, the THB-PDOL based full cell exhibited stable cycling at 1C, maintaining a reversible capacity of 136 mA h g−1 even after 200 cycles, and the polymerized electrolyte inhibited the degradation of the ether chain more effectively due to its conversion of the volatile liquid electrolyte into a crosslinked structure, which improved the performance of the cell at high-temperature conditions.
Except for LiFePO4 cathode, THB-PDOL can also be used for high potential NCM523 cathode materials (Fig. 5g and S19†). It was observed that for the NCM523 system, the initial discharge capacity of the THB-PDOL-based full cell after in situ polymerization was 153.1 mA h g−1, with a capacity retention rate of 80.19% after 180 cycles, a CE of 99.45%. In contrast, the initial discharge capacity of the LE-based cell was 144.9 mA h g−1, and its capacity retention decreased to less than 80% after only 33 cycles. These results suggest that THB-PDOL exhibits excellent ionic transport and good interfacial compatibility with electrodes, leading to outstanding rate performance and favorable cycling stability. Therefore, THB-PDOL emerges as a promising candidate for polymeric LMBs due to its superior performance characteristics.
4. Conclusions
In this work, a film-forming Lewis acid additive (THB) is demonstrated not only to catalyze the polymerization of DOL, but also promote the formation of a stable fluorine- and boron-rich SEI to improve the interfacial stability and suppress the Li dendrite growth. Moreover, the THB additive promotes the dissociation of LiTFSI and release more Li+ while the electron-deficient boron center effectively anchors the TFSI− anion, thereby increasing the Li+ transfer number. These beneficial effects enhance the stability of the Li metal interface as well as the battery's long cycle life and high temperature performance. Overall, this strategy of incorporating small amounts of additives that serve as initiators and film-forming agents not only demonstrates good compatibility with lithium-metal batteries but also offers a promising approach for the widespread application of PDOL-based LMBs.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Ting Li: conceptualization, visualization, methodology, writing – original draft. Anjun Hu: investigation, writing – review & editing. Borui Yang: conceptualization, visualization. Kun Li: conceptualization, visualization. Bin Li: validation. Miao He: formal analysis, validation. Liu Yang: formal analysis, validation. Jianping Long: writing – review & editing, funding acquisition, project administration, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors acknowledge the National Natural Science Foundation of China (21805018; 52002039), the Natural Science Foundation of Sichuan Province (2024NSFSC1016), and Scientific Research Startup Foundation of Chengdu University of Technology (10912-KYQD2023-10240). The authors extend their gratitude to Mr. Li Julong from Shiyanjia Lab (https://www.shiyanjia.com) for SEM tests and SCI-GO (https://www.sci-go.com) for FTIR tests. And we appreciate Phadcalc (https://www.phadcalc.com) for the molecular dynamics calculation.References
- S. Tan, W. Wang, Y. Tian, S. Xin and Y. Guo, Adv. Funct. Mater., 2021, 31, 2105253 CrossRef CAS.
- R. Li, Y. Fan, C. Zhao, A. Hu, B. Zhou, M. He, J. Chen, Z. Yan, Y. Pan and J. Long, Small Methods, 2023, 7, 2201177 CrossRef CAS PubMed.
- A. Hu, W. Chen, X. Du, Y. Hu, T. Lei, H. Wang, L. Xue, Y. Li, H. Sun, Y. Yan, J. Long, C. Shu, J. Zhu, B. Li, X. Wang and J. Xiong, Energy Environ. Sci., 2021, 14, 4115–4124 RSC.
- C. Zhao, Z. Yan, B. Zhou, Y. Pan, A. Hu, M. He, J. Liu and J. Long, Angew. Chem., 2023, 135, e202302746 CrossRef.
- A. Hu, F. Li, W. Chen, T. Lei, Y. Li, Y. Fan, M. He, F. Wang, M. Zhou, Y. Hu, Y. Yan, B. Chen, J. Zhu, J. Long, X. Wang and J. Xiong, Adv. Energy Mater., 2022, 12, 2202432 CrossRef CAS.
- T. Dong, G. Xu, B. Xie, T. Liu, T. Gong, C. Sun, J. Wang, S. Zhang, X. Zhang, H. Zhang, L. Huang and G. Cui, Adv. Mater., 2024, 2400737, DOI:10.1002/adma.202400737.
- D. Zhang, R. Gu, Y. Yang, J. Ge, J. Xu, Q. Xu, P. Shi, M. Liu, Z. Guo and Y. Min, Angew. Chem., Int. Ed., 2024, 63, e202315122 CrossRef CAS PubMed.
- S. Xia, C. Yang, Z. Jiang, W. Fan, T. Yuan, Y. Pang, H. Sun, T. Chen, X. Li and S. Zheng, Adv. Compos. Hybrid Mater., 2023, 6, 198 CrossRef CAS.
- C. Chen, Z. Li, X. Du, Q. Zhou, P. Han and G. Cui, eTransportation, 2024, 20, 100318 CrossRef.
- Z. Lin, X. Guo, Z. Wang, B. Wang, S. He, L. A. O'Dell, J. Huang, H. Li, H. Yu and L. Chen, Nano Energy, 2020, 73, 104786 CrossRef CAS.
- S. Chai, Z. Chang, Y. Zhong, Q. He, Y. Wang, Y. Wan, M. Feng, Y. Hu, W. Li, W. Wei and A. Pan, Adv. Funct. Mater., 2023, 33, 2300425 CrossRef CAS.
- J. Zhang, H. Wu, X. Du, H. Zhang, L. Huang, F. Sun, T. Liu, S. Tian, L. Zhou, S. Hu, Z. Yuan, B. Zhang, J. Zhang and G. Cui, Adv. Energy Mater., 2023, 13, 2202529 CrossRef CAS.
- F. Jiang, X. Cheng, S. Yang, J. Xie, H. Yuan, L. Liu, J. Huang and Q. Zhang, Adv. Mater., 2023, 35, 2209114 CrossRef CAS PubMed.
- K. An, M. J. Joo, Y. H. T. Tran, S. Kwak, H. G. Kim, C. S. Jin, J. Suk, Y. Kang, Y. J. Park and S.-W. Song, Adv. Funct. Mater., 2023, 33, 2301755 CrossRef CAS.
- C. Zhao, Y. Pan, R. Li, A. Hu, B. Zhou, M. He, J. Chen, Z. Yan, Y. Fan, N. Chen, M. Liu and J. Long, Chem. Eng. J., 2023, 463, 142386 CrossRef CAS.
- B. Yang, Y. Pan, T. Li, A. Hu, K. Li, B. Li, L. Yang and J. Long, Energy Storage Mater., 2024, 65, 103124 CrossRef.
- A. Hu, W. Chen, F. Li, M. He, D. Chen, Y. Li, J. Zhu, Y. Yan, J. Long, Y. Hu, T. Lei, B. Li, X. Wang and J. Xiong, Adv. Mater., 2023, 35, 2304762 CrossRef CAS PubMed.
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, 1902029 CrossRef CAS PubMed.
- Q. Wang, J. Yang, X. Huang, Z. Zhai, J. Tang, J. You, C. Shi, W. Li, P. Dai, W. Zheng, L. Huang and S. Sun, Adv. Energy Mater., 2022, 12, 2103972 CrossRef CAS.
- S. Huo, L. Sheng, W. Xue, L. Wang, H. Xu, H. Zhang and X. He, InfoMat, 2023, 5, e12394 CrossRef CAS.
- C. Ma, W. Cui, X. Liu, Y. Ding and Y. Wang, InfoMat, 2022, 4, e12232 CrossRef CAS.
- D. Zhou, D. Shanmukaraj, A. Tkacheva, M. Armand and G. Wang, Chem, 2019, 5, 2326–2352 CAS.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- H. Yang, M. Jing, L. Wang, H. Xu, X. Yan and X. He, Nano-Micro Lett., 2024, 16, 127 CrossRef PubMed.
- H. Xu, J. Zhang, H. Zhang, J. Long, L. Xu and L. Mai, Adv. Energy Mater., 2023, 13, 2204411 CrossRef CAS.
- Y. Zhao, T. Zhou, D. Baster, M. El Kazzi, J. W. Choi and A. Coskun, ACS Energy Lett., 2023, 8, 3180–3187 CrossRef CAS.
- H. Yang, M. Jing, L. Wang, H. Xu, X. Yan and X. He, Nano-Micro Lett., 2024, 16, 127 CrossRef PubMed.
- H. Wu, B. Tang, X. Du, J. Zhang, X. Yu, Y. Wang, J. Ma, Q. Zhou, J. Zhao, S. Dong, G. Xu, J. Zhang, H. Xu, G. Cui and L. Chen, Adv. Sci., 2020, 7, 2003370 CrossRef CAS PubMed.
- J. Ma, X. Feng, Y. Wu, Y. Wang, P. Liu, K. Shang, H. Jiang, X. Hou, D. Mitlin and H. Xiang, J. Energy Chem., 2023, 77, 290–299 CrossRef CAS.
- W. Li, J. Gao, H. Tian, X. Li, S. He, J. Li, W. Wang, L. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202114805 CrossRef CAS PubMed.
- W. Li, L. Ma, S. Liu, X. Li, J. Gao, S. Hao and W. Zhou, Angew. Chem., 2022, 134, e202209169 CrossRef.
- Z. Li, W. Tang, Y. Deng, M. Zhou, X. Wang, R. Liu and C. Wang, J. Mater. Chem. A, 2022, 10, 23047–23057 RSC.
- Z. Wang, Y. Wang, L. Shen, Z. Jin, H. M. Law, A. Wang, W. Wang and F. Ciucci, Energy Environ. Sci., 2023, 16, 4084–4092 RSC.
- H. Yang, B. Zhang, M. Jing, X. Shen, L. Wang, H. Xu, X. Yan and X. He, Adv. Energy Mater., 2022, 12, 2201762 CrossRef CAS.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2022, 12, 2102932 CrossRef CAS.
- J. Zhao, M. Li, H. Su, Y. Liu, P. Bai, H. Liu, L. Ma, W. Li, J. Sun and Y. Xu, Small Methods, 2023, 7, 2300228 CrossRef CAS PubMed.
- Q. Zhao, X. Liu, S. Stalin, K. Khan and L. A. Archer, Nat. Energy, 2019, 4, 365–373 CrossRef CAS.
- K. Mu, D. Wang, W. Dong, Q. Liu, Z. Song, W. Xu, P. Yao, Y. Chen, B. Yang, C. Li, L. Tian, C. Zhu and J. Xu, Adv. Mater., 2023, 35, 2304686 CrossRef CAS PubMed.
- W. Li, J. Gao, H. Tian, X. Li, S. He, J. Li, W. Wang, L. Li, H. Li, J. Qiu and W. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202114805 CrossRef CAS PubMed.
- J. Xiang, Y. Zhang, B. Zhang, L. Yuan, X. Liu, Z. Cheng, Y. Yang, X. Zhang, Z. Li, Y. Shen, J. Jiang and Y. Huang, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- J. Yu, X. Lin, J. Liu, J. T. T. Yu, M. J. Robson, G. Zhou, H. M. Law, H. Wang, B. Z. Tang and F. Ciucci, Adv. Energy Mater., 2022, 12, 2102932 CrossRef CAS.
- W. Ren, C. Ding, X. Fu and Y. Huang, Energy Storage Mater., 2021, 34, 515–535 CrossRef.
- J. Xiang, Y. Zhang, B. Zhang, L. Yuan, X. Liu, Z. Cheng, Y. Yang, X. Zhang, Z. Li, Y. Shen, J. Jiang and Y. Huang, Energy Environ. Sci., 2021, 14, 3510–3521 RSC.
- R. Rojaee, S. Cavallo, S. Mogurampelly, B. K. Wheatle, V. Yurkiv, R. Deivanayagam, T. Foroozan, M. G. Rasul, S. Sharifi-Asl, A. H. Phakatkar, M. Cheng, S. Son, Y. Pan, F. Mashayek, V. Ganesan and R. Shahbazian-Yassar, Adv. Funct. Mater., 2020, 30, 1910749 CrossRef CAS.
- Y. Meng, D. Zhou, R. Liu, Y. Tian, Y. Gao, Y. Wang, B. Sun, F. Kang, M. Armand, B. Li, G. Wang and D. Aurbach, Nat. Energy, 2023, 8, 1023–1033 CrossRef CAS.
- Y. Wang, J. Lu, Y. Qiao, W. Dai, H. Xia, C. Yu, Y. Hu, Z. Ma and Z. Fu, J. Power Sources, 2023, 580, 233402 CrossRef CAS.
- Y. Du, L. Zhao, C. Xiong, Z. Sun, S. Liu, C. Li, S. Hao, W. Zhou and H. Li, Energy Storage Mater., 2023, 56, 310–318 CrossRef.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. (David) Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- Z. Li, L. Wang, X. Huang and X. He, Small, 2023, 20, 2305429 CrossRef PubMed.
- J. Liu, X. Li, J. Huang, G. Yang and J. Ma, Adv. Funct. Mater., 2024, 34, 2312762 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02010c |
| This journal is © The Royal Society of Chemistry 2024 |