
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLate-stage peptide modification and macrocyclization enabled by tertiary amine catalyzed tryptophan allylation†
Yuyang
Liu‡
abc,
Guofeng
Li‡
 c,
Wen
Ma
b,
Guangjun
Bao
b,
Yiping
Li
b,
Zeyuan
He
b,
Zhaoqing
Xu
*b,
Rui
Wang
*abc and
Wangsheng
Sun
*ab
c,
Wen
Ma
b,
Guangjun
Bao
b,
Yiping
Li
b,
Zeyuan
He
b,
Zhaoqing
Xu
*b,
Rui
Wang
*abc and
Wangsheng
Sun
*ab
aResearch Unit of Peptide Science (2019RU066), Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China
bKey Laboratory of Preclinical Study for New Drugs of Gansu Province, School of Basic Medical Sciences, Lanzhou University, Lanzhou 730000, China. E-mail: sunws@lzu.edu.cn; zqxu@lzu.edu.cn; wangrui@lzu.edu.cn
cSchool of Pharmacy, Shenzhen University Medical School, Shenzhen University, Shenzhen, 518055, China
First published on 14th June 2024
Abstract
Late-stage modification of peptides could potentially endow peptides with significant bioactivity and physicochemical properties, and thereby provide novel opportunities for peptide pharmaceutical studies. Since tryptophan (Trp) bears a unique indole ring residue and plays various critical functional roles in peptides, the modification methods for tryptophan were preliminarily developed with considerable progress via transition-metal mediated C–H activation. Herein, we report an unprecedented tertiary amine catalyzed peptide allylation via the SN2′–SN2′ pathway between the N1 position of the indole ring of Trp and Morita–Baylis–Hillman (MBH) carbonates. Using this method that proceeds under mild conditions, we demonstrated an extremely broad scope of Trp-containing peptides and MBH carbonates to prepare a series of peptide conjugates and cyclic peptides. The reaction is amenable to either solid-phase (on resin) or solution-phase conditions. In addition, the modified peptides can be further conjugated with other biomolecules at Trp, providing a new handle for bioconjugation.
Introduction
Peptides are highly promising bioactive molecules that have been extensively studied in recent decades as important potential therapeutic agents and biomaterials,1–3 chemical biology probes,4 and a platform for targeted therapeutics.5 In contrast to their flourishing successes in academics, however, native peptides that can often suffer from innate limitations such as poor proteolytic stability and low membrane permeability,3 are hindered from further application and transformation as clinical drugs, which can be mitigated by introducing structural modifications and cyclizations.6–9 In addition, the introduction of electrophilic “warheads” that are beyond canonical amino acids into peptides enables them to covalently bind to their target of interest to further increase their potency, which has been demonstrated by the success of proteasome inhibitor carfilzomib.10,11 In this context, chemical manipulation of peptides has been in urgent demand and emerged as a major tool for structurally sound arrangement of peptides, leading to an arsenal of late-stage peptide bioconjugation and modification.12–18 Despite the fruitful progress, alternatives for peptide late-stage chemical manipulation are still highly desired and attracted intensive attention from the chemical community.Tryptophan (Trp), the rarest native proteinogenic amino acid accounting for only ∼1% abundance while existing in approximately 90% of all proteins,19 is an ideal site of interest for peptide and protein modification. The side chain of Trp, in particular, bears a unique indole ring that showcases a huge potential in late-stage modification due to its variable chemical reactivity. As a consequence, a variety of methods leveraging Trp modification have been developed and applied to peptides and proteins (Scheme 1a).20,21 Take the last 3 years for instance, transition-metal-catalyzed C–H activation provides distinct peptide late-stage modification with good chemoselectivity, which has been explored by several groups.22–30 However, the reported methods require harsh reaction conditions, such as high temperatures, oxidant additives, auxiliary directing groups, and complex reaction conditions, which restrict their application prospects in bioactive peptide research. With the development of photochemistry in recent years, radical-medicated C2 amination,31–33 fluoroalkylation,34,35 alkylation,36,37 and thiation38,39 on the indole ring of tryptophan have been explored for the functionalization of peptides. In sharp contrast, modification on the NH position of the indole ring of Trp is much less explored due to the lower nucleophilicity and basicity of indole N–H of Trp among the multiple nucleophilic sites in peptide. A copper-promoted N(in)-arylation of the indole ring of Trp with triarylbismuthine species has been reported by Gagnon and coworkers.40 More recently, Paixão and coworkers have disclosed the dual photoredox/metal catalysis mediated N(in)-arylation of tryptophan-containing peptide on resin.41 Our group has developed a chiral phosphoric acid-catalyzed chemospecific enantioselective functionalization of tryptophan at the N1 position, enabling direct access to chiral propargyl aminal derivatives with good yields and stereoselectivities.42 Studer and coworker have reported the visible-light-mediated N1 position hemiaminal modification of Trp-containing peptides through the N–H insertion of siloxycarbenes photochemically generated from acylsilanes.43 Very recently, Spring and coworkers reported a tryptophan-mediated multicomponent Petasis reaction for peptide stapling and late-stage functionalization.44 Nevertheless, the development of Trp modification on its N1 position of the indole ring is still in its infancy. Thus, it is essential to introduce novel strategies to enrich the toolbox for peptide modification.
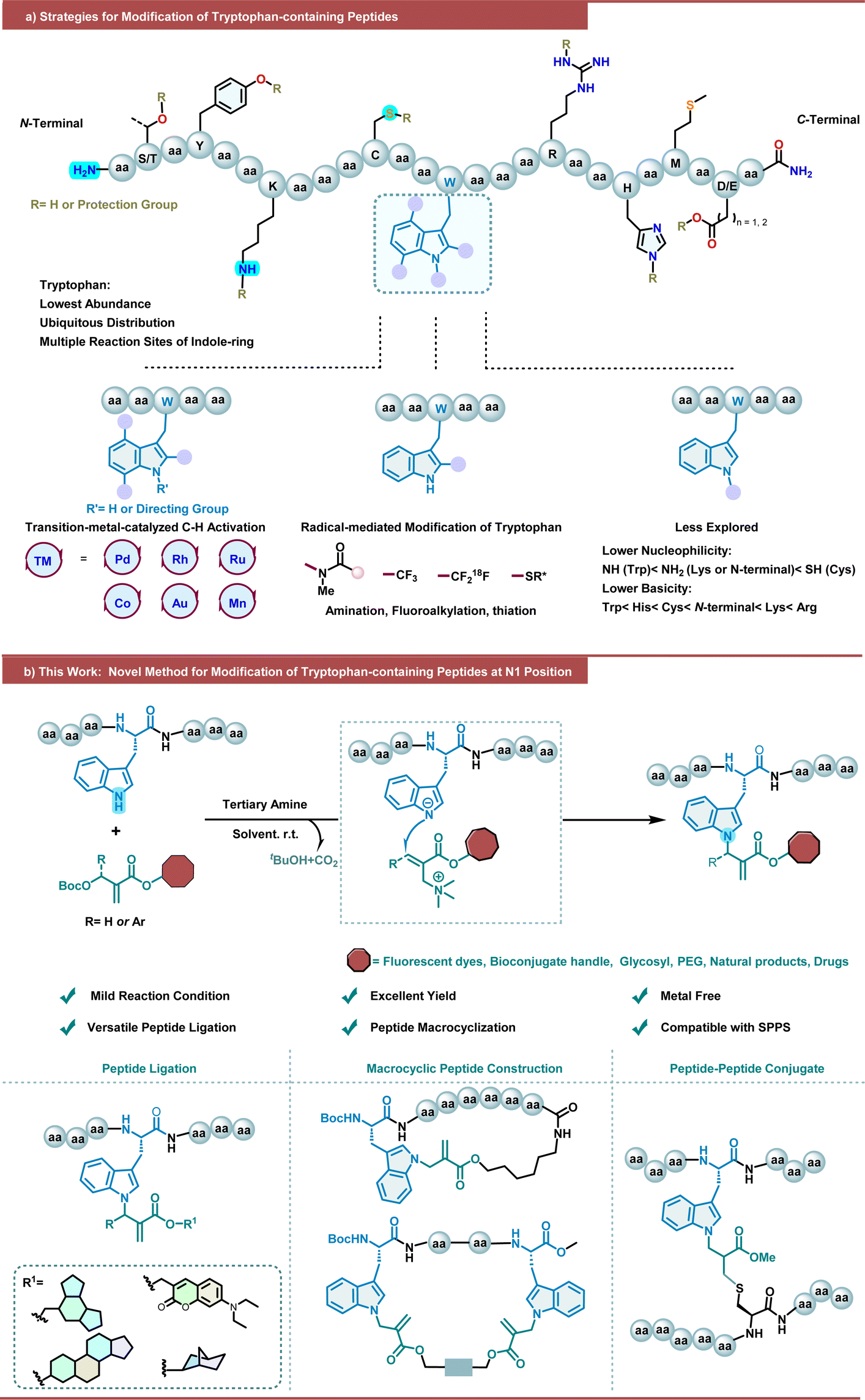 | ||
| Scheme 1 Late-stage modification of peptides containing the Trp side chain. | ||
In line with our ongoing interest in peptide chemistry, including those Trp modifications,33,39,42,45–49 we have been trying to explore more mild, easy-to-manipulate alternatives for peptide modification/functionalization to enrich the scenario. Promoted by our previous work on organocatalytic indole functionalization50,51 and Morita–Baylis–Hillman (MBH) carbonate based allylation,52–54 and inspired by Chen's N-allylic alkylation of indoles with MBH carbonates,55 we sought to explore the reaction between Trp and MBH carbonates to develop a novel strategy for peptide functionalization. Interestingly, in 2018, Ackermann and coworkers accomplished MnBr(CO)5-catalyzed C2–H activation allylation of the indole ring of Trp with an auxiliary pyridyl directing group using MBH carbonates for oligopeptide modification and cyclization.56 Herein, we envisioned that MBH carbonates could be utilized to achieve the indole N1-allylation of Trp via the SN2′–SN2′ pathway under tertiary amine catalysis, ultimately enabling the metal-free late-stage functionalization and macrocyclization of peptides under mild conditions, with electrophilic acrylate “warheads” introduced into peptides of interest (Scheme 1b). However, several challenges must be taken into account from the precision perspective: (1) the existence of various competitive nucleophilic functional groups in peptides, such as –SH (Cys), –NH2 (Lys), –OH (Tyr, Ser, and Thr) and others, makes it a daunting task to control the chemoselectivity; (2) the multiple competitive reactive sites, particularly the very electron-rich C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C3 region, on the indole ring of Trp make its regioselective control extremely challenging; (3) potential stereoselectivity control is also a challenge.
C3 region, on the indole ring of Trp make its regioselective control extremely challenging; (3) potential stereoselectivity control is also a challenge.
Results and discussion
To verify our hypothesis, we initiated our investigation using the model reaction of Boc–Trp–OMe (1a) with an MBH carbonate (2a) catalyzed by different tertiary amines (10 mol%) in DCE at room temperature overnight (Table S1, entries 1–4†). Fortunately, the DABCO-catalyzed reaction afforded only one product, an N-allylation product, in excellent yields (Table S1, entry 1,† 96% yield). Encouraged by this result, we further investigated different inorganic bases and tertiary phosphine catalysts (Table S1, entries 5–9†) and found that DABCO exhibited the highest catalytic activity. Further parameters were then screened to optimize the reaction. The choice of solvent had a significant effect on the yield (Table S1, entries 10–17†), with DCM proving to be the optimal solvent. Pleasingly, when the catalyst loading was increased to 20 mol%, the reaction time was reduced to 1 h without any loss of yield. Furthermore, we conducted an investigation of the chemoselectivity between tryptophan and other natural amino acids with nucleophilic residues (see the ESI†). This reaction demonstrated exclusive chemoselectivity with Trp, Tyr, and His, but unsatisfactory selectivity with Lys, Arg, Ser and Cys.With the optimized conditions in hand, we explored the scope of the proposed method. First, we investigated the generality of our allylation regime for a broad range of Trp-containing peptides (Fig. 1). Gratifyingly, all the peptides with Trp residues reacted readily with 2a, affording the corresponding products in high isolated yields (up to 99%). All di- and tripeptides with protected residues, such as lysine and tyrosine, were converted to produce allylation products in excellent yields (3b–3i). This successful application encouraged the exploration of more complex peptide structures. To our delight, this approach proved to be robust for the conversion of tetrapeptides, pentapeptides, and heptapeptides (3j–3n). In addition, the position of the Trp residue in the peptide sequence can be divided into an N-terminus, an internal segment, and a C-terminus; however, the results suggest that positioning has a negligible effect on conversion (3d, 3f, and 3h). In contrast, the composition of amino acids in peptide sequences has a more significant effect on conversion, presumably because these peptides have low solubility in DCM (3j, 3l, and 3m). More importantly, these findings demonstrate the utility of our approach in directly accessing the modification of bioactive peptides (endomorphin-1, 3k, and spinorphin, 3n) without relying on long de novo syntheses based on prefunctionalized building blocks.
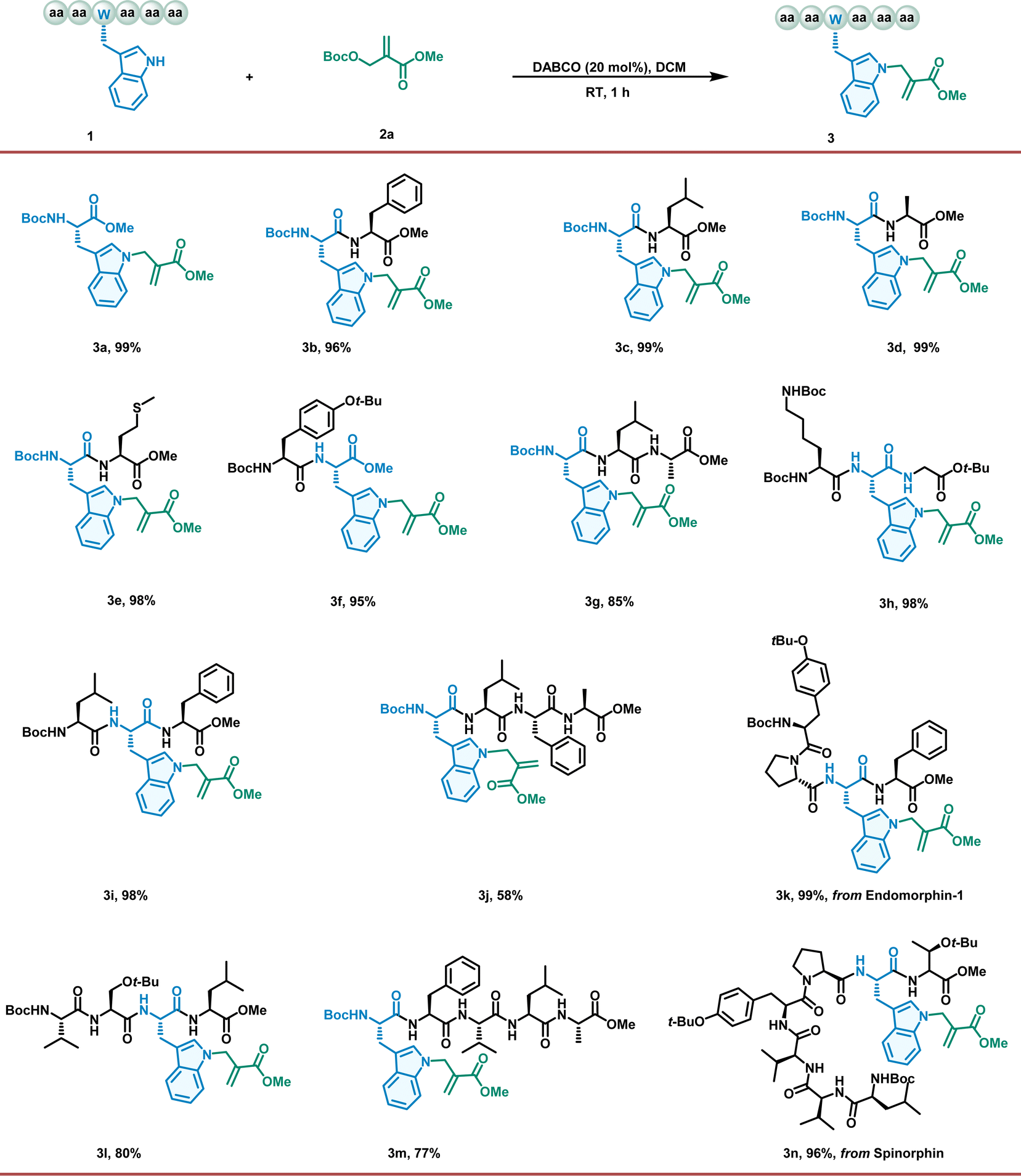 | ||
| Fig. 1 Alkylation of tryptophan-containing peptides with MBH carbonate. Unless otherwise stated, the reaction was carried out with 1 (0.1 mmol), 2a (0.2 mmol, 2 eq.), catalyst (0.02 mmol, 20 mol%) in 1 mL DCM at room temperature for 1 hour. Yields are isolated yields after column chromatography. | ||
Next, we turned our attention to investigating the applicability of this allylation strategy through the smooth ligation of a tripeptide with a wide variety of MBH carbonates (Fig. 2). To our delight, all the MBH carbonate derivatives reacted smoothly with the Boc–Leu–Trp–Phe–OMe tripeptide, furnishing the desired products in good-to-excellent yields (up to 94%). As shown in Fig. 2, the reaction with different acrylates, including ethyl, tert-butyl, and benzyl esters, proceeded efficiently, producing the corresponding allylation products in good yields (4a–4c). This success indicates that allylation provides a unique opportunity to regulate the physical and biological properties of peptides by introducing various substituents with different characteristics. The lipidated and PEGylated motifs can assemble with the tripeptide to regulate the hydrophilic and hydrophobic properties in good yields (4d and 4e). The PEGylation of peptides is also attractive for enhancing their pharmacological properties for application in drug delivery. Similarly, the biorthogonal alkyne handle was ligated with a peptide for further click chemistry, resulting in excellent yields (4f, 94%). This allylation strategy enabled position-specific fluorescent labelling of peptides by assembling them with a coumarin fluorescent dye (4g, 68%). The versatile N-allylation of Trp-containing peptide manifolds has also proved amenable to the ligation of bioactive molecules. Glycosylation can modulate the properties and functions of the peptides and proteins involved in various biological processes. Under our experimental conditions, the masked galactose efficiently conjugated to the Trp residue to produce glycosylated peptides in good yields (4h, 83%). Furthermore, several natural products and drugs, such as piperonyl alcohol, (L)-menthol, L(−)-borneol, epiandrosterone, and cholesterol, were also efficiently conjugated to the Trp residue of the tripeptide in moderate-to-good yields under standard conditions (4i–4m). These results demonstrate the potential of this chemistry for the construction of peptide conjugates. We further explored the scope of sterically hindered MBH carbonates bearing benzene rings (Fig. 2). After meticulous screening of chiral tertiary amine catalysts, we found that the bifunctional catalyst beta-isocupreidine (β-ICD) readily catalyzed the allylation of Boc–Trp–OMe and afforded the product in excellent yields (98%) with 7.1:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 d.r., indicating that properly distributed H-bonds in the transition state may play an essential role in conveying stereochemical information (see the ESI†). In addition, the reaction with the Boc–Leu–Trp–Phe–OMe tripeptide proceeded smoothly, affording the allylation product in 99% yield and 5.3:1 d.r. (4n). We then investigated a series of MBH carbonates bearing halogen substituents on the benzene rings. All MBH carbonates provided the corresponding products in good yields and moderate diastereoselectivities (4o–4q). When an MBH carbonate containing strong electron-withdrawing groups (such as NO2) was employed, the reaction proceeded smoothly and afforded 4r in moderate yields (53%). The allylation also proceeded smoothly using the MBH carbonate with an aliphatic benzene and a naphthyl substituent (4s and 4t). These results demonstrate the great application potential of organic asymmetric catalysis for late-stage modification of peptides.
1 d.r., indicating that properly distributed H-bonds in the transition state may play an essential role in conveying stereochemical information (see the ESI†). In addition, the reaction with the Boc–Leu–Trp–Phe–OMe tripeptide proceeded smoothly, affording the allylation product in 99% yield and 5.3:1 d.r. (4n). We then investigated a series of MBH carbonates bearing halogen substituents on the benzene rings. All MBH carbonates provided the corresponding products in good yields and moderate diastereoselectivities (4o–4q). When an MBH carbonate containing strong electron-withdrawing groups (such as NO2) was employed, the reaction proceeded smoothly and afforded 4r in moderate yields (53%). The allylation also proceeded smoothly using the MBH carbonate with an aliphatic benzene and a naphthyl substituent (4s and 4t). These results demonstrate the great application potential of organic asymmetric catalysis for late-stage modification of peptides.
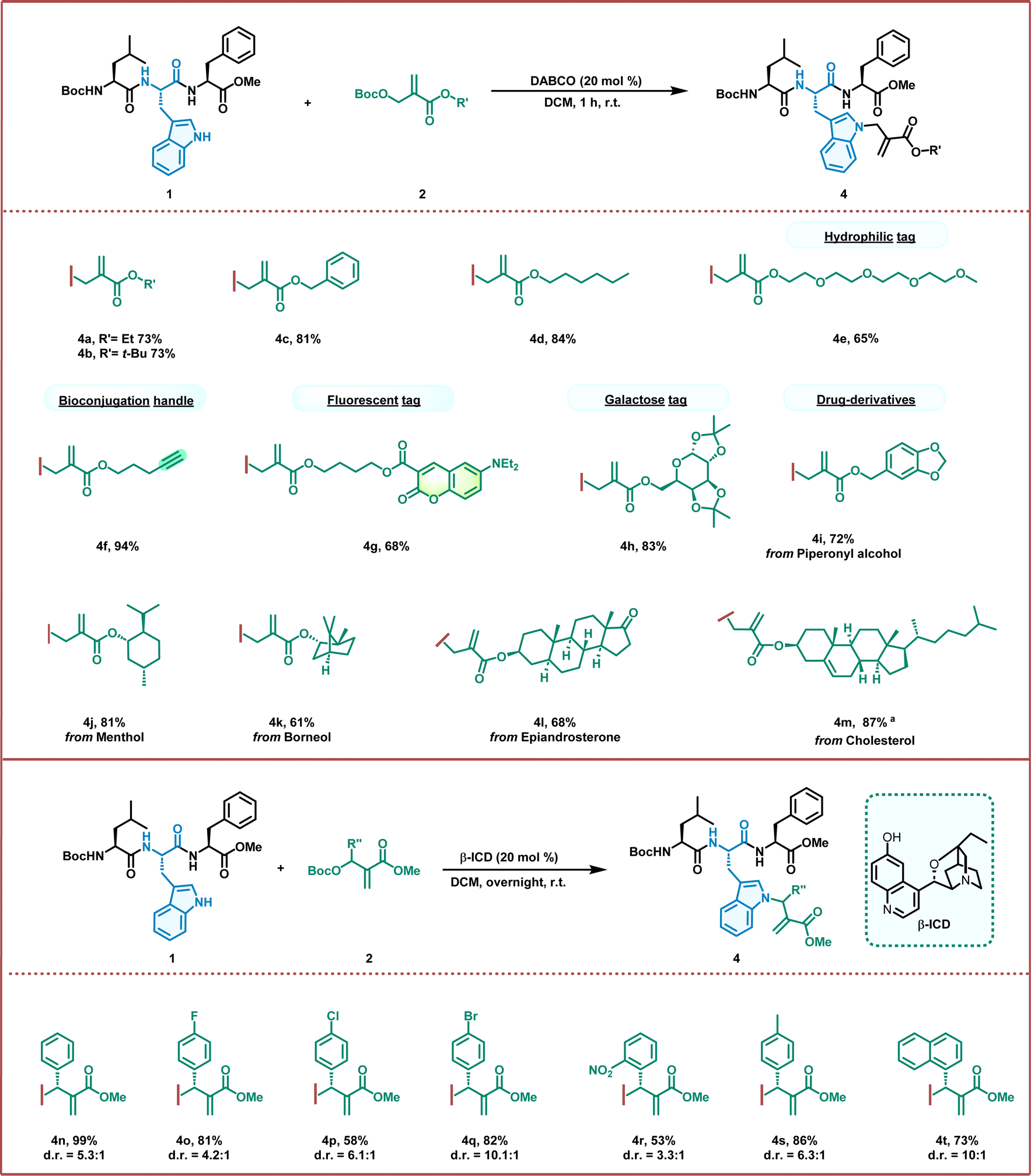 | ||
| Fig. 2 Allylation of tryptophan-containing peptides with MBH carbonates. Unless otherwise stated, the reaction was carried out with 1 (0.1 mmol, 1.0 eq.), 2 (0.2 mmol, 2.0 eq.), and DABCO or β-ICD (0.01 mmol, 20 mol%) in 1 mL DCM at room temperature for 1 hour or overnight. Yields are isolated yields after column chromatography. d.r. was determined by 1H NMR. aThe reaction was carried out for 24 h. | ||
As macrocyclic peptides can significantly enhance the cellular uptake, metabolic stability, and binding affinity of targets, it is essential to develop strategies to construct macrocyclic peptides.8,9,57 We investigated the practicality of the intramolecular Trp allylation method for preparing macrocyclic peptides (Fig. 3A). Allylation-modified macrocyclic peptides were obtained in moderate-to-good yields by intramolecular methods using alkyl MBH carbonate cross-linkers. To improve the yield of such intramolecular macrocyclic peptides and avoid detrimental di- and oligomerization, we diluted the reaction mixture after a simple screening of the reaction concentration. Gratifyingly, under the dilute conditions, a series of linear peptides were smoothly cyclized with excellent chemoselectivity and good yields, furnishing 17- to 35-membered macrocycles (5a–5f). Various amino acids (such as Asp and Ser) can be tolerated under these conditions. The functional side chain of the natural amino acid (Glu) can also be used to tether the MBH carbonate moiety. Thus, cyclic peptide (5e) was readily obtained in moderate yields (49%). The yield did not decrease with increasing linear peptide length. When the linear peptide sequence contained a β-hairpin motif sequence, the cyclization yield becomes slightly higher than that without β-hairpin motif sequences (5f). Peptide stapling is an important strategy for the synthesis of macrocyclic peptides with α-helical conformations.58,59 We further investigated the practicality of the intermolecular Trp allylation method for the construction of stapling peptides (Fig. 3B). Three macrocyclic peptides containing 30–34 members were obtained by stapling different lengths of alkyl MBH carbonates with tetrapeptides via intermolecular allylation (5g–5i). This stapling method can introduce more diverse staple linkages with the inherent amino acids in peptides, avoiding unnatural amino acid synthesis and incorporation.
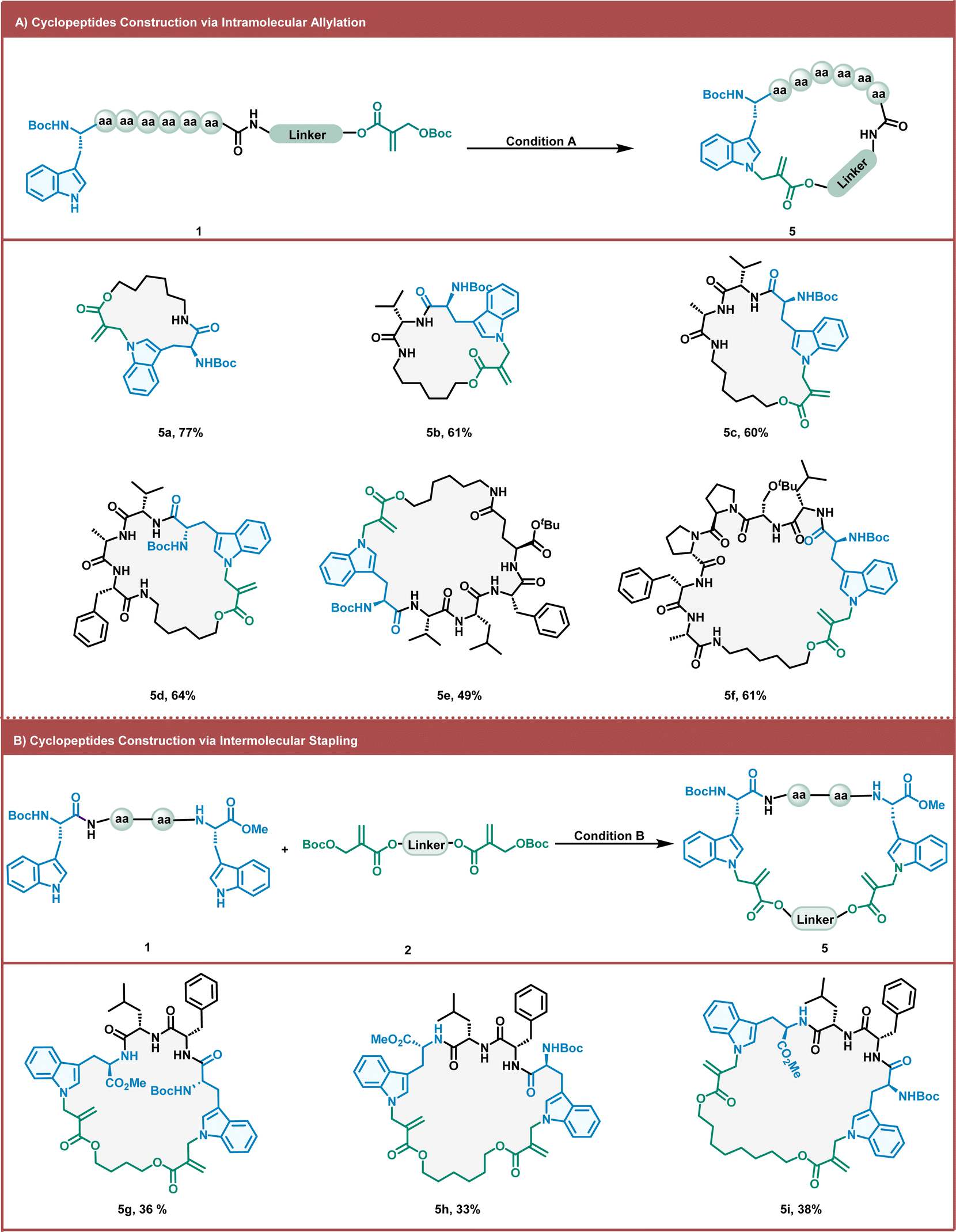 | ||
| Fig. 3 Macrocyclic peptide construction via intramolecular or intermolecular allylation of Trp. Reaction conditions (A) 0.1 mmol 1, DABCO (0.02 mmol, 20 mol%) in 10 mL DCM at room temperature for 1 h. Yields are isolated yields after column chromatography. Reaction conditions (B) 0.1 mmol 1, 0.12 mmol 2, DABCO (0.02 mmol, 20 mol%) in 10 mL DCM at room temperature for 1 h. Yields are isolated yields after column chromatography. | ||
Encouraged by these outstanding results in the Trp residue allylation regime, we further investigated the peptide allylation to obtain the desired modified bioactive peptides and fragments with good chemoselectivity and yields. Notably, this powerful organocatalytic allylation successfully occurred on a solid support despite the limitations of heterogeneous reaction conditions (Fig. 4). For this purpose, we first incorporated unprotected Trp residues into the peptides during SPPS. After the synthesis of the resin-supported peptides, we explored allylation to obtain the desired modified bioactive peptides and fragments with good chemoselectivity and yields (6a–6f). This may provide a rapid and efficient alternative for the preparation of diverse peptide lead analogues.
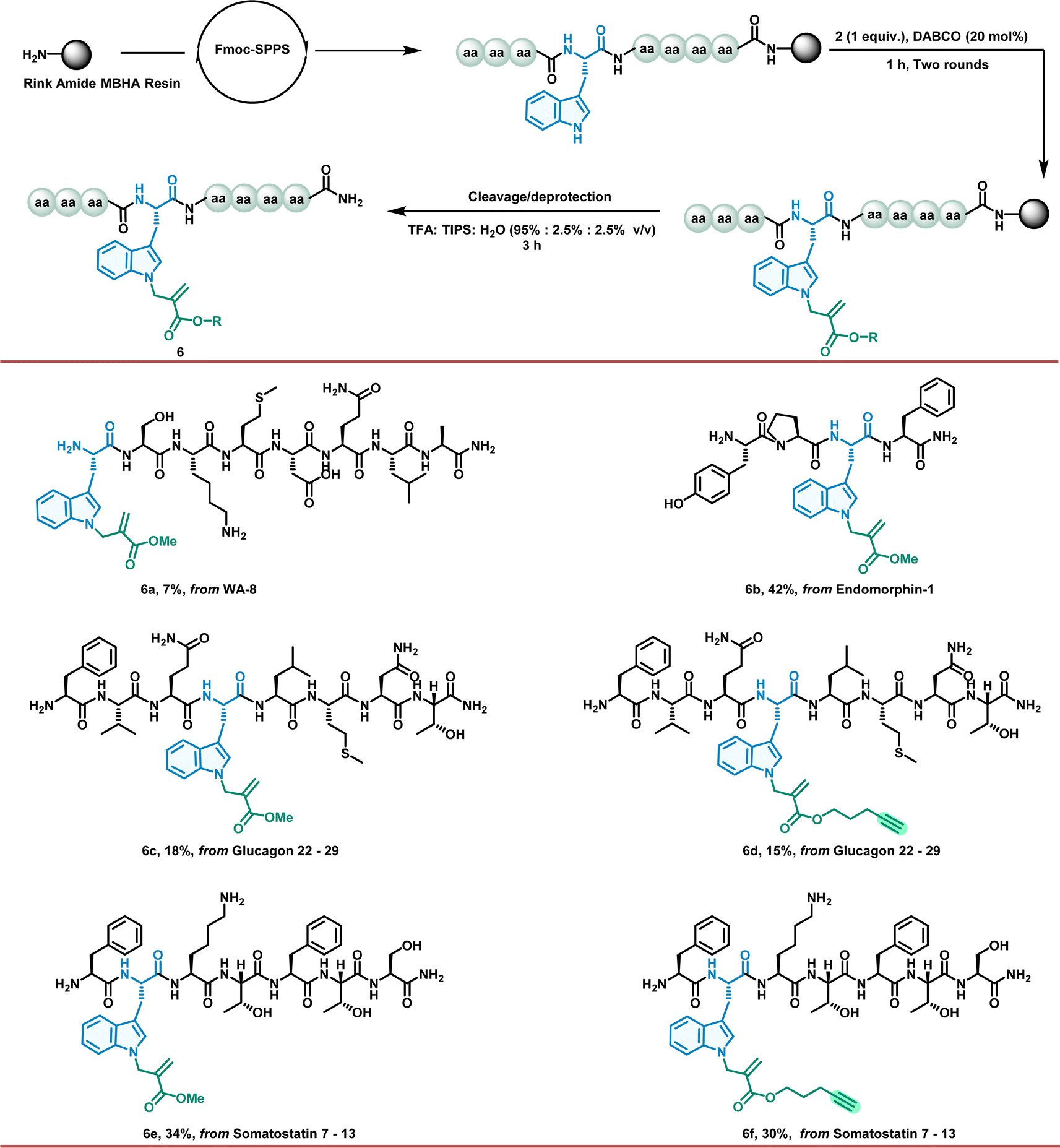 | ||
| Fig. 4 Application of the allylation modification on SPPS. | ||
To further extend the capabilities of this chemistry, we constructed peptide–peptide conjugates via the thia-Michael addition reaction utilizing the Michael receptor incorporated into the peptide and the thiol group of a cysteine residue in the peptide. Next, we performed peptide–peptide conjugation (Fig. 5). The modified peptide (6e) prepared by solid support allylation was reacted with the oligopeptide (P-10: AP02702; containing 13 amino acids within 1 cysteine) at 37 °C and pH 8.35 for 1 h, and the result was analysed by LC-MS/MS. To our delight, the peptide–peptide conjugate was readily constructed with high efficiency under mild conditions. The success of this peptide–peptide conjugate may support further applications of our work in the field of peptide–protein conjugates or peptide inhibitors.
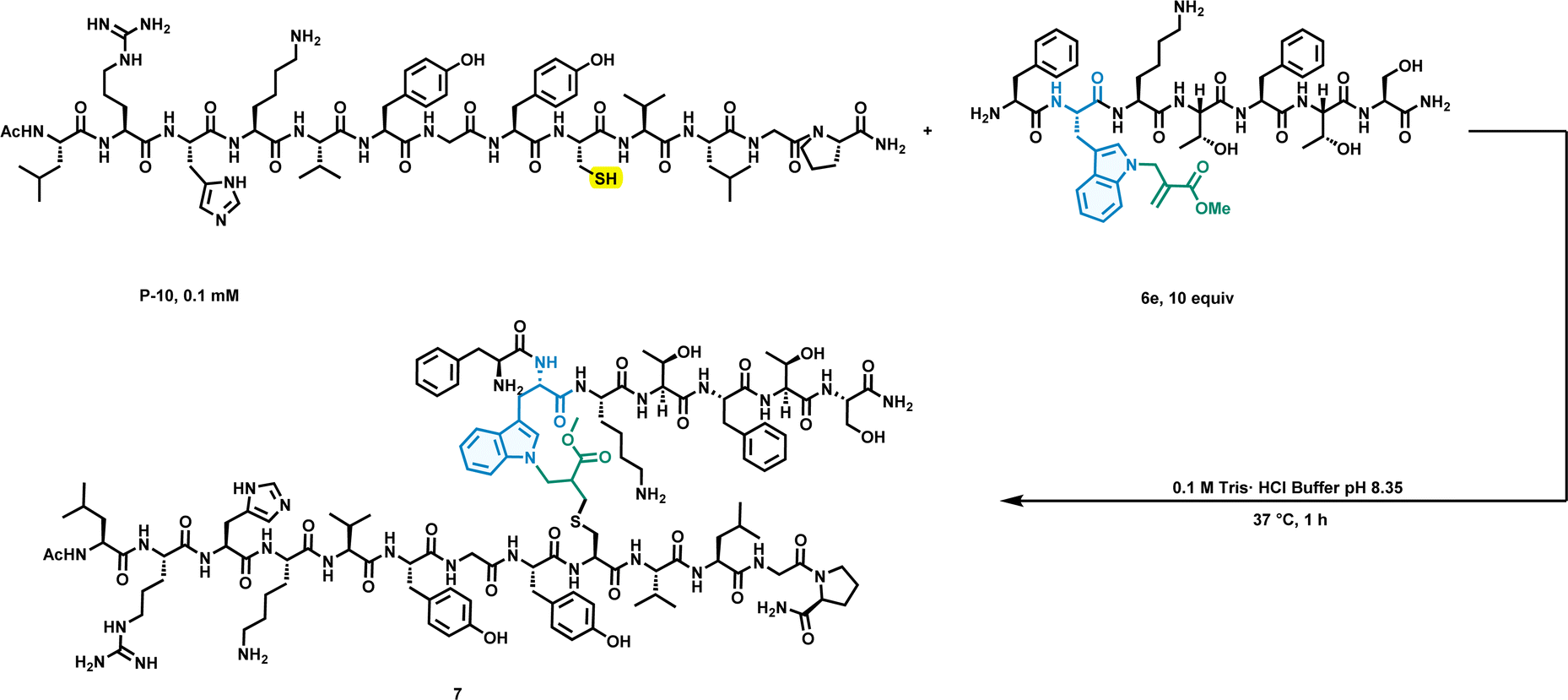 | ||
| Fig. 5 Peptide–peptide conjugate by the thia-Michael addition reaction. | ||
Conclusions
We have developed a practical and efficient strategy for the late-stage functionalization of Trp-containing peptides via organocatalysis. This is the first example of organocatalyzed allylation of peptides at the indole NH site of the Trp residue with various acrylate substituents, using MBH carbonates as reaction counterparts. To further demonstrate the application of this strategy, various functional molecules, including fluorophores, lipids, hydrophilic tags, saccharides, and natural products, were incorporated into the peptides. Significantly, this allylation enables the construction of cyclopeptides via intramolecular cyclization and intermolecular stapling under mild conditions. In addition, this reaction allows the peptide allylation on resin by solid-phase peptide synthesis (SPPS), granting access to structurally intricate peptide molecules. Ultimately, the modified peptides can be conjugated to other biomolecules via an electrophilic warhead generated by the reaction. Hence, this strategy provides an important toolkit for peptide modification and peptide–drug conjugate design. We anticipate that this toolkit will have broad applications in peptide chemical biology and peptide drug discovery.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
W. S. and R. W. conceived the project and designed the experiments. Y. L., W. M. and Z. H. conducted the experiments. Y. L., G. L., G. B., Y. L., and W. S. conducted the data analysis and wrote the manuscript. All authors discussed the results and commented on and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the CAMS Innovation Fund for Medical Sciences (CIFMS) (2019-I2M-5-074, 2021-I2M-1-026, and 2021-I2M-3-001), National Natural Science Foundation of China (U23A20524, 22271126, and 22307052), the Program for the Ministry of Education “Peptide Drugs” Innovation Team (IRT_15R27).Notes and references
- I. W. Hamley, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 CrossRef CAS PubMed.
- C. L. Charron, J. L. Hickey, T. K. Nsiama, D. R. Cruickshank, W. L. Luyt and L. G. Turnbull, Nat. Prod. Rep., 2016, 33, 761–800 RSC.
- B. M. Cooper, J. Iegre, D. H. O’Donovan, M. Ö. Halvarsson and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1480–1494 RSC.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- M. Erak, B. Bellmann-Sickert, S. Els-Heindl and A. G. Beck-Sickinger, Bioorg. Med. Chem., 2018, 26, 2759–2765 CrossRef CAS PubMed.
- L. Reguera and D. G. Rivera, Chem. Rev., 2019, 119, 9836–9860 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- V. Y. Berdan, P. C. Klauser and L. Wang, Bioorg. Med. Chem., 2021, 29, 115896 CrossRef CAS PubMed.
- M. Gehringer and S. A Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775–8806 CrossRef CAS PubMed.
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CrossRef CAS PubMed.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS PubMed.
- H.-R. Tong, B. Li, G. Li, G. He and G. Chen, CCS Chem., 2020, 2, 1797–1820 Search PubMed.
- A. S. Mackay, R. J. Payne and L. R. Malins, J. Am. Chem. Soc., 2022, 144, 23–41 CrossRef CAS PubMed.
- V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2022, 122, 1752–1829 CrossRef CAS PubMed.
- C. Wang, R. Qi, R. Wang and Z. Xu, Acc. Chem. Res., 2023, 56, 2110–2125 CrossRef CAS PubMed.
- D. Gilis, S. Massar, N. J. Cerf and M. Rooman, Genome Biol., 2001, 2, research0049.1 CrossRef PubMed.
- J.-J. Hu, P.-Y. He and Y.-M. Li, J. Pept. Sci., 2021, 27, e3286 CrossRef CAS PubMed.
- H. Gruß and N. Sewald, Chem.–Eur. J., 2020, 26, 5328–5340 CrossRef PubMed.
- W. Wang, J. Wu, R. Kuniyil, A. Kopp, R. N. Lima and L. Ackermann, Chem, 2020, 6, 3428–3439 CAS.
- N. Kaplaneris, F. Kaltenhäuser, G. Sirvinskaite, S. Fan, T. D. Oliveira, L.-C. Conradi and L. Ackermann, Sci. Adv., 2021, 7, eabe6202 CrossRef CAS PubMed.
- N. Kaplaneris, A. Puet, F. Kallert, J. Pöhlmann and L. Ackermann, Angew. Chem., Int. Ed., 2023, 62, e202216661 CrossRef CAS PubMed.
- Z. Bai, C. Cai, W. Sheng, Y. Ren and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 14686–14692 CrossRef CAS PubMed.
- L. Liu, X. Fan, B. Wang, H. Deng, T. Wang, J. Zheng, J. Chen, Z. Shi and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e202206177 CrossRef CAS PubMed.
- J. Liu, P. Wang, W. Zeng, Q. Lu and Q. Zhu, Chem. Commun., 2021, 57, 11661–11664 RSC.
- P. Wang, J. liu, X. Zhu, Kenry, Z. Yan, J. Yan, J. Jiang, M. Fu, J. Ge, Q. Zhu and Y. Zheng, Nat. Commun., 2023, 14, 3973 CrossRef CAS PubMed.
- J. Li, J. Sun, X. Zhang, R. Zhang, Q. Wang, L. Zhang, L. Xie, C. Li, Y. Zhou, J. Wang, G. Xiao, F. Bai and H. Liu, Chem. Commun., 2023, 59, 868–871 RSC.
- M. Nuruzzaman, B. M. Colella, C. P. Uzoewulu, A. E. Meo, E. J. Gross, S. Ishizawa, S. Sana, H. Zhang, M. E. Hoff, B. T. W. Medlock, E. C. Joyner, S. Sato, E. A. Ison, Z. Li and J. Ohata, J. Am. Chem. Soc., 2024, 146, 6773–6783 CrossRef CAS PubMed.
- S. T. Tower, W. J. Hetcher, T. E. Myers, N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2020, 142, 9112–9118 CrossRef CAS PubMed.
- C. R. Hoopes, F. J. Garcia, A. M. Sarkar, N. J. Kuehl, D. T. Barkan, N. L. Collins, G. E. Meister, T. R. Bramhall, C.-H. Hsu and M. D. Jones, J. Am. Chem. Soc., 2022, 144, 6227–6236 CrossRef CAS PubMed.
- C. Yu, R. E, X.-W. Zhang, W.-Q. Hu, G. Bao, Y. Li, Y. Liu, Z. He, J. Li, W. Ma, L.-Y. Mou, R. Wang and W. Sun, ChemMedChem, 2023, 18, e2022006 CrossRef PubMed.
- C. W. Kee, O. Tack, F. Guibbal, C. Wilson, P. G. Isenegger, M. Imiolek, S. Verhoog, M. Tilby, G. Boscutti, S. Ashworth, J. Chupin, R. Kashani, A. W. J. Poh, J. K. Sosabowski, S. Machooll, C. Plisson, B. Cornelissen, M. C. Willis, J. Passchier, B. G. Davis and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 1180–1185 CrossRef CAS PubMed.
- N. J. Kuehl and M. T. Taylor, J. Am. Chem. Soc., 2023, 145, 22878–22884 CrossRef CAS PubMed.
- B. Laroche, X. Tang, G. Archer, R. D. Sanza and P. Melchiorre, Org. Lett., 2021, 23, 285–289 CrossRef CAS PubMed.
- R. N. Lima, J. A. C. Delgado, D. I. Bernardi, R. G. S. Berlinck, N. Kaplaneris, L. Ackermann and M. W. Paixão, Chem. Commun., 2021, 57, 5758–5761 RSC.
- L. R. Malins, K. M. Cergol and R. J. Payne, Chem. Sci., 2014, 5, 260–266 RSC.
- G. Bao, P. Wang, J. Li, X. Song, T. Yu, J. Zhang, Y. Li, Z. He, R. E, X. Miao, J. Xie, J. Ni, R. Wang and W. Sun, CCS Chem., 2024, 6, 1547–1556 CrossRef CAS.
- A. Le Roch, H.-C. Chan and A. Gagnon, Eur. J. Org Chem., 2020, 5815–5819 CrossRef CAS.
- J. A. C. Dolgado, Y.-M. Tian, M. Marcon, B. König and M. W. Paixão, J. Am. Chem. Soc., 2023, 145, 26452–26462 CrossRef PubMed.
- J. Yang, Z. He, L. Hong, W. Sun and R. Wang, Org. Biomol. Chem., 2020, 18, 4169–4173 RSC.
- J. Reimler and A. Studer, Chem.–Eur. J., 2021, 27, 15392–15395 CrossRef CAS PubMed.
- S. Krajcovicova and D. R. Spring, Angew. Chem., Int. Ed., 2023, 62, e202307782 CrossRef CAS PubMed.
- G. Bao, P. Wang, G. Li, C. Yu, Y. Li, Y. Liu, Z. He, T. Zhao, J. Rao, J. Xie, L. Hong, W. Sun and R. Wang, Angew. Chem., Int. Ed., 2021, 60, 5331–5338 CrossRef CAS PubMed.
- Y. Liu, Z. He, W. Ma, G. Bao, Y. Li, C. Yu, J. Li, R. E, Z. Xu, R. Wang and W. Sun, Org. Lett., 2022, 24, 9248–9253 CrossRef CAS PubMed.
- G. Bao, P. Wang, X. Guo, Y. Li, Z. He, X. Song, R. E, T. Yu, J. Xie and W. Sun, Org. Lett., 2023, 25, 8338–8343 CrossRef CAS PubMed.
- Q. Zuo, Y. Li, X. Lai, G. Bao, L. Chen, Z. He, X. Song, R. E, P. Wang, Y. Shi, H. Luo, W. Sun and R. Wang, Adv. Sci., 2024, 11, 2308491 CrossRef CAS PubMed.
- C. Yu, R. E, Y. An, X. Guo, G. Bao, Y. Li, J. Xie and W. Sun, Org. Lett., 2024, 26, 4767–4772 CrossRef CAS PubMed.
- G. Zhu, G. Bao, Y. Li, W. Sun, J. Li, L. Hong and R. Wang, Angew. Chem., Int. Ed., 2017, 56, 5332–5335 CrossRef CAS PubMed.
- J. Yang, Z. Wang, Z. He, G. Li, L. Hong, W. Sun and R. Wang, Angew. Chem., Int. Ed., 2020, 59, 642–647 CrossRef CAS PubMed.
- W. Sun, L. Hong, C. Liu and R. Wang, Org. Lett., 2010, 12, 3914–3917 CrossRef CAS PubMed.
- W. Sun, X. Ma, L. Hong and R. Wang, J. Org. Chem., 2011, 76, 7826–7833 CrossRef CAS PubMed.
- G. Zhu, J. Yang, G. Bao, M. Zhang, J. Li, W. Sun, L. Hong and R. Wang, Chem. Commun., 2016, 52, 7882–7885 RSC.
- H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737–5740 CrossRef CAS PubMed.
- N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2018, 58, 3476–3480 CrossRef PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- Y. H. Lau, P. de Andrade, Y. Wu and D. R. Spring, Chem. Soc. Rev., 2015, 44, 91–102 RSC.
- X. Li, S. Chen, W.-D. Zhang and H.-G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01244e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |