
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeconvoluting nitric oxide–protein interactions with spatially resolved multiplex imaging†
Yi
Li‡
 a,
Kaijun
Pan‡
b,
Yanan
Gao
b,
Jia
Li
be,
Yi
Zang
*ce and
Xin
Li
*ad
a,
Kaijun
Pan‡
b,
Yanan
Gao
b,
Jia
Li
be,
Yi
Zang
*ce and
Xin
Li
*ad
aCollege of Pharmaceutical Sciences, National Key Laboratory of Advanced Drug Delivery and Release Systems, Zhejiang University, 866 Yuhangtang Street, Hangzhou 310058, China. E-mail: lixin81@zju.edu.cn
bJiangsu Key Laboratory of Drug Screening, China Pharmaceutical University, Nanjing 210009, China
cLingang Laboratory, Shanghai 201203, China. E-mail: yzang@lglab.ac.cn
dFuture Health Laboratory, Innovation Center of Yangtze River Delta, Zhejiang University, Jiashan 314100, China
eState Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medical, Chinese Academy of Sciences, Shanghai 201203, China
First published on 8th April 2024
Abstract
Simultaneous imaging of nitric oxide (NO) and its proximal proteins should facilitate the deconvolution of NO–protein interactions. While immunostaining is a primary assay to localize proteins in non-genetically manipulated samples, NO imaging probes with immunostaining-compatible signals remain unexplored. Herein, probe NOP-1 was developed with an NO-triggered proximal protein labeling capacity and fluorogenic signals. The trick is to fuse the native chemical ligation of acyl benzotriazole with the protein-conjugation-induced fluorogenic response of Si-rhodamine fluorophore. NOP-1 predominantly existed in the non-fluorescent spirocyclic form. Yet, its acyl o-phenylenediamine moiety was readily activated by NO into acyl benzotriazole to conjugate proximal proteins, providing a fluorogenic response and translating the transient cellular NO signal into a permanent stain compatible with immunostaining. NOP-1 was utilized to investigate NO signaling in hypoglycemia-induced neurological injury, providing direct evidence of NO-induced apoptosis during hypoglycemia. Mechanistically, multiplex imaging revealed the overlap of cellular NOP-1 fluorescence with immunofluorescence for α-tubulin and NO2-Tyr. Importantly, α-tubulin was resolved from NOP-1 labeled proteins. These results suggest that NO played a role in hypoglycemia-induced apoptosis, at least in part, through nitrating α-tubulin. This study fills a crucial gap in current imaging probes, providing a valuable tool for unraveling the complexities of NO signaling in biological processes.
Introduction
Nitric oxide (NO) is an intriguing gaseous signaling molecule vital for various physiological processes, such as vasodilation, neurotransmission, and immunity.1–4 Endogenously generated from L-arginine by nitric oxide synthases (NOSs),5,6 NO exerts chemical signaling functions by binding to soluble guanylate cyclase (sGC) to enhance its enzymatic activity,7,8 or by nitrosylating proteins post-translationally to modulate their functions.9,10 Aberrant NO levels have been associated with multiple disorders, including cardiovascular diseases, neurological diseases, metabolic disorders, and cancer.11–17 Though NO was identified as a biological messenger almost three decades ago,18 novel insights into its mechanisms in various pathophysiology have been continually revealed.4,19,20 Consequently, there emerges an assay demand to explore its implications in diverse biological processes. However, such an assay remains challenging due to the transient nature of NO. Currently, the assay is generally carried out by immunostaining NOSs alongside the proteins of interest.21–25 Direct measurement of NO alongside its potential interacting proteins should provide more accurate information to elucidate NO-related pathophysiology.Given the importance of NO, there have been many activity-based fluorescent probes reported for directly measuring cellular NO levels.26–31 These probes are generally fluorophores derivatized with o-phenylenediamine serving to trap NO and to quench the fluorescence through the mechanism of photo-induced electron transfer. o-Phenylenediamine is readily transformed into a non-quenching benzotriazole moiety after reacting with NO, generating fluorogenic signals for detection (Fig. 1A). While these probes have allowed for direct imaging of NO in live cells and even live mice, they readily diffuse out of cells and are incompatible with immunostaining which typically necessitates membrane permeability and harsh wash-out procedures.32 Since immunostaining is a primary assay for identifying proteins in a spatially resolved way,33–36 it should hold promise for studying NO-protein interactions. In this context, a novel NO-sensing probe that can be activated by NO to covalently label proximal proteins is advocated, which should translate the transient cellular NO signal into a permanent one compatible with immunostaining, and link cellular NO levels with various biological processes.
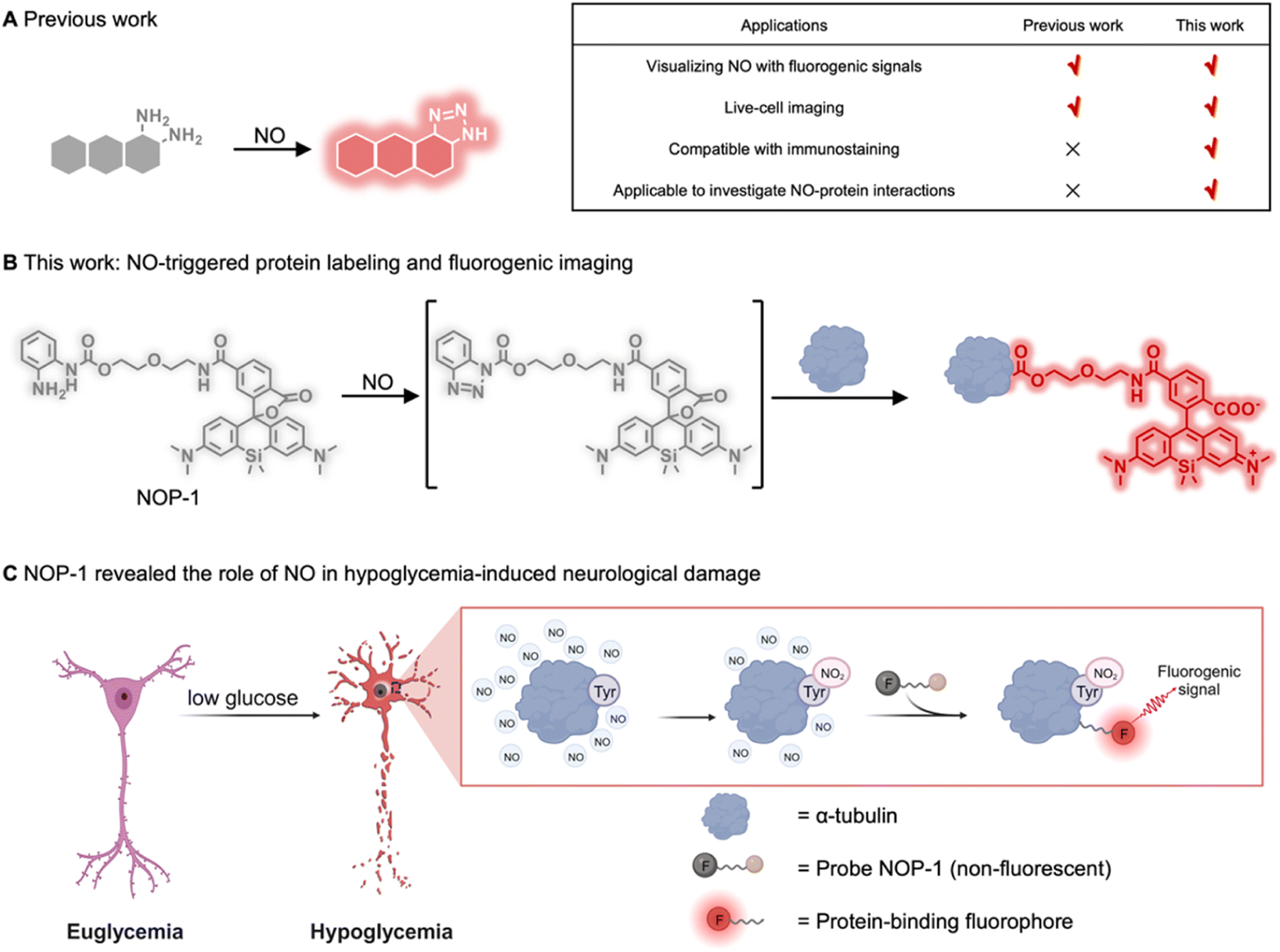 | ||
| Fig. 1 Illustration of NO-triggered protein labeling for studying the mechanism of hypoglycemia-induced neurological damage. (A) Mechanism of previous probes for imaging NO. (B) Chemical mechanism of probe NOP-1 for NO-triggered protein labeling and fluorogenic imaging. (C) Probe NOP-1 revealed the role of NO in hypoglycemia-induced neurological damage by nitrating α-tubulin. | ||
The strategy of analyte-triggered protein labeling has been proposed to interrogate analyte-regulated signaling pathways.37–40 However, integrating this strategy with activity-based sensing to provide dual-functional probes capable of fluorogenically imaging NO and covalently labeling proteins remains challenging, which requires a unique group that is intrinsically inert but can be specifically activated by NO to label proteins, and that a fluorogenic signal should be yielded accompanying this labeling event. Herein, we reported probe NOP-1 fulfilling these requirements. The probe was developed by combining the native chemical ligation reaction of acyl benzotriazole and the protein-ligation-induced fluorogenic response of the Si-rhodamine fluorophore. It bears an acyl o-phenylenediamine to trap NO and subsequently label proximal proteins. The Si-rhodamine fluorophore in NOP-1 exists in the non-fluorescent spirocyclic form in the absence of NO but readily switches to the fluorescent zwitterionic form upon ligating on proteins. In this way, NOP-1 can image NO with a fluorogenic signal that is permanent and compatible with immunostaining (Fig. 1B). NOP-1 displayed superior performance to the classic commercial NO probe DAF-FM DA in terms of immunostaining compatibility. Facilitated by NOP-1, we observed an upregulation of NO in cells under hypoglycemic stress, and for the first time revealed that NO contributed to hypoglycemia-induced neurological injury by nitrating proteins, especially α-tubulin (Fig. 1C). These observations were realized only with the aid of the probe NOP-1 which was entailed to sense NO and subsequently label proteins, producing both fluorogenic and immunostaining-compatible signals. These results highlight the versatility of NOP-1 and promise its potential for exploring NO-related pathophysiology in various biological processes.
Results and discussion
Designing probes with NO-triggered protein labeling and fluorogenic response
To realize NO-triggered protein labeling, the native chemical ligation between acyl benzotriazoles and protein cysteines is inspiring, which is biocompatible, fast, and efficient.41 In this regard, it is straightforward to envision that any fluorophore could be derivatized with a carbonyl acid group and then conjugated with o-phenylenediamine. The resulting acyl o-phenylenediamine should then readily sense NO and be activated into acyl benzotriazole to label proximal proteins. However, the simultaneous achieving of a desirable fluorogenic signal is challenging. Reported probes for fluorogenically imaging NO usually contain o-phenylenediamine as part of their fluorophore cores (Fig. 1A). In this way, their baseline fluorescence is effectively quenched by the mechanism of photo-induced electron transfer. Moving o-phenylenediamine away from the fluorophore core should greatly compromise its fluorescence-quenching ability, given that the efficacy of photo-induced electron transfer is heavily distance-dependent.42 In this context, alternative fluorescence tuning principles should be explored to design probes achieving NO-triggered protein labeling while preserving fluorogenic signals.We unexpectedly observed that the derivatization of SiR-COOH with a hydrophobic chain would shift it from the fluorescent zwitterionic state to the non-fluorescent spirocyclic state in both solids and high-polar solutions. We attributed this phenomenon to the result of the intramolecular interaction between the fluorophore skeleton and the hydrophobic chain. We envisioned that binding with proteins should decrease the negative effect of the hydrophobic chain on fluorescence intensity, contributing to a fluorogenic response. Actually, protein-binding induced fluorogenic response of SiR-COOH derivatives has been reported.43–45 With these considerations, this observation inspired us to the possibility of achieving protein-labeling-induced fluorogenic response by employing this spirocyclization–zwitterion equilibrium. To verify this hypothesis, we synthesized three alkyl amide derivatives of SiR-COOH (Fig. S1a and Scheme S1†). Compared to SiR-COOH appearing as a dark blue solid, the derivatives had a greenish color that dimmed as hydrophobicity increased. When dissolved in dimethyl sulfoxide (DMSO) at 5 mM, the SiR-COOH solution was greenish, while the other solutions were almost colorless (Fig. S1b†). This observation suggested that SiR-COOH tended to exist in the zwitterionic form, whereas its hydrophobic derivatization would shift the equilibrium in favor of the non-fluorescent spirocyclic form. To make a quantitative comparison, the absorption and emission spectra of the compounds were recorded at 6 μM in aqueous solutions (Fig. S1c†). SiR-COOH exhibited an intense absorption band with a peak at 645 nm, whereas SiR-N4C showed a 50% decrease in absorption, and SiR-N8C and SiR-N12C were nearly non-absorptive in this range. Similar results were observed in the fluorescence spectra (Fig. S1d†). These results provide further evidence that the hydrophobic derivatives of SiR-COOH tend to exist in the non-fluorescent spirocyclic form. Interestingly, if the colorless aqueous solution of SiR-N8C (6 μM) was added with the surfactant sodium dodecyl sulfate (SDS) to decrease the negative effect from the hydrophobic chains, even a trace amount of SDS could shift the equilibrium to the zwitterionic state, as shown by the increase of the absorption at 645 nm which intensified in an SDS-concentration-dependent manner. Similar results were observed for the fluorescence intensity at 672 nm (Fig. S1e–g†). This result promised that the covalent binding of the hydrophobic derivatives of SiR-COOH to proteins would yield fluorogenic signals.
After identifying the unique characteristic of the SiR-COOH fluorophore switching from the spirocyclic form to the zwitterionic form upon protein-binding, we then proceeded to develop NO-imaging probes with the desirable protein-labeling ability and fluorogenic response. This could be straightforwardly realized by tethering o-phenylenediamine to SiR-COOH with an acyl linker. We currently used a carbamate group to link the o-phenylenediamine. However, an amide group may also be an alternative. To tune the protein-labeling efficiency, alkyl or ether linkers were incorporated. Finally, four candidate probes (NOP-1 to NOP-4) were designed (Fig. 2A). These probes were facilely synthesized by conjugating SiR-COOH with o-phenylenediamine-attached acyl linkers (Scheme S2†). Unlike SiR-COOH, which appeared dark blue in solid form and greenish in a DMSO solution (5 mM), all four NOPs were greenish in solid form and colorless in DMSO solutions (5 mM), indicating their preference for the spirocyclic form (Fig. 2B). This was corroborated by their weak fluorescence in aqueous solutions (Fig. 2C). However, when the probes were treated with NO in the presence of bovine serum albumin (BSA), significant fluorogenic responses were observed, suggesting NO-triggered protein labeling which converted the spirocyclic fluorophore into the zwitterionic form. Noteworthy, we confirmed that the presence of protein was necessary for the fluorogenic response, as NO itself couldn't lighten up the fluorescence of the probes (Fig. 2C and D). This observation is in accord with the proposed mechanism of protein-binding induced switching to the zwitterionic form of the SiR fluorophore. Furthermore, the covalent labeling of BSA by the NOPs under the activation of NO was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and in-gel fluorescence analysis, which showed a prominent fluorescence band corresponding to BSA in the presence of NO donor DEA·NONOate while no fluorescence band was evident in the absence of NO (Fig. 2E and F). Among the four candidates, NOP-1 exhibited the most efficient protein labeling. Furthermore, we assessed the NO-imaging performance of the probes in cells (Fig. 2G and H). Remarkably, NOP-1 demonstrated the highest proficiency in labeling, and was therefore selected for a systematic investigation of its NO-triggered protein labeling ability.
 | ||
| Fig. 2 Probe NOPs for NO-triggered protein labeling and fluorogenic imaging. (A) Structures of NOPs. (B) Images of SiR-COOH and NOPs either dissolved in DMSO (5 mM) or as solids. (C) Fluorescence spectra of NOPs (5 μM) in PBS before (green) and after (orange) NO treatment, or after NO treatment in the presence of BSA (blue). NOPs (50 μM) in PBS were first incubated with solvent control, DEA·NONOate (500 μM), or DEA·NONOate (500 μM) plus BSA (25 mg mL−1) at 37 °C for 60 min, and then diluted to 5 μM to record the fluorescence spectra. (D) Normalized fluorescent intensity of NOPs at 670 nm in (C). Intensity was normalized to the data of NOPs incubated with solvent control. Error bars represent the standard deviation (SD) (n = 3). (E) In-gel fluorescence analysis of BSA (0.5 mg mL−1) incubated with NOPs (1 μM) in the presence or absence of DEA·NONOate (50 μM) at 37 °C for 60 min. The upper panel showed an image of the Coomassie Brilliant Blue (CBB)-stained gel while the lower panel showed an image of the in-gel fluorescence. (F) Quantification of labeling yield of bands in (F). Signal intensity was normalized to protein concentration as measured by CBB. Error bars represent the SD (n = 2). (G) Confocal fluorescence imaging of 293T cells stimulated by different concentrations of DEA·NONOate and then stained with NOPs. 293T cells were pretreated with DEA·NONOate (0, 100 μM) for 30 min, followed by incubation with NOPs (1 μM) for 20 min. IF signals of NOPs: red; Hoechst: blue. Scale bar = 50 μm. (H) Normalized red fluorescence intensity output in (G). Data were normalized to the control group. Error bars represented SD (n = 3). **p < 0.01. | ||
NO-triggered protein labeling ability of NOP-1
To verify if NO could activate the acyl o-phenylenediamine group in NOP-1 into the readily conjugatable acyl benzotriazole, we used liquid chromatography-mass spectrometry (LC-MS) to monitor the reaction between NOP-1 and NO. As expected, upon NO treatment, NOP-1 was transformed into the benzotriazole derivative, accompanied by its further hydrolysis or its conjugation with methanol to the carbonate (Fig. 3A, B and S2†). To test if the acyl benzotriazole derivative would covalently label proteins, BSA was used as the model protein, and its molecular weight was measured before and after the treatment of NOP-1 and NO by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry (Fig. 3C). It was observed that the treatment of NOP-1 and NO shifted the mass peak of BSA from 66.5 kDa to 67.1 kDa. This increase of ca. 0.6 kDa well agreed with the molecular weight of the protein-conjugatable part of NOP-1, suggesting that activated NOP-1 covalently labeled BSA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio under this condition.
:1 ratio under this condition.
 | ||
| Fig. 3 NO-triggered protein labeling of NOP-1. (A) LC-MS analysis of NOP-1 (50 μM) before and after 30 min treatment with DEA·NONOate (500 μM) in PBS (0.25 mM, pH 7.4) containing 20% methanol. The blue star represents NOP-1, while the green diamond, yellow circle, and red square represent the hydrolyzed product, the methyl carbonate, and the benzotriazole derivative, respectively. (B) Mass spectra of the peaks at 8.787 min and 9.362 min which were attributed to NOP-1 and the benzotriazole derivative, respectively. (C) Mass spectra of BSA treated with NOP-1 (150 μM) alone or treated with NOP-1 (150 μM) in the presence of DEA·NONOate (600 μM) at 37 °C for 120 min. Spectra were obtained via MALDI-TOF mass spectrometry (matrix: saturated sinapinic acid in 50% ethanol/50% MeCN/0.1% TFA). (D) In-gel fluorescence analysis of BSA (0.5 mg mL−1) incubated with NOP-1 (1 μM) and DEA·NONOate (0–50 μM) at 37 °C for 60 min. (E) In-gel fluorescence analysis of BSA (0.5 mg mL−1) incubated with NOP-1 (1 μM) and DEA·NONOate (50 μM) at 37 °C for 0–120 min. (F) Quantification of labeling yields of bands in (D). Signal intensity was normalized to protein concentration as measured by CBB and to the data point of the 2 μM DEA·NONOate group. The black dashed line represented the fitted linear relationship between the relative labeling yield and concentration of DEA·NONOate. (G) Quantification of labeling yield of bands in (E). Signal intensity was normalized to protein concentration as measured by CBB and to the data point of the 5 min treatment group. The black dashed curve represented the fitted relationship between the relative labeling yield and incubation time. (H) In-gel fluorescence analysis of BSA (0.5 mg mL−1) incubated with NOP-1 (1 μM) and various species at 37 °C for 60 min. (I) Quantification of labeling yield of bands in (H). Signal intensity was normalized to protein concentration as measured by CBB and to the data point of the NO group. NOP-1 demonstrated superb selectivity towards NO over other biologically relevant reactive species. The upper panels show images of the CBB-stained gels while the lower panels show images of in-gel fluorescence scanning. Error bars represent the SD (n = 3). | ||
Then, the NO-dependent protein labeling capability of NOP-1 was examined in detail by SDS-PAGE in combination with in-gel fluorescence analysis. We first used BSA as the model protein. BSA (0.5 mg mL−1) was incubated with NOP-1 (1 μM) and DEA·NONOate (0–50 μM) for 60 min. Quantification of the fluorescence intensity of the labeled band revealed a positive linear correlation with the NO donor concentrations (Fig. 3D and F). This result was consistent with the NO-activated protein labeling mechanism. Similar results were observed in the time course of protein labeling efficacy. DEA·NONOate (50 μM) was added to a mixture of BSA (0.5 mg mL−1) and NOP-1 (1 μM), and aliquots were taken in a time-lapsed manner and then analyzed (Fig. 3E and G). The fluorescence intensity of the bands gradually intensified over time and reached saturation at approximately 90 min. Finally, we confirmed that NO specifically triggered the labeling event among biologically relevant reactive species. As shown in Fig. 3H and I, no significant fluorescently labeled bands were observed in samples treated with species other than NO. This result is in line with the established selectivity of o-phenylenediamine towards NO.
In addition to BSA as a model protein, we verified that NOP-1 retained its efficacy to label proteins upon NO activation in more complicated systems, such as in fetal bovine serum (FBS) (Fig. S3–S5†) and cell lysates (Fig. S6 and S7†). Interestingly, in the NO-triggered protein labeling experiment conducted in HepG2 cell lysates, some bands could be visible only by in-gel fluorescence scanning but almost invisible by Coomassie Brilliant Blue (CBB) staining, highlighting the sensitivity of NOP-1 imparted by its fluorescent signals (Fig. S7†). This sensitivity is promising to facilitate the biological-process elucidation by labeling and lightening up NO-signaling relevant proteins.
NOP-1 imaged NO in cells
To assess NOP-1's ability to image NO in cells, human embryonic kidney-derived 293T cells were pretreated with DEA·NONOate of various doses for 30 min and then stained with NOP-1 (1 μM). The cellular probe fluorescence intensity was observed positively correlated with DEA·NONOate doses (Fig. S8†), indicating NOP-1's capability to image cellular NO. Noteworthy, we observed an asymmetric distribution of cellular probe fluorescence between the cytoplasm and nuclei (Fig. S8A†), suggesting increased sensitivity of cytoplasmic proteins to NO signaling. Given the brain's susceptibility to nitrative stress,46 we further evaluated the fluorogenic performance of NOP-1 in murine hippocampal HT22 cells following NO stimulation. Consistent with previous results in 293T cells, we observed a NO dose-dependent increase in intracellular probe fluorescence, accompanied by an asymmetric distribution (Fig. S9†). These results collectively demonstrate the NO-activated protein labeling and fluorogenic visualization of the labeled proteins by NOP-1 in different cells.NOP-1 imaging revealed the implication of NO signaling in hypoglycemia-induced neurological damage
After confirming NOP-1's ability to image NO in cells and its capacity for covalently labeling proteins post-NO activation, we employed NOP-1 to investigate the pathology of hypoglycemia, a common adverse effect of diabetes mellitus treatment associated with an increased risk of neuropathy and cognitive impairment.47,48 Previous studies from our group have implicated NO signaling in various pathological processes.49–51 To explore the potential modulation of neurological damage under hypoglycemic conditions by NO signaling, HT22 cells were cultured in a 1 mM glucose medium for 6 h to mimic hypoglycemia.52 Cells were then stained either with NOP-1 or the commercial NO probe DAF-FM DA. It was observed that either probe consistently exhibited weak fluorescence in high-glucose-treated cells but strong fluorescence in low-glucose-treated cells (Fig. S10†), suggesting hypoglycemia-induced upregulation of NO in HT22 cells. To interrogate the effect of hypoglycemia duration on cellular NO levels, HT22 cells were cultured in a 1 mM glucose medium for 0, 3, 6, 9, and 12 h, followed by staining with NOP-1 (1 μM). The cellular probe fluorescence intensity increased within the first 6 h, peaked at the 6th hour, and then decreased from 6 to 12 h, revealing a distinct NO signaling pattern in response to hypoglycemia (Fig. 4A and B). To validate this relation, HT22 cells were pretreated with 1 mM glucose medium and then stained with DAF-FM DA (1 μM) (Fig. 4C). Consistent with NOP-1 staining, a noticeable peak of DAF-FM DA fluorescence intensity was observed after 6 h of hypoglycemic treatment (Fig. 4D), further confirming the reliability of NOP-1 in imaging cellular NO levels and suggesting an association between elevated NO signaling and hypoglycemia-induced neurological damage.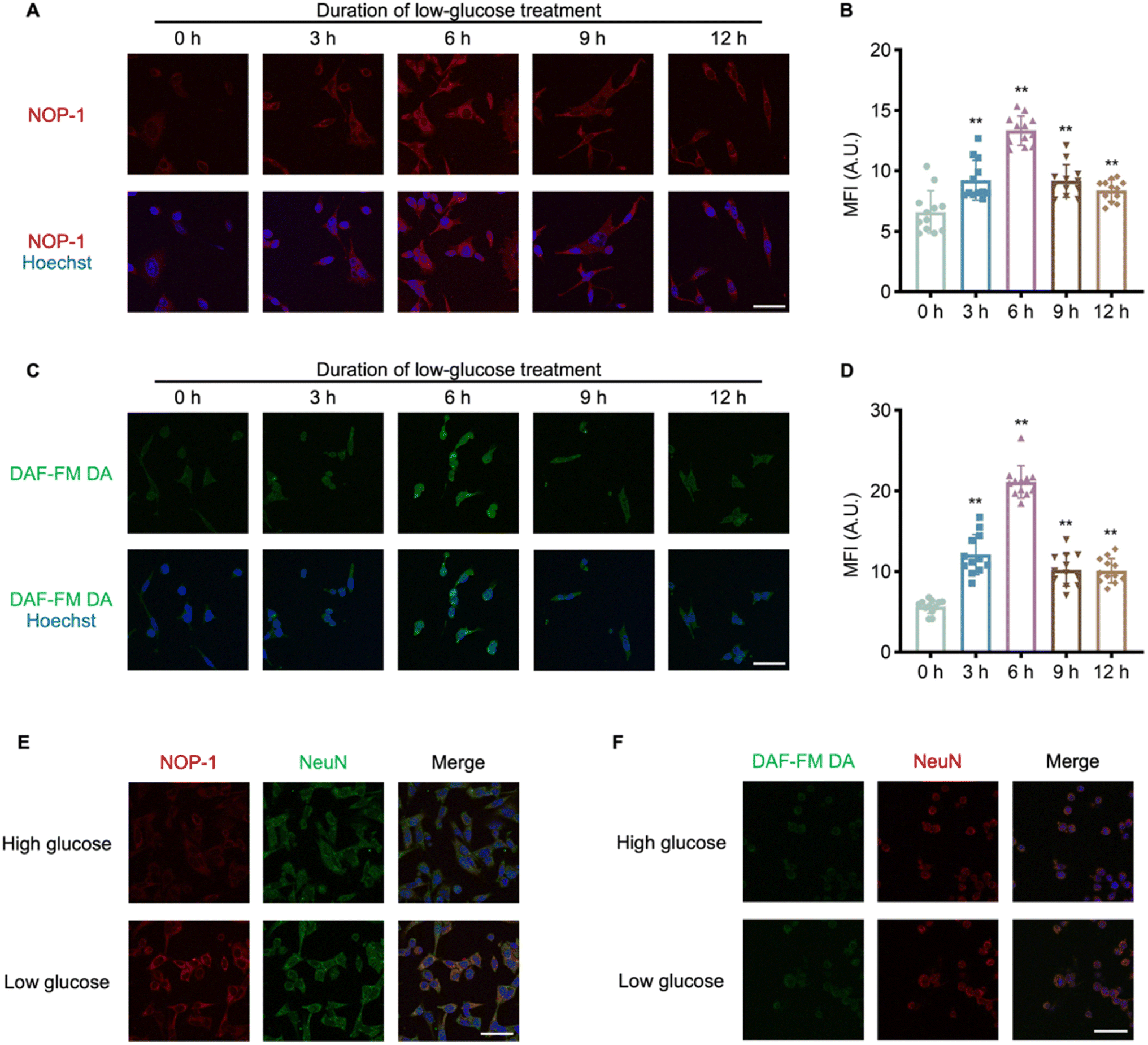 | ||
| Fig. 4 Dynamic visualization of NO fluctuations in HT22 cells in low-glucose conditions mimicking the hypoglycemic process. (A) Fluorescence images of HT22 cells stained with NOP-1 revealed the temporal changes of cellular NO levels. HT22 cells were stimulated with 1 mM glucose for 0, 3, 6, 9, and 12 h, followed by incubation with NOP-1 (1 μM) for 20 min. IF signals of NOP-1: red; Hoechst: blue. Scale bar = 50 μm. (B) Quantification of the red fluorescence intensity in (A) indicating relative NO levels. (C) Fluorescence images of HT22 cells stained with DAF-FM DA. HT22 cells were stimulated with 1 mM glucose for 0, 3, 6, 9, and 12 h, followed by incubation with DAF-FM DA (1 μM) for 20 min. IF signals of DAF-FM DA: green; Hoechst: blue. Scale bar = 50 μm. (D) Quantification of the green fluorescence intensity in (C). Error bars represent SD. *p < 0.05; **p < 0.01. (E and F) The comparison between NOP-1 and DAF-FM DA in multiplex staining in combination with immunostaining. (E) IF signals of NOP-1: red; NeuN: green; Hoechst: blue. (F) IF signals of DAF-FM DA: green; NeuN: red; Hoechst: blue. Scale bar = 50 μm. The cell experiments were repeated three times. | ||
To assess the compatibility of NOP-1 fluorescence with immunostaining, HT22 cells without or with low-glucose pretreatment were first stained with NOP-1, subjected to 0.5% Triton X-100 penetration, and then subjected to immunostaining with NeuN (a marker of neuron) antibody and corresponding secondary antibody. After extensive washing, both the probe fluorescence and the immunofluorescence from the antibody were recorded under microscopy. Cells without low-glucose pretreatment presented only NeuN immunofluorescent signals, while cells subjected to hypoglycemia demonstrated both NOP-1 and immunofluorescence (Fig. 4E). This suggested that hypoglycemia-induced NO caused a permanent probe NOP-1 stain in the cells, consistent with the NO-activated protein labeling mechanism of the probe. In sharp contrast, when similar procedures were used but cells were stained with DAF-FM DA, only NeuN immunofluorescent signals were observed, regardless of low-glucose pretreatment (Fig. 4F). This is in accord with the immunostaining-incompatible signal of DAF-FM DA. To further confirm the NO-activated protein covalent labeling ability of NOP-1, HT22 cells subjected to high or low glucose stimulation were stained either with NOP-1 or the commercial NO probe DAF-FM DA. Cells were then lysed, and the proteins were resolved by SDS-PAGE and observed by in-gel fluorescence. As shown in Fig. S11,† obvious in-gel fluorescence was observed only in the low glucose plus NOP-1 staining group; while no significant signal was observed in the low glucose plus DAF-FM DA staining group. Since we have confirmed the upregulation of NO in the low glucose stimulation group, this result further emphasizes the superiority of NOP-1 for its protein-labeling ability and its immunostaining-compatible signals in imaging cellular NO.
Multiplex imaging revealed the contribution of α-tubulin nitration to hypoglycemia-induced neuronal apoptosis
After confirming NOP-1's immunostaining-compatible signals, we employed it to investigate the mechanism of NO signaling in hypoglycemia-induced neurological damage. It has been reported that glucose deprivation leads to ATP depletion and apoptotic pathway activation.52 This prompted us to analyze the effect of NO signaling on apoptosis in HT22 cells. Cultured in 1 mM glucose for 6 h, HT22 cells exhibited a significant increase in apoptotic cells, as revealed by the TUNEL assay,53 indicating a close association between NO signaling and hypoglycemia-induced apoptosis (Fig. 5A and B). To confirm this relationship, HT22 cells were treated with L-NMMA, a NOS inhibitor, which effectively suppressed apoptotic cell death under low-glucose conditions, suggesting that the upregulation of NO-signaling in hypoglycemia contributed to neuronal apoptosis.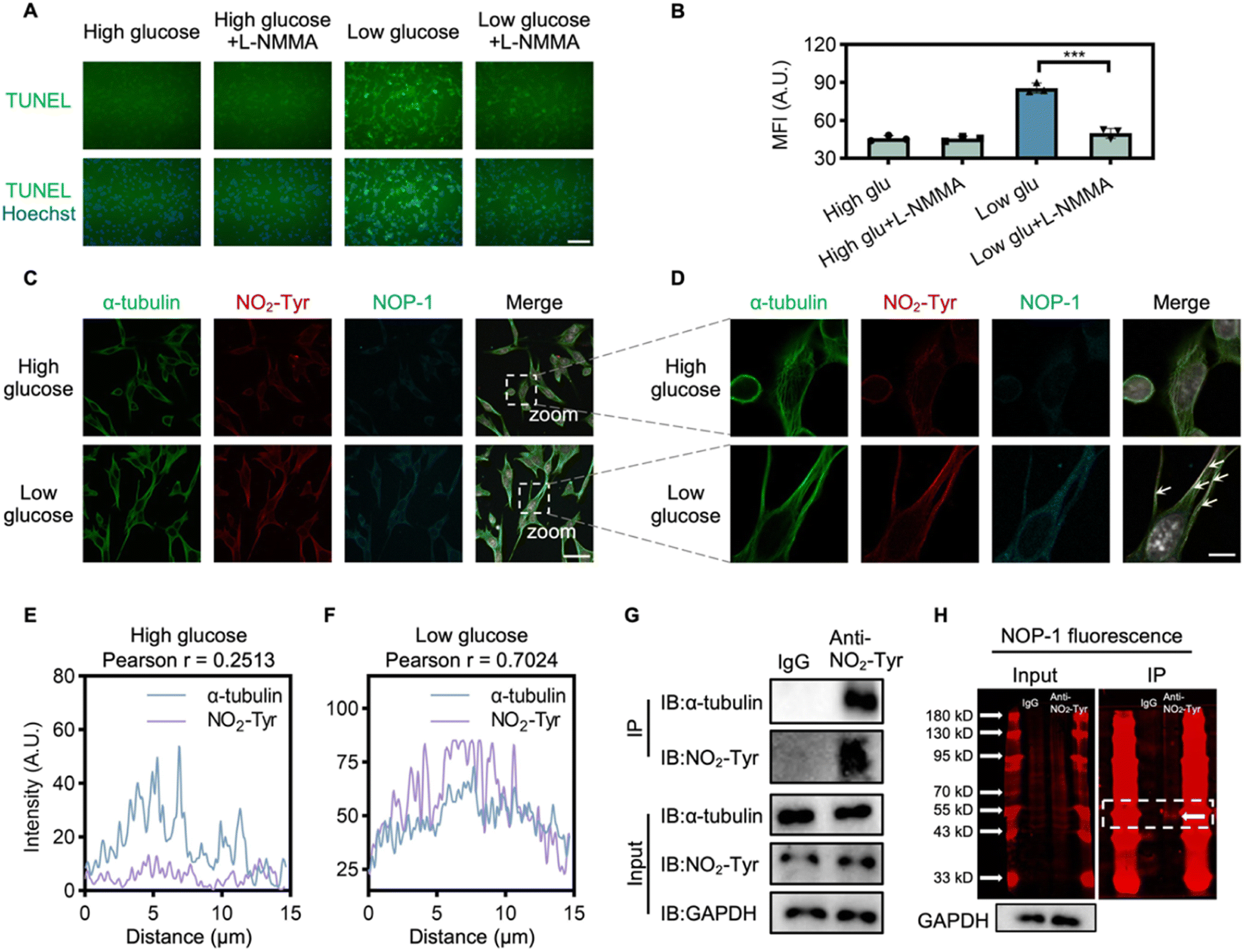 | ||
| Fig. 5 Investigation into the NO-related mechanism in hypoglycemia-induced neurological injury. (A) Immunofluorescence of TUNEL revealed neuronal apoptosis in HT22 cells. HT22 cells were treated with or without L-NMMA (100 μM) in low-glucose (1 mM) or high-glucose conditions, and TUNEL staining was conducted. IF signals of TUNEL: green; Hoechst: blue. Scale bar = 100 μm. (B) Quantification of TUNEL fluorescent intensity in (A). *p < 0.05; **p < 0.01; ***p < 0.001. (C) Multiplex immunofluorescence revealed an increase in NO2-Tyr-modification accompanied by elevated NOP-1 fluorescence in HT22 cells under low-glucose treatment. HT22 cells were cultured in low-glucose (1 mM) or high-glucose conditions for 6 h, subsequently stained with NOP-1, and then immunostaining for α-tubulin and NO2-Tyr was conducted. IF signals of α-tubulin: green; NO2-Tyr: red; NOP-1: cyan; Hoechst: blue. Scale bar = 50 μm. (D) Ultra-high-resolution microscopic imaging and deconvolution analysis revealed the collocation of NO2-Tyr-modification with α-tubulin. IF signals of α-tubulin: green; NO2-Tyr: red; NOP-1: cyan; Hoechst: blue. Scale bar = 10 μm. (E and F) Analysis of colocalization between α-tubulin and NO2-Tyr under euglycemic (E) and hypoglycemic (F) conditions. (G) Immunoprecipitation assay identified NO2-Tyr-α-tubulin in low-glucose treated HT22 cells. (H) In-gel fluorescence revealed α-tubulin labeled by NOP-1 in immunoprecipitated proteins by NO2-Tyr antibody. IF signals of NOP-1: red. λex = 647 nm. The cell experiments were repeated three times except for the IP assay. | ||
Moving forward, we explored the mechanism of NO-mediated apoptosis through multiplex imaging. NO, known to modify amino acids covalently, can lead to 3-nitrotyrosine (NO2-Tyr) formation.54,55 Considering that NO2-Tyr-modified α-tubulin has been linked to cellular apoptosis,54,56 we investigated whether NO2-Tyr-modified α-tubulin is involved in neuronal apoptosis under hypoglycemic stress. HT22 cells were treated with low glucose and stained first with NOP-1, followed by immunofluorescence staining with both the α-tubulin antibody and the NO2-Tyr antibody. Low-glucose treatment significantly elevated the level of NO2-Tyr-modification in HT22 cells, accompanied by an increase in NOP-1 fluorescence (Fig. 5C and S12† for enlarged version). Ultra-high-resolution microscopy and deconvolution analysis of HT22 cells revealed a good colocalization of α-tubulin and NO2-Tyr-modified protein, along with microtubule disorganization (Fig. 5D and S13† for enlarged version), indicating a potential link between NO signaling and NO2-Tyr-modified α-tubulin. Conversely, in cells from the high-glucose group, no significant co-localization was observed (Fig. 5E and F).
Further supporting our findings, immunoprecipitation (IP) demonstrated the pulldown of α-tubulin by the NO2-Tyr antibody from low-glucose-treated cell lysates (Fig. 5G). Additionally, SDS-PAGE analysis of the immunoprecipitated proteins by the NO2-Tyr antibody revealed a NOP-1 labeled fluorescent band corresponding to the molecular weight of α-tubulin (55 kDa), suggesting the labeling of α-tubulin by NOP-1 in cells under hypoglycemia stimulation and further highlighting the advantage of NOP-1 in trapping protein (Fig. 5H). This confirmed NOP-1 as a tool to bridge cellular NO levels with NO-modified proteins, facilitating the interrogation of the mechanism of NO signaling. In summary, our results suggest that hypoglycemia-induced neurological damage is closely related to NO2-Tyr-modified α-tubulin. It should be noted that when whole-cell lysis was analyzed by SDS-PAGE for NOP-1 signal in parallel with western blot for NO2-Tyr signal, some bands showed much more dramatic NOP-1 labeling signals than NO2-Tyr signals, and vice versa (Fig. S14†). Since NOP-1 labeling is the direct result of NO activation while NO2-Tyr antibody detects proteins with nitrated tyrosine residues, we reason that NOP-1 and NO2-Tyr antibody should complement each other to study NO signaling with improved accuracy.
Conclusion
In conclusion, we have successfully developed the dual-functional activity-based probe NOP-1, capable of imaging NO in cells and covalently labeling proximal proteins. The key innovation lies in integrating the native chemical ligation reaction of acyl benzotriazole with the specific fluorescent properties of Si-rhodamine fluorophores, transitioning from a non-fluorescent spirocyclic form to a fluorescent zwitterionic form upon binding to proteins. NOP-1 incorporates an acyl o-phenylenediamine moiety for NO sensing, with its NO-triggered transformation to an acyl benzotriazole moiety activating the protein-labeling ability of the probe. This unique design translates transient cellular NO signals into a permanent fluorescent stain, compatible with immunostaining and multiplex imaging, thereby bridging cellular NO levels with diverse biological processes. Notably, our study using NOP-1 revealed the previously unexplored contribution of NO upregulation to hypoglycemia-induced neurological damage. Mechanistically, the overlap of cellular NOP-1 fluorescence with immunofluorescence for α-tubulin and NO2-Tyr, coupled with the identification of α-tubulin from NOP-1 labeled proteins, strongly suggests that NO exerts its damaging role by nitrating α-tubulin, ultimately leading to apoptosis. We envision NOP-1 as a versatile tool with broad applications for studying NO-related pathophysiology across various conditions, offering valuable insights and potential avenues for further research and therapeutic development.Data availability
The data underlying this study are available in the published article and its ESI.†Author contributions
X. Li and Y. Zang conceived and designed the project. Y. Li synthesized the compounds, characterized the photophysical properties of the probes, and conducted the SDS-PAGE experiments. K. Pan performed the cell imaging and immunoprecipitation experiments under the guidance of J. Li. Y. Gao performed part of the cell imaging experiments. Y. Li and K. Pan analyzed the data and drafted the manuscript. X. Li and Y. Zang revised it. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by grants from the National Natural Science Foundations of China (22077112, 22377106, 82151219), the National Natural Science Foundations of Zhejiang province (LZ23H300001) and Natural Science Foundation of Shanghai (22ZR1415200). X. L. was supported by the National Program for Support of Top-notch Young Professionals (grant 2021). Y. Z. was supported by the Program of Shanghai Academic Research Leader (23XD1450800). We appreciate Dr Jianyang Pan for helping us perform the NMR experiments.References
- R. M. Palmer, A. G. Ferrige and S. Moncada, Nature, 1987, 327, 524–526 CrossRef CAS PubMed.
- O. Arancio, M. Kiebler, C. J. Lee, V. Lev-Ram, R. Y. Tsien, E. R. Kandel and R. D. Hawkins, Cell, 1996, 87, 1025–1035 CrossRef CAS PubMed.
- R. Urbano, J. E. Karlinsey, S. J. Libby, P. T. Doulias, H. Ischiropoulos, H. I. Warheit-Niemi, D. H. Liggitt, A. R. Horswill and F. C. Fang, Cell Host Microbe, 2018, 23, 594–606 CrossRef CAS PubMed.
- J. O. Lundberg and E. Weitzberg, Cell, 2022, 185, 2853–2878 CrossRef CAS PubMed.
- D. S. Bredt, P. M. Hwang, C. E. Glatt, C. Lowenstein, R. R. Reed and S. H. Snyder, Nature, 1991, 351, 714–718 CrossRef CAS PubMed.
- S. P. Janssens, A. Shimouchi, T. Quertermous, D. B. Bloch and K. D. Bloch, J. Biol. Chem., 1992, 267, 14519–14522 CrossRef CAS PubMed.
- W. P. Arnold, C. K. Mittal, S. Katsuki and F. Murad, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 3203–3207 CrossRef CAS PubMed.
- R. Liu, Y. Kang and L. Chen, Nat. Commun., 2021, 12, 5492 CrossRef CAS PubMed.
- R. Yang, Y. Gao, H. Li, W. Huang, D. Tu, M. Yang, X. Liu, J. S. Hong and H. M. Gao, Cell Rep., 2022, 40, 111330 CrossRef CAS PubMed.
- K. R. Kim, E. J. Cho, J. W. Eom, S. S. Oh, T. Nakamura, C. K. Oh, S. A. Lipton and Y. H. Kim, Cell Death Differ., 2022, 29, 2137–2150 CrossRef CAS PubMed.
- G. G. Schiattarella, F. Altamirano, D. Tong, K. M. French, E. Villalobos, S. Y. Kim, X. Luo, N. Jiang, H. I. May, Z. V. Wang, T. M. Hill, P. P. A. Mammen, J. Huang, D. I. Lee, V. S. Hahn, K. Sharma, D. A. Kass, S. Lavandero, T. G. Gillette and J. A. Hill, Nature, 2019, 568, 351–356 CrossRef CAS PubMed.
- J. Erdmann, K. Stark, U. B. Esslinger, P. M. Rumpf, D. Koesling, C. de Wit, F. J. Kaiser, D. Braunholz, A. Medack, M. Fischer, M. E. Zimmermann, S. Tennstedt, E. Graf, S. Eck, Z. Aherrahrou, J. Nahrstaedt, C. Willenborg, P. Bruse, I. Brænne, M. M. Nöthen, P. Hofmann, P. S. Braund, E. Mergia, W. Reinhard, C. Burgdorf, S. Schreiber, A. J. Balmforth, A. S. Hall, L. Bertram, E. Steinhagen-Thiessen, S. C. Li, W. März, M. Reilly, S. Kathiresan, R. McPherson, U. Walter, J. Ott, N. J. Samani, T. M. Strom, T. Meitinger, C. Hengstenberg and H. Schunkert, Nature, 2013, 504, 432–436 CrossRef CAS PubMed.
- J. G. Spiers, H. C. Chen, J. M. Bourgognon and J. R. Steinert, Free Radical Biol. Med., 2019, 134, 468–483 CrossRef CAS PubMed.
- R. Shankar, J. S. Zhu, B. Ladd, D. Henry, H. Q. Shen and A. D. Baron, J. Clin. Invest., 1998, 102, 1403–1412 CrossRef CAS PubMed.
- J. L. Heinecke, L. A. Ridnour, R. Y. Cheng, C. H. Switzer, M. M. Lizardo, C. Khanna, S. A. Glynn, S. P. Hussain, H. A. Young, S. Ambs and D. A. Wink, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6323–6328 CrossRef CAS PubMed.
- M. Fujita, V. Somasundaram, D. Basudhar, R. Y. S. Cheng, L. A. Ridnour, H. Higuchi, K. Imadome, J. H. No, G. Bharadwaj and D. A. Wink, Redox Biol., 2019, 22, 101158 CrossRef CAS PubMed.
- Z. Huang, P. L. Huang, N. Panahian, T. Dalkara, M. C. Fishman and M. A. Moskowitz, Science, 1994, 265, 1883–1885 CrossRef CAS PubMed.
- F. Murad, Angew Chem. Int. Ed. Engl., 1999, 38, 1856–1868 CrossRef CAS PubMed.
- J. Kim and S. N. Thomas, Pharmacol. Rev., 2022, 74, 1146–1175 CrossRef CAS PubMed.
- M. Carlström, Nat. Rev. Nephrol., 2021, 17, 575–590 CrossRef PubMed.
- V. Garcia, E. J. Park, M. Siragusa, F. Frohlich, M. Mahfuzul Haque, J. V. Pascale, K. R. Heberlein, B. E. Isakson, D. J. Stuehr and W. C. Sessa, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9497–9507 CrossRef CAS PubMed.
- H. Wu, L. Lu, J. Chen, C. Zhang, W. Liu and S. Zhuang, Environ. Sci. Technol., 2020, 54, 2922–2930 CrossRef CAS PubMed.
- C. H. Switzer, H. J. Cho, T. R. Eykyn, P. Lavender and P. Eaton, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2200022119 CrossRef CAS PubMed.
- K. S. Aulak, J. W. Barnes, L. Tian, N. E. Mellor, M. M. Haque, B. Willard, L. Li, S. C. Comhair, D. J. Stuehr and R. A. Dweik, Redox Biol., 2020, 36, 101625 CrossRef CAS PubMed.
- J. Zhang, J. Huang, Y. Gu, M. Xue, F. Qian, B. Wang, W. Yang, H. Yu, Q. Wang, X. Guo, X. Ding, J. Wang, M. Jin and Y. Zhang, Cell. Mol. Immunol., 2021, 18, 1476–1488 CrossRef CAS PubMed.
- W. L. Jiang, Y. Li, H. W. Liu, D. Y. Zhou, J. Ou-Yang, L. Yi and C. Y. Li, Talanta, 2019, 197, 436–443 CrossRef CAS PubMed.
- M. S. Jani, J. Zou, A. T. Veetil and Y. Krishnan, Nat. Chem. Biol., 2020, 16, 660–666 CrossRef CAS PubMed.
- Y. Tang, Y. Li, Z. Wang, W. Huang, Q. Fan and B. Liu, ACS Nano, 2023, 17, 18299–18307 CrossRef CAS PubMed.
- Z. He, D. Liu, Y. Liu, X. Li, W. Shi and H. Ma, Anal. Chem., 2022, 94, 10256–10262 CrossRef CAS PubMed.
- L. Y. Lin, X. Y. Lin, F. Lin and K. T. Wong, Org. Lett., 2011, 13, 2216–2219 CrossRef CAS PubMed.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS PubMed.
- Y. Chen, Nitric Oxide, 2020, 98, 1–19 CrossRef CAS PubMed.
- M. Yang, X. Wu, J. Hu, Y. Wang, Y. Wang, L. Zhang, W. Huang, X. Wang, N. Li, L. Liao, M. Chen, N. Xiao, Y. Dai, H. Liang, W. Huang, L. Yuan, H. Pan, L. Li, L. Chen, L. Liu, L. Liang and J. Guan, J. Hepatol., 2022, 76, 1138–1150 CrossRef CAS PubMed.
- S. M. Lüttgenau, C. Emming, T. Wagner, J. Harms, J. Guske, K. Weber, U. Neugebauer, R. Schröter, O. Panichkina, Z. Pethő, F. Weber, A. Schwab, A. K. Wege, P. Nedvetsky and M. P. Krahn, Mol. Cancer, 2021, 20, 74 CrossRef PubMed.
- S. K. Pirooznia, C. Yuan, M. R. Khan, S. S. Karuppagounder, L. Wang, Y. Xiong, S. U. Kang, Y. Lee, V. L. Dawson and T. M. Dawson, Mol. Neurodegener., 2020, 15, 17 CrossRef CAS PubMed.
- J. Feng, Y. Li, Y. Li, Q. Yin, H. Li, J. Li, B. Zhou, J. Meng, H. Lian, M. Wu, Y. Li, K. Dou, W. Song, B. Lu, L. Liu, S. Hu and Y. Nie, Circulation, 2024, 149, 1004–1015, DOI:10.1161/circulationaha.123.066298.
- T. Miki, M. Awa, Y. Nishikawa, S. Kiyonaka, M. Wakabayashi, Y. Ishihama and I. Hamachi, Nat. Methods, 2016, 13, 931–937 CrossRef CAS PubMed.
- H. Zhu, T. Tamura, A. Fujisawa, Y. Nishikawa, R. Cheng, M. Takato and I. Hamachi, J. Am. Chem. Soc., 2020, 142, 15711–15721 CrossRef CAS PubMed.
- A. T. Pezacki, C. D. Matier, X. Gu, E. Kummelstedt, S. E. Bond, L. Torrente, K. L. Jordan-Sciutto, G. M. DeNicola, T. A. Su, D. C. Brady and C. J. Chang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202736119 CrossRef CAS PubMed.
- S. Uchinomiya, T. Nagaura, M. Weber, Y. Matsuo, N. Zenmyo, Y. Yoshida, A. Tsuruta, S. Koyanagi, S. Ohdo, N. Matsunaga and A. Ojida, J. Am. Chem. Soc., 2023, 145, 8248–8260 CrossRef CAS PubMed.
- J. X. Wang, G. M. Fang, Y. He, D. L. Qu, M. Yu, Z. Y. Hong and L. Liu, Angew Chem. Int. Ed. Engl., 2015, 54, 2194–2198 CrossRef CAS PubMed.
- T. P. Le, J. E. Rogers and L. A. Kelly, J. Phys. Chem. A, 2000, 104, 6778–6785 CrossRef CAS.
- G. Lukinavičius, K. Umezawa, N. Olivier, A. Honigmann, G. Yang, T. Plass, V. Mueller, L. Reymond, I. R. Corrêa Jr, Z. G. Luo, C. Schultz, E. A. Lemke, P. Heppenstall, C. Eggeling, S. Manley and K. Johnsson, Nat. Chem., 2013, 5, 132–139 CrossRef PubMed.
- G. Lukinavičius, L. Reymond, E. D'Este, A. Masharina, F. Göttfert, H. Ta, A. Güther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H. D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef PubMed.
- L. Wang, M. Tran, E. D'Este, J. Roberti, B. Koch, L. Xue and K. Johnsson, Nat. Chem., 2020, 12, 165–172 CrossRef CAS PubMed.
- M. Muriach, M. Flores-Bellver, F. J. Romero and J. M. Barcia, Oxid. Med. Cell. Longevity, 2014, 2014, 102158 Search PubMed.
- A. Nakhleh and N. Shehadeh, World J. Diabetes, 2021, 12, 2036–2049 CrossRef PubMed.
- A. J. Graveling, I. J. Deary and B. M. Frier, Diabetes Care, 2013, 36, 3240–3246 CrossRef CAS PubMed.
- F. Wang, X. Jiang, H. Xiang, N. Wang, Y. Zhang, X. Yao, P. Wang, H. Pan, L. Yu, Y. Cheng, Y. Hu, W. Lin and X. Li, Biosens. Bioelectron., 2021, 172, 112756 CrossRef CAS PubMed.
- X. Li, R. R. Tao, L. J. Hong, J. Cheng, Q. Jiang, Y. M. Lu, M. H. Liao, W. F. Ye, N. N. Lu, F. Han, Y. Z. Hu and Y. H. Hu, J. Am. Chem. Soc., 2015, 137, 12296–12303 CrossRef CAS PubMed.
- X. Jiang, M. Li, Y. Wang, C. Wang, Y. Wang, T. Shen, L. Shen, X. Liu, Y. Wang and X. Li, Nat. Commun., 2023, 14, 1401 CrossRef CAS PubMed.
- C. Li, X. Chai, J. Pan, J. Huang, Y. Wu, Y. Xue, W. Zhou, J. Yang, X. Zhu and S. Zhao, J. Mol. Neurosci., 2022, 72, 923–938 CrossRef CAS PubMed.
- K. Kyrylkova, S. Kyryachenko, M. Leid and C. Kioussi, Methods Mol. Biol., 2012, 887, 41–47 CrossRef CAS PubMed.
- F. Wang, Q. Yuan, F. Chen, J. Pang, C. Pan, F. Xu and Y. Chen, Front. Cell Dev. Biol., 2021, 9, 742483 CrossRef PubMed.
- E. S. Dremina, V. S. Sharov and C. Schöneich, J. Neurochem., 2005, 93, 1262–1271 CrossRef CAS PubMed.
- J. P. Eiserich, A. G. Estévez, T. V. Bamberg, Y. Z. Ye, P. H. Chumley, J. S. Beckman and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6365–6370 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Compound synthesis and structure characterization, photophysical property measurement methods, sodium dodecyl sulfate-polyacrylamide gel electrophoresis methods, cell culture and imaging methods, supplementary figures and schemes, and compound NMR and MS traces. See DOI: https://doi.org/10.1039/d4sc00767k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |