
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnisotropic electronic coupling in three-dimensional assembly of CsPbBr3 quantum dots†
Kazushi
Enomoto
 *a,
Retno
Miranti
a,
Jianjun
Liu
a,
Rinkei
Okano
a,
Daishi
Inoue
a,
DaeGwi
Kim
*b and
Yong-Jin
Pu
*a
*a,
Retno
Miranti
a,
Jianjun
Liu
a,
Rinkei
Okano
a,
Daishi
Inoue
a,
DaeGwi
Kim
*b and
Yong-Jin
Pu
*a
aRIKEN Center for Emergent Matter Science (CEMS), Wako, Saitama 351-0198, Japan. E-mail: kazushi.enomoto@riken.jp; yongjin.pu@riken.jp
bDepartment of Physics and Electronics, Osaka Metropolitan University, Osaka 558-8585, Japan. E-mail: kimtegi@omu.ac.jp
First published on 15th July 2024
Abstract
Cesium lead halide (CsPbX3, X = Cl, Br, or I) perovskite quantum dots (PeQDs) show promise for next-generation optoelectronics. In this study, we controlled the electronic coupling between PeQD multilayers using a layer-by-layer method and dithiol linkers of varying structures. The energy shift of the first excitonic peak from monolayer to bilayer decreases exponentially with increasing interlayer spacer distance, indicating the resonant tunnelling effect. X-ray diffraction measurements revealed anisotropic inter-PeQD distances in multiple layers. Photoluminescence (PL) analysis showed lower energy emission in the in-plane direction due to the electronic coupling in the out-of-plane direction, supporting the anisotropic electronic state in the PeQD multilayers. Temperature-dependent PL and PL lifetimes indicated changes in exciton behaviour due to the delocalized electronic state in PeQD multilayers. Particularly, the electron–phonon coupling strength increased, and the exciton recombination rate decreased. This is the first study demonstrating controlled electronic coupling in a three-dimensional ordered structure, emphasizing the importance of the anisotropic electronic state for high-performance PeQDs devices.
Introduction
Cesium lead halide (CsPbX3, X= Cl, Br, and I) perovskite nanocrystals have been widely studied because of their superior photophysical properties. In addition to the general advantages of metal chalcogenide quantum dots (QDs),1 these attributes include the high photoluminescence quantum yield (PLQY), rapid radiative decay rate, controllable emission color by halogen composition, and defect tolerance.2 Particularly, perovskite quantum dots (PeQDs) with diameters smaller than the exciton Bohr size show fine bandgap tunability depending on their size,3–5 resulting in great potential for application in optoelectronic devices, such as light-emitting diodes, photovoltaics, photodetectors, and field-effect transistors.6–8 The hot injection method is typically used to synthesize uniform and stably dispersed PeQDs in hydrophobic media, in which long-alkyl chain oleic acid and oleylamine are used as surface ligands.3 Colloidally stable PeQDs possess considerable potential for implementation in solution-processed devices.QDs possess molecular-like discrete electronic states that arise from the quantum confinement effect. Emerging electronic states, called minibands, caused by short-range electronic coupling of wave functions among closely packed QDs have been reported.9 Minibands are expected to offer superior optoelectronic features, such as reduced trap-assisted recombination, enhanced charge transfer, multi-exciton generation, and hot excitons.10–15 However, long alkyl ligands enveloping the QDs act as insulators and have a high energy barrier, thereby preventing electronic coupling. Hence, researchers have proposed ligand exchange with shorter ligands to lower the potential barrier and enable electronic coupling.10,16–19 Previous studies reported miniband formation in periodically aligned QD assemblies (superlattices) of metal chalcogenides.9,10,20–29 The ordering of superlattices is highly sensitive to miniband formation,30 necessitating the precise control of the inter-QD interaction based on dimensional tailoring of the assembly. QD superlattices have been prepared using various approaches, such as solvent destabilization, slow evaporation, and liquid/air interface. Recently, a layer-by-layer (LbL) assembly method has been reported to fabricate CdTe QD superlattices.31 LbL assembly, which allows for the preparation of precisely controlled QD multi-layer structures, is a simple, useful, and promising approach. The distance between each QD layer can be adjusted with single-nanometer accuracy using a linker molecule or polymer spacer.
Multiple studies have reported the formation of minibands in CsPbX3 PeQD superlattices.32–38 These superlattices exhibited synergistic photophysical characteristics such as low energy-shifted narrower emission and high charge mobility.32,34,35 However, these exceptional properties were only observed in 3D supercrystals and solution-coated 2D monolayers. For practical applications, it is crucial to create a multi-layer PeQD superlattice in a film state with an electronic coupling that is precisely controlled. In solar cell production, multi-layered PeQD structures were created through sequential spin-coating of a perovskite nanocrystal solution, linker solution, and washing solvent.39–47 This process, however, inevitably led to significant variations in the thickness of the QD layers and incomplete ligand exchange.48 In several other studies, the electronic coupling of cadmium chalcogenide QD was controlled by adjusting the interparticle distance in the solution.48–50 Nonetheless, no studies have precisely controlled PeQD multi-layer structures and electronic coupling between QDs.
In this study, we successfully produced a PeQD multi-layer superlattice through a precisely controlled LbL process. We observed anisotropic electronic coupling between PeQD layers, as depicted in Fig. 1. The PeQD superlattice was formed by alternating the immersion of the substrate into PeQD and dithiol linker solutions. This sequence substituted the long-alkyl ligands on the PeQD surfaces with dithiol linkers. By altering the linker length, we could regulate the electronic coupling strength in the PeQD superlattice. Additionally, we evaluated this strength by analyzing the temperature dependence of the PL and PL lifetimes. X-ray diffraction (XRD) and the angular dependence of the PL demonstrated the anisotropic structure and electronic coupling towards the out-of-plane direction in the three-dimensional PeQD superlattice.
 | ||
| Fig. 1 Schematic illustration of layer-by-layer (LbL) assembly of perovskite quantum dots (PeQDs) and coupling strength controlled by inter-PeQD distance with different linker lengths. | ||
Results and discussion
CsPbBr3 QDs were synthesized using a typical hot-injection method51 and treated with ammonium thiocyanate to achieve a high PLQY (80%). Their average size was 6.8 ± 0.5 nm, estimated using transmission electron microscopy (TEM) (Fig. S1a and c†). Fig. S1b† shows a high-resolution TEM image of the CsPbBr3 QD. The periodic constant (0.58 nm) indicates the (100) plane in the cubic crystal structure.52 Fig. S1d† shows the absorption and PL spectra of the CsPbBr3 QD-toluene solution. A sharp band edge absorption peak at 2.477 eV, green PL with a peak at 2.446 eV and a narrow full width at half maximum (FWHM) of 77 meV were observed, indicating the monodispersity in the size of the QDs and the well-electronically confined features.Fabrication of PeQD first layer
In the LbL process for CsPbBr3 QDs, polyethyleneimine (PEI) was chosen as the pre-coated underlayer on the substrate. PEI contains repeating units of secondary amine groups and facilitates the adsorption of CsPbBr3 QDs onto the PEI underlayer, forming the first PeQD monolayer. The density of PeQDs in the monolayer was controlled using the withdrawal speed of the substrate from the PeQD solution. Fig. 2a shows the absorption spectra of PeQD monolayers prepared using different withdrawal speeds. The density of the PeQD monolayer was estimated from the optical density (OD) of the first absorption peak of the PeQD film. As the withdrawal speed decreased, the OD value of the monolayer increased, and the peak top energy remained constant at 2.473 ± 0.002 eV, equivalent to that of the solution (Fig. 2b and S1†). The constant peak top energy indicated the absence of electronic coupling among the PeQDs in the in-plane direction. The QDs were coated with long-alkyl-chain ligands, namely oleic acid and oleylamine, which were insulating and of sufficient length to impede electronic coupling between the QDs.27 | ||
| Fig. 2 a) Absorbance spectra of perovskite quantum dot (PeQD) monolayer prepared with different withdrawal speeds. (b) Dependence of withdrawal speed on the optical density (OD) and first excitonic peak position of the PeQD monolayer. | ||
We could improve the wettability of the PeQD solution on the substrate by incorporating diglyme. As shown in Fig. S2a,† drop-cast PeQD films from toluene and toluene/diglyme solutions were compared. The surface of the PeQD film from toluene exhibited a rough texture due to the hydrophobic nature of toluene, which caused it to spread unevenly on the hydrophilic substrate. In contrast, adding diglyme, which is amphiphilic and has been identified as a suitable poor solvent for PeQDs without impacting their PLQY,53 resulted in a film with a smooth surface. Furthermore, the PLQY (89%) of the film from the toluene/diglyme mixture was similar to that of the toluene solution-prepared film. The absorption, PL, and transient PL (TrPL) spectra of the PeQDs from the toluene/diglyme solution were unaffected, as shown in Fig. S2b and c.† The first excitonic peak energy in the absorption spectra and the top of the PL spectra remain the same. Additionally, the TrPL decay rate did not change. As illustrated in Fig. S2d,† the TEM image reveals a closely packed PeQD monolayer in the drop-cast film from toluene/diglyme while maintaining the size and cubic structure of the QDs. These findings demonstrate that adding diglyme can improve the wettability of the PeQD solution on the substrate without affecting the optical properties of the PeQDs in the film state. Furthermore, the relationship between the volume fraction of diglyme and the OD of the PeQD monolayer dip-coated on the quartz substrate with PEI coating is shown in Fig. S2e.† An increasing volume fraction of diglyme results in an increase in the OD of the PeQD monolayer due to the improved wettability while maintaining the peak top energy of the QD constant, indicating the absence of electronic coupling in the in-plane direction. Finally, we used a mixture of diglyme/toluene (15/85 v/v) for the multi-layer LbL process.
Layer-by-layer assembly of PeQD
A bifunctional ligand was utilized as a linker for LbL assembly to prepare the multi-layer film. PeQDs have been reported to bind to various functional groups, such as amine,54 carboxylic acid/amine complex,55 ammonium halide,7,56–59 sulfonic acid,60 phosphoric acid,61–63 and zwitterion64–68 as ligands. Traditional linkers such as dicarboxylic acid and diamine molecule were tried to be used for ligand exchange reactions of QDs. However, aliphatic dicarboxylic acid was hard to soluble into hydrophobic solvent such as toluene. In addition, absorption of PeQD monolayer was disappeared after soaking into diamine solution, indicating degradation of the film. Recent studies have shown that thiol ligands can passivate Pb defects on the surface of PeQDs.69–73 The LbL assembly utilized dithiol ligands as linkers to control over the interparticle distance. The absorbance of the PeQD film gradually increases when 1,3-propane dithiol (C3DT) is applied as the linker in the LbL process (Fig. 3a). The OD value exhibits a linear increase with the number of layers N, indicating the PeQD layers were successively stacked with constant packing density (Fig. 3c). The LbL assembly of the PeQD film was accomplished using a longer alkyl dithiol linker (1,10-decane dithiol; C10DT, Fig. 3b), displaying almost the same increase in OD value. The absorption energy shifts in the LbL films with the C3DT and C10DT ligands differ remarkably. The peak top energy gradually shifts to lower energy with increased N (Fig. 3d). These low energy shifts become nearly constant at different points (−44.0 meV in C3DT and −14.2 meV in C10DT systems at N = 8), supporting that the low energy shifts can be ascribed to the electronic coupling between the PeQD layers.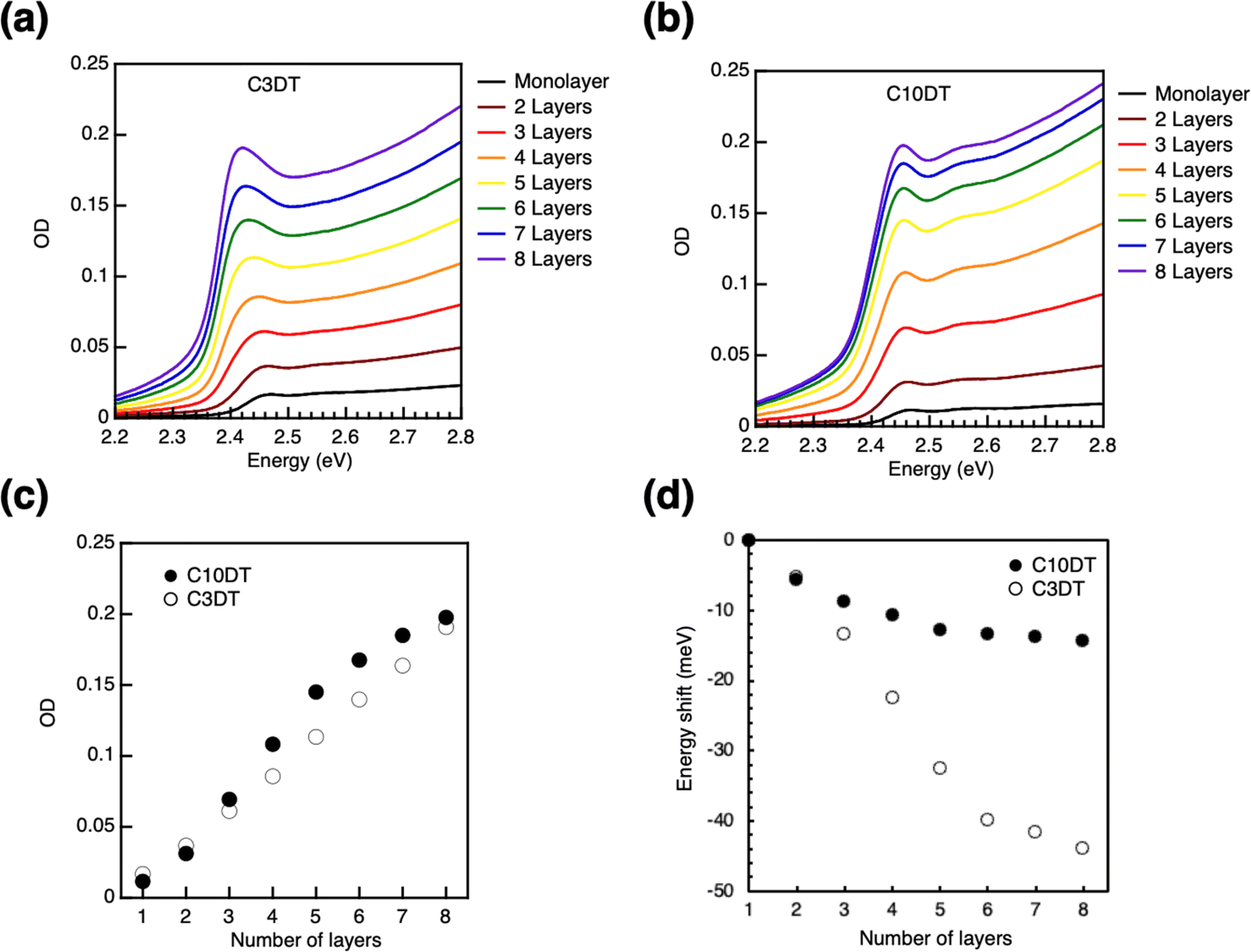 | ||
| Fig. 3 Absorbance spectra of perovskite quantum dot (PeQD) multi-layer prepared using the layer-by-layer (LbL) method using a dithiol linker with different alkyl spacers: (a) C3DT, (b) C10DT. Dependence of N on (c) optical density (OD) and (d) energy shift in PeQD multi-layers. | ||
The electronic coupling strength between the QDs is explained by eqn (1) based on the resonant tunnelling effect:25
 | (1) |
 | ||
| Fig. 4 Energy shift from monolayer to bilayers and interlayer distance determined by linker length. | ||
This study investigated the structural order of QDs in LbL-PeQD films. A cross-sectional scanning electron microscopy (SEM) image in Fig. S4a† reveals the PeQD layers (N = 20) prepared using the C3DT linker. The measured thickness was 130 ± 10 nm, in agreement with the estimated value of 147 nm, calculated using the number of layers and the average periodic distance of PeQD (7.35 nm) derived from the average diameter of PeQD (6.8 nm) and the spacer length of C3DT (0.55 nm). Fig. S4b† depicts the SEM energy-dispersive X-ray spectroscopy (SEM-EDS) mapping images and the EDS spectrum. Elemental analysis confirms the presence of Cs, Pb, Br, C, and S, indicating the successful introduction of the thiol linker into the PeQD multi-layer. Fig. S5† shows the XRD patterns of drop-cast and multi-layer (N = 30) PeQD films. The drop-cast film displayed a diffraction peak at q = 0.62 nm−1, signifying a center-to-center interparticle distance of 10.1 nm in the out-of-plane directions. This was attributed to the three-dimensional close packing of the unmodified PeQDs. In contrast, the peak in the out-of-plane direction shifted to the high q region in the multi-layer films, and the film with the shorter linker, C3DT, showed a higher q peak than the film with C10DT (C10DT: 0.78 nm−1 for 8.1 nm and C3DT: 0.82 nm−1 for 7.7 nm). These values align with the periodic distance of the PeQDs estimated from the average diameter of the PeQDs (6.8 nm) and spacer lengths of C10DT (1.46 nm) and C3DT (0.55 nm). The diffraction peak in the in-plane direction was observed at the similar q value of 0.630–0.648 nm−1, corresponding to 9.70–9.97 nm in all films, which the analysis was performed with the fitting of raw data and first and second derivatives, using combination of Gaussian and polynomial functions (Fig. S6, ESI†). This result suggests that the ligand exchange from the long oleyl ligands to the shorter dithiol linkers primarily occurs in the out-of-plane direction. The binding energy of the thiol group on the surface of PeQDs was not as strong as that of conventional metal chalcogenide QDs. Accessing the lateral side of the QDs within the layer by linker molecules can be deemed more challenging than accessing the top side of the QDs. This is due to the dense packing of PeQDs with each other in the layer. The diverse accessibilities and weak binding capacity of thiol linkers onto the PeQD facet are accountable for the distinct packing observed in the in-plane and out-of-plane directions. These results of the XRD measurements support the anisotropic electronic coupling between PeQD layers in the out-of-plane direction. To ensure the absence of electronic coupling in the in-plane direction, PeQD monolayer films with varying packing densities were immersed in a C3DT linker solution (Fig. S7†). Nevertheless, the absorption peaks remained unaffected. Further, there were no differences in absorption spectra before and after soaking PeQD monolayer with high packing density into several linker solutions. These results verify that the electronic coupling of PeQDs in the in-plane direction did not transpire in the LbL assemblies, aligning with the observation of an unchanged XRD peak in the in-plane direction in the LbL assembly.
Optical characteristics of PeQD assembly
The anisotropic feature of PL intensity on the detective angle was reported in semiconductor nanowires.74,75 The anisotropic electronic coupling in the out-of-plane direction causes a transition dipole moment perpendicular to the substrate, affecting the angle dependence on the PL intensity. Therefore, we measured the angular dependence of the PL of drop-cast, monolayer, and multi-layer films created using C3DT and C10DT to examine the anisotropic coupling in the out-of-plane direction within the LbL assembly (Fig. 5a–d). The monolayer and drop-cast films displayed similar PL peak energies and no angular dependence. The drop-cast film thickness was adjusted to match that of a six-layer film. Angle-dependent PL spectra of the drop-cast film showed slight low-energy shift with increasing angle probably because of re-absorption effect (Fig. S8†). In contrast, multi-layer films with C3DT and C10DT exhibited a distinct angular dependence, with lower energy shifts occurring towards the in-plane direction (higher angle). These findings indicate that the PeQD multi-layer electronic coupling, created through the LbL process, primarily occurred in the out-of-plane direction, forming an anisotropic electronic state in the PeQD film. | ||
| Fig. 5 Angle-dependent photoluminescence (PL) spectra of (a) monolayer, (b) drop cast film, six-layer bridged with (c) C10DT, and (d) C3DT. The angle is defined by a substrate rotation on the axis perpendicular to the plane consisting of incident light and emitting light. When the emitting light direction to the detector is perpendicular to the substrate, the angle is 0°. | ||
To better understand the miniband structure of the LbL PeQDs, we investigated the temperature dependence of the PL (Fig. S9†). The results demonstrate that the coupled multi-layer PeQD film exhibits a narrower FWHM emission, similar to that reported for the PeQD superlattice, forming a miniband. The temperature dependence of the PL peak top energy E(T) and FWHM Γ(T) are shown in Fig. S10.† These parameters are related to the exciton–phonon distribution function and exciton–phonon coupling strength, which can be described by the following equations:76–84
 | (2) |
 | (3) |
Here, E0 is the unrenormalized bandgap, ATE is the weight of thermal expansion, AEP is the electron–phonon coupling coefficient, ħω is the average optical phonon energy, Γ0 is the temperature-independent inhomogeneous broadening, γLO is the exciton–phonon coupling strength, and ELO is the LO phonon energy. Here, the acoustic phonon term was neglected because its value quite small contribution was reported in CsPbBr3 nanocrystal system (the order of µeV K−1), and it was often omitted.76,77,82,83,85–89 The parameters obtained by curve fitting are listed in Table S1.† All parameters, except ELO and γLO, aligned with the previously reported parameters for CsPbBr3 QDs, as summarized in Table S2.† The ELO value would be intrinsic to its crystal nature. Then, fitting was performed using ELO value fixed according to the reported values. Fitting results were summarized in Table S1,† changing the ELO value to 10, 20, and 30 meV. The γLO value was decreased with decreasing the ELO value, while the electronically coupled LbL samples still showed higher γLO value than that of monolayer film. On the other hand, the r2 value was also decreased, indicating worse fitting. Any γLO estimated by the different four fitting conditions was increased in the multilayers compared to that of the monolayer, and the film with the shorter C3DT ligand and more layers (N = 6) exhibited larger γLO. Generally, the increased FWHM at higher temperatures is attributed to increased exciton–phonon coupling. Increasing γLO is caused by stronger confinement in PeQD or lower dimensionality of perovskite crystals (from 3D single crystal, 2D nanosheet, to 1D nanowire) (Table S2†). The increment of ELO and γLO from the non-coupled monolayer to the coupled multi-layer PeQD films may be due to the synergistic effect of 1D-like anisotropic electron–phonon couplings between QDs and between QD and the dithiol linkers in the PeQD layers. A more detailed discussion based on further theoretical study will be necessary.
The temperature dependence of the TrPL lifetime of the PeQD films was investigated to understand the behavior of the excitons in the miniband (Fig. 6 and S11†). To observe the emission from the miniband, we set the detection wavelength to a lower energy point, which was at half the PL intensity at room temperature. Fig. 6d shows the average PL lifetime τ as the function of temperature. The TrPL decay curve was fitted using the triexponential equation: I(t) = A1 exp(−t/τ1) + A2 exp(−t/τ2) + A3 exp(−t/τ3). The decay parameters τ1, τ2, τ3, A1, A2, and A3 are the lifetimes and their corresponding amplitudes. The average lifetime is estimated using the weight average. The monolayer film exhibited a constant average lifetime independent of the temperature, which is a typical characteristic of 0D excitons.90,91 In contrast, the trilayers and six-layer films showed longer lifetimes with decreasing temperatures. The elongation of the lifetime at lower temperatures is caused by a reduction in the recombination rate of the exciton, due to the delocalization of electrons over several PeQD layers in the excited state.92,93 In the C3DT system, the six layers exhibited stronger temperature dependence than the trilayer. However, similar temperature dependence was observed for the six-layers and trilayers prepared with C10DT. These results are consistent with the energy shift observed in the absorption spectra with N (Fig. 3). The energy shift was nearly saturated in the six- and trilayer films of the C3DT and C10DT systems, respectively. Strong coupling with the shorter linker resulted in a wider delocalization of electrons. This result indicates the fine-tuning of the electron delocalization state in the PeQD superlattice.
 | ||
| Fig. 6 Temperature dependence of transient photoluminescence (TrPL) decays of (a) monolayer, (b) trilayer, and (c) six-layer prepared with C3DT. The detection energy: (a) 2.385 eV, (b) 2.362 eV, (c) 2.353 eV, respectively. (d) Temperature dependence of PL lifetime τ. | ||
Conclusion
We effectively controlled the miniband formation in PeQD multilayers through the LbL technique, utilizing dithiol molecules as linkers for layering. Our analysis indicates that the energy shift of the first excitonic peak from monolayer to bilayer follows an exponential decreasing pattern as the linker length is increased. We observed different saturation points for increasing the number of layers N, with significant energy shifts for short C3DT and small shifts for long C10DT systems. These findings provide direct evidence that the resonant tunnelling effect governs electronic coupling between PeQD layers.The XRD profiles suggest that the interparticle distance in the out-of-plane direction is shorter than in the in-plane direction. Moreover, the angle-dependent PL exhibited an anisotropically coupled electronic state in the out-of-plane direction.
Temperature-dependent PL measurements showed a substantial enhancement in the electron–phonon coupling strength in PeQD multilayers, indicating the preferable formation of electronic coupling. The PL lifetime τ of PeQD multi-layers displayed an elongated pattern with lower temperatures. This suggests that the recombination rate of excitons decreased due to the delocalised electronic state in the PeQD multi-layer.
This is the first demonstration of finely controlled anisotropic electronic coupling between PeQDs in a multilayer superlattice. The selectivity of electronic coupling in different directions can be applied to other semiconductor QDs. The anisotropic electronic state in the deposited PeQD multi-layer would affect the charge transfer rate, light outcoupling efficiency, and the radiative decay rate, which holds great promise for novel applications in PeQD optoelectronic devices.
Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
K. E., D. K., and Y.-J. P. conceptualized the project. K. E. performed synthesis, LbL, measurements, and data analysis. R. M., J. L., and R. O. supported sample preparation and measurement. D. I. supported the cross-section SEM and EDS mapping measurements. K. E. and Y.-J. P. wrote the manuscript. All authors contributed to the interpretation of the results and preparation of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported in part by JSPS KAKENHI (grant nos. 20H02554, 20K15111, and 23K13616).Notes and references
- B. J. Roman, N. M. Villegas, K. Lytle and M. Sheldon, Nano Lett., 2020, 20, 8874–8879 CrossRef CAS.
- J. Kang and L. W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- H. Liu, Z. Liu, W. Xu, L. Yang, Y. Liu, D. Yao, D. Zhang, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 14256–14265 CrossRef CAS.
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y. J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353 Search PubMed.
- J. M. Pietryga, Y. S. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS PubMed.
- D. Kim, K. Shin, S. G. Kwon and T. Hyeon, Adv. Mater., 2018, 30, e1802309 CrossRef PubMed.
- D. Kim, S. Tomita, K. Ohshiro, T. Watanabe, T. Sakai, I. Y. Chang and K. Hyeon-Deuk, Nano Lett., 2015, 15, 4343–4347 CrossRef CAS PubMed.
- Y. Liu, M. Gibbs, J. Puthussery, S. Gaik, R. Ihly, H. W. Hillhouse and M. Law, Nano Lett., 2010, 10, 1960–1969 CrossRef CAS PubMed.
- Z. Zhang, J. Sung, D. T. W. Toolan, S. Han, R. Pandya, M. P. Weir, J. Xiao, S. Dowland, M. Liu, A. J. Ryan, R. A. L. Jones, S. Huang and A. Rao, Nat. Mater., 2022, 21, 533–539 CrossRef CAS PubMed.
- J. Cui, Y. E. Panfil, S. Koley, D. Shamalia, N. Waiskopf, S. Remennik, I. Popov, M. Oded and U. Banin, Nat. Commun., 2019, 10, 5401 CAS.
- A. R. Khabibullin, A. L. Efros and S. C. Erwin, Nanoscale, 2020, 12, 23028–23035 CAS.
- V. Lesnyak, J. Phys. Chem. Lett., 2021, 12, 12310–12322 CrossRef CAS PubMed.
- M. S. Khoshkhoo, J. F. L. Lox, A. Koitzsch, H. Lesny, Y. Joseph, V. Lesnyak and A. Eychmuller, ACS Appl. Electron. Mater., 2019, 1, 1560–1569 CrossRef.
- M. T. Frederick and E. A. Weiss, ACS Nano, 2010, 4, 3195–3200 CrossRef CAS PubMed.
- M. T. Frederick, V. A. Amin, N. K. Swenson, A. Y. Ho and E. A. Weiss, Nano Lett., 2013, 13, 287–292 CrossRef CAS.
- M. T. Frederick, V. A. Amin and E. A. Weiss, J. Phys. Chem. Lett., 2013, 4, 634–640 CrossRef CAS PubMed.
- M. S. Azzaro, A. Dodin, D. Y. Zhang, A. P. Willard and S. T. Roberts, Nano Lett., 2018, 18, 3259–3270 CrossRef CAS.
- A. Dong, J. Chen, P. M. Vora, J. M. Kikkawa and C. B. Murray, Nature, 2010, 466, 474–477 CrossRef CAS PubMed.
- X. Zhang, L. Lv, L. Ji, G. Guo, L. Liu, D. Han, B. Wang, Y. Tu, J. Hu, D. Yang and A. Dong, J. Am. Chem. Soc., 2016, 138, 3290–3293 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CrossRef CAS.
- S. A. Crooker, J. A. Hollingsworth, S. Tretiak and V. I. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS PubMed.
- T. Hanrath, J. Vac. Sci. Technol., A, 2012, 30, 30802 CrossRef.
- C. R. Kagan and C. B. Murray, Nat. Nanotechnol., 2015, 10, 1013–1026 CrossRef CAS.
- S. J. Yoon, Z. Guo, P. C. Dos Santos Claro, E. V. Shevchenko and L. Huang, ACS Nano, 2016, 10, 7208–7215 CrossRef CAS.
- J. S. Lee, M. V. Kovalenko, J. Huang, D. S. Chung and D. V. Talapin, Nat. Nanotechnol., 2011, 6, 348–352 CrossRef CAS PubMed.
- E. Talgorn, Y. N. Gao, M. Aerts, L. T. Kunneman, J. M. Schins, T. J. Savenije, M. A. van Huis, H. S. J. van der Zant, A. J. Houtepen and L. D. A. Siebbeles, Nat. Nanotechnol., 2011, 6, 733–739 CrossRef CAS PubMed.
- Z. Wang, A. D. Christodoulides, L. Dai, Y. Zhou, R. Dai, Y. Xu, Q. Nian, J. Wang, J. A. Malen and R. Y. Wang, Nano Lett., 2022, 22, 4669–4676 CrossRef CAS.
- T. Lee, K. Enomoto, K. Ohshiro, D. Inoue, T. Kikitsu, K. Hyeon-Deuk, Y. J. Pu and D. Kim, Nat. Commun., 2020, 11, 5471 CrossRef CAS PubMed.
- Y. Tong, E. P. Yao, A. Manzi, E. Bladt, K. Wang, M. Doblinger, S. Bals, P. Muller-Buschbaum, A. S. Urban, L. Polavarapu and J. Feldmann, Adv. Mater., 2018, e1801117, DOI:10.1002/adma.201801117.
- E. Penzo, A. Loiudice, E. S. Barnard, N. J. Borys, M. J. Jurow, M. Lorenzon, I. Rajzbaum, E. K. Wong, Y. Liu, A. M. Schwartzberg, S. Cabrini, S. Whitelam, R. Buonsanti and A. Weber-Bargioni, ACS Nano, 2020, 14, 6999–7007 CrossRef CAS.
- C. Zhou, Y. Zhong, H. Dong, W. Zheng, J. Tan, Q. Jie, A. Pan, L. Zhang and W. Xie, Nat. Commun., 2020, 11, 329 CrossRef CAS PubMed.
- D. Baranov, S. Toso, M. Imran and L. Manna, J. Phys. Chem. Lett., 2019, 10, 655–660 CrossRef CAS.
- Y. Tang, D. Poonia, M. van der Laan, D. Timmerman, S. Kinge, L. D. A. Siebbeles and P. Schall, ACS Appl. Energy Mater., 2022, 5, 5415–5422 CrossRef CAS PubMed.
- H. Huang, M. W. Feil, S. Fuchs, T. Debnath, A. F. Richter, Y. Tong, L. Z. Wu, Y. O. Wang, M. Doblinger and B. Nickel, Chem. Mater., 2020, 32, 8877–8884 CrossRef CAS.
- X. T. Tang, D. Rossi, J. Cheon and D. H. Son, Chem. Mater., 2022, 34, 7181–7189 CrossRef CAS.
- S. B. Shivarudraiah, M. Ng, C. H. A. Li and J. E. Halpert, ACS Appl. Energy Mater., 2020, 3, 5620–5627 CrossRef CAS.
- Y. H. Chen, T. Chen and L. M. Dai, Adv. Mater., 2015, 27, 1053–1059 CrossRef CAS.
- J. Z. Song, J. H. Li, L. M. Xu, J. H. Li, F. J. Zhang, B. N. Han, Q. S. Shan and H. B. Zeng, Adv. Mater., 2018, 30, 1800764 CrossRef.
- X. L. Zhang, Y. L. Qian, X. F. Ling, Y. Wang, Y. N. Zhang, J. W. Shi, Y. Shi, J. Y. Yuan and W. L. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 27307–27315 CrossRef CAS PubMed.
- Q. Y. Li, Y. T. Zhang, Y. Yu, Z. L. Chen, L. F. Jin, Y. F. Li, T. T. Li, Y. Yang, H. L. Zhao, J. Li, H. T. Dai, J. B. Yang and J. Q. Yao, Nanotechnology, 2019, 30, 37LT01 CrossRef CAS.
- N. K. Kumawat, A. Swarnkar, A. Nag and D. Kabra, J. Phys. Chem. C, 2018, 122, 13767–13773 CrossRef CAS.
- Q. Zhao, A. Hazarika, X. H. Chen, S. P. Harvey, B. W. Larson, G. R. Teeter, J. Liu, T. Song, C. X. Xiao, L. Shaw, M. H. Zhang, G. R. Li, M. C. Beard and J. M. Luther, Nat. Commun., 2019, 10, 2842 CrossRef PubMed.
- L. M. Xu, J. H. Li, B. Cai, J. Z. Song, F. J. Zhang, T. Fang and H. B. Zeng, Nat. Commun., 2020, 11, 3902 CrossRef CAS.
- B. B. Li, X. M. Li, X. Li, H. L. Liu, Z. N. Li, G. H. Xiang, Y. H. Liu, T. J. Zhou, X. Fang and Z. Y. Zhang, RSC Adv., 2018, 8, 27201–27206 RSC.
- M. C. Weidman, K. G. Yager and W. A. Tisdale, Chem. Mater., 2015, 27, 474–482 CrossRef CAS.
- M. C. Weidman, Q. Nguyen, D. M. Smilgies and W. A. Tisdale, Chem. Mater., 2018, 30, 807–816 CrossRef CAS.
- K. Mukai, F. Suetsugu and K. Niwa, IEEE Trans. Nanotechnol., 2017, 16, 600–605 CAS.
- J. Pan, L. N. Quan, Y. Zhao, W. Peng, B. Murali, S. P. Sarmah, M. Yuan, L. Sinatra, N. M. Alyami, J. Liu, E. Yassitepe, Z. Yang, O. Voznyy, R. Comin, M. N. Hedhili, O. F. Mohammed, Z. H. Lu, D. H. Kim, E. H. Sargent and O. M. Bakr, Adv. Mater., 2016, 28, 8718–8725 CrossRef CAS.
- A. Ghorai, S. Mahato, S. K. Srivastava and S. K. Ray, Adv. Funct. Mater., 2022, 32, 2202087 CrossRef CAS.
- K. Hoshi, T. Chiba, J. Sato, Y. Hayashi, Y. Takahashi, H. Ebe, S. Ohisa and J. Kido, ACS Appl. Mater. Interfaces, 2018, 10, 24607–24612 CrossRef CAS PubMed.
- Q. Zhong, M. Cao, Y. Xu, P. Li, Y. Zhang, H. Hu, D. Yang, Y. Xu, L. Wang, Y. Li, X. Zhang and Q. Zhang, Nano Lett., 2019, 19, 4151–4157 CrossRef CAS.
- J. De Roo, M. Ibanez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CAS.
- A. Stelmakh, M. Aebli, A. Baumketner and M. V. Kovalenko, Chem. Mater., 2021, 33, 5962–5973 CrossRef CAS PubMed.
- Y. Shynkarenko, M. I. Bodnarchuk, C. Bernasconi, Y. Berezovska, V. Verteletskyi, S. T. Ochsenbein and M. V. Kovalenko, ACS Energy Lett., 2019, 4, 2703–2711 CrossRef CAS PubMed.
- H. Wu, Y. Zhang, M. Lu, X. Zhang, C. Sun, T. Zhang, V. L. Colvin and W. W. Yu, Nanoscale, 2018, 10, 4173–4178 RSC.
- M. Imran, P. Ijaz, L. Goldoni, D. Maggioni, U. Petralanda, M. Prato, G. Almeida, I. Infante and L. Manna, ACS Energy Lett., 2019, 4, 819–824 CrossRef CAS.
- D. Yang, X. Li, W. Zhou, S. Zhang, C. Meng, Y. Wu, Y. Wang and H. Zeng, Adv. Mater., 2019, 31, e1900767 CrossRef PubMed.
- J. Shamsi, D. Kubicki, M. Anaya, Y. Liu, K. Ji, K. Frohna, C. P. Grey, R. H. Friend and S. D. Stranks, ACS Energy Lett., 2020, 5, 1900–1907 CrossRef CAS PubMed.
- H. Wang, N. Sui, X. Bai, Y. Zhang, Q. Rice, F. J. Seo, Q. Zhang, V. L. Colvin and W. W. Yu, J. Phys. Chem. Lett., 2018, 9, 4166–4173 CrossRef CAS.
- B. W. Zhang, L. Goldon, J. Zito, Z. Y. Dang, G. Almeida, F. Zaccaria, J. de Wit, I. Infante, L. De Trizio and L. Manna, Chem. Mater., 2019, 31, 9140–9147 CrossRef CAS.
- F. Krieg, P. C. Sercel, M. Burian, H. Andrusiv, M. I. Bodnarchuk, T. Stoferle, R. F. Mahrt, D. Naumenko, H. Amenitsch, G. Raino and M. V. Kovalenko, ACS Cent. Sci., 2021, 7, 135–144 CrossRef CAS.
- F. Krieg, Q. K. Ong, M. Burian, G. Raino, D. Naumenko, H. Amenitsch, A. Suess, M. J. Grotevent, F. Krumeich, M. I. Bodnarchuk, I. Shorubalko, F. Stellacci and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 19839–19849 CrossRef CAS PubMed.
- S. Guo, H. Liu, H. He, W. Wang, L. Jiang, X. Xiong and L. Wang, Langmuir, 2020, 36, 6775–6781 CrossRef CAS.
- F. Krieg, S. T. Ochsenbein, S. Yakunin, S. ten Brinck, P. Aellen, A. Suess, B. Clerc, D. Guggisberg, O. Nazarenko, Y. Shynkarenko, S. Kumar, C. J. Shih, I. Infante and M. V. Kovalenko, ACS Energy Lett., 2018, 3, 641–646 CrossRef CAS.
- T. A. Cohen, Y. P. Huang, N. A. Bricker, C. S. Juhl, T. J. Milstein, J. D. MacKenzie, C. K. Luscombe and D. R. Gamelin, Chem. Mater., 2021, 33, 3779–3790 CrossRef CAS.
- J. Cao, J. Yin, S. Yuan, Y. Zhao, J. Li and N. Zheng, Nanoscale, 2015, 7, 9443–9447 RSC.
- L. Ruan, W. Shen, A. Wang, A. Xiang and Z. Deng, J. Phys. Chem. Lett., 2017, 8, 3853–3860 CrossRef CAS PubMed.
- L. Ruan, W. Shen, A. Wang, Q. Zhou, H. Zhang and Z. Deng, Nanoscale, 2017, 9, 7252–7259 RSC.
- L. Liu, L. Deng, S. Huang, P. Zhang, J. Linnros, H. Zhong and I. Sychugov, J. Phys. Chem. Lett., 2019, 10, 864–869 CrossRef CAS.
- S. Park, H. Cho, W. Choi, H. Zou and D. Y. Jeon, Nanoscale Adv., 2019, 1, 2828–2834 RSC.
- M. B. Rota, A. S. Arneruddin, H. A. Fonseka, Q. Gao, F. Mura, A. Polimeni, A. Miriametro, H. H. Tan, C. Jagadish and M. Capizzi, Nano Lett., 2016, 16, 5197–5203 CrossRef CAS PubMed.
- D. Tedeschi, H. A. Fonseka, E. Blundo, A. G. del Aguila, Y. N. Guo, H. H. Tan, P. C. M. Christianen, C. Jagadish, A. Polimeni and M. De Luca, ACS Nano, 2020, 14, 11613–11622 CrossRef CAS.
- J. Yi, X. Y. Ge, E. X. Liu, T. Cai, C. J. Zhao, S. C. Wen, H. Sanabria, O. Chen, A. M. Rao and J. B. Gao, Nanoscale Adv., 2020, 2, 4390–4394 RSC.
- X. Y. Zhang, X. Gao, G. T. Pang, T. C. He, G. C. Xing and R. Chen, J. Phys. Chem. C, 2019, 123, 28893–28897 CrossRef CAS.
- S. Rudin, T. L. Reinecke and B. Segall, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 11218–11231 CrossRef CAS.
- S. M. Lee, C. J. Moon, H. Lim, Y. Lee, M. Y. Choi and J. Bang, J. Phys. Chem. C, 2017, 121, 26054–26062 CrossRef CAS.
- J. Serrano, C. Schweitzer, C. T. Lin, K. Reimann, M. Cardona and D. Frohlich, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 125110 CrossRef.
- J. Bhosale, A. K. Ramdas, A. Burger, A. Munoz, A. H. Romero, M. Cardona, R. Lauck and R. K. Kremer, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 195208 Search PubMed.
- X. Y. Zhang, G. T. Pang, G. C. Xing and R. Chen, Mater. Today Phys., 2020, 15, 100259 CrossRef.
- K. Wei, Z. J. Xu, R. Z. Chen, X. Zheng, X. G. Cheng and T. Jiang, Opt. Lett., 2016, 41, 3821–3824 CrossRef CAS.
- C. L. Yu, Z. Chen, J. J. Wang, W. Pfenninger, N. Vockic, J. T. Kenney and K. Shum, J. Appl. Phys., 2011, 110, 63526 Search PubMed.
- Z. Liu, Q. Y. Shang, C. Li, L. Y. Zhao, Y. Gao, Q. Li, J. Chen, S. Zhang, X. F. Liu, Y. S. Fu and Q. Zhang, Appl. Phys. Lett., 2019, 114, 101902 CrossRef.
- C. Wolf and T. W. Lee, Mater. Today Energy, 2018, 7, 199–207 Search PubMed.
- Z. Zhao, M. Y. Zhong, W. C. Zhou, Y. H. Peng, Y. L. Yin, D. S. Tang and B. S. Zou, J. Phys. Chem. C, 2019, 123, 25349–25358 CrossRef CAS.
- M. Gramlich, C. Lampe, J. Drewniok and A. S. Urban, J. Phys. Chem. Lett., 2021, 12, 11371–11377 CAS.
- Y. S. Guo, O. Yaffe, T. D. Hull, J. S. Owen, D. R. Reichman and L. E. Brus, Nat. Commun., 2019, 10, 1175 CrossRef.
- J. Li, L. H. Luo, H. W. Huang, C. Ma, Z. Z. Ye, J. Zeng and H. P. He, J. Phys. Chem. Lett., 2017, 8, 1161–1168 CAS.
- D. Rosales, T. Bretagnon, B. Gil, A. Kahouli, J. Brault, B. Damilano, J. Massies, M. V. Durnev and A. V. Kavokin, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 125437 CrossRef.
- F. Minami, K. Hirata, K. Era, T. Yao and Y. Masumoto, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 2875–2878 CrossRef CAS.
- M. Nakayama, J. Lumin., 2000, 87–9, 15–19 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01769b |
| This journal is © The Royal Society of Chemistry 2024 |