
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDestabilized reporters for background-subtracted, chemically-gated, and multiplexed deep-tissue imaging†
Jason
Yun‡
a,
Yimeng
Huang‡
a,
Austin D. C.
Miller
b,
Brandon L.
Chang
c,
Logan
Baldini
d,
Kaamini M.
Dhanabalan
d,
Eugene
Li
d,
Honghao
Li
a and
Arnab
Mukherjee
 *abd
*abd
aDepartment of Chemistry, University of California, Santa Barbara, CA 93106, USA. E-mail: arnabm@ucsb.edu
bBiomolecular Science and Engineering Graduate Program, University of California, Santa Barbara, CA 93106, USA
cDepartment of Molecular, Cell, and Developmental Biology, University of California, Santa Barbara, CA 93106, USA
dDepartment of Chemical Engineering, University of California, Santa Barbara, CA 93106, USA
First published on 4th June 2024
Abstract
Tracking gene expression in deep tissues requires genetic reporters that can be unambiguously detected using tissue penetrant techniques. Magnetic resonance imaging (MRI) is uniquely suited for this purpose; however, there is a dearth of reporters that can be reliably linked to gene expression with minimal interference from background tissue signals. Here, we present a conceptually new method for generating background-subtracted, drug-gated, multiplex images of gene expression using MRI. Specifically, we engineered chemically erasable reporters consisting of a water channel, aquaporin-1, fused to destabilizing domains, which are stabilized by binding to cell-permeable small-molecule ligands. We showed that this approach allows for highly specific detection of gene expression through differential imaging. In addition, by engineering destabilized aquaporin-1 variants with orthogonal ligand requirements, it is possible to distinguish distinct subpopulations of cells in mixed cultures. Finally, we demonstrated this approach in a mouse tumor model through differential imaging of gene expression with minimal background.
Introduction
Genetic reporters, commonly based on fluorescent and bioluminescent proteins, allow gene expression to be monitored with outstanding specificity in living systems. However, optical imaging in vertebrates is challenging because of absorption and scattering of light by thick tissues. In contrast to optical methods, magnetic resonance imaging (MRI) provides unfettered depth access and enables wide-field imaging with fairly high spatial resolution. To make MRI signals fully genetically encodable, we recently introduced metal-free reporters based on the human water channel aquaporin 1 (Aqp1).1 Aqp1 expression increases the rate of water exchange across the cell membrane, which permits its detection using an MRI technique known as diffusion-weighted imaging. This technique applies a weighting factor (b) determined by magnetic field gradients, such that the MRI signal intensity (S) scales with the diffusivity of water molecules (D) as S ∝ e (−bD).2,3 Cells expressing Aqp1 allow for faster diffusion of water, making them appear hypointense (darker) compared to background tissue. Recent studies have applied Aqp1 in conjunction with diffusion-weighted MRI to trace neural connectivity,4 observe tumor gene activity,5 and image astrocytes on a brain-wide scale in live mice.6The inherent sensitivity of cellular water diffusion to aspects of tissue microstructure such as cell size, volume fraction, tissue geometry, and subcellular architecture7,8 requires careful consideration of the specificity of signals obtained using Aqp1 as a gene reporter. For instance, changes in tissue microstructure, such as those caused by cell death or swelling, could alter water diffusivity in ways that compete with or cancel the diffusion enhancement induced by Aqp1. Furthermore, air-tissue interfaces and calcified tissue may also lead to signal darkening in MRI, making it challenging to distinguish Aqp1-derived contrast from other sources of background noise. To achieve specific, accurate, and reliable imaging with Aqp1, it is necessary to develop an approach that allows Aqp1 signals to be clearly resolved from the tissue background and other non-specific factors that affect tissue water diffusion.
One method to distinguish reporter signals from tissue background in MRI is difference imaging. This technique involves subtracting images acquired with the reporter in an “on” state from those obtained when the reporter is “off” to create a background-free hotspot that reflects signals specific to reporter gene expression. For example, reporters based on chemical exchange saturation transfer (CEST) are visualized as difference signals obtained by saturating the probe at a unique resonance frequency (offset from that of bulk water) and subtracting pre and post-saturation images.9–13 MRI reporters based on gas vesicles can also be visualized through difference imaging by subtracting images acquired with intact vesicles from images obtained after acoustically collapsing the vesicles, which destroys their contrast.14 While the MRI signals from CEST reporters and gas vesicles can be dynamically modulated for background-subtracted imaging, these reporter classes have important limitations. CEST provides limited sensitivity, while gas vesicles involve large multi-gene clusters that are challenging to express in mammalian cells. In contrast, Aqp1 is a single-gene, human-origin reporter that is a highly sensitive but lacks a built-in modulation mechanism to differentiate its signals from nonspecific events.
An ideal approach to modulate Aqp1 signals should achieve a sufficiently large on-off amplitude, operate in multiple cell types and in vivo, exhibit a favorable safety profile, and avoid off-target effects, particularly on endogenously expressed aquaporins. To this end, we sought to modulate Aqp1 stability by engineering its fusion with ligand-binding destabilizing domains (DDs). DDs are genetically engineered protein motifs designed to be unstable. When fused to a protein of interest, DDs result in ubiquitination and degradation of the fused protein.15,16 Ligand-binding DDs17 offer the ability to control protein levels using small-molecule ligands. Binding of these ligands can either stabilize the DD and the fused protein partner by blocking degron-ubiquitin ligase interactions18–21 or promote the recruitment of E3 ubiquitin ligase, leading to the degradation of the protein fusion.22–27 Ligand-binding DDs are extensively utilized in basic research, drug discovery,28 and synthetic biology,29–33 as they enable rapid, reversible, post-translational regulation of protein function. Here, we describe a strategy that involves the incorporation of ligand-binding DDs at the C-terminus of Aqp1, which, when combined with cell-permeable small molecules, enables the post-translational biochemical modulation of Aqp1 expression and MRI signals. We demonstrate the use of Aqp1-DD fusion proteins for biochemically resolved background-subtracted imaging in multiple cell types and subcutaneous tumors in mice. Additionally, we show that Aqp1-DDs can be used to develop two capabilities of considerable interest for reporter gene applications: drug-gated monitoring of transcriptional activity, and imaging of two distinct cell types in a mixed population, using MRI.
Results
Aqp1-DDs enable background-subtracted reporter gene imaging through differential MRI
To achieve the regulation of Aqp1 through the use of DDs, we sought ligand-DD pairs that fulfilled four key criteria. First, ligand binding should result in a significant change in Aqp1 levels, allowing for a substantial MRI response amplitude. Second, the modulation should be reversible to permit serial imaging of Aqp1-labeled cells for longitudinal studies. Third, the small-molecule ligands should be non-toxic and suitable for in vivo applications in rodent models. Fourth, the ligand should be bioorthogonal, minimizing off-target binding with native cellular proteins. Based on these criteria, we selected and tested seven ligand-DD pairs for their ability to either trigger ligand-activated degradation or ligand-induced stabilization of the fused Aqp1 reporter (ESI Fig. 1†). To assess the ligand-activated degradation system, we fused Aqp1 to a mutated mammalian prolyl isomerase, FKBP12 (FKBP12F36V) and full-length and truncated forms of the auxin-binding plant protein IAA17.24,34 In the presence of their cognate ligands, d-TAG13 (ref. 26) and the auxin-family hormone, indole-3-acetic acid (ESI Fig. 1†), these DDs form a complex with E3 ubiquitin ligase, which leads to the degradation of the fused protein. Additionally, we tested a fusion between Aqp1 and a modified FKBP12F36V domain that incorporates a cryptic 19-amino acid degron.27 This degron is displaced and exposed upon binding to the FKBP12F36V ligand shield-1 (ESI Fig. 1c†), resulting in the degradation of the fused protein. To assess the efficacy of ligand-induced stabilization systems, we fused Aqp1 to DDs derived from the directed evolution of three distinct proteins: ligand-binding domain of the estrogen receptor (ER),20 bacterial dihydrofolate reductase (DHFR),19 and FKBP12F36V/L106P.18 These DDs are stabilized by their respective cell-permeable ligands: 4-hydroxytamoxifen, trimethoprim, and shield-1 (ESI Fig. 1c–e†), which permit off-to-on modulation of the fused protein of interest.We used lentiviral transduction to stably express the aforementioned fusion constructs in Chinese hamster ovary cell lines (CHO). We treated the engineered cells with their respective small-molecule ligands and used diffusion-weighted MRI to quantify the percent change in the diffusivities of ligand-treated cells relative to the vehicle-treated controls (ΔD/Do). The ligand-activated DDs showed negligible to moderate ligand-dependent decrease in diffusivity, with the largest response (ΔD/Do = - 21 ± 2%, mean ± s.d., n = 4, P = 1.5 × 10−4, 2-sided t-test) observed in cells expressing Aqp1 fused to the truncated variant of the auxin-inducible degradation domain derived from IAA17 (ref. 34) (Fig. 1a, ESI Fig. 2a†). In contrast, the ligand-stabilized DDs generated a larger change in diffusivity, with the largest response measured in cells expressing the Aqp1- FKBP12-DD fusion construct18,35 (Fig. 1b, ESI Fig. 2b–d†). Specifically, treatment of Aqp1-FKBP12-DD expressing cells with the cognate ligand shield-1 elicited an 80 ± 3% increase in diffusivity (n = 6, P < 10−6, 2-sided t-test) relative to vehicle-treated cells, with half-maximum activation occurring in under 6 hours (Fig. 1c) and with no noticeable toxic effects (P > 0.05, n = 3, 2-sided t-test) based on ATP and MTT viability assays (Fig. 1d). Modulation by shield-1 was specific to Aqp1-FKBP12-DD as we observed no change in the diffusivities (P > 0.05, n = 3, 2-sided t-test) of wild-type cells and erythrocytes, which natively express aquaporin-4 (ESI Fig. 3a†) or in cells engineered to express Aqp1 devoid of FKBP12-DD (ESI Fig. 3b†).
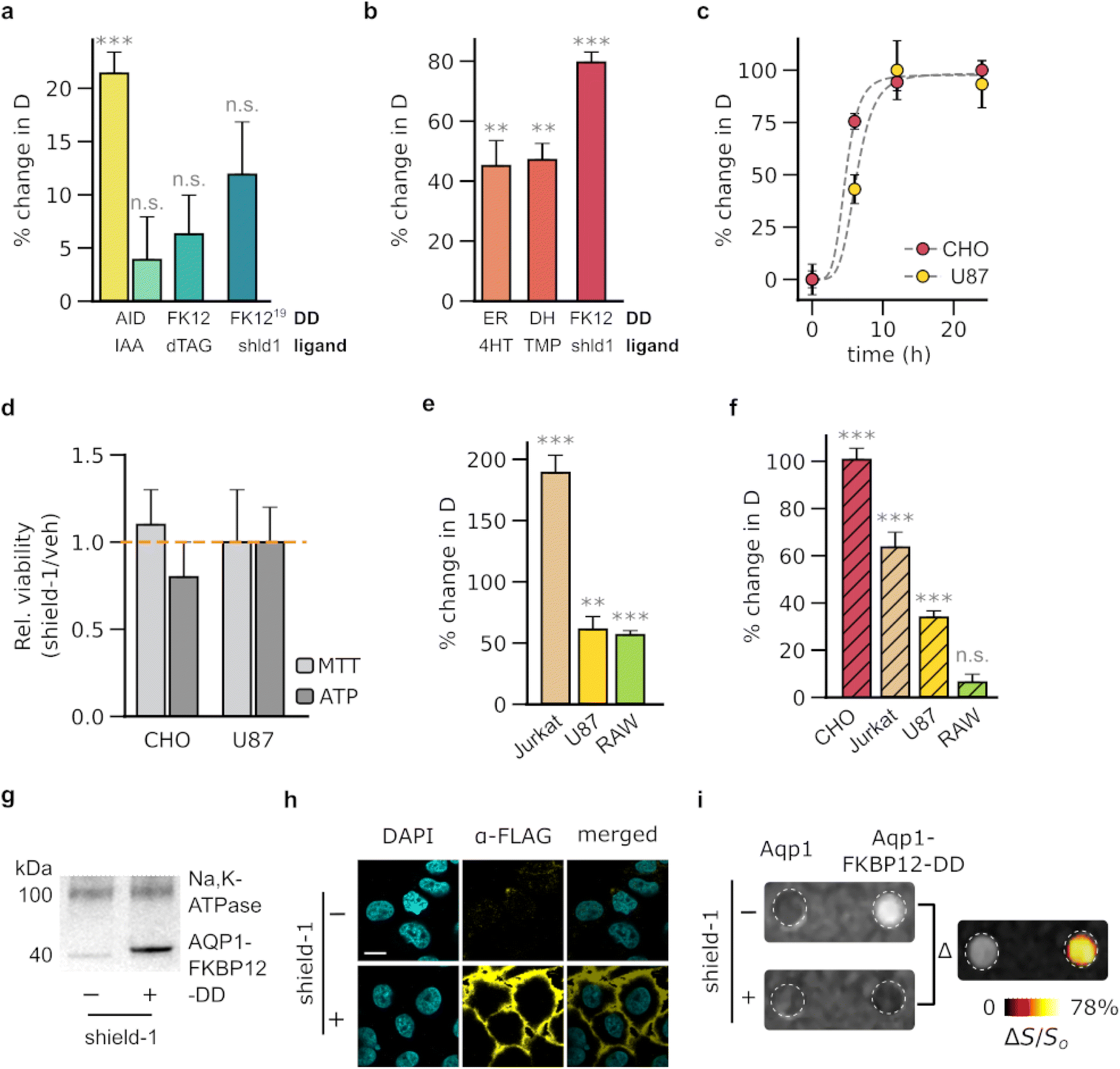 | ||
| Fig. 1 Biochemical modulation of Aqp1 for background-subtracted imaging. (a) Percentage change in the diffusivities of CHO cells engineered to express ligand-activated Aqp1-DDs, following a 24 hour incubation with the respective ligand. Ligands include indole-3-acetic acid (IAA), dTAG-13, and shield-1 (shld1) which bind to degrons based on full-length (228 amino acids) and truncated (68 amino acids) forms of the plant protein, IAA17; the F36V mutant of the mammalian prolyl isomerase, FKBP12 (denoted as FK12 for brevity); and FKBP12F36V modified to incorporate a 19-amino acid cryptic degron (denoted as FK12 (ref. 19)). (b) Percentage change in the diffusivities of CHO cells that have been engineered to express ligand-stabilized Aqp1-DDs, following a 24 hour incubation with the respective ligand. Ligands include 4-hydroxytamoxifen (4HT), trimethoprim (TMP), and shield-1, which bind to DDs based on the estrogen receptor ligand-binding domain (ER), e. coli dihydrofolate reductase (DH), and the F36V/L106P double mutant of FKBP12 (FK12). (c) Time-dependent increase in the diffusivity of Aqp1-FKBP12-DD transduced CHO and U87 cells upon exposure to shield-1. (d) Relative viability of cells following 24 hour incubation with 1 μM shield-1. (e) Percent change in the diffusivities of various cell lines engineered to express Aqp1-FKBP12-DD, following a 24 hour incubation with 1 μM shield-1. (f) Percent change in the diffusivities of cell lines engineered to express Aqp1 N-terminally fused to FKBP12-DD, following a 24 hour incubation with 1 μM shield-1. (g) Western blotting of cell extracts obtained from Aqp1-FKBP12-DD expressing CHO cells in the absence or presence of shield-1 treatment. Aqp1 was detected using antibodies targeting a FLAG epitope inserted at its N-terminus. The Na+/K+-ATPase pump (100 kDa) served as a loading control. (h) Immunofluorescence imaging of Aqp1-FKBP12-DD expressing CHO cells in the presence and absence of shield-1 treatment. Scale bar is 5 μm. Detection was performed using antibodies targeting a FLAG epitope inserted at the N-terminus of Aqp1. (i) Representative image of CHO cells expressing DD-free Aqp1 and Aqp1-FKBP12-DD that have undergone background subtraction. The difference image was produced by subtracting voxel-wise diffusion-weighted datasets (effective b-value ∼3 ms μm−2) acquired with and without shield-1 incubation, denoising the resulting image using a median filter, and displaying it as a pseudo colored “hotspot.” Error bars represent the standard deviation (n ≥ 3). * denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001, and n.s. is non-significant (P ≥ 0.05). P-values were determined using unpaired, 2-sided t-test. For the time-series data, P-values were computed through one-way ANOVA followed by Tukey's HSD test. | ||
Next, we examined the ability of shield-1 to modulate Aqp1-FKBP12-DD in various cell-types, including Jurkat (T lymphocyte), U87 (glioblastoma), and RAW264.7 (macrophage). As before, we used lentiviral transduction to stably express Aqp1-FKBP12-DD in each cell line and measured shield-1 dependent changes in the diffusion coefficients. We observed a significant response in all cell types, with ΔD/Do values of 57 ± 3%, 61 ± 11%, and 189 ± 14% in RAW264.7, U87, and Jurkat cells respectively (n = 4, P < 0.001, 2-sided t-test) (Fig. 1e, ESI Fig. 4a–c†). Quantitative RT-PCR using primers specific for the Aqp1 transgene (Table S2†) revealed no statistically significant disparities in expression levels (ESI Fig. S5†) among the transduced cell lines. This suggests that the observed variations in ΔD/Do values are probably attributable to factors unrelated to transduction efficiency, such as cell type-specific differences in Aqp1 trafficking, cell surface expression, and/or baseline diffusion characteristics.
Given that protein stability may be influenced by the terminus at which the DD is attached, we further investigated whether moving the FKBP12-DD to the N-terminus of Aqp1 would confer a similar shield-1 dependent change in diffusivity. We found that N-terminal fusions resulted in smaller changes in diffusivity than C-terminal fusions in all cell lines, except for CHO cells, where the fold-change was similar between both fusion types (Fig. 1f). Western blotting and qRT-PCR (ESI Fig. 6 and 7†) did not reveal any substantial decrease in the expression of N-terminal fusions compared to that of C-terminal fusions. Hence, we speculate that additional factors other than transgene expression, such as the trafficking of Aqp1 from the endoplasmic reticulum to the plasma membrane and cell surface expression, may account for the observed variations in ΔD/Do between the two types of fusions.
To confirm that shield-1 modulation of diffusion corresponds to the stabilization of Aqp1-FKBP12-DD, we introduced a FLAG epitope at the N-terminus of Aqp1 and used an anti-FLAG antibody to immunoblot membrane fractions prepared from shield-1 and vehicle-treated cells. In agreement with the diffusivity measurements, we observed a prominent band in the extracts prepared from shield-1 treated cells, which was absent or very faint in the control cells (Fig. 1g, ESI Fig. 7†). To further investigate the shield-1 dependent changes in membrane-localized Aqp1-FKBP12-DD, we performed confocal immunofluorescence microscopy. Given that both termini of Aqp1 are cytoplasmic, we inserted a FLAG epitope in an extracellular loop between Gln43 and Thr44, which enabled us to probe Aqp1-FKBP12-DD without having to permeabilize the cells. Consistent with the western blotting data, immunofluorescence imaging also revealed membrane-localized Aqp1 specifically in shield-1 treated cell lines (Fig. 1h).
Based on the favorable characteristics of Aqp1-FKBP12-DD, we hypothesized that this construct would permit the differential imaging of engineered cells via shield-1 based modulation to subtract background signals. To validate this hypothesis, we acquired diffusion-weighted images of Aqp1-FKBP12-DD transduced CHO cells alongside cells expressing unmodified Aqp1. We placed the cells in an agarose phantom, which mimics a high-diffusion background, and performed voxel-wise subtraction of diffusion-weighted images acquired with and without shield-1 incubation. Through this approach, we were able to clearly distinguish the Aqp1-FKBP12-DD cells from both the diffusive background and native Aqp1 signals (Fig. 1i, ESI Fig. 8a†). This approach can also be employed to detect Aqp1-FKBP12-DD expression in terms of a differential diffusion map, where each voxel is colored based on the difference in diffusivity values obtained with and without shield-1 incubation (ESI Fig. 8b†).
Aqp1-FKBP12-DD allows chemically-gated imaging of transcriptional activity
Having established the feasibility of shield-1 modulation of Aqp1-FKBP12-DD, we sought to determine whether this approach could be utilized for chemically-gated imaging of transcriptional activity. Reporters whose output can be gated by a small-molecule drug are highly desirable for monitoring gene expression coincident with user-defined experimental epochs, such as a particular therapeutic regime or behavioral task.36–38 The design of such reporters typically exploits logical AND operation to generate an output only when promoter activity is accompanied by an external drug applied by the experimenter39,40 (Fig. 2a, ESI Fig. 9a†). To test whether Aqp1-FKBP12-DD could be used for drug-gated imaging of transcriptional activity, we created a construct in which Aqp1-FKBP12-DD expression was driven by a doxycycline-inducible minimal CMV promoter. We delivered this construct into CHO cells using lentiviral infection, treated stable cells with both shield-1 and doxycycline, as well as each compound individually, and acquired whole-cell diffusion measurements as previously described. In contrast to Aqp1, where promoter activity (induced by doxycycline) alone is sufficient to generate a reporter gene output (Fig. 2b), both promoter induction and shield-1 were required to elicit maximum response in Aqp1-FKBP12-DD (ΔD/Do = 95 ± 15% P < 0.001, n = 6, one-way ANOVA, Tukey's HSD test) (Fig. 2c). In agreement with these results, differential imaging revealed a reporter signal only when promoter activity was also accompanied by shield-1 application (Fig. 2d, ESI Fig. 9b†). Taken together, these findings provide a proof-of-concept for the potential of using Aqp1-FKBP12-DD as a drug-gated MRI reporter to record transcriptional activity accumulated during a user-defined window determined by the addition and washout of shield-1.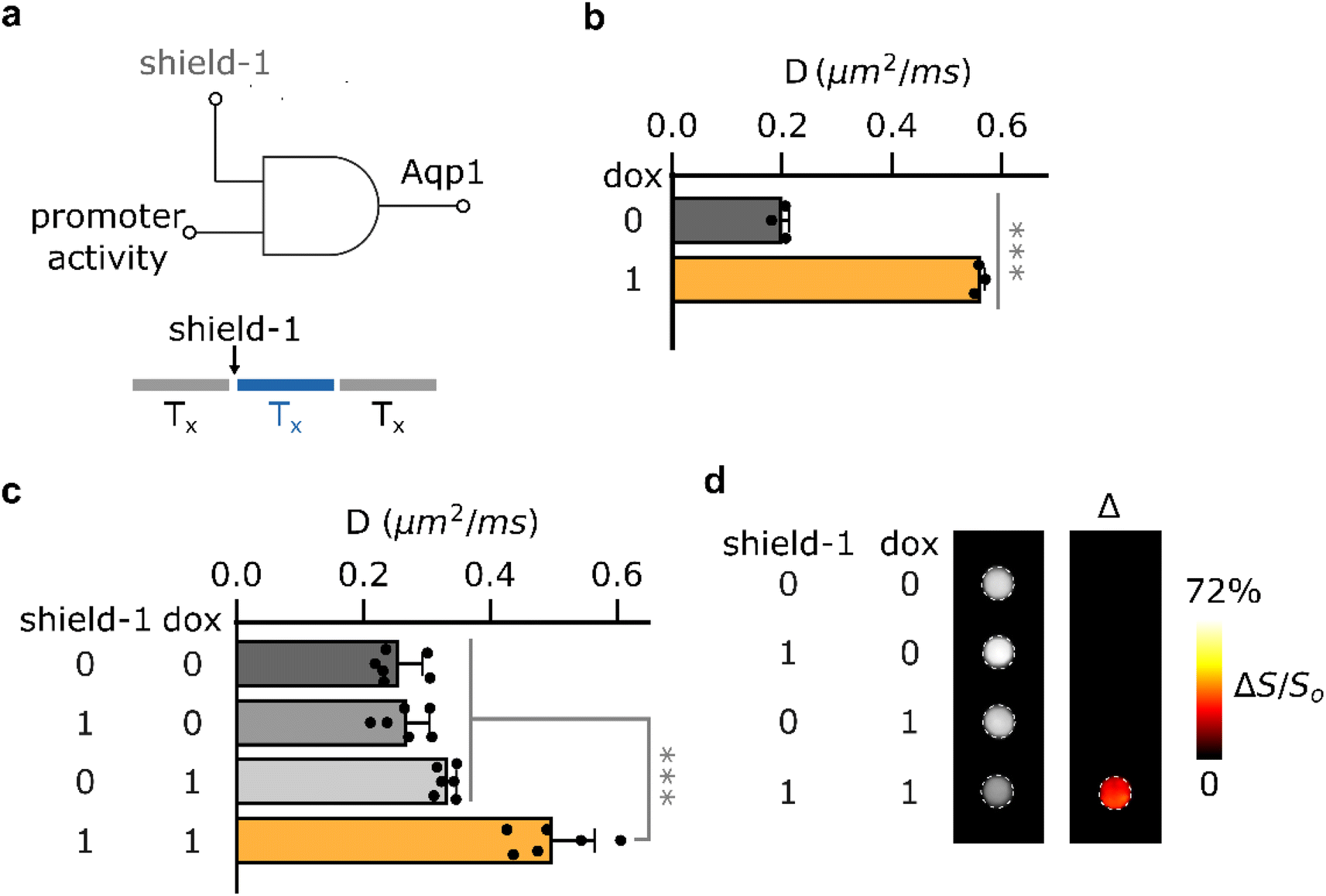 | ||
| Fig. 2 Biochemically-gated imaging of transcriptional activity. (a) Gated reporters leverage genetic AND logic to generate an output only when transcriptional activity (Tx) is accompanied by an externally added drug (in this case, shield-1). (b) Doxycycline-dependent change in diffusivities of CHO cells expressing conventional DD-free Aqp1 under the control of a doxycycline-inducible minimal CMV promoter. (c) Doxycycline-driven changes in diffusivities of CHO cells expressing Aqp1-FKBP12-DD under the control of a doxycycline-inducible minimal CMV promoter following incubation with shield-1, doxycycline, and both ligands. (d) Representative difference image showing shield-1 gated imaging of transcriptional activity. The difference image was obtained through voxel-wise subtraction of diffusion-weighted datasets (effective b-value ∼1.6 ms μm−2) acquired in the presence of shield-1, doxycycline, or both from a dataset acquired in their absence. The resulting image was subsequently denoised using a median filter and presented as a pseudo-colored “hotspot.” Error bars represent the standard deviation (n = 3–6). *** denotes P < 0.001. P-values were computed based on 2-sided, unpaired t-test (2b) or using one-way ANOVA followed by Tukey's HSD test (2c). | ||
Orthogonal Aqp1-DD pairs allow detection of cells in mixed populations
Many applications in biomedicine would benefit from the ability to use MRI-based reporter genes to simultaneously map multiple biological features, such as distinct cell types or promoter activities. Although proton MRI is fundamentally a “monochromatic” technique, we hypothesized that it would be possible to use orthogonal pairs of Aqp1-DDs to image cells in multiplex by stabilizing one reporter at a time and performing difference imaging. For this approach to work, the Aqp1-DD constructs should exhibit non-overlapping ligand-binding preference, thereby permitting stepwise modulation of diffusivity with addition of each ligand (Fig. 3a). Conceptually, this approach is inspired by substrate-resolved imaging in bioluminescence41–43 and CEST,13 where reporter signals are separated based on their selectivity for orthogonal ligands. We first established that the three ligand-stabilized Aqp1-DD constructs identified in our initial screening, namely Aqp1-FKBP12-DD, Aqp1-DHFR-DD, and Aqp1-ER-DD were mutually orthogonal, that is, cells transduced with either of the three reporters did not respond to non-cognate ligands (Fig. 3b, ESI Fig. 10a–c†). Next, to demonstrate “2-color” imaging, we prepared a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CHO cells transduced with Aqp1-FKBP12-DD and Aqp1-DHFR-DD. We obtained whole-cell diffusion measurements on untreated cells, cells treated with shield-1 only, and cells treated with both shield-1 and trimethoprim. We observed an expected stepwise increase in the diffusion coefficient of mixed cells with the addition of ligands (Fig. 3c, ESI Fig. 11†). Finally, we performed pairwise subtraction of diffusion-weighted images obtained after treating cells with one or both ligands, which allowed the two reporter-expressing cell types to be separately detected in the mixed population (Fig. 3d). To verify that the above approach is generalizable to other cell types and degron-tagged constructs, we also demonstrated multiplex imaging of a U87 + Jurkat cell mixture engineered to express orthogonal Aqp1-DD reporter constructs (ESI Fig. 12†).
:1 mixture of CHO cells transduced with Aqp1-FKBP12-DD and Aqp1-DHFR-DD. We obtained whole-cell diffusion measurements on untreated cells, cells treated with shield-1 only, and cells treated with both shield-1 and trimethoprim. We observed an expected stepwise increase in the diffusion coefficient of mixed cells with the addition of ligands (Fig. 3c, ESI Fig. 11†). Finally, we performed pairwise subtraction of diffusion-weighted images obtained after treating cells with one or both ligands, which allowed the two reporter-expressing cell types to be separately detected in the mixed population (Fig. 3d). To verify that the above approach is generalizable to other cell types and degron-tagged constructs, we also demonstrated multiplex imaging of a U87 + Jurkat cell mixture engineered to express orthogonal Aqp1-DD reporter constructs (ESI Fig. 12†).
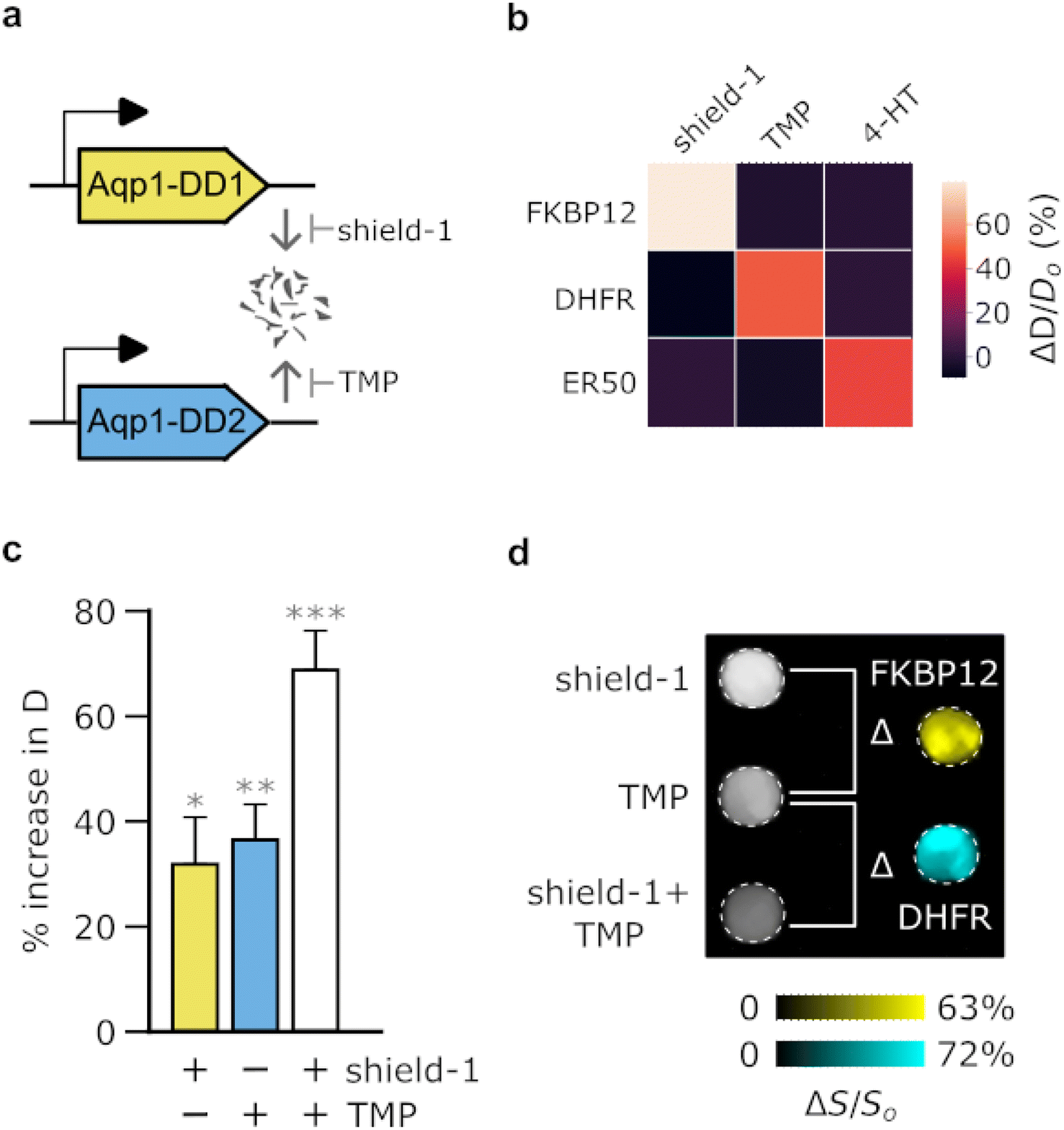 | ||
| Fig. 3 Biochemical unmixing of Aqp1-DDs for imaging in multiplex. (a) Illustration of approach for modulating diffusivity signals using orthogonal Aqp1-DD reporters that are stabilized by non-overlapping small-molecule ligands. (b) Matrix showing the mean response of various Aqp1-DDs to either cognate (diagonal) or non-cognate ligands (off-diagonal). (c) Ligand-dependent increase in the diffusivity of a mixed population comprising equal numbers of Aqp1-FKBP12-DD and Aqp1-DHFR-DD transduced CHO cells. Ligand addition permits the modulation of diffusivity in a stepwise manner by stabilizing one or both Aqp1-DD constructs. (d) The components of the mixed-cell population were resolved through difference imaging. Subtraction of diffusion-weighted images of untreated cells from images acquired after treatment with shield-1 reveals the Aqp1-FKBP12-DD labeled population. Subtraction of diffusion-weighted images (effective b-value ∼3 ms μm−2) of shield-1 treated cells from those obtained after treatment with both shield-1 + trimethoprim (TMP) revealed the Aqp1-DHFR-DD labeled population. The images were denoised by median filtering and pseudo-colored to distinguish the two sub-populations. Error bars represent standard deviation (n = 2–4). P-values were computed using one-way ANOVA followed by Tukey's HSD test.* denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001. | ||
Aqp1-FKBP12-DD enables diffusion-weighted difference imaging in vivo
In order to evaluate the in vivo utility of Aqp1-FKBP12-DD, we used the reporter in a mouse tumor xenograft model, which provides a common platform for testing new tissue-penetrant reporter gene techniques. We created transgenic tumors by injecting CHO cells bilaterally into the hind flanks of mice, with one set of cells engineered to express Aqp1-FKBP12-DD and the other set expressing Aqp1, without a DD (Fig. 4a). Once the tumors reached a volume of 100–300 mm3, we acquired two sets of diffusion-weighted images: the first before injecting shield-1 and the next 6–8 hours post-injection. In agreement with the trends observed in vitro, the Aqp1-FKBP12-DD tumors showed a significant increase in the diffusion coefficient following the injection of shield-1 (ΔD/Do = 56 ± 17% n = 6, P = 0.003, paired t-test), while the diffusivity of the Aqp1 tumors was not altered (D/Do = 0.98 ± 0.31, n = 6, P = 0.85, paired t-test) (Fig. 4b). Accordingly, differential imaging based on shield-1 modulation allowed for the specific visualization of Aqp1-FKBP12-DD expression as a background-subtracted ‘hotspot’ overlaid on a higher-resolution anatomical image (Fig. 4c). As before, the difference images can also be depicted quantitatively as diffusion maps, where each voxel is represented by its diffusivity differential (ESI Fig. 13†).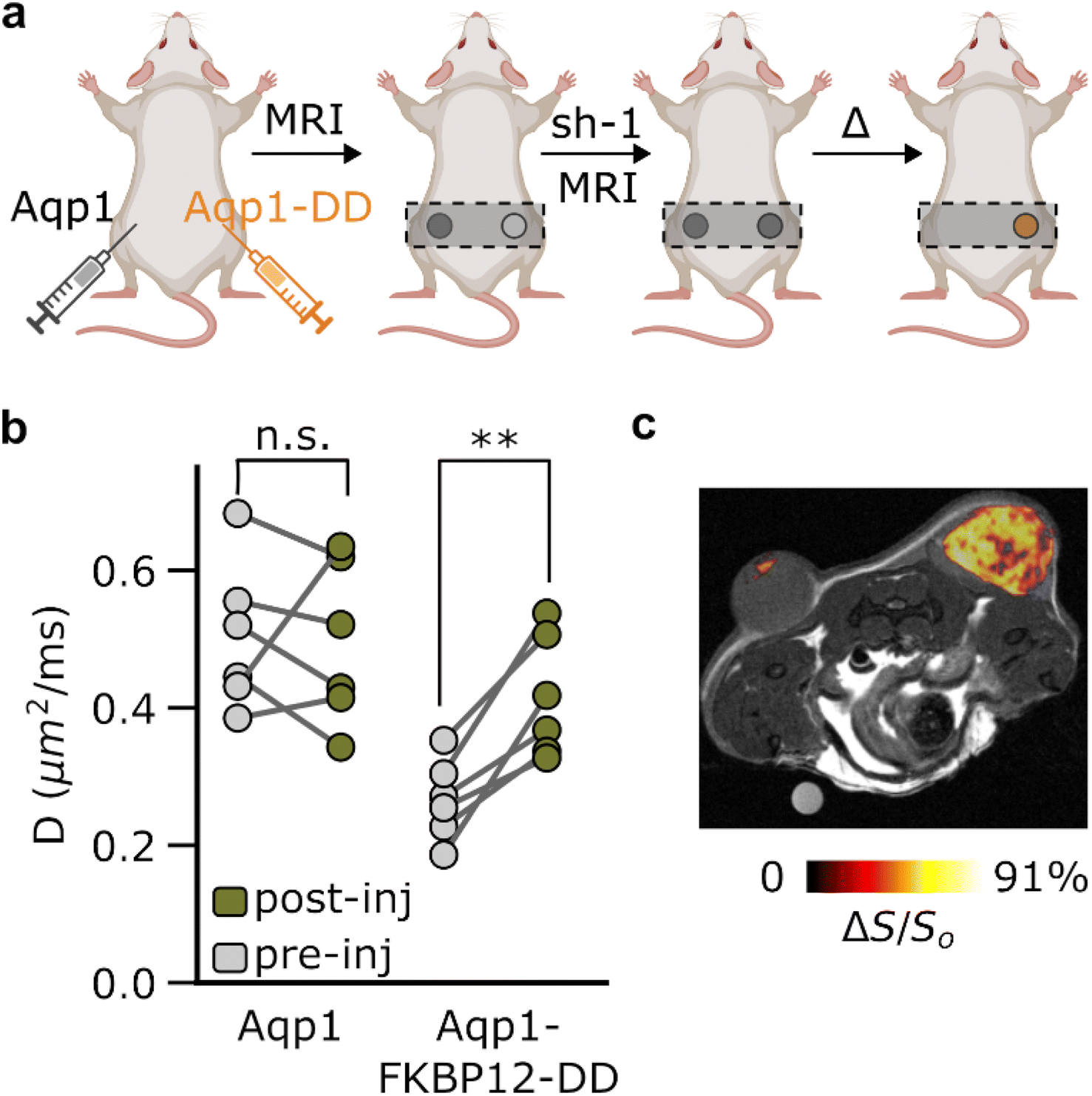 | ||
| Fig. 4 In vivo imaging using background subtraction. (a) Schematic outline of the in vivo study. (b) Diffusivities of transgenic tumors engineered to express Aqp1-FKBP12-DD or DD-free Aqp1 (n = 6) measured before and after intraperitoneal injection of shield-1 (10 mg kg−1). (c) Background subtracted image of the Aqp1-FKBP12-DD labeled tumor. The difference image was generated by subtracting voxel-wise diffusion-weighted images (effective b-value = 3.4 ms μm−2) of the tumors acquired before and after intraperitoneal injection of shield-1 (10 mg kg−1). This difference image was then overlaid on an anatomical image to provide spatial context. Error bars represent the standard deviation. P-values were computed using paired, 2-tailed t-test (2-sided). ** denotes P < 0.01, and n.s. is non-significant (P ≥ 0.05). | ||
Discussion
Our work establishes ligand-induced stabilization of Aqp1 as a conceptually new approach for imaging gene expression using MRI. The best-performing construct, Aqp1-FKBP12-DD, enables small-molecule modulation of diffusion signals using a safe, cell-permeable, and bioorthogonal chemical, shield-1. Differential imaging minimizes interference from signals unrelated to the reporter, thereby making Aqp1 expression visible as a background-subtracted “hotspot”. Aqp1-FKBP12-DD also permits the detection of promoter activity via genetic AND circuits, paving the way for time-locked monitoring of transcriptional signals in user-defined experimental windows. Finally, we showed that orthogonal Aqp1-DDs may be paired for multiplex imaging of gene expression by chemical unmixing of diffusion-weighted signals.We anticipate that Aqp1-FKBP12-DD will be broadly useful for applications requiring gene expression to be accurately tracked in non-transparent animals. Notably, the in vivo safety and biodistribution of shield-1 is already established in various rodent models44–46 and furthermore, shield-1 was recently shown to cross the blood–brain barrier.47 Accordingly, Aqp1-FKBP12-DD can be used to monitor viable cells and map gene expression in tumor tissue by unambiguously separating reporter signals from factors like necrosis, apoptosis, and edema, which cause nonspecific diffusion increases in the tumor microenvironment. Aqp1-FKBP12-DD also offers the advantage of tracking transient changes in transcriptional activity, which would be missed by conventional reporters with long half-lives.48 Additionally, the ability to post-translationally gate reporter output in MRI using shield-1 will be useful for neuroscience applications to accurately correlate brain-wide gene expression patterns with behavior and neuromodulation paradigms. Recent research has demonstrated that Aqp1, devoid of the DD motif, serves as a feasible reporter in the mouse brain,4,6 offering a promising prospect for expanding Aqp1-FKBP12-DD in large-scale neural mapping studies. Additionally, this approach could be applied to tracking the distribution of immune cells and stem cell-based therapies within the body. To this end, the mammalian origin of both Aqp1 and FKBP12-DD provides an important advantage in addressing potential immunogenicity concerns associated with in vivo applications of other classes of background-free deep-tissue reporters, such as gas vesicles.
We recognize that the present technology has certain limitations that also create opportunities for future advances. First, although the ligand-controlled changes in signals obtained using Aqp1-DDs were on the order of (or larger than) signal amplitudes typically detected with existing 1H MRI reporters, these changes were weaker than those observed for DD fusions to other reporter systems, such as fluorescent and bioluminescent proteins. This difference was not entirely surprising, given that the tested DDs were engineered to operate in the context of cytoplasmic reporters, not Aqp1 or any other membrane protein. However, the reduced fold-change suggests that the differential datasets obtained using Aqp1-DDs are susceptible to noise and imaging artifacts. To address this issue, further increasing ΔD/Do – for instance, by enhancing Aqp1-DD degradation in the ligand-free state or stabilizing the fusion protein in the ligand-bound state – would improve robustness and detection sensitivity. Second, although the kinetics of Aqp1-FKBP12-DD modulation align with numerous biological processes, such as gene expression and cell trafficking, faster modulation would allow access to a broader range of dynamic biological functions. Third, while this work provides an initial proof-of-concept for distinguishing cells labeled with orthogonal Aqp1-DDs via chemical unmixing, considerable optimization is necessary to apply this approach for imaging distinct cell-types and transcriptional signals in vivo. In particular, in vivo application of chemical unmixing will require careful characterization of the pharmacokinetic properties of the injected ligands to ensure that signals from a previously stabilized Aqp1-DD construct are completely reset before administration of the second ligand. Additionally, the fact that our method is based on detecting changes in ligand-dependent image intensity, as opposed to directly identifying differences in spectral signatures (such as CEST and bioluminescence-based multiplexing), presents a practical challenge (in terms of sensitivity to noise) when attempting to resolve mixed cultures, particularly those with sparsely labeled cells. To address this issue, protein engineering techniques such as directed evolution can be employed to optimize Aqp1-DDs, as has been demonstrated in the context of several cytosolic reporters. Although the low throughput of MRI presents a practical challenge for designing large-scale screening experiments, our ability to detect the biochemical modulation of Aqp1-DDs by FLAG epitope tagging (Fig. 1i) offers a potential strategy for screening mutant libraries using high-throughput methods such as fluorescence- or magnetic-activated cell sorting. Finally, it should be noted that differential images are generated from two separately acquired scans and are therefore sensitive to motion artifacts and registration errors between pre- and post-injection images. To mitigate this problem, the development of faster strategies to modulate Aqp1 activity may enable pre- and post-injection images to be acquired in rapid succession without moving the animal between scans. Additionally, the use of respiratory or cardiac gating and applying Aqp1-DDs in organs such as the brain, wherein reduced motion and standardized anatomical atlases can enhance imaging accuracy, will further mitigate the effects of registration errors and artifacts. Finally, it should be emphasized that MRI is widely used for longitudinal studies that span multiple days or even months, which has led to the development of several techniques for registering longitudinally acquired datasets. In the future, these techniques can be integrated with the Aqp1-DD imaging workflow to improve registration accuracy.
In summary, our methodology provides a conceptually new approach to unambiguously track gene expression in intact organisms by combining the biophysics of cellular water diffusion with chemogenetic tools to manipulate diffusion. We expect that the development of Aqp1-DDs will synergize with technological advances in diffusion-weighted MRI,49 the advent of complementary reporter gene modalities,50,51 and existing functional MRI paradigms such as metabolic imaging,52 to bring us closer to the goal of exploring complex biological functions on large spatial scales and with molecular and cell-type specificity in intact vertebrates.
Methods
Reagents
Reagents for PCR amplification and Gibson assembly were purchased from New England Biolabs (Ipswich, MA, USA). Polyethyleneimine (linear, 25 kDa) was purchased from Polysciences (Warrington, PA, USA). Lenti-X concentrator was purchased from Takara Bio (Kusatsu, Shiga, Japan). Polybrene was purchased from Santa Cruz Biotechnology (Dallas, TX, US). Dulbecco's Modified Eagle Media (DMEM), sodium pyruvate, doxycycline hyclate, penicillin-streptomycin (104 units per mL penicillin and 10 mg mL−1 streptomycin), beta-mercaptoethanol, MG-132, chloroquine diphosphate, polyethylene glycol 400, Tween®-80, and N,N-dimethylformamide were purchased from Sigma-Aldrich (St. Louis, MO, USA). Roswell Park Memorial Institute media (RPMI 1640), Gibco™ fetal bovine serum (FBS), Pierce™ BCA Protein Assay Kit, and DAPI (4′,6-diamidino-2-phenylindole) lactate salt were purchased from Thermo Fisher Scientific (Waltham, MA, USA). MycoAlert® Plus Mycoplasma Detection Assay was purchased from Lonza. U87 (ATCC#: HTB-14™), RAW264.7 (ATCC#: TIB-71™), and Jurkat cells (ATCC#: TIB-152™) were purchased from the American Type Culture Collection (ATCC). CHO-TetON cells were obtained from Takara Bio (San Jose, CA, USA). Mouse CD1 red blood cells were purchased from Fisher Scientific. Trimethoprim and 4-hydroxytamoxifen were purchased from Fisher Scientific; shield-1 was obtained from Aobious (Gloucester, MA, USA); indole-3-acetic acid from Neta Scientific (Hainesport, NJ, USA); dTAG-13 was purchased from Tocris Bioscience (Bristol, UK). USP-grade doxycycline hyclate was purchased from Fresenius Kabi USA. Reagents for denaturing polyacrylamide gel electrophoresis, including pre-cast gels, Laemmli buffer, Tris-buffered saline with 0.1% Tween®-20 (TBS-T), non-fat dried milk, and Clarity™ Western ECL substrate were purchased from Bio-Rad (Hercules, CA, USA). Ladders for western blotting were purchased from LI-COR Biosciences (Lincoln, NE, USA) – WesternSure Chemiluminescent pre-stained ladder or GoldBio (St. Louis, MO, USA) – BLUEstain™ Protein ladder (11–245 kDa). ProteoExtract® native membrane protein extraction kit and 10 kDa Amicon® Ultra-15 centrifugal filters were purchased from Millipore Sigma (USA). Assays for measuring cell toxicity were purchased from Promega (Madison, WI, USA). Anti-FLAG primary antibody was purchased from Sigma-Aldrich (#F1804). Horseradish peroxidase-conjugated (#TL280988) and Alexa Fluor 647 (#A21235) conjugated goat anti-mouse IgG secondary antibodies were purchased from Thermo Fisher. Type F immersion oil was purchased from Leica Microsystems (Deerfield, IL, USA).Molecular biology
Plasmids harboring the various degron sequences – DHFR-DD (Addgene 29326), ER-DD (Addgene 37261), miniIAA7 (Addgene 129721), and FKBP12 (Addgene 17416) were amplified using Q5 High-Fidelity 2× Master Mix and cloned by Gibson assembly in a lentiviral transfer vector at the C or N-terminus of the aquaporin-1 reporter (Aqp1) under the control of either a constitutive promoter, EF1α (Addgene 60058) or a doxycycline-inducible minimal CMV promoter (Addgene 26431). An EGFP reporter was co-expressed with Aqp1 using an internal ribosome entry site (IRES) to allow selection of stably transduced cell by fluorescence-activated cell sorting (FACS). All constructs (Table S1†) were verified by Sanger DNA sequencing (Genewiz, San Diego, CA, USA) or whole-plasmid nanopore sequencing (Plasmidsaurus, Eugene, OR, USA).Cell culture and engineering
Cells were routinely cultured at 37 °C in a humidified 5% CO2 incubator using DMEM (CHO-TetON, U87, RAW264.7) or RPMI (Jurkat) supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. For cells harboring an inducible reporter gene cassette, doxycycline hyclate (1 μg mL−1) was added to the growth media at the time of seeding. Cells were periodically checked for Mycoplasma contamination using a bioluminescent assay (MycoAlert® Plus). Lentivirus was packaged using a three plasmid system, comprising a packaging plasmid, an envelope plasmid encoding the vesicular stomatitis virus G protein (to pseudo type lentivirus for broad tropism), and the transfer plasmids constructed above. The three plasmids (22 μg each of the packaging and transfer plasmid; 4.5 μg of envelope plasmid) were delivered to 293T cells by transient transfection using polyethyleneimine. Approximately 17–24 h post transfection, the 293T cells were treated with sodium butyrate (10 mM) to promote expression of viral genes. Viral production was allowed to proceed for another 72 h before precipitating the cell-free supernatant using a commercial polyethylene glycol formulation (Lenti-X concentrator) to harvest lentivirus. The resulting viral particles were resuspended in 200 μL DMEM and immediately used to transduce the recipient cell lines, including CHO Tet ON, Jurkat, U87, and RAW 264.7. For viral transduction, cell lines were first grown to 70–80% confluency in a six-well plate format, clarified by aspiration or gentle centrifugation (300×g, 5 min) to remove spent media, and treated with purified virus resuspended in 800 μL DMEM containing 8 μg mL−1 polybrene. The cells were spinfected by centrifuging the six-well plates at 1048×g for 90 min at 30 °C before returning to the 37 °C CO2 incubator for another 48 h. The AQP1-miniIAA17 (truncated) and AQP1-IAA17 (full-length) expressing cell lines were transduced a second time with lentivirus encoding AtAFB2 (Addgene 129718) and OsTIR1 (Addgene 72834) auxin receptor F-box proteins, which are required to bind the auxin degron tag and form a complex with ubiquitin E3 ligase for degradation. To enable sorting of doubly-transduced cells, the above genes were co-expressed with an mCherry reporter. Following lentiviral transduction, stably transduced cells were enriched by fluorescence activated cell sorting (FACS) using a Sony SH800 cell sorter and stored as cryo-stocks until further use. Using the above protocol, we are consistently able to produce cell lines that exhibit stable reporter transgene activity (measured in terms of diffusivity) for extended periods of time ranging from several months to years (ESI Fig. 14†).Cell viability assay
To assess potential toxicity resulting from shield-1 treatment, cells were assayed for MTT reduction (CellTiter 96® Non-radioactive Cell Proliferation Assay) and intracellular ATP content (CellTiter-Glo®). For each assay, cells were grown to 80–90% confluency in 96-well plates and incubated with the assay reagents as per the manufacturer's protocol. Absorbance (MTT reduction) and luminescence readings (ATP content) were obtained using a plate reader (Tecan Spark). Integration time for luminescence measurements was set to 1 s.Western blotting
In preparation for western blotting, cells were lysed using RIPA Lysis Buffer System® (Santa Cruz Biotechnology, Dallas, TX, USA). Total protein concentration was estimated using BCA assay to ensure that similar amounts of total sample were probed by western blotting. Next, 20 μL of cell lysate was denatured by mixing with an equal volume of 2× Laemmli buffer supplemented with 5% beta-mercaptoethanol and incubated at room temperature for 16 h. Next, the samples were boiled at 95 °C for 5 minutes immediately before loading in a gel. The denatured extract was resolved by electrophoresis in an SDS-PAGE gel at 90 V for 90 min at 4 °C and transferred to a polyvinylidene difluoride membrane using a Biorad Trans-Blot® Turbo™ transfer system. The membrane was blocked for 1 h in 50 mL of Tris-Buffered Saline containing 0.1% (w/v) Tween® 20 (TBS-T) and supplemented with 5% (w/v) non-fat dried milk. Subsequently, the membrane was incubated overnight at 4 °C in 50 mL of TBS-T containing 1 μg mL−1 mouse anti-FLAG primary antibody (Sigma-Aldrich, F1804), 0.025 μg mL−1 rabbit anti-Na+/K+-ATPase (Abcam, ab76020) and 5% (w/v) non-fat dried milk. The membrane was washed three times using 50 mL of TBS-T with an incubation time of 15 min per wash. The membrane was then treated with 50 mL TBS-T containing 0.5 μg mL−1 horseradish peroxidase-conjugated goat anti-mouse IgG secondary antibodies, 0.025 μg mL−1 horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibodies, and 5% (w/v) non-fat dried milk for 1 h under room temperature. Subsequently, the membrane was washed with 50 mL of TBS-T three times as before. Finally, the membrane was incubated with Clarity™ Western ECL Substrate and imaged using an iBright™ FL1500 imaging system (Thermo Fisher Scientific).Quantitative RT-PCR
qRT-PCR was performed as described in our earlier work using primers that recognize the FLAG sequence incorporated in our Aqp1 transgene (Table S2†). Briefly, transgene expressing cells were grown in six-well plates for 24 hours and lysed in Buffer RLT (Qiagen) supplemented with 1% β-mercaptoethanol. The lysate was then centrifuged in QIAshredder columns to shear genomic DNA. Total RNA was extracted according to the manufacturer's (Qiagen) protocol and reverse-transcribed using iScript™ cDNA synthesis reagents. The cDNA was diluted 10-fold in nuclease-free water and qRT-PCR was performed using Power Up™ SYBR™ Green Master Mix in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Cycle threshold values (Cq) were determined using CFX Maestro Software (Bio-Rad). Expression was quantified using the 2−ΔΔCq method with beta-actin serving as the housekeeping gene.Immunofluorescence imaging
LSAqp1-transduced CHO cells were grown in 35 mm #1.5 glass bottom dishes (Ibidi, Cat. no. 81218-200) to a confluency of 70–80%. After aspirating the medium, cells were fixed by incubating in 1 mL 4% paraformaldehyde for 15 minutes at room temperature inside a fume hood. Cells were rinsed three using 2 mL sterile phosphate buffered saline (PBS) with an incubation time of 5 minutes per wash. The cells were then blocked by incubation with blocking solution (2% BSA, 5% goat serum in PBS) for 1 hour. Subsequently, the cells were incubated for 18 hours at 4 °C in a rotary shaker (set at 200 r.p.m) with 1 mL blocking solution containing 5 μg mL−1 mouse anti-FLAG primary antibody solution. The cells were washed three times using 2 mL PBS with an incubation time of 5 minutes for each wash. Finally, the cells were incubated with 1 mL of PBS containing Alexa Fluor 647 conjugated goat anti-mouse IgG secondary antibody (2 μg mL−1) and 2% bovine serum albumin (BSA). The plate was foil wrapped, incubated in a rotary shaker (200 r.p.m) at room temperature for 2 hours, and washed two times using 2 mL PBS with an incubation time of 5 minutes at each wash step. Next, the cells were incubated for 5 minutes at room temperature with DAPI (4′,6-diamidino-2-phenylindole) lactate salt (2.5 μg mL−1 in PBS) to stain the nucleus, washed twice with 1 mL PBS (3 minutes incubation per wash step), and finally overlaid with 1 mL PBS in preparation for confocal imaging.Confocal microscopy was performed using a Leica Dmi8 SP8 resonant scanning microscope equipped with an HC PL APO 63×/1.40 oil-immersion objective using Type F immersion oil. A 405 nm laser line was used for DAPI excitation and the emitted light was detected between 410 nm and 483 nm. A 653 nm laser line was used for Alexa Fluor 647 excitation and the emitted light was detected between 665 nm and 779 nm. Leica Application Suite X (LAS X) was used for image acquisition and Fiji was used for image analysis.
In vitro MRI and difference imaging
In preparation for MRI, cells were seeded such that they reached confluency in approximately 48 h. 24 h post-seeding, cells were treated with a small-molecule ligand matched to the degron-type expressed by the cell: 10 μM trimethoprim (DHFR-DD), 1 μM 4-hydroxytamoxifen (ER-DD), 1 μM shield-1 (FKBP12F36V/L106P-DD and FKBP12F36V-19-amino acid degron), 1 μM dTAG-13 (FKBP12F36V), and 500 μM indole-3-acetic acid (full-length and truncated IAA17-based DDs). Control cohorts were prepared by treating cells with the respective vehicle solution, i.e. ethanol (for indole-3-acetic acid) and 0.1% dimethyl sulfoxide (for all the other compounds). Adherent cells were harvested by trypsinization, centrifuged at 350×g, resuspended in 200 μL sterile PBS and transferred to 0.2 mL tubes. For the mixed cell experiments, the two cell-types were cultured separately, counted using a hemocytometer, mixed by gentle pipetting, centrifuged as before, and transferred to 0.2 mL tubes. The tubes were centrifuged at 500×g for 5 min to form a cell pellet and placed in water-filled agarose (1% w/v) molds housed in a 3D-printed MRI phantom. Imaging was performed using a 66 mm diameter coil in a Bruker 7T vertical-bore MRI scanner. Stimulated echo diffusion-weighted images of cell pellets were acquired in the axial plane using the following parameters: echo time, TE = 18 ms, repetition time, TR = 1000 ms, gradient duration, δ = 5 ms, gradient separation, Δ = 300 ms, matrix size = 128 × 128, field of view (FOV) = 5.08 × 5.08 cm2, slice thickness = 1–2 mm, and number of averages = 5. Diffusion-sensitizing gradients were applied in the readout direction (dorsal–ventral axis) in the plane orthogonal to the axial slice-selection gradient using nominal b-values of 0–0.8 ms μm−2, which correspond to effective b-values (viz., b-values corrected to include the contribution of imaging gradients) in the range of 1–3 ms μm−2. Diffusion-weighted intensity at a given b-value was estimated by computing the average intensity of all voxels inside a region of interest (ROI) encompassing the axial cross section of a cell pellet. The slope of the logarithmic decay in mean signal intensity as a function of b-value was used to calculate the apparent diffusivity (D). In separate experiments, we verified that the diffusivity measurements obtained in Aqp1-expressing cells by employing diffusion-weighting gradients along the three primary axes of our vertical-bore MRI were comparable (ESI Fig. 15†). Therefore, to reduce acquisition time, we typically acquired diffusivity measurements in a single direction (dorsal-ventral). To generate a diffusion map, apparent diffusivity was computed for each voxel in an ROI. Least-squares regression fitting was performed using the fitnlm function in Matlab (R2022b).Differential imaging was performed by voxel-wise subtraction of diffusion-weighted images or diffusion maps of cells belonging to various treatment groups (e.g. shield-1, shield-1 + doxycycline, shield-1 + trimethoprim) from corresponding images of vehicle-treated cells. The difference images were denoised using a median filter and pseudo colored according to an 8 bit color scale. Image subtraction was performed in Matlab (R2022b), while median filtering and pseudo coloring were implemented in Fiji.
Kinetic studies
For the kinetic studies, pellets of LSAqp1-transduced CHO cells were collected at multiple time points (t = 0, 6, 12, and 24 h) following treatment with shield-1. In vitro MRI (i.e. imaging of cell pellets) is an end-point technique. Therefore, out of practical necessity, cells corresponding to each time point were harvested from different plates, which were all seeded in a similar manner from cryo-stocks. Diffusion-weighted imaging and whole-cell diffusivity measurements were performed exactly as described above.Mouse tumor xenograft
All experiments involving animals were performed following protocols approved by the Institutional Animal Care and Use Committee (IACUC protocol number 946.1) of the University of California, Santa Barbara. CHO cells expressing LSAqp1 or Aqp1-only (viz. without a degron) were grown to 80–90% confluency, harvested by trypsinization, centrifuged at 300×g for 5 min, and resuspended in 100 μL sterile PBS. The resuspended cells were mixed with an equal volume of Matrigel®, loaded into 1 mL sterile syringes fitted with 25G needles, and subcutaneously injected in the left and right hind limbs of 5–7 weeks old female NOD/SCID/γ-mice (Jackson Labs Strain no. 005557). Tumors were measured daily using calipers and tumor volume was calculated as 0.52 × (short axis)2 × (long axis)2. All protocols were approved by the Institutional Animal Care and Use Committee of the University of California, Santa Barbara.In vivo MRI
Once the subcutaneous tumors reached a volume of 100–300 mm3 (typically 17–21 days post-implantation), mice were imaged using a Bruker 7T vertical-bore MRI scanner. Mice were first anesthetized using 2–3% isoflurane in medical-grade oxygen and secured in an animal cradle with medical tape to ensure proper positioning of tumors and to minimize motion-induced artifacts. An ophthalmic ointment (Puralube) was applied on the eyes in order to prevent corneal drying. Mice were then subcutaneously injected with sterile saline (20 mL kg−1 body weight) to ensure adequate fluid levels are maintained during the MRI session. Next, the cradle was inserted in the MRI scanner concentrically with a 40 mm diameter imaging coil. Respiration rate and body temperature were monitored throughout the imaging session using a pneumatic cushion (Biopac, Goleta, CA, USA) and a fiber optic rectal probe (Opsens) respectively. Body temperature was maintained at 36.5–38 °C using warm air flow with feedback temperature control. Respiration rate was maintained at 40 ± 5 breaths per minute. To induce Aqp1 and Aqp1-FKBP12-DD expression in the tumor cells, mice were intraperitoneally injected with 50 mg kg−1 doxycycline approximately 24 h before imaging.After positioning the mouse in the MRI scanner, the location of the tumors was estimated by acquiring localizer scans in the three principal anatomical orientations: head-foot, dorsal-ventral, and left-right. The localizer scans were implemented using a FLASH sequence with the following parameters: echo time, TE = 3 ms, repetition time, TR = 100 ms, flip angle = 30°, matrix size = 256 × 256, field of view (FOV) = 6 × 6 cm2, slice thickness = 2 mm, number of averages = 10, and number of slices = 3, resolution = 0.234 × 0.234 mm2. Based on the localizer scan, we then acquired a conventional anatomical image of the tumor in the axial direction (head-foot) using a RARE sequence with the following parameters: echo time, TE = 14.22 ms, repetition time, TR = 660.2 ms, RARE factor = 4, matrix size = 256 × 256, field of view (FOV) = 3.5 × 3.5 cm2, slice thickness = 1 mm, number of averages = 10, and number of slices = 10, resolution = 0.137 × 0.137 mm2. Diffusion-weighted images were subsequently obtained using a stimulated echo pulse sequence with the following parameters: echo time, TE = 18 ms, repetition time, TR = 1000 ms, gradient duration, δ = 5 ms, gradient interval, Δ = 300 ms, matrix size = 96 × 96, field of view (FOV) = 3.5 × 3.5 cm2, number of averages = 18, resolution = 0.365 × 0.365 mm2, and slice thickness = 1 mm. Diffusion-sensitizing gradients were applied in the readout direction (viz., dorsal–ventral axis) in the plane orthogonal to the axial slice-selection gradient using nominal b-values of 0 and 1 ms μm−2, which correspond to effective b-values (viz., b-values corrected to include the contribution of imaging gradients) of 1.3 and 3.4 ms μm−2. Fat suppression and respiratory gating were not implemented in our acquisition protocol.
Next, mice were intraperitoneally injected with 10 mg kg−1 shield-1 dissolved in a mixture of 50% N,N-dimethylformamide and 50% 9:1 PEG-400: Tween®-80. Approximately 6 h after injecting shield-1, the mouse was returned to the imaging cradle. The post-injection scan was acquired using the same procedure as before, taking care to position the axial slice to align as closely as possible with the pre-injection tumor plane. While manual alignment of pre- and post-injection slices is challenging, we note that this step can be greatly simplified through the use of commercially available automated mouse positioning systems. Immediately after MRI, mice were moved to a clear chamber and monitored for complete recovery from anesthesia before returning to the home cage.
To calculate apparent diffusivity, ROI(s) were drawn in the tumor volume using Fiji software. Mean intensities were calculated for each ROI and diffusivity was estimated from the decrease in signal intensity versus b-value as described previously. Differential images were generated by overlaying pre- and post-injection diffusion-weighted images of the tumor ROIs, using Matlab's (R2022b) imregister function followed by voxel-wise subtraction of signals. The ensuing difference image was pseudo colored and spatially displayed by overlaying on the anatomical image acquired in roughly the same plane as the diffusion-weighted image.
Statistical analysis
Data are summarized by their mean and standard deviation obtained from multiple (n ≥ 3) biological replicates and compared using a 2-tailed t-test (for two groups) or one-way ANOVA followed by Tukey's HSD test (for > 2 groups) as indicated in the figure captions. A paired t-test was used to compare diffusivities of bilateral tumors in the same subject. The quality of model fitting for estimating diffusivity is assessed based on the regression coefficient. A P-value of less than 0.05 is considered statistically significant.Abbreviations
| MRI | magnetic resonance imaging |
| Aqp1 | aquaporin-1 |
| CEST | chemical exchange saturation transfer |
| DD | destabilizing domain |
| FKBP12 | 12 kDa FK506 Binding Protein |
| DHFR | Dihydrofolate reductase |
| ER | estrogen receptor |
| TMP | trimethoprim |
| 4HT | 4-hydroxytamoxifen |
| dTAG | degradation tag |
| CHO | chinese hamster ovary cell line |
| RAW264.7 | immortalized mouse macrophage cell line |
| U87 | immortalized human glioblastoma cell line |
| Jurkat | immortalized human T lymphocyte cell line |
Materials transfer agreement
All plasmid constructs used in this work can be provided by the corresponding author pending scientific review and a completed material transfer agreement. Requests for plasmids should be submitted to: Arnab Mukherjee (E-mail: arnabm@ucsb.edu).Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.Author contributions
Jason Yun: methodology, investigation, validation, formal analysis, writing - original draft, writing - review & editing. Yimeng Huang: methodology, investigation, validation, formal analysis. Austin D. C. Miller, Brandon L. Chang, Kaamini M. Dhanabalan, Logan Baldini, Eugene Li, Honghao Li: investigation, validation. Arnab Mukherjee: conceptualization, methodology, formal analysis, resources, data curation, writing - original draft, writing - review & editing, visualization, supervision, project administration, funding acquisition.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
We thank members of the Mukherjee lab for helpful discussions. Dr Jerry Hu (UC, Santa Barbara) is gratefully acknowledged for assistance with setting up the MRI workflow. This research was supported by the National Institutes of Health (R35-GM133530, R21-EB033989, R03-DA050971, and R01-NS128278 to A. M.), the U.S. Army Research Office via the Institute for Collaborative Biotechnologies cooperative agreement W911NF-19-D-0001-0009 (A. M.), and a NARSAD Young Investigator Award from the Brain & Behavior Research Foundation (A. M.). This project has been made possible in part by a grant from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation. J. Y. gratefully acknowledges support from the Chair's Fellowship from UC Santa Barbara (Department of Chemistry). L. B. acknowledges support from the National Science Foundation Graduate Research Fellowships Program. All MRI experiments were performed at the Materials Research Laboratory (MRL) at UC, Santa Barbara. The research reported here made use of the shared facilities of the Materials Research Science and Engineering Center (MRSEC) at UC Santa Barbara: NSF DMR-2308708. The UC Santa Barbara MRSEC is a member of the Materials Research Facilities Network (https://www.mrfn.org). We acknowledge the use of the Neuroscience Research Institute-MCDB Microscopy Facility and the Resonant Scanning Confocal supported by NSF MRI grant 1625770.References
- A. Mukherjee, D. Wu, H. C. Davis and M. G. Shapiro, Non-invasive imaging using reporter genes altering cellular water permeability, Nat. Commun., 2016, 7, 13891 CrossRef CAS PubMed.
- K.-D. Merboldt, W. Hanicke and J. Frahm, Self-diffusion NMR imaging using stimulated echoes, J. Magn. Reson., 1969, 64, 479–486 Search PubMed.
- E. O. Stejskal and J. E. Tanner, Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient, J. Chem. Phys., 2004, 42, 288–292 CrossRef.
- N. Zheng, M. Li, Y. Wu, C. Kaewborisuth, Z. Li, Z. Gui, J. Wu, A. Cai, K. Lin, K.-P. Su, H. Xiang, X. Tian, A. Manyande, F. Xu and J. Wang, A novel technology for in vivo detection of cell type-specific neural connection with AQP1-encoding rAAV2-retro vector and metal-free MRI, NeuroImage, 2022, 258, 119402 CrossRef PubMed.
- L. Zhang, M. Gong, S. Lei, C. Cui, Y. Liu, S. Xiao, X. Kang, T. Sun, Z. Xu, C. Zhou, S. Zhang and D. Zhang, Targeting visualization of malignant tumor based on the alteration of DWI signal generated by hTERT promoter–driven AQP1 overexpression, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 2310–2322 CrossRef CAS PubMed.
- M. Li, Z. Liu, Y. Wu, N. Zheng, X. Liu, A. Cai, D. Zheng, J. Zhu, J. Wu, L. Xu, X. Li, L.-Q. Zhu, A. Manyande, F. Xu and J. Wang, In vivo imaging of astrocytes in the whole brain with engineered AAVs and diffusion-weighted magnetic resonance imaging, Mol. Psychiatry, 2022, 1–8 Search PubMed.
- D. L. Bihan, Molecular diffusion, tissue microdynamics and microstructure, NMR Biomed., 1995, 8, 375–386 CrossRef PubMed.
- H. Li, X. Jiang, J. Xie, J. C. Gore and J. Xu, Impact of transcytolemmal water exchange on estimates of tissue microstructural properties derived from diffusion MRI, Magn. Reson. Med., 2017, 77, 2239–2249 CrossRef CAS PubMed.
- A. Bar-Shir, G. Liu, K. W. Y. Chan, N. Oskolkov, X. Song, N. N. Yadav, P. Walczak, M. T. McMahon, P. C. M. van Zijl, J. W. M. Bulte and A. A. Gilad, Human Protamine-1 as an MRI Reporter Gene Based on Chemical Exchange, ACS Chem. Biol., 2014, 9, 134–138 CrossRef CAS PubMed.
- A. Bar-Shir, J. W. M. Bulte and A. A. Gilad, Molecular Engineering of Nonmetallic Biosensors for CEST MRI, ACS Chem. Biol., 2015, 10, 1160–1170 CrossRef CAS PubMed.
- I. Minn, A. Bar-Shir, K. Yarlagadda, J. W. M. Bulte, P. B. Fisher, H. Wang, A. A. Gilad and M. G. Pomper, Tumor-specific expression and detection of a CEST reporter gene, Magn. Reson. Med., 2015, 74, 544–549 CrossRef CAS PubMed.
- A. A. Gilad, M. T. McMahon, P. Walczak, P. T. Winnard, V. Raman, H. W. M. van Laarhoven, C. M. Skoglund, J. W. M. Bulte and P. C. M. van Zijl, Artificial reporter gene providing MRI contrast based on proton exchange, Nat. Biotechnol., 2007, 25, 217–219 CrossRef CAS PubMed.
- H. Allouche-Arnon, O. Khersonsky, N. D. Tirukoti, Y. Peleg, O. Dym, S. Albeck, A. Brandis, T. Mehlman, L. Avram, T. Harris, N. N. Yadav, S. J. Fleishman and A. Bar-Shir, Computationally designed dual-color MRI reporters for noninvasive imaging of transgene expression, Nat. Biotechnol., 2022, 40, 1143–1149 CrossRef CAS PubMed.
- G. J. Lu, A. Farhadi, J. O. Szablowski, A. Lee-Gosselin, S. R. Barnes, A. Lakshmanan, R. W. Bourdeau and M. G. Shapiro, Acoustically modulated magnetic resonance imaging of gas-filled protein nanostructures, Nat. Mater., 2018, 17, 456–463 CrossRef CAS PubMed.
- T. B. Faust, K. A. Donovan, H. Yue, P. P. Chamberlain and E. S. Fischer, Small-Molecule Approaches to Targeted Protein Degradation, Annual Review of Cancer Biology, 2021, 5, 181–201 CrossRef.
- J. Trauth, J. Scheffer, S. Hasenjäger and C. Taxis, Synthetic Control of Protein Degradation during Cell Proliferation and Developmental Processes, ACS Omega, 2019, 4, 2766–2778 CrossRef CAS.
- D. P. Bondeson and C. M. Crews, Targeted Protein Degradation by Small Molecules, Annu. Rev. Pharmacol. Toxicol., 2017, 57, 107–123 CrossRef CAS PubMed.
- L. A. Banaszynski, L. Chen, L. A. Maynard-Smith, A. G. L. Ooi, T. J. Wandless and A. Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules, Cell, 2006, 126, 995–1004 CrossRef CAS PubMed.
- M. Iwamoto, T. Björklund, C. Lundberg, D. Kirik and T. J. Wandless, A general chemical method to regulate protein stability in the mammalian central nervous system, Chem. Biol., 2010, 17, 981–988 CrossRef CAS PubMed.
- Y. Miyazaki, H. Imoto, L. Chen and T. J. Wandless, Destabilizing Domains Derived from the Human Estrogen Receptor, J. Am. Chem. Soc., 2012, 134, 3942–3945 CrossRef CAS PubMed.
- H. D. Lau, J. Yaegashi, B. W. Zaro and M. R. Pratt, Precise Control of Protein Concentration in Living Cells, Angew. Chem., Int. Ed., 2010, 49, 8458–8461 CrossRef CAS PubMed.
- V. Koduri, S. K. McBrayer, E. Liberzon, A. C. Wang, K. J. Briggs, H. Cho and W. G. Kaelin, Peptidic degron for IMiD-induced degradation of heterologous proteins, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2539–2544 CrossRef CAS PubMed.
- S. Li, X. Prasanna, V. T. Salo, I. Vattulainen and E. Ikonen, An efficient auxin-inducible degron system with low basal degradation in human cells, Nat. Methods, 2019, 16, 866–869 CrossRef CAS PubMed.
- K. Nishimura, T. Fukagawa, H. Takisawa, T. Kakimoto and M. Kanemaki, An auxin-based degron system for the rapid depletion of proteins in nonplant cells, Nat. Methods, 2009, 6, 917–922 CrossRef CAS PubMed.
- H. K. Chung, C. L. Jacobs, Y. Huo, J. Yang, S. A. Krumm, R. K. Plemper, R. Y. Tsien and M. Z. Lin, Tunable and reversible drug control of protein production via a self-excising degron, Nat. Chem. Biol., 2015, 11, 713–720 CrossRef CAS PubMed.
- B. Nabet, J. M. Roberts, D. L. Buckley, J. Paulk, S. Dastjerdi, A. Yang, A. L. Leggett, M. A. Erb, M. A. Lawlor, A. Souza, T. G. Scott, S. Vittori, J. A. Perry, J. Qi, G. E. Winter, K.-K. Wong, N. S. Gray and J. E. Bradner, The dTAG system for immediate and target-specific protein degradation, Nat. Chem. Biol., 2018, 14, 431–441 CrossRef CAS PubMed.
- K. M. Bonger, L. Chen, C. W. Liu and T. J. Wandless, Small-molecule displacement of a cryptic degron causes conditional protein degradation, Nat. Chem. Biol., 2011, 7, 531–537 CrossRef CAS PubMed.
- A. C. Lai and C. M. Crews, Induced protein degradation: an emerging drug discovery paradigm, Nat. Rev. Drug Discovery, 2017, 16, 101–114 CrossRef CAS PubMed.
- E. P. Tague, H. L. Dotson, S. N. Tunney, D. C. Sloas and J. T. Ngo, Chemogenetic control of gene expression and cell signaling with antiviral drugs, Nat. Methods, 2018, 15, 519–522 CrossRef CAS PubMed.
- E. P. Tague, J. B. McMahan, N. Tague, M. J. Dunlop and J. T. Ngo, Controlled Protein Activities with Viral Proteases, Antiviral Peptides, and Antiviral Drugs, ACS Chem. Biol., 2023, 18(5), 1228–1236 CrossRef CAS PubMed.
- H.-S. Li, D. V. Israni, K. A. Gagnon, K. A. Gan, M. H. Raymond, J. D. Sander, K. T. Roybal, J. K. Joung, W. W. Wong and A. S. Khalil, Multidimensional control of therapeutic human cell function with synthetic gene circuits, Science, 2022, 378, 1227–1234 CrossRef CAS PubMed.
- H.-S. Li, N. M. Wong, E. Tague, J. T. Ngo, A. S. Khalil and W. W. Wong, High-performance multiplex drug-gated CAR circuits, Cancer Cell, 2022, 40, 1294–1305.e4 CrossRef CAS PubMed.
- C. Grohmann, C. M. Magtoto, J. R. Walker, N. K. Chua, A. Gabrielyan, M. Hall, S. A. Cobbold, S. Mieruszynski, M. Brzozowski, D. S. Simpson, H. Dong, B. Dorizzi, A. V. Jacobsen, E. Morrish, N. Silke, J. M. Murphy, J. K. Heath, A. Testa, C. Maniaci, A. Ciulli, G. Lessene, J. Silke and R. Feltham, Development of NanoLuc-targeting protein degraders and a universal reporter system to benchmark tag-targeted degradation platforms, Nat. Commun., 2022, 13, 2073 CrossRef CAS PubMed.
- T. Kubota, K. Nishimura, M. T. Kanemaki and A. D. Donaldson, The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication, Mol. Cell, 2013, 50, 273–280 CrossRef CAS PubMed.
- L. A. Banaszynski, M. A. Sellmyer, C. H. Contag, T. J. Wandless and S. H. Thorne, Chemical control of protein stability and function in living mice, Nat. Med., 2008, 14, 1123–1127 CrossRef CAS PubMed.
- L. G. Reijmers, B. L. Perkins, N. Matsuo and M. Mayford, Localization of a Stable Neural Correlate of Associative Memory, Science, 2007, 317, 1230–1233 CrossRef CAS PubMed.
- L. A. DeNardo, C. D. Liu, W. E. Allen, E. L. Adams, D. Friedmann, L. Fu, C. J. Guenthner, M. Tessier-Lavigne and L. Luo, Temporal Evolution of Cortical Ensembles Promoting Remote Memory Retrieval, Nat. Neurosci., 2019, 22, 460–469 CrossRef CAS PubMed.
- C. J. Guenthner, K. Miyamichi, H. H. Yang, H. C. Heller and L. Luo, Permanent Genetic Access to Transiently Active Neurons via TRAP: Targeted Recombination in Active Populations, Neuron, 2013, 78, 773–784 CrossRef CAS PubMed.
- L. DeNardo and L. Luo, Genetic strategies to access activated neurons, Curr. Opin. Neurobiol., 2017, 45, 121–129 CrossRef CAS PubMed.
- N. V. Barykina, M. M. Karasev, V. V. Verkhusha and D. M. Shcherbakova, Technologies for large-scale mapping of functional neural circuits active during a user-defined time window, Prog. Neurobiol., 2022, 216, 102290 CrossRef CAS PubMed.
- M. A. Moroz, J. Zurita, A. Moroz, E. Nikolov, Y. Likar, K. Dobrenkov, J. Lee, L. Shenker, R. Blasberg, I. Serganova and V. Ponomarev, Introducing a new reporter gene, membrane-anchored Cypridina luciferase, for multiplex bioluminescence imaging, Mol. Ther.–Oncolytics, 2021, 21, 15–22 CrossRef CAS PubMed.
- C. K. Brennan, Z. Yao, A. A. Ionkina, C. M. Rathbun, B. Sathishkumar and J. A. Prescher, Multiplexed bioluminescence imaging with a substrate unmixing platform, Cell Chem. Biol., 2022, 29, 1649–1660.e4 CrossRef CAS PubMed.
- C. M. Rathbun, A. A. Ionkina, Z. Yao, K. A. Jones, W. B. Porterfield and J. A. Prescher, Rapid Multicomponent Bioluminescence Imaging via Substrate Unmixing, ACS Chem. Biol., 2021, 16, 682–690 CrossRef CAS PubMed.
- K. Lohith, S. Kwon, P. Padakanti, S. Thanna, C. Hou, C.-C. Weng, M. Sellmyer, R. Mach and M. Farwell, Radiosynthesis and in vitro evaluation of [11C]Shield-1, a PET probe for imaging CAR T cells that express the iCasp9 suicide gene, J. Nucl. Med., 2018, 59, 61 Search PubMed.
- M. A. Sellmyer, S. H. Thorne, L. A. Banaszynski, C. H. Contag and T. J. Wandless, A General Method for Conditional Regulation of Protein Stability in Living Animals, Cold Spring Harb Protoc, 2009, pdb.prot5173 CrossRef PubMed.
- R. Sa, W. Lc, M. Ek, K. Ur, A. Sm and M. Mc, Ligand-Induced Degradation of a CAR Permits Reversible Remote Control of CAR T Cell Activity In Vitro and In Vivo, Mol. Ther., 2020, 28, 1600–1613 CrossRef PubMed.
- E. Auffenberg, A. Jurik, C. Mattusch, R. Stoffel, A. Genewsky, C. Namendorf, R. M. Schmid, G. Rammes, M. Biel, M. Uhr, S. Moosmang, S. Michalakis, C. T. Wotjak and C. K. Thoeringer, Remote and reversible inhibition of neurons and circuits by small molecule induced potassium channel stabilization, Sci. Rep., 2016, 6, 19293 CrossRef CAS PubMed.
- L. He, R. Binari, J. Huang, J. Falo-Sanjuan and N. Perrimon, In vivo study of gene expression with an enhanced dual-color fluorescent transcriptional timer, eLife, 2019, 8, e46181 CrossRef PubMed.
- G. A. Johnson, Y. Tian, D. G. Ashbrook, G. P. Cofer, J. J. Cook, J. C. Gee, A. Hall, K. Hornburg, Y. Qi, F.-C. Yeh, N. Wang, L. E. White and R. W. Williams, Merged magnetic resonance and light sheet microscopy of the whole mouse brain, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2218617120 CrossRef CAS PubMed.
- R. C. Hurt, M. T. Buss, M. Duan, K. Wong, M. Y. You, D. P. Sawyer, M. B. Swift, P. Dutka, P. Barturen-Larrea, D. R. Mittelstein, Z. Jin, M. H. Abedi, A. Farhadi, R. Deshpande and M. G. Shapiro, Genomically mined acoustic reporter genes for real-time in vivo monitoring of tumors and tumor-homing bacteria, Nat. Biotechnol., 2023, 1–13, 919–931 CrossRef PubMed.
- S. Ghosh, N. Li, M. Schwalm, B. B. Bartelle, T. Xie, J. I. Daher, U. D. Singh, K. Xie, N. DiNapoli, N. B. Evans, K. Chung and A. Jasanoff, Functional dissection of neural circuitry using a genetic reporter for fMRI, Nat. Neurosci., 2022, 25, 390–398 CrossRef CAS PubMed.
- H. M. De Feyter, K. L. Behar, Z. A. Corbin, R. K. Fulbright, P. B. Brown, S. McIntyre, T. W. Nixon, D. L. Rothman and R. A. de Graaf, Deuterium metabolic imaging (DMI) for MRI-based 3D mapping of metabolism in vivo, Sci. Adv., 2018, 4, eaat7314 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00377b |
| ‡ Denotes equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |