
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacet-dependent synthesis of H2O2 from H2 and O2 over single Pt atom-modified Pd nanocrystal catalysts†
Ying
Zhang
a,
Qingdi
Sun
a,
Ziyue
Wang
a,
Guanghui
Guo
a,
Hao
Liu
a,
Xiaohui
He
 *ac and
Hongbing
Ji
*ab
*ac and
Hongbing
Ji
*ab
aKey Laboratory of Bioinorganic and Synthetic Chemistry of Ministry of Education, Fine Chemical Industry Research Institute, School of Chemistry, IGCME, Sun Yat-sen University, Guangzhou 510275, China. E-mail: jihb@mail.sysu.edu.cn; hexiaohui@mail.sysu.edu.cn
bState Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, Institute of Green Petroleum Processing and Light Hydrocarbon Conversion, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou, 310014, P. R. China
cGuangdong Technology Research Center for Synthesis and Separation of Thermosensitive Chemicals, China
First published on 22nd May 2024
Abstract
Hydrogen peroxide (H2O2) is one of the most valuable clean energy sources with a rapidly growing requirement in industry and daily life. The direct synthesis of H2O2 from hydrogen and oxygen is considered to be an economical and environmentally friendly manufacturing route to replace the traditional anthraquinone method, although it remains a formidable challenge owing to low H2O2 selectivity and production. Here, we report a catalyst consisting of Pd(111) nanocrystals on TiO2 modified with single Pt atoms (Pt1Pd(111)/TiO2), which displays outstanding reactivity, producing 1921.3 μmol of H2O2, a H2 conversion of 62.2% and H2O2 selectivity of 80.3% over 30 min. Kinetic and isotope experiments confirm that the extraordinary catalytic properties are due to stronger H2 activation (the rate-determining step). DFT calculations confirm that Pt1Pd(111) exhibits lower energy barriers for H2 dissociation and two-step O2 hydrogenation, but higher energy barriers for side reactions than Pt1Pd(100), demonstrating clear facet dependence and resulting in greater selectivity and amount of H2O2 produced.
Introduction
Hydrogen peroxide (H2O2) is considered to be one of the most crucial chemicals in the world. It has been widely used in various fields, including the synthesis of chemicals and pharmaceuticals, environmental protection, sterilization, and bleaching, because of its strong oxidation capability of which the exclusive byproduct is water.1–4 The environmental friendliness and effectiveness of H2O2 have caused a continuous increase in its use and production.5 Currently, more than 95% of industrial H2O2 production is highly dependent on the anthraquinone process, which is a large-scale multi-step process involving intense energy input, hazardous organic compounds, and highly concentrated H2O2 solutions.6–10 However, it is important to note that more than two-thirds of end-use application demand is for low concentration H2O2 (less than 9 wt%).11–13 Under such circumstances, environmental and economic factors have led to a strong interest in low-scale and low-energy processes to synthesize hydrogen peroxide.14,15Compared with conventional processes, the direct synthesis of H2O2 (DSHP) is considered to be a green and appealing alternative clean manufacturing route that can potentially produce H2O2 in a ready-to-use, eco-friendly manner.16–18 Despite the above-mentioned advantages, the low yield of H2O2 owing to its thermodynamically favorable side reactions (including the dissociation of O2 to form H2O, H2O2 hydrogenation, and H2O2 decomposition, as shown in Scheme 1) means that the DSHP remains a tremendous challenge.19 Pd is regarded as a credible catalytic component for the DSHP owing to its superior hydrogen activation ability, especially at low operating temperatures. However, pure Pd is not only active in the synthesis of H2O2 but also in the aforementioned side reactions. Therefore, researchers improved the DSHP catalytic performance by incorporating a second component and found that the reaction path could be regulated by finely tuning the electronic environment around the Pd species.13,17,18,20–23 For example, Hutchings and co-workers reported a PdSn/TiO2 catalyst with a layer of tin oxide wrapped around the surface of small Pd-rich particles synthesized via an appropriate heat treatment cycle, which could shut down the successive hydrogenation and decomposition reactions, thus improving the catalytic performance of Pd in the DSHP.17 Lewis and co-workers reported that the introduction of base metals into a AuPd catalyst improved the catalytic performance towards the DSHP, attributing the improvement to the modification of the electronic environment of Pd.24 Additionally, single-atom species, due to their unique electronic structures, can also be used as electronic promoters to modify the electronic structure of the active sites.25,26 For example, Wang and co-workers reported that single Pt atoms can operate as electronic promoters to effectively modify the electronic structure of catalysts, thus improving the catalytic performance in the water-gas shift reaction.27 However, the lack of a complete understanding of the inherent mechanism of the Pd-catalyzed DSHP has seriously hindered its practical application.28 Therefore, more investigations are desired to further understand the structure–activity relationships in order to obtain more active, selective and stable catalysts for the DSHP.
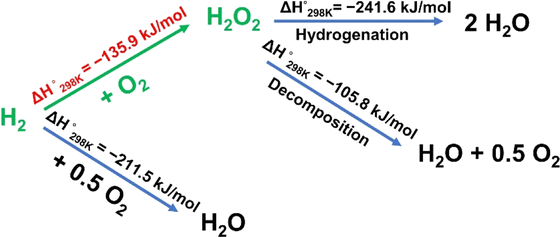 | ||
| Scheme 1 Reactions and corresponding enthalpies in the direct synthesis of H2O2. | ||
It is also noteworthy that the surface morphology of the active sites plays an essential role in regulating the catalytic activity, as the properties of the active sites and the energetics of the reaction are ultimately determined by the exposed facets.29–31 Nanocrystals with facet-controlled shapes can provide an accessible platform to reveal the underlying mechanisms of catalytic reactions, since the atomic arrangements and well-documented electronic configurations of specific facets are beneficial for revealing structure–activity relationships.32–36
Inspired by the above discussion, we aim to develop a DSHP catalyst with high performance while providing an in-depth understanding of the reaction mechanism. Herein, we employ single Pt atoms as promoters to modify the surface interface structure of Pd nanocrystals with different exposed facets for the DSHP. Interestingly, Pt1Pd nanocrystal catalysts with Pd(111) surfaces (Pt1Pd(111)/TiO2) not only showed a prominent facet-dependent effect but also exhibited outstanding catalytic activity when compared to catalysts with Pd(100) surfaces (Pt1Pd(100)/TiO2). The amount of generated H2O2 reached 1921.3 μmol for 30 min. Kinetic experiments and theoretical calculations revealed the possible mechanism by which the catalysts with Pt1Pd(111) surfaces can effectively reduce the H2 dissociation and O2 reduction energy barriers, thus facilitating the H2O2 synthesis process.
Results and discussion
Synthesis of catalysts and catalytic performance
Single Pt atom were introduced as promoters into Pd nanocrystals using a simple solvothermal method, as shown in Fig. S1,† according to a slightly modified previous report.37 The detailed procedure is given in the catalyst preparation section in the Materials and methods. The synthesis of the Pt1Pd(111) nanocrystals was conducted in an aqueous solution containing K2PdCl4, H2PtCl6·6H2O, PVP, citric acid, and L-ascorbic acid at 120 °C for 3 h. The purified Pt1Pd(111) nanocrystals were collected as a colloidal dispersion in water. The transmission electron microscopy (TEM) images displayed mainly monodisperse octahedron-shaped nanocrystals with a size of 8.9 ± 1.0 nm (Fig. 1a–c). High-resolution TEM images (Fig. 1d–f) revealed the individual crystalline character of the nanocrystals and that they have a lattice spacing of 2.25 Å, which was in good agreement with the Pd metal phase (2.26 Å of PDF#87-0643 for Pd(111)). The successful preparation of the Pt1Pd(111) nanocrystals also was certified by the X-ray diffraction (XRD) results (Fig. S2a†). Next, Pt1Pd(111)/TiO2 was obtained by impregnating the Pt1Pd(111) nanocrystals onto TiO2. The inductively coupled plasma optical emission spectrometry (ICP-OES) results indicated that the Pd loading was 3.3 wt% and the Pt loading was 0.09 wt% (Table S1†), which were close to the nominal loadings. The dark- and bright-field scanning transmission electron microscopy (STEM) images and the XRD results (Fig. 1g and S2†) suggested that the Pt1Pd(111) nanocrystals were uniformly dispersed on the TiO2 and still retained their octahedral shape. A detailed characterization of the dispersion of Pt and Pd atoms in Pt1Pd(111)/TiO2 was obtained via aberration-corrected high-angle annular dark-field STEM (AC HAADF-STEM). Fig. 1h showed that the individual Pt1Pd(111) nanocrystals were supported on TiO2. Single Pt atoms on the Pd(111) surface were clearly observed and are highlighted by yellow circles in Fig. 1h. Furthermore, the elemental analysis mapping (Fig. 1i) and elemental line–scan profiles (Fig. S3†) confirmed a few Pt atoms were uniformly distributed on the surface of Pd(111).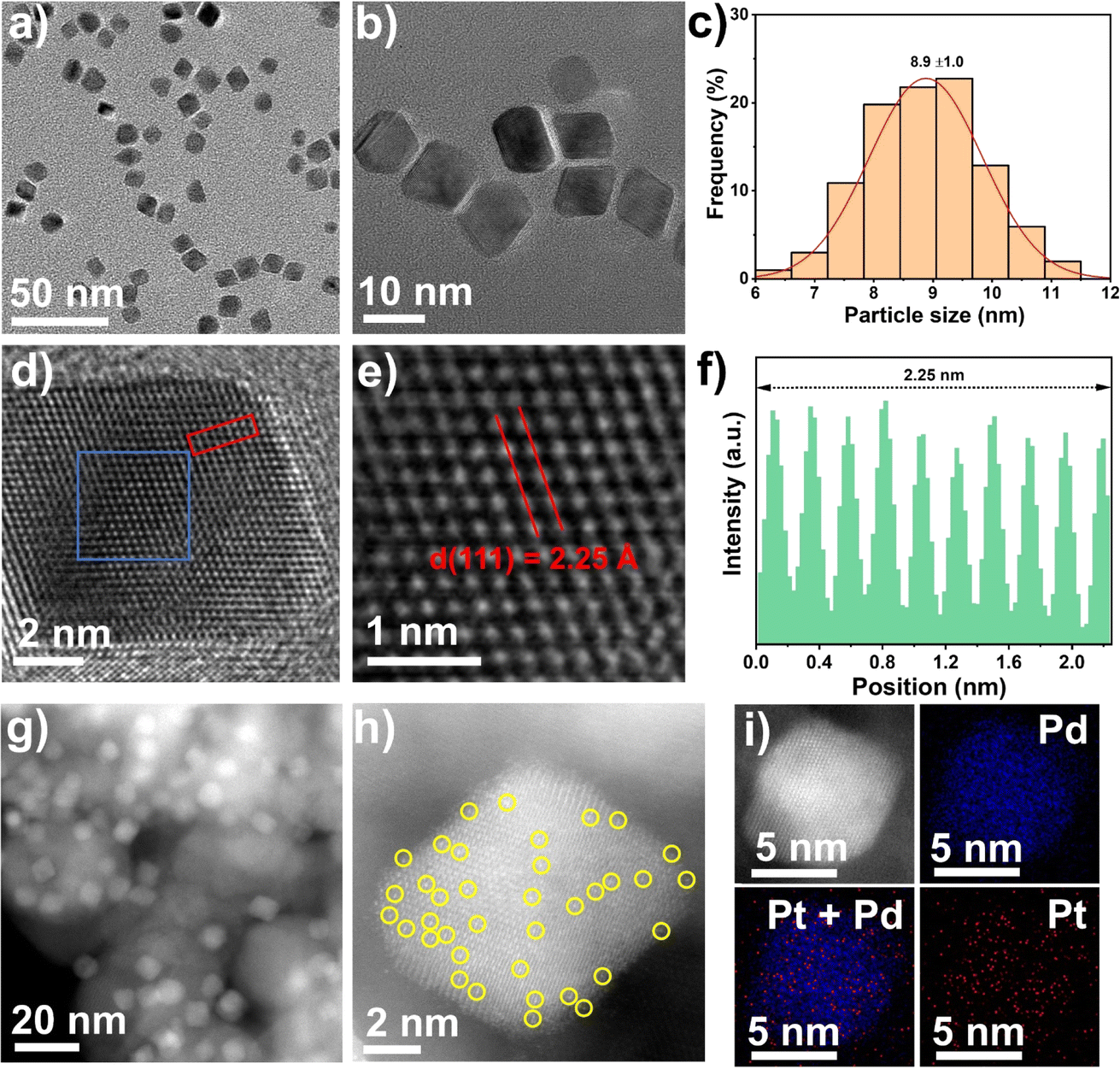 | ||
| Fig. 1 Morphology and structure of the monodispersed Pt1Pd(111) nanocrystals. (a) Representative TEM image. (b and d) High-resolution TEM images. (c) Pt1Pd(111) nanocrystals particle size distribution. (e) Magnified TEM image of the blue box from (d). (f) Intensity profile of the red box from (d). (g) The dark-field STEM image of the Pt1Pd(111) nanocrystals supported on TiO2 (Pt1Pd(111)/TiO2). (h) AC HAADF-STEM images of Pt1Pd(111)/TiO2, single Pt atoms are highlighted by the yellow circles. (i) EDS mapping of Pt1Pd(111)/TiO2. | ||
In order to investigate the influence of the exposed facets on the DSHP, unsupported Pt1Pd(100) nanocrystals and Pt1Pd(100)/TiO2 (Pd loading of 3.4 wt% and Pt loading of 0.08 wt%, Table S1†) were synthesized via a similar method using KBr as a capping agent at 80 °C, as shown in Fig. S1.† The detailed structural characterization is shown in Fig. S4–6.† The TEM images showed that the cubic shaped Pt1Pd(100) nanocrystals with a size of 10.5 ± 1.8 nm and lattice spacing of 1.95 Å (1.95 Å of PDF#87-0643 for Pd(200)) were successfully prepared. STEM images and XRD indicated that the Pt1Pd(100) nanocrystals were uniformly dispersed on the TiO2 and still retained their cubic shape. AC HAADF-STEM and elemental analysis mapping confirmed the uniform distribution of the individual Pt atoms on the Pd(100) surface. For comparison, Pd(111)/TiO2 (Pd loading of 3.2 wt% and particle size of 7.8 ± 1.1 nm) and Pd(100)/TiO2 (Pd loading of 3.6 wt% and particle size of 10.8 ± 1.8 nm) were also successfully synthesized by the same method without adding H2PtCl6·6H2O, as evidenced by TEM, STEM images and XRD (Table S1 and Fig. S7–10†).
In a typical reaction, the catalytic test of the DSHP was conducted in a 100 mL stainless-steel autoclave at 0 °C employing methanol as the solvent. Initially, Pd(100)/TiO2 showed low H2 conversion (16.8%), H2O2 selectivity (49.3%), productivity (2.0 mol gPd−1 h−1), and amount of generated H2O2 (360.0 μmol) (Table 1, entry 1). After adding the single Pt atoms to the Pd(100) surface, the catalyst (Pt1Pd(100)/TiO2) showed higher activity (H2 conversion of 38.2%, H2O2 selectivity of 60.0%, productivity of 5.8 mol gPd−1 h−1, and generated H2O2 of 985.0 μmol) (Table 1, entry 3) than Pd(100)/TiO2. Interestingly, the catalytic performance (H2 conversion of 24.4%, H2O2 selectivity of 49.5%, productivity of 3.3 mol gPd−1 h−1, generated H2O2 of 525.0 μmol on Pd(111)/TiO2, and H2 conversion of 62.2%, H2O2 selectivity of 80.3%, productivity of 11.8 mol gPd−1 h−1, generated H2O2 of 1921.3 μmol on Pt1Pd(111)/TiO2) (Table 1, entries 2 and 4) were obviously enhanced by adjusting the exposed Pd(100) surface to Pd(111) surface, implying a facet-dependent synthesis of H2O2 from H2 and O2. Moreover, the above results also implied that the presence of the Pt single atoms improved the selectivity and activity of the H2O2 synthesis. To the best of our knowledge, the amount of generated H2O2 on Pt1Pd(111)/TiO2 was notable compared to previous reports1,7,14,17,21,38,39 under similar reaction conditions (Table 1, entries 5–11). In addition, the hydrogenation and decomposition of H2O2 were carried out. As shown in Fig. S11,† the H2O2 hydrogenation and decomposition rates on the Pd(111) surfaces were lower than those on the Pd(100) surfaces, indicating that H2O2 hydrogenation and decomposition were inhibited on the Pd(111) surfaces, in line with the observed higher H2O2 selectivity on Pt1Pd(111)/TiO2.
| Entry | Catalyst | H2 conversion (%) | H2O2 selectivity (%) | Productivity (mol gPd−1 h−1) | Amount of H2O2 (μmol) | Time (min) | Reference |
|---|---|---|---|---|---|---|---|
| a The reaction conditions of H2O2 synthesis: 10 mL CH3OH, 0.02 M HCl, 2.9 MPa 5% H2/N2, 1.1 MPa 25% O2/N2, T = 0 °C, catalyst weight: 10 mg, stirring: 1200 rpm, reaction time: 30 min. | |||||||
| 1 | Pd(100)/TiO2 | 16.8 | 49.3 | 2.0 | 360.0 | 30 | This worka |
| 2 | Pd(111)/TiO2 | 24.4 | 49.5 | 3.3 | 525.0 | 30 | This worka |
| 3 | Pt1Pd(100)/TiO2 | 38.2 | 60.0 | 5.8 | 985.0 | 30 | This worka |
| 4 | Pt1Pd(111)/TiO2 | 62.2 | 80.3 | 11.8 | 1921.3 | 30 | This worka |
| 5 | Pd6Pb NRs/TiO2-H-A | 40.0 | 56.7 | 5.7 | 447.7 | 30 | 1 |
| 6 | 0.1%O–Pd/TiO2 | 3.9 | >99 | 115 | 143.8 | 30 | 7 |
| 7 | PdL/PdSn-NW | 22.1 | 95.3 | 12.8 | 656.0 | 15 | 14 |
| 8 | 3 wt% Pd-2 wt% Sn/TiO2 | 9.0 | 96.0 | 2.0 | 300.0 | 30 | 17 |
| 9 | AuPd@HZSM-5 | 28.0 | 90.0 | 1.1 | 396.0 | 30 | 21 |
| 10 | Pd-HHDMA/C | 9.7 | 79.8 | 12.8 | 108.8 | 30 | 38 |
| 11 | Pd-Sb/TiO2 | 13.9 | 73.0 | 1.6 | 388.0 | 10 | 39 |
Fig. 2a–c and S12† show the catalytic activity as a function of reaction time for Pd(111)/TiO2, Pd(100)/TiO2, Pt1Pd(111)/TiO2, and Pt1Pd(100)/TiO2. The H2 conversion (Fig. 2a) increased linearly with reaction time for all four catalysts. For example, the H2 conversion over Pt1Pd(111)/TiO2 increased from 30.7% to 81.4% as the reaction time was extended from 10 to 60 min, which was higher than that of Pt1Pd(100)/TiO2 (16.0% to 57.2%), Pd(100)/TiO2 (12.5% to 23.0%), and Pd(111)/TiO2 (13.7% to 38.2%). Fig. 2b reveals that Pt1Pd(111)/TiO2 provided the highest H2O2 selectivity (∼80%) with almost no degradation after 60 min of reaction. Fig. 2c and S12† show that the amount of generated H2O2, H2O2 concentration and H2O2 yield increased with reaction time for all four catalysts. An H2O2 concentration of 1.10% and H2O2 yield of 66.9% were achieved after 60 min of reaction over Pt1Pd(111)/TiO2. The amount of generated H2O2 was 2575.0 μmol in 60 min, which was 3.6, 5.0, and 1.8 times that of Pd(111)/TiO2 (710.0 μmol), Pd(100)/TiO2 (510.0 μmol), and Pt1Pd(100)/TiO2 (1402.5 μmol), respectively. In another set of experiments, the effect of the H2/O2 ratio on the DSHP was examined. As shown in Fig. 2d–f and S13,† the catalytic performance of the four catalysts, including the H2 conversion, amount of H2O2, and H2O2 concentration, gradually increased when the ratio of H2/O2 was increased from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 1:2. However, the H2O2 selectivity gradually decreased as the H2/O2 ratio increased, and when the H2/O2 ratio was lower than 1:8, the H2O2 selectivity was capable of reaching 100%. Moreover, the optimal H2O2 yield could be obtained when the H2/O2 ratio was 1:3. The performance on the Pd(111) surface was always better than on the Pd(100) surface, and the performance of the catalysts with single Pt atoms was always greater than those without single Pt atoms. All the above results indicate that the DSHP catalytic activity displays a facet-dependence and that it is more favorable on Pd(111) than Pd(100).
:10 to 1:2. However, the H2O2 selectivity gradually decreased as the H2/O2 ratio increased, and when the H2/O2 ratio was lower than 1:8, the H2O2 selectivity was capable of reaching 100%. Moreover, the optimal H2O2 yield could be obtained when the H2/O2 ratio was 1:3. The performance on the Pd(111) surface was always better than on the Pd(100) surface, and the performance of the catalysts with single Pt atoms was always greater than those without single Pt atoms. All the above results indicate that the DSHP catalytic activity displays a facet-dependence and that it is more favorable on Pd(111) than Pd(100).
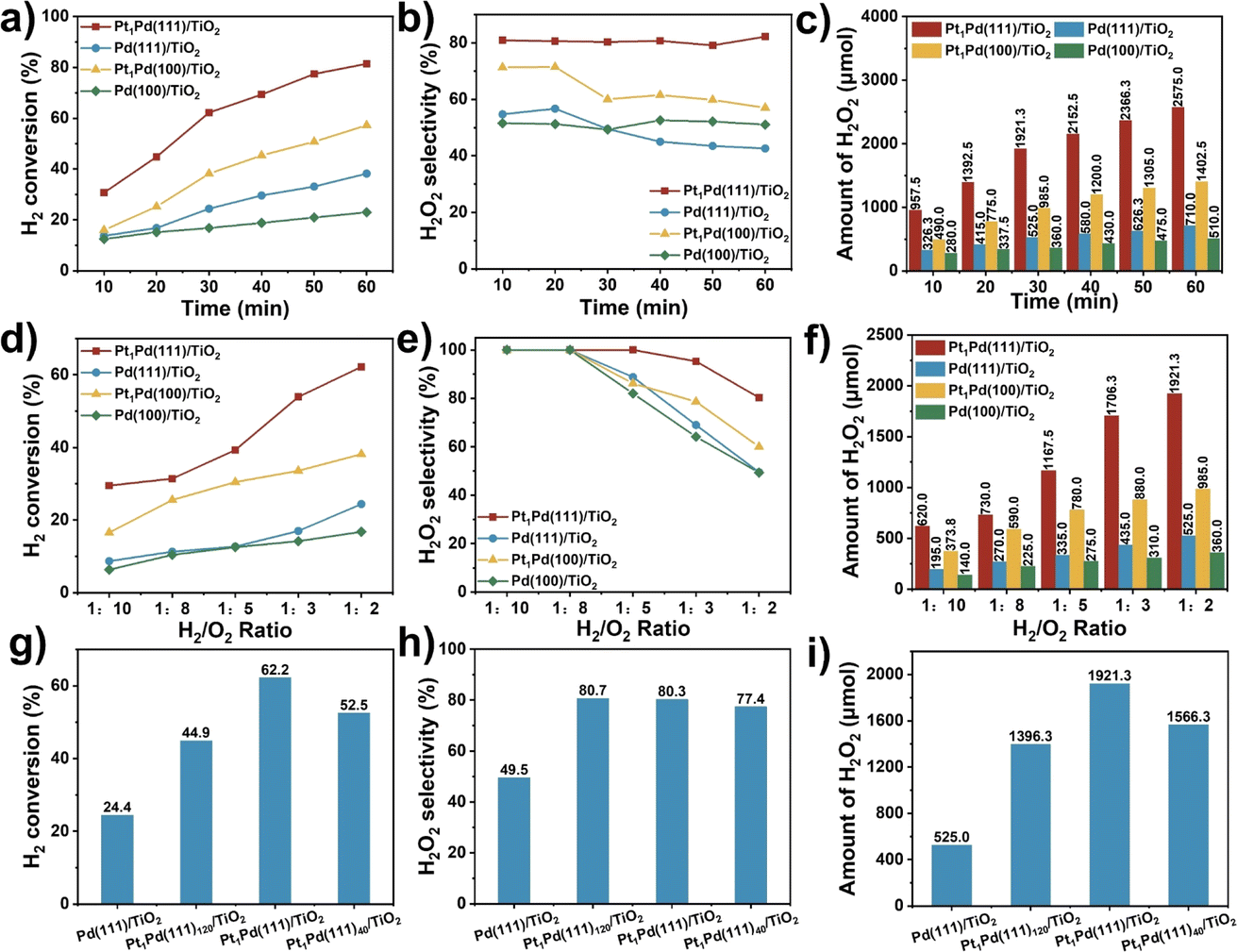 | ||
| Fig. 2 Comparison of (a) H2 conversion, (b) H2O2 selectivity and (c) amount of H2O2 as a function of H2O2 synthesis reaction time. Comparison of (d) H2 conversion, (e) H2O2 selectivity and (f) amount of H2O2 at different H2/O2 ratios. Comparison of (g) H2 conversion, (h) H2O2 selectivity and (i) amount of H2O2 at different Pd/Pt atomic ratios. Reaction conditions: 10 mL CH3OH, 0.02 M HCl, 2.9 MPa 5% H2/N2, 1.1 MPa 25% O2/N2, T = 0 °C, catalyst weight: 10 mg, stirring: 1200 rpm, reaction time: 30 min. | ||
In order to evaluate the influence of the Pt content on the reaction, catalysts with similar Pd content (∼3.5 wt%) and different Pt contents (0.07 to 0.17 wt%) were prepared, and each sample was characterized accordingly using ICP-OES and XRD (Table S1 and Fig. S14†). The DSHP activity measurements across the range of Pt content showed a volcano-like trend (Fig. 2g–i and S15a†). The optimal catalytic performance was obtained at a Pt/Pd ratio of 1:80, while the H2O2 selectivity remained almost the same throughout (∼80%), implying that the addition of single Pt atoms produced a large increase in the catalytic activity and selectivity. However, the results of H2O2 hydrogenation indicated that the hydrogenation rate increased (from 1.1 to 8.9 mol gPd−1 h−1) as the Pt content increased (Fig. S15b†), while Fig. S15c† shows that the addition of Pt can suppress the decomposition of H2O2. Furthermore, the DSHP activity of the Pt1Pd(111)/TiO2 catalyst in four different solvents was investigated, including H2O, CH3OH, C2H5OH, and 30% H2O + 70% CH3OH. The results in Fig. S16† show that alcohols are beneficial for improving the DSHP catalytic activity, with methanol being the best solvent and H2O resulting in the lowest activity due to the fact that short-chain alcohols facilitate the mass transfer of H2.40 Eventually, the stability of Pt1Pd(111)/TiO2 was tested whilst the H2 conversion was maintained at ∼30%. As show in Fig. S17,† the catalytic activity remained almost unchanged after five cycles of testing, and the amount of generated H2O2 remained at ∼1000 μmol, showing extraordinary stability. The morphology (Fig. S18†) and metal content (Table S1†) of Pt1Pd(111)/TiO2 remained almost unchanged after the reaction.
Finally, utilizing the H2O2 solution in the Fenton system, pollutants (e.g., rhodamine B) can be effectively removed. Rhodamine B was added to the H2O2 solution together with FeCl2. Afterwards, the rhodamine B was totally degraded, as evidenced by color changes and UV-visible spectroscopy (Fig. S19 and S20†).
Spectroscopy of catalysts
To understand the enhancement mechanism in detail, the CO binding strength of the catalysts with different exposed facets was investigated at ambient pressure using IR spectroscopy (Fig. 3). The signals in Fig. 3a–d were basically steady after purging for 30 minutes, and the peaks of CO absorption on the four catalysts at ∼1978 and ∼1886 cm−1 were ascribed to the bridged (CO–Pd2) and threefold hollow (CO–Pd3) configurations, respectively,41,42 indicating that the Pd species mainly exist as metallic Pd. However, a weak peak at 2078 cm−1 across Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 was observed and can be assigned to linearly absorbed CO on the Pt atoms.43,44 This peak was not observed on Pd(111)/TiO2 and Pd(100)/TiO2 (Fig. 3e–i), suggesting that the Pt species exist as single atoms on Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2.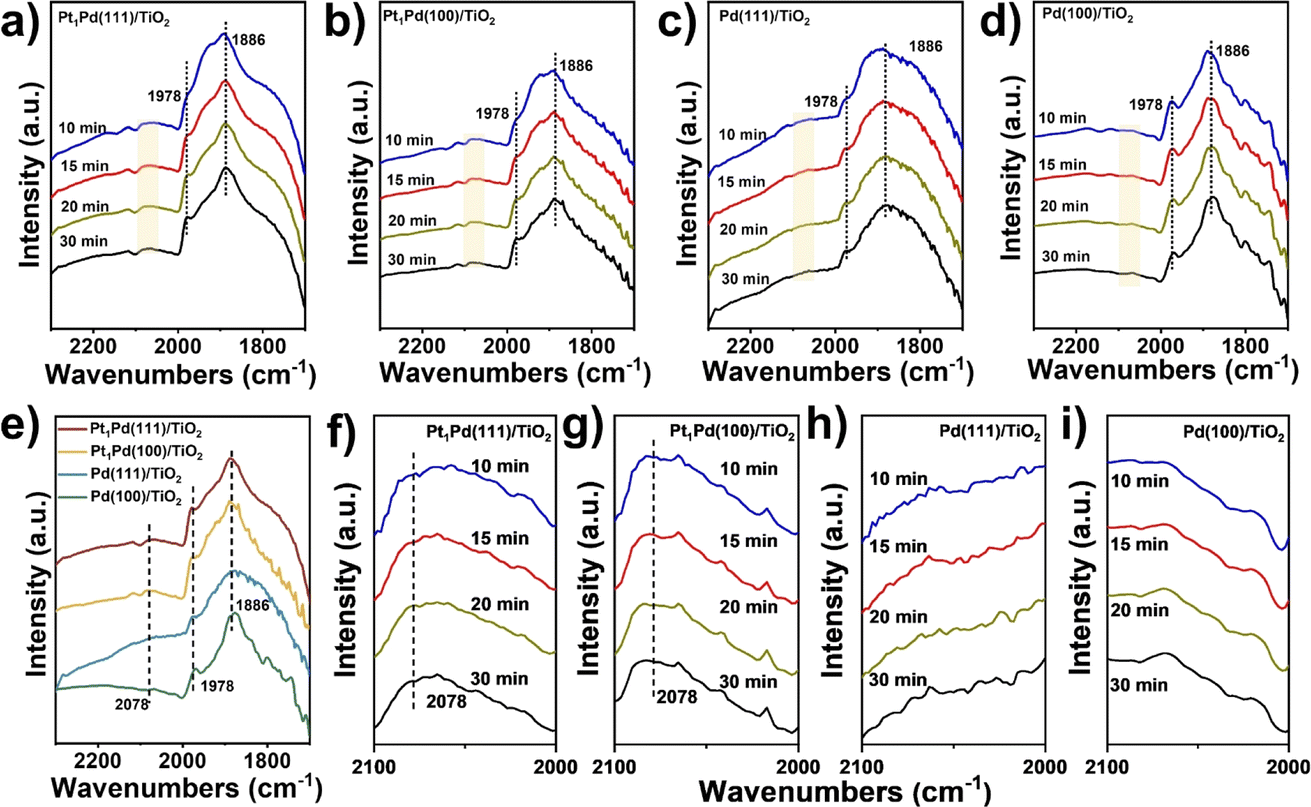 | ||
| Fig. 3 DRIFT spectra of CO adsorption on (a) Pt1Pd(111)/TiO2, (b) Pt1Pd(100)/TiO2, (c) Pd(111)/TiO2 and (d) Pd(100)/TiO2 at room temperature at different time points. (e) Comparison of CO-DRIFT spectra at 30 min. (f–i) Magnified CO-DRIFT spectra between 2000 and 2100 cm−1 from (a), (b), (c) and (d), respectively. | ||
To probe the local electronic states and coordination structures of the Pt and Pd atoms in the different exposed facet catalysts, X-ray adsorption spectroscopy (XAS) was carefully performed. As shown in Fig. 4a, the Pd K-edge X-ray absorption near edge structure (XANES) of Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 was similar to that of Pd foil, indicating that the Pd species exist in a metallic state. In addition, the near-edge features of Pt1Pd(111)/TiO2 shifted to a lower valence state compared to that of Pt1Pd(100)/TiO2, indicating the existence of slightly electron-rich Pd species, which is beneficial for H2O2 synthesis.45Fig. 4b shows the Fourier-transform extended X-ray absorption fine structure (EXAFS) spectra of Pd foil, Pt1Pd(111)/TiO2, and Pt1Pd(100)/TiO2, with the main peak at 2–3 Å arising from Pd–Pd/Pt bonding (yellow region in Fig. 4b).46 The results were further confirmed by the K-space spectra (Fig. S21†) and wavelet transform (WT) for the Pd K-edge (Fig. 4c). The Pt L3-edge XANES and EXAFS spectra were consistent with that of Pd. The Pt L3-edge XANES in Fig. 4d shows that the valence state of the Pt species in Pt1Pd(111)/TiO2 is higher than that in Pt1Pd(100)/TiO2. The EXAFS of Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 in Fig. 4e have two peaks at 2.2 and 3.1 Å, which were assigned to Pt–Pd bonding and Pt–Pd–Pt bonding, respectively,47 which was further confirmed by the K-space spectra (Fig. S22†) and wavelet transform (WT) (Fig. 4f). The fitting parameters (Fig. S23, S24 and Tables S2, S3†), including the coordination number and bond length, suggested a stronger interaction exists between the Pd and Pt species on Pd(111) than on Pd(100), indicating a greater amount of electron transfer occurs in Pt1Pd(111)/TiO2.
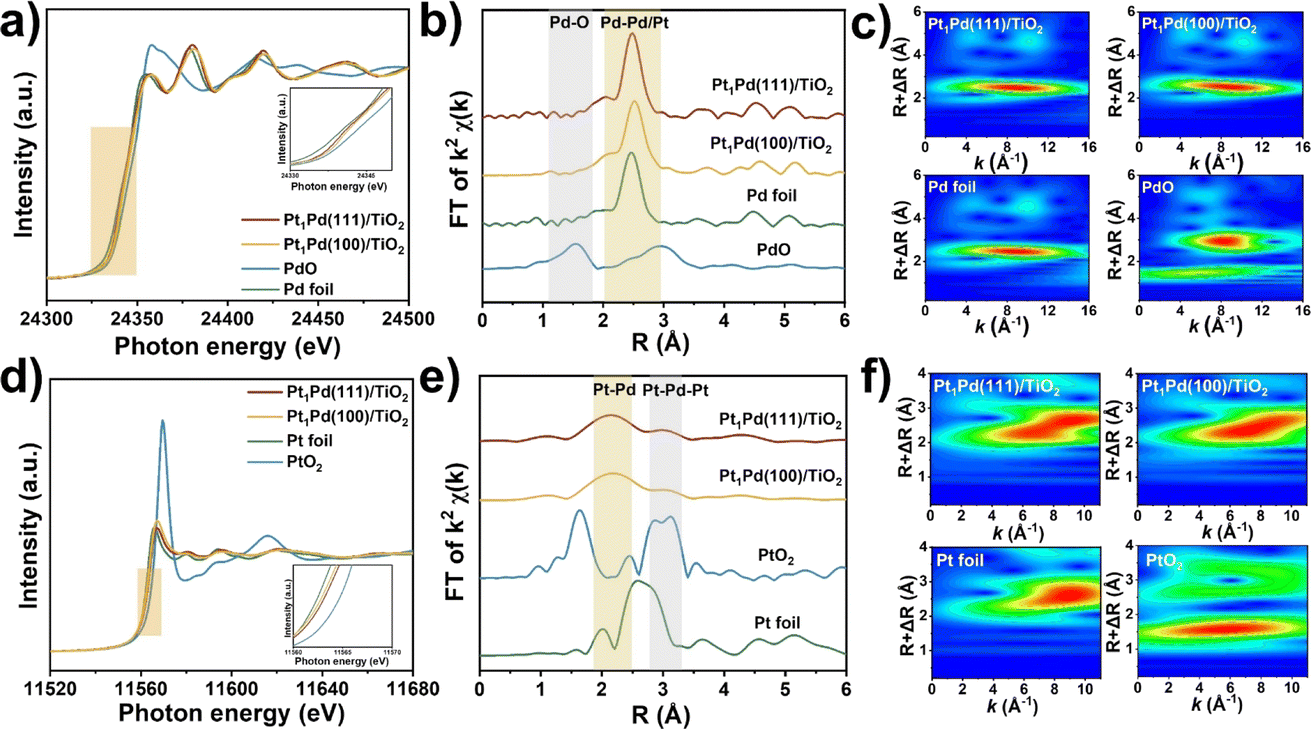 | ||
| Fig. 4 (a) Pd K-edge XANES profiles in R space. (b) Pd extended X-ray absorption fine structure (EXAFS) spectra. (c) Wavelet transform (WT) for the Pd K-edge of Pt1Pd(111)/TiO2, Pt1Pd(100)/TiO2, Pd foil, and PdO. (d) Pt L3-edge XANES profiles in R space. (e) Pt extended X-ray absorption fine structure (EXAFS) spectra. (f) Wavelet transform (WT) for the Pt L3-edge of Pt1Pd(111)/TiO2, Pt1Pd(100)/TiO2, Pt foil, and PtO2. | ||
X-ray photoelectron spectroscopy (XPS) was carried out on Pd 3d, as shown in Fig. S25.† The peaks at 340.5 and 335.3 eV of Pd(111)/TiO2 assigned to the Pd 3d3/2 and Pd 3d5/2 orbitals shift to a lower binding energy than is observed in Pd(100)/TiO2 (340.6 and 335.4 eV, respectively). Similarly, after adding the single Pt atoms, the peaks Pd 3d3/2 and Pd 3d5/2 of Pt1Pd(111)/TiO2 (340.3 and 335.1 eV, respectively) still appear at lower energy than those of Pt1Pd(100)/TiO2 (340.5 and 335.3 eV, respectively). In summary, the aforementioned results indicated that the Pd(111) and Pd(100) surfaces featured different electronic environments, with more electrons enriched on the Pd(111) surfaces, which is more favorable for dissociation of H2, as discussed below.48 Moreover, the addition of single Pt atoms enabled the peaks of the Pd species to shift to lower binding energies, favoring H2O2 synthesis.
Kinetics and isotope experiments
Herein, the effects of stirring rate, catalyst weight, reaction time, temperature, and H2 pressure on the DSHP in the critical kinetic region were systematically evaluated. Control experiments were performed by varying stirring rate and catalyst weight to exclude the impacts of external diffusion. Fig. S26† shows that the H2O2 concentration was basically unchanged (∼0.8 wt%) when the stirring rate was higher than 1200 rpm (1200 rpm was selected in our experiments). Furthermore, Fig. S27† shows that the H2 conversion is linearly correlated to the catalyst weight. Thus, the above results imply that the DSHP reaction was kinetically controlled and was not dependent on external diffusion.As shown in Fig. 5a, the curve of ln(c(H2) mol L−1) as a function of reaction time (min) gave a straight line, indicating a pseudo first-order reaction with respect to the DSHP. The Pt1Pd(111)/TiO2 (0.027 min−1) and Pd(111)/TiO2 (0.007 min−1) catalysts showed higher apparent rate constants than Pt1Pd(100)/TiO2 (0.014 min−1) and Pd(100)/TiO2 (0.003 min−1), indicating that the catalysts with Pd(111) facets exhibited higher activity than those with Pd(100) facets. The apparent rate constant for the DSHP using D2 was obtained for Pt1Pd(111)/TiO2, as shown in Fig. S28.† An obvious kinetic isotope effect (KIE) of kH2/kD2 at 3.0 was identified, suggesting that H2 activation contributes to the rate-determining step. The Arrhenius plots in Fig. 5b show that the apparent activation energy values (Ea) for Pt1Pd(111)/TiO2 (11.1 kJ mol−1) and Pd(111)/TiO2 (21.4 kJ mol−1) were lower than those for Pt1Pd(100)/TiO2 (14.0 kJ mol−1) and Pd(100)/TiO2 (22.1 kJ mol−1). Notably, a preferential H2O2 synthesis activity (H2 reaction order of 1.7 for Pt1Pd(111)/TiO2 and 2.1 for Pd(111)/TiO2, Fig. 5c) was achieved over the catalysts with the Pd(111) facets, which was lower than those achieved over the catalysts with Pd(100) facets (H2 reaction order of 1.8 for Pt1Pd(100)/TiO2 and 2.2 for Pd(100)/TiO2). This implies that the DSHP on the catalysts with Pd(111) facets were more favorable than on those with Pd(100) facets. Fig. 5d illustrates that the four catalysts share a similar H2 reaction order (∼1.6), meaning that the hydrogenation of H2O2 is not a dominant step affecting the H2O2 selectivity. H2–D2 exchange experiments were also conducted to investigate the ability of H2 activation on Pt1Pd(111)/TiO2, Pt1Pd(100)/TiO2, Pd(111)/TiO2, and Pd(100)/TiO2. As shown in Fig. 5e, the HD peaks of Pt1Pd(111)/TiO2 and Pd(111)/TiO2 were much higher than those of Pt1Pd(100)/TiO2 and Pd(100)/TiO2, indicating that H2 activation on the Pd(111) surface was more favorable than on the Pd(100) surface, leading to a higher H2 conversion rate on catalysts with Pd(111) facets. Furthermore, the above kinetics and isotope experiments also indicated that the catalysts with single Pt atoms were favorable for H2O2 synthesis.
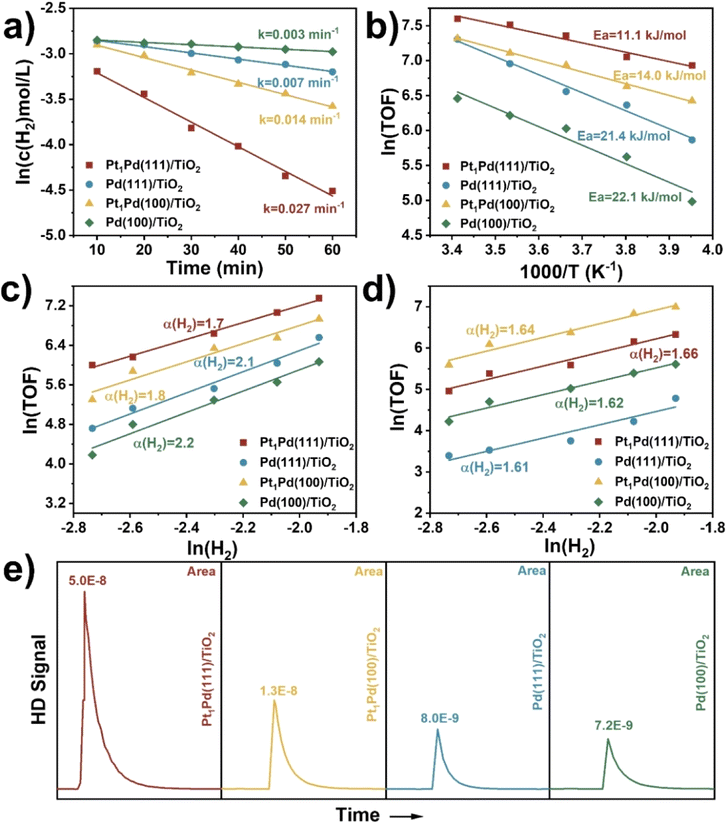 | ||
| Fig. 5 (a) The apparent rate constant and (b) the experimental Arrhenius plots for H2O2 synthesis. H2 reaction orders of (c) H2O2 synthesis and (d) H2O2 hydrogenation. (e) HD signal in H2-D2 exchange experiments at room temperature. 5% H2/N2 (20 mL min−1) as carrier and D2 (10 mL min−1) as pulse gas, 100 μL quantitative ring, catalysts: 50 mg. The apparent rate constant (k) is the slope of the corresponding fitted curve. The TOF was the number for moles of H2 converted on one mole of Pd per unit time. The H2 reaction orders (α) is the slope of the corresponding fitted curve. The Ea is the slope of the corresponding fitted curve multiplied by −8.314. | ||
Density-functional theory calculations
Based on the above reactivity and characterization results, we have developed Pt1Pd(111)/TiO2 catalysts with superior catalytic activity relating to the exposed facets. To better understand the how the different exposed facets, Pd(111) and Pd(100), affect the reaction mechanism density-functional theory (DFT) calculations were performed. The simulation models (Fig. S29†) of Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 were built according to the reflections obtained from the AC HAADF STEM and EXAFS analyses. To determine the H2 adsorption site, the adsorption energies of H2 at different Pd sites over Pt1Pd(111)/TiO2 were calculated (the corresponding adsorption models are shown in Fig. S30a–c).† Fig. S31a† shows that the adsorption energies of H2 at Pd0 (the Pd site connected to the Pt atom), Pd1 (the Pd site away from the Pt atom), and Pd1+ (the Pd site further away from the Pt atom) were −0.45, −0.31, and −0.20 eV, respectively, indicating that H2 was preferentially adsorbed on the Pd sites connected to the Pt atom. Moreover, H2 was also preferentially adsorbed on the Pd0 site over O2 (adsorption energies of −0.30 eV), as shown in Fig. S30d and S31b.† Thus the possible elementary steps and mechanism of the DSHP on Pt1Pd(111)/TiO2 are proposed in Fig. 6a. Firstly, H2 is adsorbed on the Pt1Pd(111)/TiO2 surface (* + H2 → *H2) followed by dissociation into 2*H absorbed on a Pd site (*H2 → *H + *H). Then O2 is adsorbed on the Pt1Pd(111)/TiO2 surface (O2 + *H → *O2 + *H) reacting with the adjacent *H to form *OOH (*O2 + *H → *OOH). Subsequently, *OOH combines with another *H to form * H2O2 (*OOH + *H → *H2O2), and finally H2O2 is desorbed from the Pt1Pd(111)/TiO2 surface (*H2O2 → * + H2O2).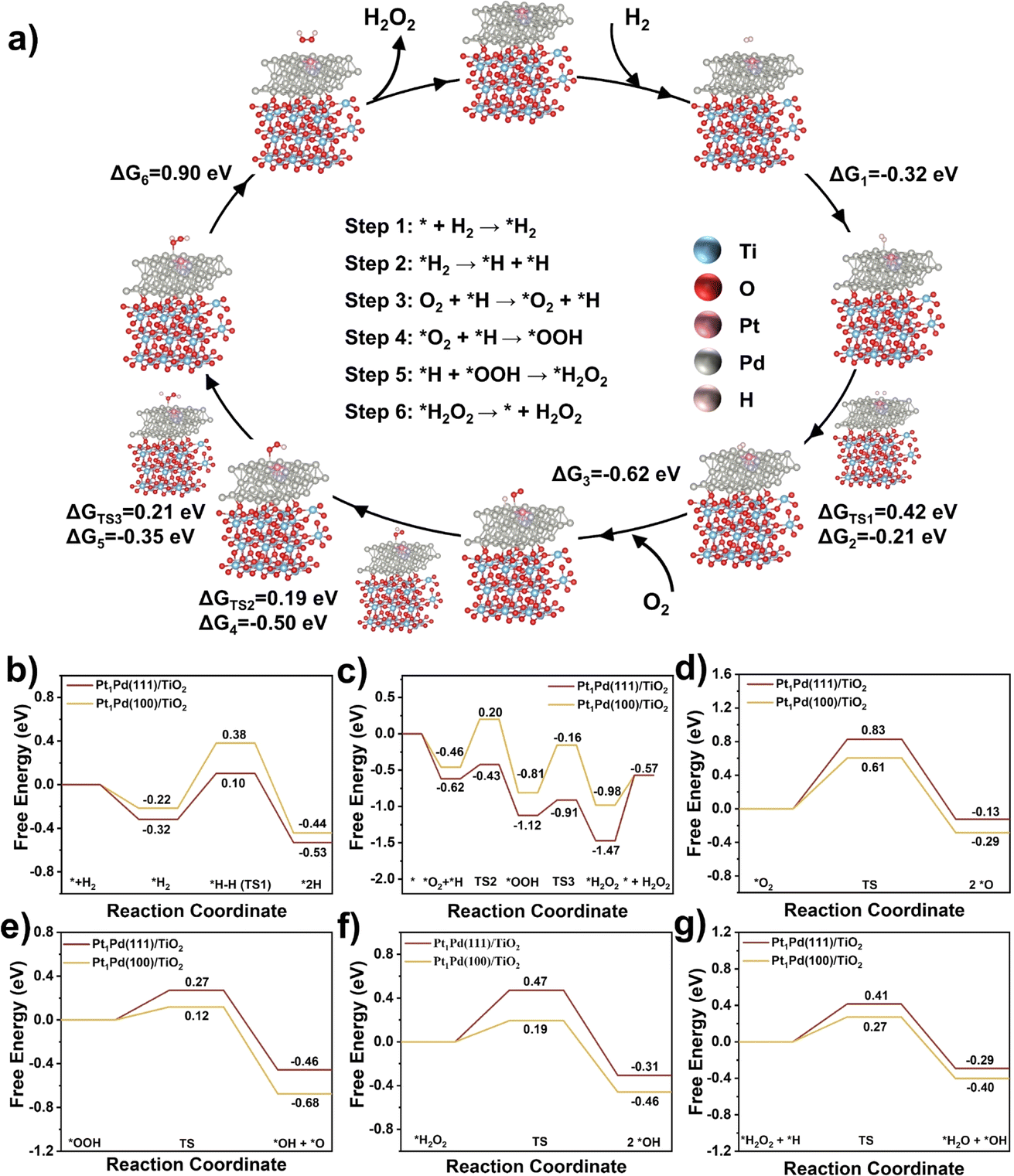 | ||
| Fig. 6 DFT calculations and proposed mechanism. (a) The possible elementary steps and catalytic mechanism over Pt1Pd(111)/TiO2 for the DSHP. (b) Free energy profiles for (a) H2 dissociation, (c) O2 hydrogenation, (d) *O2 dissociation, (e) *OOH dissociation, (f) *H2O2 dissociation, and *H2O2 hydrogenation on the Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 surfaces. TS: transition state. | ||
The corresponding free energies on the surface of Pt1Pd(111)/TiO2 and Pt1Pd(100)/TiO2 were investigated. The results from Fig. 6a, b, and S32† show that H2 was more readily adsorbed on the Pt1Pd(111)/TiO2 (−0.32 eV) surface than on the Pt1Pd(100)/TiO2 (−0.22 eV). On the Pt1Pd(111)/TiO2 surface, the cleavage of the H–H bond has to overcome an energy barrier of 0.42 eV, which is lower than that on Pt1Pd(100)/TiO2 (0.60 eV). Therefore, H2 dissociated more readily on Pt1Pd(111)/TiO2, providing a hydrogen source for the subsequent O2 hydrogenation. The two-step O2 hydrogenation to produce H2O2 is described in Fig. 6a, c, and S33.† The energy barriers for the first (*O2 + *H → *OOH) and second (*OOH + *H → *H2O2) hydrogenation of O2 on Pt1Pd(100)/TiO2 were found to be 0.66 and 0.65 eV, respectively. In contrast, the energy barriers of 0.19, 0.21 eV, respectively, on Pt1Pd(111)/TiO2 were lower. The corresponding reaction energies were also lower than on Pt1Pd(100)/TiO2 (−0.50 vs. −0.35 eV and −0.35 vs. −0.17 eV, respectively). However, the free energy of H2O2 desorption on the Pt1Pd(111)/TiO2 (0.90 eV) surface was higher than that of the Pt1Pd(100)/TiO2 (0.41 eV) surface. The above investigations of the transition states demonstrated that the whole DSHP process had a lower energy barrier on Pt1Pd(111)/TiO2 with H2 dissociation assumed to be the rate determining step, in agreement with the experimental observations. Based on the DFT calculations, it could be concluded that Pt1Pd(111)/TiO2 presents an elevated ability to cleave the H–H bond in H2 and O2 hydrogenation, which strongly promoted the formation of H2O2, leading to enhanced DSHP performance with an obvious facet dependence.
Next, we simulated the four main side reactions, including *O2 dissociation (*O2 → 2 *O), *OOH dissociation (*OOH → *OH + *O), *H2O2 dissociation (*H2O2 → 2 *OH), and *H2O2 hydrogenation (*H2O2 + *H → *H2O + *OH). The corresponding free energies, energy barriers, and structures are depicted in Fig. 6d–g and S34–37.† On Pt1Pd(111)/TiO2, the energy barriers of the four side reactions were 0.83, 0.27, 0.47, and 0.41 eV, respectively, which were greater than those of the corresponding competitive reactions (two-step O2 hydrogenation to H2O2), indicating high H2O2 selectivity on Pt1Pd(111)/TiO2. In addition, Pt1Pd(100)/TiO2 showed lower energy barriers than Pt1Pd(111)/TiO2 for the four side reactions (0.61, 0.12, 0.19, and 0.27 eV), which implies that the H2O2 selectivity on Pt1Pd(111)/TiO2 could be higher than that on Pt1Pd(100)/TiO2, in agreement with the experimental results showing an obvious facet dependence.
Conclusions
The selective production of H2O2 in high amounts is a challenge in the DSHP. In this contribution, we propose a single Pt atom-modified Pd nanocrystal catalyst to efficiently synthesize H2O2. The as-prepared Pt1Pd(111)/TiO2 catalyst presents outstanding reactivity, producing 1921.3 μmol of H2O2 (the highest amount of H2O2 compared to previous reports), a H2 conversion of 62.2%, and a H2O2 selectivity of 80.3% during the DSHP for 30 min. These results are better than those obtained for Pt1Pd(100)/TiO2, showing that the reaction exhibits a prominent facet dependence. Kinetic and isotopic results confirm that the remarkable activity of Pt1Pd(111)/TiO2 is attributed to the electron-rich structure of the Pd(111) surface, which contributes to H2 activation, the rate-determining step in the DSHP process. The introduction of single Pt atoms also contributes to the synthesis of H2O2. DFT calculations confirm that the energy barriers for H2 dissociation and the two-step O2 hydrogenation on the Pt1Pd(111) surface are lower than on the Pt1Pd(100) surface. Furthermore, the energy barriers of the four side reactions (*O2 dissociation, *OOH dissociation, *H2O2 dissociation, and *H2O2 hydrogenation) on the Pt1Pd(111) surface are larger than on Pt1Pd(100), resulting in higher selectivity and the production of greater amounts of H2O2 on Pt1Pd(111)/TiO2. This work provides a promising strategy to design and develop highly active DSHP catalysts, and deepens our understanding of the reaction mechanism.Materials and methods
Chemicals
Potassium tetrachloropalladate (K2PdCl4), chloroplatinic acid (H2PtCl6·6H2O), and methanol (HPLC grade) were purchased from Energy Chemical. Hydrogen peroxide (30%) and acetone were purchased from Guangzhou Chemical Reagent Factory. L-Ascorbic acid, citric acid, potassium bromide (KBr), and polyvinyl pyrrolidone (PVP) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Titanium oxide (TiO2, P25) and ferroin indicator were purchased from Shanghai Macklin Biochemical Co. Ltd. Cerous sulfate (Ce(SO4)2, 0.1 mol L−1) was purchased from Tan–Mo Technology Co. Ltd.Catalyst preparation
Synthesis of the Pt1Pd(111) nanocrystals: the Pt1Pd(111) nanocrystals were synthesized according to a slightly modified previous report.37 105 mg of PVP, 60 mg of citric acid, and 60 mg of L-ascorbic acid were added into a 3-neck flask containing 8 mL of deionized water at room temperature. Then the 3-neck flask equipped with a reflux condenser was heated to 120 °C and stirred for 5 min in an oil bath. Meanwhile, 65 mg of K2PdCl4 and 10 μL of H2PtCl6·6H2O solution (0.1 g mL−1) were dissolved in 3 mL of deionized water. The mixed solution was injected into the 3-necked flask via a peristaltic pump at 5 mL h−1. Finally, the reaction was continued and stirred in air for 3 h at 120 °C. After the reaction, the samples were washed with acetone/deionized water three times to remove the excess molecules by centrifugation. Thus the Pt1Pd(111) nanocrystals were obtained.Synthesis of Pt1Pd(111)/TiO2: the pre-prepared Pt1Pd(111) nanocrystals were suspended in 5 mL of deionized water and then dropped into 10 mL of deionized water containing 0.5 g of TiO2 under vigorous stirring at room temperature. After stirring for 4 h, the temperature was increased to 80 °C in order to evaporate the deionized water. Finally, the samples were obtained after calcining.
Synthesis of Pt1Pd(100) nanocrystals: 105 mg of PVP, 300 mg of KBr, and 60 mg of L-ascorbic acid were added into a 3-neck flask containing 8 mL of deionized water at room temperature. Then the 3-neck flask equipped with a reflux condenser was heated to 80 °C and stirred for 5 min in an oil bath. Meanwhile, 65 mg of K2PdCl4 and 10 μL of H2PtCl6·6H2O solution (0.1 g mL−1) were dissolved in 3 mL of deionized water. The mixed solution was then added into the 3-necked flask and stirred for 3 h at 80 °C. After the reaction, the samples were washed with acetone/deionized water three times to remove the excess molecules by centrifugation. Thus the Pt1Pd(100) nanocrystals were obtained.
Synthesis of Pt1Pd(100)/TiO2: the preparation process was similar to that of Pt1Pd(111)/TiO2. Pd(111)/TiO2, Pt1Pd(111)40/TiO2, and Pt1Pd(111)120/TiO2 were synthesized by the same method as Pt1Pd(111)/TiO2. Pd(100)/TiO2 was synthesized by the same method as Pt1Pd(100)/TiO2.
Catalyst characterization
Inductively coupled plasma optical emission spectrometry was used to determine the metal loadings of all catalysts (ICP-OES, Agilent 7700). X-ray photoelectron spectroscopy (Thermo Fisher Nexsa) was used to evaluate the valence states of the catalysts, and the data was processed using the XPSPEAK software. The X-ray diffraction patterns were produced on a LIFM-X-ray powder diffractometer (Smart Lab) using s Cu Kα (λ = 0.15432 nm) beam, and the data was processed and compared with PDF cards using the MDI Jade 6 software. Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) images were captured on a FEI Tecnai G2 F30 transmission electron microscope using an acceleration voltage of 300 kV. The high-resolution TEM, aberration-corrected high-angle annular darkfield STEM, and energy dispersive X-ray (EDX) spectrometry mapping were produced using a JEM-ARM200F transmission electron microscope equipped with double spherical aberration correctors. Pd K-edge and Pt L3-edge extended X-ray absorption fine structure spectrometry (EXAFS) were performed at the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility. The incident beam was monochromatized using a Si (311) double-crystal monochromator while the high harmonics of the monochromatic beam were diminished. EXAFS data analysis was carried out on the Demeter software employing the conventional processes: background removal, normalization and Fourier transformation of the EXAFS oscillations. A Shimadzu UV2600 spectrophotometer with wavelength precision of 0.3 nm was used to collect the UV-Vis spectra. The in situ diffuse reflectance infrared Fourier-transform spectra (DRIFTS) of CO absorption were measured on a Nicolet iS50 (Thermo Science) with an in situ transmission cell. The catalysts were reduced at 200 °C in a 20% H2/N2 atmosphere for 1 hour before the test. The background was collected under N2 after reduction. At this point, 10% CO/N2 was introduced for 30 minutes, and the adsorption spectrum was recorded until there was no change in the spectrum. Finally, N2 was employed to sweep away the physically adsorbed CO species, while the spectrum was obtained. H2-D2 exchange experiments were performed on a continuous flow fixed bed at room temperature and atmospheric pressure. The catalysts were loaded in a quartz tube with an inner diameter of 7 mm. Pure D2 with a flow rate of 10 mL min−1 was used as the pulse gas with a 100 μL quantitative ring, and 5% H2/N2 with a flow rate of 20 mL min−1 was used as the carrier gas. HD signals (m/z = 3) were recorded online using mass spectrometry (Hiden Analytical HPR-20 QIC benchtop Gas Analysis System). The H2 reaction orders were calculated using the turnover frequency (TOF) values obtained at different H2 partial pressures. The Arrhenius plots were calculated using the turnover frequency (TOF) values obtained at different temperatures (−20, −10, 0, 10, and 20 °C). The apparent rate constants were calculated using the H2 concentration after reaction at the corresponding reaction time (10, 20, 30, 40, 50, and 60 min).Catalytic performance test
The direct synthesis of H2O2 was carried out in a stainless-steel autoclave with a nominal volume of 100 mL and a digital pressure gauge. A solution, containing 10 mL of methanol, 0.02 M HCl, and 10 mg of catalyst, was encapsulated in the autoclave to assess the catalytic performance of the catalysts for H2O2 synthesis. The autoclave was sealed and then charged with 1.1 MPa 25% O2/N2 (7.30 mmol O2) and 2.9 MPa 5% H2/N2 (3.85 mmol H2) at room temperature, following purging with 0.7 MPa 5% H2/N2three times. After cooling to 0 °C in an ice-water bath, the reaction mixture was stirred at 1200 rpm for 30 minutes. The hydrogen content before and after the reaction was calculated using a gaseous chromatograph (GC) equipped with a thermal conductivity detector (TCD) and a TDX-01 column. The amount of H2O2 was determined by titrating aliquots of the final solution with acidified cerium sulfate standard solution (Ce(SO4)2, 0.01 mol L−1) in the presence of three drops of ferroin indicator solution. The H2 conversion, H2O2 selectivity, and H2O2 productivity were calculated as follows: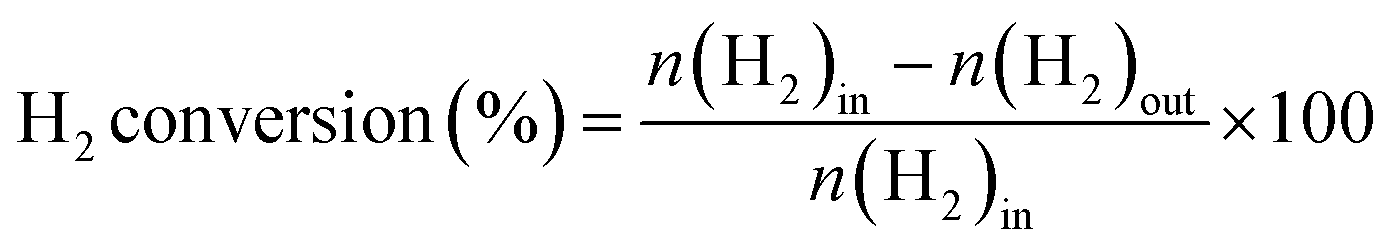 | (1) |
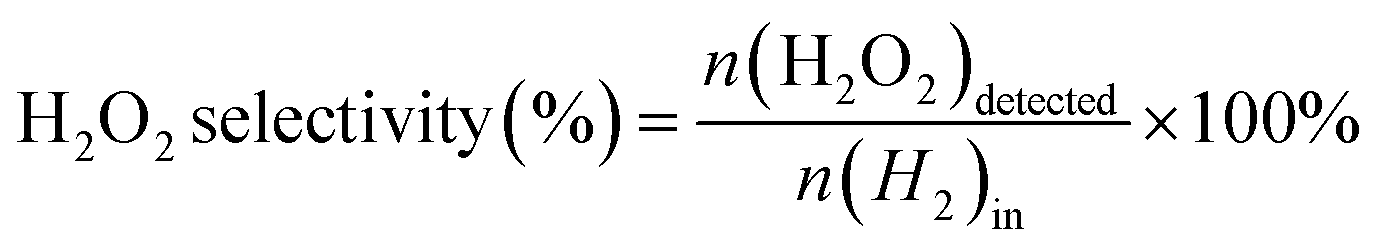 | (2) |
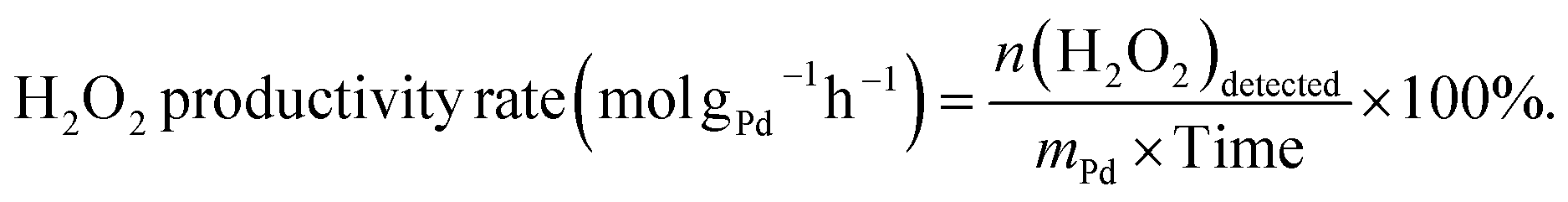 | (3) |
The hydrogenation of H2O2 was performed in a stainless-steel autoclave using the same procedure as for the synthesis of H2O2. 10 mL of methanol, 250 μL of H2O2 (∼30 wt%, 2.56 mmol), 0.02 M HCl, and 10 mg of catalyst were encapsulated in the autoclave. The sealed autoclave was then charged with 2.9 MPa 5% H2/N2 after purging with 0.7 MPa 5% H2/N2 three times at room temperature. The reaction mixture was stirred for 30 minutes at 1200 rpm after being cooled in an ice-water bath to 0 °C. The hydrogenation of H2O2 was determined by titrating aliquots of the final solution with acidified cerium sulfate standard solution (Ce(SO4)2, 0.002 mol L−1) in the presence of three drops of ferroin indicator solution after the reaction. The H2O2 hydrogenation rate was calculated as follows:
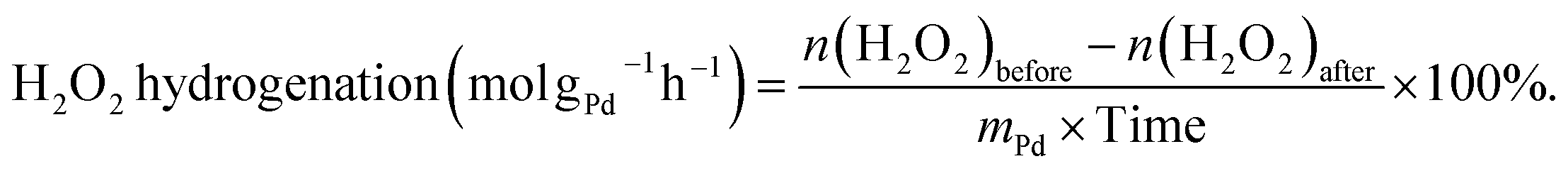 | (4) |
The decomposition of H2O2 was evaluated using a similar method as used for the hydrogenation of H2O2, except that 2.9 MPa 5% H2/N2 was replaced with 4 MPa N2. The H2O2 decomposition rate was calculated as follows:
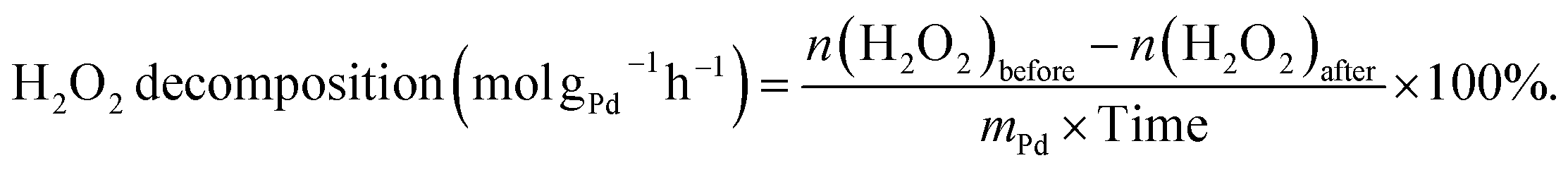 | (5) |
Computational methods
The Vienna Ab Initio Package (VASP)49 was employed to perform all the density-functional theory (DFT) calculations within the generalized gradient approximation (GGA) using the PBE50 formulation. The projected augmented wave (PAW) potentials were chosen to describe the ionic cores and take the valence electrons into account using a plane wave basis set with a kinetic energy cutoff of 520 eV. Partial occupancies of the Kohn–Sham orbitals were allowed using the Gaussian smearing method and a width of 0.05 eV. The electronic energy was considered self-consistent when the energy change was smaller than 10−5 eV. A geometry optimization was considered convergent when the force change was smaller than 0.03 eV Å−1 and the spin polarization effect was included in the whole calculation. Grimme's DFT-D3 methodology was used to describe the dispersion interactions. The vacuum spacing perpendicular to the plane of the structure is 18 Å. The Brillouin zone integral uses the surface structures of 2 × 2 × 1 Monkhorst pack K-point sampling. Finally, the adsorption energies (Eads) are calculated as Eads = Ead/sub − Ead − Esub, where Ead/sub, Ead and Esub are the optimized adsorbate/substrate system, the adsorbate in the structure and the clean substrate respectively. The free energy is calculated as follows:| G = E + ZPE − TS, | (6) |
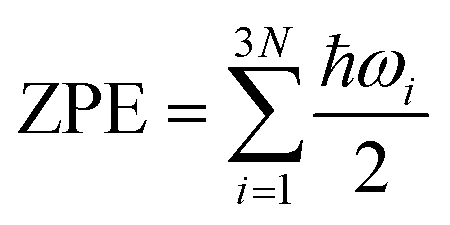 , where ℏ is Planck's constant and ωi is the frequency of the ith vibrational mode of the adsorbed molecule. The TiO2(101) and Pd(111), Pd(100) surfaces were used in our calculations.
, where ℏ is Planck's constant and ωi is the frequency of the ith vibrational mode of the adsorbed molecule. The TiO2(101) and Pd(111), Pd(100) surfaces were used in our calculations.
Data availability
Additional details regarding experimental and calculation data are given in the ESI.†Author contributions
The manuscript was written through the contributions of all authors. Xiaohui He and Ying Zhang developed the concept. Ying Zhang designed these experiments and analyzed experimental data. Ziyue Wang and Guanghui Guo contributed to the catalyst synthesis. Qingdi Sun contributed to the theoretical calculations. Hao Liu performed the EXAFS measurements and analyzed the data. Ying Zhang and Xiaohui He wrote the paper. Xiaohui He and Hongbing Ji directed the project. All the authors discussed the results and commented on the paper. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program Nanotechnology Specific Project (No. 2020YFA0210900), Guangdong Natural Science Funds for Distinguished Young Scholar (2022B1515020035), the National Natural Science Foundation of China (22078371, U22A20428, 21961160741), the special fund for Science and Technology Innovation Teams of Shanxi Province (202304051001007), The authors thank the Shanghai Synchrotron Radiation Facility of China for the XAFS spectra measurements at the BL14W1 beamline, Guangdong Basic Research Center of Excellence for Functional Molecular Engineering, and the Chemistry and Chemical Engineering Guangdong Laboratory (Grant No. 1922010). Also, thanks go to eceshi (https://www.eceshi.com/s) for collecting the data of XPS and DRIFTS CO chemisorption.References
- K. Cao, H. Yang, S. Bai, Y. Xu, C. Yang, Y. Wu, M. Xie, T. Cheng, Q. Shao and X. Huang, ACS Catal., 2021, 11, 1106–1118 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, e2104561 CrossRef PubMed.
- R. Svensson and H. Gronbeck, J. Am. Chem. Soc., 2023, 145, 11579–11588 CrossRef CAS PubMed.
- A. Huang, R. S. Delima, Y. Kim, E. W. Lees, F. G. L. Parlane, D. J. Dvorak, M. B. Rooney, R. P. Jansonius, A. G. Fink, Z. Zhang and C. P. Berlinguette, J. Am. Chem. Soc., 2022, 144, 14548–14554 CrossRef CAS PubMed.
- G. H. Han, S. H. Lee, S. Y. Hwang and K. Y. Lee, Adv. Energy Mater., 2021, 11, 2003121 CrossRef CAS.
- Z. Yang, Z. Wei, S. Zhou, B. Bao, S. Zhao and F. Gong, Chem. Eng. J., 2023, 456, 140915 CrossRef CAS.
- S. Yu, X. Cheng, Y. Wang, B. Xiao, Y. Xing, J. Ren, Y. Lu, H. Li, C. Zhuang and G. Chen, Nat. Commun., 2022, 13, 4737 CrossRef CAS PubMed.
- R. J. Lewis, M. Koy, M. Macino, M. Das, J. H. Carter, D. J. Morgan, T. E. Davies, J. B. Ernst, S. J. Freakley, F. Glorius and G. J. Hutchings, J. Am. Chem. Soc., 2022, 144, 15431–15436 CrossRef CAS PubMed.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. Qin, T. E. Davies, D. J. Morgan, A. Stenner, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, L. Chen, Y. Yamamoto and G. J. Hutchings, ACS Catal., 2023, 13, 1934–1945 CrossRef CAS.
- G. V. Fortunato, E. Pizzutilo, I. Katsounaros, D. Gohl, R. J. Lewis, K. J. J. Mayrhofer, G. J. Hutchings, S. J. Freakley and M. Ledendecker, Nat. Commun., 2022, 13, 1973 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- S. Shaybanizadeh, R. Luque and A. Najafi Chermahini, Green Chem., 2022, 24, 5524–5534 RSC.
- H. C. Li, Q. Wan, C. Du, J. Zhao, F. Li, Y. Zhang, Y. Zheng, M. Chen, K. H. L. Zhang, J. Huang, G. Fu, S. Lin, X. Huang and H. Xiong, Nat. Commun., 2022, 13, 6072 CrossRef CAS PubMed.
- T. Richards, J. H. Harrhy, R. J. Lewis, A. G. R. Howe, G. M. Suldecki, A. Folli, D. J. Morgan, T. E. Davies, E. J. Loveridge, D. A. Crole, J. K. Edwards, P. Gaskin, C. J. Kiely, Q. He, D. M. Murphy, J.-Y. Maillard, S. J. Freakley and G. J. Hutchings, Nat. Catal., 2021, 4, 575–585 CrossRef CAS.
- G. M. Lari, B. Puertolas, M. Shahrokhi, N. Lopez and J. Perez-Ramirez, Angew. Chem., Int. Ed., 2017, 56, 1775–1779 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- Z. Yang, Z. Hao, S. Zhou, P. Xie, Z. Wei, S. Zhao and F. Gong, ACS Appl. Mater. Interfaces, 2023, 15, 23058–23067 CrossRef CAS PubMed.
- S. Wang, G. Jiang, Z. Yang, L. Mu, T. Ji, X. Lu and J. Zhu, ACS Sustain. Chem. Eng., 2022, 10, 13750–13758 CrossRef CAS.
- R. J. Lewis, K. Ueura, X. Liu, Y. Fukuta, T. E. Davies, D. J. Morgan, L. Chen, J. Qi, J. Singleton, J. K. Edwards, S. J. Freakley, C. J. Kiely, Y. Yamamoto and G. J. Hutchings, Science, 2022, 376, 615–620 CrossRef CAS PubMed.
- Z. Jin, Y. Liu, L. Wang, C. Wang, Z. Wu, Q. Zhu, L. Wang and F.-S. Xiao, ACS Catal., 2021, 11, 1946–1951 CrossRef CAS.
- T. Ricciardulli, S. Gorthy, J. S. Adams, C. Thompson, A. M. Karim, M. Neurock and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 5445–5464 CrossRef CAS PubMed.
- V. R. Naina, S. Wang, D. I. Sharapa, M. Zimmermann, M. Hähsler, L. Niebl-Eibenstein, J. Wang, C. Wöll, Y. Wang, S. K. Singh, F. Studt and S. Behrens, ACS Catal., 2021, 11, 2288–2301 CrossRef CAS.
- A. Barnes, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catal. Sci. Technol., 2022, 12, 1986–1995 RSC.
- Y. Wang, M. Zheng, Y. Li, C. Ye, J. Chen, J. Ye, Q. Zhang, J. Li, Z. Zhou, X. Z. Fu, J. Wang, S. G. Sun and D. Wang, Angew. Chem., Int. Ed., 2022, 61, e202115735 CrossRef CAS PubMed.
- M. Ma, G. Li, W. Yan, Z. Wu, Z. Zheng, X. Zhang, Q. Wang, G. Du, D. Liu, Z. Xie, Q. Kuang and L. Zheng, Adv. Energy Mater., 2022, 12, 2103336 CrossRef CAS.
- J. Li, L. Sun, Q. Wan, J. Lin, S. Lin and X. Wang, J. Phys. Chem. Lett., 2021, 12, 11415–11421 CrossRef CAS PubMed.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- H. Zhang, X. Qiu, Y. Chen, S. Wang, S. E. Skrabalak and Y. Tang, Small, 2020, 16, e1906026 CrossRef PubMed.
- T. H. Yang, Y. Shi, A. Janssen and Y. Xia, Angew. Chem., Int. Ed., 2020, 59, 15378–15401 CrossRef CAS PubMed.
- G. Wang, Z. Yang, Y. Du and Y. Yang, Angew. Chem., Int. Ed., 2019, 58, 15848–15854 CrossRef CAS PubMed.
- M. Zhao, Z. Chen, Z. Lyu, Z. D. Hood, M. Xie, M. Vara, M. Chi and Y. Xia, J. Am. Chem. Soc., 2019, 141, 7028–7036 CrossRef CAS PubMed.
- G. Liu, W. Zhou, Y. Ji, B. Chen, G. Fu, Q. Yun, S. Chen, Y. Lin, P. F. Yin, X. Cui, J. Liu, F. Meng, Q. Zhang, L. Song, L. Gu and H. Zhang, J. Am. Chem. Soc., 2021, 143, 11262–11270 CrossRef CAS PubMed.
- W. Zhang, Y. Shi, Y. Yang, J. Tan and Q. Gao, Chin. J. Catal., 2022, 43, 3116–3125 CrossRef CAS.
- G. Fang, W. Li, X. Shen, J. M. Perez-Aguilar, Y. Chong, X. Gao, Z. Chai, C. Chen, C. Ge and R. Zhou, Nat. Commun., 2018, 9, 129 CrossRef PubMed.
- Q. Sun, X. Wang, H. Wang, H. Zhang, Q. He, Y. Zhang, Y. Cheng, X. Zhang, S. Shi, L. Tao, X. He and H. Ji, J. Mater. Chem. A, 2022, 10, 10837–10843 RSC.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS PubMed.
- L. F. de L. e Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- D. Ding, X. Xu, P. Tian, X. Liu, J. Xu and Y.-F. Han, Chin. J. Catal., 2018, 39, 673–681 CrossRef CAS.
- A. Akram, G. Shaw, R. J. Lewis, M. Piccinini, D. J. Morgan, T. E. Davies, S. J. Freakley, J. K. Edwards, J. A. Moulijn and G. J. Hutchings, Catal. Sci. Technol., 2020, 10, 8203–8212 RSC.
- D. Jiang, G. Wan, C. E. García-Vargas, L. Li, X. I. Pereira-Hernández, C. Wang and Y. Wang, ACS Catal., 2020, 10, 11356–11364 CrossRef CAS.
- L. Wang, S. Deo, A. Mukhopadhyay, N. A. Pantelis, M. J. Janik and R. M. Rioux, ACS Catal., 2022, 12, 12927–12941 CrossRef CAS.
- X. Li, X. I. Pereira-Hernandez, Y. Chen, J. Xu, J. Zhao, C. W. Pao, C. Y. Fang, J. Zeng, Y. Wang, B. C. Gates and J. Liu, Nature, 2022, 611, 284–288 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- K. Guo, X. Han, S. Wei, J. Bao, Y. Lin, Y. Li and D. Xu, Nano Lett., 2023, 23, 1085–1092 CrossRef CAS PubMed.
- L. Zhang, H. Liu, S. Liu, M. Norouzi Banis, Z. Song, J. Li, L. Yang, M. Markiewicz, Y. Zhao, R. Li, M. Zheng, S. Ye, Z.-J. Zhao, G. A. Botton and X. Sun, ACS Catal., 2019, 9, 9350–9358 CrossRef CAS.
- Y. Zhang, Y. Cheng, X. Wang, Q. Sun, X. He and H. Ji, ACS Catal., 2022, 12, 15091–15096 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01560f |
| This journal is © The Royal Society of Chemistry 2024 |