
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe third strategy: modulating emission colors of organic light-emitting diodes with UV light during the device fabrication process†
Yuanhui
Sun
 a,
Shipan
Xu
a,
Huaiteng
Hang
a,
Jun
Xi
b,
Hua
Dong
b,
Bo
Jiao
b,
Guijiang
Zhou
*a and
Xiaolong
Yang
*a
a,
Shipan
Xu
a,
Huaiteng
Hang
a,
Jun
Xi
b,
Hua
Dong
b,
Bo
Jiao
b,
Guijiang
Zhou
*a and
Xiaolong
Yang
*a
aSchool of Chemistry, Xi'an Key Laboratory of Sustainable Energy Material Chemistry, Engineering Research Center of Energy Storage Materials and Devices, Ministry of Education, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: zhougj@xjtu.edu.cn; xiaolongyang@xjtu.edu.cn
bSchool of Electronic Science and Engineering, Xi'an Jiaotong University, Xi'an 710049, China
First published on 30th April 2024
Abstract
The modulation of emission color is one of the most critical topics in the research field of organic light-emitting diodes (OLEDs). Currently, only two ways are commonly used to tune the emission colors of OLEDs: one is to painstakingly synthesize different emitters with diverse molecular structures, the other is to precisely control the degree of aggregation or doping concentration of one emitter. To develop a simpler and less costly method, herein we demonstrate a new strategy in which the emission colors of OLEDs can be continuously changed with UV light during the device fabrication process. The proof of concept is established by a chromene-based Ir(III) complex, which shows bright green emission and yellow emission before and after UV irradiation, respectively. Consequently, under different durations of UV irradiation, the resulting Ir(III) complex is successfully used as the emitter to gradually tune the emission colors of related solution-processed OLEDs from green to yellow. Furthermore, the electroluminescent efficiencies of these devices are unaffected or even increased during this process. Therefore, this work demonstrates a distinctive point of view and approach for modulating the emission colors of OLEDs, which may prove great inspiration for the fabrication of multi-colored OLEDs with only one emitter.
Introduction
Organic light-emitting diodes (OLEDs) have been widely regarded as the next generation of display technology because devices can be flexible, transparent, and even stretchable.1–4 In addition, OLED displays can exhibit an impressively high resolution and large color gamut.5–9 Therefore, modulation of the emission colors of OLEDs is of great importance, but to date it has generally been limited to two ways. The most common way is to use different emitters with diverse molecular structures, which can emit different colors of light; thus, a lot of chemical structure design and synthesis work need to be done.10–15 For example, by changing the structures of cyclometalating ligands, the emission colors of related Ir(III) complexes can be tuned from blue to near infrared.16–21 Another less used way is to utilize the aggregation or concentration effects of one emitter, i.e., doping the same emitter at diverse concentrations, but this needs precise control over the degree of aggregation or concentration.22–25 It is apparent that both methods are somewhat inconvenient for manipulating the emission colors of OLEDs.Herein, we propose a novel strategy to modulate the emission colors of OLEDs without the need for designing and synthesizing different emitters or relying on molecular aggregation or concentration effects. Therefore, this approach, which marks the third strategy, differs from the conventional two and has been effectively demonstrated using our newly developed chromene-based Ir(III) complex that exhibited impressive photochromic performance. The chromene-based Ir(III) complex showed efficient green emission with wavelength peak (λem) at 523 nm and a high photoluminescence quantum yield (PLQY) of 0.54 in CH2Cl2 solution. After irradiation with 365 nm UV light, the emission color of the solution gradually switched to pure yellow of λem = 554 nm with a PLQY of 0.46. In addition, the yellow emission intensity showed good stability. Therefore, solutions containing the chromene-based Ir(III) complex and 4,4′-bis(9-carbazolyl)biphenyl (CBP) were first irradiated with 365 nm UV light for different durations, and then, OLEDs with a simple conventional device structure could be fabricated through the solution-processed method by spin-coating UV-irradiated solutions to form emissive layers. As expected, resultant OLEDs exhibited emission colors that changed seamlessly from green to pure yellow, along with even enhanced efficiencies. The highest EQE of the yellow OLED based on the UV-irradiated complex reached 15.64%, which was an increase of ca. 50% compared with that of the device based on the complex without UV irradiation. Unambiguously, these results demonstrated the success of our strategy to modulate the emission colors of solution-processed OLEDs with UV light during the device fabrication process (Scheme 1).
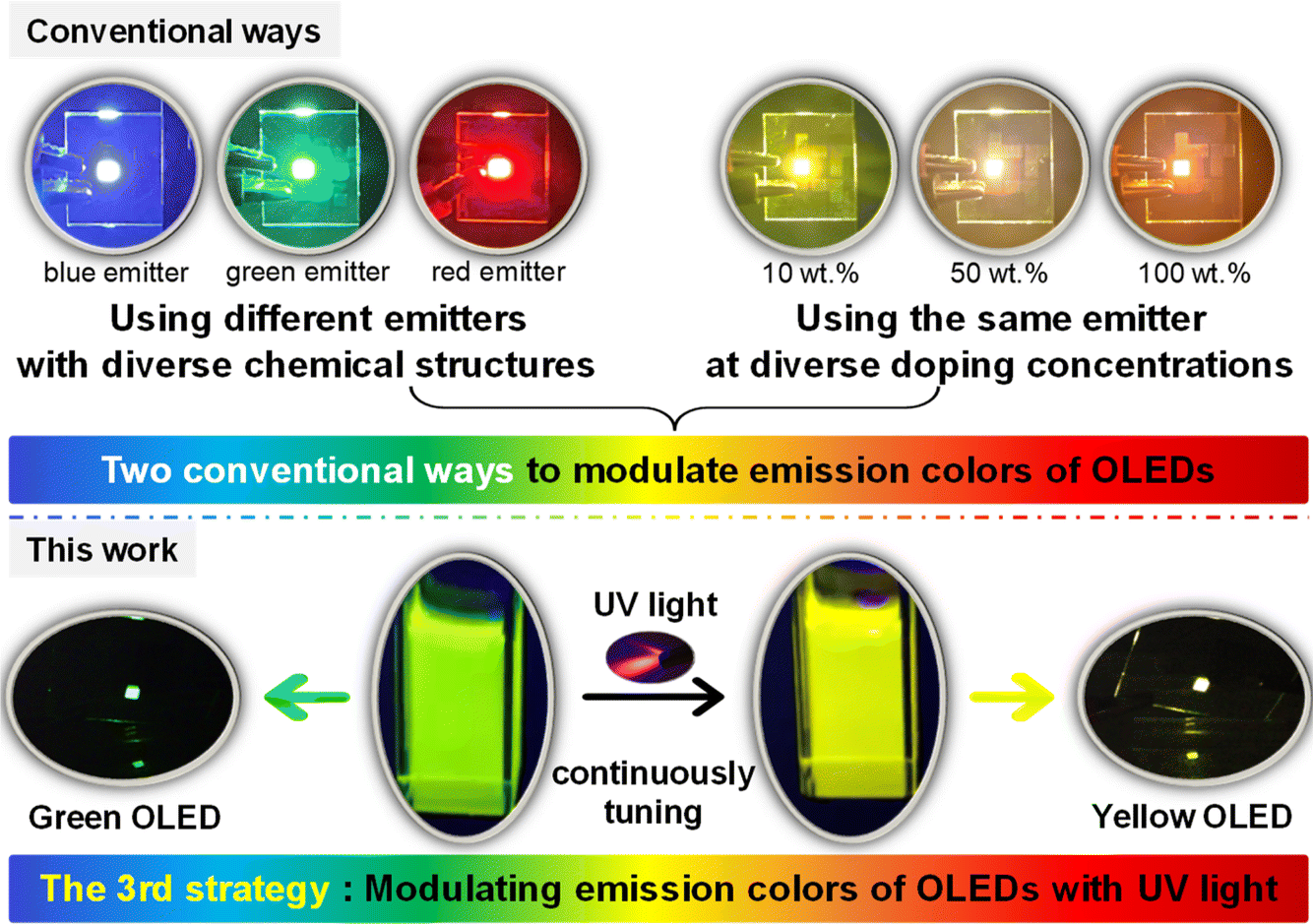 | ||
| Scheme 1 Strategies to modulate the emission colors of OLEDs. | ||
Results and discussion
To change the emission colors of OLEDs without using different emitters or using one emitter at diverse doping concentrations, we thought of using external stimuli to change the emission color of the emitters. It has been reported that stimuli such as solvents, heat, force or light could change the emission colors of specific emitters.26–30 However, these external stimuli work well only when the emitters are in solution or solid state (powder or doped film). Because of the sandwich-like characteristics of the solid-state structure of OLEDs, neither solvents nor external forces are present, and the devices are also unable to withstand external heating. Consequently, these external stimuli will undoubtedly become ineffective in OLEDs or during the device fabrication process. External light may penetrate the transparent anode of a bottom-emitting OLED or transparent cathode of a top-emitting OLED to change the emission colors of the emitters, but this will not work well because the process of change in the solid state is usually very slow, and the external light intensity will be attenuated due to the multi-layer structure of the device. Therefore, we realized that we could manipulate the emission color of the emitter before integrating it into a device. Then, we completed the device fabrication process using the emitter with the already altered emission color.To find an emitter which met the needs of the above-mentioned strategy, we examined and synthesized lots of photochromic materials, including organic molecules and organometallic complexes based on dithienylethenes, spiropyrans, and azobenzenes.31,32 However, according to the literature, photochromic materials typically exhibit quenching behavior upon UV light irradiation or have very low PLQY values.33 Therefore, this could be one reason why no studies to date have reported OLEDs based on these photochromic materials.34,35 Finally, we noticed that chromene (benzopyran) derivatives could show photochromic properties due to UV-promoted pyran ring-opening behavior.36–40 However, currently reported chromene-based photochromic materials are pure organic small molecules or polymers, which generally show very fast (over a few seconds per minutes) and reversible color change back to their initial state in the dark as well as in daylight.36–40 This property is good for applications in some fields such as photochromic lenses, but it is extremely unfavorable for preparing OLEDs because the emitter should be settled to keep the performance of a stable, including the emission color. In addition, these chromene-based pure organic small molecules or polymers commonly show very low PLQYs and exhibit serious quenching behavior under UV irradiation. Therefore, we considered that the heavy-metal Ir(III) ion could be used to optimize photochromic properties. As a result, the photochromic behavior of this chromene-based Ir(III) complex was in sharp contrast to other photochromic organometallic complexes, which usually displayed strong quenching after UV irradiation.33 Furthermore, the bright yellow emission could last over 3600 s in dark and daylight conditions, indicating the greatly improved stability of the chromene-based Ir(III) complex after UV irradiation. The impressively high stability was very favorable for the preparation of OLEDs.
The preparation of the chromene-based Ir(III) complex chromene–Ir-CF was started from the synthesis of the bromo-substituted chromene unit chromene–Br (Schemes 2 and S1 in the ESI†).41 Then, a pyridine ring was introduced to the chromene unit through a Stille cross-coupling reaction to obtain the cyclometalating ligand chromene–L. Finally, the desired photochromic cyclometalated Ir(III) complex was synthesized in a classical two-step procedure via a cyclometalated Ir(III) μ-chloro-bridged dimer.42 The chromene units in the freshly obtained complex chromene–Ir-CF were in closed form (CF). Under UV irradiation, the chromene unit will go through a ring-open reaction to give the complex in open form (OF). The chemical structure of chromene–Ir-CF was confirmed by the high-resolution mass spectrum and NMR spectroscopy (Fig. S1 and S2†).
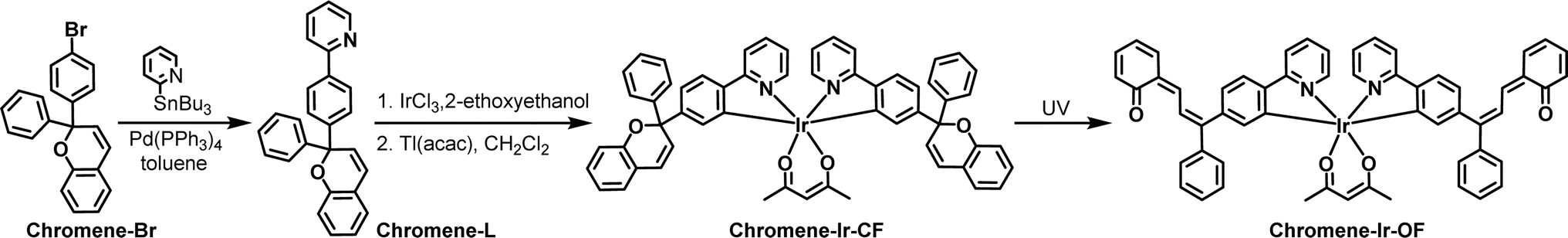 | ||
| Scheme 2 Synthesis of chromene–Ir-CF and chromene–Ir-OF. | ||
The thermal stability of chromene–Ir-CF was investigated by thermal gravimetric analysis (TGA), which revealed that the decomposition temperature (Td) was ca. 301 °C (Fig. S3†). The electrochemical property of chromene–Ir-CF was studied by cyclic voltammetry (CV) in acetonitrile solution, which exhibited only one oxidation peak at a potential of ca. 0.43 V against a ferrocene/ferrocenium (Fc/Fc+) couple (Fig. S4†). Therefore, the energy level of the highest occupied molecular orbital (HOMO) was estimated to be −5.23 eV. No clear reduction process for chromene–Ir-CF was observed during the cathodic scan; thus, the energy level of its lowest unoccupied molecular orbital (LUMO) was tentatively determined from EHOMO and the optical band gap (Egap) to be −2.75 eV.
As depicted in Fig. 1a, the UV-vis absorption of chromene–Ir-CF in CH2Cl2 solution displayed two main bands. The strong absorption with a wavelength shorter than 350 nm could be assigned to the singlet ligand centered π–π* transition, while the weak absorption around 468 nm could be attributed to a mixture of the triplet ligand centered π–π* transition and metal-to-ligand charge transfer (MLCT) transition.42 This attribution was supported by theoretical calculation. As the calculation result suggested, the electron density of the HOMO was located mainly on the Ir(III) center and two phenyl rings chelated to the Ir(III) center, while the electron density of the LUMO was contributed mainly by the phenyl pyridinyl segment of one cyclometalating ligand (Fig. S5†). When the CH2Cl2 solution of chromene–Ir-CF was irradiated with 365 nm UV light, the UV-vis absorption was red-shifted and a new absorption band at longer wavelength appeared quickly (Fig. 1a). This photochromic phenomenon could be attributed to the ring-open behavior of the chromene units in chromene–Ir-CF, resulting in the opened form of the chromene-based Ir(III) complex, chromene–Ir-OF.39 As theoretical calculation results revealed, besides the notable contribution from the Ir(III) center and two phenyl rings chelated to the Ir(III) center, the HOMO of chromene–Ir-OF consisted of a significant contribution from one of the ring-opened chromene segments, i.e., the 6-(buta-1,3-dien-1-yl)cyclohexa-2,4-dien-1-one moiety (Fig. S5†), indicating that this group had a certain electron-donating property. In addition, unlike that of chromene–Ir-CF, the LUMO distribution of chromene–Ir-OF was dominated by the other ring-opened chromene segments, and the contribution to the LUMO from two pyridyl rings was negligible, which suggested the strong electron-accepting ability of the 6-(buta-1,3-dien-1-yl)cyclohexa-2,4-dien-1-one moiety. Therefore, the low energy absorption of chromene–Ir-OF should result mainly from the MLCT process mixed with some ligand-to-ligand charge transfer (LLCT) transition.
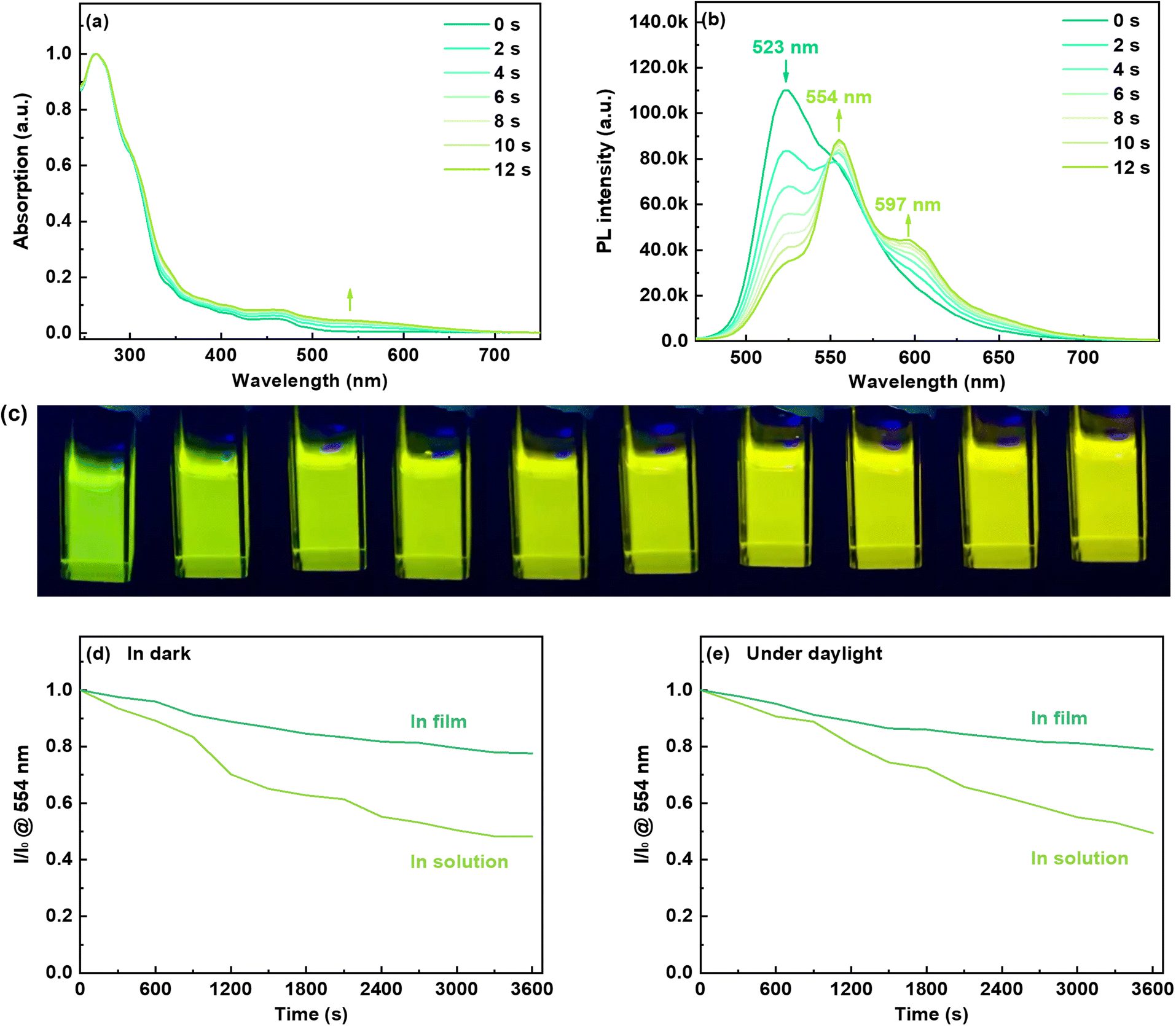 | ||
| Fig. 1 Changes in (a) absorption and (b) emission of chromene–Ir-CF in CH2Cl2 at r.t. before and after 365 nm UV light irradiation. (c) Photographs of emissions of chromene–Ir-CF in CH2Cl2 at r.t. during irradiation by 365 nm UV light. The changes in emission intensity at 554 nm as a function of time (d) in dark and (e) under daylight (I0 is the initial intensity for chromene–Ir-CF in doped CBP film or CH2Cl2 solution at r.t. after 365 nm UV light irradiation, and I is the intensity at different times after 365 nm UV light irradiation). | ||
Excited with 365 nm UV light at room temperature (r.t.), a CH2Cl2 solution of chromene–Ir-CF displayed bright green emission with a peak at 523 nm (Fig. 1b). The observed emission lifetime (τp) was 0.09 μs (Table 1), and the emission intensity could be enhanced by purging the CH2Cl2 solution with N2, indicating a triplet characteristic. Theoretical investigation of natural transition orbitals (NTO) revealed that the hole-to-particle orbital transition made a 94.5% contribution to the lowest triplet state. The hole orbital was mainly contributed by the Ir(III) center and two phenyl rings chelated to the Ir(III) center with a small contribution from one of the pyridyl rings, while the particle orbital was mainly contributed by the phenyl pyridinyl segment of this one cyclometalating ligand (Fig. 2). The chromene units made a negligible contribution to either hole or particle orbitals. Therefore, the phosphorescent emission of chromene–Ir-CF originated mainly from a mixture of 3MLCT and 3π–π* transitions. Upon UV irradiation, the emission intensity at 523 nm observed for chromene–Ir-CF in the CH2Cl2 solution dropped rapidly, while new emission bands with peaks at 554 and 597 nm appeared and were enhanced at the same time (Fig. 1b). Based on changes in the emission intensity of the PL spectrum at 523 nm with different UV irradiation times, the photochromic kinetics of chromene–Ir-CF could be described by the monoexponential function y = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−x/t1) + y0, in which A1 = 0.78, t1 = 5.9, and y0 = 0.21 with R2 = 0.998 (Fig. S6†). Therefore, the photochromic rate of chromene–Ir-CF was determined to be 0.169 s−1.
exp(−x/t1) + y0, in which A1 = 0.78, t1 = 5.9, and y0 = 0.21 with R2 = 0.998 (Fig. S6†). Therefore, the photochromic rate of chromene–Ir-CF was determined to be 0.169 s−1.
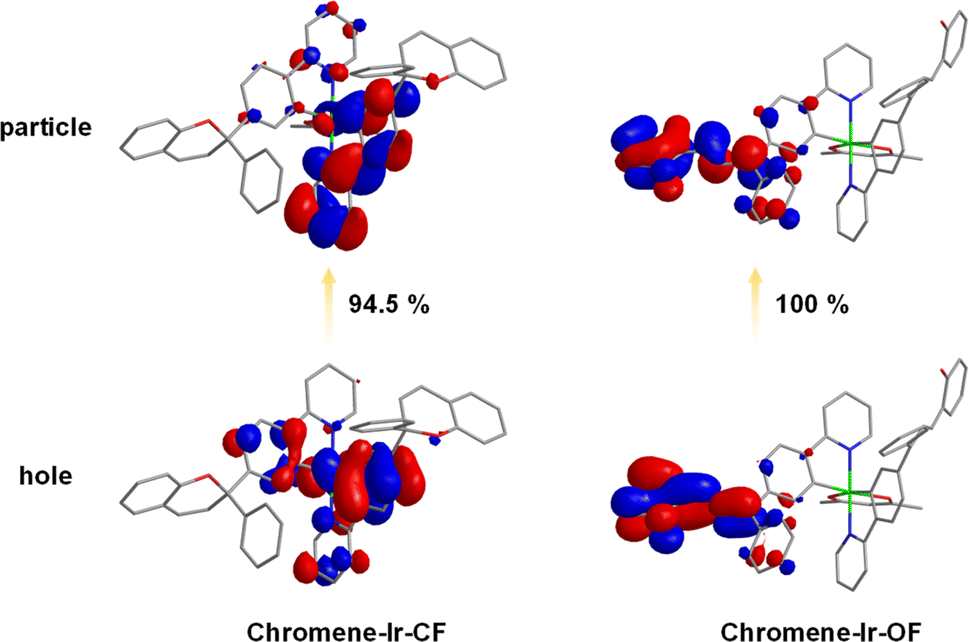 | ||
| Fig. 2 Calculated NTO distributions of hole and particle orbitals based on optimized T1 geometries. | ||
It is worth noting that mass spectra of chromene–Ir-CF before and after UV light irradiation displayed strong (M + Na)+m/z signals of ca. 1035.37 (Fig. S1†), which demonstrated that no degradation reaction occurred during the switch from chromene–Ir-CF to chromene–Ir-OF. The interesting change in emission color from green to yellow (Fig. 1c) was also in good agreement with the red-shift of the UV-vis absorption. Besides, τp for this yellow emission was 0.32 μs, which was notably longer than that of chromene–Ir-CF. In addition, the emission peak of chromene–Ir-OF was only slightly blue-shifted by 4 nm when the temperature was cooled from r.t. to 77 K (λem = 550 nm at 77 K vs. λem = 554 nm at r.t.) (Fig. S7†), while a larger blue-shift of 11 nm was recorded for chromene–Ir-CF (λem = 512 nm at 77 K vs. λem = 523 nm at r.t.). To gain an insight into the results, an NTO theoretical investigation was performed for the T1 state. The calculation results showed that the ring-opened chromene segment of one cyclometalating ligand made dominant contributions to hole and particle orbitals, while the Ir(III) center and phenyl pyridinyl segments were basically not involved in the hole → particle transition (Fig. 2). Therefore, the emission of chromene–Ir-OF resulted from a predominantly 3π–π* state, which could reasonably explain why chromene–Ir-OF exhibited the structured emission spectrum with an extended lifetime and a small blue-shift.42 Since the emissions of organometallic photochromic systems upon UV irradiation are usually significantly quenched,43–51 it was exciting to note that the PLQY of the yellow emission from chromene–Ir-OF remained as high as 0.46, which made it possible to realize our idea of modulating the emission colors of OLEDs with UV light alone. To investigate the stability of the yellow emission, two chromene–Ir-CF CH2Cl2 solution samples were prepared. After UV irradiation, one sample was kept in the dark, and the other sample was kept in daylight. The emission intensity at 554 nm of both solutions was monitored every 300 s. As depicted in Fig. 1c and d, yellow emissions could retain ca. 50% of their initial intensities after 3600 s. Since the complex would be used in the solid state in OLEDs, PL spectra as well as the stability of the emission intensity of films prepared from mixed solutions of chromene–Ir-CF and CBP were also measured. As shown in Fig. S8,† films showed a clear emission color change behavior similar to solutions. More importantly, the yellow emissions peaking at 554 nm could retain ca. 80% of their initial intensities after 3600 s, indicating that the stability of the emission intensity in films had been significantly increased compared to that in solution (Fig. 1d and e). The remarkably enhanced emission stability was very reasonable since changes in the chemical structure frame would be restricted within the solid matrix. Therefore, compared with other organic chromene derivatives whose ring-opened products could usually only last for several seconds or minutes in the dark or in daylight, the stability of the yellow emission of chromene–Ir-OF had been greatly improved, which was beneficial for fabricating solid-state devices. In ground and excited states, large differences in the distribution of frontier molecular orbitals between closed and opened forms might be the reason for the greatly improved stability of chromene–Ir-OF. In brief, we realized for the first time a chromene-based Ir(III) complex with improved stability and high PLQY in the opened form, which gave us the inspiration for controlling the emission colors of OLEDs by simply irradiating the chromene-based complex with UV light during the device fabrication process.
To test the above idea, a proof of concept was provided by fabricating OLED devices through the solution-processed method with a simple structure of ITO/PEDOT:PSS (30 nm)/8 wt% Ir complex:CBP (40 nm)/TPBI (40 nm)/Cs2CO3 (2 nm)/Al (100 nm). Compounds such as poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), CBP and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBI) were used as the hole-injection layer, host material and electron-transporting layer, respectively. During the device preparation, the mixtures of chromene–Ir-CF and CBP in chlorobenzene were first irradiated with 365 nm UV light and were further spin-coated on the PEDOT:PSS layer to form emissive layers. Because the concentrations of chromene–Ir-CF in chlorobenzene solution were much higher than 10−5 M, irradiation times at the same light intensity were extended to 0, 20, 30, 70 and 90 seconds (corresponding to devices A, B, C, D and E, respectively). EL spectra, current density–voltage–luminance (J–V–L) characteristics and efficiencies versus luminance curves of these devices are provided in Fig. 3, and key EL data for these devices is summarized in Table 2.
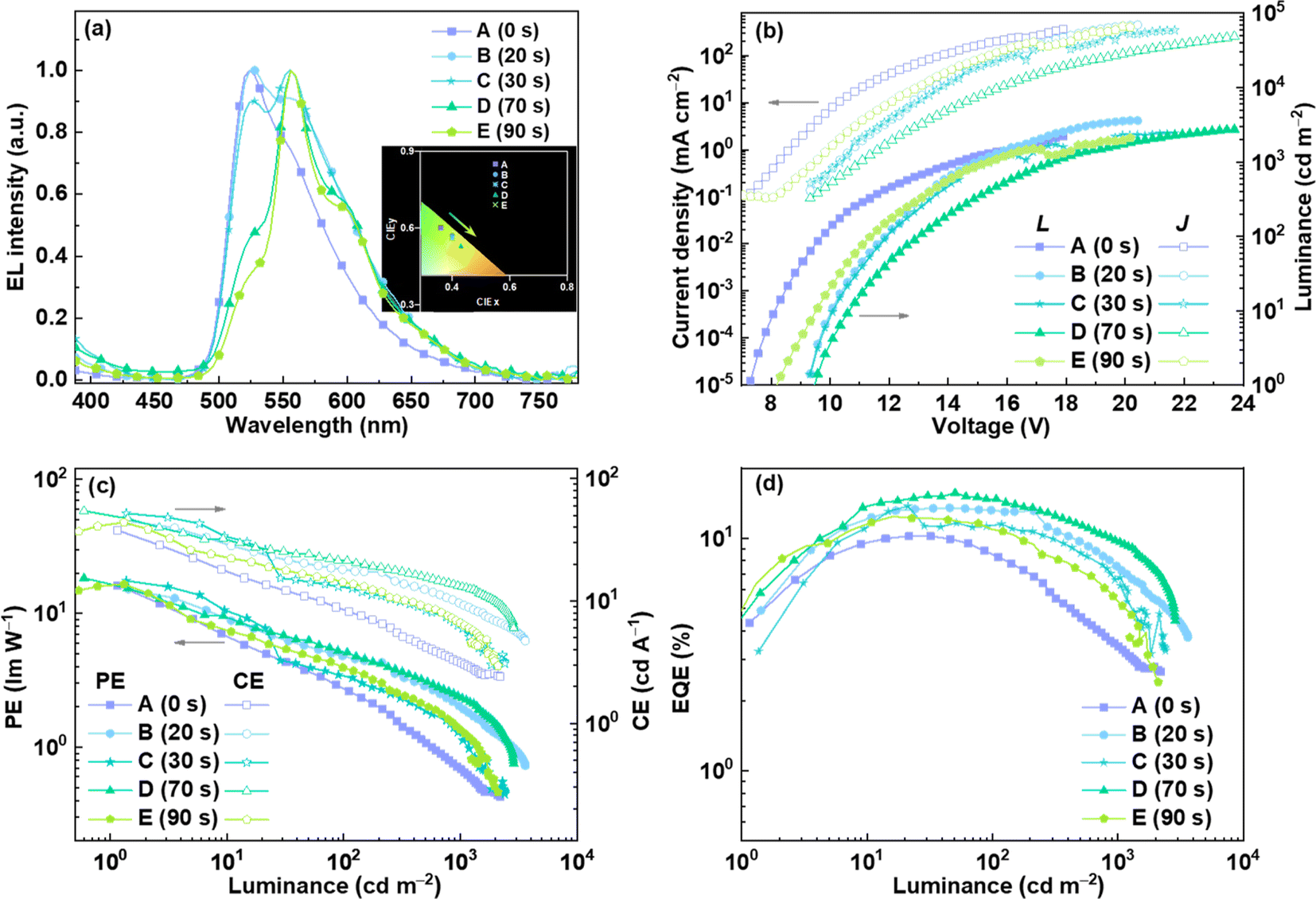 | ||
| Fig. 3 EL performance of OLEDs based on a chromene–Ir complex: (a) EL spectra, (b) J–V–L characteristics, (c) curves of EQE vs. luminance, and (d) curves of CE and PE vs. luminance. | ||
| Device | V turn-on (V) | Luminance Lmax (cd m−2) | EQEmax (%) | CEmax (cd A−1) | PEmax (lm W−1) | λ EL (nm) | CIE |
|---|---|---|---|---|---|---|---|
| A | 7.2 | 2190 | 10.21 | 37.73 | 16.21 | 524 | (0.36, 0.60) |
| B | 8.9 | 3601 | 13.50 | 44.98 | 15.14 | 528 | (0.40, 0.57) |
| C | 9.0 | 2398 | 13.72 | 51.98 | 17.50 | 556 | (0.40, 0.56) |
| D | 9.4 | 2740 | 15.64 | 54.39 | 18.31 | 556 | (0.43, 0.53) |
| E | 8.2 | 2100 | 12.41 | 43.81 | 16.53 | 556 | (0.45, 0.53) |
In the absence of UV irradiation on the emitter, device A displayed green emission with a peak at 524 nm, which was almost identical to that of the PL spectrum of chromene–Ir-CF. Device A also showed moderate EL performance with maximum EQE, PE, and CE of 10.21%, 37.73 cd A−1, and 16.21 lm W−1, respectively. When the emitters were irradiated by UV light, the corresponding EL spectra gradually changed with the appearance and enhancement of new emissions peaking at ca. 556 nm, which also vividly reproduced PL emission changes observed for the chromene–Ir-CF solution when it was irradiated with UV light. The CBP solution was irradiated with 365 nm UV light under same conditions as those used for the mixture of chromene–Ir-CF and CBP in chlorobenzene. The emissions of the CBP solution showed peaks at 387 nm and barely changed, which indicated that CBP had no influence on the emission color change process under 365 nm UV light (Fig. S9†). As shown in Fig. 3a, Commission Internationale de l'éclairage (CIE) chromaticity coordinates gradually migrated from the green to the pure yellow region. Obviously, the color of EL emission had been successfully modulated from green to yellow by simply irradiating the emitter with UV light during the device fabrication process. In addition, the EL performance of devices B–E based on UV-irradiated emitters were comparable to or even better than that of device A. Among all these solution-processed OLEDs, device D showed the best EL performance with maximum EQE, PE, and CE increasing to 15.64%, 54.39 cd A−1, and 18.31 lm W−1, respectively. These results demonstrated that there would be no harmful effects on device efficiencies when tuning the emission color with UV light. Furthermore, regardless of whether it was exposed to UV light or not, the emitter demonstrated that the EL spectra, particularly EL peaks, remained unchanged or exhibited only minor variations when the driving voltages of the devices were increased (Fig. S10†). This behavior revealed the good EL spectral stability of the emitter. The operational lifetimes of unencapsulated devices lasted only a few minutes before their luminance dropped to 50% of their initial values. The stabilities of these devices were not very good, which might be related to the relatively less stable chromene–Ir-CF emitter, as the TGA investigation suggested. Although the efficiencies and stabilities of these chromene–Ir-based devices cannot compete with those of today's high-performance OLEDs, these devices are the first examples whose emission colors were distinctly tuned by simply irradiating the emitter with UV light during the device fabrication process, indicating that we can prepare devices with emission colors being continuously regulated, i.e., we could theoretically achieve infinite numbers of emission colors, by varying only the duration of UV irradiation on emitters. Undoubtedly, this UV-light-modulated emission color tuning strategy will greatly simplify the preparation process and cost of multicolor OLED devices.
Conclusions
In conclusion, for the first time, an efficient phosphorescent Ir(III) complex showing a unique photochromic property was developed based on the chromene unit. Under UV irradiation, the emission color of this Ir(III) complex in the CH2Cl2 solution gradually switched from green to yellow due to the ring-open behavior of the chromene unit. More importantly, the yellow emission from the opened form could show a high PLQY up to 0.46 with greatly improved stability compared with other chromene-based molecules. Therefore, this Ir(III) complex was irradiated with UV light with different durations to fabricate solution-processed OLEDs. Excitingly, the emission colors of the resultant devices could be continuously regulated from green to pure yellow. The EQEs of the devices even increased during the color change process, indicating that the color change process did not adversely affect the device efficiencies. These results clearly demonstrated that electroluminescent colors could be effectively manipulated with UV light alone during the device fabrication process. Therefore, this work not only reports the first chromene-based phosphorescent organometallic complex with impressive photochromic performance but also provides a distinctive strategy to modulate the emission colors of OLEDs more finely and easily. The reported strategy might become the third solution to control the emission colors of OLEDs compared with the two traditional means of changing the emission colors by synthesizing different emitters or varying the concentrations of one emitter.Data availability
All relevant data are presented in the paper and ESI.†Author contributions
Y. Sun, S. Xu, J. Xi, H. Dong and B. Jiao: investigation, formal analysis, validation, and visualization. Y. Sun, G. Zhou and X. Yang: conceptualization, funding acquisition, writing – original draft and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22375158, 52161145411, 22175137 and 51803163), Natural Science Foundation of Shaanxi Province (2023-JC-QN-0144), and Fundamental Research Funds for the Central Universities (xzy012022020 and xzy012023039). The characterization assistance from the Instrument Analysis Center of Xi'an Jiaotong University is also acknowledged.References
- W. Liu, C. Zhang, R. Alessandri, B. T. Diroll, Y. Li, H. Liang, X. Fan, K. Wang, H. Cho, Y. Liu, Y. Dai, Q. Su, N. Li, S. Li, S. Wai, Q. Li, S. Shao, L. Wang, J. Xu, X. Zhang, D. V. Talapin, J. J. de Pablo and S. Wang, Nat. Mater., 2023, 22, 737–745 CrossRef CAS PubMed
.
- C. Keum, C. Murawski, E. Archer, S. Kwon, A. Mischok and M. C. Gather, Nat. Commun., 2020, 11, 6250 CrossRef CAS PubMed
- J.-H. Kim and J.-W. Park, Sci. Adv., 2021, 7, eabd9715 CrossRef CAS PubMed
- L. Liu, K. Cao, S. Chen and W. Huang, Adv. Opt. Mater., 2020, 8, 2001122 CrossRef CAS
- W.-J. Joo, J. Kyoung, M. Esfandyarpour, S.-H. Lee, H. Koo, S. Song, Y.-N. Kwon, S. H. Song, J. C. Bae, A. Jo, M.-J. Kwon, S. H. Han, S.-H. Kim, S. Hwang and M. L. Brongersma, Science, 2020, 370, 459–463 CrossRef CAS PubMed
- Y.-Y. Jing, N. Li, X. Cao, H. Wu, J. Miao, Z. Chen, M. Huang, X. Wang, Y. Hu, Y. Zou and C. Yang, Sci. Adv., 2023, 9, eadh8296 CrossRef CAS PubMed
- Y. Fu, H. Liu, B. Z. Tang and Z. Zhao, Nat. Commun., 2023, 14, 2019 CrossRef CAS PubMed
- X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photonics, 2023, 17, 280–285 CrossRef CAS
- B. Sun, L. Ding, X. Wang, Z.-L. Tu and J. Fan, Chem. Eng. J., 2023, 476, 146511 CrossRef CAS
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484–8575 RSC
- D. Chen, W. Li, L. Gan, Z. Wang, M. Li and S.-J. Su, Mater. Sci. Eng., R, 2020, 142, 100581 CrossRef
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef
- J. Chen, X. Wu, H. Liu, N. Qiu, Z. Liu, D. Yang, D. Ma, B. Z. Tang and Z. Zhao, CCS Chem., 2023, 5, 598–606 Search PubMed
- J. He, Y. Xu, S. Luo, J. Miao, X. Cao and Y. Zou, Chem. Eng. J., 2023, 471, 144565 Search PubMed
- X. Hu, Y. Qin, Z. Li, H. Gao, T. Gao, G. Liu, X. Dong, N. Tian, X. Gu, C.-S. Lee, P. Wang and Y. Wang, Chin. Chem. Lett., 2022, 33, 4645–4648 CrossRef CAS
- X. Yang, X. Zhou, Y.-X. Zhang, D. Li, C. Li, C. You, T.-C. Chou, S.-J. Su, P.-T. Chou and Y. Chi, Adv. Sci., 2022, 9, 2201150 CrossRef CAS PubMed
- J.-M. Kim, K. Y. Hwang, S. Kim, J. Lim, B. Kang, K. H. Lee, B. Choi, S.-Y. Kwak and J. Y. Lee, Adv. Sci., 2022, 9, 2203903 CrossRef CAS PubMed
- X.-F. Luo, Z.-Z. Qu, H.-B. Han, J. Su, Z.-P. Yan, X.-M. Zhang, J.-J. Tong, Y.-X. Zheng and J.-L. Zuo, Adv. Opt. Mater., 2021, 9, 2001390 CrossRef CAS
- P. Gnanasekaran, Y. Yuan, C.-S. Lee, X. Zhou, A. K. Y. Jen and Y. Chi, Inorg. Chem., 2019, 58, 10944–10954 CrossRef CAS
- X. Chang, K. Lu, S. Zeng, D. Liu, J. Huang, B. Ma, L. Wang, X. Gan, J. Yu, Y. Wang, S. Su and W. Zhu, Chem. Eng. J., 2023, 475, 146031 CrossRef CAS
- X. Yang, S. Xu, Y. Zhang, C. Zhu, L. Cui, G. Zhou, Z. Chen and Y. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309739 CrossRef CAS
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS
- Q. Li and Z. Li, Sci. China Mater., 2020, 63, 177–184 CrossRef
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed
- H. Liu, H. Liu, J. Fan, J. Guo, J. Zeng, F. Qiu, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2020, 8, 2001027 CrossRef CAS
- K. Zhang, X. Zhou, S. Li, L. Zhao, W. Hu, A. Cai, Y. Zeng, Q. Wang, M. Wu, G. Li, J. Liu, H. Ji, Y. Qin and L. Wu, Adv. Mater., 2023, 35, 2305472 CrossRef CAS PubMed
- Z. Wang, Z. Ding, Y. Yang, L. Hu, W. Wu, Y. Gao, Y. Wei, X. Zhang and G. Jiang, Chem. Eng. J., 2023, 457, 141293 CrossRef CAS
- J.-C. Yang, Z. Fu, H. Ma, T. Wang, Q. Li, K. Wang, L. Wu, P. Chen, H.-T. Feng and B. Z. Tang, ACS Mater. Lett., 2023, 5, 1441–1449 CrossRef CAS
- F. Gu, Y. Li, T. Jiang, J. Su and X. Ma, CCS Chem., 2022, 4, 3014–3022 CrossRef CAS
- Z. Wei, K. Zhang, C. K. Kim, S. Tan, S. Wang, L. Wang, J. Li and Y. Wang, Chin. Chem. Lett., 2021, 32, 493–496 CrossRef CAS
- J. Zhang, Q. Zou and H. Tian, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed
- L. Ma, G. Wang, B. Ding and X. Ma, CCS Chem., 2022, 4, 2080–2089 CrossRef CAS
- C.-C. Ko and V. W.-W. Yam, Acc. Chem. Res., 2018, 51, 149–159 CrossRef CAS
- Y. Ru, Z. Shi, J. Zhang, J. Wang, B. Chen, R. Huang, G. Liu and T. Yu, Mater. Chem. Front., 2021, 5, 7737–7758 RSC
- O. Galangau, L. Norel and S. Rigaut, Dalton Trans., 2021, 50, 17879–17891 RSC
- R. S. Becker and J. Michl, J. Am. Chem. Soc., 1966, 88, 5931–5933 CrossRef CAS
- S. Han and Y. Chen, J. Mater. Chem., 2011, 21, 4961–4965 RSC
- K. Arai, Y. Kobayashi and J. Abe, Chem. Commun., 2015, 51, 3057–3060 RSC
- C. M. Sousa, J. Pina, J. S. de Melo, J. Berthet, S. Delbaere and P. J. Coelho, Org. Lett., 2011, 13, 4040–4043 CrossRef CAS
- N. Malic, J. A. Campbell and R. A. Evans, Macromolecules, 2008, 41, 1206–1214 CrossRef CAS
- W. Zhao and E. M. Carreira, Org. Lett., 2003, 5, 4153–4154 CrossRef CAS
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed
- C.-T. Poon, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 13992–13993 CrossRef CAS
- W. Zhu, X. Meng, Y. Yang, Q. Zhang, Y. Xie and H. Tian, Chem.–Eur. J., 2010, 16, 899–906 CrossRef CAS
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690–12705 CrossRef CAS PubMed
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, W.-T. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 11504–11508 CrossRef CAS
- S. Chen, Z. Guo, S. Zhu, W.-e. Shi and W. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 5623–5629 CrossRef CAS
- S. Pu, Z. Tong, G. Liu and R. Wang, J. Mater. Chem. C, 2013, 1, 4726–4739 RSC
- J. Guerin, A. Léaustic, S. Delbaere, J. Berthet, R. Guillot, C. Ruckebusch, R. Metivier, K. Nakatani, M. Orio and M. Sliwa, Chem.–Eur. J., 2014, 20, 12279–12288 CrossRef CAS PubMed
- G. Lv, B. Cui, H. Lan, Y. Wen, A. Sun and T. Yi, Chem. Commun., 2015, 51, 125–128 RSC
- J.-X. Wang, C. Li and H. Tian, Coord. Chem. Rev., 2021, 427, 213579 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01812e |
| This journal is © The Royal Society of Chemistry 2024 |