
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-activatable charge transfer in gold nanoclusters†
Hao-Hua
Deng‡
 a,
Kai-Yuan
Huang‡
a,
Yu
Zhong
a,
Ye
Li
a,
Hong-Xiang
Huang
a,
Xiang-Yu
Fang
a,
Wei-Ming
Sun
*a,
Qiaofeng
Yao
b,
Wei
Chen
*a and
Jianping
Xie
*c
a,
Kai-Yuan
Huang‡
a,
Yu
Zhong
a,
Ye
Li
a,
Hong-Xiang
Huang
a,
Xiang-Yu
Fang
a,
Wei-Ming
Sun
*a,
Qiaofeng
Yao
b,
Wei
Chen
*a and
Jianping
Xie
*c
aHigher Educational Key Laboratory for Nano Biomedical Technology of Fujian Province, Department of Pharmaceutical Analysis, Fujian Medical University, Fuzhou 350004, China. E-mail: sunwm@fjmu.edu.cn; weichen@fjmu.edu.cn
bTianjin Key Laboratory of Molecular Optoelectronic Sciences, Key Laboratory of Organic Integrated Circuits, Ministry of Education, Department of Chemistry, School of Science, Tianjin University, Tianjin 300072, China
cDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: chexiej@nus.edu.sg
First published on 14th May 2024
Abstract
Surface-protecting ligands, as a major component of metal nanoclusters (MNCs), can dominate molecular characteristics, performance behaviors, and biological properties of MNCs, which brings diversity and flexibility to the nanoclusters and largely promotes their applications in optics, electricity, magnetism, catalysis, biology, and other fields. We report herein the design of a new kind of water-soluble luminescent gold nanoclusters (AuNCs) for enzyme-activatable charge transfer (CT) based on the ligand engineering of AuNCs with 6-mercaptopurine ribonucleoside (MPR). This elaborately designed cluster, Au5(MPR)2, can form a stable intramolecular CT state after light excitation, and exhibits long-lived color-tunable phosphorescence. After the cleavage by purine nucleoside phosphorylase (PNP), the CT triplet state can be easily directed to a low-lying energy level, leading to a bathochromic shift of the emission band accompanied by weaker and shorter-lived luminescence. Remarkably, these ligand-engineered AuNCs show high affinity towards PNP as well as decent performance for analyzing and visualizing enzyme activity and related drugs. The work of this paper provides a good example for diversifying physicochemical properties and application scenarios of MNCs by rational ligand engineering, which will facilitate future interest and new strategies to precisely engineer solution-based nanocluster materials.
Introduction
As a crucial intermediate excited state in numerous molecules and nanostructures, charge-transfer (CT) states are involved in both the generation and conversion of luminescent states, resulting in a tunable emission band.1–3 This phenomenon is especially intriguing as it can be potentially applied in many areas, including probes,4,5 photovoltaics,6,7 encrypted information storage,8 and cellular imaging.9 However, despite the substantial advance made in investigating the CT states of materials, it remains a great challenge to translate the accumulated theoretical and experimental insights into design strategies for high-quality nanoluminophores toward different sets of application scenarios.10,11 This probably arises from both ill-defined structure–function relationships and unusual electron–hole coupling of the conventional luminescent nanomaterials.12,13Over the past two decades, ligand-protected gold nanoclusters (AuNCs) as unique materials have attracted considerable attention from fundamental aspects to potential applications.14–19 In ligand-protected AuNCs, the ligand layer capped on the surface of the gold core is an important component because it will dramatically influence both molecular characteristics and performance behaviours of AuNCs.20,21 Apart from offering good stabilization for AuNCs in solution, the ligand shell is also critical to the chemical and physical properties of ligand-protected AuNCs. For example, it has been reported that the photoluminescence (PL) of AuNCs can be readily modulated by the anchoring point,22,23 chain length,24,25 functional group,26,27 and flexibility degree of the protecting ligands.28,29 More intriguingly, the design and choice of a ligand shell can also dominate the biological properties of AuNCs. By using a certain biocompatible ligand (e.g., amino acid, peptide, protein, DNA, and biomimetic substrate),30–34 the native function (e.g., catalytic activity and recognition ability) of biomolecules may be retained after the synthesis of AuNCs,35,36 which provides huge opportunities for the fabrication of multifunctional platforms for diverse applications in biomedical fields.
The advent of AuNCs has unlocked the possibility for elucidating CT behaviours in luminescent nanomaterials on the atomic scale. The CT luminescence state of AuNCs is derived from the molecule-like charge transfer between the semiring ligands and Au core, which can be readily modulated by twist degree, electron-donating strength, and chain length of the protecting ligands.37–39 This peculiar property endows AuNCs with the ability to combine the advantages of both conventional nanoluminophores (e.g., long-lived luminescence and high resistance to photobleaching) and organic molecules (e.g., controllable dual-emission centres).40,41 Hitherto, unique intramolecular CT luminescence behaviours of AuNCs have only been studied in several bitetrahedral kernel structures,41,42 and they still need to be comprehensively understood.
Herein, we described an enzyme-catalysed approach for activating the intramolecular CT state in ligand-engineered AuNCs. The well-designed Au5(SR)2 (SR: thiolate) NCs exhibit intense long-lived luminescence in aqueous solution. The PL origin of these AuNCs was fully unravelled using a combination of different spectroscopic technologies, and an intramolecular CT-mediated triplet state was identified. Moreover, we found that after the cleavage by enzyme, the CT triplet state in Au5(SR)2 NCs can be easily directed to a low-lying energy level, leading to a weaker red-shifted PL emission. Finally, we translated these findings into the development of an efficient probe for enzyme activity monitoring, cell imaging, and drug screening.
Results and discussion
AuNC synthesis and characterization
The key to our design strategy is the selection of an appropriate ligand to produce specific surface-engineered AuNCs. We adopted 6-mercaptopurine ribonucleoside (MPR) as a ligand for the preparation of enzyme-activatable charge-transfer AuNCs due to the following reasons (Fig. 1a): (1) the thiol group of MPR (anchoring point) can strongly interact with the Au atom through the sulfur–Au bond; (2) the purine ring structure of MPR (ligand body) possesses unique electronic properties, which participate in the hybridization of the luminous energy level and tune the charge-transfer process; and (3) the glycosidic bond in MPR (functional group) can be recognized and cleaved by purine nucleoside phosphorylase (PNP). Besides, owing to the significant mixing of excited states between purine and ribose moieties, the ribose substituent in MPR can regulate the electron-accepting ability of the purine ring, thereby apparently affecting the charge-transfer process.43 | ||
| Fig. 1 Optical and structural characterization of MPR-AuNCs. (a) Illustration of the role of the MPR ligand on the surface of AuNCs. (b) UV-vis absorption spectrum of MPR-AuNCs. Inset: photographs of MPR-AuNCs under room light (left) and 302 nm UV light (right). (c) Two-dimensional fluorescence spectra of MPR-AuNCs. (d) ESI-MS spectrum of MPR-AuNCs. The inset shows a comparison between the experimental data (blue line) and the calculated isotope pattern (orange line) of [Au5(MPR)2 − 2H]−. (e) XPS spectrum of MPR-AuNCs. (f) High resolution O(1s) peak of MPR-AuNCs. (g) Comparison of the emission bands of MPR-AuNCs in water, dimethyl sulfoxide (DMSO), and dimethylformamide (DMF). (h) DLS measurements of MPR-AuNCs in water, DMSO, and DMF. (i) The viscosity-dependent PL spectra of MPR-AuNCs. The lines labelled with colors changing from orange to blue correspond to the PL spectra of MPR-AuNCs in the presence of 0%, 40%, 50%, 55%, 60%, 70%, and 80% glycerol, respectively. The inset shows photographs of MPR-AuNCs in glycerol solution with different concentrations under a 302 nm UV lamp. | ||
The fabrication of AuNCs was carried out in aqueous solution at room temperature based on a facile one-pot approach by employing MPR as the reducing-cum-protecting agent (more details can be found in Experimental). The optical properties of the as-synthesized AuNCs were evaluated by UV-vis absorption spectroscopy and fluorescence spectroscopy. The formed aqueous solution of AuNCs coated with MPR (MPR-AuNCs) appears faint yellow under room light and glows yellow-orange under UV irradiation (Fig. 1b inset). The UV-vis absorption spectrum of the obtained MPR-AuNCs is depicted in Fig. 1b, which reveals an onset at approximately 500 nm without any clear characteristic absorption peaks. The visible absorption feature may arise from a charge transfer state.37–39Fig. 1c shows the excitation/emission 2-dimensional contour plot of MPR-AuNCs. The plot illustrates that the maximum excitation and emission wavelengths of these AuNCs are located at 420 nm and 590 nm, respectively. The zeta potential of MPR-AuNCs was measured to be −24.4 ± 1.2 mV (n = 3).
To gain insight into the cluster formula, we analysed the samples by electrospray ionization mass spectrometry (ESI-MS) in negative mode. The ESI-MS analysis manifested a relatively clean spectrum (Fig. 1d). An intense mass peak at m/z = 1549.13 was observed, with the spacing of its isotope pattern being 1, hence indicating a 1− charge. Therefore, the as-prepared AuNCs should be assigned as [Au5(MPR)2 − 2H]−, and the accuracy of our assignment was exemplified by the good match between experimental and simulated isotope patterns (Fig. 1d inset).
Detailed investigations using X-ray photoelectron spectroscopy (XPS) and Fourier-transform infrared spectroscopy (FTIR) validated that AuNCs are successfully engineered with MPR and the glycosidic bond in MPR is well preserved after cluster synthesis. The full-survey XPS spectrum (Fig. 1e) revealed the existence of Au (Au 4f), carbon (C 1s), nitrogen (N 1s), oxygen (O 1s), and sulfur (S 2p). The spectrum of high-resolution Au 4f (Fig. S1†) showed that the Au 4f5/2 binding energy appears at 84.3 eV, which suggested the presence of both Au(0) and Au(I).44 An enlarged view of the S 2p binding energy region is displayed in Fig. S2,† wherein the dominant peak at 162.3 eV can be attributable to the sulfur atom of MPR bound to the Au surface.45 Peaks at binding energies associated with O 1s were fitted as two components corresponding to C–O–C bonding and C–O–H bonding (Fig. 1f), respectively.46 The FTIR spectrum of AuNCs exhibited the characteristics of the ligand MPR (Fig. S3†). A band at 2671 cm−1 due to the S–H stretching vibration disappeared in MPR-AuNCs while a strong absorption band at around 1200 cm−1 originating from the C–O stretching vibration of the ribose structure was retained, supporting that MPR is bound to the Au surface by the 6-SH group.47,48 In addition, several stretching C–H bands in the 3000–3200 cm−1 became inconspicuous after the formation of AuNCs. Meanwhile, the intensities of the bands at 2850 and 2920 cm−1 were strengthened, which can be attributed to the transformation of the different tautomers present in the free MPR molecule to the thiol species in the adsorbed state.47
To unveil the nature of charge transfer in MPR-AuNCs, their PL properties in different environments were first investigated. As the solvent polarity increases, the emission peak of MPR-AuNCs showed a strong bathochromic shift (Fig. 1g). A similar variation tendency can be seen in their corresponding absorption spectra (Fig. S4†). The almost identical size distribution of MPR-AuNCs in the tested solvents can rule out that the solvent-dependent emission peaks are generated by aggregation-induced quenching (Fig. 1h). In addition to polarity, the luminescence of MPR-AuNCs was also found to be highly correlated with the solvent viscosity. Significant enhancement of the PL intensity and noticeable hypsochromic shift of the emission band were detected with increasing concentration of glycerol (Fig. 1i). The luminescence QY of MPR-AuNCs determined in 80% glycerol is nearly 20 times higher than that in water (Fig. S5†). The above findings are reminiscent of the intramolecular CT model that has been frequently observed in organic compounds.49
Enzyme-activatable charge transfer in MPR-AuNCs
Having identified the charge-transfer feature of these ligand-engineered AuNCs, PNP was then introduced to testify the feasibility of our design concept. PNP is a crucial enzyme in the purine degradation and salvage pathway, which can catalyse the phosphorolytic cleavage of the glycosidic bond of MPR to produce 6-mercaptopurine (MP) and ribose-1-phosphate (RP) (Fig. 2a). As shown in Fig. 2b, when MPR-AuNCs were mixed with PNP in phosphate buffer (PB) solution, an apparent decrease in PL intensity was noticed, along with a red-shift in the emission maximum. We found that by replacing PB solution with glycine-NaOH buffer solution (their pH values were both kept at 8.0), the PNP-caused PL quenching was almost completely inhibited (Fig. 2c). This phenomenon is plausible because the PNP-catalysed hydrolysis reaction requires inorganic orthophosphate (Pi) as a second substrate. After adding Pi to glycine-NaOH buffer solution, PNP is capable of suppressing the luminescence of MPR-AuNCs again. These results verified that the quenched PL of MPR-AuNCs by PNP indeed stems from an enzymatic reaction, which was further evidenced by ESI-MS and 1H NMR measurements. Two newly emerging peaks with a spacing of 1 at 1325.90 Da and 1347.88 Da were observable in the positive-mode ESI-MS spectrum of the PNP/MPR-AuNC mixture (Fig. 2d), which are consistent with [Au5(MP)2 + K]+ and [Au5(MP)2 + K + Na − H]+, respectively. Additionally, a new peak with a spacing of 1 that represents [2RP − H]− was clarified at m/z 461 Da in the negative-mode ESI-MS spectrum of the PNP/MPR-AuNC mixture (Fig. S6†). As can be seen from the 1H NMR spectra shown in Fig. 2e, a broad peak at ∼13.0 ppm that belongs to the proton of the N(9)-H group was present in MPR-AuNCs upon the addition of PNP.50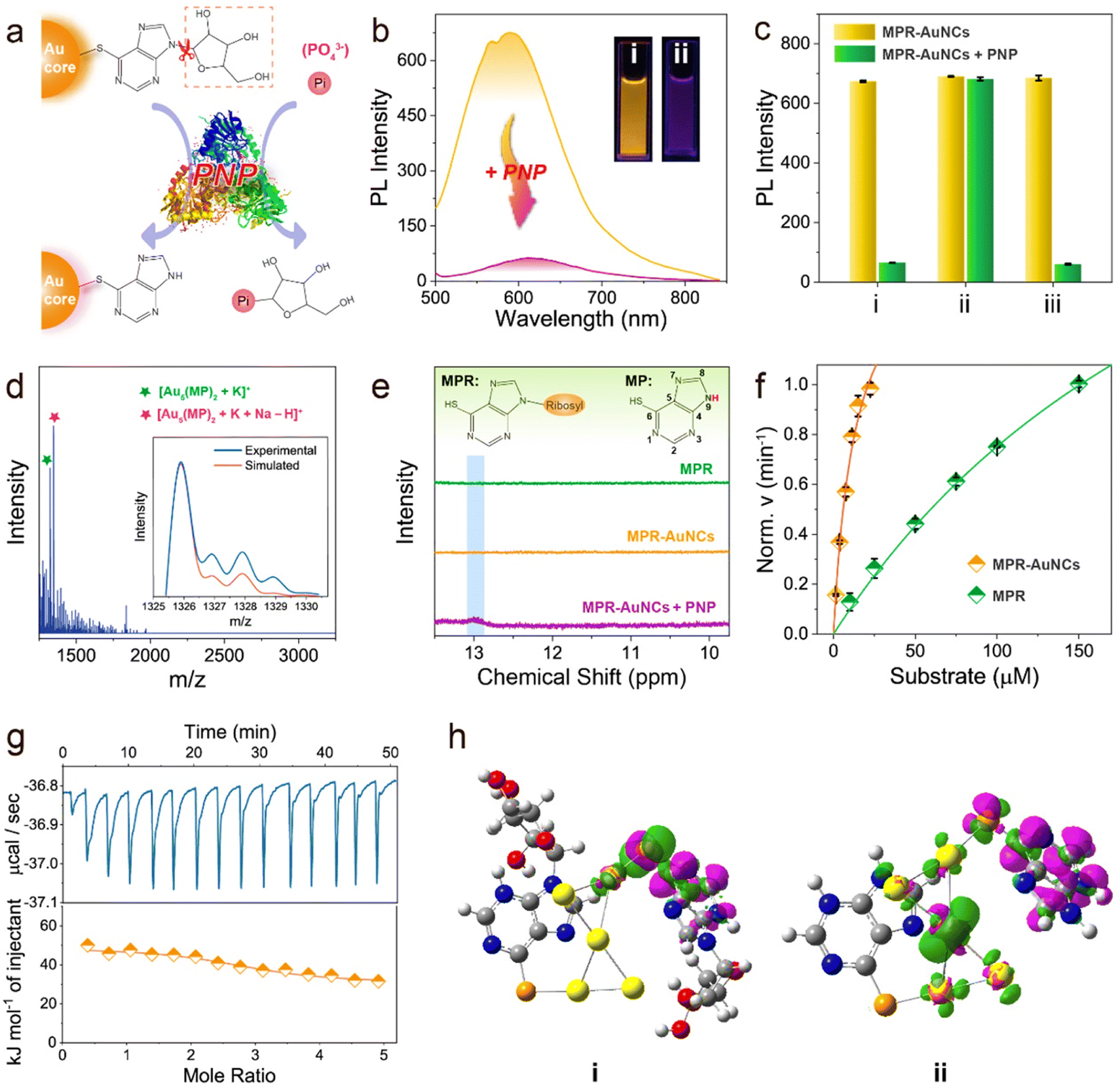 | ||
| Fig. 2 PNP-activated intramolecular CT states in MPR-AuNCs. (a) Schematic representation of the catalytic hydrolysis of MPR-AuNCs by PNP. (b) Emission spectra of MPR-AuNCs before and after the addition of 50 U L−1 PNP. Inset: the photographs of MPR-AuNCs in the (i) absence and (ii) presence of 50 U L−1 PNP under a 302 nm UV lamp. (c) PNP-induced PL quenching of MPR-AuNCs in different buffers: (i) PB (20 mM); (ii) Gly-NaOH (50 mM); and (iii) a Gly-NaOH buffer containing 10 mM Pi. The pH values of the tested buffers were kept at 8.0. (d) ESI-MS spectrum of MPR-AuNCs after the reaction with PNP (50 U L−1). Inset shows the experimental (orange line) and simulated (blue line) isotope patterns of [Au5(MP)2 + K]+. (e) 1H NMR spectra of MPR, MPR-AuNCs, and the PNP/MPR-AuNC mixture in d6-DMSO. (f) Michaelis–Menten plot of the initial velocities of PNP reaction versus MPR-AuNCs and MPR concentrations. Error bars denote standard deviation over three independent measurements. (g) Typical data obtained from isothermal titration calorimetry (ITC) measurements of MPR-AuNCs titrated with PNP. Top: corrected heat rate of the titration. Bottom: integrated normalized heats from each titration step corrected by the heats of dilution (filled rhombus) together with a fit corresponding to an independent binding model (straight line). (h) Electron density differences (EDDs) between triplet and singlet states of the most stable (i) MPR-AuNC and (ii) MP-AuNC with an isovalue of 0.01 a.u. Green and purple areas represent electron density increases and decreases, respectively. | ||
It is worth mentioning that PNP exhibits high catalytic activity towards MPR-AuNCs. The Michaelis constant (Km) values of PNP for the substrates MPR and MPR-AuNCs were 260 and 14 μM (Fig. 2f), respectively. The electrostatic interaction between PNP and Au5(MPR)2 NCs can be ruled out first due to the negative surface charge of both PNP (isoelectric point: 4.2 (ref. 51)) and MPR-AuNCs. Hence, the much stronger affinity of MPR-AuNCs towards PNP should be ascribed to the tighter binding of MPR-AuNCs than MPR to PNP driven by hydrophobic interactions (Fig. 2g, S7,† and Table 1).52 The obtained Km value in MPR-AuNCs was also found to be lower than or comparable to those of other substrates.53–55
The distinctive redshifted emission peak and absorption band (Fig. S8†) remind us that a strengthened intramolecular CT process probably occurs after converting MPR-AuNCs into MP-AuNCs by the PNP-catalysed reaction.56 Dynamic light scattering (DLS) measurements manifested that there is no formation of aggregates in MP-AuNCs (Fig. S9†). The PL lifetime decreases from 4.37 μs to 0.92 μs with a drop of the PLQY from 3.5% to 0.4% (Fig. S10†). Based on the PLQY and PL lifetime data, the nonradiative rates of MPR-AuNCs and MP-AuNCs were calculated to be 2.2 × 105 s−1 and 1.1 × 107 s−1,26 respectively. The accelerated nonradiative rate of MP-AuNCs is in good agreement with the characteristics of an enhanced intramolecular CT process.49,57 The long luminescence lifetimes on the microsecond timescale imply that the PL of both MPR-AuNCs and MP-AuNCs stems from a charge transfer-mediated triplet (3CT) state, which can be confirmed by the O2 quenching experiments (Fig. S11†). Because of their smaller driving force of Dexter energy transfer,58 the emission of MP-AuNCs was found to be less sensitive to O2. This result provides a clear proof for the lower 3CT state in MP-AuNCs and supports the occurrence of an enhanced intramolecular CT process.
The enhanced intramolecular CT process was further analysed through the density functional theory (DFT) calculations on the electron distributions of MPR-AuNCs and MP-AuNCs. The optimized isomeric structures of MPR-AuNCs are shown in Fig. S12.† For the global minimum of MPR-AuNC (isomer A), the electron densities in the HOMO and LUMO were mainly localized on the Au5 moiety (electron donor) and the purine ring group (electron acceptor), respectively (Fig. S13†). Meanwhile, the decrease in the electron density of the Au5 core and the increase of electron density of the MPR ligand under the excited state can also be found in the visualized electron density difference (EDD) figure of the most stable MPR-AuNC between triplet and singlet states (Fig. 2h(i)). This demonstrates that a clear intramolecular CT from the Au5 core to the MPR ligand occurs when the MPR-AuNCs were excited. The increased redistribution of electrons from the Au5 core in the HOMO to the MP upon excitation (LUMO) manifests that an enhanced intramolecular CT can be found for MP-AuNCs (Fig. S14†). This result was further verified by the EDD of the most stable MP-AuNC (Fig. 2h(ii)). By the natural bonding orbital (NBO) calculations, it is found that the electron transfer from the Au5 core to the MPR ligand is 0.091 e for MPR-AuNC, which is lower than that (0.696 e) for MP-AuNCs (Table S1†). This confirms that the charge transfer from Au5 to the ligand is enhanced by removing the glucoside group.
Unveiling the role of the enhanced intramolecular CT on the luminescence behaviours
To follow the dynamics of excited-stated states in MPR-AuNCs and MP-AuNCs, femtosecond transient absorption (fs-TA) spectroscopy measurements were conducted with excitation at 350 nm. The 2D fs-TA spectra of MPR-AuNCs exhibit two strong excited-state absorption (ESA) bands at 460 nm and 610 nm (Fig. 3a). Considering that MPR shows negligible absorbance at 350 nm (Fig. S15†) and has quite disparate TA features,59 the electron-pumping process of the ground state (S0) of the Au5 core, rather than the MPR ligand, is responsible for the observed fs-TA signals. With a closer look at the early-stage evolution of fs-TA spectra of MPR-AuNCs, a growth of a new ESA band centred at 610 nm following a decay of the ESA peak at 460 nm can be found (Fig. 3b). The ESA band centred at 610 nm quite resembles the absorbance of the purine ribonucleoside radical anion,60 strongly indicating that the intramolecular CT process occurs between the Au5 core and the MPR ligand. Hence, the ESA bands at 460 nm and 610 nm can be assigned to the electronic transitions of the first excited singlet state (S1) and charge-transfer singlet state (1CT) in MPR-AuNCs, respectively. The best fitting of the kinetics traces of the ESA peak at 460 nm yielded a rise time constant of 0.11 ps (Fig. S16†), corresponding to the internal conversion (IC) from the higher singlet state (Sn) to the S1 state due to the molecule-like properties of AuNCs. The ESA species recorded at 610 nm had a rise time constant of 0.66 ps, and decayed with time constants of 59 ps and 1370 ps. The rise time constant can be assigned to the rate of intramolecular CT from the S1 state to the 1CT state. Because the absorbance of the 3CT state will appear at a longer timescale and cannot be observed in the fs-TA spectra of MPR-AuNCs, the shorter decay time component is the rate of dark charge recombination (CR) from the 1CT state to the ground state (S0) and the longer decay time constant is the rate of ISC between the 1CT state and the 3CT state. The ISC efficiency in MPR-AuNC, which depends on competitive rates of ISC and dark CR,61 can therefore be determined as 4.2%. | ||
| Fig. 3 Femtosecond transient absorption (fs-TA) measurements. (a) 2D fs-TA contour map of MPR-AuNCs. (b) The fs-TA spectra evolution of MPR-AuNCs at the early stage. (c) The decay profile of MPR-AuNCs at 610 nm. (d) 2D fs-TA contour map of MP-AuNCs. (e) The fs-TA spectra evolution of MP-AuNCs at the early stage. (f) The decay profile of MP-AuNCs at 600 nm. | ||
After treatment with PNP, the TA spectra display a similar broad ESA feature in the visible region, but all ESA peaks decay faster (Fig. 3d). Owing to the ultrafast dynamics of the intramolecular CT process in luminated MP-AuNCs, the intramolecular CT character becomes not so conspicuous as that recorded in MPR-AuNCs (Fig. 3e). The shape and early-stage evolution of the short-wavelength ESA band of MP-AuNCs at 460 nm were similar to those of MPR-AuNCs at the long-term stage (Fig. S17†). Meanwhile, the intramolecular CT absorption band of MPR-AuNCs at 610 nm showed a slight blue shift to 600 nm after incubation with PNP (Fig. S17†), corresponding to the spectral feature of the purine radical anion.62 Therefore, the observed positive bands centred at 460 nm and 600 nm can be safely assigned to the absorbances of the S1 state and new 1CT state of MP-AuNCs, respectively. The ESA band species recorded at 460 nm gave an increased time constant of 0.12 ps (Fig. S18†) which corresponds to the rate of IC from the Sn state to the S1 state. The similar IC rate in MPR-AuNCs and MP-AuNCs indicates that the transition of the gold core is too short-lived to effectively participate in the intramolecular CT reaction event.41 The absorbance of the 1CT state monitored at 600 nm rose with a time constant of 0.30 ps, and decayed with time constants of 8.2 ps and 476 ps (Fig. 3f). Based on the above established decay model in MRP-AuNCs, such time components can be attributable to the rates of intramolecular CT from the S1 state to the 1CT state, dark CR of the 1CT state, and ISC between the 1CT state and the 3CT state, respectively. The increase in the intramolecular CT rate originates from a larger energy gap between the S1 state and the 1CT state,63 suggesting a lower-lying energy level of the 1CT state in MP-AuNCs, as confirmed by ultraviolet photoelectron spectroscopy (Fig. S19 and Table S2†). This result offers direct evidence for the strengthened intramolecular CT in MP-AuNCs. Of particular note, a 2.9-fold increase of ISC rate was found in MP-AuNCs, which suggests a smaller energy gap between the 1CT state and the 3CT state (ΔE1CT−3CT) according to Fermi's golden rule (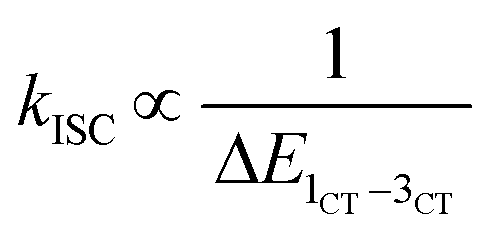 ).64 However, despite that, the ISC efficiency of MP-AuNCs still decreases to 1.2% due to a 7.2-fold increase of the dark CR rate of the 1CT state.
).64 However, despite that, the ISC efficiency of MP-AuNCs still decreases to 1.2% due to a 7.2-fold increase of the dark CR rate of the 1CT state.
Apart from ISC efficiency, the dark CR of 3CT excitons is also a factor affecting the phosphorescence QY.65 Obviously, a 3.5-fold decrease of ISC efficiency is still much lower than an 8.75-fold drop of PLQY in luminated MP-AuNCs, and thus the investigation on the dark nonradiative decay of the 3CT state in photoexcited MP-AuNCs is necessary. Fundamentally, the activation energy (Ea) of the nonradiative relaxation channel of the 3CT state is a thermally accelerated electron-phonon process.66 Temperature-dependent steady-state PL measurements showed that the emission peaks of MPR-AuNCs and MP-AuNCs were both blue-shifted with the decrease of test temperature from 300 K to 80 K (Fig. 4a and b), suggesting that the intramolecular rotation of the dark CR of the 3CT state is effectively suppressed in a rigid environment. The PL intensities of MPR-AuNCs and MP-AuNCs were found to increase by 12 times and 58 times, respectively, when temperature dropped from 300 K to 80 K, which indicates that the PL is more favourable in MP-AuNCs than in MPR-AuNCs at low temperatures. The temperature-dependent data were extracted and analysed by the Arrhenius expression (Fig. 4c). The fitted results demonstrated that the activation energies of the nonradiative decay of the 3CT state in photoexcited MPR-AuNCs and MP-AuNCs were found to be 125 meV and 72 meV, respectively. Similar to the evolution of the PL intensity, an increase of luminescence lifetime with decreasing temperature was also observed (Fig. 4d and e). The PL decays were fitted with a bi-exponential function, and the total decay rates obtained from the analysed average lifetimes were fitted by a distortional Arrhenius expression. The Ea values for MPR-AuNCs and MP-AuNCs were determined to be 144 meV and 87 meV (Fig. 4f), respectively, in correspondence with the results from temperature-dependent steady-state PL measurements. The declined activation energy suggests that the nonradiative deexcitation of the 3CT state in MPR-AuNCs is significantly enhanced upon the addition of PNP.
 | ||
| Fig. 4 Temperature-dependent emission spectra and lifetime measurements. (a) Temperature-dependent emission spectra of MPR-AuNCs. (b) Temperature-dependent emission spectra of MP-AuNCs. (c) Arrhenius plots of the thermal quenching of PL intensity. (d) PL decay traces of MPR-AuNCs at different temperatures. (e) PL decay traces of MP-AuNCs at different temperatures. (f) The observed total decay rates of MPR-AuNCs and MP-AuNCs as a function of temperature. | ||
Combined with fluorescence measurements, fs-TA spectroscopy measurements, and temperature-dependent steady-state and time-resolved fluorescence measurements, the underlying mechanism of this enzyme-activatable CT process is delineated in Fig. 5. When the ground state (S0) electrons of MPR-AuNCs are pumped, the Sn-state excitons in MPR-AuNCs undergo an ultrafast relaxation to the S1 state. Subsequently, the S1-state excitons rapidly transfer to the 1CT state by an intramolecular CT process, and then convert to triplet excitons through the ISC between the 1CT state and the 3CT state. Finally, these triplet excitons decay to the S0 state and generate bright phosphorescence emission. After the incubation with PNP, the ribose group of the surface capped MPR ligand is cleaved and the driving force of charge transfer between the S1 state and the 1CT state is enlarged, leading to an enhanced intramolecular CT process. The accelerated dark CR rate of the 1CT state competes with the 1CT-3CT transition and suppresses the formation of triplet excitons, which accounts for the decreased ISC efficiency. Moreover, due to the lower-lying 3CT state, the nonradiative loss of triplet excitons was greatly promoted. As a result, MP-AuNCs emit a weak longer-wavelength light with a low luminescence QY.
 | ||
| Fig. 5 Schematic diagram illustrating the energy-migration pathways and excited-state dynamics of MPR-AuNCs and MP-AuNCs. Arrows represent the electron transitions between different states. S0, ground state; Sn, high singlet state; S1, lowest singlet state; IC, internal conversion; 1CT, charge transfer singlet state; 3CT, charge transfer triplet state; ISC, intersystem crossing; CR, charge recombination; λmax, maximum emission wavelength; τave, average photoluminescence lifetime; QY, photoluminescence quantum yield. The conjoined pellets denote the electronic orbit, and the black arrows represent the spin direction of electrons. | ||
Analysing and visualizing PNP activity and its inhibitor using MPR-AuNCs as luminescent probes
PNP is essential for cell function and survival while its abnormal expression is closely related to the occurrence of various diseases.67,68 In addition, PNP is a vital target for drug discovery and its inhibitors have been extensively investigated due to their promising uses as specific immunosuppressive, antiviral, and anticancer agents.69 It is therefore highly desirable to design and develop facile methods to determine the activity of PNP as well as evaluate the efficiency of its inhibitor. The good spectral (long-wavelength excitation and emission, high luminescence QY, and prolonged PL lifetime) and substrate properties (strong binding affinity) of MPR-AuNCs encouraged us to further develop a label-free detection approach for such tasks. Under the optimized experimental conditions (Fig. S20†), the sensing performance of this probe for the determination of PNP activity was examined. As presented in Fig. 6a, the PL intensity gradually decreased as the enzymatic activity of PNP was increased and a good linear correlation was observed in the range of 5–30 U L−1. The limit of detection (LOD), defined as 3σ/slope, was calculated to be 0.6 U L−1, demonstrating the high sensitivity of this PNP sensing method. The constructed detection system displayed high selectivity and anti-interference ability to PNP over other potential interfering substances (Fig. S21†). As shown in Fig. 6b, the inhibition efficiency rises quickly with the increasing concentration of forodesine, a potent inhibitor of PNP. The maximum inhibition efficiency is 95.4% with a half-maximal inhibition (IC50) value of 0.26 μM. Such a high inhibition efficiency is in accordance with previous reports.70,71 | ||
| Fig. 6 Utilizing MPR-AuNCs as a visualization tool for PNP activity monitoring and drug screening. (a) Emission spectra of MPR-AuNCs upon the addition of different enzyme activities of PNP. From the top line to bottom line: 0, 5, 10, 15, 20, 25, 30, and 40 U L−1. The inset shows ΔI as a function of the PNP activity. ΔI = I0 − I, where I0 and I are the PL intensities of MPR-AuNCs at 590 nm in the absence and presence of PNP, respectively). (b) Dose-dependent inhibition of PNP activity (20 U L−1) by forodesine using MPR-AuNCs as luminescent probes. The inset shows structure of forodesine. (c) Toxicity of MPR-AuNCs to H9C2 cells. (d) Confocal fluorescence images of H9C2 cells treated with PB, MPR-AuNCs (30 μM), MPR-AuNCs (30 μM) + PNP (0.25 U mL−1), and MPR-AuNCs (30 μM) + PNP (0.25 U mL−1) + forodesine (20 μM). | ||
Our strategy can also be well applicable to visualize PNP activity and screen the PNP inhibitor in living cells. The cell counting kit-8 (CCK-8) assay demonstrates the high biocompatibility of MPR-AuNCs (Fig. 6c). The observation of brightly yellow luminescence from nonfluorescent H9C2 cells shows that MPR-AuNCs have been translocated into the cells and provide emission (control group and probe group in Fig. 6d). Consistent with the expectation of the PL quenching experiment, the labelled cells treated with PNP exhibit significant supersession of the luminescence signal (probe + PNP group in Fig. 6d), manifesting that the probe can be used to monitor cellular PNP activity. Moreover, upon the addition of forodesine, the observed quenched luminescence was completely recovered (probe + PNP + inhibitor group in Fig. 6d), confirming that the MPR-AuNC-based probe can be utilized as a simple visualization tool for inhibitor screening in the field of drug discovery.
Conclusions
In summary, we put forward an efficient strategy to modulate the CT properties of AuNCs through ligand engineering. The newly designed Au5(MPR)2 cluster can form a stable intramolecular CT state upon light excitation, which gives rise to an orange-red luminescence with high PLQY and a long PL lifetime. When incubating with PNP, the intramolecular CT process of these clusters was found to be enhanced, and the CT-type excitons were moved to a low-lying energy level, leading to significant suppression of phosphorescence with a red-shifted emission peak. This enzyme-activatable CT in MPR-AuNCs will not only shed fundamental light on the design principles of ligand-engineered MNCs but also provide a fresh perspective on deciphering the PL origin of AuNCs, which is one of the major topics in current nanoscience. The observed enzyme-activatable CT in MPR-AuNCs was further employed to construct luminescent probes to analyse and visualize PNP activity and its inhibitor. Owing to the good spectral and substrate properties of MPR-AuNCs, this label-free sensing strategy displayed satisfactory analytical performance. This approach can not only overcome the UV excitation and complicated modification processes of previously reported PNP fluorescence sensing methods,53,72 but also will provide guidance for the rational design of next-generation sensing platforms for diverse applications.Experimental
Chemicals and reagents
MPR was brought from Shanghai Acmec Biochemical Co., Ltd (Shanghai, China). HAuCl4·3H2O, FeCl2, FeCl3, glucose, urea, and glucose oxidase were obtained from the Aladdin Reagent Co., Ltd (Shanghai, China). PNP, alkaline phosphatase, α-glucosidase, β-glucuronidase, xanthine oxidase, and trypsin were provided by Sigma-Aldrich (Shanghai, China). Forodesine was brought from MedChemExpress (Shanghai, China). Bovine serum albumin, NaOH, ZnCl2, NaH2PO4, and Na2HPO4 were purchased from Shanghai Chemical Reagent Co., Ltd (Shanghai, China). Rat myocardial H9C2 cells were obtained from China Centre for Type Culture Collection (Shanghai, China). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), trypsin and penicillin-streptomycin (PS) were purchased from Gibco (New York, USA). Phosphate buffer solution (PBS, pH 7.4) was purchased from Hyclone (Utah, USA). Cell counting kit-8 (CCK-8) was obtained from Beyotime (Shanghai, China). Other reagents were all of analytical grade and used freshly without further purification. Ultrapure water was used to prepare all the solutions.Synthesis of MPR-AuNCs
A volume of 20 mL of MPR (10 mM), 2 mL of HAuCl4 (50 mM), and 0.8 mL of NaOH (1 M) were added in sequence to 78 mL of water. The mixed solution was reacted at room temperature for 30 min. After that, the resulting reaction solution was purified by dialysis in a dialysis membrane for over 12 h against ultrapure water to remove all small-molecular impurities. The obtained solution of MPR-AuNCs was stored in the dark at 4 °C before analysis.Determination of kinetic constants of the enzymatic reactions
The kinetics of PNP toward MPR was determined using the following procedures: 50 μL of PNP (2 U mL−1) and 200 μL of xanthine oxidase (2 U mL−1) were added in sequence to 1.75 mL of phosphate buffer (PB, 20 mM, pH 8.0) containing different concentrations of MPR. The resulting solutions were incubated at 45 °C for 30 min, and the absorbance at 260 nm was recorded.The kinetics of PNP toward MPR-AuNCs was determined using the following procedures: a volume of 50 μL of PNP (2 U mL−1) was added to 1.95 mL of phosphate buffer (PB, 20 mM, pH 8.0) containing different concentrations of MPR-AuNCs. The mixtures were incubated at 45 °C for 30 min, and the changes in luminescence intensity of MPR-AuNCs before and after incubation with PNP were measured.
Isothermal titration calorimetry assay
The MPR-AuNCs, MPR and PNP were dissolved in 0.02 M phosphate buffer (pH 8.0). All solutions were filtered by using a 0.22 μm nuclepore membrane filter and degassed for at least 5 min prior to titration. The PNP solution (0.0077 mM) was placed in the sample cell and titrated with MPR-AuNCs (0.3 mM) under continuous stirring at 125 rpm, 355 K. During the measurement, 14 injections of 2.5 μL titrant were performed with self-balance after the first 0.5 μL of titrant. MPR was titrated with the same procedures.Sensing PNP activity
A volume of 100 μL of the as-prepared MPR-AuNCs (300 μM) and 100 μL of PNP with different enzyme activities were added in sequence to 1.8 mL of phosphate buffer (PB, 20 mM, pH 8.0). The mixture was incubated at 45 °C for 60 min. After that, the emission spectrum was recorded with an excitation wavelength of 420 nm.Screening the PNP inhibitor
A volume of 100 μL of the as-prepared MPR-AuNCs (300 μM), 100 μL of forodesine with different concentrations, and 100 μL of PNP (200 U L−1) were added in sequence to 1.7 mL of phosphate buffer (PB, 20 mM, pH 8.0). The mixture was incubated at 45 °C for 60 min. The emission spectrum was then recorded with an excitation wavelength of 420 nm.Cell culture and treatment
H9C2 cells were maintained in 89% DMEM supplemented with 10% FBS and 1% PS and grown in a 5% CO2 atmosphere at 37 °C. When H9C2 cells covered 80–90% of the bottom of the culture flask, they were digested, centrifuged and counted in turn, resulting in a cell density of 1 × 105 cells per mL. Subsequently, 1 mL cell suspension was extracted and dropped onto the glass bottom cell culture dish, followed by incubation for 24 h. After attachment, the supernatant was replaced with PBS. The cells on dishes with different treatments were divided into four groups, involving the control group (culturing in PBS for 1 h), AuNC group (incubating with 17 μM MPR-AuNCs for 1 h), AuNCs + PNP group (culturing with 0.5 U mL−1 PNP for 1 h after treating with 17 μM MPR-AuNCs for 1 h) and AuNCs + PNP + drug group (incubating with 17 μM MPR-AuNCs for 1 h, 0.5 U mL−1 PNP for 1 h and 20 μM drug for 1 h, sequentially). Then, the morphology and AuNC labelling of H9C2 cells were observed using a laser scanning confocal microscope.Cytotoxicity assay
The biocompatibility of MPR-AuNCs was measured through the CCK-8 assay. H9C2 cells were seeded into 96-well plates with 1.5 × 104 cells per well and cultured overnight. After attachment, the cells were incubated with 0, 0.0034, 0.034, 0.34, 3.4 and 34 μM MPR-AuNCs (prepared in complete medium) for 24 h, respectively. Then, 10 μL CCK-8 solution was added to each well and incubated for 3 h at 37 °C. Subsequently, the absorbance of each well was measured at 450 nm using a microplate spectrophotometer.Michaelis constants of the PNP-enzymatic reactions
The Michaelis constants (Km) of PNP toward MPR and MPR-AuNCs were fitted by:73where ν, νmax, and [S] are the initial velocity, the maximum velocity, and concentration of substrates, respectively.

Efficiency of intersystem crossing
The yield of intersystem crossing (ΦISC) between the 1CT state and 3CT sate can be measured by:74where kISC and kCR are the rates of intersystem crossing and dark charge recombination of the 1CT state, respectively.

Arrhenius expression
The relation between the activation energy of the nonradiative relaxation channel (Ea) and the observed temperature-dependent fluorescence intensity (IT) can be expressed as:75where I0, kB,β, and T are a constant determined by excitation, Boltzmann constant, the ratio of radiative and nonradiative probabilities, and absolute temperature, respectively.

The relation between Ea and the observed total decay rate ( ) can be expressed as:75
) can be expressed as:75

Data availability
Data are available from the authors upon reasonable request.Author contributions
W. C. and J. X. co-supervised this work. W. C., J. X., H. D., and K. H. conceived the idea, and designed the experiments, whereas H. D. and K. H. carried out the experiments and characterization. W. S. performed the DFT calculations. Y. Z. carried out the cell imaging. H. D. and H. K. wrote the manuscript. Q. Y., Y. L., H. H., and X. F. discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support of the National Natural Science Foundation of China (22374022 to W. C., 22071174 to J. X., 21804021 to H. D., and 21603032 to W. S.), the Program for Innovative Leading Talents in Fujian Province (2016B016 to W. C.), the Program for Innovative Research Team in Science and Technology in Fujian Province University (2018B033 to W. C.), and Startup Fund for Scientific Research, Fujian Medical University (2021QH2011 to K. H.). J. X. acknowledges the financial support from the Ministry of Education, Singapore, Academic Research Fund (AcRF, Grant No. A-0009186-01-00 and A-8000054-01-00). H. D. thanks the China Scholarship Council (No. 20200835004) for financial support to visit National University of Singapore.References
- C. Lin, T. Kim, J. D. Schultz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2022, 14, 786–793 CrossRef CAS PubMed.
- K. R. Parenti, R. Chesler, G. He, P. Bhattacharyya, B. Xiao, H. Huang, D. Malinowski, J. Zhang, X. Yin, A. Shukla, S. Mazumdar, M. Y. Sfeir and L. M. Campos, Nat. Chem., 2023, 15, 339–346 CrossRef CAS PubMed.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS PubMed.
- W. Chi, Q. Qiao, R. Lee, W. Liu, Y. S. Teo, D. Gu, M. J. Lang, Y.-T. Chang, Z. Xu and X. Liu, Angew. Chem., Int. Ed., 2019, 58, 7073–7077 CrossRef CAS PubMed.
- S. Samanta, S. Halder and G. Das, Anal. Chem., 2018, 90, 7561–7568 CrossRef CAS PubMed.
- S. Li, L. Zhan, N. Yao, X. Xia, Z. Chen, W. Yang, C. He, L. Zuo, M. Shi, H. Zhu, X. Lu, F. Zhang and H. Chen, Nat. Commun., 2021, 12, 4627 CrossRef CAS PubMed.
- K. Zeng, Y. Lu, W. Tang, S. Zhao, Q. Liu, W. Zhu, H. Tian and Y. Xie, Chem. Sci., 2019, 10, 2186–2192 RSC.
- S. Feng, L. Zhu, D. Wang, C. Li, Y. Chen, X. Chen, J. Liu, W. Huang, Y. Ling and W. Huang, Adv. Mater., 2022, 34, 2201337 CrossRef CAS PubMed.
- H. Chen, L. Liu, K. Qian, H. Liu, Z. Wang, F. Gao, C. Qu, W. Dai, D. Lin, K. Chen, H. Liu and Z. Cheng, Sci. Adv., 2022, 8, eabo3289 CrossRef CAS PubMed.
- S. Sasaki, G. P. C. Drummen and G.-i. Konishi, J. Mater. Chem. C, 2016, 4, 2731–2743 RSC.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656–12678 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- H. Zhu, Y. Yang, K. Wu and T. Lian, Annu. Rev. Phys. Chem., 2016, 67, 259–281 CrossRef CAS PubMed.
- Q. Yao, L. Liu, S. Malola, M. Ge, H. Xu, Z. Wu, T. Chen, Y. Cao, M. F. Matus, A. Pihlajamäki, Y. Han, H. Häkkinen and J. Xie, Nat. Chem., 2022, 15, 230–239 CrossRef PubMed.
- J. Wang, F. Xu, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, Angew. Chem., Int. Ed., 2022, 61, e202207492 CrossRef CAS PubMed.
- W. D. Si, Y. Z. Li, S. S. Zhang, S. Wang, L. Feng, Z. Y. Gao, C. H. Tung and D. Sun, ACS Nano, 2021, 15, 16019–16029 CrossRef CAS PubMed.
- X. S. Han, X. Luan, H. F. Su, J. J. Li, S. F. Yuan, Z. Lei, Y. Pei and Q. M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS PubMed.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- R. R. Nasaruddin, T. Chen, N. Yan and J. Xie, Coord. Chem. Rev., 2018, 368, 60–79 CrossRef CAS.
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS PubMed.
- H. Li, F. Song, D. Zhu, Y. Song, C. Zhou, F. Ke, L. Lu, X. Kang and M. Zhu, J. Am. Chem. Soc., 2022, 144, 4845–4852 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. E. Crawford, C. M. Andolina, A. M. Smith, L. E. Marbella, K. A. Johnston, P. J. Straney, M. J. Hartmann and J. E. Millstone, J. Am. Chem. Soc., 2015, 137, 14423–14429 CrossRef CAS PubMed.
- H. Deng, K. Huang, L. Xiu, W. Sun, Q. Yao, X. Fang, X. Huang, H. A. A. Noreldeen, H. Peng, J. Xie and W. Chen, Nat. Commun., 2022, 13, 3381 CrossRef CAS PubMed.
- L. Zanetti-Polzi, P. Charchar, I. Yarovsky and S. Corni, ACS Nano, 2022, 16, 20129–20140 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- H. H. Deng, X. Q. Shi, F. F. Wang, H. P. Peng, A. L. Liu, X. H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- X. Yuan, B. Zhang, Z. Luo, Q. Yao, D. T. Leong, N. Yan and J. Xie, Angew. Chem., Int. Ed., 2014, 53, 4623–4627 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- H. H. Deng, H. P. Peng, K. Y. Huang, S. B. He, Q. F. Yuan, Z. Lin, R. T. Chen, X. H. Xia and W. Chen, ACS Sens., 2019, 4, 344–352 CrossRef CAS PubMed.
- S. Chakraborty, S. Babanova, R. C. Rocha, A. Desireddy, K. Artyushkova, A. E. Boncella, P. Atanassov and J. S. Martinez, J. Am. Chem. Soc., 2015, 137, 11678–11687 CrossRef CAS PubMed.
- H. H. Deng, K. Y. Huang, S. B. He, L. P. Xue, H. P. Peng, D. J. Zha, W. M. Sun, X. H. Xia and W. Chen, Anal. Chem., 2020, 92, 2019–2026 CrossRef CAS PubMed.
- F. Wen, Y. Dong, L. Feng, S. Wang, S. Zhang and X. Zhang, Anal. Chem., 2011, 83, 1193–1196 CrossRef CAS PubMed.
- C. L. Liu, H. T. Wu, Y. H. Hsiao, C. W. Lai, C. W. Shih, Y. K. Peng, K. C. Tang, H. W. Chang, Y. C. Chien, J. K. Hsiao, J. T. Cheng and P. T. Chou, Angew. Chem., Int. Ed., 2011, 50, 7056–7060 CrossRef CAS PubMed.
- S. Zhou, B. Peng, Y. Duan, K. Liu, O. Ikkala and R. H. A. Ras, Angew. Chem., Int. Ed., 2022, 61, e202210808 CrossRef CAS PubMed.
- K. Y. Huang, L. F. Xiu, X. Y. Fang, M. R. Yang, H. A. A. Noreldeen, W. Chen and H. H. Deng, J. Phys. Chem. Lett., 2022, 13, 9526–9533 CrossRef CAS PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962 CrossRef CAS PubMed.
- Y. Li, Y. Song, X. Zhang, T. Liu, T. Xu, H. Wang, D. e. Jiang and R. Jin, J. Am. Chem. Soc., 2022, 144, 12381 CrossRef CAS PubMed.
- Q. Li, D. Zhou, J. Chai, W. Y. So, T. Cai, M. Li, L. A. Peteanu, O. Chen, M. Cotlet, X. Wendy Gu, H. Zhu and R. Jin, Nat. Commun., 2020, 11, 2897 CrossRef CAS PubMed.
- M. Zhou, S. Vdović, S. Long, M. Zhu, L. Yan, Y. Wang, Y. Niu, X. Wang, Q. Guo, R. Jin and A. Xia, J. Phys. Chem. A, 2013, 117, 10294–10303 CrossRef CAS PubMed.
- R. Improta and V. Barone, Theor. Chem. Acc., 2008, 120, 491–497 Search PubMed.
- P. Zhang, J. Phys. Chem. C, 2014, 118, 25291–25299 CrossRef CAS.
- L. Fabris, S. Antonello, L. Armelao, R. L. Donkers, F. Polo, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2006, 128, 326–336 CrossRef CAS PubMed.
- H. Ding, F. Du, P. Liu, Z. Chen and J. Shen, ACS Appl. Mater. Interfaces, 2015, 7, 6889–6897 CrossRef CAS PubMed.
- A. J. Viudez, R. Madueño, T. Pineda and M. Blázquez, J. Phys. Chem. B, 2006, 110, 17840–17847 CrossRef CAS PubMed.
- S. Zhu, T. Li, W. B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- W. S. Sheldrick and P. Bell, Inorg. Chim. Acta, 1987, 137, 181–188 CrossRef CAS.
- R. Haag and R. A. Lewis, Mol. Cell. Biochem., 1994, 135, 129–136 CrossRef CAS PubMed.
- G. Zolotnitsky, U. Cogan, N. Adir, V. Solomon, G. Shoham and Y. Shoham, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11275–11280 CrossRef CAS PubMed.
- R. K. Harijan, I. Zoi, D. Antoniou, S. D. Schwartz and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6209–E6216 CrossRef CAS PubMed.
- A. Stachelska-Wierzchowska and J. Wierzchowski, Anal. Chim. Acta, 2020, 1139, 119–128 CrossRef CAS PubMed.
- M. D. Erion, K. Takabayashi, H. B. Smith, J. Kessi, S. Wagner, S. Hönger, S. L. Shames and S. E. Ealick, Biochemistry, 1997, 36, 11725–11734 CrossRef CAS PubMed.
- S. Garain, S. N. Ansari, A. A. Kongasseri, B. Chandra Garain, S. K. Pati and S. J. George, Chem. Sci., 2022, 13, 10011–10019 RSC.
- G. J. Zhao, F. Yu, M. X. Zhang, B. H. Northrop, H. Yang, K.-L. Han and P. J. Stang, J. Phys. Chem. A, 2011, 115, 6390–6393 CrossRef CAS PubMed.
- X. Luo, G. Liang, Y. Han, Y. Li, T. Ding, S. He, X. Liu and K. Wu, J. Am. Chem. Soc., 2020, 142, 11270–11278 CrossRef CAS PubMed.
- L. A. Ortiz-Rodríguez, G. Ortiz-Zayas, M. Pollum, S. J. Hoehn, S. Jockusch and C. E. Crespo-Hernández, Photochem. Photobiol., 2022, 98, 617–632 CrossRef PubMed.
- J. Ma, F. Wang, S. A. Denisov, A. Adhikary and M. Mostafavi, Sci. Adv., 2017, 3, e1701669 CrossRef PubMed.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2019, 5, 138–155 CAS.
- P. N. Moorthy and E. Hayon, J. Am. Chem. Soc., 1975, 97, 3345–3350 CrossRef CAS PubMed.
- H. H. Deng, K. Y. Huang, C. T. Zhu, J. F. Shen, X. P. Zhang, H. P. Peng, X. H. Xia and W. Chen, J. Phys. Chem. Lett., 2021, 12, 876–883 CrossRef CAS PubMed.
- A. K. Pati, O. El Bakouri, S. Jockusch, Z. Zhou, R. B. Altman, G. A. Fitzgerald, W. B. Asher, D. S. Terry, A. Borgia, M. D. Holsey, J. E. Batchelder, C. Abeywickrama, B. Huddle, D. Rufa, J. A. Javitch, H. Ottosson and S. C. Blanchard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24305–24315 CrossRef CAS PubMed.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- J. A. Steele, P. Puech, B. Monserrat, B. Wu, R. X. Yang, T. Kirchartz, H. Yuan, G. Fleury, D. Giovanni, E. Fron, M. Keshavarz, E. Debroye, G. Zhou, T. C. Sum, A. Walsh, J. Hofkens and M. B. J. Roeffaers, ACS Energy Lett., 2019, 4, 2205–2212 CrossRef CAS.
- L. A. Birder and E. K. Jackson, Nat. Rev. Urol., 2022, 19, 681–687 CrossRef PubMed.
- E. R. Abt, K. Rashid, T. M. Le, S. Li, H. R. Lee, V. Lok, L. Li, A. L. Creech, A. N. Labora, H. K. Mandl, A. K. Lam, A. Cho, V. Rezek, N. Wu, G. Abril-Rodriguez, E. W. Rosser, S. D. Mittelman, W. Hugo, T. Mehrling, S. Bantia, A. Ribas, T. R. Donahue, G. M. Crooks, T.-T. Wu and C. G. Radu, J. Clin. Invest., 2022, 132, e160852 CrossRef CAS PubMed.
- M. Rabuffetti, F. Rinaldi, A. Lo Bianco, G. Speranza, D. Ubiali, M. C. de Moraes, L. C. Rodrigues Pereira da Silva, G. Massolini, E. Calleri and A. Lavecchia, ChemMedChem, 2021, 16, 1325–1334 CrossRef CAS PubMed.
- K. Balakrishnan, R. Nimmanapalli, F. Ravandi, M. J. Keating and V. Gandhi, Blood, 2006, 108, 2392–2398 CrossRef CAS PubMed.
- G. B. Evans, P. C. Tyler and V. L. Schramm, ACS Infect. Dis., 2018, 4, 107–117 CrossRef CAS PubMed.
- J. Wierzchowski, M. Ogiela, B. Iwańska and D. Shugar, Anal. Chim. Acta, 2002, 472, 63–74 CrossRef CAS.
- S. U. Hettiarachchi, B. Prasai and R. L. McCarley, J. Am. Chem. Soc., 2014, 136, 7575–7578 CrossRef CAS PubMed.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- Z. Zhang, K. T. V. Grattan and A. W. Palmer, Phys. Rev. B, 1993, 48, 7772–7778 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary methods, Fig. S1–S21, and Tables S1–S2. See DOI: https://doi.org/10.1039/d4sc01509f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |