
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances and challenges in the electrochemical reduction of carbon dioxide
Jingyi
Han
,
Xue
Bai
,
Xiaoqin
Xu
,
Xue
Bai
,
Anaer
Husile
,
Siying
Zhang
,
Luoluo
Qi
and
Jingqi
Guan
 *
*
Institute of Physical Chemistry, College of Chemistry, Jilin University, Changchun 130021, PR China. E-mail: guanjq@jlu.edu.cn
First published on 2nd May 2024
Abstract
The electrocatalytic carbon dioxide reduction reaction (ECO2RR) is a promising way to realize the transformation of waste into valuable material, which can not only meet the environmental goal of reducing carbon emissions, but also obtain clean energy and valuable industrial products simultaneously. Herein, we first introduce the complex CO2RR mechanisms based on the number of carbons in the product. Since the coupling of C–C bonds is unanimously recognized as the key mechanism step in the ECO2RR for the generation of high-value products, the structural–activity relationship of electrocatalysts is systematically reviewed. Next, we comprehensively classify the latest developments, both experimental and theoretical, in different categories of cutting-edge electrocatalysts and provide theoretical insights on various aspects. Finally, challenges are discussed from the perspectives of both materials and devices to inspire researchers to promote the industrial application of the ECO2RR at the earliest.
1. Introduction
In the late 2023, the 28th United Nations Climate Change Conference (COP28) conducted the first “global carbon inventory” of the world's efforts to address climate change.1 The parties unanimously decided to accelerate the pace of reducing greenhouse gas emissions by 2030,2 calling on governments to advance the effective implementation of carbon reduction actions to achieve the ultimate goal of the Paris Agreement as soon as possible,3i.e., in addition to natural autonomous pathways such as bioenergy with carbon capture and storage (BECCS), industrial technologies of carbon capture, utilization and storage (CCUS) are to be eagerly promoted alternatively.4 With the depletion of traditional fossil fuels, the energy crisis has become an extreme urgency.5As an ideal solution, the electrocatalytic carbon dioxide reduction reaction (ECO2RR) can not only end the artificial carbon cycle, but also convert renewable power such as solar energy and wind energy into beneficial chemical energy products, including clean fuels and industrial raw materials to achieve a virtuous cycle of turning waste into treasure as a whole.6–9 However, as depicted in Fig. 1, to date, the related research in the field of ECO2RRs is still in the emerging stage, which is far from mature compared with other electrochemical technologies such as hydrogen production through the electrolysis of water, and the prerequisite for its practical application is a robust catalyst with high electrochemical activity and high selectivity toward the target product.10–13 Noteworthily, the complexity of the reaction mechanism of the CO2RR should not be underestimated, and a large number of studies have confirmed that the C–C coupling step is crucial for the advancement of the ECO2RR and has a high degree of structural sensitivity.14–17 Therefore, the in-depth exploration of the influence of the electronic structure of materials on the ECO2RR catalytic performance has always been the main direction of researchers.
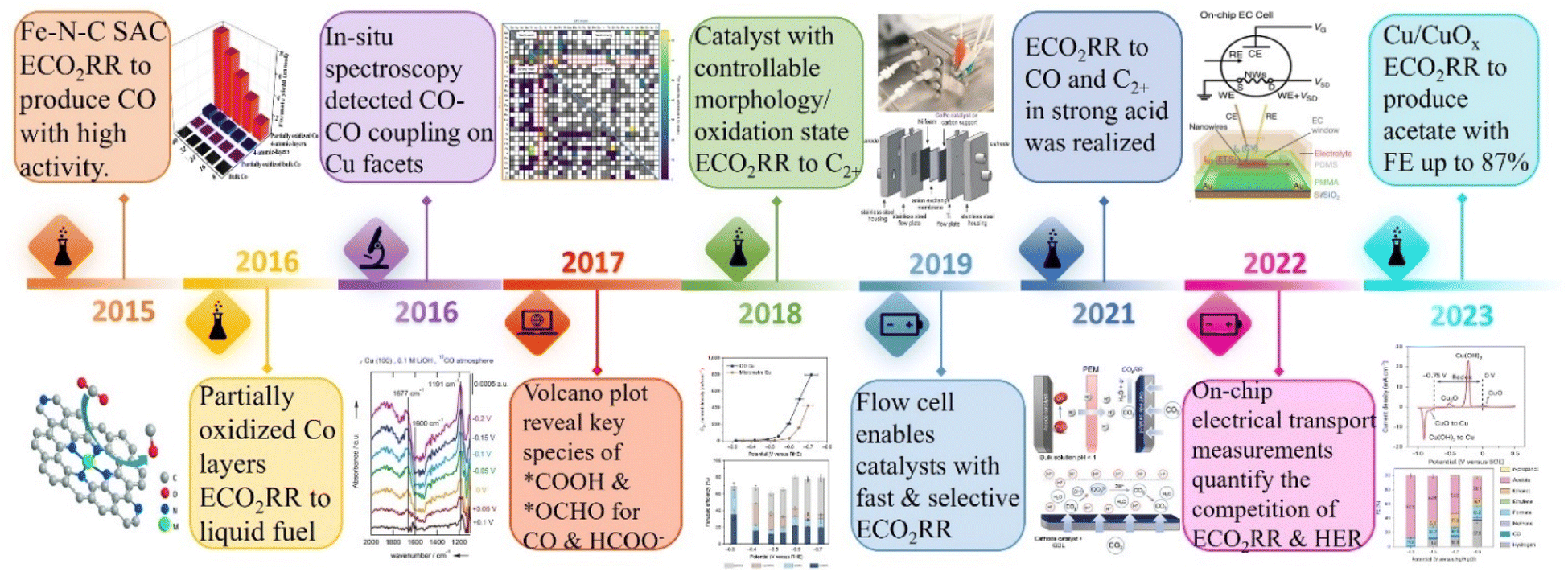 | ||
| Fig. 1 Timeline of the major developments in the ECO2RR. | ||
In this review, the reaction mechanisms of the ECO2RR are first classified based on the number of carbon atoms in the products. On this basis, we thoroughly discuss the profound insights gained on the structure–activity relationship, and the most cutting-edge catalysts reported in recent years have been comprehensively reviewed according to the detailed categories, including atomically-dispersed catalysts, alloys, metal–organic frameworks (MOFs) and covalent organic frameworks (COFs). In terms of theoretical calculations, authoritative opinions are representatively classified. Finally, we analyze the remaining challenges from both the material and device perspectives, with a view to inspire researchers to help pave a new stage in the field of ECO2RR.
2. ECO2RR mechanisms
In the process of carbon dioxide reduction, due to the extremely high stability of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O chemical bonds and the competition of hydrogen evolution reaction (HER), the carbon dioxide reduction process faces major challenges, which seriously hinders the effective implementation.18–20 Therefore, it is urgent to design an efficient catalyst to overcome the high activation energy barrier. However, although there are more and more experimental and theoretical studies on various catalysts, the available catalysts need to have a large overpotential for specific products, and their selectivity is usually very low. On different catalysts under different conditions, different products will be produced, such as CO, formate, methane and ethanol.21 In addition, the electrochemical CO2RR process involves multi-electron transfer processes, and the product is very complex, making the subsequent product separation and purification processes very difficult. Therefore, it is important to understand the reaction mechanism and reaction intermediates, which is related to the design and modification of catalysts.22Table 1 lists the half-reactions that may occur on the cathode surface during the electrochemical reduction of CO2 and their equilibrium potentials relative to the standard hydrogen electrode (SHE) in electrolytes at pH 7.19
O chemical bonds and the competition of hydrogen evolution reaction (HER), the carbon dioxide reduction process faces major challenges, which seriously hinders the effective implementation.18–20 Therefore, it is urgent to design an efficient catalyst to overcome the high activation energy barrier. However, although there are more and more experimental and theoretical studies on various catalysts, the available catalysts need to have a large overpotential for specific products, and their selectivity is usually very low. On different catalysts under different conditions, different products will be produced, such as CO, formate, methane and ethanol.21 In addition, the electrochemical CO2RR process involves multi-electron transfer processes, and the product is very complex, making the subsequent product separation and purification processes very difficult. Therefore, it is important to understand the reaction mechanism and reaction intermediates, which is related to the design and modification of catalysts.22Table 1 lists the half-reactions that may occur on the cathode surface during the electrochemical reduction of CO2 and their equilibrium potentials relative to the standard hydrogen electrode (SHE) in electrolytes at pH 7.19
| CO2 reduction half reaction | The equilibrium potential relative to SHE at pH 7 |
|---|---|
| CO2 + 2H+ + 2e− → HCOOH + H2O | E 0redox = −0.610 V |
| CO2 + 2H+ + 2e− → CO + H2O | E 0redox = −0.530 V |
| 2CO2 + 2H+ + 2e− → H2C2O4 | E 0redox = −0.913 V |
| CO2 + 4H+ + 4e− → HCHO + H2O | E 0redox = −0.480 V |
| CO2 + 6H+ + 6e− → CH3OH + H2O | E 0redox = −0.380 V |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | E 0redox = −0.240 V |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | E 0redox = −0.349 V |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | E 0redox = −0.329 V |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | E 0redox = −0.270 V |
| 3CO2 + 18H+ + 18e− → C3H7OH + H2O | E 0redox = −0.310 V |
| 2H+ + 2e− → H2 | E 0redox = −0.42 V |
Theoretical calculations and modeling can provide information such as binding energy, free energy and activation energy of the intermediates, which is helpful to evaluate the feasibility of each reaction step of the synthesized catalyst and screen out the key intermediates conducive to the reaction.23 Electrochemical reduction method is one of the key means to determine the reaction intermediates. Pure metals such as Al, Fe, Ni, Pt and Ti can reduce CO2 to CO, but the HER rate during the reduction process is much higher than the reduction rate of CO2, while Ag, Au and Zn will produce CO with high current efficiency.24 Formate is the main product of the catalytic reduction on metals such as Hg, In and Sn.25 Cu-based catalysts can reduce CO2 to hydrocarbons and alcohols. Fig. 2 shows the mechanism of CO2RR on metal electrocatalysts.26 This section introduces the main mechanisms of electroreduction of CO2 to HCOOH/HCOO−, CO, HCHO, CH3OH, CH4, C2H4, CH3CH2OH and CH3CH2CH2OH.
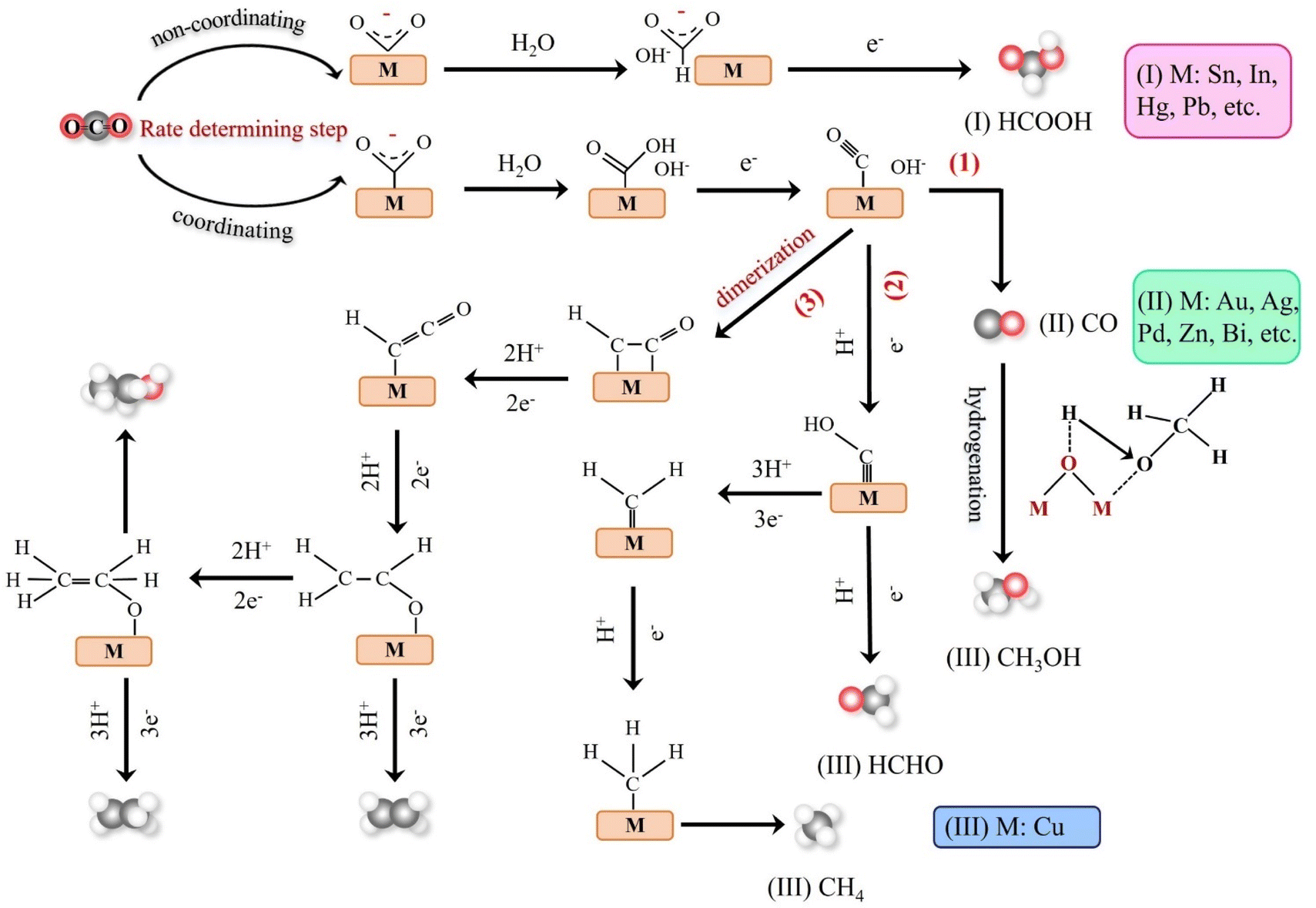 | ||
| Fig. 2 Schematic mechanism of various metal electrocatalysts for the CO2RR. | ||
2.1. Reduction of CO2 to C1 products
The products of CO2 reduction to C1 include HCOOH/HCOO−, CO, HCHO, CH3OH and CH4.27–31 The formation of HCOOH/HCOO− can be divided into three steps. First, CO2 molecules are inserted into the metal–hydrogen bond and then combined with one or two oxygen atoms to the metal electrode, followed by further protonation to form formate (Fig. 3a).32 Second, the surface hydroxyl group reacts with CO2 to form a carbonate intermediate, thereby forming HCOO− (Fig. 3b).33 Finally, *CO2− radicals react with adjacent water molecules to form formate/formate salts (Fig. 3c).34 The reduction of CO2 to CO is carried out by forming *COOH intermediates. The *COOH intermediate is formed by the proton coupling of *COO−, electron transfer or single electron transfer, and then protonation. Further, proton-coupled electrons are transferred to the adsorbed *COOH intermediate, resulting in dissociation to form CO (Fig. 3d). Hansen et al. found that *COOH was reduced to *CO, but it was inhibited by the first step of weak COOH binding.35 The formation of HCHO, CH3OH and CH4 is carried out by the transfer of 4, 6 and 8 electrons, respectively. The initial *CO is converted to *CHO, *CH2O and *CH3O by hydrogenation, and *CH3O is further hydride oxygenated to CH4,36 which can also be produced by the multi-step hydrogenation of *COH. *CH2O is desorbed to form HCHO, and *CH3O is hydrogenated to CH3OH (Fig. 3e).37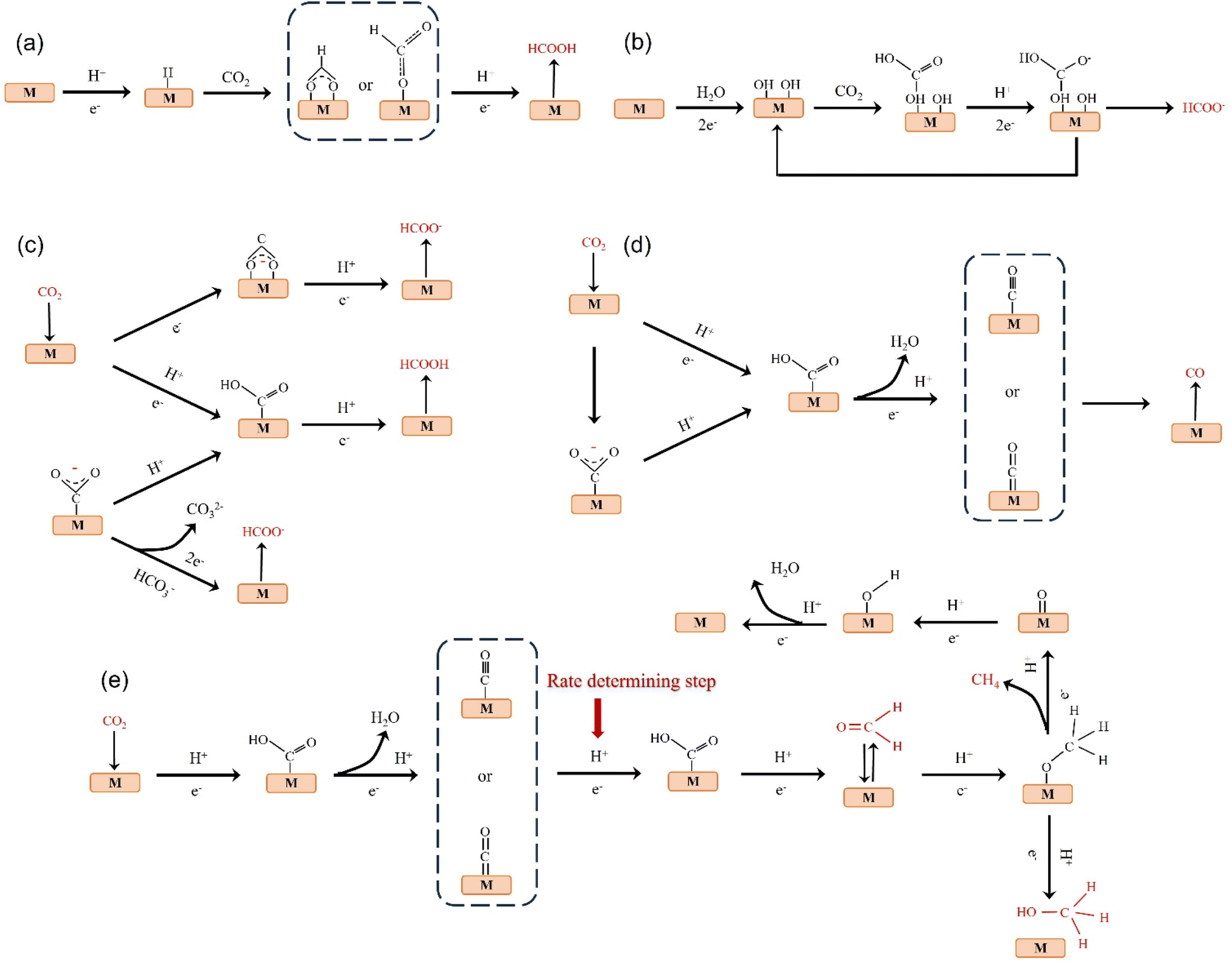 | ||
| Fig. 3 (a) Monodentate or bidentate intermediate route for HCOOH formation. (b) Surface-bound carbonate intermediate route for the formation of HCOOH/formate. (c) *CO2− radical intermediate route for the formation of formate. (d) Possible reaction pathway of CO2 reduction to CO. (e) Possible reaction pathways for the reduction of CO2 to HCHO, CH3OH and CH4. | ||
2.2. Reduction of CO2 to C2 products
C2 products mainly include C2H4, CH3CHO and CH3CH2OH.38–40 At low negative potentials, the formation of C2 products is a complex process with multiple reaction pathways. Nie et al. proposed that the formation of C2H4 is the result of the coupling of two *CH2 intermediates or the insertion of CO in the Fischer–Tropsch-like step.41 Another pathway is that the *CO intermediate is dimerized to form the *C2O2 intermediate, which is then transformed into *COCHO by electron transfer and finally hydride oxygenated to form C2H4.42 The formation of CH3CH2OH has the same reaction intermediate as C2H4. Researchers proposed that *CHO dimerization is the main pathway of ethanol on Cu (100) at low potentials.43 The intermediate of vinyl alcohol can be further reduced to C2H4 or CH3CHO and C2H5OH (Fig. 4).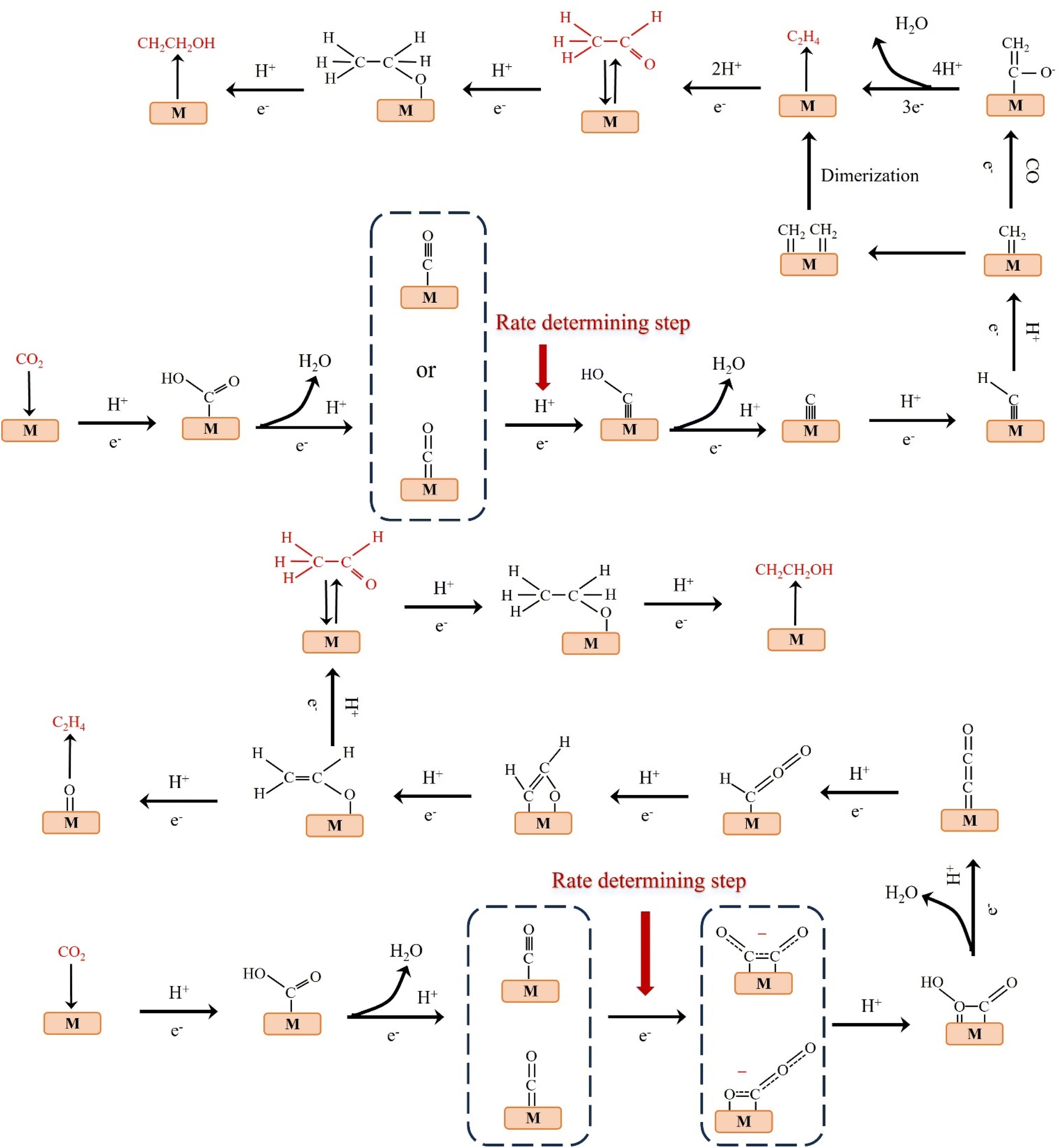 | ||
| Fig. 4 Possible reaction paths for electrocatalytic CO2 reduction to produce C2H4, CH3CHO, and C2H5OH. | ||
2.3. Reduction of CO2 to C3 products
The reduction of CO2 to C3 long-chain products (such as propanol) has always been a difficult problem to overcome.44 When CO2 is reduced to propanol, the following steps may be carried out. The adsorbed C2 intermediate undergoes a C–C coupling reaction with adjacent C1 intermediate, followed by proton or electron transfer to produce propionaldehyde, which is finally reduced to propanol (Fig. 5).45 Although C2+ products are eagerly awaited due to their peak economic value, the generation of C2+ by ECO2RR remains challenging at this stage. Table 2 lists the formation rates and FEs obtained so far for different C2+ products to provide a more comprehensive overview.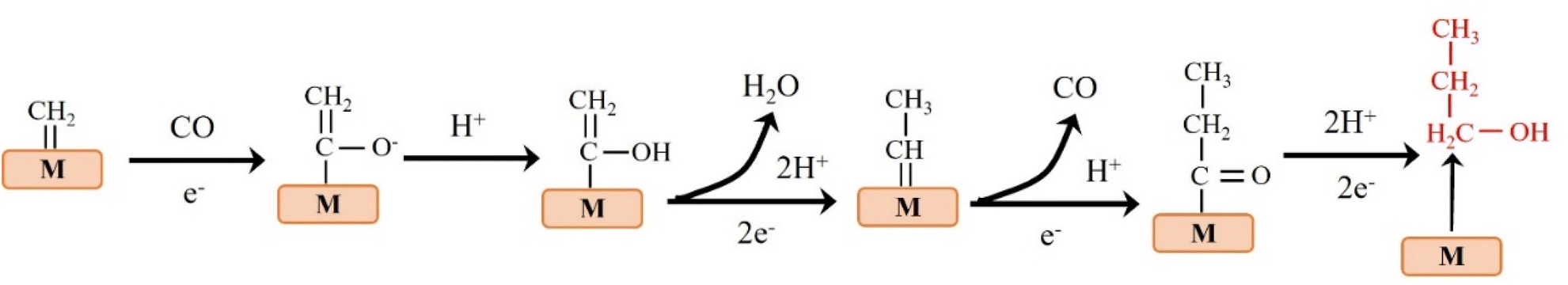 | ||
| Fig. 5 Possible reaction pathway for electrocatalytic CO2 reduction to C3H7OH. | ||
| Catalyst | FE of C2+ products (%) | Formation rate of C2+rC2+ (mmol cm−2 h−1) | Geometric area normalized jC2+ (mA cm−2) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| Cu/Pd-1% | 66.2 | 1.45 | 463.2 | 1 M KOH | 46 |
| CuO NS | 85.6 | 0.74 | 237 | 1 M KOH | 47 |
| CuAg-wire-6% | 85.1 | 0.8 | 264.5 | 1 M KOH | 48 |
| dCu2O/Ag2.3% | 82.1 | 2.06 | 656.8 | 1 M KOH | 49 |
| Li2−xCuO2 | 90.6 | 0.65 | 200 | 1 M KOH | 50 |
| Cu3Nx | 81.7 | 0.99 | 307 | 1 M KOH | 51 |
| Cu(OH)2-D | 86.8 | 0.65 | 217 | 1 M KOH | 52 |
| Cu–CuI | 71 | 1.13 | 381 | 1 M KOH | 53 |
| Nanoporous Cu | 62 | 1.25 | 411 | 1 M KOH | 54 |
| CuPb-0.7/C | 73.6 | 1.01 | 294.4 | 1 M KOH | 55 |
| CuO NS | 58.6 | 1.32 | 410.2 | 1 M KHCO3 | 56 |
| Ag–Cu/GDY | 55.1 | — | 24.98 | 1 M KHCO3 | 57 |
| N-doped graphene quantum dots | 47.75 | — | ∼90 | 1 M KOH | 58 |
| N-functionalized GO | 45.3 | — | ∼1.3 | 0.1 M KHCO3 | 59 |
| N-functionalized GO | 45 | — | ∼50 | 1 M KOH | 60 |
| SnO2 nanoparticles | 10.4 | — | 150 | 1 M KHCO3 | 61 |
3. Structure–activity relationships
The electronic structure of the catalyst is key to optimizing the catalytic processes that fundamentally dominate their various electrocatalytic properties, including activity, selectivity, and stability.62–65 Therefore, the structure–performance relationship of the catalyst as the core of electrocatalysis is of great significance. However, the factors affecting the electronic state of the catalytic center are complicated. The combination of active metal elements, defect site engineering and coordination environment optimization between adjacent atoms have been developed as the main means to effectively regulate the electronic structure.66–68 The in-depth understanding about the structure–performance relationship not only broadens the way for the rational development of new catalytic materials with more potential and pertinence but also provides a strong theoretical guidance for revealing the mechanism of electrocatalytic reactions. Furthermore, theoretical modulation of electrocatalysts can effectively adjust the local hindrance and electronic structure to manipulate the entire workflow of carbon dioxide in the electroreduction process, including local migration, adsorption, activation, hydrogenation, and desorption of the products, thus providing great possibilities and versatility for the realization of carbon dioxide electroreduction. Therefore, reasonable theoretical modulation strategy is the key to improve the catalyst performance. Electrocatalytic reactions generally occur at the interface or surface of the catalyst, and differences in the catalyst structure can affect the strength between the catalyst surface and the adsorbate. At the same time, the catalyst geometry or electronic structure engineering all produce synergistic effects, influencing the catalytic activity, stability, and selectivity. In addition, changes in the electronic structure of catalysts can directly affect the spin change and change the way catalysts bind to the adsorbent, leading to a decrease in the reaction energy and ultimately an increase in the catalytic activity.69–72 Therefore, we also combine theoretical modulation to conduct a more profound and comprehensive analysis of the structure activity relationship of ECO2RR catalysts.3.1. Metal centers
Metal sites play an important role as the main foundation for adsorbing CO2 and activating it into different products.73–76 Since various metal elements have their distinct state of electron clouds in different shell orbitals, they congenitally have significant differences in the CO2RR electrocatalytic activity, including current density, stability, and even the types and selectivity of generated products. To date, researchers have researched single-atom catalysts (SACs) covering most of the transition metal elements like Mo, Fe, Co, Ni, Cu, and Zn, precious metal elements like Au, Ag, Pt, and Ir, and p-block elements like Bi, Sn, and Sb to explore their intrinsic CO2RR electrocatalytic properties (Fig. 6).77 As far as single metal elements are concerned, it commonly possesses the ability to convert CO2 into CO (g), especially transition metals. Notably, certain metals can reduce CO2 to produce different products simultaneously.78 For example, in addition to CO, Co-SACs and Sn-SACs can yield second liquid product CH3OH and HCOOH, respectively.79 However, the determinants of product selectivity remain unclear. The well-designed Zn-N4-C structure can generate CH4 with high economic value.80 This Zn SAC exhibited remarkable CO2RR activity with high faradaic efficiency of 85% toward CH4 and can maintain this level up to 35 hours. Density functional theory (DFT) calculations revealed that the Zn site follows a unique CO2RR mechanism, where the initial activation of CO2 is more inclined to the O-binding intermediate *OCHO rather than the C-binding intermediate *COOH, which is conducive to the hydrogenation of CO2 to CH4. It has been reported that Co, Cu, Mo, In, Sn and Sb can rarely reduce CO2 to liquid C1 products including HCOOH and CH3OH.81 For example, atomically-dispersed Bi sites follow another reaction mechanism in which the energy barrier advantage of forming *COOH intermediate is obvious, making the Bi center have higher activity and selectivity for converting CO2 into HCOOH.82 Another element used to produce HCOOH is In, which also follows a similar reaction pathway.83 Interestingly, for the N-C structure, high concentrations of In atoms have been verified to be more inclined to generate HCOOH, while highly dispersed In sites have the ability to accelerate the formation of CO. Unlike In, the selectivity of HCOOH at atomic Sb sites differs slightly from that of pure metal, while the activity advantage of the former is significant.84 Theoretical calculations revealed that the intermediate species formed on the Sb-N4 structure favors forming HCOO* rather than COOH*, thus providing a pathway to generate formate with FEformate up to 94% at −0.8 V vs. RHE. In particular, due to the dense electron distribution with rich energy of the d and f orbitals in the inner layer, Cu has a strong effect of localization and exchange interaction, thus exhibiting remarkable ability of reducing CO2 to C2, C3 and even more carbon products.85 Interestingly, the type and yield of products on the same metal-based material are closely related to the morphology. For instance, Irabien et al. systematically focused on the effect of particle size in the range of 25–80 nm on the conversion efficiency and selectivity of continuous ECO2RR on Cu-based nanoparticles.20 They found that when the particle size was reduced to 25 nm, the ethylene yield and Faraday efficiency with high economic value reached the peak, which were 1148 μmol m−2 s−1 and 92.8%, respectively. Simultaneously, the competitive HER was weakened, which strongly verified the inextricable relationship between the material morphology like particle size and the electrocatalytic activity of ECO2RR. | ||
| Fig. 6 The known product distribution of the reported elements for the ECO2RR. | ||
Building on single metal sites, introducing additional metal sites to construct heteronuclear active centers can combine the property advantages of various metals.86 Multinuclear electrocatalysts have a variety of active centers including top and bridging sites, thus providing more diversified adsorption modes for CO2 and intermediates.87 Moreover, the coordination between various metal atoms and adjacent ligands is rather flexible, and the d-band center can be regulated through the interaction of electron orbitals, thereby optimizing the adsorption energy of the intermediate species so that it possesses properties particular for SACs to effectively break their inevitable linear scale barrier.88 The asymmetric multinuclear active centers not only have a significantly enhanced charge density gradient but also locally generate torque, which is conducive to the efficient overlap of atomic orbitals when activating active linear molecules, resulting in the leap of the overall electrocatalytic CO2RR performance and showing more potential for practical applications. For example, due to the superior intrinsic activity of Ni and Fe at high and low potentials, respectively, Ren et al. designed a Ni/Fe-N-C bimetallic material, which can maintain a high selectivity of CO at a wide potential span from − 0.4 V to −1 V, and a high FECO of 98% can be obtained at −0.7 V.89 Gong et al. screened Zn and Co elements,90 in which the Co site is beneficial for the formation of *COOH, while the Zn site is beneficial for the desorption of *COOH. The bimetallic site of Zn/Co-N-C can simultaneously reduce the energy barrier of both the processes to greatly promote the conversion of CO2 to CO.
3.2. Vacancy defects
Vacancies not only have unique electronic properties but also generate a fresh coordination structure to cooperatively optimize the electronic and band structure of the active center to upgrade the chemical properties.91 Furthermore, vacancy defects provide additional adsorption and active sites for reactive species and moderate the free energy of CO2 activation and product desorption. Additionally, the defect sites play a crucial role in the electrochemical performance and stability. Due to their high controllable preparation characteristics, vacancy defect engineering is widely used in geometric/electronic structure modulation. In general, vacancies applied in CO2RR can be classified into cationic and anionic vacancies.92 Due to the relatively low energy required to generate anionic vacancies such as O, S, and N, they have been extensively explored combined with theoretical calculations.93–95 For example, a Cu–Sn diatom-site electrocatalyst stabilized by double oxygen vacancies on CeO2−x was constructed by Chen et al. Theoretical calculations emphasized the importance of the synergistic effect of copper and tin in lowering the activation energy and promoting the formation of the intermediate *OCHO, thus explaining its high formate selectivity (Fig. 7a and b).96 Besides, a sacrificial protection strategy to stabilize the interfacial crystalline CuO by embedding active amorphous SnO2 (c-CuO/a-SnO2) was reported by Guo et al. DFT calculations demonstrated that the loss of oxygen in the heterostructure mainly originates from amorphous SnO2, avoiding the reduction of c-CuO. Meanwhile, the production of Ov in c-SnO2 leads to off-domain s-orbital electrons around the tin atom, which provides additional electrons in higher orbitals than the unoccupied d-orbitals of Cu2+ (Fig. 7c and d). Therefore, a-SnO2 can effectively retain Cu2+ as a reservoir for oxygen species, thus avoiding oxygen loss from c-CuO, greatly improving the electrocatalytic activity, selectivity, and stability.97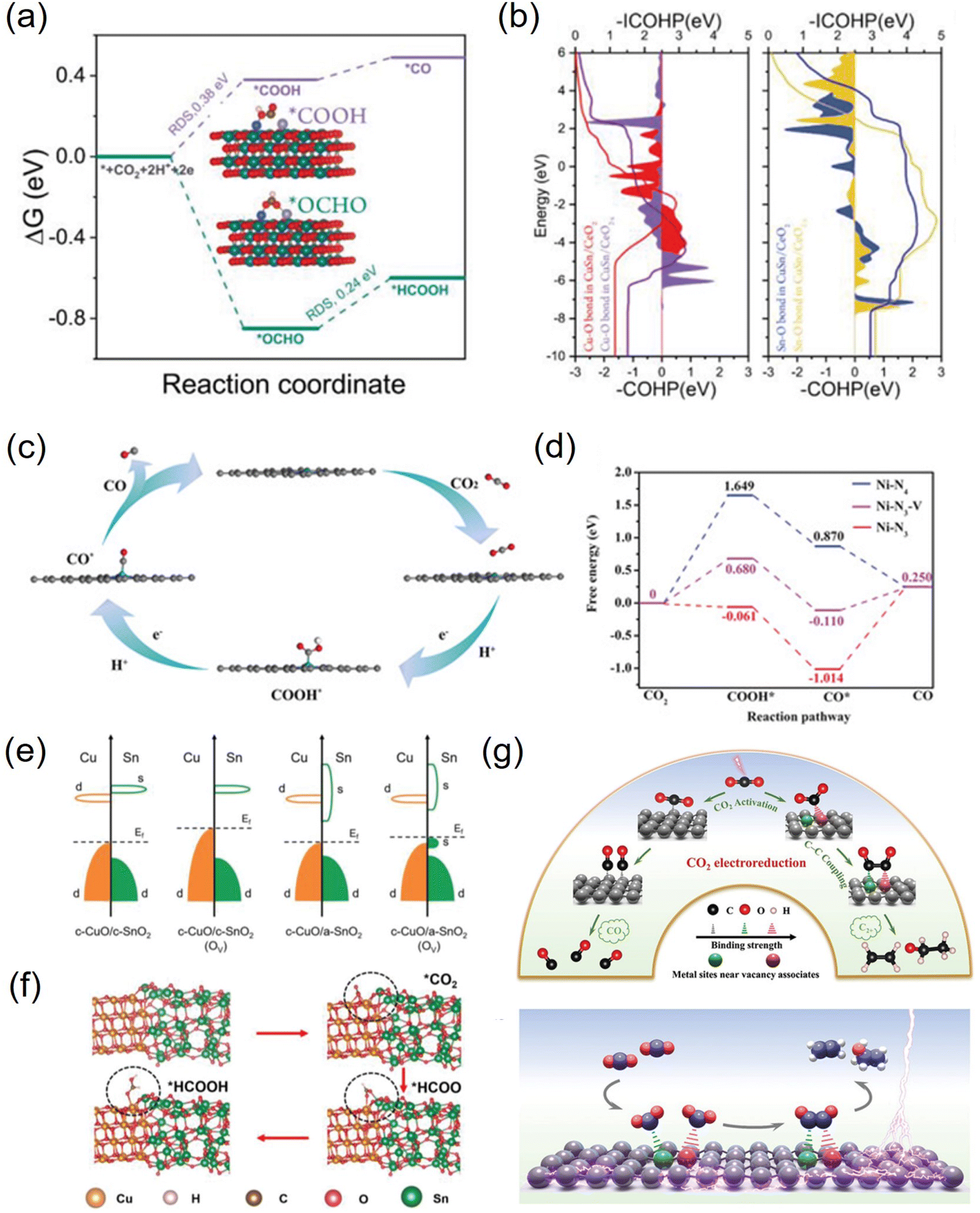 | ||
| Fig. 7 (a) Free energy profiles of the CO2RR on CuSn/CeO2−x. (b) The COHP and ICOHP of Cu–O and Sn–O bonds in CuSn/CeO2 and CuSn/CeO2−x. (c) Band structures before and after the formation of Ov for c-CuO/c-SnO2 and c-CuO/a-SnO2. (d) The typical adsorption process of formate on c-CuO/a-SnO2. (e) The proposed reaction paths of Ni-N3-V for CO2 electroreduction to CO. (f) Calculated free-energy diagram for the conversion of CO2 to CO. (g) Facilitating CO2 electroreduction to C2+ production through reconstructed Cu vacancy associates. (a and b) Reproduced with permission.96 Copyright 2023, Wiley-VCH Verlag. (c and d) Reproduced with permission.97 Copyright 2023, Wiley-Blackwell. (e and f) Reproduced with permission.99 Copyright 2019, John Wiley and Sons Ltd. (g) Reproduced with permission.100 Copyright 2024, Wiley-Blackwell. | ||
The cation vacancies of various metal atoms that act as shallow transport acceptors outperform in catalytic processes, which can effectively tune the band structure, thus giving them unexpected properties without creating new intermediate species.98 For instance, Lu et al. synthesized Ni-N3-V single-atom catalysts with vacancy defects at the Ni site. DFT calculations showed that the presence of vacancy defects in Ni-N3-V SACs could significantly improve their electrocatalytic activity for CO2 reduction (Fig. 7e and f).99 Besides, Xie et al. synthesized dynamically reconstructed tri-copper vacancy aggregates confined in copper nanowires. Theoretical calculations showed that triple copper vacancies lead to charge transfer from the surroundings to locations near the vacancies, increasing their local charge density. The charge density difference between the two *CO on the triple-copper vacancy copper sheet and the pristine copper sheet shows that the different charge distributions of different copper atoms near the defect clusters lead to different charge distributions of the bonded neighboring *CO, which alleviate the electrostatic repulsion and promote the C–C coupling (Fig. 7g). As a result, the triple copper vacancy binding enriches and redistributes the local negative charge, enhances the adsorption of *CO intermediates, and lowers the energy barrier for CO–CO coupling, thereby increasing the C2+ yield.100 Furthermore, Hu et al. designed a Se vacancy-containing bimetallic electrocatalyst denoted as CuInSe2. The Se vacancies facilitate electron delocalization, promote electron redistribution, slightly adjust the energy band structure, and optimize the CO2RR pathway of the bimetallic selenium compound. As a result, the Se-deficient CuInSe2 possessed a highly selective CO generation ability in aqueous solution, and the V-CuInSe2 cathode exhibited satisfactory performance in zinc–CO2 batteries in water. The CO Faraday efficiency was as high as 91%. In addition, water-based zinc–CO2 cells based on V-CuInSe2 cathodes reached a discharge voltage of ≈0.55 V and had a good lifetime.62 Although many achievements have been made at the theoretical level, the studies on cation vacancy defects are still limited and need to be further enriched.101
O bond is stretched to activate the CO2 molecule, and the energy barrier of the intermediate species is stabilized to a low value.103 For example, Gao et al. prepared an atomic-layered CO3O4 material and deliberately created oxygen vacancies on the surface to conduct targeted investigation on its role, revealing that the VO-rich material surpasses the contrast sample without VO in all the catalytic properties (Fig. 8a–e).104 The current density of the former is double that of the latter; the FEHCOOH is drastically increased from 67.3% to 87.6%, and it can sustain the high FEHCOOH for up to 40 hours (Fig. 8c and d). Revealed by the CO2 adsorption isotherm, the remarkable upgradation can be attributed to the abundant CO2 adsorption sites additionally provided by the VO (Fig. 8e). Combined with DFT calculations, they found that VO has the effect of stabilizing HCOO− radicals, reducing the energy barrier to initiate the rate-limiting step (the electron transfer from HCO3−) from 0.51 eV to 0.4 eV (Fig. 8a and b). Zeng et al. introduced VO on the surface of ZnO nanosheets via H2 plasma etching, and the current density (16.1 mA cm−2) and FECO (83%) of the obtained VO-ZnO nanosheets were 5 and 1.9 times higher than that of untreated ZnO, respectively, which could operate enduringly without deactivation.105 Combined with the calculation results, they concluded that VO increases the number of charges near the valence band of ZnO, improves the capture capacity of CO2, and switches the rate-determining step to the desired surface reaction (Fig. 8f–j).
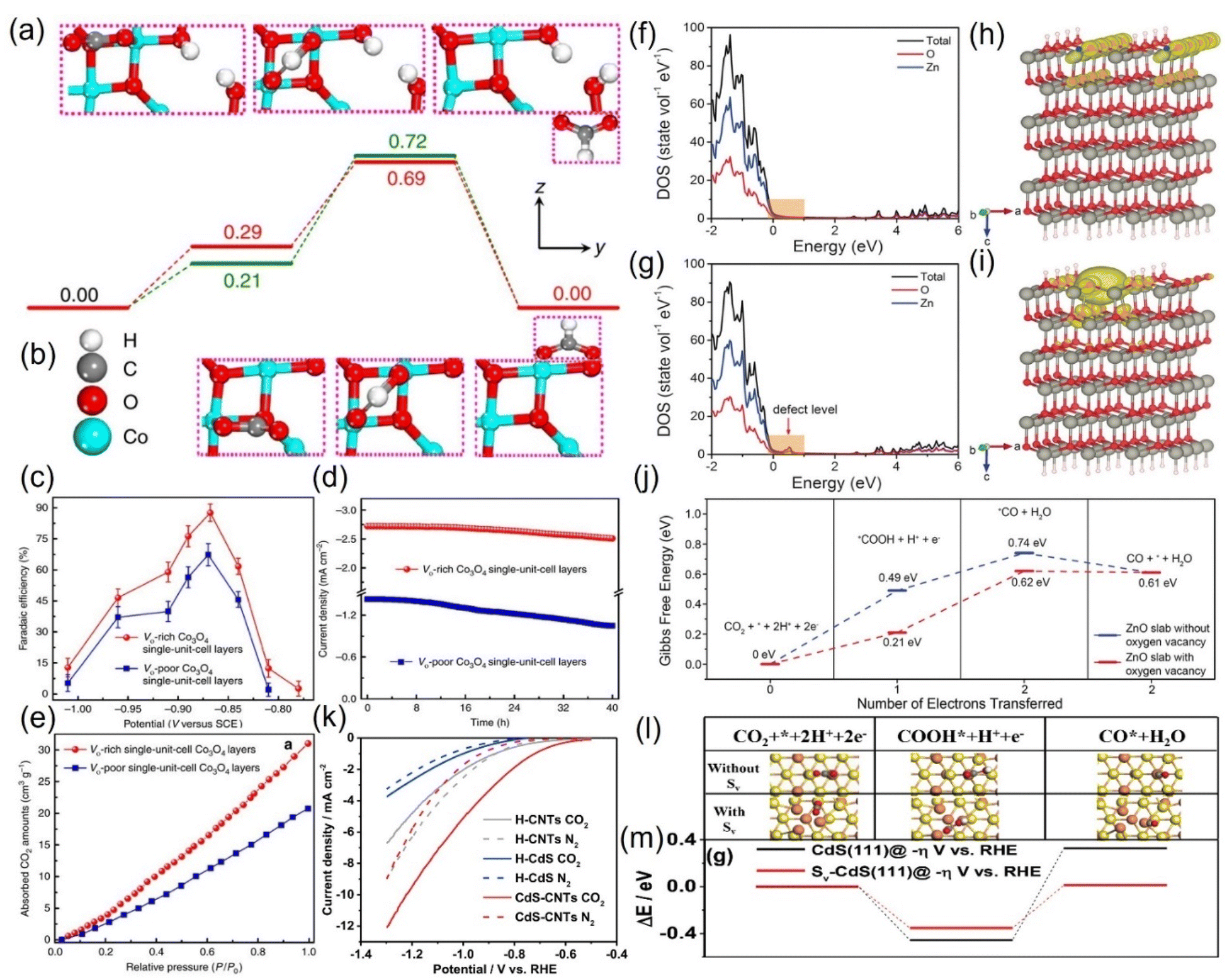 | ||
| Fig. 8 Gibbs free energy diagrams of (a) VO-rich and (b) VO-poor single-unit-cell Co3O4 layers. (c) FEHCOOH at different applied potentials. (d) Chronoamperometry tests and (e) CO2 adsorption isotherms of VO-rich/poor single-unit-cell Co3O4 layers. (f and g) DOS. (h and i) Charge density distribution and (j) Gibbs free energy diagrams of (f and h) ZnO slab and (g and i) ZnO slab with oxygen vacancy. (k) LSV curves. (l) Models of different intermediates and (m) DFT calculation results of corresponding intermediates during CO2RR on CdS without/with S-vacancy (Sv). (a–e) Reproduced with permission.104 Copyright 2017, Springer Nature. (f–j) Reproduced with permission.105 Copyright 2018, John Wiley and Sons Ltd. (k–m) Reproduced with permission.107 Copyright 2020, Royal Society of Chemistry. | ||
Besides oxygen vacancies, other anion vacancies have been discovered.106 For example, Li and colleagues synthesized CdS with ample S vacancies (VS) by pyrolyzing the mixtures of cadmium chloride and thiourea with serial ratios.107 At the optimal VS concentration, the current density of VS-CdS at 1 V vs. RHE can be as high as 20.5 mA cm−2, and the FECO can reach 95% with an error less than 0.5% (Fig. 8k). In contrast, the activity of the compared traditional CdS is much lower, indicating that the VS is the main contributor to the performance improvement. The VS was theoretically revealed to be more prone to adsorb the key intermediate species *COOH, essentially reducing the difficulty of CO generation (Fig. 8l and m).
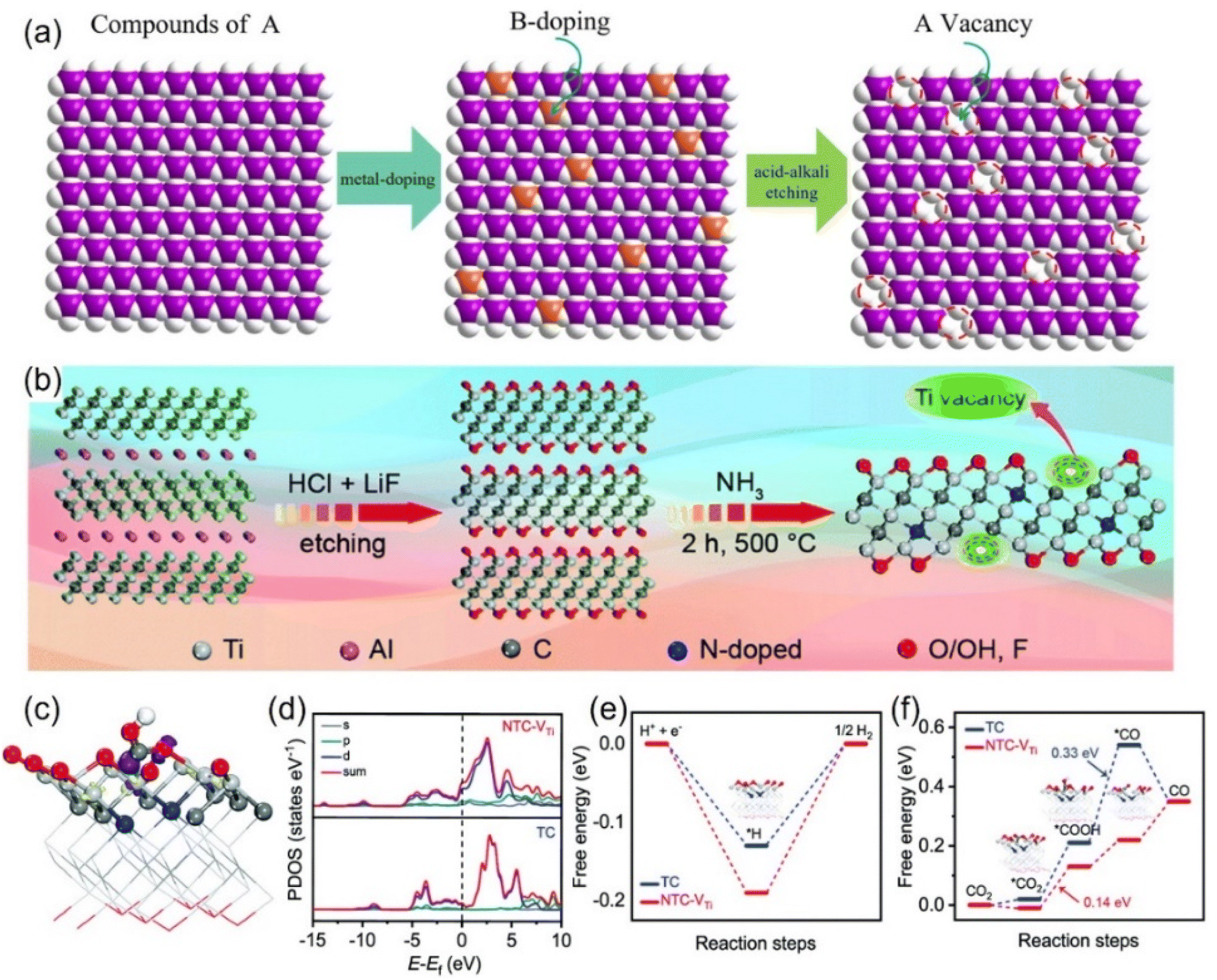 | ||
| Fig. 9 Schematic diagrams of preparing vacancies through (a) strong acid alkali for targeted-etching and (b) NH3-assisted etch-pyrolysis. (c) Electron density model (purple: electron accumulation; yellow: electron depletion). (d) Calculated DOS diagrams of *COOH adsorbed on NTC-VTi. Free energy diagrams of (e) the HER and (f) CO2RR. (a) Reproduced with permission.101 Copyright 2023, the Royal Society of Chemistry. (b–f) Reproduced with permission.109 Copyright 2020, the Royal Society of Chemistry. | ||
3.3. Strain effect
The Sabatier principle points out that the difficulty of combining the key reaction species and active sites has a direct and profound effect on the reaction rate, and a moderate binding energy barrier will significantly increase the yield and selectivity of the target product.116 In addition, through theoretical modeling, the location of the d-band center has a significant impact on the adsorption strength of species, i.e., the decrease in the d-band center position will weaken the adsorption between the active site and the intermediate species, and vice versa.117Strain engineering is a highly suitable means to regulate the central position of the d-band, and a variety of CO2RR active metals have intense strain response. Furthermore, the strain effect has the unique advantage of breaking through the linear scaling barrier, which will break the original equilibrium space queue between catalyst atoms and cause it to deviate. This will directly fluctuate the binding energy barrier of the key intermediate, which is conducive to the adsorption and activation of CO2 and the desorption of products and greatly reduces the overall reaction difficulty.118 These advantages make strain engineering an urgent optimization strategy to be developed. For example, changing the thickness of the Cu sheet on the upper layer of Pt resulted in significant differences in the cell parameters of Cu (3.61 Å) and Pt (3.92 Å), which directly led to the geometric tensile strain of the surface lattice, thus obtaining a more reactive surface with dramatically enhanced the adsorption intensity of the *CO species, even profoundly altering the optimal reactive pathway.119 Similarly, Wang et al. prepared Ag@Cu NPs with Ag as the core coated with Cu shells with a series of thicknesses by controlling the heating time in the synthesis method of polyol reduction. Ag@Cu-20 with a moderate thickness of Cu layer obtained by heating for 20 min shows the highest selectivity to ethylene, which is much better than that of the corresponding SACs (Fig. 10a).120 This can be attributed to the surface lattice tensile strain effect, which optimizes the surface activity of the material to hydrocarbons and the adsorption strength of *CO. Unfortunately, as the shell thickens, the tensile strain decreases due to its short-range properties, resulting in a decrease in the Faraday efficiency of C2H4 after the peak. In another similar core–shell system of Cu coated with In2O3, both the current density and the Faraday efficiency of the by-product H2 and the target product CO can be adjusted by tuning the shell thickness (Fig. 10b).121 It has been reported that the synergistic effect of compressive strain and Cu doping effect promotes the formation of *COOH rather than the HCOO* intermediate, thus boosting the precipitation of CO (Fig. 10c–e). Interestingly, Sun et al. reported another Cu-based material wrapped by SnO2. When the thickness of SnO2 is 1.8 nm, the material is more inclined to exhibit the activity of Sn with the higher selectivity of HCOOH, while when it becomes thinner than 0.8 nm, the FECO overwhelmingly reaches 93% (Fig. 10f and g).122 Combined with various characterization and theoretical calculation results, this unique phenomenon is attributed to the lattice mismatch between SnO2 and Cu, resulting in a large compressive strain of up to 10% in the shell (Fig. 10h–k).
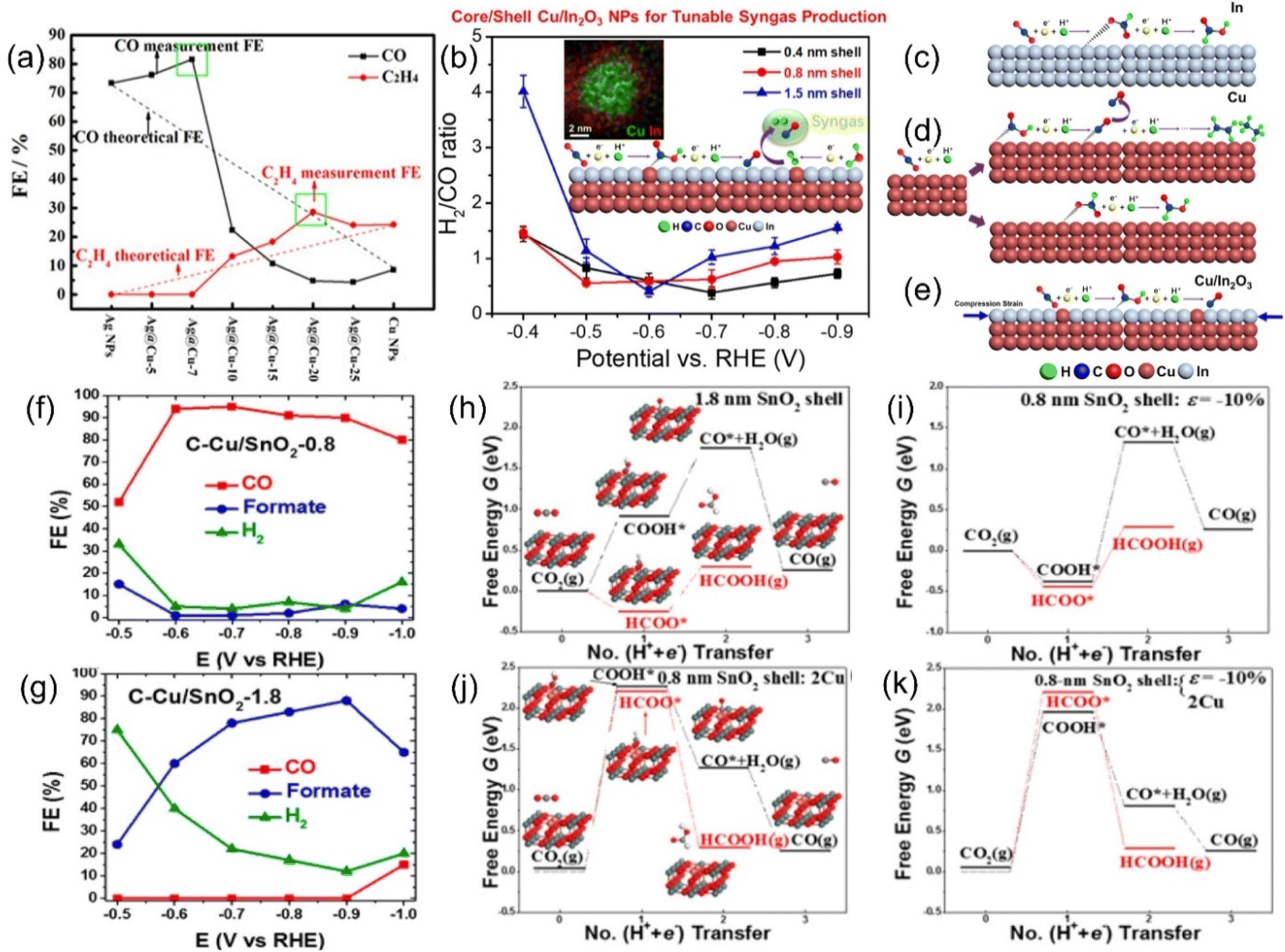 | ||
| Fig. 10 Theoretical linear “dilution effects” and test values of FEs of CO (black) and C2H4 (red). (b) H2/CO ratios generated by C–Cu/In2O3 catalysts. Schematic diagrams of the possible ECO2RR pathways on (c) In foil, (d) Cu NPs and (e) Cu/In2O3 NPs. FEs of various products for the ECO2RR of (f) C–Cu/SnO2-0.8 and (g) C–Cu/SnO2-1.8. Free energy diagrams of two ECO2RR pathways on SnO2 shells of different thickness with 10% uniaxial compression of (h) 1.8 nm and (i) 0.8 nm; and 0.8 nm SnO2 shells with (j) both surface Cu atoms and (k) 10% uniaxial compression. (a) Reproduced with permission.120 Copyright 2017, American Chemical Society. (b–e) Reproduced with permission.121 Copyright 2018, American Chemical Society. (f–k) Reproduced with permission.122 Copyright 2017, American Chemical Society. | ||
Furthermore, the electrocatalytic reduction of carbon dioxide to multi-carbon products involves complex multiple proton and C–C coupled electron transfers, which are not only determined by the intrinsic reactivity of the catalyst but are also related to the spin state of the electrons. Yao et al. observed the electrocatalytic activity of spin-enhanced carbon dioxide reduction over oxide-derived copper (OD-Cu) catalysts (Fig. 11a). Due to the magnetic nature of the Cu site, the Faraday efficiency of CO2RR could be increased by 15% using the OD-Cu catalyst. In addition, the stray field of copper polarizes the electron spins transferred from the neighboring copper, facilitating the conversion of the C–C coupling reaction on the Cu–Cu site to C2 products (e.g., ethane), thereby reducing the generation overpotential of the C2 products.123 Metal-nonmetal hybridized diatomic catalysts were designed and synthesized by An et al. Combining spin-polarized DFT and hydrogen electrode computational models, the hybridized M–B diatomic centers are superior to single and double M centers in driving CO2RR, especially C–C coupling. The electronic spin state of the metal may be particularly important in regulating the selectivity of CO2RR and the C–C coupling potential. Under ambient conditions, the high spin moments of the M–B diatomic site M favor the promotion of C–C coupling and protonation in CO2RR (Fig. 11b and c). The enhanced spin state of the metal led the authors to predict Fe@B-C2N as an active catalyst for the conversion of CO2RR to C2+.124 Chen et al. investigated the effect of oxidation states of Fe ions with different spins in FeN4C on the electrochemical mechanism of CO2 conversion to CO/HCOOH. Potential energy analysis revealed that among the two oxidation states with different spins, the rate-limiting step of CO/HCOOH generation catalyzed by intermediate-spin FeIIN4CM has lower potential barriers (0.72/0.52 eV). In addition, the overpotentials (−0.03/0.29 V) on FeIIN4CM for *CO/*HCOOH generation are also significantly lower than that of other cases. Both confirm that Fe2+ ions with intermediate spins in FeN4C are favorable for the conversion of CO2 to CO/HCOOH.125
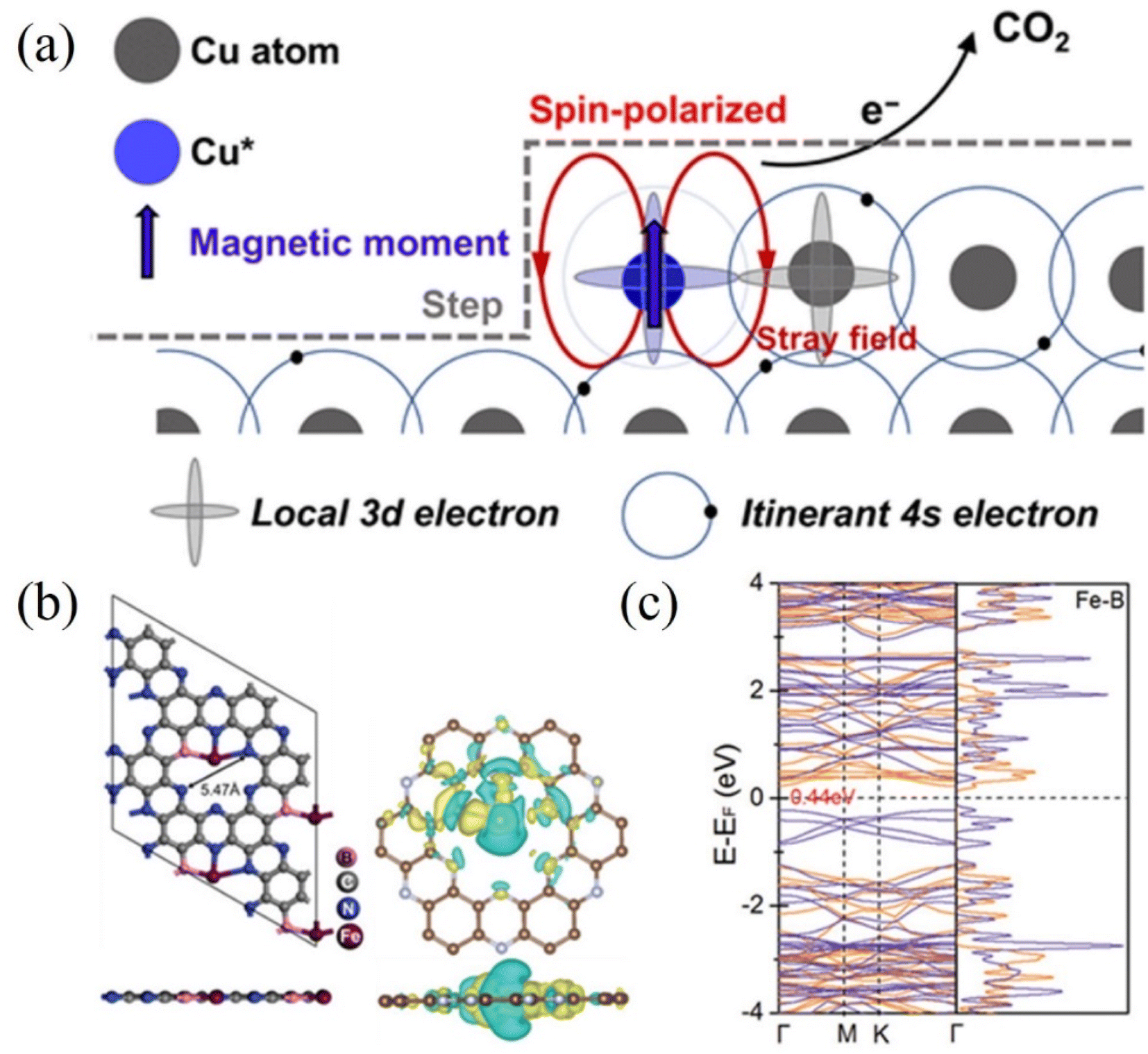 | ||
| Fig. 11 (a) Spin-related catalysis for the CO2RR in the OD-Cu catalyst. (b) Optimized structure of Fe@B-C2N along with the iso-surface of charge density difference. Yellow: charge accumulation; cyan: charge depletion. (c) DFT-calculated electronic band structure and density of states of Fe@B-C2N near the Fermi level using the HSE06 functional. (a) Reproduced with permission.123 Copyright 2023, Chinese Chemical Society. (b and c) Reproduced with permission.124 Copyright 2021, Wiley-VCH Verlag. | ||
3.4. Coordination environment
The coordination environment composed of metal active sites as the core and adjacent bonding atoms is considered to be the universal knob for regulating the electrocatalytic performance. The spatial site of these atoms will create a variety of three-dimensional structures that have a fatal impact on the interaction strength between the CO2 molecules/intermediate species and the reaction site. Moreover, the lattice distortion and charge transfer between the atoms in the coordination environment directly affect the geometry and electronic structure of the active site, thus endowing the material with a wide range of catalytic properties and even fundamentally changes the reaction pathway of electrocatalytic CO2RR.126 Therefore, the in-depth analysis of the coordination environment to clarify the key factors affecting its configuration and the essential way in which it impacts the catalytic process is of great significance, which is helpful to promote the precise regulation of the coordination environment and open up the way for the rational design of a new generation of high-performance industrial grade catalysts.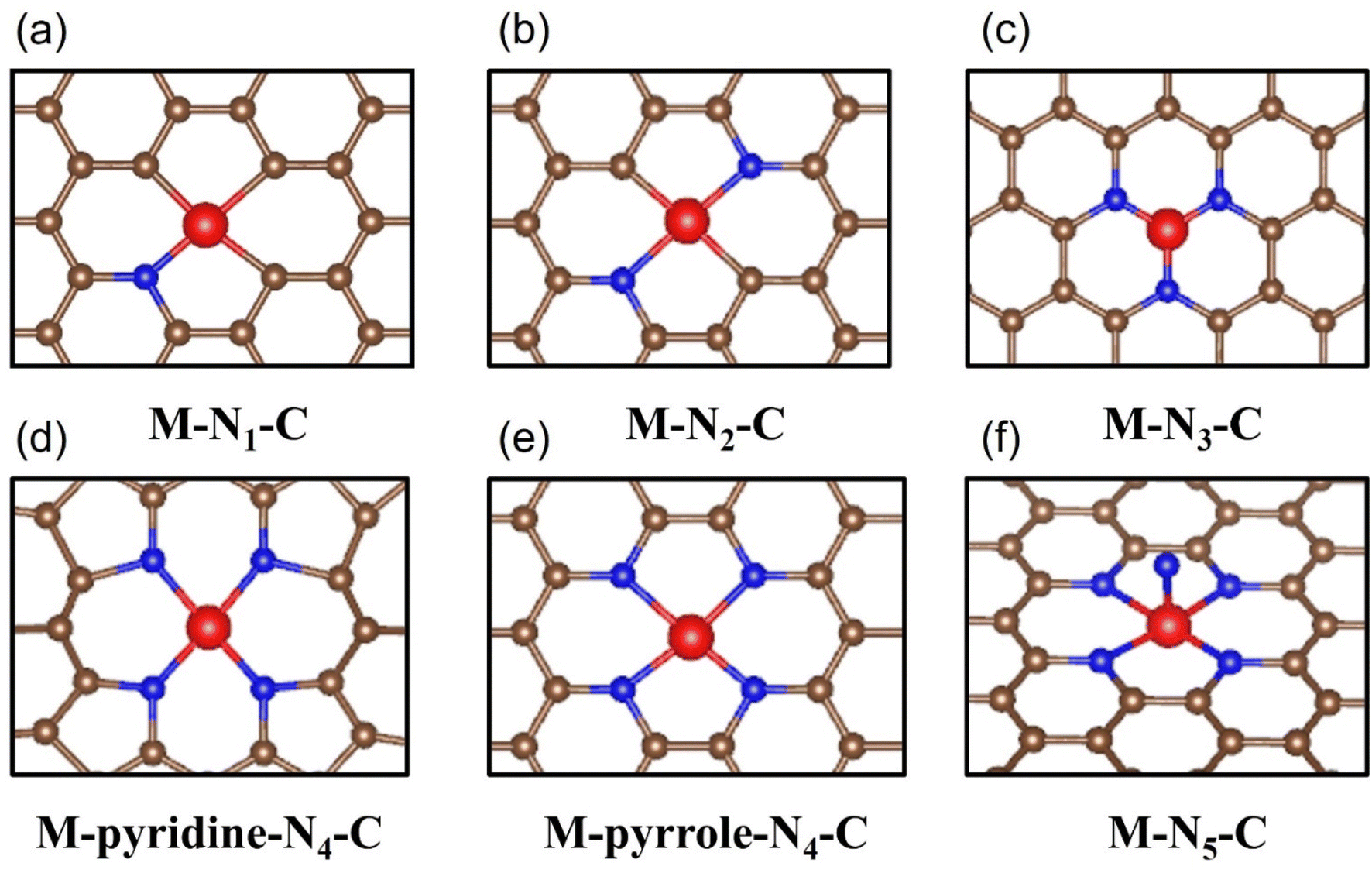 | ||
| Fig. 12 Schematic diagrams of all reported M-N-C configuration types including (a) M-N1-C, (b) M-N2-C, (c) M-N3-C, (d) M-pyridine-N4-C, (e) M-pyrrole-N4-C and (f) M-N5-C. | ||
At the theoretical level, DFT calculations predicted that the Ni-N-C3 configuration has the best electroactivity of CO2RR.128 Compared with the Ni-C4 structure, the introduction of an N atom increases the overall charge capacity, enhancing the reduction ability of CO2 molecules, optimizing the binding energy of CO*, and thus reducing the desorption difficulty of CO. However, Ni-N4 has a lower work function in the charge-neutral state, which makes it more difficult to replenish its charge to the same Fermi level. In another calculation model, when selecting different rate-determining steps, the structure with the highest catalytic potential for CO2RR was determined to be highly coordinated M-N4/N5.129 For example, Luo et al. selected grand canonical potential kinetics (GCP-K) as the basic algorithm for theoretical prediction to obtain the result of Ni-N4 exhibiting the best catalytic performance (Fig. 13a).130 This conclusion has also been verified from the experimental results that under −1.05 V vs. RHE, such a structure can convert CO2 to CO with nearly 100% selectivity, and jCO is as high as 40 mA cm−2, which is superior to low coordination configuration with 2N or 3N. The coordination environment of other metals also follows a similar rule. For instance, Li et al. reported that the FECO and jCO in the Co-N4 configuration in the wider potential range from −0.5 V to −1.0 V vs. RHE always occupy one place in the group of Co-N4−x configurations.131 As shown in Fig. 13d and e, the same M-N4 configuration while using diverse N-containing ligands will result in differences in the final coordination motifs with vastly different intrinsic activity. For example, when pyrrolic N is used as the ligand, Fe3+ is bonded ultra-firmly to achieve the rapid adsorption of CO2, while in the case of pyridinic N, the catalytic performance is exactly the opposite and the electron cloud of Fe3+ is regulated more actively, causing it to be more inclined to absorb electrons and be reduced to Fe2+ (Fig. 13b–f).132
 | ||
| Fig. 13 (a) Schematic representation of calculated free energies with GCP-K as the basic algorithm. Partial charge density of (b) the O-C-Fe-N plane adsorbing CO within the energy range from −1.56 to −0.82 eV for FeN4 model and (c) the O-C-Fe-N-pyrrolic N plane adsorbing CO within the energy range from −3.31 to −0.99 eV for the FeN5 model. (d and e) Calculated DOS diagrams of the (d) FeN4 model and (e) FeN5 model adsorbing CO and (f) free energy diagram for the ECO2RR to CO. Schematic diagrams of (g) N-heteroatom co-doped M-N2X2-C configuration and (h) axially-coordinated M-N4X1-C configuration. (a) Reproduced with permission.130 Copyright 2020, Springer Nature. (b–f) Reproduced with permission.132 Copyright 2019, John Wiley and Sons Ltd. | ||
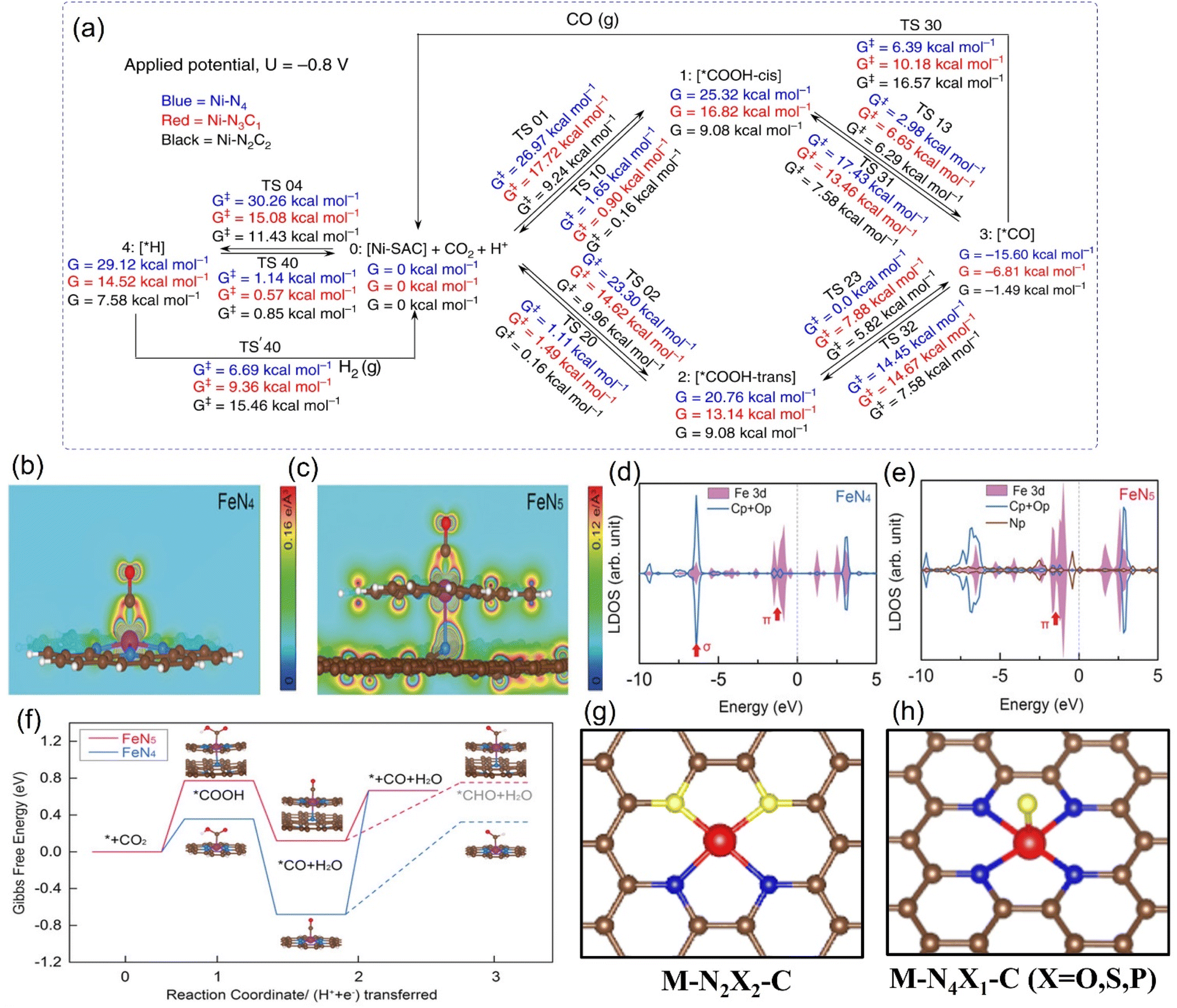
The experimental evaluation reveals that M-Nx has better electrocatalytic CO2RR ability than M-N4 because the former has a higher Gibbs free energy for side reaction, leading to HER with less probability, while the formation energy barrier of the active *COOH species is lower.133 For example, in a series of N-coordinated configurations of Co metal atoms (Co-N2-C, Co-N3-C, and Co-N4-C), Co-N2-C has prominent CO2RR activity, with a TOF value of up to 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 h−1.134 Similarly, the catalytic performance of Cu-N2 with FECO is up to 81%, better than that of Cu-N4 configuration when applying a small potential of −0.5 V vs. RHE (Fig. 14a).135 As explained by the difference in the geometric structure of different coordination modes, the chemical bond coordinated by double N atoms is not as long as that of four N atoms, which makes the electron transfer of metal sites and adsorbed intermediates faster, thus accelerating the activation of key *CO2 species (Fig. 14b). In addition, some metals show the advantage of catalyzing CO2RR in the three-N coordination environment. For example, the rate-limiting step of Ni-N3, the process of activating CO2 to a *COOH intermediate, has an easier barrier to cross than that of Ni-N4.136 Combined with various in situ characterizations, the electrons in the metal center of the Mn-N3-C3N4 coordination structure constructed on three-dimensional carbon nanotubes will rearrange after adsorbing CO2 molecules, resulting in an increase in the oxidation state.137 After a round of CO2RR cycle, the oxidation state will be reduced back to the initial state, which verifies the efficient CO2RR on this configuration. The FECO and the corresponding current density can reach 98.8% and 14 mA cm−2 at a low bias of 0.44 V vs. RHE, respectively. The local density of states analysis demonstrates that the d-band center of Mn-N3-C3N4 is closer to the Fermi level than that of the Mn-N4 configuration supported on two-dimensional graphene, which greatly reduces the difficulty of CO2 capture and activation (Fig. 14c and d). Interestingly, on the basis of the planar configuration of M-N4, the structure of M-N5 formed by bonding an additional N atom in the axial direction has been developed (Fig. 14f).132 For example, the oxidation state of the typical Fe-N5 group obtained by the synchrotron X-ray absorption spectroscopy (XAS) is significantly higher than that of the common Fe-N4, thus effectively promoting the process of catalyzing CO2RR.132 This conclusion has been reasonably explained via theoretical calculations, i.e., the d-electron transfer between the Fe site and the additional N atom is more active, which weakens the bonding energy of the former and the *CO species and makes it easier for CO to detach from the output (Fig. 14e and f). It is concluded that even small differences in the coordination environment can have an unpredictable and crucial effect on the electrocatalytic activity of CO2RR.
200 h−1.134 Similarly, the catalytic performance of Cu-N2 with FECO is up to 81%, better than that of Cu-N4 configuration when applying a small potential of −0.5 V vs. RHE (Fig. 14a).135 As explained by the difference in the geometric structure of different coordination modes, the chemical bond coordinated by double N atoms is not as long as that of four N atoms, which makes the electron transfer of metal sites and adsorbed intermediates faster, thus accelerating the activation of key *CO2 species (Fig. 14b). In addition, some metals show the advantage of catalyzing CO2RR in the three-N coordination environment. For example, the rate-limiting step of Ni-N3, the process of activating CO2 to a *COOH intermediate, has an easier barrier to cross than that of Ni-N4.136 Combined with various in situ characterizations, the electrons in the metal center of the Mn-N3-C3N4 coordination structure constructed on three-dimensional carbon nanotubes will rearrange after adsorbing CO2 molecules, resulting in an increase in the oxidation state.137 After a round of CO2RR cycle, the oxidation state will be reduced back to the initial state, which verifies the efficient CO2RR on this configuration. The FECO and the corresponding current density can reach 98.8% and 14 mA cm−2 at a low bias of 0.44 V vs. RHE, respectively. The local density of states analysis demonstrates that the d-band center of Mn-N3-C3N4 is closer to the Fermi level than that of the Mn-N4 configuration supported on two-dimensional graphene, which greatly reduces the difficulty of CO2 capture and activation (Fig. 14c and d). Interestingly, on the basis of the planar configuration of M-N4, the structure of M-N5 formed by bonding an additional N atom in the axial direction has been developed (Fig. 14f).132 For example, the oxidation state of the typical Fe-N5 group obtained by the synchrotron X-ray absorption spectroscopy (XAS) is significantly higher than that of the common Fe-N4, thus effectively promoting the process of catalyzing CO2RR.132 This conclusion has been reasonably explained via theoretical calculations, i.e., the d-electron transfer between the Fe site and the additional N atom is more active, which weakens the bonding energy of the former and the *CO species and makes it easier for CO to detach from the output (Fig. 14e and f). It is concluded that even small differences in the coordination environment can have an unpredictable and crucial effect on the electrocatalytic activity of CO2RR.
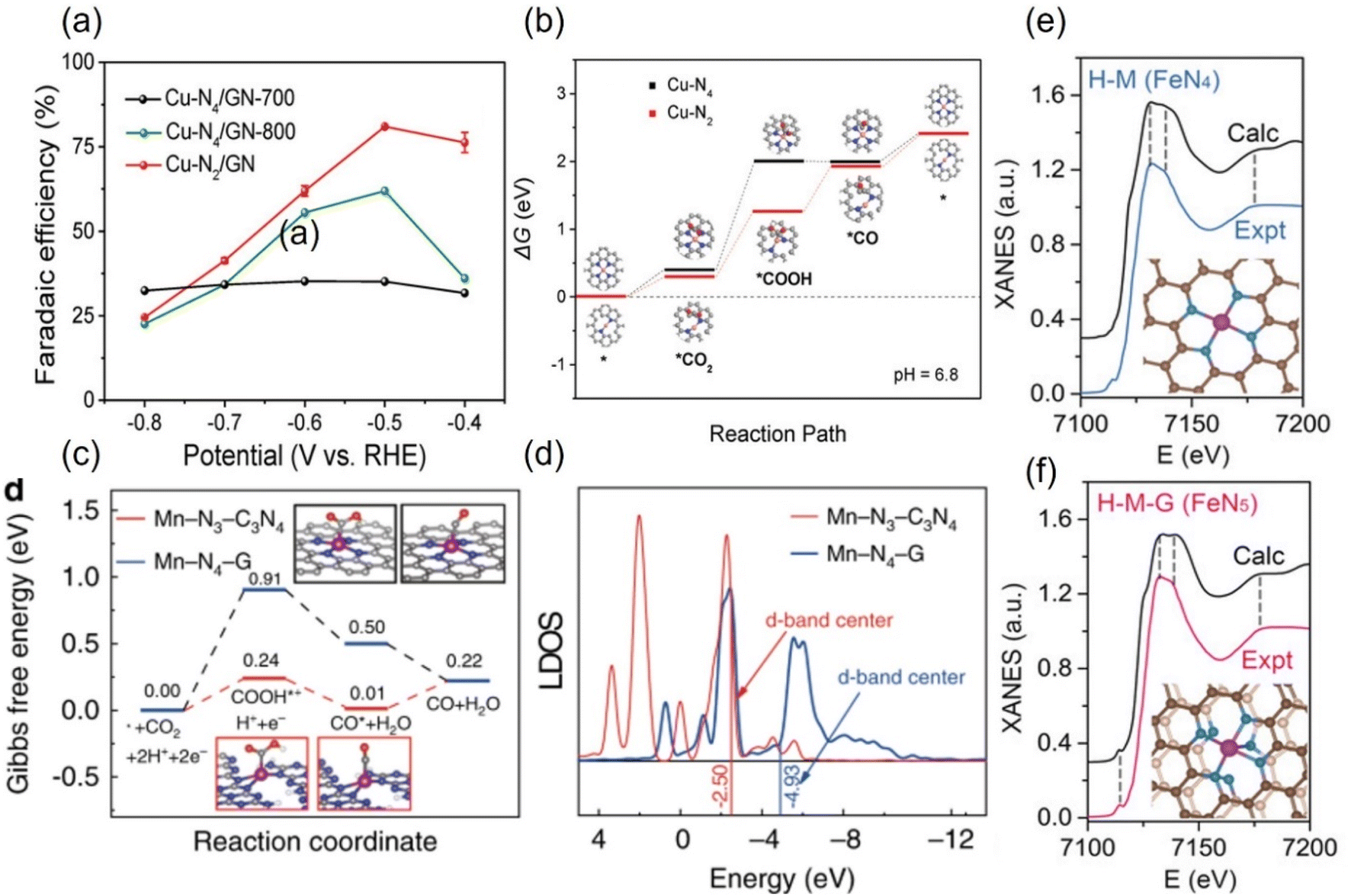 | ||
| Fig. 14 (a) FECO diagrams and (b) calculated free energy diagrams for the ECO2RR of Cu in Cu-N4 and Cu-N2. (c) Calculated free energy diagrams for the ECO2RR and (d) calculated DOS diagrams of Mn in Mn-N3-C3N4 and Mn-N4-G. Comparison of the experimental with the calculated XANES data of (e) FeN4 and (f) FeN5. (a, and b) Reproduced with permission.135 Copyright 2019, Wiley-VCH Verlag. (c and d) Reproduced with permission.137 Copyright 2020, Springer Nature. (e and f) Reproduced with permission.132 Copyright 2019, John Wiley and Sons Ltd. | ||
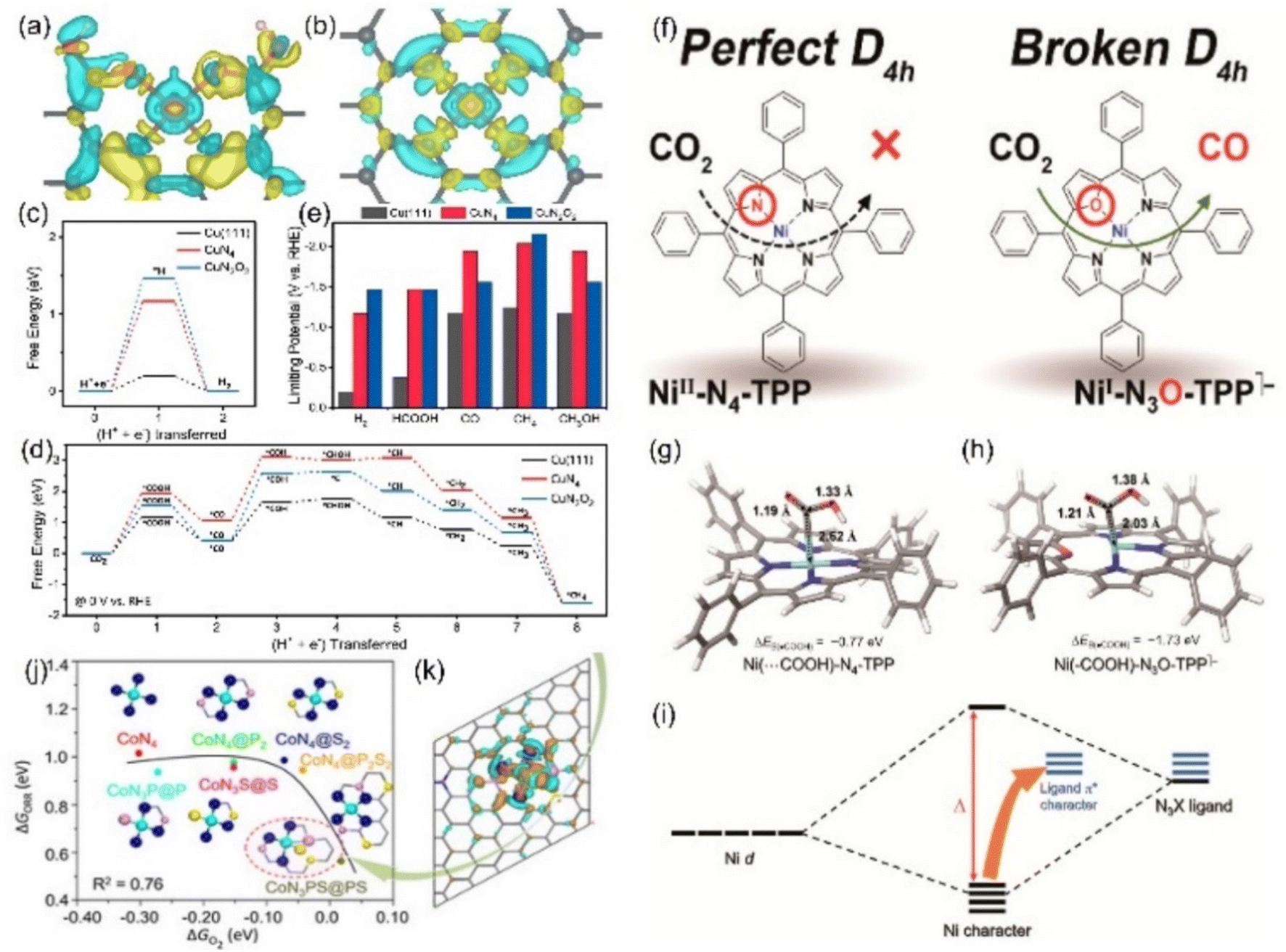 | ||
Fig. 15 Charge densities of (a) CuN2O2 and (b) CuN4 (yellow: increase in the number of electrons; blue: the depletion zone). Free energy diagram of (c) the HER and (d) ECO2RR to CH4 and (e) summary of the limiting potentials of various products. (f) Schematic diagrams of Ni-N4-TPP and Ni-N3O-TPP. DFT-optimized structures of adsorbing *COOH in (g) Ni-N4-TPP and (h) Ni-N3O-TPP. (i) Schematic diagram of the molecular orbital. (j) The linear relationship for the overpotential as a function of the  adsorption energy and (k) charge density difference of the Co site of CoN3PS. (a–e) Reproduced with permission.139 Copyright 2021, Springer Nature. (f–i) Reproduced with permission.140 Copyright 2021, American Chemical Society. (j and k) Reproduced with permission.142 Copyright 2020, John Wiley and Sons Ltd. adsorption energy and (k) charge density difference of the Co site of CoN3PS. (a–e) Reproduced with permission.139 Copyright 2021, Springer Nature. (f–i) Reproduced with permission.140 Copyright 2021, American Chemical Society. (j and k) Reproduced with permission.142 Copyright 2020, John Wiley and Sons Ltd. | ||
Other elements except N can also be axially doped to construct the same coordination configuration as M-N5, as discussed in the previous section (Fig. 11h). For example, Han's team reported the Cd-N4S1 structure that exhibits completely different CO2RR catalytic properties in comparison with the Cd-N5 configuration.143 Although both the axial S and N atom play a significant role in activity optimization, the S atom reduces the Gibbus free energy threshold of the rate-limiting step due to its extremely high spin density state with charge delocalization effect (Fig. 16a–c). In addition to the fact that the Cd atom is instinctively inclined to form this configuration due to its large radius, the beneficial effect of the fifth axial heteroatom also acts on other metals. For instance, Xin et al. chose organic diphenyl sulfide as the S source to axially incorporate the fifth S atom to enhance the electron transfer capacity (Fig. 16d and e). The TOF value of Co-N4S1 (2.4 s−1) at −0.87 V is 1.5 times that of Co-N4 (Fig. 16f).144 In addition, when O is used as the axial ligand of the Fe center, it exhibits excellent stability at low potentials. This can be attributed to the fact that the adsorption energy of *CO species on the Fe-N4O configuration is much lower than that of Fe-N4 or Fe-N5, which makes the precipitation process smoother (Fig. 16g and h).145 More and more related studies have shown that it is infinitely possible to design different types of coordination environments to develop new areas of electrocatalytic CO2RR. More in-depth research in this direction will be dominant in the future.
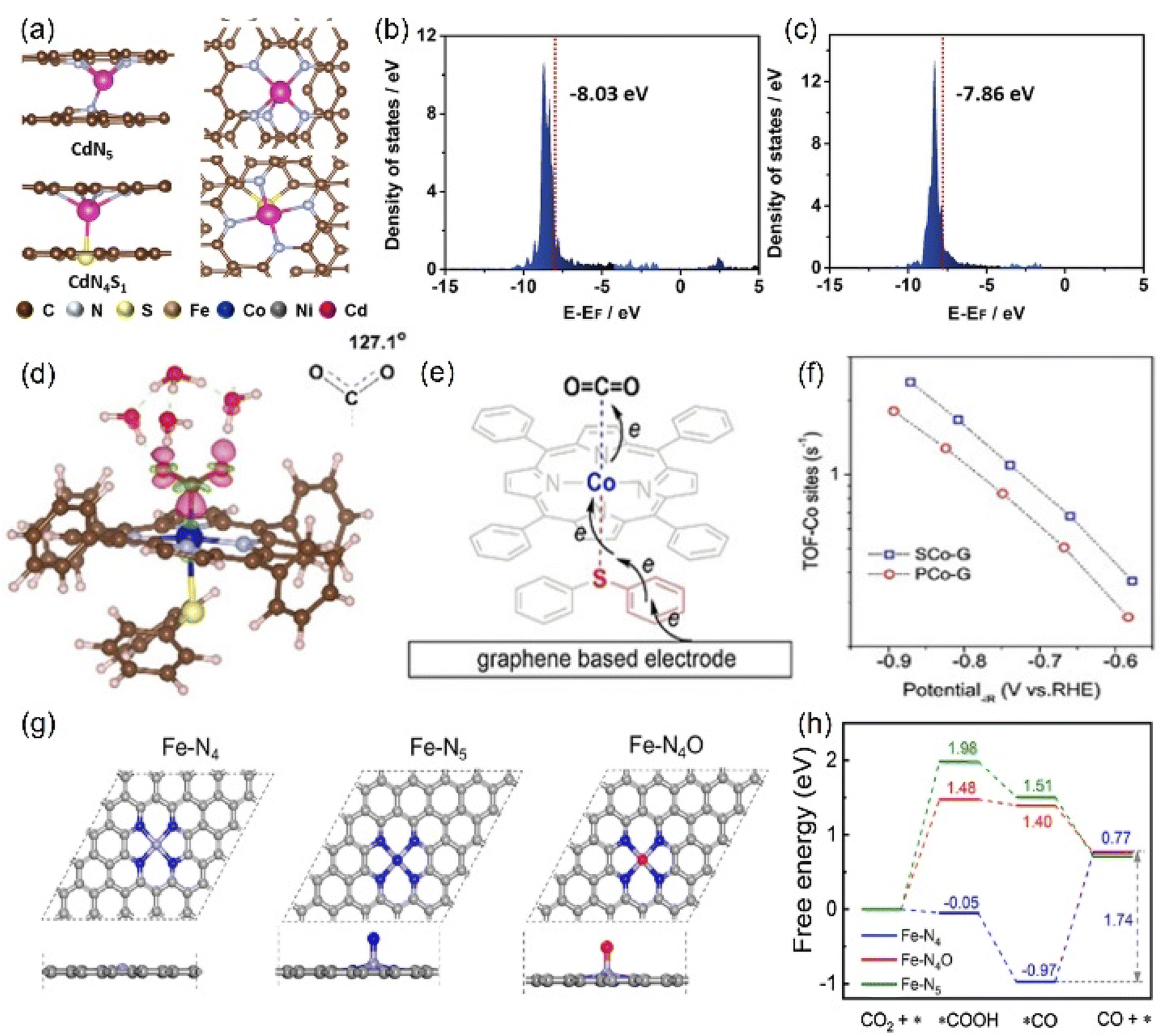 | ||
| Fig. 16 (a) The side and top structures and (b) the partial density of states of Cd site of CdN4S1 and CdN5 configurations (red line: the d-band center). (d) Charge density difference diagram exhibiting CO2 activation on the Co atom in Co-N4S1. (e) Schematic diagram of the possible electron transfer pathways of the CO2 activation from graphene, Co-N4S1 to CO2. (g) The side and top structures and (h) Gibbs free energy diagram of the ECO2RR to CO of Fe-N4, Fe-N5 and Fe-N4O configurations. (a–c) Reproduced with permission.143 Copyright 2021, John Wiley and Sons Ltd. (d–f) Reproduced with permission.144 Copyright 2020, John Wiley and Sons Ltd. (g and h) Reproduced with permission.145 Copyright 2020, Elsevier. | ||
4. Categories of catalysts for ECO2RR
Thanks to the indefatigable efforts of researchers, great achievements have been achieved in recent decades in improving the activity of ECO2RR catalysts. Herein, the most advanced and capable ECO2RR catalysts are representatively classified as atomically-dispersed catalysts, alloys, MOFs and COFs, providing a comprehensive overview of their inherent characteristics and achievements in electrocatalytic performance. Herein, we present more intuitive summaries and comparisons of the cutting-edge ECO2RR catalysts in Table 3 in combination with structure–activity optimization strategies to provide researchers with the most frontier research level and design inspiration.| Catalyst | Optimization strategy | FE (%) | Current density (mA cm−2) | Applied potential (V vs. RHE) | Electrolyte | Device | Ref. |
|---|---|---|---|---|---|---|---|
| B-CuO | Modulating the electronic structure of the active center | C2+ (62.1) | −139 | −0.62 | 1 M KOH | Flow cell | 146 |
| Cu(B) | C2H4 (52) | −70 | −1.1 | 0.1 M KOH | H-cell | 147 | |
| PDI@Cu(Cu-C) | CH4 (38.9) | −145 | −1.0 | 0.1 M KHCO3 | Flow cell | 148 | |
| Cu1.5/GDY | CH4 (57.3) | −312 | −1.7 | 1 M KOH | Flow cell | 149 | |
| Cu-N-C-900 | CH4 (38.6) | −38.3 | −1.6 | 0.1 M KHCO3 | H-cell | 150 | |
| PSB-CuN3 | HCOO− (97.9) | −127.7 | −1.1 | 0.5 M KHCO3 | Flow cell | 151 | |
| Cu-on-Cu3N | C2H4 (39) | −23 | −0.95 | 0.1 M KHCO3 | H-cell | 152 | |
| C3H7OH (6) | |||||||
| Cu3Nx | C2H4 (56) | −307 | −1.15 | 1 M KOH | Flow cell | 153 | |
| Cu3P | HCOO− (8) | −0.5 | −0.2 | 0.5 M KHCO3 | H-cell | 154 | |
| P0.075-Cu | C2H4 (30.7) | −17.57 | −1.6 | 0.1 M KHCO3 | H-cell | 155 | |
| P-doped Cu | C2H4 (40) | −210 | — | 1 M KOH | Flow cell | 156 | |
| Oxide-derived Cu | C2+ (60.1) | −11 | −1.0 | 0.1 M KHCO3 | H-cell | 157 | |
| CuO-derived Cu | C2+ (7) | −400 | — | 1 M KOH | Flow cell | 158 | |
| CuSx-DSV | C3H7OH (15.4) | −20 | −1.05 | 0.1 M KHCO3 | H-cell | 159 | |
| Cu2S-Cu-V | C2+ alcohol (32) | −400 | — | 1 M KOH | Flow cell | 160 | |
| SNC@Cu1.96S | CO (85.2) | −21 | −0.84 | 0.5 M KHCO3 | H-cell | 161 | |
| Smx-CuSy | HCOOH (92.1) | −300 | — | 1 M KOH | Flow cell | 162 | |
| Cu2-CuN3 | C2H5OH (51) | −28.2 | −1.1 | 0.1 M KHCO3 | H-cell | 163 | |
| Cu-N-C | C2H5OH (55) | −16.2 | −1.2 | 0.1 M CsHCO3 | Flow cell | 164 | |
| Cu2P2O7 | Constructing vacancy defects, including anionic and cationic vacancies, altering the lattice ordering and electronic structure to optimize electron transport pathways and energy barriers for critical species | C2H4 (41.8) | −200 | — | 0.1 M KOH | MEA-based | |
| VSe-Cu2−xSe | C2H5OH (68.1) | −10.96 | −0.8 | 0.5 M KHCO3 | H-cell | 165 | |
| Cu1.63Se(1/3) | CH3OH (77.6) | −41.5 | −2.1 | [Bmim]PF6 (30 wt.)/CH3CN/H2O (5 wt.) | H-cell | 166 | |
| BiCuSeO | HCOOH (93.4) | −267 | −1.1 | 1 M KOH | Flow cell | 167 | |
| Cu2Se | CH3CH2OH (84) | −50 | −0.6 | 0.3 M NaHCO3 | H-cell | 168 | |
| Cu2Te | CH4 (63) | −300 | — | 1 M KHCO3 | Flow cell | 169 | |
| CuOx-Vo | C2H4 (63) | −30 | −1.4 | 0.1 M KHCO3 | H-cell | 170 | |
| O2, 10 min-plasma Cu | C2+ (60) | — | — | 0.1 M CsHCO3 | H-cell | 171 | |
| e-CuOHFCl | C2H4 (36.3) | −18.5 | −1.0 | 0.1 M KHCO3 | H-cell | 172 | |
| Cu2OCl | C3–C4 (10) | −7 | −1.6 | 0.1 M KCl | H-cell | 173 | |
| Cu_KI | C2H4 (44.5) | −40 | −1.15 | 0.1 M KHCO3 | H-cell | 174 | |
| O2 plasma-activated Cu foil | C2H4 (60) | −2 | −0.9 | 0.1 M KHCO3 | H-cell | 175 | |
| Vo-rich ZnO nanosheets | CO (83) | −16.1 | −1.1 | 0.1 M KHCO3 | H-cell | 105 | |
| Nb4N5-NO/NC | CO (91) | −2.8 | −0.8 | 0.1 M KHCO3 | H-cell | 176 | |
| Ni-N3-V | CO (90) | −65 | −0.9 | 0.5 M KHCO3 | H-cell | 177 | |
| Bi2S3–Bi2O3@rGO | HCOOH (90.1) | — | −0.9 | 0.1 M KHCO3 | H-cell | 178 | |
| VO-SnOx/CF | HCOOH (86) | −30 | −1 | 0.1 M KHCO3 | H-cell | 179 | |
| H-InOx NRs | HCOOH (91.7) | — | −0.7 | 0.5 M KHCO3 | H-cell | 180 | |
| SnOx | HCOOH (88.6) | −15 | −0.98 | 0.5 M KHCO3 | H-cell | 181 | |
| CdS | CO (100 ± 0.5) | −20.5 | −1.1 | 0.5 M KHCO3 | H-cell | 182 | |
| Cu/CeO2−x HDs | CH4 (54) | — | −1.2 | 0.1 M KHCO3 | H-cell | 183 | |
| CdS-CNTs | CO (92) | −11.4 | −1.2 | 0.1 M KHCO3 | H-cell | 106 | |
| De-Au3Cu | CO (94.3) | — | −0.43 | 0.5 M KHCO3 | H-cell | 184 | |
| Cuv-Cu2O | C2H4 (51) | −15.7 | −0.76 | 0.1 M KHCO3 | H-cell | 185 | |
| Bi/Bi2O3-CP | HCOOH (90.4) | — | −0.87 | 0.5 M KHCO3 | H-cell | 186 | |
| Vo-rich SnO2 | CO (92.5) | −44.2 | −1.1 | 0.5 M KHCO3 | H-cell | 187 | |
| Iodide-mediated Cu | Strain engineering can break the queue of the original equilibrium between atoms to optimize the energy barrier of key reactive species | C2H4 and oxygenates (25) | — | — | 0.1 M KHCO3 | H-cell | 188 |
| Cu–CuI | C2+ (71) | −832.4 | −1.0 | 1 M KOH | Flow cell | 189 | |
| Cu/CuxOF | CH3COOH (27) | −15 | −0.3 | 1 M KOH | Flow cell | 190 | |
| Ag,S-Cu2O/Cu | CH3OH (67.4) | −122.7 | −1.18 | BMImBF4/H2O (molar ratio 1:3) |
H-cell | 191 | |
| Cu2O@Cu-MOF | CH4 (63.2) | −14 | −1.71 | 0.1 M KHCO3 | H-cell | 192 | |
| Cu(OH)2/CuO | C2+ (64.5) | −26.2 | −0.9 | 0.1 M KHCO3 | H-cell | 173 | |
| 3D dendritic Cu-Cu2O/Cu | C2 (80.7) | −11.5 | −0.4 | 0.1 M KCl | H-cell | 193 | |
| Ag-Cu2OPB | C2H5OH (34.15) | −4 | −1.2 | 0.2 M KCl | H-cell | 194 | |
| Tin-based oxides SnOx/Sn GDE | HCOO− (64) | −3 | −1.399 | 0.1 M KHCO3 | H-cell | 195 | |
| Pd/SnO2 NSs | CH3OH (55) | −1.45 | −0.24 | 0.1 M NaHCO3 | H-cell | 196 | |
| SnO2-0.8/C-Cu | CO (93) | −4.6 | −0.7 | 0.5 M KHCO3 | H-cell | 122 | |
| Cu/SnOx-CNT (6.2 SnOx) | CO (89) | −11.3 | −0.99 | 0.1 M KHCO3 | H-cell | 197 | |
| Cu/SnOx-CNT (30.2 SnOx) | HCOOH (77) | −4 | −0.99 | 0.1 M KHCO3 | H-cell | 197 | |
| Zn2SnO4/SnO2 | HCOOH (77) | −5.77 | −1.08 | 0.1 M KHCO3 | H-cell | 198 |
4.1. Atomically-dispersed catalysts
Atomically-dispersed metal catalysts (ADMCs) are known to have the highest atomic utilization among common catalytic systems. Compared with bulk metal structures, ADMCs exhibit new or optimized electrocatalytic properties due to their unique geometric, electronic properties, and uniform active sites, having been widely used in electrocatalytic CO2RR. However, owing to high cohesion energy and large surface tension, dispersed metal atoms tend to agglomerate and lose their catalytic activity.199 The strong fixation of the conductive carrier is essential for maximizing the activity and stability of ADMCs.Due to the non-delocalization characteristic of the 3s orbital, CO is less adsorbed on Mg than other transition metals (Fe, Co, etc.), which is beneficial for the high selectivity of CO.201 According to the simulation of CO generation path, the energy barrier of CO desorption on Mg-C3N4 was only 0.13 eV (Fig. 17a–c). The electron transfer number of Mg-C3N4 to CO is 0.1 e, which is much smaller than the corresponding Fe-C3N4 and Co-C3N4 (Fig. 17d–f). However, the FECO can reach 90% at large applied voltages. In addition, the SA Ca has almost no CO2RR activity due to the large activation barrier.
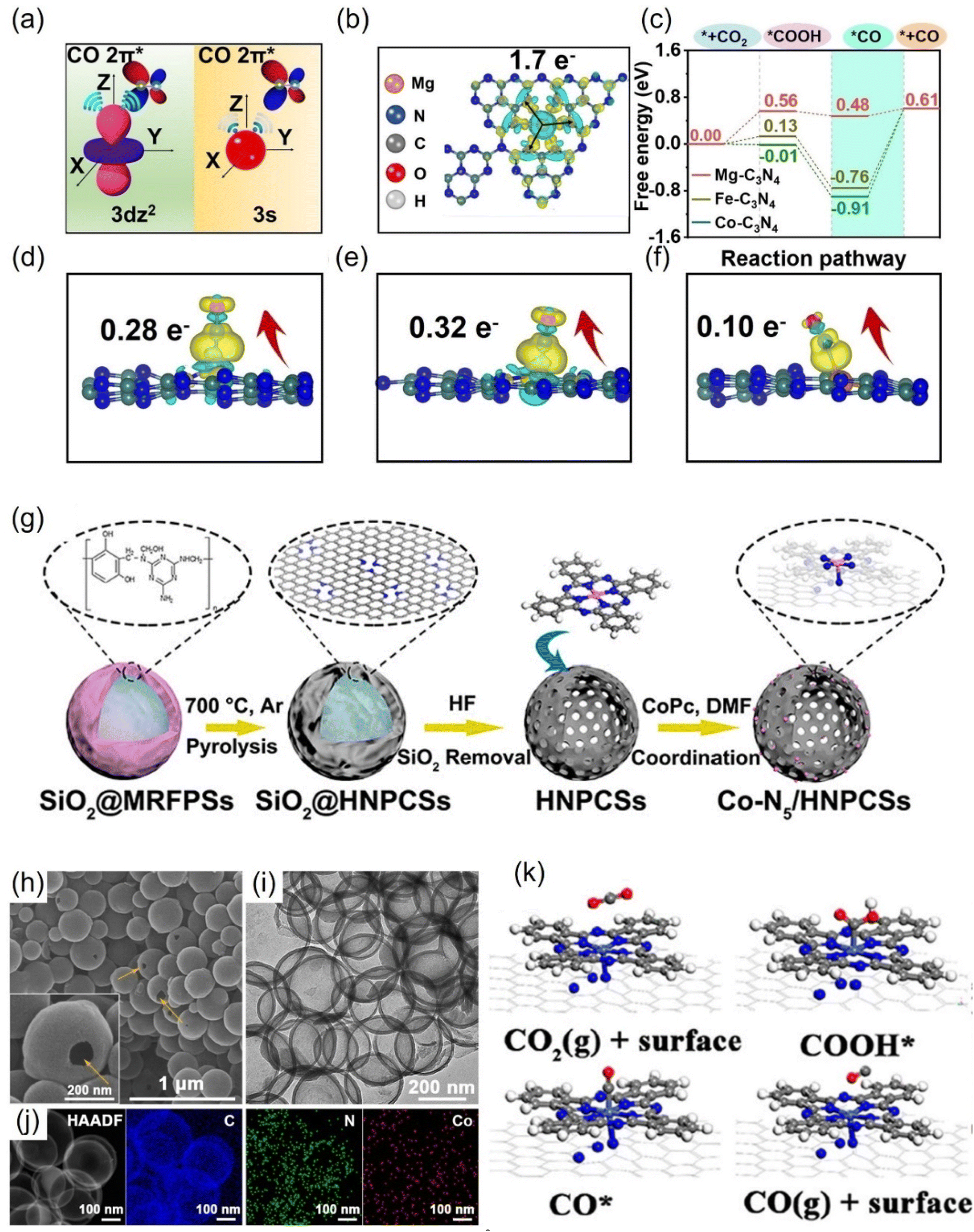 | ||
| Fig. 17 (a) Comparison of the schematic diagram for CO adsorbed on 3dz2 and 3s orbitals. (b) The charge density differences and Bader charge transfer for Mg-C3N4. (c) Free energy diagram and (d–f) charge density differences and Bader charge transfer for CO adsorbed on Co-C3N4, Fe-C3N4 and Mg-C3N4. (g) Schematic illustration, (h) SEM, (i) TEM, (j) HAADF-STEM and EDS images and (k) optimized structures for the intermediates. (a–f) Reproduced with permission.201 Copyright 2021, Wiley-VCH GmbH. (g–k) Reproduced with permission.131 Copyright 2018, American Chemical Society. | ||
Zhao et al. synthesized Ni SAs/N-C by the calcination of Ni-MOF, in which the uniformly dispersed Ni-N3C1 is the active center of electrocatalysis.202 When the voltage is −0.89 V, the FE of CO2 reduction to CO reaches a maximum of 71.9%. Further, a SAC named Ni-N3-C, where Ni is bonded to three N atoms and has a carbon defect site at an adjacent site, prepared by a two-step process of high-temperature annealing and ion exchange, showed enhanced CO selectivity.203 The obvious unsaturated coordination greatly accelerated the rate-determining step (RDS), the formation of COOH*, and the FE of CO reached 95.6%, higher than that of Ni-N4 without carbon defects.
The controllable uniform reaction site represents high reaction selectivity. The precise control of the types of coordination nitrogen atoms (pyridine nitrogen, pyrrole nitrogen, graphite nitrogen, etc.) can further improve the unity of the coordination structure of metal atoms. By self-assembly, Zheng et al. used melamine and cyanuric acid as raw materials to obtain metallic SAs coordinated with four pyrrole nitrogen.80 In the electrocatalytic CO2RR process, the FE (98.5%) of CO on pyrrole-type Ni-N4 was higher than the corresponding Fe, Cu, and Co. Pyridine-type Fe-N4 on C3N4 also displayed high CO selectivity at −0.6 V.204 Instead of pyridine nitrogen, high-valent Fe(III) is more easily stabilized by pyrrole nitrogen, exhibiting faster CO2 adsorption and weaker CO adsorption.205 During the electrocatalytic process, Fe could remain in its high valence and not be reduced due to the strong coupling effect. When the voltage was greater than −0.5 V, Fe lost a pyrrole N ligand and was reduced. The low-valence single Ni(I) atom, with 9 d electrons, exhibited high inherent CO2 reduction activity, with FECO of 97% and stability of up to 100 h at an overpotential of 0.61 V.206 During the CO2RR process, the transfer of electrons from the Ni(I) orbital to the 2p orbital of carbon formed CO2δ− species, causing the activation of CO2 and lowering the barrier for CO2 reduction. Using the template method, Li et al. synthesized a high-metal-content single-atom catalyst Co-N5/HNPCSs, in which Co was coordinated with five nitrogen and uniformly dispersed on the hollow carbon sphere (Fig. 17g–j).131 Over a wide voltage range, the catalyst produced CO with a selectivity of more than 90%, and the selectivity reached a maximum of 99.4% at −0.79 V. The improved CO selectivity was attributed to the strong adsorption of COOH* and the suitable adsorption of CO* at Co-N5 sites (Fig. 17k). As the most used MOF precursor, ZIF-8 can effectively isolate the agglomeration of single atoms during calcination.207 The Zn-N4 site reduced the reaction barrier of *COOH generation and converted CO2 to CO with an FE of 95%.208 Single Mn, Zn, Fe, or Cr atoms are all potential catalysts that can reduce CO2 to CO.
Due to the participation of multiple electrons in adsorption and desorption reactions, rare-earth-based materials will produce strong adsorption with CO2RR intermediates, showing a poor electrocatalytic activity. However, owing to the strong metal-carrier interaction, rare earth metals can be easily fixed on the carrier, facilitating the synthesis of single atoms, which gives them CO2RR activity. Due to large atomic radii and abundant electron orbitals, isolated Sc and Y tend to adsorb at large defect sites and form five- or six-coordinated configurations. Liu et al. synthesized Sc- and Y-SACs (Y1/NC and Sc1/NC), where each metal atom was simultaneously bonded to three nitrogen atoms and three carbon atoms.209 In the process of CO2RR, Y1/NC and Sc1/NC reached the highest FECO of 88.3% and 81.3% at −0.58 V and −0.68 V, respectively.
The dispersion of Pd atoms with strong CO adsorption on the Au surface in the form of diatomic Pd–Pd would adjust the CO adsorption strength to moderate and further enhance the CO selectivity (Fig. 18a).213 Pb2 DACs could be prepared by anion replacement deposition, and the distance between Pd and Pd was 1.96 Å, indicating the formation of a dual-nuclear site.214 At an applied voltage of −0.85 V, Pb2 DACs presented high selectivity for CO with an FE of 98.2%, which was much higher than that of the corresponding single atom catalyst (65%) and nanostructured catalyst (80%). The CO poisoning reaction of Pd nanoparticles was not conducive to CO2RR, resulting in a large reaction barrier. However, for Pd-based atomically-dispersed catalysts, the activation of CO2 to COOH* was the RDS, and both isolated Pd1 and  exhibited greater free energy of RDS than Pd2. The enhanced electrocatalytic activity was attributed to the electron transfer between Pd–Pd.
exhibited greater free energy of RDS than Pd2. The enhanced electrocatalytic activity was attributed to the electron transfer between Pd–Pd.
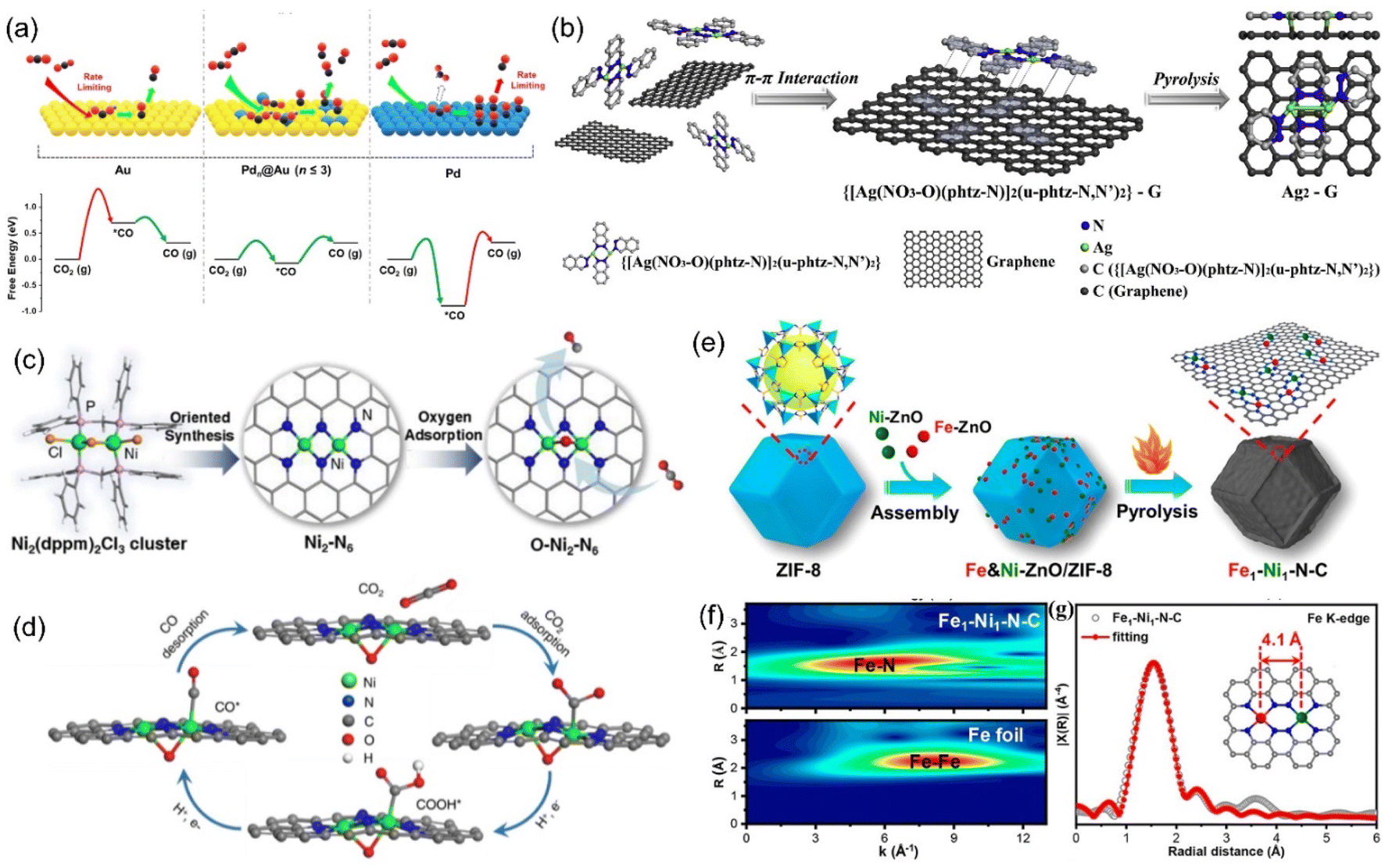 | ||
| Fig. 18 (a) Illustration of the concept using atomically-dispersed Pd sites on Au surface to enhance CO2 reduction. (b) Synthetic illustration of the Ag2-G catalyst. (c) The synthesis process of the Ni2-N6 catalyst and (d) proposed reaction pathways on O-Ni2-N6. (e) Synthetic illustration of Fe1-Ni1-N-C; (f) WT plots for Fe element. (g) Fe K-edge EXAFS spectrum. (a) Reproduced with permission.213 Copyright 2019, American Chemical Society. (b) Reproduced with permission.215 Copyright 2020, Elsevier B.V. All rights reserved. (c and d) Reproduced with permission.218 Copyright 2021, American Chemical Society. (e–g) Reproduced with permission.221 Copyright 2021, American Chemical Society. | ||
The AgN3–AgN3 sites can be obtained by fixing a binuclear Ag precursor to graphene through the π–π interaction (Fig. 18b).215 Two Ag atoms interact with C and O at the same time, stabilizing the CO* intermediate and reducing the reaction barrier for the formation of *COOH. At −0.7 V, the current density of 11.87 mA cm−2 could be achieved, and the FE of CO reached 93.4%. When metal oxides or sulfides and alloys are used as the support, dispersed atom-based catalysts are more likely to achieve high loading capacity. Jiao et al. synthesized Cu-APC, with atomic copper pair (Cu10–Cu1x+) sites by adding Cu onto Te defects of the Pd10Te3 alloy nanowires.216 In the copper pair, Cu1x+ adsorbed water molecules in the electrolyte and promoted the adsorption and activation of CO2 at the adjacent Cu10 sites. In 0.2 M NaHCO3, the FECO of 0.10%-Cu-APC was 92%, while the original APC mainly showed HER, implying that the Cu10–Cu1x+ was the active site of CO2RR.
The synergistic effect of heteronuclear metal atoms on the electronic structure is obvious. Without direct bonding, M1 and M2 are merely adjacent and are connected by bridging bonds (M1-X-M2), exhibiting enhanced CO2RR activity than the individual M1 or M2. The active sites of catalysts can change from a single atom to an atomic pair by changing the synthetic conditions. For example, when Fe-ZIF-8 was calcined in pure argon, the product was SAC Fe1-N4-C, and the iron content was 0.155 wt%.217 However, when hydrogen was introduced into argon, Fe atoms underwent a transition from single-core to dual-core. When the hydrogen content was 5%, 80% of the iron in the product was in the form of atomic pair (Fe2-N6), and the iron content was 0.313 wt%. The orbital coupling between the two Fe sites reduced the energy gap between the antibonding state and the bonding state in *CO adsorption, improving the selectivity of electrocatalytic products. Similarly, Ni2-N6 was successful synthesized using Ni2 organic molecules as the precursor, where two of the Ni atoms share two nitrogen atoms (Fig. 18c).218 In the process of CO2 adsorption, the two complex-unsaturated Ni atoms tended to adsorb oxygen in the solution to form an oxygen bridge bond (Ni–O–Ni), generating more active O–Ni2–N6 (Fig. 18d).
Due to the strong Fe–CO interaction, the desorption of CO* on Fe-SACs was the RDS for CO2RR, and the selectivity of Fe-N-C for CO was only 80%.219 With the assistance of Zn, the Ni–Fe atomic pair was successfully synthesized with ZIF-8, where the distance between Fe and Ni was 4.1 Å (Fig. 18e–g). After the introduction of Ni, the charge transfer of Fe to CO in the Fe-Ni-N6 catalyst was less than that in the corresponding catalyst without Ni. In Fe1-Ni1-N-C, the adsorption of CO2 occurred at the Fe site, while the adjacent Ni atom promoted the formation of COOH* on the iron atom through a remote electron interaction via the N atom. At a low voltage of −0.5 V, Fe1-Ni1-N-C still has 96.2% FE against CO. When the atomic pair was composed of Ni and Zn atoms, Ni was the main adsorption site. The addition of Zn at the Ni site reduced the gap between the d band center of Ni and the Fermi level, enhanced the bonding between the metal and C, and presented an obvious acceleration effect in both kinetic and thermodynamic aspects.220 The FE for CO was more than 90% in the wide voltage range (−0.5 to −1.0 V), and the maximum reached 99%.
From the view of theoretical calculations, the in-depth analysis of the nature of ECO2RR activity gain in polymetallic systems can be performed. Atomic-level control of the coordination environment and electron distribution of electrocatalysts is an effective strategy to improve the electrocatalytic performance, but there are still significant challenges.222 Previous work has shown that synergistic interactions between atoms in a catalyst can locally tune the electronic structure, weakening the adsorption energy of the reaction intermediates and effectively lowering the energy barrier of the catalytic reaction.223 Theoretical calculations showed that the co-catalytic strategy affects the adsorption or desorption of adsorbates on the catalyst surface, influencing the d-band centers of the metal atoms and thus changing the free energy barriers.220 Lu et al. used nickel–zinc diatomic coordination to change the d-state of the metal atoms, resulting in significantly enhanced synergistic effect compared to monoatomic catalysts. DFT calculations showed that this synergistic effect enhances the catalytic performance due to the reduction of the distance between the center of the d-band of Ni–Zn orbitals and Fermi energy level and the enhancement of the intermediate's electronic interactions with the active center (Fig. 19a). This lowers the free energy barrier in the thermodynamic pathway, reduces the activation energy, and strengthens the metal–carbon bond in the kinetic pathway. Ni–Zn diatomic catalyst can achieve 99% Faraday efficiency at −0.8 V.220 Wang et al. designed an efficient catalyst with CoPc traced on Fe-N-C. After the positioning of CoPc molecules, the synergistic interaction resulted in the movement of the d-band of the Fe center toward the Fermi energy level, which enhanced the reactivity of the Fe center, and this electronic interaction was reflected in the adsorption of reactive intermediate species (Fig. 19b and c). As a result, CO desorption on the catalyst's surface was promoted, and the competing HER on the Fe-N site was suppressed, realizing a high CO Faraday efficiency.224 Qiao et al. investigated the relationship between synergistic and intermetallic interactions using a combination of theoretical simulations and experiments. In the case of NiCu DACs, the electronic contribution from the Cu center is transferred through the NC substrate to the Ni center and finally reaches the *COOH intermediate. This electron transfer through the structure is likely to be the result of non-bonding synergies between neighboring DACs (Fig. 19d–f). Moreover, the synergistic effect modulates the electronic structure and promotes intermediate layer adsorption, thus improving the electroreduction activity and selectivity.225
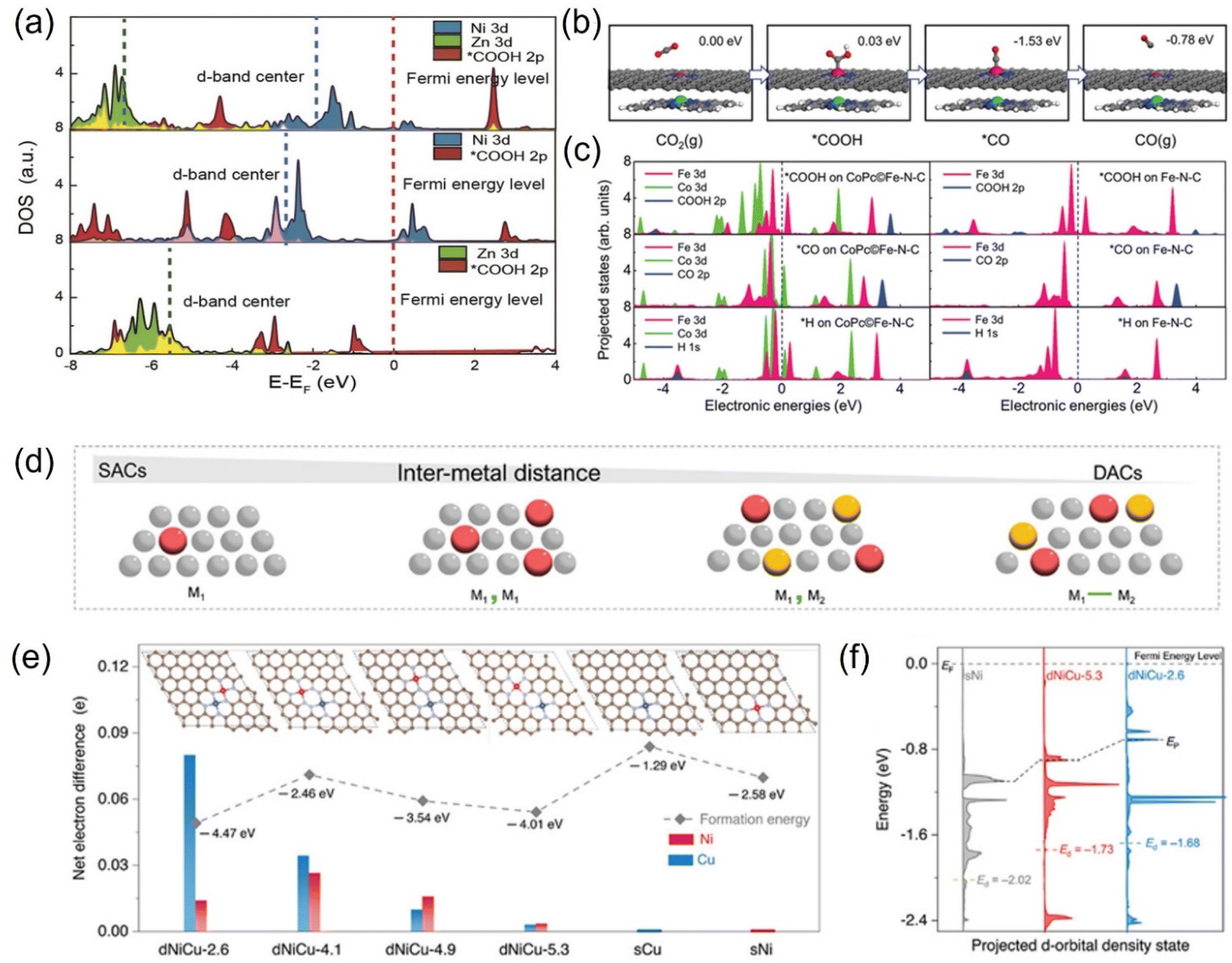 | ||
| Fig. 19 (a) Projected DOS (PDOS) for Zn/Ni 3d orbitals and oxide 2p orbitals in COOH. (b) Schematic atomic structure of the CO2RR process on the Fe site in CoPc©Fe-N-C (C: gray; N: blue; O: red; H: white; Fe: pink; Co: green). (c) PDOS of adsorption structures of *COOH, *CO, and *H on CoPc©Fe-N-C and Fe-N-C. (d) Schematic of SACs and DACs showing the change in the site proximity with different models. (e) Net electron difference of Ni and Cu sites in different dNiCu models. (f) Calculated DOS diagram of the Ni d-orbital for dNiCu-5.3, dNiCu-2.6, and sNi models. (a) Reproduced with permission.220 Copyright 2021, Wiley-Blackwell. (b and c) Reproduced with permission.224 Copyright 2019, Wiley-Blackwell. (d–f) Reproduced with permission.225 Copyright 2023, Wiley-Blackwell. | ||
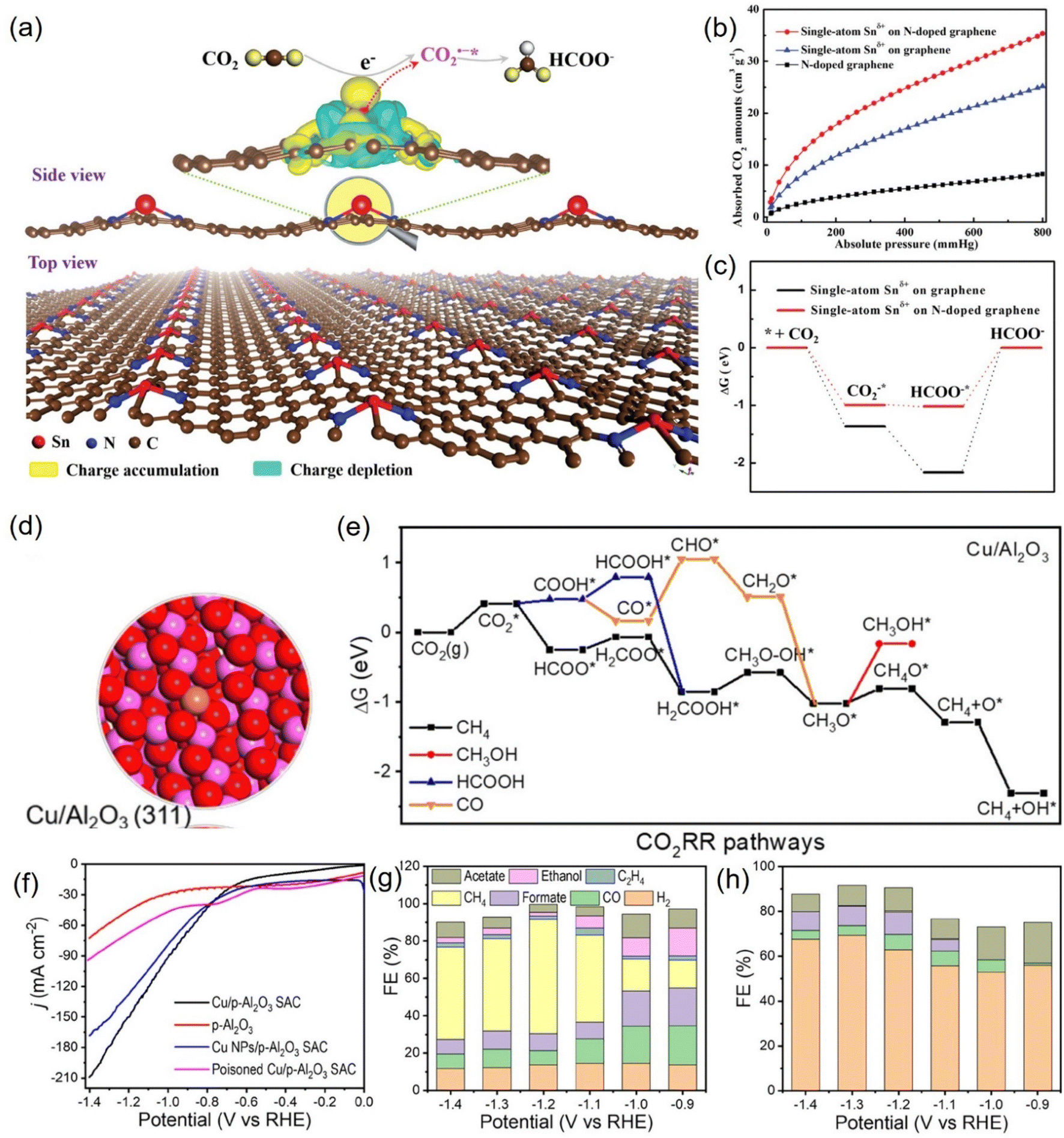 | ||
| Fig. 20 (a) 3D differential charge densities of single-atom Snδ+ on N-doped graphene for CO2R into formate. (b) CO2 adsorption isotherms and (c) calculated free energy diagrams. (d) Electronic structure of Cu/Al2O3 SAC. (e) Calculated free-energy diagrams for the CO2RR over Cu/Al2O3 SAC. (f) LSV curves and FEs of various products for (g) Cu/p-Al2O3 SAC and (h) p-Al2O3. (a–c) Reproduced with permission.227 Copyright 2019, Wiley-Blackwell. (d–h) Reproduced with permission.229 Copyright 2021, American Chemical Society. | ||
4.2. Alloys
Alloys have become one of the alternative catalysts for CO2RR due to the advantages that the constituent elements can form a mutual stabilizing mechanism, thus improving the conductivity and providing more active sites. According to the type and content of metal, alloy electrocatalysts can be classified into single-atom alloys, bimetallic/trimetallic alloys and high-entropy alloys.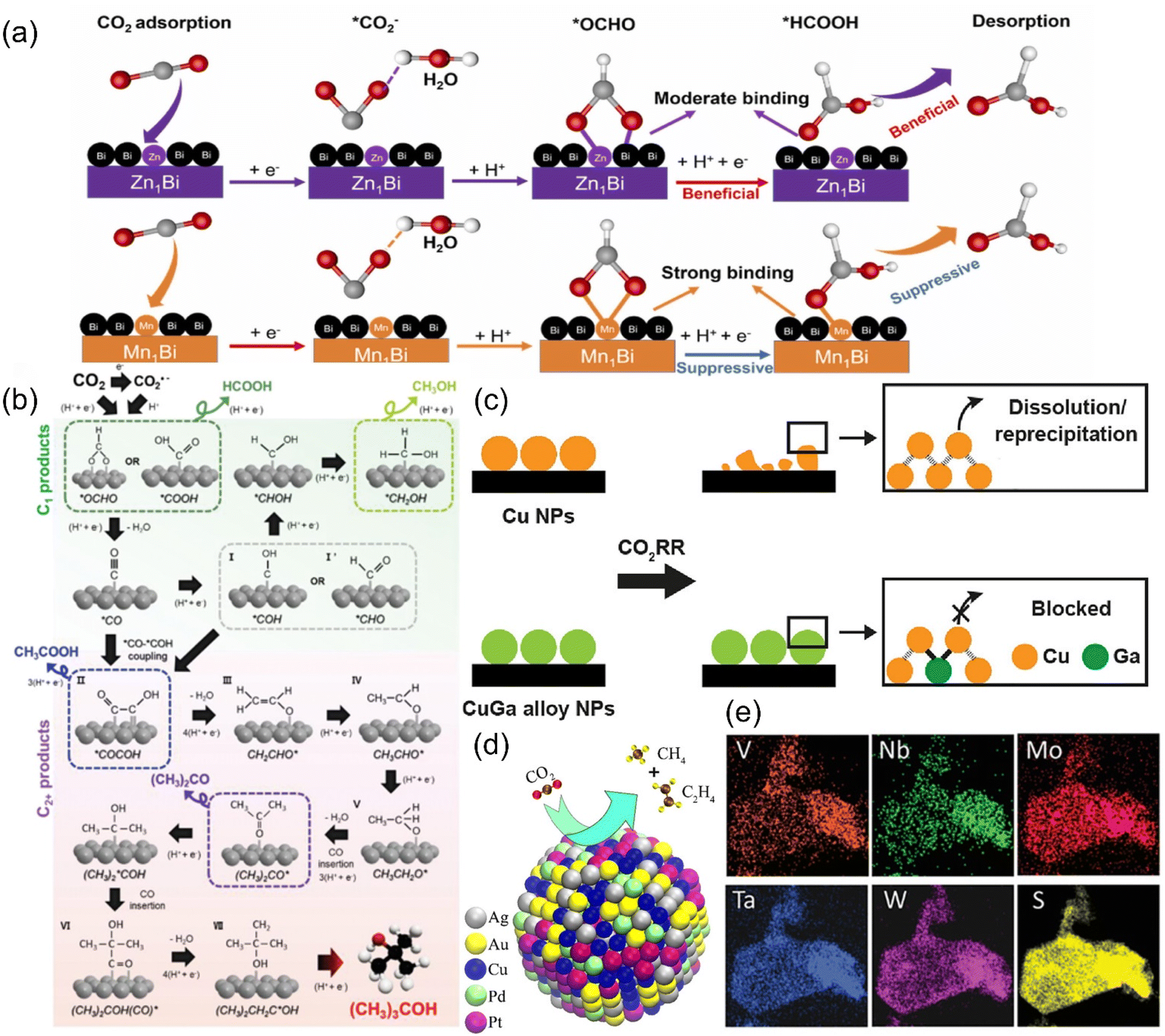 | ||
| Fig. 21 (a) A proposed reaction mechanism for CO2R to formate on SAA-Zn1Bi and SAA-Mn1Bi. (b) A probable mechanism for CO2R to (CH3)3COH. (c) Illustration of the CO2RR stabilization mechanism on CuGa alloy NPs. (d) Schematic of the catalysis reaction on AuAgPtPdCu. (e) EDS maps of (MoWVNbTa)S2. (a) Reproduced with permission.233 Copyright 2023, Elsevier Ltd. (b) Reproduced with permission.236 Copyright 2023, Wiley-VCH GmbH. (c) Reproduced with permission.237 Copyright 2023, American Chemical Society. (d) Reproduced with permission.240 Copyright 2020, American Chemical Society. (e) Reproduced with permission.241 Copyright 2021, Wiley-VCH GmbH. | ||
4.3. Metal–organic frameworks
It is well known that porous carbon-based materials with good chemical and thermal stability are favorable catalysts or supports for CO2 capture.243,244 Among porous carbon-based materials, MOFs are porous materials with well-defined crystallinity constructed from a diversity of organic linkers and feasible synthesis methods.245,246 MOFs are superb catalyst templates due to their outstanding physiochemical stability, structure diversity, highly ordered porous structure, uniform pore sizes and environments, high internal surface areas, and functional organic linkers.247,248 Benefiting from these unique physical and chemical properties, MOF derivatives exhibit the superiorities of higher stability, conductivity, and catalytic activity, which have been widely applied in CO2RR.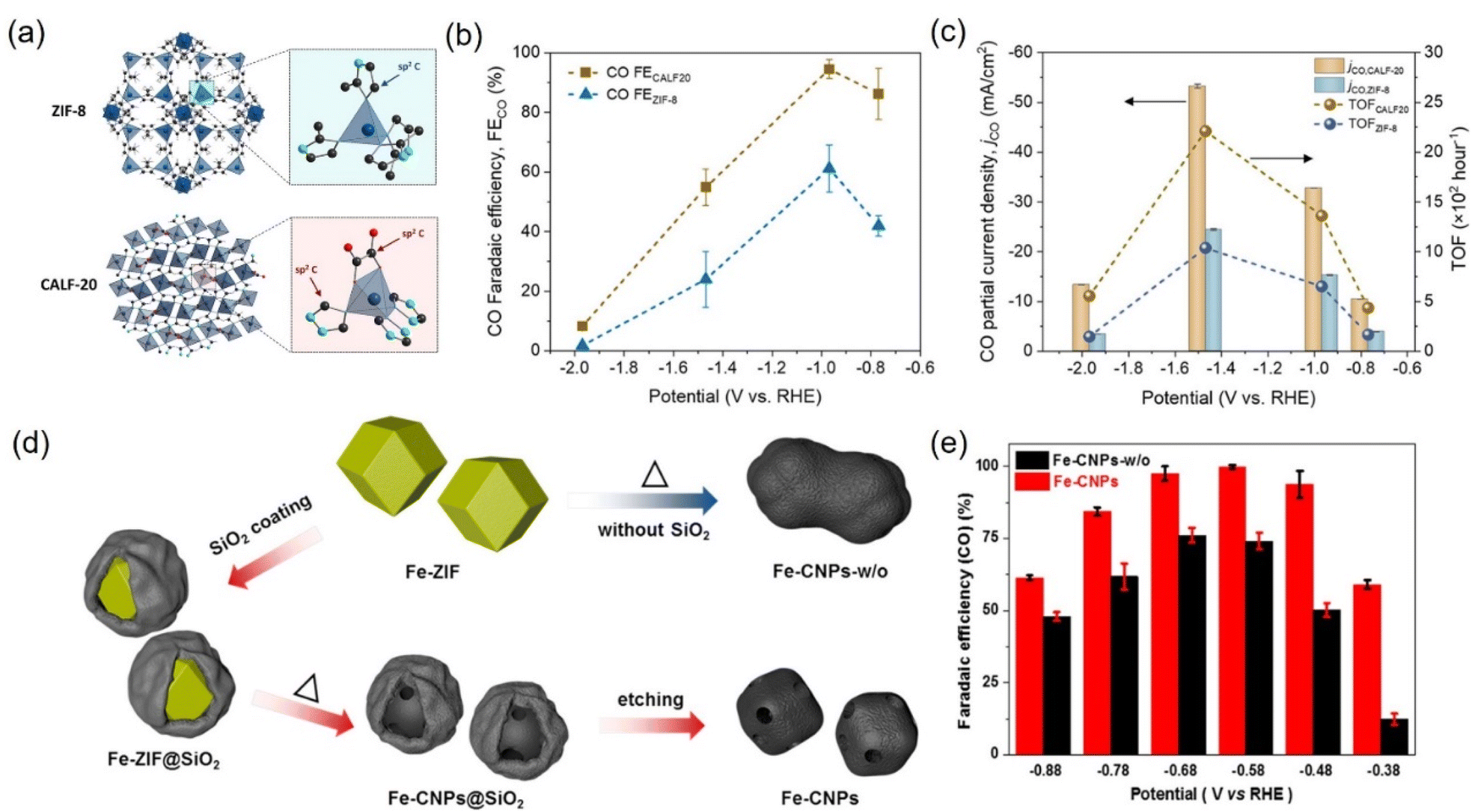 | ||
| Fig. 22 (a) Schematic illustration of the crystal structure of ZIF-8 and CALF20. (b) FECO and (c) CO partial current densities and TOF at different applied potentials for CALF20 and ZIF-8 in 1.0 M KOH. (d) Synthetic illustration of Fe-CNPs and Fe-CNPs-w/o. (e) FECO at different potentials. (a–c) Reproduced with permission.249 Copyright 2021, American Chemical Society. (d and e) Reproduced with permission.252 Copyright 2019, American Chemical Society. | ||
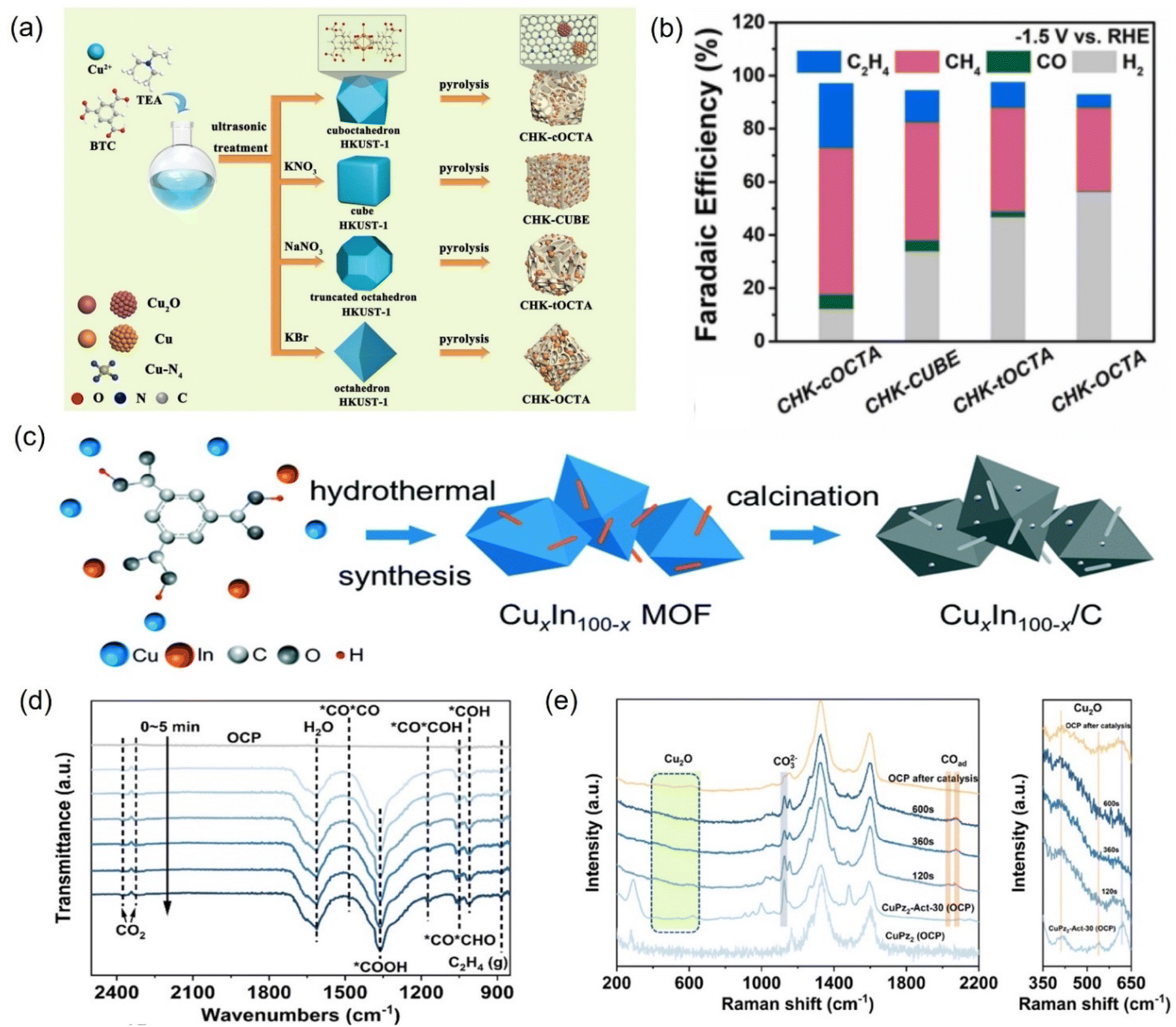 | ||
| Fig. 23 (a) Illustration of the fabrication of different Cu-based nanoreactors and (b) faradaic efficiency at −1.5 V vs. RHE. (c) Synthetic diagram of Cu–In bimetallic catalysts. (d) In situ ATR-FTIR spectra of CuPz2-Act-30 collected at −1.03 V vs. RHE and (e) in situ Raman spectra of CuPz2 (OCP), CuPz2-Act-30 (OCP), and CuPz2-Act-30. (a and b) Reproduced with permission.255 Copyright 2022, Elsevier. (c) Reproduced with permission.257 Copyright 2021, the Royal Society of Chemistry. (d and e) Reproduced with permission.258 Copyright 2022, American Chemical Society. | ||
4.4. Covalent organic frameworks
COFs are reticulated crystalline solids with a periodic structure consisting of molecular units connected by functional organic ligands.202,263,264 COFs, as promising alternative materials for electrocatalytic CO2RR, are mainly attributed to more exposed surface area and active sites, tunable structure, and the possibility of modifying a wide range of metal types (e.g., Co, Cu, and Ni) into their structure to confer the COFs with tunable catalytic centers.265 These materials allow the integration of various organic building blocks into two-dimensional (2D) or three-dimensional (3D) mesh networks. Over the past decade, hundreds of COFs have been developed and used in a variety of applications, including gas adsorption, multiphase catalysis, photonics, photoemission, and wastewater treatment.266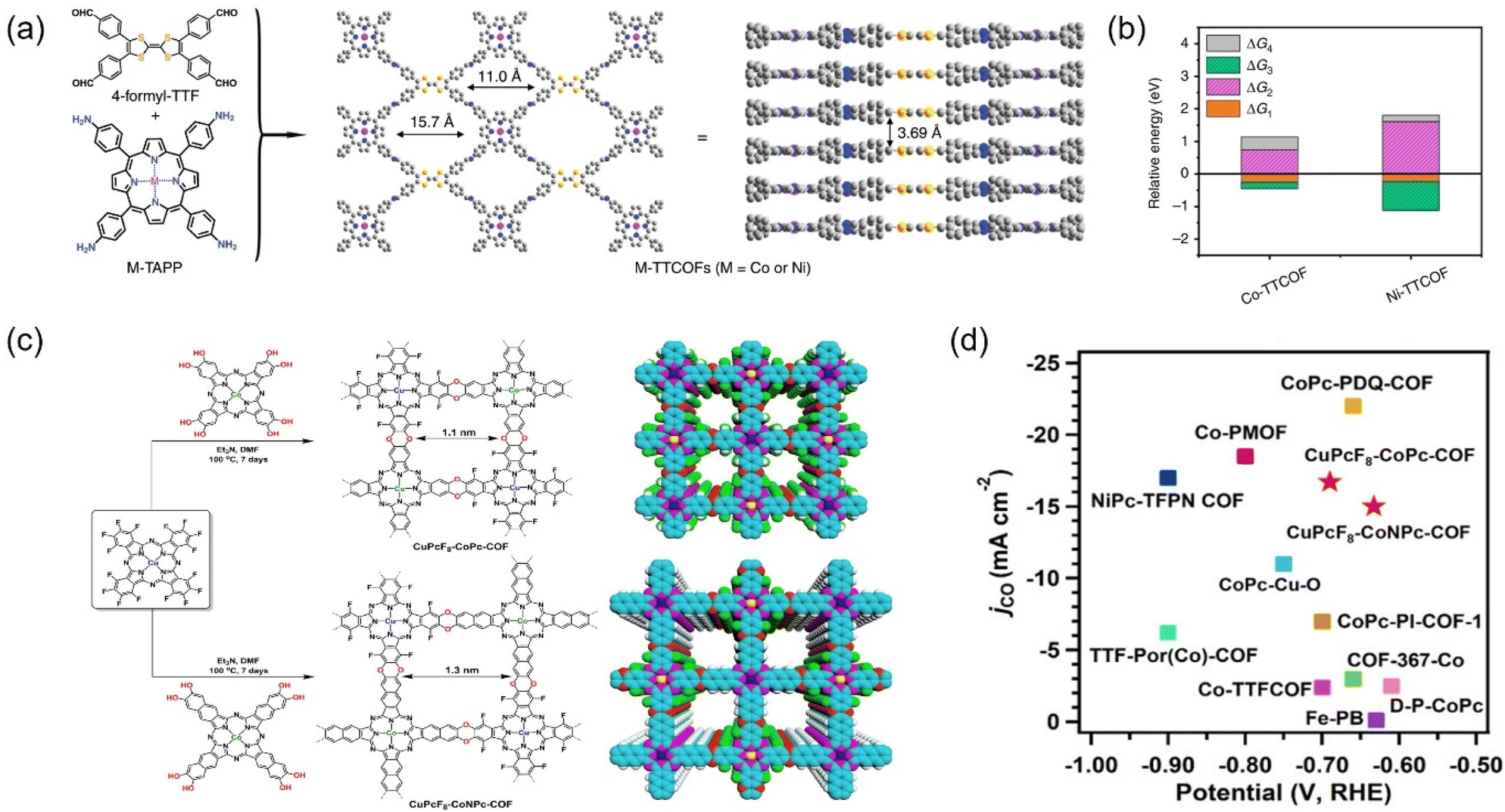 | ||
| Fig. 24 (a) The structure of M-TTCOFs obtained through the condensation of 4-formyl-TTF and M-TAPP. (b) Comparison of the relative energy of each elementary reaction (ΔG1, ΔG2, and ΔG3 represent the free energy of *CO2, *COOH, and *CO formation, respectively, and ΔG4 stands for the free energy of CO desorption process) in the electrocatalytic CO2RR for M-TTCOFs (M = Co or Ni). (c) Schematic representation for the synthesis of CuPcF8-CoPc-COF and CuPcF8-CoNPc-CO. (d) Comparison of jCO. (a and b) Reproduced with permission.265 Copyright 2020, Springer Nature. (c and d) Reproduced with permission.266 Copyright 2021, American Chemical Society. | ||
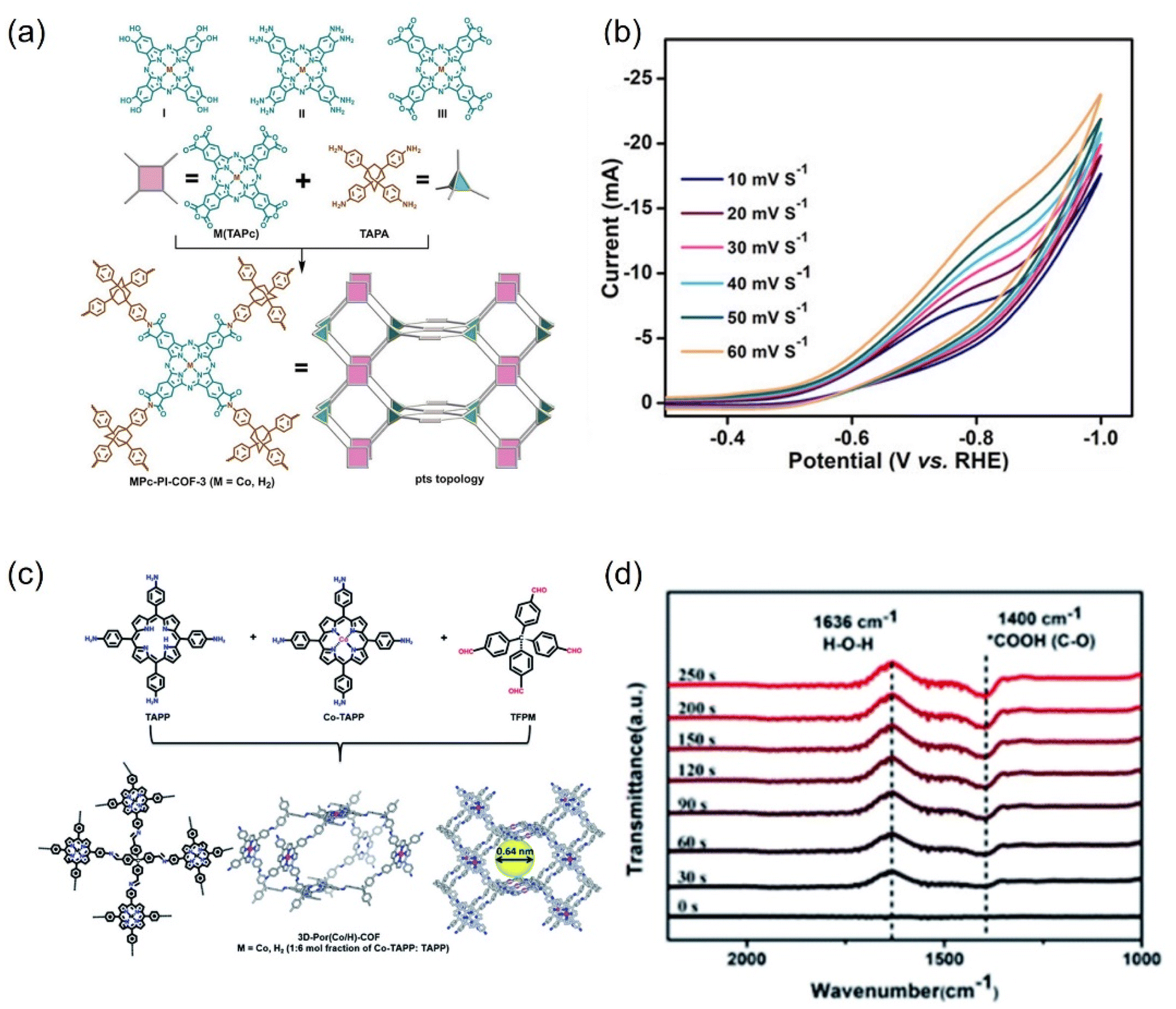 | ||
| Fig. 25 (a) Synthesis of MPc-PI-COF-3 (M = CoII or H2) and (b) scan rate dependence of cyclic voltammetry response of CoPc-PI-COF-3. (c) Schematic representation of the synthesis of 3D-Por(Co/H)-COF and the simulated network. (d) In situ ATR-IR of 3D-Por(Co/H)-COF at −0.9 V. (a and b) Reproduced with permission.274 Copyright 2022, John Wiley and Sons Ltd. (c and d) Reproduced with permission.275 Copyright 2022, Royal Society of Chemistry. | ||
5. Challenges and perspectives
Metal-based electrocatalysts have been widely studied for the CO2RR due to their controllable composition and structure, excellent chemical stability and clear catalytically active sites. In this review, we summarize the basic research and current research progress of catalyst structure regulation and carrier selection involved in CO2RR. Although significant progress has been made in electrocatalytic CO2RR, the actual related research is still in its infancy. As a promising way to close the carbon cycle and generate value-added products, CO2RR is attracting more and more attention. However, CO2RR is extremely sensitive to the reaction environment, which makes it difficult to accurately grasp the reaction process. Physical and chemical changes may occur during the catalytic process as well as morphological evolution such as surface recombination and roughening on the catalyst surface. In order to obtain efficient target products, there are still several crucial challenges to be overcome.(1) Compared with the easily available C1 product, the reduction reactions of C2 and C3 products remain to be further studied. For two-electron products, high Faraday efficiency can be achieved at a given potential, but the overpotential is still unsatisfactory compared with the theoretical value. Hence, improving the Faraday efficiency of multi-carbon products is the main direction of future research. Commercially, the reduction products are required to have high Faraday efficiency (>90%) and current density (>200 mA cm−2). Increasing the selectivity for high-value products remains a major challenge and there is still a huge room for progress.
It is necessary, on the one hand, to develop the catalytic activity and product possibilities of more metal elements, and, on the other hand, to improve the specific Faraday efficiency of metals known to produce multi-carbon products. Besides, most of the reported electrocatalysts are usually designed as powders, which may lead to the serious agglomeration of the material, thereby reducing the utilization of active sites. Furthermore, excessive pursuit of performance improvement while ignoring the cost considerations is also not conducive to practical application. Therefore, people are eager to minimize the use of expensive precursors and surfactants and simplify the synthesis processes to obtain catalytic systems with both low cost and high efficiency. On the basis of clarifying the active sites by experiments and theoretical calculations, the core of the solution needs to be considered from the perspective of the catalyst itself. Optimization strategies including heteroatom doping, defect engineering, and morphology design are useful for assisting electrocatalysts in achieving economical products with higher purity. Conceptualizing ingenious strategies is the golden key to meet the selectivity requirements in practical application. For example, as the material size is optimized for further reduction, the density of the active site along with the specific surface area will continue to expand, which can promote mass transport and improve the kinetic activity. Furthermore, the selective introduction of defects such as vacancies and grain boundaries can effectively contribute to the desired outcome. In addition, the hydrophilicity and hydrophobicity of the catalyst can be adjusted to optimize the binding energy of the intermediate, thus affecting the competitive relationship between CO2RR and HER.
(2) Compared with the success achieved in the catalytic activity of ECO2RR, the breakthrough in the stability has been more dismal. To date, almost all the reported materials have a lifetime of less than 1000 hours.14 The peak achievement is that the operating time for CO and formic acid can reach 3800 and 2400 hours, respectively.276,277 However, these existing catalysts are far from meeting the commercial threshold of more than 50000 hours for the low-temperature operation of ECO2RR.278 Therefore, on the road to the commercialization of ECO2RR, meeting the most troublesome difficulties of long-term stable operation is the core technical problem to be solved urgently. There are many factors that lead to the instability of electrocatalysis. For example, small perturbations in the electrochemical microenvironment may increase the difficulty of species mass transfer, thereby limiting the diffusion and adsorption of CO2 molecules. In this regard, it is necessary to optimize the entire electrolysis system and alleviate flooding and carbonate problems. In addition, the active sites will undergo isomeric collapse as the ECO2RR continues, resulting in the inability of electrons to be quickly transferred, and the key intermediate species cannot be effectively adsorbed, which has a devastating effect on both the stability and activity. The key to solving this thorny problem is to create more thermodynamic and kinetic barriers to atomic migration. Enhancing the bond strength of lattice atomic bonds to chemically improve the degree of bonding and wrapping canning materials to physically prevent atomic diffusion are effective strategies to avoid surface reconstruction, which can be achieved by ligand protection, doping/alloying, interface construction and protective layer encapsulation. Notably, the reasonable manipulation of operating conditions such as pulse electrolysis can also uniquely extend the durability of the material by repairing the active center and adjusting the interfacial chemistry. Specifically, pulse operation prolongs the ECO2RR process by inhibiting carbonate deposition, removing surface toxins/bubbles, and recovering active phases.
(3) In addition to the optimization of the catalyst itself, it is particularly critical to design a more suitable CO2RR electrolytic cell.279 The setting of electrolytic cells will have a profound impact on the overall performance of ECO2RR.280 With the improvement of this technical field, it is particularly critical to significantly reduce the overpotential of CO2RR in the electrolytic cell. Through the analysis and comparison of various types of electrolytic cells, in most of the current studies, the three reactors of H-type electrolytic cell,281 flow type electrolytic cell282 and membrane electrode electrolytic cell283 are widely used. The H-type electrolytic cell is separated from the left and right chambers by an ion-exchange membrane.284 The separation of electrolyte and cathode reaction gas can be achieved by exchange. However, the solubility of CO2 in the environment containing water electrolyte is relatively low. Therefore, in the H-type reactor, the mass transfer process is greatly restricted. Due to the large interfacial tension between water and air, the gas cannot enter the cathode chamber through this area, thus affecting the electrolysis reaction. In addition, continuous carbon dioxide blowing may lead to an increase in the pH value of the electrolyte, which is more detrimental to the performance of the H-type battery. In order to solve the problems existing in the H-type electrolytic cell, researchers have designed a flow electrolytic cell for CO2RR. Compared with the H-type electrolytic cell, the flow cell adds a gas chamber, and the gas diffusion layer (GDL) is used to separate the cathode electrolyte and the CO2 gas. The CO2 gas can directly pass through the GDL and react on the surface of the catalyst. The flow cell also has the ability to utilize alkaline electrolytes, which is beneficial because it can inhibit HER in a high pH environment, thereby improving the performance of CO2RR. From the perspective of commercialization, membrane electrode electrolyzers are considered to be the most suitable equipment for commercial applications. However, some organic products may cause damage to solid electrolytes and ion-exchange membranes during long-term use, thereby reducing their durability. Therefore, for a variety of different products, we need to optimize and adjust the system of solid electrolyte electrolyzers in a timely manner. The careful design of electrodes and electrolytic reactors plays a vital role in achieving catalytic activity with industrial application value.
(4) DFT calculations can explore the electronic structure and directly identify the active sites at the atomic level. It is a favorable weapon for reasonably analyzing the reaction mechanism and predicting the catalytic steps and has an indispensable inspiration and guidance for experimental preparation.285 For ECO2RR, the intrinsic properties of CO2 molecules are variable, the types of intermediates of CO2RR are multifarious, and the effects of solvation and ion on CO2RR at the solid–liquid interface are relatively complex. Theoretical calculations are increasingly being relied upon to reveal the mechanism of the tedious reaction steps and to screen for more optimized material compositions and structures. Due to the escalating difficulty, the theoretical insights of CO2RR are far less comprehensive and profound than other electrocatalytic reactions such as OER, HER, and ORR, and there is still a huge unknown space to be exploited. Typically, the calculation of the hydrogen electrode (CHE) is modeled under the assumption that the catalyst is charge-neutral to calculate the energy barrier of each intermediate step. However, ECO2RR usually occurs at the gas–liquid–solid interface with a constant electrode potential. The surface of the catalyst is charged and is balanced by electron exchange with the electrode, rather than without the electrode potential, which is a non-negligible influence factor in ECO2RR. For example, the basic step energy barrier of the proton–electron transfer of ECO2RR to CO on Cu (100) is profoundly affected by the electrode potential, and their change trend is opposite. In this regard, Wang et al. regulated the electrode potential of the system by introducing anions (Cl−) and cations (Na+) and detect their effects using constrained molecular dynamic (MD) sampling and thermodynamic integration.286 They also elucidated the mechanism of electron transfer-proton transfer (ET-PT) decoupling and hydrogen bond-assisted CO2 activation, indicating that the efficient adsorption and activation of CO2 are closely related to the electron transfer of active sites, demonstrating that the electrode potential is the key to achieving efficient capture of CO2. In addition, the hydrogen bond network between the water solvent molecules interacts strongly with the key species adsorbed on the catalyst surface, and this solvent effect almost determines the stability of ECO2RR intermediates. In summary, it is of great significance to construct a clear model of aqueous solution system and introduce the consideration of electrode potential into the calculation simultaneously. In the future, the influencing factors included in the theoretical research on ECO2RR need to be more comprehensive to fit the actual reaction in the experiment as much as possible.
Looking ahead, ECO2RR research will mainly focus on the system design of the electrolyzer and the optimization and evaluation of electrochemical performance, especially in the design of the reactor, the nature of the electrode, electrolyte, ion exchange membrane, voltage, current density, energy efficiency and the service life of the electrolyzer. These research results will provide a strong impetus and guidance for the commercial application of ECO2RR.
Author contributions
All of the authors contributed to the literature search, writing and editing of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22075099), and the Natural Science Foundation of Jilin Province (No. 20220101051JC).Notes and references
- S. Gao, Y. Zhu, M. Umar, B. Kchouri and A. Safi, Technol. Forecast. Soc., 2024, 201, 123177 CrossRef.
- R. Huang, Sci. Total Environ., 2024, 913, 169775 CrossRef CAS PubMed.
- J. Rogelj and R. D. Lamboll, Commun. Earth Environ., 2024, 5, 35 CrossRef.
- Z.-C. Zeng, T. Pongetti, S. Newman, T. Oda, K. Gurney, P. I. Palmer, Y. L. Yung and S. P. Sander, Nat. Commun., 2023, 14, 5353 CrossRef CAS PubMed.
- M. Umar, S. Farid and M. A. Naeem, Energy, 2022, 240, 122702 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef CAS.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Albo, M. Perfecto-Irigaray, G. Beobide and A. Irabien, J. CO2 Util., 2019, 33, 157–165 CrossRef CAS.
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS.
- Y. Zou and S. Wang, Adv. Sci., 2021, 8, 2003579 CrossRef CAS.
- X. Zhang, Z. Zhang, H. Li, R. Gao, M. Xiao, J. Zhu, M. Feng and Z. Chen, Adv. Energy Mater., 2022, 12, 2201461 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2016, 10, 1100–1109 CrossRef PubMed.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef CAS PubMed.
- H. Wu, A. Singh-Morgan, K. Qi, Z. Zeng, V. Mougel and D. Voiry, ACS Catal., 2023, 13, 5375–5396 CrossRef CAS PubMed.
- S. Kumar De, D.-I. Won, J. Kim and D. H. Kim, Chem. Soc. Rev., 2023, 52, 5744–5802 RSC.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- I. Merino-Garcia, J. Albo and A. Irabien, Nanotechnology, 2018, 29, 014001 CrossRef CAS PubMed.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- L. R. L. Ting and B. S. Yeo, Curr. Opin. Electrochem., 2018, 8, 126–134 CrossRef CAS.
- C. Shi, K. Chan, J. S. Yoo and J. K. Nørskov, Org. Process Res. Dev., 2016, 20, 1424–1430 CrossRef CAS.
- S. Back, M. S. Yeom and Y. Jung, ACS Catal., 2015, 5, 5089–5096 CrossRef CAS.
- A. Dutta, A. Kuzume, M. Rahaman, S. Vesztergom and P. Broekmann, ACS Catal., 2015, 5, 7498–7502 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Adv. Sci., 2017, 5, 1700275 CrossRef.
- G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed.
- J. Y. Yang, T. A. Kerr, X. S. Wang and J. M. Barlow, J. Am. Chem. Soc., 2020, 142, 19438–19445 CrossRef CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- L. Deng, Z. Wang, X. Jiang, J. Xu, Z. Zhou, X. Li, Z. You, M. Ding, T. Shishido, X. Liu and M. Xu, Appl. Catal., B, 2023, 322, 122124 CrossRef CAS.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS.
- M. F. Baruch, J. E. Pander, J. L. White and A. B. Bocarsly, ACS Catal., 2015, 5, 3148–3156 CrossRef CAS.
- X. Min and M. W. Kanan, J. Am. Chem. Soc., 2015, 137, 4701–4708 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS.
- C. Chen, J. F. Khosrowabadi Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z.-J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed.
- C.-T. Dinh, T. Burdyny, Md G. Kibria, S. Ali, C. M. Gabardo, F. Pelayo García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- K. D. Yang, C. W. Lee, K. Jin, S. W. Im and K. T. Nam, J. Phys. Chem. Lett., 2017, 8, 538–545 CrossRef CAS PubMed.
- E. Pérez-Gallent, M. C. Figueiredo, F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 3621–3624 CrossRef PubMed.
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- X. Li, M. Qin, X. Wu, X. Lv, J. Wang, Y. Wang and H. B. Wu, Small, 2023, 19, 2302530 CrossRef CAS PubMed.
- J. Zhang, Y. Wang, Z. Li, S. Xia, R. Cai, L. Ma, T. Zhang, J. Ackley, S. Yang, Y. Wu and J. Wu, Adv. Sci., 2022, 9, 2200454 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S.-Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS.
- C. Peng, X. Zhu, Z. Xu, S. Yan, L. Y. Chang, Z. Wang, J. Zhang, M. Chen, T. K. Sham, Y. Li and G. Zheng, Small, 2021, 18, 2106433 CrossRef PubMed.
- C. Peng, G. Luo, Z. Xu, S. Yan, J. Zhang, M. Chen, L. Qian, W. Wei, Q. Han and G. Zheng, Adv. Mater., 2021, 33, 2103150 CrossRef CAS.
- D. Zhong, Z. J. Zhao, Q. Zhao, D. Cheng, B. Liu, G. Zhang, W. Deng, H. Dong, L. Zhang, J. Li, J. Li and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 4879–4885 CrossRef CAS.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- J. J. Lv, M. Jouny, W. Luc, W. Zhu, J. J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed.
- P. Wang, H. Yang, Y. Xu, X. Huang, J. Wang, M. Zhong, T. Cheng and Q. Shao, ACS Nano, 2020, 15, 1039–1047 CrossRef.
- X. Wang, K. Klingan, M. Klingenhof, T. Möller, J. Ferreira de Araújo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS.
- Q. Zhu, Y. Hu, H. Chen, C. Meng, Y. Shang, C. Hao, S. Wei, Z. Wang, X. Lu and S. Liu, Nanoscale, 2023, 15, 2106–2113 RSC.
- J. Wu, S. Ma, J. Sun, J. I. Gold, C. Tiwary, B. Kim, L. Zhu, N. Chopra, I. N. Odeh, R. Vajtai, A. Z. Yu, R. Luo, J. Lou, G. Ding, P. J. A. Kenis and P. M. Ajayan, Nat. Commun., 2016, 7, 13869 CrossRef CAS.
- J. Yuan, W.-Y. Zhi, L. Liu, M.-P. Yang, H. Wang and J.-X. Lu, Electrochim. Acta, 2018, 282, 694–701 CrossRef CAS.
- X. Zou, M. Liu, J. Wu, P. M. Ajayan, J. Li, B. Liu and B. I. Yakobson, ACS Catal., 2017, 7, 6245–6250 CrossRef CAS.
- C. Liang, B. Kim, S. Yang, Y. L. Yang Liu, C. Francisco Woellner, Z. Li, R. Vajtai, W. Yang, J. Wu, P. J. A. Kenis and P. M. Ajayan, J. Mater. Chem. A, 2018, 6, 10313–10319 RSC.
- M. Yu, P. F. Sui, X. Z. Fu, J. L. Luo and S. Liu, Adv. Energy Mater., 2022, 13, 2203191 CrossRef.
- L. Xiao, X. Bai, J. Han, T. Tang, S. Chen, H. Qi, C. Hou, F. Bai, Z. Wang and J. Guan, Nano Res., 2023, 17, 2429–2437 CrossRef.
- L. Xiao, L. Qi, J. Sun, A. Husile, S. Zhang, Z. Wang and J. Guan, Nano Energy, 2024, 120, 109155 CrossRef CAS.
- S. Chen, T. Zhang, J. Han, H. Qi, S. Jiao, C. Hou and J. Guan, Nano Res. Energy, 2024, 3, e9120106 CrossRef.
- Y. Huang, F. Rehman, M. Tamtaji, X. Li, Y. Huang, T. Zhang and Z. Luo, J. Mater. Chem. A, 2022, 10, 5813–5834 RSC.
- T. Tang, Z. Wang and J. Guan, Acta Phys.–Chim. Sin., 2022, 2208033 Search PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- X. Bai, Y. Wang, J. Han, X. Niu and J. Guan, Appl. Catal., B, 2023, 337, 122966 CrossRef CAS.
- T. Tang, Z. Wang and J. Guan, Coord. Chem. Rev., 2023, 492, 215288 CrossRef CAS.
- J. Han and J. Guan, Nano Res., 2023, 16, 1913–1966 CrossRef CAS.
- T. Zhang, J. Sun and J. Guan, Nano Res., 2023, 16, 8684–8711 CrossRef CAS.
- L. Li, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS.
- H. Yao, M.-Y. Wang, C. Yue, B. Feng, W. Ji, C. Qian, S. Wang, S. Zhang and X. Ma, Trans. Tianjin Univ., 2023, 29, 254–274 CrossRef CAS.
- T. Tang, J. Han, Z. Wang, X. Niu and J. Guan, Nano Res., 2023, 17, 3794–3800 CrossRef.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- J. Ma, C. Liu, M. Bai, Z. Fu, P. Zhao, Y. Gao, M. Zhao, Y. He, H. Xiao and J. Jia, Mol. Catal., 2023, 551, 113632 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS.
- W. Zheng, F. Chen, Q. Zeng, Z. Li, B. Yang, L. Lei, Q. Zhang, F. He, X. Wu and Y. Hou, Nano-Micro Lett., 2020, 12, 108 CrossRef CAS PubMed.
- G. Qu, K. Wei, K. Pan, J. Qin, J. Lv, J. Li and P. Ning, Nanoscale, 2023, 15, 3666–3692 RSC.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS PubMed.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- H. Xie, T. Wang, J. Liang, Q. Li and S. Sun, Nano Today, 2018, 21, 41–54 CrossRef CAS.
- D. Gao, T. Liu, G. Wang and X. Bao, ACS Energy Lett., 2021, 6, 713–727 CrossRef CAS.
- Y. Jiang, Y. Wang, R. Chen, Y. Li and C. Li, Energy Fuels, 2023, 37, 17951–17965 CrossRef CAS.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS PubMed.
- C. Jia, K. Dastafkan and C. Zhao, Curr. Opin. Electrochem., 2022, 31, 100854 CrossRef CAS.
- Z. Wu, Y. Zhao, W. Jin, B. Jia, J. Wang and T. Ma, Adv. Funct. Mater., 2020, 31, 2009070 CrossRef.
- Y. Li, M. Wen, Y. Wang, G. Tian, C. Wang and J. Zhao, Angew. Chem., Int. Ed., 2020, 60, 910–916 CrossRef PubMed.
- Z. Niu, X. Gao, S. Lou, N. Wen, J. Zhao, Z. Zhang, Z. Ding, R. Yuan, W. Dai and J. Long, ACS Catal., 2023, 13, 2998–3006 CrossRef CAS.
- Z. Li, A. Cao, Q. Zheng, Y. Fu, T. Wang, K. T. Arul, J. L. Chen, B. Yang, N. M. Adli, L. Lei, C. L. Dong, J. Xiao, G. Wu and Y. Hou, Adv. Mater., 2020, 33, 2005113 CrossRef PubMed.
- H. Wang, N. Wen, Y. Wang, X. Jiao, Y. Xia and D. Chen, Adv. Funct. Mater., 2023, 33, 2303473 CrossRef CAS.
- B. Jia, L. Li, C. Xue, J. Kang, L. M. Liu, T. Guo, Z. Wang, Q. Huang and S. Guo, Adv. Mater., 2023, 35, 2305587 CrossRef CAS PubMed.
- Y. He, H. Rao, K. Song, J. Li, Y. Yu, Y. Lou, C. Li, Y. Han, Z. Shi and S. Feng, Adv. Funct. Mater., 2019, 29, 1905153 CrossRef CAS.
- X. Rong, H. J. Wang, X. L. Lu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2019, 59, 1961–1965 CrossRef PubMed.
- R. Chen, X. Zu, J. Zhu, Y. Zhao, Y. Li, Z. Hu, S. Wang, M. Fan, S. Zhu, H. Zhang, B. Ye, Y. Sun and Y. Xie, Adv. Mater., 2024, 2314209, DOI:10.1002/adma.202314209.
- W. Ding, S. Yuan, Y. Yang, X. Li and M. Luo, J. Mater. Chem. A, 2023, 11, 23653–23682 RSC.
- H. Yu, F. Chen, X. Li, H. Huang, Q. Zhang, S. Su, K. Wang, E. Mao, B. Mei, G. Mul, T. Ma and Y. Zhang, Nat. Commun., 2021, 12, 4594 CrossRef CAS PubMed.
- Y. Liang, C. Wu, S. Meng, Z. Lu, R. Zhao, H. Wang, Z. Liu and J. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 30262–30271 CrossRef CAS PubMed.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang, S. Wei and Y. Xie, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- L. Cheng, Y. Li, A. Chen, Y. Zhu and C. Li, Chem. Commun., 2020, 56, 563–566 RSC.
- D. Yan, C. Xia, W. Zhang, Q. Hu, C. He, B. Y. Xia and S. Wang, Adv. Energy Mater., 2022, 12, 2202317 CrossRef CAS.
- D. Qu, X. Peng, Y. Mi, H. Bao, S. Zhao, X. Liu and J. Luo, Nanoscale, 2020, 12, 17191–17195 RSC.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS.
- S. Dou, L. Tao, R. Wang, S. El Hankari, R. Chen and S. Wang, Adv. Mater., 2018, 30, 1705850 CrossRef.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 RSC.
- C. Kong, F. Zhang, Y. Wang and J. Huang, J. Alloys Compd., 2021, 876, 160165 CrossRef CAS.
- H. Bi, S. Zhu, Y. Liang, H. Jiang, Z. Li, S. Wu, H. Wei, C. Chang and Z. Cui, Chem. Eng. J., 2022, 442, 136146 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- F. Wang, W. Zhang, H. Wan, C. Li, W. An, X. Sheng, X. Liang, X. Wang, Y. Ren, X. Zheng, D. Lv and Y. Qin, Chin. Chem. Lett., 2022, 33, 2259–2269 CrossRef CAS.
- S. S. A. Shah, M. Sufyan Javed, T. Najam, C. Molochas, N. A. Khan, M. A. Nazir, M. Xu, P. Tsiakaras and S.-J. Bao, Coord. Chem. Rev., 2022, 471, 214716 CrossRef CAS.
- Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS.
- R. Reske, M. Duca, M. Oezaslan, K. J. P. Schouten, M. T. M. Koper and P. Strasser, J. Phys. Chem. Lett., 2013, 4, 2410–2413 CrossRef CAS.
- Z. Chang, S. Huo, W. Zhang, J. Fang and H. Wang, J. Phys. Chem. C, 2017, 121, 11368–11379 CrossRef CAS.
- H. Xie, S. Chen, F. Ma, J. Liang, Z. Miao, T. Wang, H.-L. Wang, Y. Huang and Q. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36996–37004 CrossRef CAS PubMed.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J.-j. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- J. Hao, S. Xie, Q. Huang, Z. Ding, H. Sheng, C. Zhang and J. Yao, CCS Chem., 2023, 5, 2046–2058 CrossRef CAS.
- M. He, W. An, Y. Wang, Y. Men and S. Liu, Small, 2021, 17, 2104445 CrossRef CAS PubMed.
- Y. Xie, N. Liu, X. Li, X. Qin, X. Luo, Y. Wang and X. Chen, J. Phys. Chem. C, 2021, 125, 21460–21470 CrossRef CAS.
- Z. Wang, R. Shi, S. Lu, K. Zhang and T. Zhang, Rep. Prog. Phys., 2022, 85, 026501 CrossRef CAS.
- Z. L. Wang, J. Choi, M. Xu, X. Hao, H. Zhang, Z. Jiang, M. Zuo, J. Kim, W. Zhou, X. Meng, Q. Yu, Z. Sun, S. Wei, J. Ye, G. G. Wallace, D. L. Officer and Y. Yamauchi, ChemSusChem, 2020, 13, 929–937 CrossRef CAS PubMed.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4201–4215 RSC.
- Md Delowar Hossain, Y. Huang, T. H. Yu, W. A. Goddard and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang, J. Lu and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- W. Zheng, J. Yang, H. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. Li, B. Yang, L. Lei, C. Yuan, Q. He, M. Qiu and X. Feng, Adv. Funct. Mater., 2019, 30, 1907658 CrossRef.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57, 14095–14100 CrossRef CAS.
- J. Feng, H. Gao, L. Zheng, Z. Chen, S. Zeng, C. Jiang, H. Dong, L. Liu, S. Zhang and X. Zhang, Nat. Commun., 2020, 11, 4341 CrossRef.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K.-S. Lee, S.-P. Cho, J. S. Yoo, Y.-E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- Y. Cai, J. Fu, Y. Zhou, Y.-C. Chang, Q. Min, J.-J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 586 CrossRef CAS PubMed.
- H. Kim, D. Shin, W. Yang, D. H. Won, H.-S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae, J. Y. Ryu, J. Lee, S. J. Cho, J. Seo, H. Kim and C. H. Choi, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed.
- S. Cao, S. Wei, X. Wei, S. Zhou, H. Chen, Y. Hu, Z. Wang, S. Liu, W. Guo and X. Lu, Small, 2021, 17, 2100949 CrossRef CAS PubMed.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2020, 60, 3212–3221 CrossRef PubMed.
- Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma, Y. Huang and B. Han, Angew. Chem., Int. Ed., 2021, 60, 20803–20810 CrossRef CAS PubMed.
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed.
- X. Wang, Y. Pan, H. Ning, H. Wang, D. Guo, W. Wang, Z. Yang, Q. Zhao, B. Zhang, L. Zheng, J. Zhang and M. Wu, Appl. Catal., B, 2020, 266, 118630 CrossRef CAS.
- K. K. Patra, S. Park, H. Song, B. Kim, W. Kim and J. Oh, ACS Appl. Energy Mater., 2020, 3, 11343–11349 CrossRef CAS.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. Gui, L. Li, B. Yu, D. Wang, B. Yang, Q. Gu, Y. Zhao, Y. Zhu and Y. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 25516–25523 CrossRef CAS PubMed.
- W. Rong, H. Zou, W. Zang, S. Xi, S. Wei, B. Long, J. Hu, Y. Ji and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 466–472 CrossRef CAS PubMed.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- J. Dong, Y. Liu, J. Pei, H. Li, S. Ji, L. Shi, Y. Zhang, C. Li, C. Tang, J. Liao, S. Xu, H. Zhang, Q. Li and S. Zhao, Nat. Commun., 2023, 14, 6849 CrossRef CAS PubMed.
- Z.-Q. Liang, T.-T. Zhuang, A. Seifitokaldani, J. Li, C.-W. Huang, C.-S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P.-L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S.-C. Lo, L.-J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed.
- C. Peng, G. Luo, Z. Xu, S. Yan, J. Zhang, M. Chen, L. Qian, W. Wei, Q. Han and G. Zheng, Adv. Mater., 2021, 33, 2103150 CrossRef CAS PubMed.
- C. A. Downes, N. J. Libretto, A. E. Harman-Ware, R. M. Happs, D. A. Ruddy, F. G. Baddour, J. R. Ferrell III, S. E. Habas and J. A. Schaidle, ACS Appl. Energy Mater., 2020, 3, 10435–10446 CrossRef CAS PubMed.
- H. Chen, Z. Wang, X. Wei, S. Liu, P. Guo, P. Han, H. Wang, J. Zhang, X. Lu and B. Wei, Appl. Surf. Sci., 2021, 544, 148965 CrossRef CAS.
- X. Kong, C. Wang, H. Zheng, Z. Geng, J. Bao and J. Zeng, Sci. China: Chem., 2021, 64, 1096–1102 CrossRef CAS.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2018, 57, 551–554 CrossRef CAS PubMed.
- A. M. Asiri, J. Gao, S. B. Khan, K. A. Alamry, H. M. Marwani, M. S. J. Khan, W. A. Adeosun, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Phys. Chem. Lett., 2022, 13, 345–351 CrossRef CAS PubMed.
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T.-K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed.
- T.-T. Zhuang, Z.-Q. Liang, A. Seifitokaldani, Y. Li, P. De Luna, T. Burdyny, F. Che, F. Meng, Y. Min, R. Quintero-Bermudez, C. T. Dinh, Y. Pang, M. Zhong, B. Zhang, J. Li, P.-N. Chen, X.-L. Zheng, H. Liang, W.-N. Ge, B.-J. Ye, D. Sinton, S.-H. Yu and E. H. Sargent, Nat. Catal., 2018, 1, 421–428 CrossRef CAS.
- S. Li, H. Duan, J. Yu, C. Qiu, R. Yu, Y. Chen, Y. Fang, X. Cai and S. Yang, ACS Catal., 2022, 12, 9074–9082 CrossRef CAS.
- J. Liu, P. Li, J. Bi, Y. Wang, Q. Zhu, X. Sun, J. Zhang, Z. Liu and B. Han, Chin. J. Chem., 2023, 41, 1443–1449 CrossRef CAS.
- X. Su, Z. Jiang, J. Zhou, H. Liu, D. Zhou, H. Shang, X. Ni, Z. Peng, F. Yang, W. Chen, Z. Qi, D. Wang and Y. Wang, Nat. Commun., 2022, 13, 1322 CrossRef CAS PubMed.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- H. Wang, X. Bi, Y. Yan, Y. Zhao, Z. Yang, H. Ning and M. Wu, Adv. Funct. Mater., 2023, 33, 2214946 CrossRef CAS.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- J. Duan, T. Liu, Y. Zhao, R. Yang, Y. Zhao, W. Wang, Y. Liu, H. Li, Y. Li and T. Zhai, Nat. Commun., 2022, 13, 2039 CrossRef CAS PubMed.
- A. Saxena, W. Liyanage, J. Masud, S. Kapila and M. Nath, J. Mater. Chem. A, 2021, 9, 7150–7161 RSC.
- S. Das, S. Senapati, G. K. Pradhan, S. Varadharajanperumal and R. Naik, ACS Appl. Nano Mater., 2023, 6, 5298–5312 CrossRef CAS.
- Z. Gu, N. Yang, P. Han, M. Kuang, B. Mei, Z. Jiang, J. Zhong, L. Li and G. Zheng, Small Methods, 2019, 3, 1800449 CrossRef.
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard III and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS.
- M. Li, Y. Ma, J. Chen, R. Lawrence, W. Luo, M. Sacchi, W. Jiang and J. Yang, Angew. Chem., Int. Ed., 2021, 60, 11487–11493 CrossRef CAS PubMed.
- S. Lee, D. Kim and J. Lee, Angew. Chem., Int. Ed., 2015, 54, 14701–14705 CrossRef CAS PubMed.
- T. Kim and G. T. R. Palmore, Nat. Commun., 2020, 11, 3622 CrossRef CAS PubMed.
- H. Mistry, A. S. Varela, C. S. Bonifacio, I. Zegkinoglou, I. Sinev, Y.-W. Choi, K. Kisslinger, E. A. Stach, J. C. Yang, P. Strasser and B. R. Cuenya, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- J. Fu, H. Bao, Y. Liu, Y. Mi, Y. Qiu, L. Zhuo, X. Liu and J. Luo, Small, 2020, 16, 1905825 CrossRef CAS PubMed.
- X. Rong, H.-J. Wang, X.-L. Lu, R. Si and T.-B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed.
- X. Yang, P. Deng, D. Liu, S. Zhao, D. Li, H. Wu, Y. Ma, B. Y. Xia, M. Li, C. Xiao and S. Ding, J. Mater. Chem. A, 2020, 8, 2472–2480 RSC.
- Y. Ma, M. Chen, H. Geng, H. Dong, P. Wu, X. Li, G. Guan and T. Wang, Adv. Funct. Mater., 2020, 30, 2000561 CrossRef CAS.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- T. Gao, A. Kumar, Z. Shang, X. Duan, H. Wang, S. Wang, S. Ji, D. Yan, L. Luo, W. Liu and X. Sun, Chin. Chem. Lett., 2019, 30, 2274–2278 CrossRef CAS.
- L. Cheng, Y. Li, A. Chen, Y. Zhu and C. Li, Chem. Commun., 2020, 56, 563–566 RSC.
- S. B. Varandili, J. Huang, E. Oveisi, G. L. De Gregorio, M. Mensi, M. Strach, J. Vavra, C. Gadiyar, A. Bhowmik and R. Buonsanti, ACS Catal., 2019, 9, 5035–5046 CrossRef CAS.
- W. Zhu, L. Zhang, P. Yang, C. Hu, H. Dong, Z.-J. Zhao, R. Mu and J. Gong, ACS Energy Lett., 2018, 3, 2144–2149 CrossRef CAS.
- X. Ren, X. Zhang, X. Cao and Q. Wang, J. CO2 Util., 2020, 38, 125–131 CrossRef CAS.
- D. Wu, G. Huo, W. Chen, X.-Z. Fu and J.-L. Luo, Appl. Catal., B, 2020, 271, 118957 CrossRef CAS.
- H. He, K. Liu, K. Liang, A. Mustapha, Z. Wang, L. Wu, C. Yang, L. Deng, S. Guo and Y.-N. Liu, J. Catal., 2020, 385, 246–254 CrossRef CAS.
- A. Yoon, J. Poon, P. Grosse, S. W. Chee and B. R. Cuenya, J. Mater. Chem. A, 2022, 10, 14041–14050 RSC.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- R. Cai, M. Sun, J. Ren, M. Ju, X. Long, B. Huang and S. Yang, Chem. Sci., 2021, 12, 15382–15388 RSC.
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang and B. Han, Nat. Commun., 2022, 13, 1965 CrossRef CAS PubMed.
- X. Tan, C. Yu, C. Zhao, H. Huang, X. Yao, X. Han, W. Guo, S. Cui, H. Huang and J. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 9904–9910 CrossRef CAS PubMed.
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef.
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS.
- J. Wu, F. G. Risalvato, S. Ma and X.-D. Zhou, J. Mater. Chem. A, 2014, 2, 1647–1651 RSC.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong, B. Wu and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS PubMed.
- S. Huo, Z. Weng, Z. Wu, Y. Zhong, Y. Wu, J. Fang and H. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 28519–28526 CrossRef CAS PubMed.
- K. Wang, D. Liu, P. Deng, L. Liu, S. Lu, Z. Sun, Y. Ma, Y. Wang, M. Li, B. Y. Xia, C. Xiao and S. Ding, Nano Energy, 2019, 64, 103954 CrossRef CAS.
- X. Bai, J. Han, S. Chen, X. Niu and J. Guan, Chin. J. Catal., 2023, 54, 212–219 CrossRef CAS.
- X. Bai, J. Han, X. Niu and J. Guan, Nano Res., 2023, 16, 10796–10802 CrossRef CAS.
- Q. Wang, K. Liu, J. Fu, C. Cai, H. Li, Y. Long, S. Chen, B. Liu, H. Li, W. Li, X. Qiu, N. Zhang, J. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- X. Li, S. Xi, L. Sun, S. Dou, Z. Huang, T. Su and X. Wang, Adv. Sci., 2020, 7, 2001545 CrossRef CAS PubMed.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- X. Chen, D.-D. Ma, B. Chen, K. Zhang, R. Zou, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- T. Tang, Y. Wang, J. Han, Q. Zhang, X. Bai, X. Niu, Z. Wang and J. Guan, Chin. J. Catal., 2023, 46, 48–55 CrossRef CAS.
- J. Han and J. Guan, Chin. J. Catal., 2023, 47, 1–31 CrossRef CAS.
- T. Tang, Z. Duan, D. Baimanov, X. Bai, X. Liu, L. Wang, Z. Wang and J. Guan, Nano Res., 2023, 16, 2218–2223 CrossRef CAS.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, J. Am. Chem. Soc., 2019, 141, 16635–16642 CrossRef CAS.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS PubMed.
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal., B, 2020, 268, 118747 CrossRef CAS.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- T. Ding, X. Liu, Z. Tao, T. Liu, T. Chen, W. Zhang, X. Shen, D. Liu, S. Wang, B. Pang, D. Wu, L. Cao, L. Wang, T. Liu, Y. Li, H. Sheng, M. Zhu and T. Yao, J. Am. Chem. Soc., 2021, 143, 11317–11324 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H.-S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS.
- Y. Li, B. Wei, M. Zhu, J. Chen, Q. Jiang, B. Yang, Y. Hou, L. Lei, Z. Li, R. Zhang and Y. Lu, Adv. Mater., 2021, 33, 2102212 CrossRef CAS PubMed.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Z. Pei, H. Zhang, Z.-P. Wu, X. Feng Lu, D. Luan and X. W. D. Lou, Sci. Adv., 2023, 9, eadh1320 CrossRef CAS PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS PubMed.
- D. Yao, C. Tang, X. Zhi, B. Johannessen, A. Slattery, S. Chern and S. Z. Qiao, Adv. Mater., 2023, 35, 2209386 CrossRef CAS PubMed.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef PubMed.
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef CAS.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y.-Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS.
- Y. Wang, Z. Chen, P. Han, Y. Du, Z. Gu, X. Xu and G. Zheng, ACS Catal., 2018, 8, 7113–7119 CrossRef CAS.
- J.-C. Jiang, J.-C. Chen, M.-D. Zhao, Q. Yu, Y.-G. Wang and J. Li, Nano Res., 2022, 15, 7116–7123 CrossRef CAS.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C.-W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- H. Shen, T. Wang, H. Jiang, P. Zhao, Z. Chen, Y. Feng, Y. Cao, Y. Guo, Q. Zhang and H. Zhang, Appl. Catal., B, 2023, 339, 123140 CrossRef CAS.
- D. Wang, R. Cao, S. Hao, C. Liang, G. Chen, P. Chen, Y. Li and X. Zou, Green Energy Environ., 2023, 8, 820–830 CrossRef CAS.
- W. Wu, J. Zhu, Y. Tong, S. Xiang and P. Chen, Nano Res., 2024, 17, 3684–3692 CrossRef CAS.
- M. G. Kim, J. Park, Y. Choi, H. C. Song, S. H. Kim, K. M. Bang, H. C. Ham, N. K. Kim, D. H. Won, B. K. Min, S. J. Yoo and W. Kim, Adv. Energy Mater., 2023, 13, 2300749 CrossRef CAS.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 CrossRef CAS PubMed.
- Y. Chen, N. Lyu, J. Zhang, S. Yan, C. Peng, C. Yang, X. Lv, C. Hu, M. Kuang and G. Zheng, Small, 2023, 2308004, DOI:10.1002/smll.202308004.
- L. Xiao, Z. Wang and J. Guan, Chem. Sci., 2023, 14, 12850–12868 RSC.
- S. Nellaiappan, N. K. Katiyar, R. Kumar, A. Parui, K. D. Malviya, K. G. Pradeep, A. K. Singh, S. Sharma, C. S. Tiwary and K. Biswas, ACS Catal., 2020, 10, 3658–3663 CrossRef CAS.
- J. Cavin, A. Ahmadiparidari, L. Majidi, A. S. Thind, S. N. Misal, A. Prajapati, Z. Hemmat, S. Rastegar, A. Beukelman, M. R. Singh, K. A. Unocic, A. Salehi-Khojin and R. Mishra, Adv. Mater., 2021, 33, 2100347 CrossRef CAS PubMed.
- D. Roy, S. C. Mandal and B. Pathak, J. Phys. Chem. Lett., 2022, 13, 5991–6002 CrossRef CAS PubMed.
- H. Tian, T. Wang, F. Zhang, S. Zhao, S. Wan, F. He and G. Wang, J. Mater. Chem. A, 2018, 6, 12816–12841 RSC.
- Z. Zhang, Z. P. Cano, D. Luo, H. Dou, A. Yu and Z. Chen, J. Mater. Chem. A, 2019, 7, 20985–21003 RSC.
- M. K. Aslam, K. Yang, S. Chen, Q. Li and J. Duan, EES Catal., 2023, 1, 179–229 RSC.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- J. Li, H. Luo, B. Li, J.-G. Ma and P. Cheng, Mater. Chem. Front., 2023, 7, 6107–6129 RSC.
- Z. Li, M. Song, W. Zhu, W. Zhuang, X. Du and L. Tian, Coord. Chem. Rev., 2021, 439, 213946 CrossRef CAS.
- T. A. Al-Attas, N. N. Marei, X. Yong, N. G. Yasri, V. Thangadurai, G. Shimizu, S. Siahrostami and M. G. Kibria, ACS Catal., 2021, 11, 7350–7357 CrossRef CAS.
- Y. Guo, H. Yang, X. Zhou, K. Liu, C. Zhang, Z. Zhou, C. Wang and W. Lin, J. Mater. Chem. A, 2017, 5, 24867–24873 RSC.
- Y. Ye, F. Cai, H. Li, H. Wu, G. Wang, Y. Li, S. Miao, S. Xie, R. Si, J. Wang and X. Bao, Nano Energy, 2017, 38, 281–289 CrossRef CAS.
- C. Hu, S. Bai, L. Gao, S. Liang, J. Yang, S.-D. Cheng, S.-B. Mi and J. Qiu, ACS Catal., 2019, 9, 11579–11588 CrossRef CAS.
- C. W. Lee, K. D. Yang, D. H. Nam, J. H. Jang, N. H. Cho, S. W. Im and K. T. Nam, Adv. Mater., 2018, 30, 1704717 CrossRef PubMed.
- J. Wang, Y. Chen, S. Zhang, C. Yang, J. Y. Zhang, Y. Su, G. Zheng and X. Fang, Small, 2022, 18, 2202238 CrossRef CAS PubMed.
- W. Xiong, D. Si, J. Yi, Y. Huang, H. Li and R. Cao, Appl. Catal., B, 2022, 314, 121498 CrossRef CAS.
- Y. J. Jang, J. Lee, J. H. Kim, B. J. Lee and J. S. Lee, J. Power Sources, 2018, 378, 412–417 CrossRef CAS.
- X. Ma, J. Tian, M. Wang, X. Jin, M. Shen and L. Zhang, Catal. Sci. Technol., 2021, 11, 6096–6102 RSC.
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- L. Li, Z. Jiang, Y. Li, F. Li, Y. Pan, X. Zhang, Y. Liang and Z. Zheng, Small Methods, 2022, 7, 2201213 CrossRef PubMed.
- Q.-H. Zheng, C. Chen, S.-M. Cao, M.-T. Peng, B.-X. Dong and Y.-L. Teng, Chin. Chem. Lett., 2023, 34, 107273 CrossRef CAS.
- I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha and J. T. Hupp, ACS Catal., 2015, 5, 6302–6309 CrossRef CAS.
- C.-W. Kung, C. O. Audu, A. W. Peters, H. Noh, O. K. Farha and J. T. Hupp, ACS Energy Lett., 2017, 2, 2394–2401 CrossRef CAS.
- P. C. Adrien, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- L. Xiao, Z. Wang and J. Guan, Adv. Funct. Mater., 2024, 34, 2310195 CrossRef CAS.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- Y. Yue, P. Cai, K. Xu, H. Li, H. Chen, H.-C. Zhou and N. Huang, J. Am. Chem. Soc., 2021, 143, 18052–18060 CrossRef CAS PubMed.
- X.-F. Qiu, J.-R. Huang, C. Yu, Z.-H. Zhao, H.-L. Zhu, Z. Ke, P.-Q. Liao and X.-M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202206470 CrossRef CAS PubMed.
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC.
- M. Liu, S. Liu, Q. Xu, Q. Miao, S. Yang, S. Hanson, G. Z. Chen, J. He, Z. Jiang and G. Zeng, Carbon Energy, 2023, 5, 1–12 Search PubMed.
- S. B. Alahakoon, K. Tan, H. Pandey, S. D. Diwakara, G. T. McCandless, D. I. Grinffiel, A. Durand-Silva, T. Thonhauser and R. A. Smaldone, J. Am. Chem. Soc., 2020, 142, 12987–12994 CrossRef CAS PubMed.
- M. Li, B. Han, S. Li, Q. Zhang, E. Zhang, L. Gong, D. Qi, K. Wang and J. Jiang, Small, 2024, 2310147, DOI:10.1002/smll.202310147.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- R. Matheu, E. Gutierrez-Puebla, M. A. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. K. Pei, N. Hanikel, B. Zhang, P. D. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 17081–17085 CrossRef CAS PubMed.
- B. Han, Y. Jin, B. Chen, W. Zhou, B. Yu, C. Wei, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202114244 CrossRef CAS PubMed.
- S.-Y. Chi, Q. Chen, S.-S. Zhao, D.-H. Si, Q.-J. Wu, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2022, 10, 4653–4659 RSC.
- H. Shin, K. U. Hansen and F. Jiao, Nat. Sustain., 2021, 4, 911–919 CrossRef.
- W. Lai, Y. Qiao, J. Zhang, Z. Lin and H. Huang, Energy Environ. Sci., 2022, 15, 3603–3629 RSC.
- I. E. L. Stephens, K. Chan, A. Bagger, S. W. Boettcher, J. Bonin, E. Boutin, A. K. Buckley, R. Buonsanti, E. R. Cave, X. Chang, S. W. Chee, A. H. M. da Silva, P. de Luna, O. Einsle, B. Endrődi, M. Escudero-Escribano, J. V. Ferreira de Araujo, M. C. Figueiredo, C. Hahn, K. U. Hansen, S. Haussener, S. Hunegnaw, Z. Huo, Y. J. Hwang, C. Janáky, B. S. Jayathilake, F. Jiao, Z. P. Jovanov, P. Karimi, M. T. M. Koper, K. P. Kuhl, W. H. Lee, Z. Liang, X. Liu, S. Ma, M. Ma, H.-S. Oh, M. Robert, B. R. Cuenya, J. Rossmeisl, C. Roy, M. P. Ryan, E. H. Sargent, P. Sebastián-Pascual, B. Seger, L. Steier, P. Strasser, A. S. Varela, R. E. Vos, X. Wang, B. Xu, H. Yadegari and Y. Zhou, JPhys Energy, 2022, 4, 042003 CrossRef CAS.
- C. Ampelli, F. Tavella, D. Giusi, A. M. Ronsisvalle, S. Perathoner and G. Centi, Catal. Today, 2023, 421, 114217 CrossRef CAS.
- N. Fujinuma, A. Ikoma and S. E. Lofland, Adv. Energy Mater., 2020, 10, 2001645 CrossRef CAS.
- S. Liang, N. Altaf, L. Huang, Y. Gao and Q. Wang, J. CO2 Util., 2020, 35, 90–105 CrossRef CAS.
- Q. Chen, X. Wang, Y. Zhou, Y. Tan, H. Li, J. Fu and M. Liu, Adv. Mater., 2023, 36, 2303902 CrossRef PubMed.
- D. A. Salvatore, C. M. Gabardo, A. Reyes, C. P. O'Brien, S. Holdcroft, P. Pintauro, B. Bahar, M. Hickner, C. Bae, D. Sinton, E. H. Sargent and C. P. Berlinguette, Nat. Energy, 2021, 6, 339–348 CrossRef CAS.
- D. Gao, P. Wei, H. Li, L. Lin, G. Wang and X. Bao, Acta Phys.–Chim. Sin., 2020, 2009021 Search PubMed.
- Y. Meng, H. Huang, Y. Zhang, Y. Cao, H. Lu and X. Li, Front. Chem., 2023, 11, 1172146 CrossRef CAS PubMed.
- H. Cao, Z. Zhang, J.-W. Chen and Y.-G. Wang, ACS Catal., 2022, 12, 6606–6617 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |