
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEasy access to polyhalogenated biaryls: regioselective (di)halogenation of hypervalent bromines and chlorines†
Daniel
Carter Martos
 a,
Maxime
de Abreu
a,
Pascal
Hauk
a,
Philipp
Fackler
b and
Joanna
Wencel-Delord‡
*a
a,
Maxime
de Abreu
a,
Pascal
Hauk
a,
Philipp
Fackler
b and
Joanna
Wencel-Delord‡
*a
aLaboratoire d'Innovation Moléculaire et Applications (LIMA, UMR CNRS 7042), Université de Strasbourg/Université de Haute Alsace, ECPM, 67087 Strasbourg, France. E-mail: wenceldelord@unistra.fr
bMerck Electronics KGaA, 64293 Darmstadt, Germany
First published on 9th April 2024
Abstract
Polyhalogenated biaryls are unique motifs offering untapped potential as versatile building blocks for the expedient synthesis of complex biaryl compounds. Overcoming the limitations of conventional syntheses, we introduce a novel, metal-free, operationally simple and one-pot approach to regioselectively (di)halogenate biaryl compounds under mild conditions using cyclic biaryl hypervalent bromine and chlorine substrates as masked arynes. Through chemoselective post-functionalizations, these valuable products can expand the toolbox for synthesizing biaryl-containing scaffolds, addressing a critical gap in the field.
Introduction
Polyhalogenated biaryls are valuable motifs with applications in many fields such as agrochemicals,1 pharmaceuticals,2 natural products,3 and organic materials4 (Scheme 1A). Moreover, polyhalogenated biaryls are uniquely promising as privileged building blocks for the assembly of complex biaryl structures through well-established chemoselective modifications, streamlining the synthesis of value-added compounds.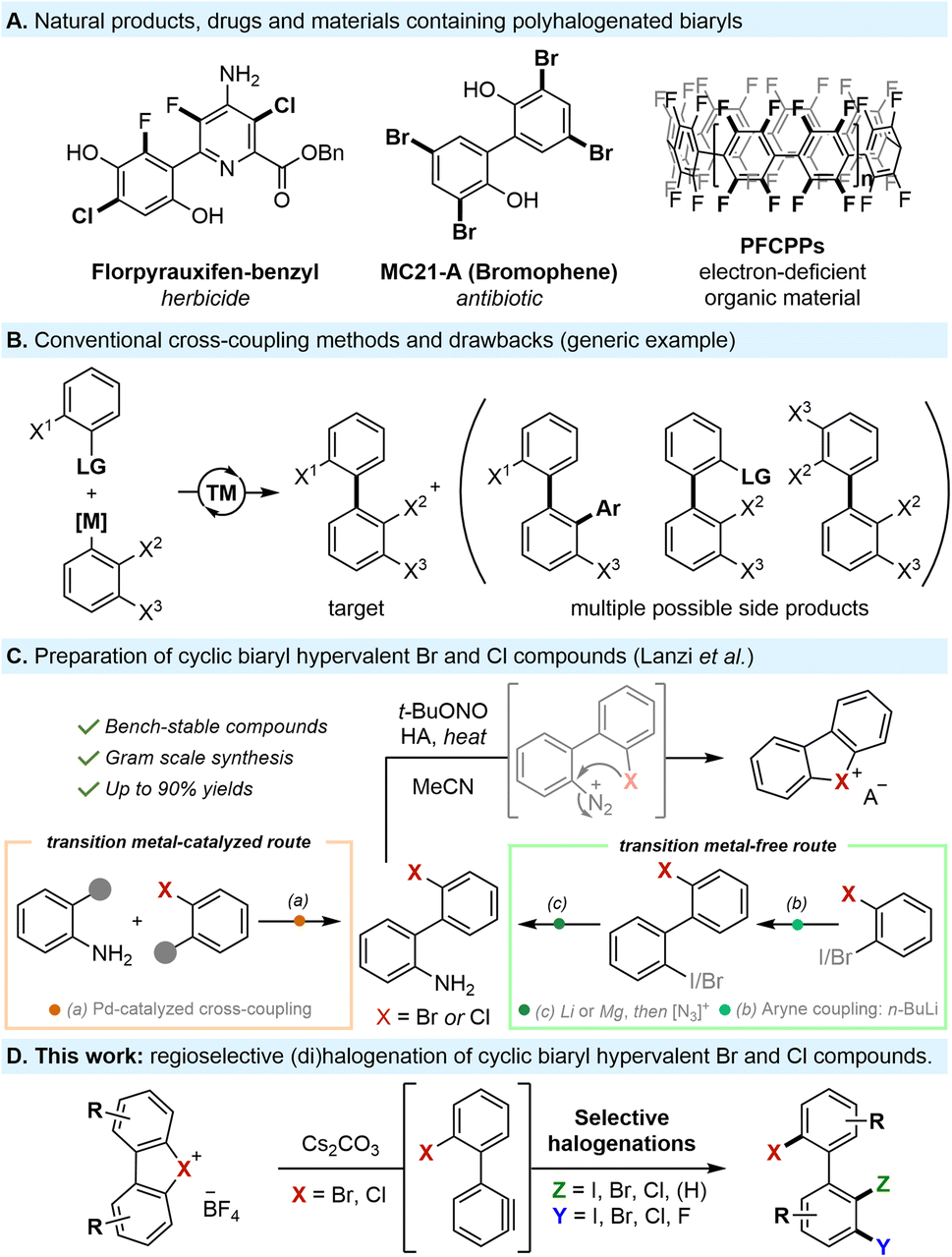 | ||
| Scheme 1 Importance and synthesis of polyhalogenated biaryl compounds. | ||
Despite their importance, constructing polyhalogenated biaryls remains a significant challenge. Conventional transition metal-catalyzed cross-couplings are, by far, the most common approach to prepare such compounds. However, not only do these methods require harsh conditions, expensive catalysts, and pre-installed functionalities, but they typically face significant chemo- and regioselectivity issues when multiple halogens are present, leading to undesired side products, over-functionalizations and polymerizations (Scheme 1B).5
Attempts to tackle these challenges through late-stage halogenations, such as Sandmeyer reactions,6 pyrylium C–N activations,7 electrophilic aromatic substitutions (SEAr),8 or C–H functionalizations,9,10 have their own distinct disadvantages and result in extended and complex synthetic sequences. Alternative approaches to construct polyhalogenated biaryls using lithium bases have also been explored, but are frequently substrate-specific and utilize harsh conditions or hazardous reagents.11
Notably, the aforementioned methods are incompatible with the synthesis of biaryls bearing highly reactive iodine and bromine substituents at specific positions. As a result, numerous polyhalogenated biaryls are commercially unavailable, prohibitively expensive, or yet undiscovered. This highlights the need for more innovative, versatile and efficient protocols to selectively introduce halogen motifs on biaryl scaffolds.
Recently, our group has delved into investigating the chemistry of bench-stable cyclic biaryl hypervalent bromine and chlorine reagents, which remain rare and poorly explored. These unusual reagents, which are safe to handle and easy to prepare (Scheme 1C), undergo unexpected metal-free cycloadditions and meta-functionalizations with C-, O- and N-nucleophiles though in situ aryne formation at room temperature.12
Based on this, we hypothesized that harnessing the unique reactivity of such hypervalent bromine and chlorine compounds as masked biaryl arynes provides a conceptually different strategy to prepare a wide array of polyhalogenated biaryl compounds in a mild and straightforward manner.
It is worth noting that several copper-catalyzed methodologies for the halogenation of analogous cyclic diaryliodonium salts have been reported.13 Nevertheless, this approach diverges in its mechanism, yielding selectively ortho-functionalized products, while meta-functionalization of hypervalent iodines remains extremely rare and requires high reaction temperatures and drastic conditions.14
In clear contrast, the use of hypervalent bromine and chlorine precursors, which are accessible either through standard Suzuki coupling or metal-free conditions, opens up the possibility of generating biaryl aryne intermediates, which have the potential to undergo vicinal difunctionalization through the concerted addition of both nucleophilic and electrophilic species under extremely mild reaction conditions (Scheme 1C). Such a double functionalization presents however an important challenge. Indeed, while 1,2-difunctionalization of arynes is widely reported, mainly through cycloadditions or reactions with ambiphilic reagents,15 versatile 3-component strategies remain uncommon.16 Furthermore, to the best of our knowledge, the 1,2-dihalogenation of arynes is mostly limited to diiodo, dibromo or dichlorinations, and is scarcely reported with mixed halogens.17 Moreover, the electrophilic intermolecular trapping of arynes originating from cyclic diaryl-bromonium or chloronium salts remains unaccomplished.18 In this work, we report the first example of a concerted nucleophilic and electrophilic intermolecular 1,2-difunctionalization of our biaryl aryne precursors, allowing the direct, mild and metal-free installation of customized halogen motifs in a fully regioselective manner (Scheme 1D).
Results and discussion
Meta-selective monohalogenations
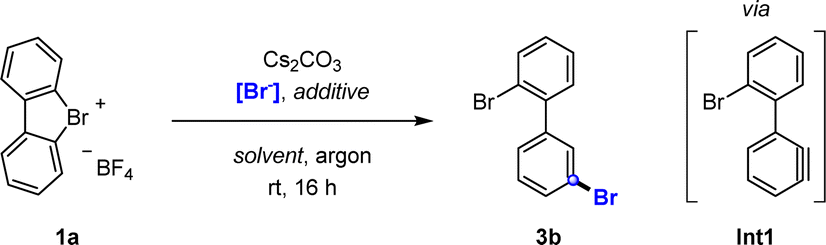
Our investigation began with the bromination of hypervalent compound 1a to synthesize product 3b. Addition of Cs2CO3 as mild base prompted simultaneous deprotonation and ring opening of 1a to generate aryne intermediate Int1. In situ attack of Int1 with a nucleophilic bromide source should thus achieve the targeted meta-selective bromination.Preliminary experiments using alkali metal bromides (KBr, NaBr, and LiBr) showed promising results, particularly when employed alongside the corresponding crown ether additives (Table 1, entries 1–4). Increased temperature did not lead to higher yields (entry 5), and swapping solvents (entries 6, and 7) or reagents (entry 8, and 9) led to varying results, frequently producing mixtures of meta and ortho bromination. Remarkably, the most favourable outcomes were obtained using tetra-n-butylammonium bromide (TBAB), furnishing the desired product in quantitative yields and full meta![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ortho selectivities (entries 11–13).
:ortho selectivities (entries 11–13).
|
|
||||
|---|---|---|---|---|
| Entry | [Br−] | Additive | Solvent | Yield, %b |
| a Standard conditions: 1a (0.10 mmol), Cs2CO3 (2.0 equiv.), [Br−] nucleophile (2.0 equiv.), additive (2.0 equiv.), solvent (2.0 mL), argon, room temperature, 16 h. b Yields determined by 1H NMR analysis using CH2Br2 as internal standard; isolated yields in parentheses. c 10 equiv. KBr. d 40 °C. e Undesired ortho-bromination. | ||||
| 1 | KBr | — | DCM | (42)/(54)c |
| 2 | KBr | 18-Crown-6 | DCM | (71) |
| 3 | NaBr | 15-Crown-5 | DCM | 56 |
| 4 | LiBr | 12-Crown-4 | DCM | 99 (87) |
| 5 | LiBr | — | DCM | 67/43d |
| 6 | LiBr | — | MeCN | 99 (77) |
| 7 | LiBr | — | MeCN/H2O 1:1 |
40 + 40e |
| 8 | NH4Br | — | DCM/H2O 1:1 |
3 + 34e |
| 9 | TBAB | — | DCM | 30 + 24e |
| 10 | TBAB | — | MeCN | 72 |
| 11 | TBAB | — | MeCN/H2O 1:1 |
99 (99) |
| 12 | TBAB | — | THF | 99 (99) |
| 13 | TBAB | — | AcOEt | 99 |
To broaden the generality of this methodology, other halogenation reactions, namely iodination, chlorination and fluorination, were subsequently investigated. Re-evaluation of the most promising conditions in Table 1 showed that tetra-n-butylammonium halide salts in THF provided the optimal global reaction conditions for all types of halogenations (see ESI†).
With the optimized conditions in hand, the scope of this transformation was investigated (Scheme 2). We initially explored all possible halogen permutations, achieving quantitative yields for most combinations except fluorination products 3d and 4d (82% and 88%, respectively), and iodination product 4a (50%). Electron donating substituents, including methyl (3e, 4e), methoxy (3h), and tert-butyl (3j), afforded the corresponding products in moderate to excellent yields (49–90%). Likewise, electron withdrawing groups such as –CF3 (3k) and –CO2Me (3l) led to good yields, and push–pull type structures (3f, 3g, 3i) yielded the respective halogenations in moderate to high yields (55–92%).
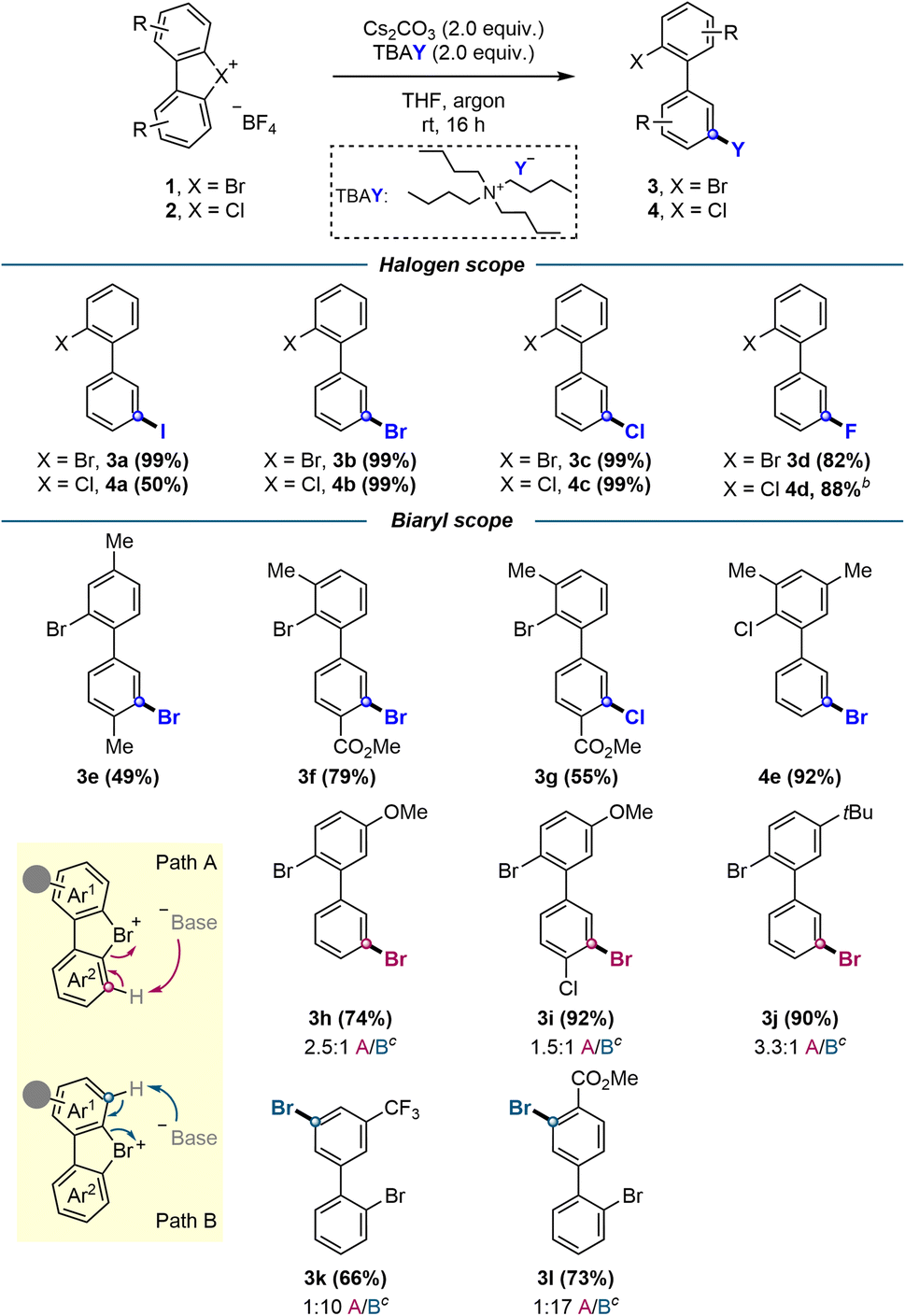 | ||
| Scheme 2 Scope of meta-selective halogenations. aStandard conditions: hypervalent bromine 1 or chlorine 2 (0.10 mmol), Cs2CO3 (2.0 equiv.), tetra-n-butylammonium halide TBAY (2.0 equiv.), THF (2.0 mL), argon, room temperature, 16 h. Isolated yields are reported unless otherwise stated. bYield determined by 1H NMR analysis using CH2Br2 as internal standard. Volatile product, isolated yield: 30%. cThe isolated yield encompasses the total yield of both isomers, irrespective of their regiochemical pathway A/B. The subsequent ratio indicates the proportion of this total obtained through pathway A relative to pathway B, and was determined via1H NMR analysis. | ||
While the nucleophilic attack on the aryne occurs selectively at the meta position, the initial deprotonation and generation of the aryne species itself is less regioselective. In non-symmetrical substrates where the aryne formation was possible on both aromatic rings, mixtures of regioisomers resulted from their subsequent meta-selective halogenations. Despite this, a preference towards deprotonation and the resulting halogenation on the most electron-poor aromatic ring was observed (Scheme 2, path A vs. path B, compounds 3h–l).
Extension to 1,2-dihalogenations
With the monohalogenation conditions established, we aimed to further expand the methodology to 1,2-dihalogenations by treating the aryne intermediate with both nucleophilic and electrophilic halogen sources in a one-pot procedure. We hypothesized that a meta-selective nucleophilic attack on the aryne moiety would generate a highly reactive aryl carbanion species on the adjacent ortho position, which, under appropriate conditions, could be trapped by suitable halogen electrophiles (Scheme 3). It must be highlighted that, for the monohalogenation protocol, this reactive carbanion is likely quenched by deprotonating another molecule of starting material in an autocatalytic fashion.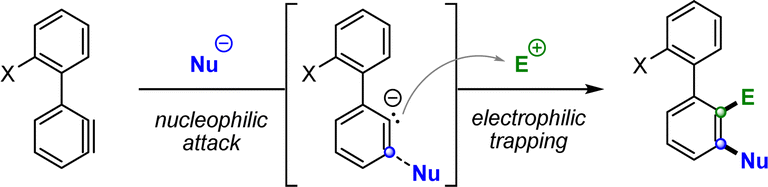 | ||
| Scheme 3 Predicted aryl carbanion species and subsequent electrophilic trapping. | ||
To develop such a rare dihalogenation protocol, we began by screening various electrophilic iodine reagents alongside the optimized monobromination conditions of TBAB in THF (Table 2). Initial attempts yielded limited success with common iodine electrophiles such as NIS, I2 or I–Cl (entries 1–5). However, the use of nonafluoro-1-iodobutane resulted in a comparatively remarkable outcome, furnishing the desired dihalogenated biaryl 5b in 86% yield (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Z | [Z+] (equiv.) | T, °C | Yieldb, % |
| a Standard conditions: 1a (0.10 mmol), Cs2CO3 (2.0 equiv.), TBAB (2.0 equiv.), [Z+] electrophile, THF (2.0 mL), argon, Temperature, 16 h. b Yields determined by 1H NMR analysis using CH2Br2 as internal standard; isolated yields in parentheses. c No reaction. | ||||
| 1 | I (5b) | I2 (1.2) | rt | < 5 |
| 2 | NIS (3.0) | rt | < 5 | |
| 3 | I–Cl (3.0) | rt | < 5 | |
| 4 | CHI3 (3.0) | rt | nrc | |
| 5 | nC6F13–I (3.0) | rt | nr | |
| 6 | nC 4 F 9 –I (5.0) | rt | (86) | |
| 7 | Br (5e) | CBr4 (5.0) | rt | < 5 |
| 8 | NBS (5.0) | rt | < 5 | |
| 9 | nC6F13–Br (5.0) | rt | 6 | |
| 10 | C6F5–Br (20) | rt | 17 | |
| 11 | nC8F17–Br (15) | 50 | (16) | |
| 12 | C 6 F 5 –Br (15) | 50 | (91) | |
| 13 | Cl (5h) | CCl4 (5.0) | rt | < 5 |
| 14 | CCl4 (1.0 mL) | rt | nr | |
| 15 | NCS (5.0) | rt | nr | |
| 16 | C6F5–Cl (5.0) | rt | < 5 | |
| 17 | C6F5–Cl (15) | 50 | (30) | |
| 18 | CCl 4 (15) | 50 | (53) | |
Motivated by this breakthrough, we set out to expand the protocol to bromane and chlorane reagents. However, the screening of Br and Cl electrophiles proved more challenging due to their decreased reactivity compared to their iodine counterparts. While some modest improvements were made using perfluoroalkyl and perfluoroaryl halides as the electrophile sources, the yields remained low (Table 2, entries 9, 10 and 16). Ultimately, temperature was found to be a key factor, as the best results for trapping with Br (entry 12), and Cl (entry 18), were observed when performing the reaction at 50 °C.
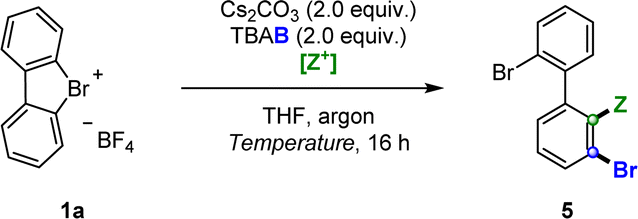
Having established the conditions for the regioselective dihalogenations, the scope of the reaction was surveyed for both hypervalent bromine and chlorine substrates (Scheme 4). Biaryls containing halogens in several different arrangements were synthesized in moderate to excellent yields, including ortho-iodinated products 5a–d and 6a–b (45–98%), ortho-brominated products 5e–f (88–91%), and ortho-chlorinated products 5g–h and 6c (53–74%). We likewise examined the compatibility of the procedure with electron donating groups such as –Me (5j, 5k, 6d) and –OMe (6e); and electron withdrawing groups such as –CO2Me 5l, all of which resulted in modest to high yields (56–75%). We were also able to dihalogenate binaphthyl-derived substrate 5i in 69% yield, a particularly interesting result given the potential of such compounds as building blocks for non-C2 symmetrical privileged ligands. Crucially, the potential for this methodology to be extended to other electrophiles was demonstrated via the electrophilic trapping with MeOD to furnish deuterated product 5m in 87% yield and 92% deuterium incorporation. Furthermore, compound 5m offered additional proof that our electrophiles were indeed functionalizing the desired ortho position. X-ray structures were also obtained for selected dihalogenated products, unequivocally confirming the predicted regioselectivity of this transformation.
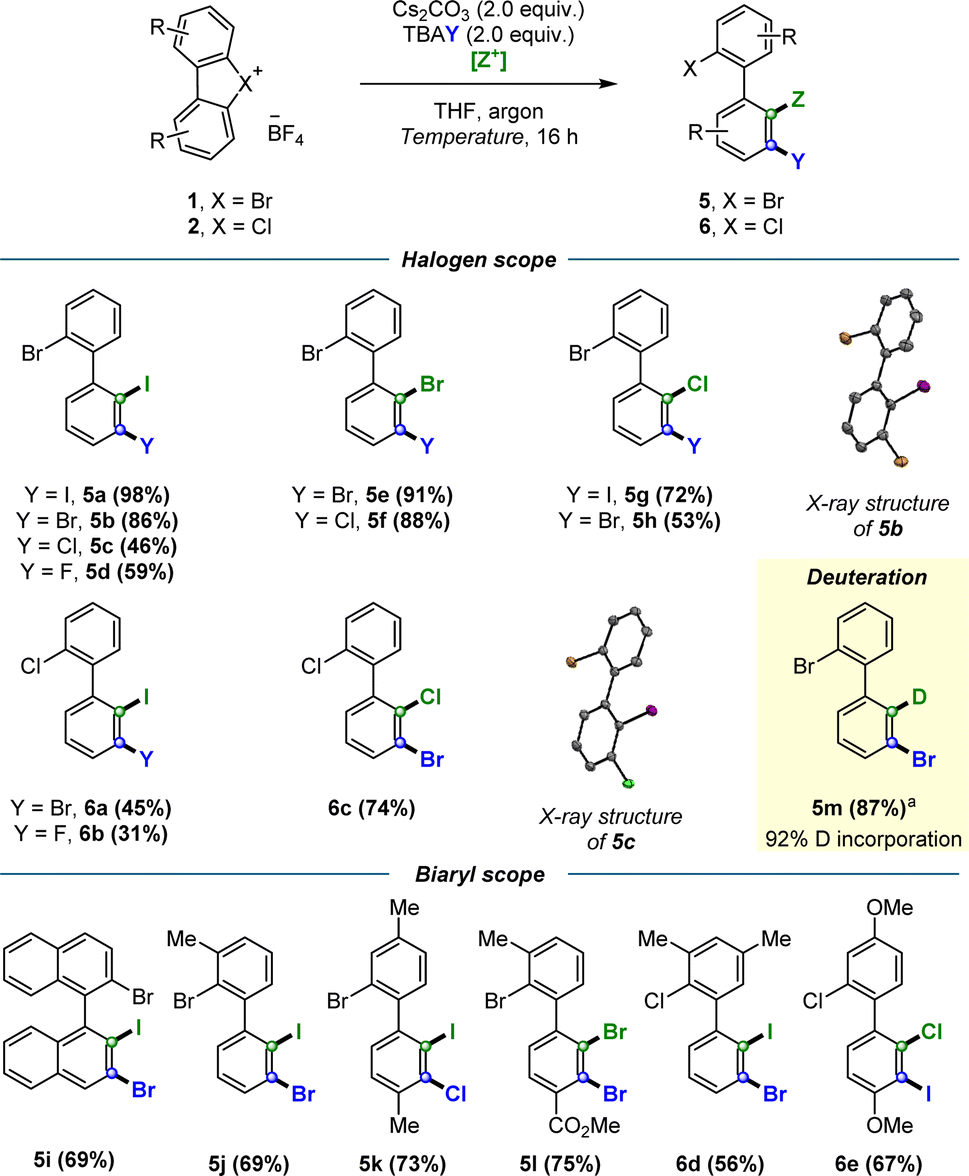 | ||
Scheme 4 Scope of the regioselective dihalogenations. Standard conditions: hypervalent bromine 1 or chlorine 2 (0.10 mmol), Cs2CO3 (2 equiv.), tetra-n-butylammonium halide TBAY (2.0 equiv.), [Z+] electrophile [Z![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) I: nonafluoro-1-iodobutane nC4F9–I (5.0 equiv.); ZBr: bromopentafluorobenzene C6F5–Br (15 equiv.); ZCl: carbon tetrachloride CCl4 (15 equiv.)], THF (2.0 mL), argon, temperature [ZI: room temperature, ZBr/Cl: 50 °C], 16 h. Isolated yields are reported unless otherwise stated. aFollowing standard conditions, electrophile: MeOD (30 equiv.), temperature: 50 °C. I: nonafluoro-1-iodobutane nC4F9–I (5.0 equiv.); ZBr: bromopentafluorobenzene C6F5–Br (15 equiv.); ZCl: carbon tetrachloride CCl4 (15 equiv.)], THF (2.0 mL), argon, temperature [ZI: room temperature, ZBr/Cl: 50 °C], 16 h. Isolated yields are reported unless otherwise stated. aFollowing standard conditions, electrophile: MeOD (30 equiv.), temperature: 50 °C. | ||
Chemoselective post-functionalizations
The potential of 5 and 6 as new and unique building blocks was then investigated via chemoselective and sequential post-functionalizations on products 5c and 5g (Scheme 5).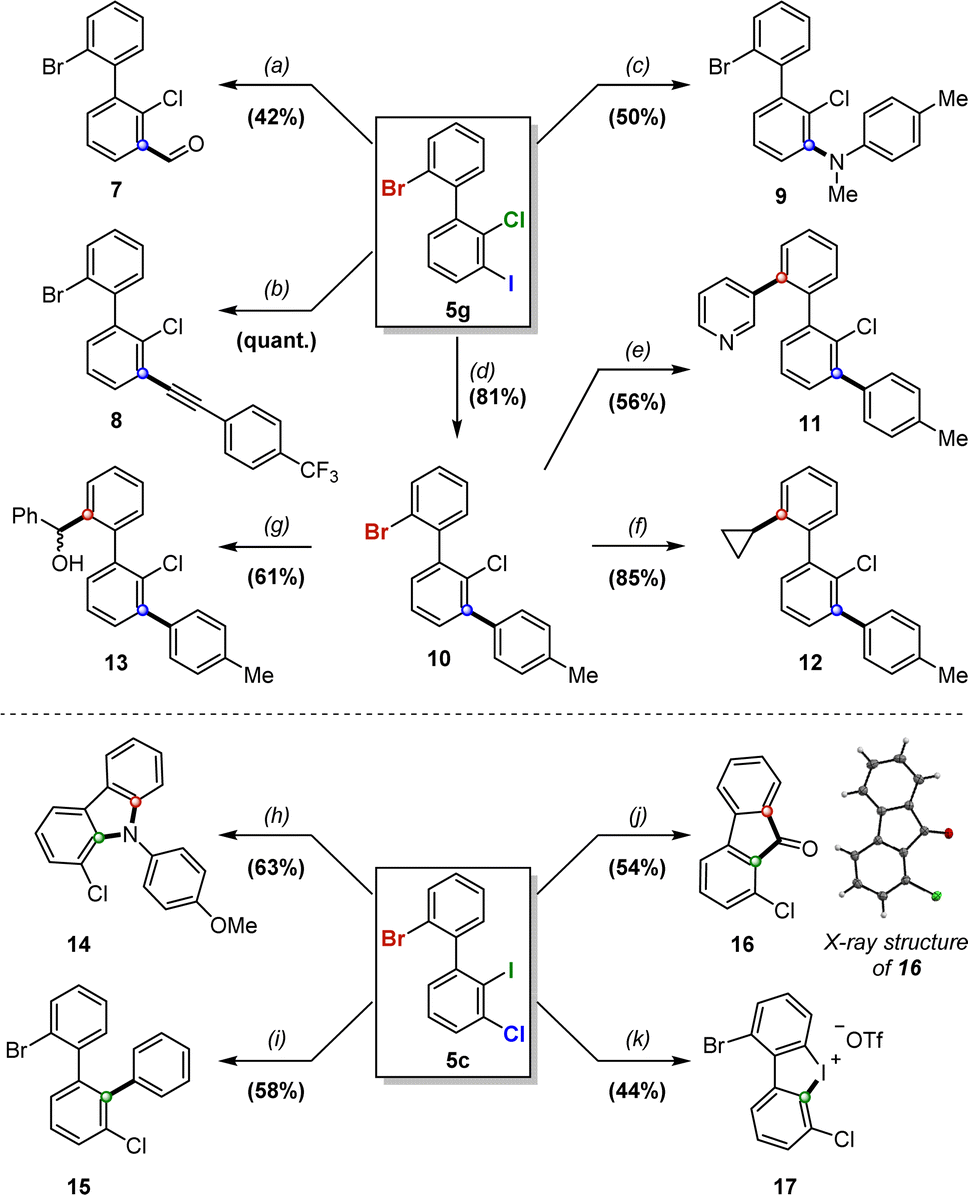 | ||
| Scheme 5 Chemoselective post-functionalizations of 5g and 5c. Conditions: (a) i-PrMgCl, THF, 0 °C then DMF, 0 °C to rt. (b) p-(trifluoromethyl)phenylacetylene, Pd(PPh3)2Cl2, K2CO3, CuI, DME/H2O, 100 °C. (c) p-TolNHMe, Pd(OAc)2, DPEphos, NaOtBu, toluene, 100 °C. (d) p-TolB(OH)2, Pd(PPh3)2Cl2, K2CO3, DME/H2O, 100 °C. (e) 3-pyridylB(OH)2, Pd(PPh3)4, K2CO3, dioxane/H2O, 100 °C. (f) cyclopropylB(OH)2, Pd(OAc)2, PCy3, K3PO4, toluene/H2O, 100 °C. (g) n-BuLi, THF, −78 °C then PhCHO, −78 °C to rt. (h) p-anisidine, Pd(dppf)Cl2, NaOtBu, dioxane, 120 °C. (i) PhB(OH)2, Pd(OAc)2, PPh3, K2CO3, THF/H2O, 80 °C. (j) t-BuLi, THF, −78 °C then DMF, −30 °C. (k) m-CPBA, TfOH, DCM, 0 °C to rt. | ||
Compound 5g was first subjected to several iodine-selective transformations. Grignard formation with i-PrMgCl followed by treatment with DMF furnished benzaldehyde 7 in moderate but non-optimized 42% yield, and Pd-catalyzed Sonogashira, Buchwald-Hartwig and Suzuki–Miyaura cross-couplings yielded the respective functionalized biaryls 8–10 in moderate to quantitative yields, with little to no presence of over-functionalization on bromine.
Selective functionalization on bromine over chlorine can also be achieved, as illustrated with compound 10via Suzuki–Miyaura cross-couplings with 3-pyridylboronic acid and cyclopropylboronic acid, affording products 11 and 12 respectively in moderate to high yields. Additionally, selective lithium–bromine exchange on 10 was realized, followed by trapping with benzaldehyde, which furnished atropostable biaryl 13 in 61% yield as a mixture of atropodiastereomers.
Compound 5c proved more challenging to functionalize selectively due to the increased steric hindrance about the biaryl axis. Buchwald–Hartwig amination using p-anisidine led to substituted carbazole 14 in 63% yield as a single product, while Suzuki–Miyaura arylation successfully furnished 15 in 58% yield. Interestingly, fluorene derivative 16 was obtained in 54% yield by treatment of 5c with tert-butyllithium, followed by addition of DMF, and its structure was confirmed by X-ray analysis. Finally, we were also able to achieve a selective oxidation on iodine to form appealing cyclic diaryliodinium compound 17 in a considerable 44% yield, which could serve as a unique platform for subsequent transformations.
Conclusions
In conclusion, we have developed a mild, metal-free and highly efficient regioselective (di)halogenation procedure by harnessing the reactivity of cyclic biaryl hypervalent bromine and chlorine compounds. By integrating mild base-mediated aryne formation with in situ nucleophilic attack, employing readily available and environmentally benign tetra-n-butylammonium halides, we successfully furnished a wide variety of meta-halogenated 2-chloro- and 2-bromobiaryls in up to quantitative yields at room temperature. More significantly, we expanded the scope of our methodology to vicinal dihalogenations by achieving an unprecedented electrophilic intermolecular trapping the reactive biaryl carbanion intermediates, effortlessly preparing a range of rare 2,2′,3′-trihalogenated biaryl compounds with perfect site selectivity in a simple, mild, one-pot process. Through our innovative and conceptually distinct (di)halogenation protocol, we unlock the opportunity to efficiently and flexibly synthesize a diverse library of tailor-made polyhalogenated biaryls stemming from only a handful of common hypervalent compounds as precursors. This approach thus circumvents the need for complex, multi-step de novo syntheses which would be required to produce such a wide variety of rare and valuable compounds. Furthermore, some compounds synthesized utilizing our method may be altogether inaccessible through traditional approaches, underscoring the importance of our protocol in expanding the space of polyhalogenated biaryls. Moreover, the unique potential of these polyhalogenated biaryls as powerful synthetic building blocks was subsequently demonstrated through several chemoselective post-functionalization reactions, thus providing a valuable toolkit for the streamlined synthesis of polyfunctionalized biaryl scaffolds.Data availability
Detailed experimental procedures, characterization data and crystallographic data are available in the ESI.†Author contributions
J. W.-D. and P. F. conceived and directed the project. D. C. M. and M. d. A. developed the method and performed the synthetic experiments. D. C. M and P. H. carried out the post-functionalization studies. D. C. M. and J. W.-D. wrote the manuscript with assistance from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W.-D., D. C. M., P. H. and P. F. thank the European Union's H2020 research and innovation programme for funding this project under the Marie Skłodowska-Curie Grant Agreement No. 860762. This work was financially supported by the CNRS (Centre National de la Recherche Scientifique), the Université de Strasbourg, the Ministère de l'Education Nationale et de la Recherche (France) and Merck Electronics KGaA (Darmstadt, Germany). J. W. D. and M. d. A. are grateful to the European Commision for the ERC-Starting Grant “ALCHIMIE” No. 949804. The authors thank Dr Emeric Wasielewski (NMR service) and Matthieu Chessé (analytical facilities) from LIMA (UMR CNRS 7042); Dr Lydia Karmazin, Dr Nathalie Gruber and Dr Corinne Bailly for X-ray diffraction analyses (Service de Radiocrystallographie, Fédération de Chimie “Le Bel” FR2010, Université de Strasbourg); and Dr Hélène Nierengarten and Dr Stéphanie Coutin for High Resolution Mass Spectrometry analyses (Service de Spectrométrie de Masse, Fédération de Chimie “Le Bel” FR2010, Université de Strasbourg). The authors are also thankful to Prof. Françoise Colobert, Dr Sabine Choppin, Dr Mark M. Maturi and Dr Tomas J. Saiegh for helpful discussions.Notes and references
- Illustrative examples may be found in: P. Jeschke, Eur. J. Org Chem., 2022, 2022, e202101513, DOI:10.1002/ejoc.202101513.
- For selected examples, see: (a) D. Benedetto Tiz, L. Bagnoli, O. Rosati, F. Marini, L. Sancineto and C. Santi, Molecules, 2022, 27, 1643 CrossRef CAS PubMed; (b) Z.-W. Zhang, H. Tu, M. Jiang, S. Vanan, S. Y. Chia, S.-E. Jang, W.-T. Saw, Z.-W. Ong, D.-R. Ma, Z.-D. Zhou, J. Xu, K.-H. Guo, W.-P. Yu, S.-C. Ling, R. A. Margolin, D. G. Chain, L. Zeng and E.-K. Tan, Sci. Signaling, 2022, 15, eabk3411 CrossRef CAS PubMed; (c) Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS PubMed; (d) M. Bruncko, T. K. Oost, B. A. Belli, H. Ding, M. K. Joseph, A. Kunzer, D. Martineau, W. J. McClellan, M. Mitten, S.-C. Ng, P. M. Nimmer, T. Oltersdorf, C.-M. Park, A. M. Petros, A. R. Shoemaker, X. Song, X. Wang, M. D. Wendt, H. Zhang, S. W. Fesik, S. H. Rosenberg and S. W. Elmore, J. Med. Chem., 2007, 50, 641–662 CrossRef CAS PubMed; (e) T. S. Haque, R. L. Martinez, V. G. Lee, D. G. Riexinger, M. Lei, M. Feng, B. Koplowitz, C. Mapelli, C. B. Cooper, G. Zhang, C. Huang, W. R. Ewing and J. Krupinski, Peptides, 2010, 31, 1353–1360 CrossRef CAS PubMed; (f) R. R. Shah, J. M. Redmond, A. Mihut, M. Menon, J. P. Evans, J. A. Murphy, M. A. Bartholomew and D. M. Coe, Bioorg. Med. Chem., 2020, 28, 115326 CrossRef CAS PubMed.
- Selected examples: (a) A. Isnansetyo and Y. Kamei, Antimicrob. Agents Chemother., 2003, 47, 480–488 CrossRef CAS PubMed; (b) S. Adak and B. S. Moore, Nat. Prod. Rep., 2021, 38, 1760–1774 RSC; (c) C. Wagner, M. El Omari and G. M. König, J. Nat. Prod., 2009, 72, 540–553 CrossRef CAS PubMed.
- For illustrative examples, see: (a) M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446–455 CrossRef CAS; (b) B. Park, H. Bae, J.-W. Ha, C. Lee, J. Lee, Y. Heo, B. Kim, S. C. Yoon, H. Choi and S.-J. Ko, Org. Electron., 2023, 113, 106717 CrossRef CAS; (c) H. Shudo, M. Kuwayama, M. Shimasaki, T. Nishihara, Y. Takeda, N. Mitoma, T. Kuwabara, A. Yagi, Y. Segawa and K. Itami, Nat. Commun., 2022, 13, 3713 CrossRef CAS PubMed; (d) Y.-M. Liu, H. Hou, Y.-Z. Zhou, X.-J. Zhao, C. Tang, Y.-Z. Tan and K. Müllen, Nat. Commun., 2018, 9, 1901 CrossRef PubMed; (e) H. Tamaoki, R. Katoono, K. Fujiwara and T. Suzuki, Angew. Chem., Int. Ed., 2016, 55, 2582–2586 CrossRef CAS PubMed; (f) Y. Sakamoto, T. Suzuki, A. Miura, H. Fujikawa, S. Tokito and Y. Taga, J. Am. Chem. Soc., 2000, 122, 1832–1833 CrossRef CAS; (g) S. Komatsu, Y. Sakamoto, T. Suzuki and S. Tokito, J. Solid State Chem., 2002, 168, 470–473 CrossRef CAS; (h) M. Ikai, S. Tokito, Y. Sakamoto, T. Suzuki and Y. Taga, Appl. Phys. Lett., 2001, 79, 156–158 CrossRef CAS; (i) Y. Liu, Y. Huang, S. Liu, D. Chen, C. Tang, Z. Qiu, J. Zhu and Y. Tan, Angew. Chem., Int. Ed., 2019, 58, 13276–13279 CrossRef CAS PubMed; (j) M. Mahl, K. Shoyama, J. Rühe, V. Grande and F. Würthner, Chem.–Eur. J., 2018, 24, 9409–9416 CrossRef CAS PubMed.
- Selected reviews: (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (b) A. Biffis, P. Centomo, A. del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (c) J. Pérez Sestelo and L. A. Sarandeses, Molecules, 2020, 25, 4500 CrossRef PubMed.
- Representative examples: (a) F.-C. Favre-Besse, O. Poirel, T. Bersot, E. Kim-Grellier, S. Daumas, S. El Mestikawy, F. C. Acher and N. Pietrancosta, Eur. J. Med. Chem., 2014, 78, 236–247 CrossRef CAS PubMed; (b) V. Akhmetov, M. Feofanov, S. Troyanov and K. Amsharov, Chem.–Eur. J., 2019, 25, 7607–7612 CrossRef CAS PubMed; (c) K. Kantarod, D. Soorukram, C. Kuhakarn, P. Surawatanawong, W. Wattanathana, V. Reutrakul and P. Leowanawat, Chem. Commun., 2022, 58, 9468–9471 RSC.
- C. Ghiazza, T. Faber, A. Gómez-Palomino and J. Cornella, Nat. Chem., 2022, 14, 78–84 CrossRef CAS PubMed.
- (a) P. B. D. de la Mare, Electrophilic Halogenation: Reaction Pathways Involving Attack by Electrophilic Halogens on Un- Saturated Compounds; Cambridge Chemistry Texts, Cambridge University Press, Cambridge, 1976 Search PubMed; (b) B. Galabov, D. Nalbantova, P. V. R. Schleyer and H. F. Schaefer, Acc. Chem. Res., 2016, 49, 1191–1199 CrossRef CAS PubMed; (c) W. Wang, X. Yang, R. Dai, Z. Yan, J. Wei, X. Dou, X. Qiu, H. Zhang, C. Wang, Y. Liu, S. Song and N. Jiao, J. Am. Chem. Soc., 2022, 144, 13415–13425 CrossRef CAS PubMed.
- Selected reviews on the topic: (a) J. Wang, C. Liu, Q. Zheng and G. Rao, Eur. J. Org Chem., 2020, 2020, 3737–3765 CrossRef CAS; (b) R. Das and M. Kapur, Asian J. Org. Chem., 2018, 7, 1524–1541 CrossRef CAS; (c) D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003–8104 CrossRef CAS PubMed.
- Additional examples: (a) C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871–13875 CrossRef CAS PubMed; (b) S. T. Linde, V. Corti, V. H. Lauridsen, J. N. Lamhauge, K. A. Jørgensen and N. M. Rezayee, Chem. Sci., 2023, 14, 3676–3681 RSC.
- (a) V. Mamane, E. Aubert, P. Peluso and S. Cossu, J. Org. Chem., 2013, 78, 7683–7689 CrossRef CAS PubMed; (b) F. Leroux and M. Schlosser, Angew. Chem., Int. Ed., 2002, 41, 4272–4274 CrossRef CAS; (c) L. Bonnafoux, R. Gramage-Doria, F. Colobert and F. R. Leroux, Chem.–Eur. J., 2011, 17, 11008–11016 CrossRef CAS PubMed; (d) F. R. Leroux, L. Bonnafoux, C. Heiss, F. Colobert and D. A. Lanfranchi, Adv. Synth. Catal., 2007, 349, 2705–2713 CrossRef CAS.
- For benchmark chemistry on synthesis and functionalization of hypervalent bromine and chlorine reagents from our research group, see: (a) M. Lanzi, Q. Dherbassy and J. Wencel-Delord, Angew. Chem., Int. Ed., 2021, 60, 14852–14857 CrossRef CAS PubMed; (b) M. Lanzi, R. A. Ali Abdine, M. De Abreu and J. Wencel-Delord, Org. Lett., 2021, 23, 9047–9052 CrossRef CAS PubMed; (c) M. Lanzi, T. Rogge, T. S. Truong, K. N. Houk and J. Wencel-Delord, J. Am. Chem. Soc., 2023, 145, 345–358 CrossRef CAS PubMed; (d) M. de Abreu, T. Rogge, M. Lanzi, T. Saiegh, K. N. Houk and J. Wencel-Delord, Angew. Chem., Int. Ed., 2024, e202319960, DOI:10.1002/anie.202319960; (e) M. Lanzi and J. Wencel-Delord, Chem. Sci., 2024, 15, 1557–1569 RSC.
- (a) B. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2015, 54, 8736–8739 CrossRef CAS PubMed; (b) J. Ke, B. Zu, Y. Guo, Y. Li and C. He, Org. Lett., 2021, 23, 329–333 CrossRef CAS PubMed; (c) K. Zhu, Z. Song, Y. Wang and F. Zhang, Org. Lett., 2020, 22, 9356–9359 CrossRef CAS PubMed.
- H. Jiang, M. Liu, Z. Ye, Z. Song, Y. Wu and F. Zhang, Org. Chem. Front., 2023, 10, 3856–3860 RSC.
- Selected reviews and articles: (a) X. Yang and G. Chit Tsui, Chem. Sci., 2018, 9, 8871–8875 RSC; (b) A. Nilova, P. A. Sibbald, E. J. Valente, G. A. González-Montiel, H. C. Richardson, K. S. Brown, P. H. Cheong and D. R. Stuart, Chem.–Eur. J., 2021, 27, 7168–7175 CrossRef CAS PubMed; (c) B. Baire, D. Niu, P. H. Willoughby, B. P. Woods and T. R. Hoye, Nat. Protoc., 2013, 8, 501–508 CrossRef CAS PubMed; (d) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3557 CrossRef CAS PubMed; (e) S. Cho and Q. Wang, Org. Lett., 2020, 22, 1670–1674 CrossRef CAS PubMed; (f) J. He, D. Qiu and Y. Li, Acc. Chem. Res., 2020, 53, 508–519 CrossRef CAS PubMed; (g) C. Ni, L. Zhang and J. Hu, J. Org. Chem., 2008, 73, 5699–5713 CrossRef CAS PubMed; (h) D. R. Stuart, Synlett, 2016, 28, 275–279 CrossRef; (i) A. Nilova, B. Metze and D. R. Stuart, Org. Lett., 2021, 23, 4813–4817 CrossRef CAS PubMed; (j) O. Smith, M. J. Hindson, A. Sreenithya, V. Tataru, R. S. Paton, J. W. Burton and M. D. Smith, Nat. Synth., 2024, 3, 58–66 CrossRef.
- Selected examples: (a) J. Mindner, S. Rombach and D. B. Werz, Org. Lett., 2024, 26(10), 2124–2128 CrossRef CAS PubMed; (b) J.-A. García-López, M. Çetin and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 2156–2159 CrossRef PubMed; (c) S. Cho and Q. Wang, Org. Lett., 2020, 22, 1670–1674 CrossRef CAS PubMed; (d) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955–2958 CrossRef CAS PubMed; (e) Y. Zeng and J. Hu, Synthesis, 2016, 48, 2137–2150 CrossRef CAS; (f) J. L. Henderson, A. S. Edwards and M. F. Greaney, J. Am. Chem. Soc., 2006, 128, 7426–7427 CrossRef CAS PubMed; (g) H. Yoshida, Y. Asatsu, Y. Mimura, Y. Ito, J. Ohshita and K. Takaki, Angew. Chem., Int. Ed., 2011, 50, 9676–9679 CrossRef CAS PubMed; (h) C. E. Hendrick, S. L. McDonald and Q. Wang, Org. Lett., 2013, 13, 3444–3447 CrossRef PubMed; (i) K.-P. Wang, S. Y. Yun, P. Mamidipalli and D. Lee, Chem. Sci., 2013, 4, 3205–3211 RSC; (j) S. S. Bhojgude, T. Roy, R. G. Gonnade and A. T. Biju, Org. Lett., 2016, 18, 5424–5427 CrossRef CAS PubMed; (k) C. Ni, L. Zhang and J. Hu, J. Org. Chem., 2008, 73, 5699–5713 CrossRef CAS PubMed.
- Precedents for the 1,2-dihalogenation of arynes: (a) Y. Zeng, G. Li and J. Hu, Angew. Chem., Int. Ed., 2015, 54, 10773–10777 CrossRef CAS PubMed; (b) D. Niu, T. Wang, B. P. Woods and T. R. Hoye, Org. Lett., 2014, 16, 254–257 CrossRef CAS PubMed; (c) L. Friedman and F. M. Logullo, Angew. Chem., Int. Ed. Engl., 1965, 4, 239–240 CrossRef; (d) M. A. Birkett, D. W. Knight, P. B. Little and M. B. Mitchell, Tetrahedron, 2000, 56, 1013 CrossRef CAS; (e) D. Rodríguez-Lojo, A. Cobas, D. Peña, D. Pérez and E. Guitán, Org. Lett., 2012, 14, 1363–1365 CrossRef PubMed.
- Prior attempts in our group to functionalize biaryl arynes derived from cyclic diarylbromonium and chloronium species with electrophiles have failed, despite the variety of strategies to do so which are reported with other aryne precursors, notably Kobayashi-type arynes. We suspect that the observed difficulty is attributed to steric hindrance inflicted by the adjacent aryl substituent.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data for all new compounds. X-ray data analysis for compounds 5b, 5c, 5e and 16. CCDC 2322393–2322396. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01234h |
| ‡ Present address: Institut für Organische Chemie, Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. Email: E-mail: joanna.wencel-delord@uni-wuerzburg.de |
| This journal is © The Royal Society of Chemistry 2024 |