
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly stereoselective α-glycosylation with GalN3 donors enabled collective synthesis of mucin-related tumor associated carbohydrate antigens†
Kunxiu
Shou‡
a,
Yunqin
Zhang‡
a,
Yujie
Ji‡
b,
Bin
Liu
 a,
Qingli
Zhou
a,
Qiang
Tan
a,
Fuying
Li
c,
Xiufang
Wang
c,
Gang
Lu
*b and
Guozhi
Xiao
*a
a,
Qingli
Zhou
a,
Qiang
Tan
a,
Fuying
Li
c,
Xiufang
Wang
c,
Gang
Lu
*b and
Guozhi
Xiao
*a
aState Key Laboratory of Phytochemistry and Natural Medicines, Kunming Institute of Botany, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 132 Lanhei Road, Kunming, 650201, China. E-mail: xiaoguozhi@mail.kib.ac.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, Shandong 250100, China. E-mail: ganglu@sdu.edu.cn
cDepartment of Chemistry, Kunming University, 2 Puxing Road, Kunming, 650214, China
First published on 2nd April 2024
Abstract
Mucin-related tumor-associated carbohydrate antigens (TACAs) are important and interesting targets for cancer vaccine therapy. However, efficient access to a library of mucin-related TACAs remains a challenging task. One of the key issues is the challenging construction of α-GalNAc linkages. Here, we report highly stereoselective α-glycosylation with GalN3N-phenyl trifluoroacetimidate donors, which features excellent yields, outstanding stereoselectivities, broad substrate scope and mild reaction conditions. This method is successfully applied to highly stereoselective synthesis of GalN3-α-O-Ser, which served as the common intermediate for collective synthesis of a wide range of TACAs including TN antigen, STN antigen, 2,6 STF antigen, 2,3 STF antigen, glycophorin and cores 1–8 mucin-type O-glycans. In particular, the rationale for this highly stereoselective α-glycosylation is provided for the first time using DFT calculations and mechanistic studies, highlighting the crucial roles of reagent combinations (TMSI and Ph3PO) and the H-bonding directing effect of the N3 group.
Introduction
O-Linked and N-linked glycans are two principal classes of glycans obtained from post-translational modifications of proteins via glycosylation.1,2 Compared with the unique trimannosyl chitobiosyl pentasaccharide core structure Man3GlcNAc2-α-N-Asn in all N-linked chains, O-linked chains are more diverse with such various monosaccharides connected to hydroxyl amino acids as GalNAc-α-O-Ser/Thr,3 Fuc-α-O-Ser/Thr,4 Xyl-β-O-Ser,5 GlcNAc-β-O-Ser,6 Man-α-O-Ser/Thr,7 Gal-β-O-hydroxyLys8 and Ara-β-O-hydroxyPro.9 Among these, GalNAc-α-O-Ser/Thr is the most ubiquitous form of protein O-linked glycosylation, namely, mucin-type O-glycosylation, which occurs widely in higher plants and the whole animal kingdom.3 Mucin-type O-glycans play vital roles in a variety of biological processes, such as mediating cell–cell interactions, influencing the stability, conformation and structure of proteins, serving as receptor-binding ligands, and being involved in cell adhesion and immune responses.10 Interestingly, aberrant expression of mucin-type O-glycans on the surface of cancer cells usually correlates with different stages of tumor progression and various human disorders, which can differentiate them from normal tissues.11 Therefore, TACAs are highly important and desirable targets for cancer immunotherapy.12TN antigen referred to the core structure GalNAc-α-O-Ser/Thr, which was often associated with colon and prostate carcinoma and TN syndrome.13 Substitutions at C3 and/or C6 hydroxyl groups of the core structure GalNAc-α-O-Ser 1 with Gal, GlcNAc, GalNAc and Neu5NAc sugar motifs could give rise to large collections of mucin-type O-glycans, including cores 1–8 O-glycans 2–9, STN antigen 10, 2,6 STF antigen 11, 2,3 STF antigen 12, and glycophorin 13 (Scheme 1A). Cores 1–2 O-glycans 2–3 are the most abundant, while cores 3–8 O-glycans 4–9 are more restricted and organ-characteristic in mucin expression, such as in human bronchial and colonic mucins (cores 3–4),14 fetal mucins in meconium and rectal adenocarcinomas (core 5),15 fetal mucins from gastric carcinomas (core 6),16 bovine submaxillary mucin (core 7),17 and human bronchial mucin (core 8).18 STN antigen is abundantly expressed in breast, colon, stomach and ovary epithelial tumors,19 while 2,3 STF antigen20 and 2,6 STF antigen21 are found in breast tumors and myelogenous leukemia cells, respectively. Glycophorin is observed as a motif in the erythrocyte membrane glycoprotein22 and acute myelogenous leukemia cells.21 These molecules, especially tumor associated carbohydrate antigens are interesting targets for the development of new therapeutic agents, diagnostic tools and cancer vaccine therapy, as well as structural and functional studies.11–13 However, it is time-consuming and extremely difficult to isolate these TACAs from tumor tissues in pure and well-defined forms. Therefore, development of strategies for the efficient synthesis of a library of TACAs is highly desirable and remains a challenging task in chemical synthesis.
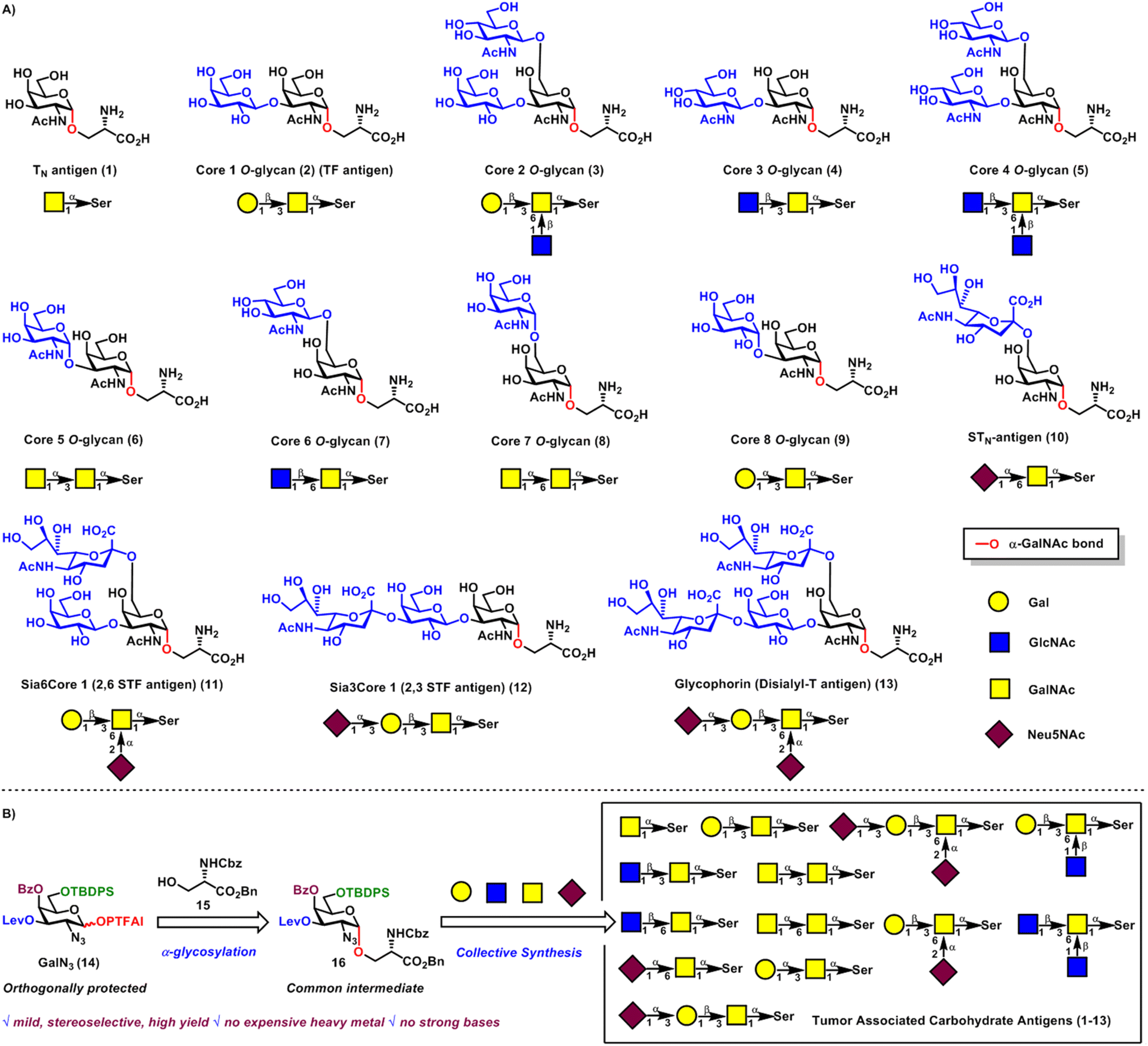 | ||
| Scheme 1 (A) Mucin-related tumor associated carbohydrate antigens 1–13; (B) collective synthesis of tumor associated carbohydrate antigens 1–13 from the common intermediate 16via a new α-glycosylation strategy. | ||
The stereoselective construction of α-GalNAc linkages is one of the key issues to be addressed for efficient access to a large library of TACAs. During the past several decades, different methods have been developed to tackle these challenges, such as glycosylation of donors with non-assisting C2-aza groups, such as azide23 and oxazolidinone,24 O-Michael addition to C2-nitro-galactals,25 4,6-di-tert-butylsilylene directed galactosylation,26 ring-opening of aziridine-2-carboxamides with O-nucleophiles of C(1)-hemiacetals,27 and nickel-catalyzed glycosylation of donors incorporated with C(2)-N-substituted benzylidene groups.28 Despite these remarkable advances, due to merits and demerits of the current state-of-the-art strategies, it remains challenging to achieve efficient, stereoselective and practical formations of α-GalNAc linkages.
Previous efforts toward syntheses of TACAs usually adopt the traditional “single-target” approach, which is not flexible enough to build large collections of these antigens. Inspired by biosynthesis of molecules in nature via the assembly of a common intermediate, a general strategy for the collective synthesis of these molecules would be ideal to expediently produce these antigens.29
Here, we report the collective synthesis of TACAs including TN antigen, STN antigen, 2,6 STF antigen, 2,3 STF antigen, glycophorin and cores 1–8 mucin-type O-glycans 1–13 from the common intermediate GalN3-α-O-Ser 16 with levulinoyl (Lev), benzoyl (Bz) and tert-butyldiphenylsilyl (TBDPS) orthogonal protecting groups at C3, C4 and C6 positions, respectively (Scheme 1B). The C6-TBDPS group could serve as a temporary protecting group for (1 → 6) branching, while the C3-Lev group could serve not only as a temporary protecting group for (1 → 3) branching, but also as a remote participating group for α-glycosylation. The C4-Bz group could serve as both a permanent protecting group and a remote participating group for stereoselective constructions of α-GalN3 linkages. The α-GalN3 linkage of the common intermediate 16 can be highly stereoselectively constructed via the newly developed merging reagent modulation and remote participation α-glycosylation30 between strategically protected GalN3N-phenyl trifluoroacetimidate31 (PTFAI) donor 14 and Cbz-protected serine amino acid 15. This α-glycosylation method features mild reaction conditions (TMSI, Ph3PO, rt), broad substrate scope, and excellent stereoselectivities and yields. DFT calculations and mechanistic studies provided rationales for this highly stereoselective α-glycosylation for the first time, uncovering important roles of TMSI and Ph3PO and the H-bonding directing effect of the N3 group.
Results and discussion
We embarked on the investigation of α-glycosylation with GalN3 PTFAI as donors 17 and HO(CH2)5NBnCbz as a strong nucleophile acceptor 18 (Scheme 2). After extensive optimization studies, when GalN3 donor 17a with both C3 and C4 Bz groups was coupled with 18 using TMSI and Ph3PO reagent combination at room temperature, glycoside 19a was obtained in excellent yield (95%) and excellent α-stereoselectivity (α/β > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (entry 1). In comparison, when donors 17b–d with no acyl groups at either the C3 or C4 position were glycosylated with 18, stereoselectivities of glycosides 19b–d were rather low (α/β = 1.3:1 to 1.7:1) (entries 2–4). Interestingly, the moderate stereoselectivity (α/β = 4:1) of glycoside 19f and good stereoselectivity (α/β = 9:1) of glycoside 19e were achieved when donor 17f with the Bz group at the C3 position and donor 17e with the Bz group at the C4 position were coupled with 18, respectively (entries 5 and 6), underlining the importance of remote participation effects of acyl groups at C3 and C4 positions.30,32 Furthermore, glycoside 19a was produced in low yield (21%) and decreased stereoselectivity (α/β = 10:1) with TMSI only (entry 7), while the low stereoselectivity (α/β = 2.8:1) of glycoside 19a was obtained with TMSOTf instead of TMSI and Ph3PO (entry 8), emphasizing the crucial role of reagent combination (TMSI and Ph3PO).30,33
:1) (entry 1). In comparison, when donors 17b–d with no acyl groups at either the C3 or C4 position were glycosylated with 18, stereoselectivities of glycosides 19b–d were rather low (α/β = 1.3:1 to 1.7:1) (entries 2–4). Interestingly, the moderate stereoselectivity (α/β = 4:1) of glycoside 19f and good stereoselectivity (α/β = 9:1) of glycoside 19e were achieved when donor 17f with the Bz group at the C3 position and donor 17e with the Bz group at the C4 position were coupled with 18, respectively (entries 5 and 6), underlining the importance of remote participation effects of acyl groups at C3 and C4 positions.30,32 Furthermore, glycoside 19a was produced in low yield (21%) and decreased stereoselectivity (α/β = 10:1) with TMSI only (entry 7), while the low stereoselectivity (α/β = 2.8:1) of glycoside 19a was obtained with TMSOTf instead of TMSI and Ph3PO (entry 8), emphasizing the crucial role of reagent combination (TMSI and Ph3PO).30,33
 | ||
| Scheme 2 Investigations of α-glycosylation with GalN3 PTFAI as donors and HO(CH2)5NBnCbz as a strong nucleophile acceptor. | ||
Next, the scope of GalN3 PTFAI donors was investigated, using 18 as a strong nucleophile and TMSI and Ph3PO as the reagent combination (entries 9–18). C3 and C4 Bz protected GalN3 PTFAI with different C6 functional groups including Lev, TBDPS, Ac and Bz were also suitable donors 17g–j, providing the corresponding glycosides 19g–j in satisfactory yields (76–90%) and outstanding α-stereoselectivities. When donors 17k–p equipped with different acyl groups at C3 and C4 were used, glycosides 19k–p were also obtained in good yields (68–85%) and excellent α-stereoselectivities (α/β = 10:1 to > 20:1).
We next examined the scope of this new α-glycosylation method using GalN317a as the donor with a large number of alcoholic nucleophiles (20a–m, 15 and 20o–ac), including primary, secondary, and tertiary alcohols and bioactive and complex natural products (Scheme 3). When reactive primary alcohols such as 1-octadecanol, benzyl alcohol, 4-penten-1-ol, 2-phenylethanol and 2,3-isopropylideneglycerol are employed, the desired glycosides 21a and 21c–f were uniformly obtained in excellent yields (91–94%) and excellent α-stereoselectivity (α/β > 20:1). It was noted that stereoselective α-glycosylation often fell short when strong nucleophilic alcohols were employed as the acceptors.30 Furthermore, coupling of primary alcohols of carbohydrate acceptors such as glucosides 20g–h, galactosides 20i–j, mannosides 20k–l and arabinofuranoside 20m with 17a proceeded smoothly, providing the desired α-glycosides 21g–m in great yields and stereoselectivities (α/β > 20:1). Of note, both higher and lower nucleophilic alcoholic acceptors (e.g.21k and 21l) are amenable to this glycosylation protocol. It was worth noting that the obtained thioglycosides 21h–i, 21m and disaccharide 21l with an anomeric para-methoxyphenyl (MP) group could be readily used as building blocks for subsequent sugar chain enlongation. Successful and stereoselective installation of α-GalN3 unit 17a to primary alcohols of L-serine 15 and 20o–p and the secondary alcohol of L-threonine 20q, including the use of different N-protecting groups, particularly, the Fmoc group (20p), indicates the promising applications of this α-glycosylation method for solid-phase synthesis of mucin glycoproteins. Besides primary alcohols, secondary alcohols of carbohydrates were also suitable acceptors, providing α-(1 → 4)-, (1 → 3)-, and (1 → 2)-glycosides 21r–t in outstanding yields and selectivities. The late stage installation of the α-GlaN3 unit on diverse bioactive natural products and drugs such as epiandrosterone 20ab, menthol 20x, cholesterol 20aa, lithocholic benzoate 21ac and estradiol benzoate 20z also successfully produced the desired glycoconjugates 21x and 21z–ac in excellent yields and stereoselectivities. Diverse functional groups such as alkene (21e and 21aa–ab), thioacetal (21h–i and 21m), acetal (21f and 21j), carbamate (21n–q) and ketone (21ab) were untouched in this α-glycosylation method, clearly demonstrating the mildness of the current protocol.
 | ||
| Scheme 3 Scope of stereoselective α-glycosylation using GalN317a as the donor. | ||
Next, application of this α-glycosylation method to the collective synthesis of mucin-type O-glycans was investigated (Scheme 4). Coupling of the strategically protected GalN3 PTFAI donor 14 with serine 15 in the presence of TMSI and Ph3PO at room temperature proceeded smoothly, furnishing the desired GalN3-α-O-Ser 16 with acceptable 60% yield and excellent stereoselectivity (α/β > 20:1) on a gram scale, which served as the common intermediate for the synthesis of all mucin-type O-glycans 1–13.
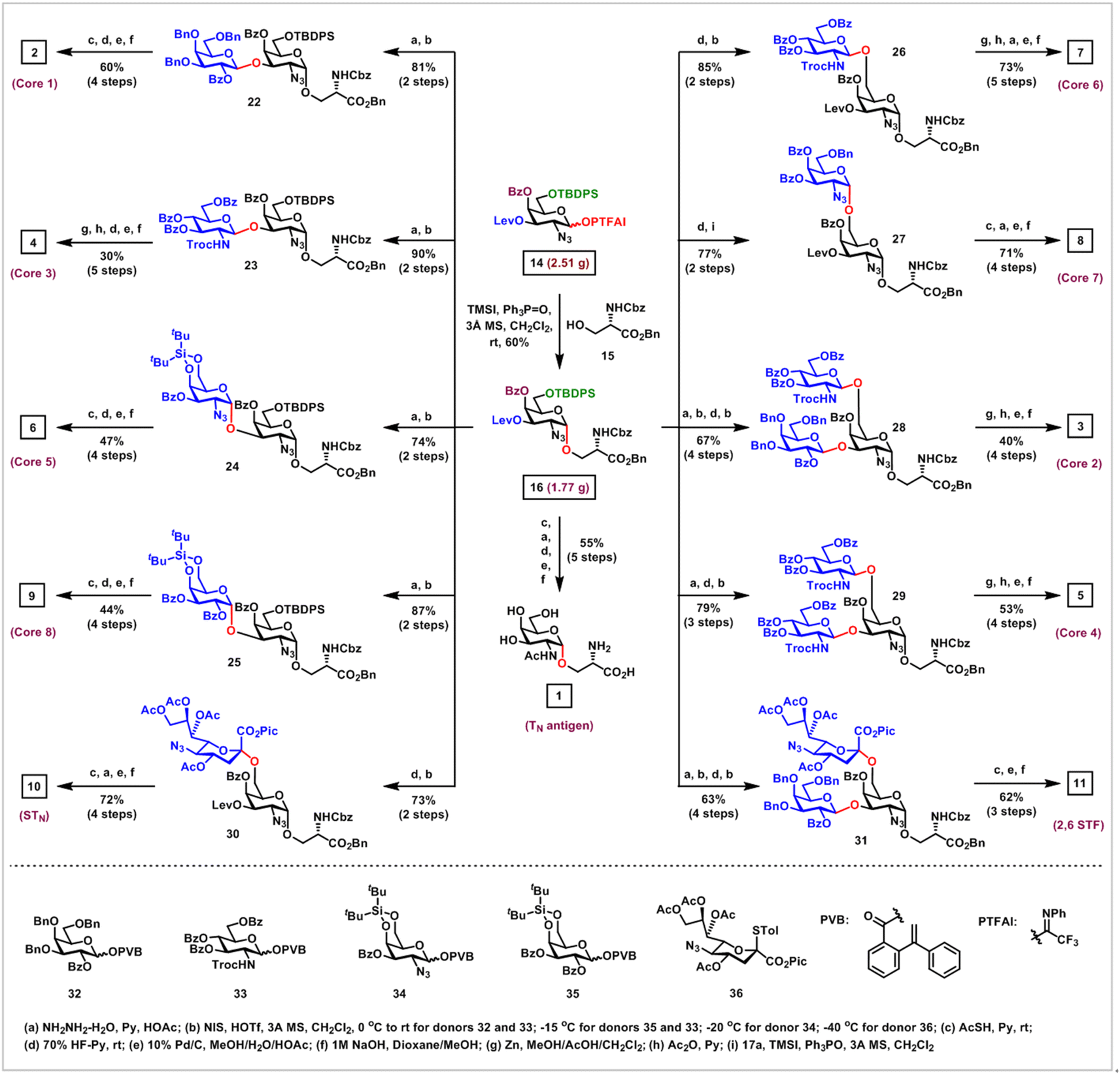 | ||
| Scheme 4 Collective synthesis of TACAs including TN antigen, STN antigen, 2,6 STF antigen and cores 1–8 mucin-type O-glycans from the common intermediate GalN3-α-O-Ser 16 via this α-glycosylation method. | ||
As for the synthesis of core 1, 3, 5 and 8 mucin-type O-glycans, selective removal of the Lev group at the C3 position of 16 afforded the C3–OH acceptor, which was glycosylated with superarmed galactosyl ortho-(1-phenylvinyl)benzoate34 (PVB) donor 32, GlcNTroc PVB donor 33, 4,6-di-tert-butylsilyl (DTBS) protected GalN3 PVB donor 34 and DTBS protected Gal PVB donor 35 in the activation of NIS and HOTf, efficiently and stereoselectively furnishing the desired glycosides 22–25 in 81%, 90%, 74% and 87% overall yields over two steps, respectively. It was noted that when superarmed PVB donor 32 was replaced with the perbenzoyl protected disarmed Gal PVB donor, the yield of the desired glycoside was low, due to the formation of significant amounts of side-products. The DTBS group ensures the highly stereoselective formation of α-(1 → 3)-GalN3 linkage in 24 and α-(1 → 3)-Gal linkage in 25.35 Interestingly, when the current method was used to install α-(1 → 3)-GalN3 linkage with GalN3 PTFAI 17a as a donor, the conversion of the reaction was low, probably due to the low reactivity of C3–OH in this GalN3 acceptor with the C4-Bz electron withdrawing group. Reductive acetylation of N3 groups to AcHN groups with thioacetic acid, followed by the first removal of silyl groups with 70% HF-Py, the second hydrogenolysis of Bn and Cbz groups, and the final saponifications of Bz groups in 22 and 24–25 successfully generated the core 1, 5 and 8 mucin-type O-glycans 2, 6 and 9 in 60%, 47% and 44% overall yields, respectively. The core 3 mucin-type O-glycan 4 was obtained in 30% overall yield over five steps via zinc-mediated deprotection of the Troc group and concomitant reduction of the N3 group in 23 to the amine groups, followed by acetylation and global deprotection.
As for the preparation of core 6 O-glycan 7, core 7 O-glycan 8 and STN antigen 10, selective removal of the TBDPS group at the C6 position in 16 afforded the C6–OH acceptor, which was coupled with GlcNTroc PVB donor 33, GalN3 PTFAI donor 17a, and 1-picolinyl-5-azido thiosialoside donor3636, affording the desired glycosides 26–27 and 30 in 85%, 77% and 73% overall yields over two steps, respectively. It was worthy of note that sialylation using Sun and Schmidt's protocol with donor 36 constructed α-(2 → 6)-sialyl linkage in 30 with excellent α-stereoselectivity (α/β > 20:1),36 while this α-glycosylation method using donor 17a highly stereoselectively assembled α-(1 → 6)-GalN3 linkage in 27. Similarly, functional group transformations and global deprotection of disaccharides 26 and 27 afforded the core 6 O-glycan 7 and core 7 O-glycan 8 in 73% and 71% overall yields, respectively. STN antigen 10 was obtained in 72% yield from 30 over the following four steps, including: (1) reductive acetylation of N3 groups to AcHN groups with thioacetic acid; (2) removal of the Lev group with NH2NH2–H2O; (3) hydrogenolysis of Bn, Cbz and Pic groups; (4) saponifications of Ac and Bz groups.
As for the synthesis of core 2 O-glycan 3 and 2,6 STF antigen 11, disaccharide 22 served as a common intermediate. Removal of the TBDPS group in 22 provided the C6–OH alcoholic acceptor, which was coupled with 33 and 36 in the presence of NIS and HOTf, successfully and stereoselectively producing the branched trisaccharides 28 and 31 in 67% and 63% overall yields via two steps, respectively. Following the above similar functional group transformation and deprotection protocol, core 2 O-glycan 3 was obtained in 40% overall yield over four steps, while 2,6 STF antigen 11 was obtained in 62% overall yield over three steps.
As for the core 4 O-glycan 5 synthesis, sequential removal of Lev and TBDPS groups in 16 gave the free C3–OH and C6–OH acceptor, which underwent double glycosylation with GlcNTroc PVB donor 33 in the activation of NIS and HOTf at −15 °C, efficiently affording the desired trisaccharide 29 in 79% overall yield over three steps. The core 4 O-glycan 5 was readily prepared in 53% overall yield over four steps via conversion of two TrocHN and azido groups to acetamido groups and global deprotection of all protecting groups.
As for TN antigen synthesis, reductive acetylation of the azido group to the acetamido group in 16, followed by global deprotection, successfully generated TN antigen 1 in 55% overall yield, which is commercially available, but very expensive to purchase ($587 per mg from Sigma-Aldrich).
Finally, we embarked on the synthesis of 2,3 STF antigen 12 and glycophorin 13via the orthogonal one-pot glycosylation strategy (Scheme 5).37 Glycosylation of sialyl phosphate donor 37 (2.5 equiv.) with 3-OH of Gal PVB 38 (1.0 equiv.) using Wong's protocol (TMSOTf, −78 °C) afforded α-NeuNAc-(2 → 3)-Gal disaccharide as a single α-isomer, which was further coupled with the poorly reactive 3-OH in 39 (0.8 equiv.) derived from 16via Lev group removal in the activation of NIS and TMSOTf at −40 °C, successfully providing the desired α-NeuNAc-(2 → 3)-β-Gal-(1 → 3)-α-GalN3-O-Ser 40 with 90% overall yield in one pot. Deprotection of the TBDPS group in 40 with 70% HF-Py afforded the trisaccharide acceptor 41, which was coupled with 1-picolinyl-5-azido thiosialoside donor 36 under the activation of NIS and HOTf at −40 °C, furnishing the desired α-NeuNAc-(2 → 3)-β-Gal-(1 → 3)-[α-NeuNAc-(2 → 6)]-α-GalN3-O-Ser 42 in satisfactory 63% yield. The final task is the removal of all protecting groups in trisaccharide 41 and tetrasaccharide 42 to generate 2,3 STF antigen 12 and glycophorin 13, which was found to be challenging due to the presence of many diverse polar groups. After extensive optimizations, the following sequences were used: (1) reductive acetylation of N3 to AcNH with thioacetic acid; (2) removal of the benzylidene group with 80% HOAc; (3) hydrogenolysis of all Bn, Pic and Cbz groups with 10% Pd/C; (4) saponifications of all esters and 5-N,4-O-carbonyl groups. 2,3 STF antigen 12 was obtained in 46% overall yield using the above optimized protocols, while glycophorin 13 was prepared in 37% overall yield using a similar protocol. It was worth noting that orthogonal one-pot glycosylation based on the pair of phosphates and PVB donors for the synthesis of 2,3 STF antigen 12 and glycophorin 13 avoided issues such as aglycone transfer inherent to orthogonal one-pot glycosylation on the basis of thioglycosides.34,38
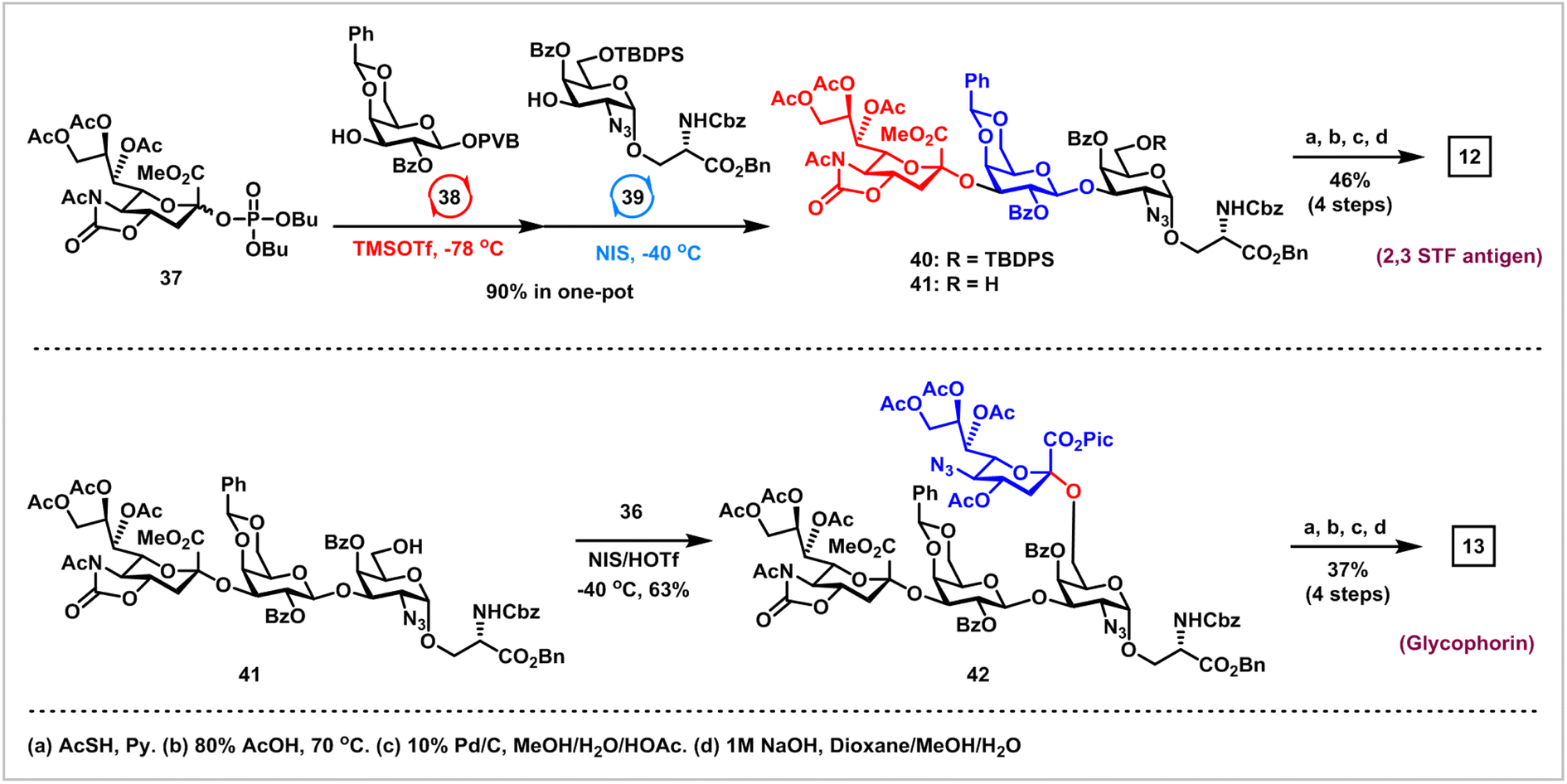 | ||
| Scheme 5 Collective synthesis of 2,3 STF antigen 12 and glycophorin 13via orthogonal one-pot glycosylation. | ||
DFT calculations were further performed to understand how exogenous reagents (TMSI and Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) modulate the process of stereoselective α-glycosylation. Based on the experimental results shown in Scheme 2, the combination of donor 17a with TMSI gave relatively low yield (21%) and decreased stereoselectivity (α/β = 10:1) (entry 7). The control experiments indicated that donor 17a can be quickly activated by TMSI, generating a mixture of α-iodide and β-iodide species (α-iodide/β-iodide = 4:1) (see Fig. S1†). This is supported by the computational results, showing that α-iodide is 1.8 kcal mol−1 more stable than β-iodide (see Fig. S3†). We thus calculated the energetics for the reaction of these iodides with the alcoholic nucleophile. As shown in Scheme 6a, the SN2 transition state (TS1) with β-iodide requires a barrier of 16.4 kcal mol−1, which is much lower than that with α-iodide (TS2, ΔG‡ = 24.6 kcal mol−1). This indicates that while all in situ formed β-iodide species are transformed into the desired α-anomer, only a small amount of α-iodide actually undergoes the nucleophilic attack by alcohol to afford the β-anomer. This is consistent with the low efficiency only in the presence of TMSI.
O) modulate the process of stereoselective α-glycosylation. Based on the experimental results shown in Scheme 2, the combination of donor 17a with TMSI gave relatively low yield (21%) and decreased stereoselectivity (α/β = 10:1) (entry 7). The control experiments indicated that donor 17a can be quickly activated by TMSI, generating a mixture of α-iodide and β-iodide species (α-iodide/β-iodide = 4:1) (see Fig. S1†). This is supported by the computational results, showing that α-iodide is 1.8 kcal mol−1 more stable than β-iodide (see Fig. S3†). We thus calculated the energetics for the reaction of these iodides with the alcoholic nucleophile. As shown in Scheme 6a, the SN2 transition state (TS1) with β-iodide requires a barrier of 16.4 kcal mol−1, which is much lower than that with α-iodide (TS2, ΔG‡ = 24.6 kcal mol−1). This indicates that while all in situ formed β-iodide species are transformed into the desired α-anomer, only a small amount of α-iodide actually undergoes the nucleophilic attack by alcohol to afford the β-anomer. This is consistent with the low efficiency only in the presence of TMSI.
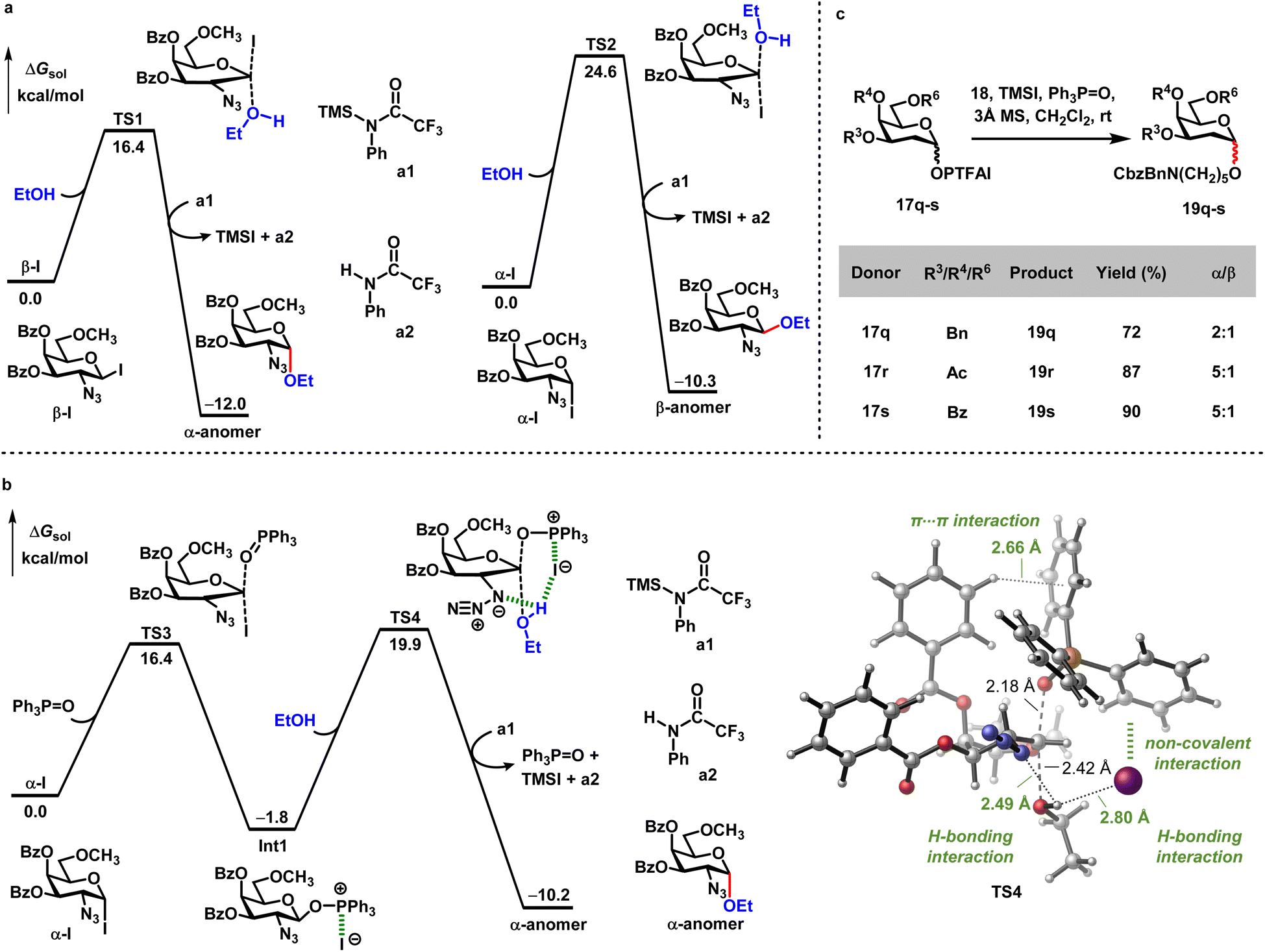 | ||
| Scheme 6 (a) Energy profiles for the SN2 reaction of the alcoholic acceptor with α-iodide and β-iodide; (b) energy profile for the Ph3PO promoted SN2 nucleophilic reaction of α-iodide with alcoholic acceptors; (c) glycosylation between 2-deoxy glycosyl PTFAI donors 17q–s and 18 with TMSI and Ph3PO. | ||
By contrast, the treatment of Ph3PO with the mixture of donor 17a and TMSI significantly enhanced both yield and stereoselectivity (95% yield, α/β > 20:1, entry 1 in Scheme 2). Since β-iodide is highly reactive towards the alcoholic acceptor (Scheme 6a), the promotion effect of Ph3PO could be attributed to its capacity for converting α-iodide into a more reactive glycosylating species. The computations indeed find that Ph3PO can replace the iodine atom of α-iodide in the SN2 fashion with a relatively low barrier (TS3, ΔG‡ = 16.4 kcal mol−1, Scheme 6b), generating the β-anomeric phosphonium iodide intermediate (Int1). The species of phosphonium iodide intermediate can be detected by ESI-MS experiment monitoring (see Fig. S2†). In Int1, there is no covalent interaction between iodine and phosphine. Instead, this intermediate is stabilized by electrostatic interactions and non-covalent interactions between I and Ph3PO moieties (see Fig. S4†). Subsequently, Int1 is attacked by the alcoholic acceptor viaTS4, delivering the α-selective glycosidic linkage. Compared with the α-iodide (TS2, ΔG‡ = 24.6 kcal mol−1), Int1 derived from α-iodide exhibits a greater reactivity to the α-face SN2 displacement (TS4, ΔG‡ = 21.7 kcal mol−1). The lower barrier of TS4 is mostly due to a series of stabilizing interactions (Scheme 6b), including the H-bonding interactions of alcohol with both N3 and I moieties, the non-covalent interactions between I and Ph3PO (Fig. S4†), and the T-shaped π⋯π interaction between C4 Bz and Ph3PO. Except for enhancing the reactivity of α-iodide, more importantly, Ph3PO also plays a critical role in transforming α-iodide into the desired α-anomer mediated by the β-anomeric phosphonium iodide species. In addition, we also studied the reaction of β-iodide with Ph3PO, which is less favorable than the direct α-face SN2 displacement by the alcoholic acceptor viaTS1 (see Fig. S5†). Taken together, the role of TMSI is to generate α-iodide and β-iodide species, and Ph3PO can further ensure the formation of α-selective glycosidic linkage. The combination of these exogenous reagents facilitates excellent yield and α/β selectivity. To further support H-bonding interactions between alcohol and N3 moieties, different 2-deoxy glycosyl PTFAI donors 17q–s were coupled with 18 in the presence of TMSI and Ph3PO. Low stereoselectivity (α/β = 5:1) of products 19r–s was obtained (Scheme 6c), highlighting the H-bonding directing role of the N3 group for this α-glycosylation.
Conclusions
In summary, highly stereoselective α-glycosylation with GalN3 PTFAI donors has been achieved, which features mild reaction conditions, broad substrate scope, and excellent stereoselectivities and yields. Furthermore, this synergistic α-glycosylation strategy was successfully applied to efficient and highly stereoselective synthesis of the common intermediate GalN3-α-O-Ser 16 with orthogonal protecting groups for collective synthesis of tumor associated carbohydrate antigens including TN antigen, STN antigen, 2,6 STF antigen, 2,3 STF antigen, glycophorin, and cores 1–8 mucin-type O-glycans 1–13. Mechanistic studies and DFT calculations uncovered the crucial roles of reagent combinations (TMSI and Ph3PO) and the H-bonding directing effect of the N3 group for achieving highly stereoselective α-glycosylation for the first time. This library of synthetic mucin-related TACAs can be used to investigate the structure–reactivity relationships of mucin-type O-glycans. We believe that the present work could facilitate and inspire advances in complex carbohydrate synthesis for the development of new carbohydrate-based therapeutics and understandings of their functions.39
Data availability
Experimental procedures, characterisation data, and NMR spectra for new compounds can be found in the ESI.†Author contributions
G. X. conceived the idea and designed the research. K. S. investigated this α-glycosylation, explored the scope, and synthesized the TN antigen. Y. Z. achieved synthesis of STN antigen, 2,6 STF antigen, 2,3 STF antigen, glycophorin, and cores 1–8 mucin-type O-glycans. Y. J. and G. L. performed DFT calculations. B. L., Q. Z., Q. T. and F. L. conducted preparation of some building blocks. G. X. G. L. and X. W. wrote the manuscript with feedback from all authors.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The financial support from the National Natural Science Foundation of China (22322110, 21973055, and 22371171), the Key Project of the Natural Science Foundation of Yunnan Province (No. 202101AS0700 30), the Young Talents Project of High-level Talent Introduction Program of Yunnan Province, the CAS Pioneer Hundred Talents Program (No. 2017-128), the Basic Research Programs of Yunnan Province (2020J0505 and 202001BA070001-065), the High-Level Talent Special Support Plans for Young Talents of Kunming City (Grant No. C202005008), and the Yunnan Province Science and Technology Department (202305AH340005) is greatly acknowledged.Notes and references
- H. Herzner, T. Reipen, M. Schultz and H. Kunz, Chem. Rev., 2000, 100, 4495–4538 CrossRef CAS PubMed.
- M. J. Grogan, M. R. Pratt, L. A. Marcaurelle and C. R. Bertozzi, Annu. Rev. Biochem., 2002, 71, 593–634 CrossRef CAS PubMed.
- (a) N. Martínez-Sáez, J. M. Peregrina and F. Corzana, Chem. Soc. Rev., 2017, 46, 7154–7175 RSC; (b) L. A. Marcaurelle and C. R. Bertozzi, Glycobiology, 2002, 12, 69R–77R CAS.
- R. J. Harris and M. W. Spellman, Glycobiology, 1993, 3, 219–224 CrossRef CAS PubMed.
- (a) J. D. Esko and L. Zhang, Curr. Opin. Struct. Biol., 1996, 6, 663–670 CrossRef CAS PubMed; (b) I. B. H. Wilson, Cell. Mol. Life Sci., 2004, 61, 794–809 CrossRef CAS PubMed; (c) B. Yang, K. Yoshida, Z. Yin, H. Dai, H. Kavunja, M. H. El-Dakdouki, S. Sungsuwan, S. B. Dulaney and X. Huang, Angew. Chem., Int. Ed., 2012, 51, 10185–10189 CrossRef CAS PubMed.
- (a) G. W. Hart, R. S. Haltiwanger, G. D. Holt and W. G. Kelly, Annu. Rev. Biochem., 1989, 58, 841–874 CrossRef CAS PubMed; (b) T. Pohl and H. Waldmann, J. Am. Chem. Soc., 1997, 119, 6702–6710 CrossRef CAS.
- (a) N. R. Smalheiser, S. M. Haslam, M. Sutton-Smith, H. R. Morris and A. Dell, J. Biol. Chem., 1998, 273, 23698–23703 CrossRef CAS PubMed; (b) J. Seifert, T. Ogawa and Y. Ito, Tetrahedron Lett., 1999, 40, 6803–6807 CrossRef CAS.
- J. Broddefalk, J. Bäcklund, F. Almqvist, M. Johansson, R. Holmdahl and J. Kihlberg, J. Am. Chem. Soc., 1998, 120, 7676–7683 CrossRef CAS.
- M. J. Kieliszewski, M. O'Neill, J. Leykam and R. Orlando, J. Biol. Chem., 1995, 270, 2541–2549 CrossRef CAS PubMed.
- F.-G. Hanisch, Biol. Chem., 2001, 382, 143–149 CAS.
- S. J. Danishefsky and J. R. Allen, Angew. Chem., Int. Ed., 2000, 39, 836–863 CrossRef CAS PubMed.
- (a) W.-H. Li and Y.-M. Li, Chem. Rev., 2020, 120, 11420–11478 CrossRef CAS PubMed; (b) R. Mettu, C.-Y. Chen and C.-Y. Wu, J. Biomed. Sci., 2020, 27, 9 CrossRef CAS PubMed; (c) M.-M. Wei, Y.-S. Wang and X.-S. Ye, Med. Res. Rev., 2018, 38, 1003–1026 CrossRef PubMed; (d) D. Feng, A. S. Shaikh and F. Wang, ACS Chem. Biol., 2016, 11, 850–863 CrossRef CAS PubMed; (e) N. Gaidzik, U. Westerlind and H. Kunz, Chem. Soc. Rev., 2013, 42, 4421–4442 RSC; (f) C. Sorieul, F. Papi, F. Carboni, S. Pecetta, S. Phogat and R. Adamo, Pharmacol. Ther., 2022, 235, 108158 CrossRef CAS PubMed.
- (a) T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed; (b) C. Pifferi, A. Ruiz-de-Angulo, D. Goyard, C. Tiertant, N. Sacristán, D. Barriales, N. Berthet, J. Anguita, O. Renaudet and A. Fernández-Tejada, Chem. Sci., 2020, 11, 4488–4498 RSC.
- (a) J. Breg, H. van Halbeek, J. F. G. Vliegenthart, A. Klein, G. Lamblin and P. Roussel, Eur. J. Biochem., 1988, 171, 643–654 CrossRef CAS PubMed; (b) D. K. Podolski, J. Biol. Chem., 1985, 260, 8262–8271 CrossRef; (c) D. K. Podolsky, J. Biol. Chem., 1985, 260, 15510–15515 CrossRef CAS PubMed.
- (a) E. F. Hounsell, A. M. Lawson, J. Feeney, H. C. Gooi, N. J. Pickering, M. S. Stoll, S. C. Lui and T. Feizi, Eur. J. Biochem., 1985, 148, 367–377 CrossRef CAS PubMed; (b) A. Kurosaka, H. Nakajima, I. Funakoshi, M. Matsuyama, T. Nagayo and I. Yamashina, J. Biol. Chem., 1983, 258, 11594–11598 CrossRef CAS PubMed.
- Y. Yamashita, Y. S. Chung, T. Sawada, Y. Kondo, K. Hirayama, A. Inui, B. Nakata, M. Okuno, R. Horie, T. Saito, K. Murayama, R. Kannagi and M. Sowa, Int. J. Cancer, 1994, 58, 349–355 CrossRef CAS PubMed.
- W. Chai, E. F. Hounsell, G. C. Cashmore, J. R. Rosankiewicz, C. J. Bauer, J. Feeney, T. Feizi and A. M. Lawson, Eur. J. Biochem., 1992, 203, 257–268 CrossRef CAS PubMed.
- H. van Halbeek, A.-M. Strang, M. Lhermitte, H. Rahmoune, G. Lamblin and P. Roussel, Glycobiology, 1994, 4, 203–209 CrossRef CAS PubMed.
- G. D. MacLean, M. A. Reddish, R. R. Koganty and B. M. Longenecker, J. Immunother., 1996, 19, 59–68 CrossRef CAS PubMed.
- K. O. Lloyd, J. Burchell, V. Kudryashov, B. W. T. Yin and J. Taylor-Papadimitriou, J. Biol. Chem., 1996, 271, 33325–33334 CrossRef CAS PubMed.
- M. Fukuda, S. R. Carlsson, J. C. Klock and A. Dell, J. Biol. Chem., 1986, 261, 12796–12806 CrossRef CAS PubMed.
- D. B. Thomas and R. J. Winzler, J. Biol. Chem., 1969, 244, 5943–5946 CrossRef CAS PubMed.
- (a) R. Kaifu and T. Osawa, Carbohydr. Res., 1977, 58, 235–239 CrossRef CAS PubMed; (b) R. M. Ratcliffe, D. A. Baker and R. U. Lemieux, Carbohydr. Res., 1981, 93, 35–41 CrossRef CAS PubMed; (c) H. Paulsen and J.-P. Hölck, Carbohydr. Res., 1982, 109, 89–107 CrossRef CAS PubMed; (d) H. Kunz and S. Birnbach, Angew. Chem., Int. Ed., 1986, 25, 360–362 CrossRef; (e) S. D. Kuduk, J. B. Schwarz, X.-T. Chen, P. W. Glunz, D. Sames, G. Ragupathi, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 12474–12485 CrossRef CAS; (f) L. Wang, Y. Zhang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codee, J. Org. Chem., 2020, 85, 15872–15884 CrossRef CAS PubMed; (g) Y. Zhang, X. Ma and L. Zhang, CCS Chem., 2023, 5, 2799–2807 CrossRef CAS PubMed.
- (a) K. Benakli, C. Zha and R. J. Kerns, J. Am. Chem. Soc., 2001, 123, 9461–9462 CrossRef CAS PubMed; (b) S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666–10667 CrossRef CAS PubMed.
- (a) K. B. Pal, A. Guo, M. Das, G. Báti and X.-W. Liu, ACS Catal., 2020, 10, 6707–6715 CrossRef CAS; (b) J.-L. Liu, Y.-T. Zhang, H.-F. Liu, L. Zhou and J. Chen, Org. Lett., 2017, 19, 5272–5275 CrossRef CAS PubMed; (c) S. Medina, M. J. Harper, E. I. Balmond, S. Miranda, G. E. M. Crisenza, D. M. Coe, E. M. McGarrigle and M. C. Galan, Org. Lett., 2016, 18, 4222–4225 CrossRef CAS PubMed; (d) R. R. Schmidt and Y. D. Vankar, Acc. Chem. Res., 2008, 41, 1059–1073 CrossRef CAS PubMed; (e) G. A. Winterfeld and R. R. Schmidt, Angew. Chem., Int. Ed., 2001, 40, 2654–2657 CrossRef CAS.
- A. Imamura, A. Kimura, H. Ando, H. Ishida and M. Kiso, Chem.–Eur. J., 2006, 12, 8862–8870 CrossRef CAS PubMed.
- D. A. Ryan and D. Y. Gin, J. Am. Chem. Soc., 2008, 130, 15228–15229 CrossRef CAS PubMed.
- (a) E. A. Mensah and H. M. Nguyen, J. Am. Chem. Soc., 2009, 131, 8778–8780 CrossRef CAS PubMed; (b) E. A. Mensah, F. Yu and H. M. Nguyen, J. Am. Chem. Soc., 2010, 132, 14288–14302 CrossRef CAS PubMed; (c) F. Yu and H. M. Nguyen, J. Org. Chem., 2012, 77, 7330–7343 CrossRef CAS PubMed; (d) M. S. McConnell, F. Yu and H. M. Nguyen, Chem. Commun., 2013, 49, 4313–4315 RSC; (e) F. Yu, M. S. McConnell and H. M. Nguyen, Org. Lett., 2015, 17, 2018–2021 CrossRef CAS PubMed.
- (a) M. R. Gadi, C. Chen, S. Bao, S. Wang, Y. Guo, J. Han, W. Xiao and L. Li, Chem. Sci., 2023, 14, 1837–1843 RSC; (b) S. Wang, C. Chen, M. R. Gadi, V. Saikam, D. Liu, H. Zhu, R. Bollag, K. Liu, X. Chen, F. Wang, P. G. Wang, P. Ling, W. Guan and L. Li, Nat. Commun., 2021, 12, 3573 CrossRef CAS PubMed; (c) M. Guberman, M. Bräutigam and P. H. Seeberger, Chem. Sci., 2019, 10, 5634–5640 RSC.
- (a) Y. Zhang, H. He, Z. Chen, Y. Huang, G. Xiang, P. Li, X. Yang, G. Lu and G. Xiao, Angew. Chem., Int. Ed., 2021, 60, 12597–12606 CrossRef CAS PubMed; (b) Y. Zhang, Y. Hu, S. Liu, H. He, R. Sun, G. Lu and G. Xiao, Chem. Sci., 2022, 13, 7755–7764 RSC; (c) P. Li, H. Fan, Q. Tan and G. Xiao, Org. Lett., 2023, 25, 2788–2792 CrossRef CAS PubMed; (d) K. Shou, S. Liu, Y. Zhang and G. Xiao, Chin. J. Chem., 2024, 42, 1593–1598 Search PubMed.
- (a) B. Yu and J. Sun, Chem. Commun., 2010, 46, 4668–4679 RSC; (b) B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- (a) A. A. Hettikankanamalage, R. Lassfolk, F. S. Ekholm, R. Leino and D. Crich, Chem. Rev., 2020, 120, 7104–7151 CrossRef CAS PubMed; (b) B. S. Komarova, Y. E. Tsvetkov and N. E. Nifantiev, Chem. Rec., 2016, 16, 488–506 CrossRef CAS PubMed.
- (a) L. Wang, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed; (b) S.-R. Lu, Y.-H. Lai, J.-H. Chen, C.-Y. Liu and K.-K. T. Mong, Angew. Chem., Int. Ed., 2011, 50, 7315–7320 CrossRef CAS PubMed; (c) S. K. Mulani, W.-C. Hung, A. B. Ingle, K.-S. Shiau and K.-K. T. Mong, Org. Biomol. Chem., 2014, 12, 1184–1197 RSC; (d) J. Zeng, R. Wang, S. Zhang, J. Fang, S. Liu, G. Sun, B. Xu, Y. Xiao, D. Fu, W. Zhang, Y. Hu and Q. Wan, J. Am. Chem. Soc., 2019, 141, 8509–8515 CrossRef CAS PubMed.
- (a) P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405 CrossRef CAS PubMed; (b) H. He, L. Xu, R. Sun, Y. Zhang, Y. Huang, Z. Chen, P. Li, R. Yang and G. Xiao, Chem. Sci., 2021, 12, 5143–5151 RSC.
- (a) A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef CAS; (b) A. Imamura, N. Matsuzawa, S. Sakai, T. Udagawa, S. Nakashima, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2016, 81, 9086–9104 CrossRef CAS PubMed.
- J. Chen, T. Hansen, Q.-J. Zhang, D.-Y. Liu, Y. Sun, H. Yan, J. D. C. Codée, R. R. Schmidt and J.-S. Sun, Angew. Chem., Int. Ed., 2019, 58, 17000–17008 CrossRef CAS PubMed.
- (a) Y. Ma, Y. Zhang, Y. Huang, Z. Chen, Q. Xian, R. Su, Q. Jiang, X. Wang and G. Xiao, J. Am. Chem. Soc., 2024, 146, 4112–4122 CrossRef CAS PubMed; (b) Y. Zhang, L. Wang, Q. Zhou, Z. Li, D. Li, C. Yin, X. Wang and G. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202301351 CrossRef CAS PubMed; (c) Y. Zhang, Z. Chen, Y. Huang, S. He, X. Yang, Z. Wu, X. Wang and G. Xiao, Angew. Chem., Int. Ed., 2020, 59, 7576–7584 CrossRef CAS PubMed; (d) Z. Chen and G. Xiao, Org. Lett., 2023, 25, 7395–7399 CrossRef CAS PubMed; (e) X. Sun, Z. Chen, R. Yang, M. Wang, X. Wang, Q. Zhang and G. Xiao, Org. Lett., 2023, 25, 7364–7368 CrossRef CAS PubMed; (f) Y. Ma, Q. Jiang, X. Wang and G. Xiao, Org. Lett., 2022, 24, 7950–7954 CrossRef CAS PubMed; (g) R. Yang, X. Sun, Y. Zhang and G. Xiao, Org. Biomol. Chem., 2022, 20, 6755–6758 RSC.
- (a) Y. Hu, K. Yu, L.-L. Shi, L. Liu, J.-J. Sui, D.-Y. Liu, B. Xiong and J.-S. Sun, J. Am. Chem. Soc., 2017, 139, 12736–12744 CrossRef CAS PubMed; (b) X. Xiao, J. Zeng, J. Fang, J. Sun, T. Li, Z. Song, L. Cai and Q. Wan, J. Am. Chem. Soc., 2020, 142, 5498–5503 CrossRef CAS PubMed.
- For selective reviews, see: (a) X. Wang and G. Xiao, Curr. Opin. Chem. Biol., 2023, 77, 102387 CrossRef CAS PubMed; (b) S. Wang, Y. Yang, Q. Zhu, G. Q. Lin and B. Yu, Curr. Opin. Chem. Biol., 2022, 69, 102154 CrossRef CAS PubMed; (c) L. Del Bino, K. E. Østerlid, D.-Y. Wu, F. Nonne, M. R. Romano, J. Codée and R. Adamo, Chem. Rev., 2022, 122, 15672–15716 CrossRef CAS PubMed; (d) P. H. Seeberger, Chem. Rev., 2021, 121, 3598–3626 CrossRef CAS PubMed; (e) D. Crich, J. Am. Chem. Soc., 2021, 143, 17–34 CrossRef CAS PubMed; (f) L. Krasnova and C.-H. Wong, J. Am. Chem. Soc., 2019, 141, 3735–3754 CrossRef CAS PubMed; (g) S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed; (h) C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed; (i) M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef CAS PubMed; (j) W.-L. Leng, H. Yao, J.-X. He and X.-W. Liu, Acc. Chem. Res., 2018, 51, 628–639 CrossRef CAS PubMed; (k) P. Peng and R. R. Schmidt, Acc. Chem. Res., 2017, 50, 1171–1183 CrossRef CAS PubMed; (l) S. J. Danishefsky, Y.-K. Shue, M. N. Chang and C.-H. Wong, Acc. Chem. Res., 2015, 48, 643–652 CrossRef CAS PubMed; (m) T. J. Boltje, T. Buskas and G.-J. Boons, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed; (n) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01348d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |