
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAll-visible-light-driven stiff-stilbene photoswitches†
Fan
Xu‡
 a,
Jinyu
Sheng‡
a,
Charlotte N.
Stindt
a,
Stefano
Crespi
b,
Wojciech
Danowski
cf,
Michiel F.
Hilbers
d,
Wybren Jan
Buma
de and
Ben L.
Feringa
*a
a,
Jinyu
Sheng‡
a,
Charlotte N.
Stindt
a,
Stefano
Crespi
b,
Wojciech
Danowski
cf,
Michiel F.
Hilbers
d,
Wybren Jan
Buma
de and
Ben L.
Feringa
*a
aCenter for System Chemistry, Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bDepartment of Chemistry-Ångström Laboratory, Uppsala University, Box 523, Uppsala, Sweden
cUniversity of Strasbourg CNRS, ISIS UMR 7006, 8 Allée Gaspard Monge, Strasbourg F-67000, France
dVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands
eInstitute for Molecules and Materials, FELIX Laboratory, Radboud University, Toernooiveld 7c, 6525 ED, Nijmegen, The Netherlands
fFaculty of Chemistry, University of Warsaw, Pasteura 1, 02-093 Warsaw, Poland
First published on 27th March 2024
Abstract
Molecular photoswitches are potent tools to construct dynamic functional systems and responsive materials that can be controlled in a non-invasive manner. As P-type photoswitches, stiff-stilbenes attract increasing interest, owing to their superiority in quantum yield, significant geometric differences between isomers, excellent thermostability and robust switching behavior. Nevertheless, the UV-light-triggered photoisomerization of stiff-stilbenes has been a main drawback for decades as UV light is potentially harmful and has low penetration depth. Here, we provided a series of para-formylated stiff-stilbenes by Rieche ortho-formylation to achieve all-visible-light-responsiveness. Additional phenolic groups provide access to late-stage chemical modification facilitating design of molecules responsive to visible light. Remarkably, the photoisomerization of aldehyde-appended stiff-stilbenes could be fully manipulated using visible light, accompanied by a high photostationary state (PSS) distribution. These features render them excellent candidates for future visible-light-controllable smart materials and dynamic systems.
Introduction
Light exhibits distinct advantages to control materials and molecular systems with high spatiotemporal precision.1–4 Photoswitches offer the opportunity to transfer energy and information from light to matter and reversibly modulate systems, structures, and functions.5–10 Nevertheless, high-energy photons usually required for photochemical reactions restrict the photoactivation of most switches to the use of light in the UV region, which suffers from low penetration depth, potential photodegradation, and being harmful to, e.g., living cells, and therefore limits their applications. Hence, visible and NIR light-activated molecular switches are much in demand.11–13 Recently, elegant molecular engineering strategies,7,11,12,14,15 such as push–pull substitution,16–18 π-extension,19–21 and energy transfer22–25 have been introduced into molecular switches, e.g. azobenzenes,14–16,25–29 diarylethenes,7,19–23,30 and overcrowded alkenes7,18,31 to achieve visible-light-responsiveness. However, most approaches, such as push–pull substitution and energy transfer, require complicated synthesis and lack design generality for late-stage modification of molecules to exploit their full potential in further applications. Additionally, approaches to all-visible-light-driven P-type (interconversion between isomers only using light) molecular switches are still less exploited compared to T-type photoswitches (interconversion can be either thermal or photochemical12,26,29,32,33). Meanwhile, new scaffolds of photoswitches have emerged in recent years responding to visible light, such as indirubin,34 (thio-)indigo-based photoswitches developed by Dube,32,35,36 imino indolinone,37 and imidazole-dimer-based photoswitches.38–40Stiff-stilbenes,41 a prominent class of photoswitches derived from stilbene that feature a fused five-membered ring, have shown distinct advantages in various research areas, such as supramolecular assemblies,42–47 molecular force probes,48–50 photoresponsive receptors,51–54 ligands,55 and light emitting materials,56 owing to their large geometric change, robust switching behavior, and excellent bi-thermostability (P-type). These alkenes are also the basis of light-driven rotary molecular motors.57–61 However, two intrinsic disadvantages of stiff-stilbenes are still challenging to address: harmful UV light-triggered switching and unfavorable photostationary state (PSS) distribution for the E–Z isomerization process (E to Z conversion).41 The low yield of the Z-isomer and short penetration depth of UV-light limit the efficiency of control over the dynamic properties of materials. A known strategy of introducing push–pull character has been applied to stiff-stilbene switches recently and has successfully led to visible-light-responsiveness.18 This system shows an interesting acid-gating feature because of the additional push–pull functionalities, which, however, also leads to extensive substitution and limits the opportunity for further modification. To date, a simple and general strategy for the development of an all-visible-light-driven stiff-stilbene system is still unexploited but greatly needed.
Here, we provide a general strategy for the development of all-visible-light-driven stiff-stilbenes. By appending bis-aldehyde functionalities at the para-positions to the olefin core via a one-step Rieche formylation, we obtained a series of stiff-stilbenes that allow visible-light modulation with improved photostationary state (PSS) distribution (Fig. 1). Furthermore, two additional phenolic groups are beneficial for potential late-stage modification toward functional structures for dynamic systems. Computational studies supported that appending bis-aldehyde groups drastically decreased the electronic transition gap of the molecules due to the extension of the π-conjugation. This straightforward strategy enables all-visible-light control of stiff-stilbenes, a unique bistable switch known and employed for the large geometric changes occurring upon isomerization. As such, these advances break new ground for increasing the application of stiff-stilbenes in smart materials and dynamic systems.
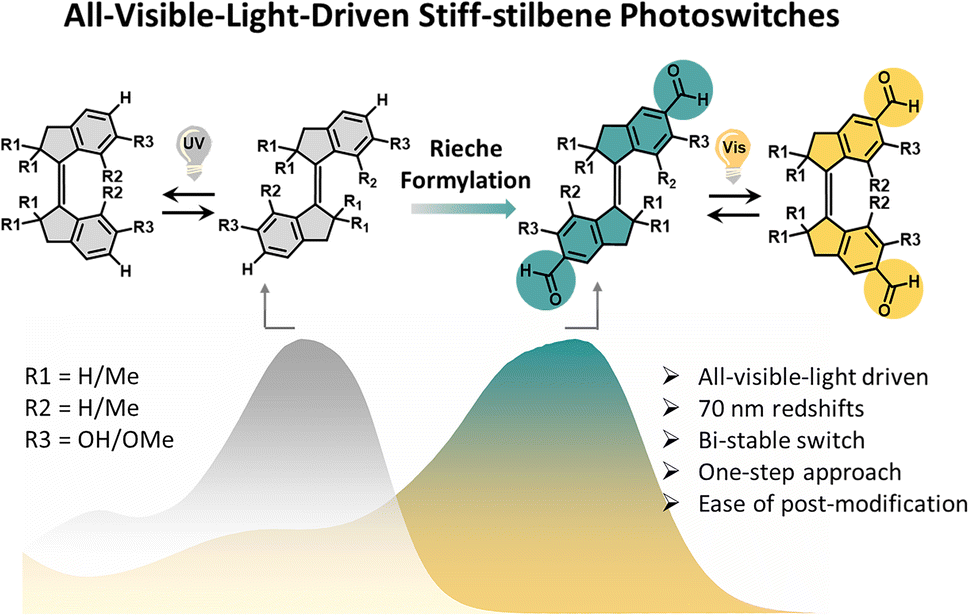 | ||
| Fig. 1 Molecular structures of the stiff-stilbenes S1–S6 controlled using UV light and visible light, respectively, and cartoon representation of the absorption band of stiff-stilbenes substituted with aldehydes (S2, S4, and S6) and those without formyl group (S1, S3, and S5). | ||
Results and discussion
Stiff-stilbenes were synthesized through McMurry coupling from the corresponding indanones, and the aldehyde derivatives were obtained by Rieche formylation inspired by our current approach for formylated molecular motor synthesis (Fig. 2a).62 The synthesis is detailed in the ESI (Fig. S1†), and the new compounds were fully characterized by NMR and HRMS (Fig. S18–S52†). Crystal structures of differently substituted stiff-stilbenes (E)-2 (methoxy group protected (E)-S1, see the ESI†), (E)-S5 and (E)-S6 were successfully obtained from X-ray diffraction analyses, providing direct evidence of the respective molecular structures (Fig. 2b). Steady-state absorption spectra of aldehyde-substituted stiff-stilbenes (E)-S2, S4 and S6 showed shifts of ∼70 nm compared with those of unsubstituted stiff-stilbenes (E)-S1, S3 and S5 (Fig. 2c).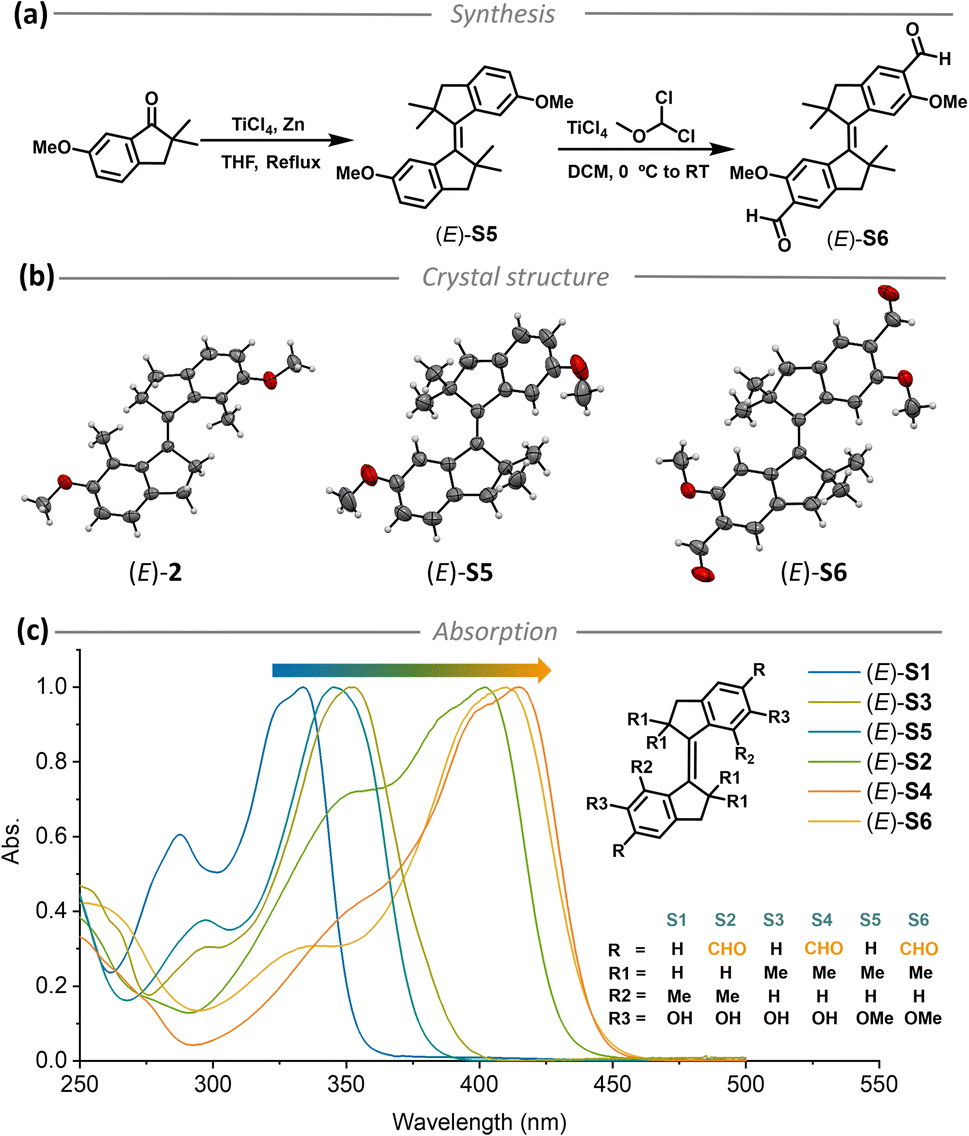 | ||
| Fig. 2 (a) The synthesis of stiff-stilbene derivatives (E)-S5 and (E)-S6 by McMurry coupling and Rieche formylation, (b) crystal structures of (E)-2, (E)-S5, and (E)-S6, (c) molecular structures of (E)-S1 to (E)-S6 and the normalized steady-state absorption spectra of (E)-S1 to (E)-S6 ((E)-S1 in DMSO and others in CH2Cl2, 298 K). | ||
We first investigated the isomerization behavior of stiff-stilbene (E)-S1 and its aldehyde derivative (E)-S2 by UV-vis and 1H NMR spectroscopy. UV-vis measurements showed that (E)-S2 displayed an absorption band at λmax = 403 nm (Fig. 3, pink spectrum), a significant bathochromic shift of ∼70 nm compared to the parent switch (E)-S1 (Fig. 3, grey spectrum), as a result of the π-system extension of the stilbene chromophore by the presence of the aldehyde groups at the para-position. E → Z photoisomerization of (E)-S1 was achieved by 340 nm light irradiation, while irradiating at 365 nm led to Z → E back-switching, as monitored by UV-vis spectroscopy (Fig. S2†). A new set of signals appeared in the 1H NMR spectrum after 340 nm light irradiation of (E)-S1 in accordance with the formation of the Z isomer (Fig. S4b†). The integration of the corresponding signals revealed a ratio of 76/24 (E/Z) at the PSS. Upon irradiation with 365 nm light, the signals of the Z-isomer decreased until a new PSS was reached with a ratio of 6/94 (Z/E), indicating the regeneration of (E)-S1 (Fig. S4c†). This photoisomerization behavior of (E)-S1 is comparable with that of other stiff-stilbenes.41,54,63
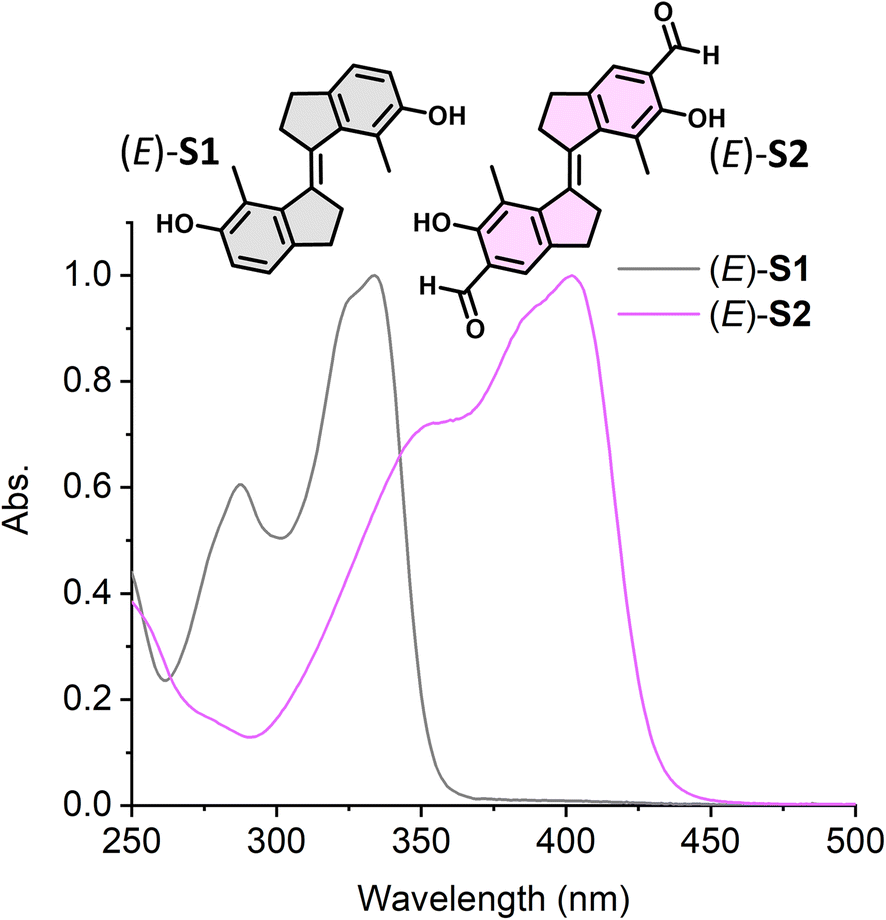 | ||
| Fig. 3 Normalized steady-state absorption spectra of (E)-S1 (grey curve) in DMSO and (E)-S2 (pink curve) in CH2Cl2 (298 K). | ||
In contrast, the photo-switching of the aldehyde-appended stiff stilbene derivative (E)-S2 can be controlled using two distinct wavelengths of visible light. Upon irradiation with 405 nm (or 395 nm) light, the absorption band at 310–410 nm decreased while the shoulder at 415–450 nm increased, indicating E → Z isomerization (Fig. 4a). Irradiating the PSS405 mixture at 455 nm resulted in an inverse spectral change, indicating Z → E isomerization (Fig. 4b). Both processes displayed an isosbestic point at 414 nm, showing the unimolecular conversion process (Fig. 4c and d). Fatigue resistance studies revealed the stability of this molecule over several irradiation cycles (Fig. 4e). In the 1H NMR spectra, the proton signals of Ha, Hb, and He shifted upfield, while Hc and Hd shifted downfield upon irradiation with 405 nm light, proving the E → Z isomerization (Fig. 4f(i) and (ii)).
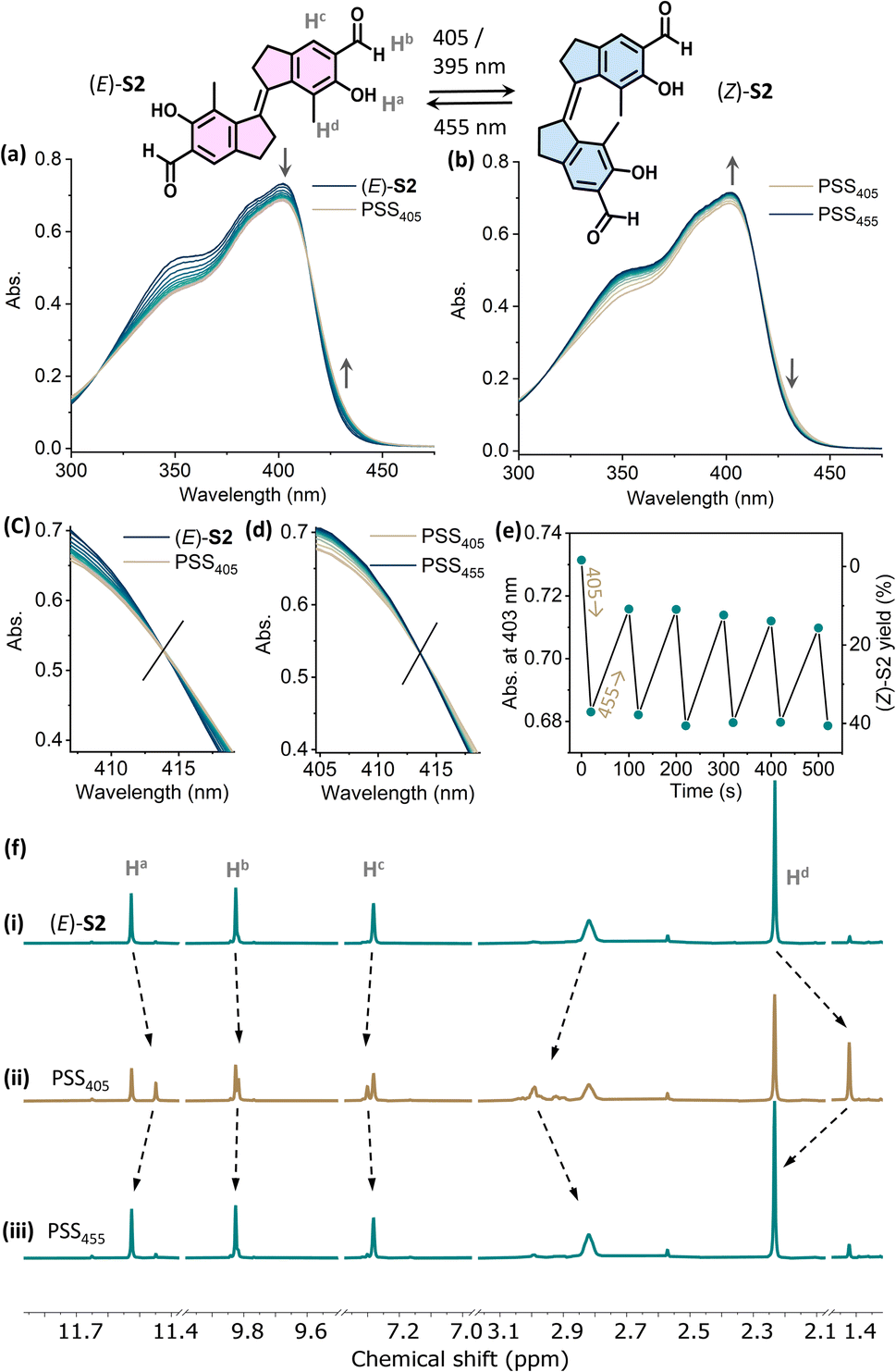 | ||
| Fig. 4 Changes in the UV-vis absorption spectra starting from (a) (E)-S2 (24 μM in CH2Cl2, 298 K) upon irradiation with 405 nm light to PSS405 and (b) upon subsequently irradiating with 455 nm light to PSS455. Enlarged spectra at the isosbestic point of (c) E to Z isomerization and (d) Z to E back-switching. (e) Plots of the absorption at λ = 403 nm upon sequential irradiation with 405 nm and 455 nm light. (f) Changes in 1H NMR spectra of (i) (E)-S2 (2.9 mM in CD2Cl2, 298 K, 500 MHz), (ii) upon irradiation with 405 nm light to PSS405, and (iii) upon subsequent irradiation with 455 nm light to PSS455. | ||
The integration of corresponding signals revealed a PSS of 37/63 (Z/E) at PSS405. A PSS distribution of 45/55 (Z/E) was achieved upon irradiation with 395 nm light (Fig. S5†). Compared with (E)-S1, the E → Z isomerization of S2 gave a higher conversion to the Z-isomer. Irradiating the PSS405 mixture with 455 nm light led to a decrease in the newly generated signals with an increase in the original ones, in line with the recovery of (E)-S2 (Fig. 4f(iii)). The ratio of the (Z)/(E)-isomers was determined to be 8/92 at PSS455 (Fig. S5†). Upon irradiation with 390 nm light, the quantum yields for the E → Z (ΦE→Z) and Z → E (ΦZ→E) processes are 31% and 50%, respectively. It should be noted that ΦE→Z of (E)-S2 (31%) is higher than that of the majority of the cases studied in the literature (around 20%).41
Encouraged by the red-shifting effect of the formyl substitution, we further explored the system with tetramethyl-stiff-stilbene.64,65 Diol-tetramethyl-stiff-stilbene (E)-S3 displayed an absorption band at λmax = 350 nm, while its aldehyde derivative (E)-S4 showed an absorption maximum at 415 nm (Fig. 5a). Compared with (E)-S1 and (E)-S2, both (E)-S3 and (E)-S4 showed more than 10 nm bathchromic shifts, respectively. As indicated above, diol-stiff-stilbenes were designed as potential intermediates for late-stage modification. We, therefore, further explored a dimethoxy-tetramethyl-stiff-stilbene as a model compound to investigate whether the formylated-tetramethyl-stiff-stilbene could be controlled using visible light after the replacement of the phenolic groups with other functional groups. The dimethoxy-substituted aldehyde tetramethyl-stiff-stilbene (E)-S6 exhibits an absorption band maximum at 410 nm, allowing the use of visible light for activation (Fig. 5b). Compared with (E)-S5 (λmax = 345 nm), (E)-S6 displays a ∼65 nm redshift due to the presence of the formyl groups.
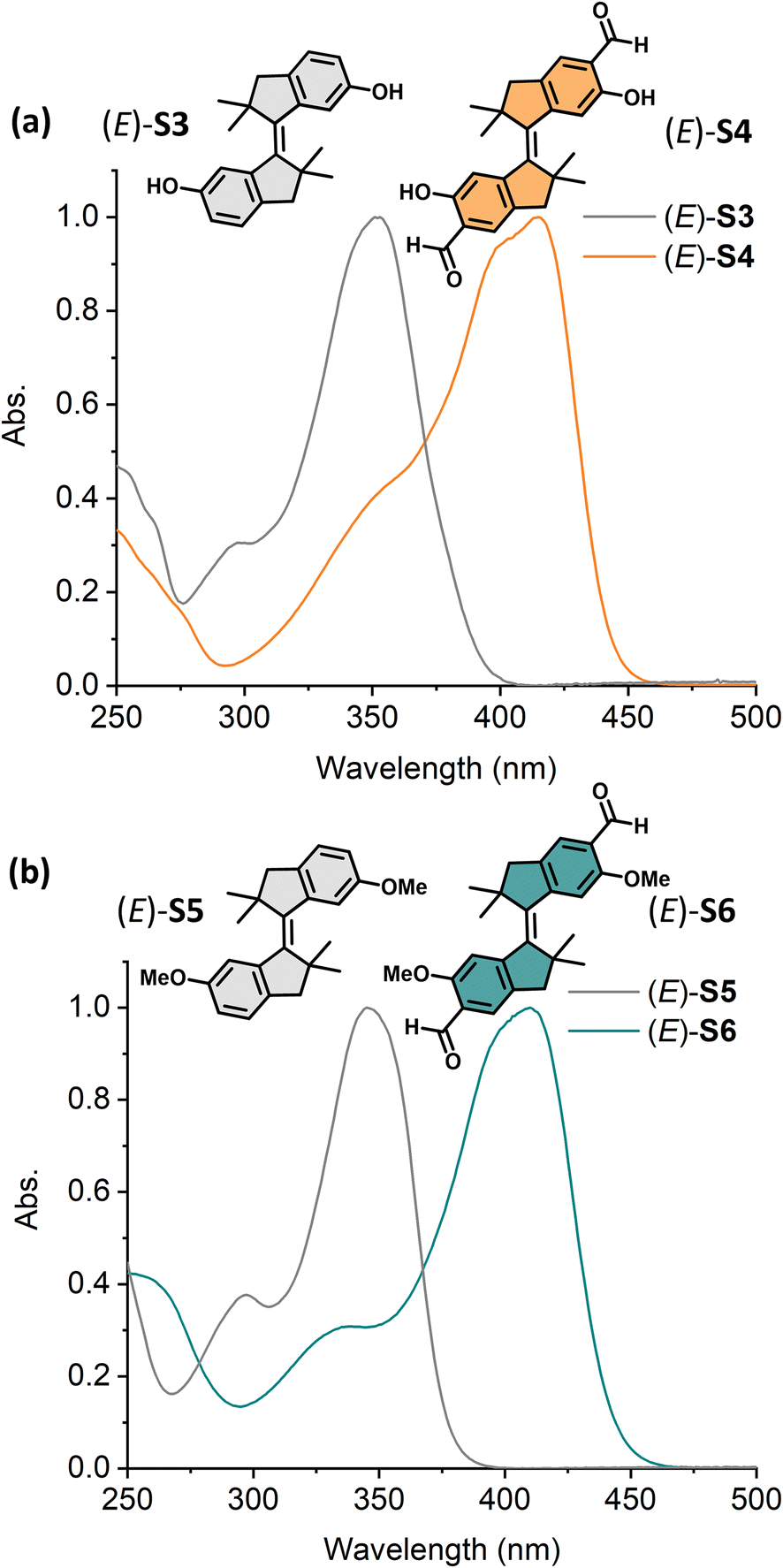 | ||
| Fig. 5 Normalized UV-vis absorption spectra (CH2Cl2, 298 K) of (a) (E)-S3 (grey curve) and (E)-S4 (orange curve) and (b) (E)-S5 (grey curve) and (E)-S6 (green curve). | ||
We further monitored the photoisomerization behavior of (E)-S5 and (E)-S6 by UV-vis and NMR spectroscopy. The reversible isomerization of (E)-S5 can be achieved by irradiation with 365 nm and 395 nm light (see the ESI for details, Fig. S3†). The integration of 1H NMR signals revealed a ratio of 52/48 (Z/E) at PSS365 and 12/88 (Z/E) at PSS395 in CD2Cl2 (Fig. S6†). Compared with (E)-S1, irradiating (E)-S5 resulted in a higher PSS, possibly due to the better separation of the absorption bands of the (E)- and (Z)-isomers (Fig. S2 and S4†). Upon 365 nm light irradiation, the quantum yields Φ for the E → Z and Z → E processes of S5 are 33% and 42%, respectively. The improved isomerization behavior of the tetramethyl-stiff-stilbene S5, compared to stiff-stilbene S1, prompted us to study its aldehyde derivative (E)-S6. Upon irradiation with 405 nm (or 420 and 395 nm) light, the absorption band centered at 410 nm decreased with an increase of a band at 425–470 nm, consistent with the (E)-S6 to (Z)-S6 isomerization (Fig. 6a). The absorption band at λmax = 410 nm was almost fully recovered upon subsequent irradiation with 455 nm light, indicating the recovery of (E)-S6 (Fig. 6b). An isosbestic point at 425 nm was observed in both processes, indicating a clean and unimolecular photoisomerization process (Fig. 6c and d). Gratifyingly, the photoisomerization processes could be manipulated for many cycles upon toggling the irradiation at 405 nm and 455 nm without any fatigue (Fig. 6e). In the 1H NMR spectra, irradiating (E)-S6 with 405 nm light led to the formation of (Z)-S6. Integrations of the related signals revealed a 73/27 (Z/E) ratio at PSS405 (Fig. S7†). The yield of the Z-isomer of E to Z conversion was significantly higher than the ones found in other commonly used stiff-stilbenes.41 Upon subsequent irradiation with 455 nm light, NMR signals of (Z)-S6 decreased until a PSS ratio of 19/81 (Z)/(E)-isomer was reached, confirming the good reversibility of the photoswitch (Fig. 6 and S7†). Kinetic studies by 1H NMR were performed with a PSS405 mixture of S6 at 298 K, and the unaltered ratio of isomers after 60 h indicates the high thermal stability of both isomers and confirms the P-type feature of the stiff-stilbene switch (Fig. S8†).41 Quantum yield data were obtained with 390 nm light irradiation, and the ΦE→Z and ΦZ→E of S6 were found to be 26% and 11%, respectively, suggesting that the efficiency of the undesired back-switching decreases after para-formylation compared with the E to Z isomerization of (E)-S5. To further understand the photoisomerization mechanism of the stiff-stilbenes and their formylated analogs, we studied S5 and S6 using nanosecond transient absorption spectroscopy. These measurements do not indicate any significant contribution of a triplet state to the photoisomerization pathways of the non-formylated stiff-stilbenes (E)-S5 and (Z)-S5 (Fig. S12 and S13†), as well as their formylated analogs (E)-S6 and (Z)-S6 (Fig. S14 and S15†). This is consistent with what has previously been reported in the literature.66,67
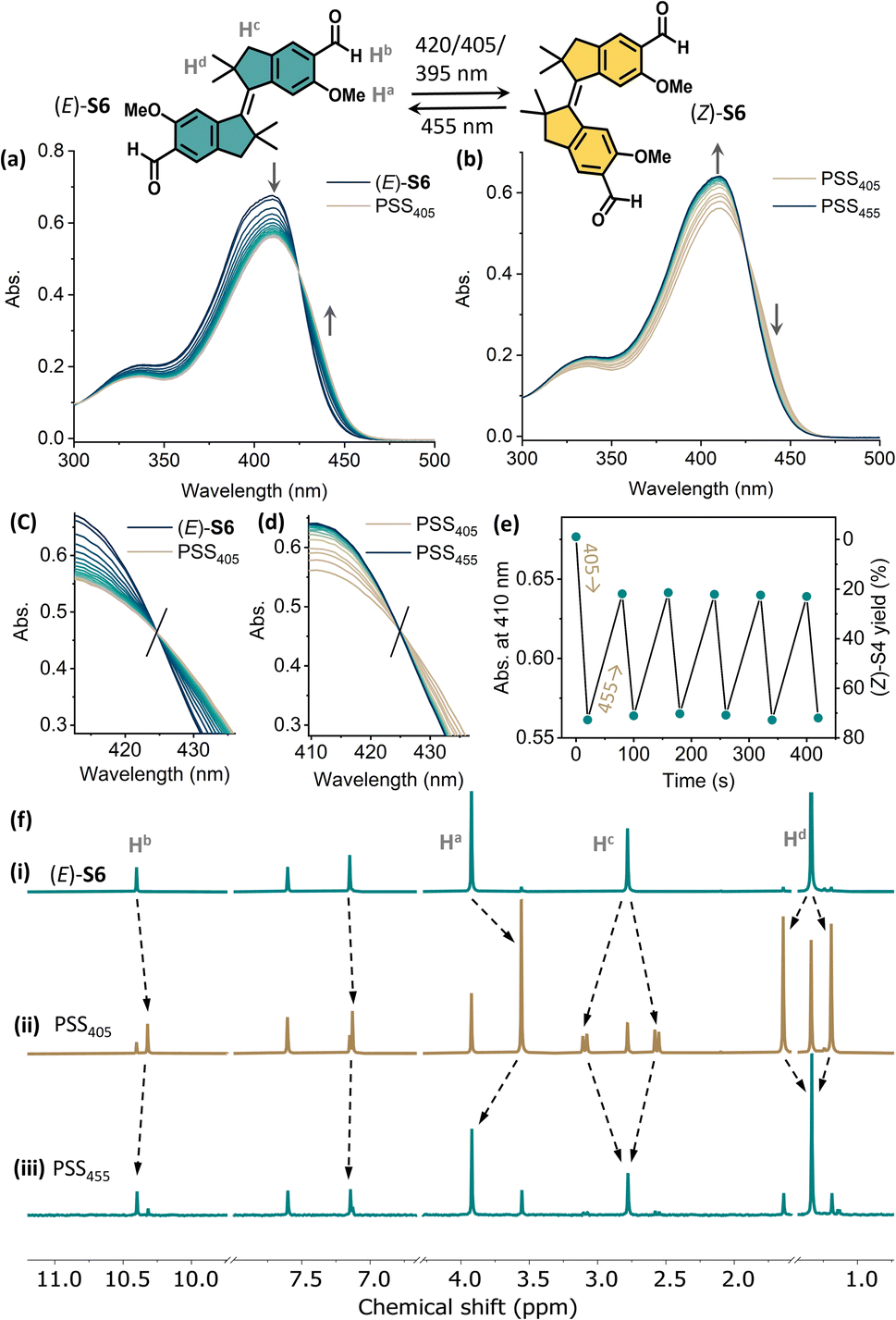 | ||
| Fig. 6 Changes in the UV-vis absorption spectra starting from (a) (E)-S6 (13 μM in CH2Cl2, 298 K) upon irradiation with 405 nm light to PSS405 and (b) upon subsequent irradiation with 455 nm light to PSS455. Enlarged spectra at the isosbestic point of (c) E to Z isomerization and (d) Z to E back-switching. (e) Plots of the absorption at λmax = 410 nm upon sequential irradiation with 405 nm and 455 nm light. (f) Changes in 1H NMR spectra of (i) (E)-S6 (6.2 mM in CD2Cl2, 298 K, 500 MHz), (ii) upon irradiation with 405 nm light to PSS405, and (iii) upon subsequent irradiation with 455 nm light to PSS455. | ||
To gain insight into the redshifted nature of these molecules by installment of the formyl group, we compared the energies of the first electronic transition in the E and Z forms of our stiff-stilbenes using density functional theory (DFT) (Fig. 7). The geometries were optimized at the r2SCAN-3c level of theory68 and single points were obtained at the PW6B95-D4/def2-QZVP69–73 level of theory (see the ESI for details and optimized structures†). The formyl group attachment to the stiff-stilbene decreases the ΔE by about 0.6 eV at both levels of theories examined. The decreased HOMO–LUMO gap contributes to the redshift of the absorption bands. The methoxy group substitution shows a very limited effect on the HOMO–LUMO gap compared to the hydroxyl group, in agreement with our experimental observations. As evident from Fig. S16 and S17,† the –CHO groups have a stabilizing effect on both orbitals. Indeed, the aldehyde groups extend the conjugation in the π scaffold and have positive overlap with the orbitals of the core of the switch both in the HOMO and LUMOs.
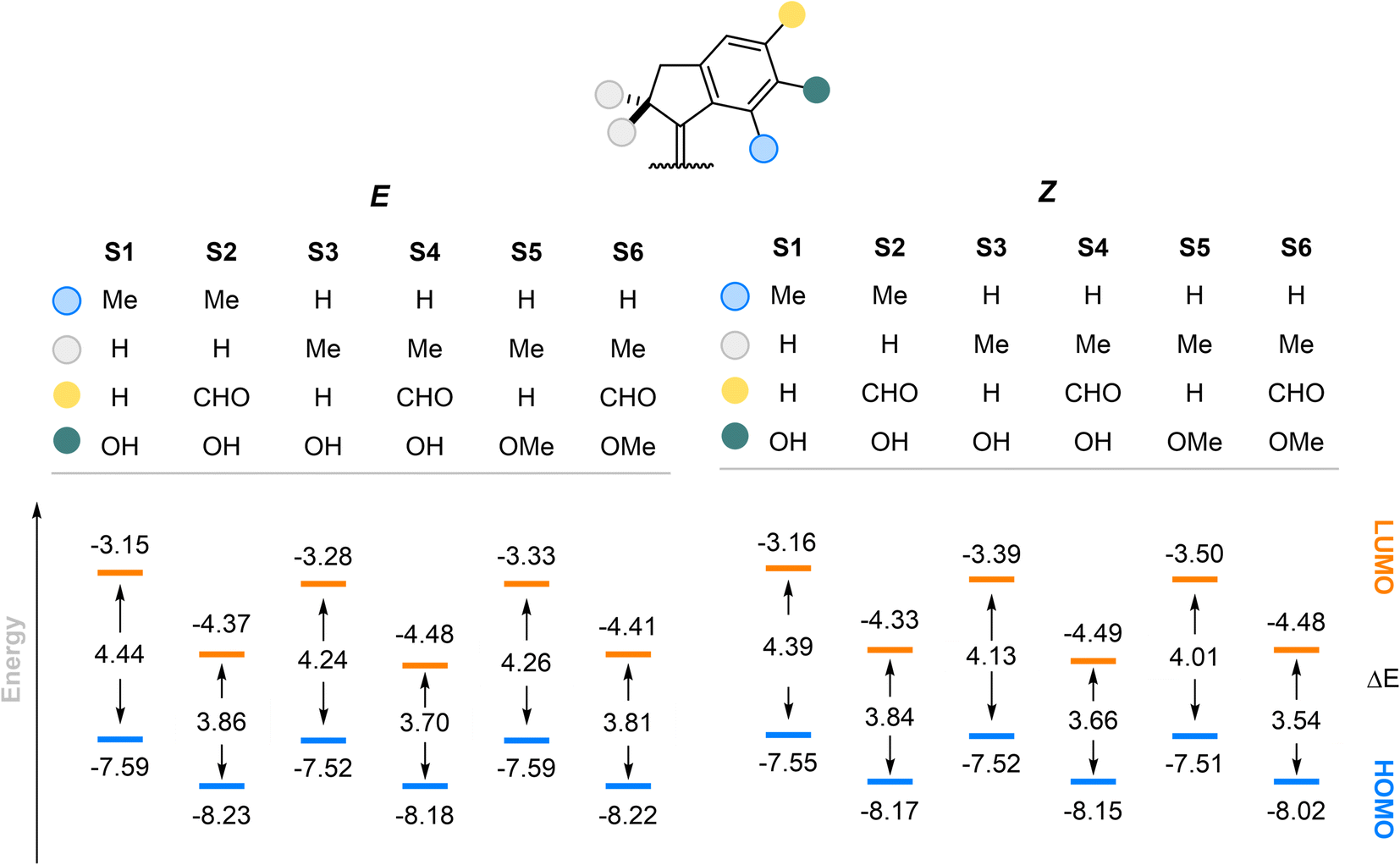 | ||
| Fig. 7 Comparison between HOMO–LUMO energies of the E and Z forms of the stilbenes at the PW6B95-D4/def2-QZVP//r2SCAN-3c level of theory. All values are in eV. | ||
Conclusions
In conclusion, we have developed a series of para-formylated stiff-stilbenes as all-visible-light-driven P-type molecular switches by a simple and general approach. This methodology not only provides a large ca. 70 nm bathochromic shift of the stilbene absorption bands but also improves their isomerization behavior without compromising their photostability. It is worth mentioning that prior to this study, neither of the stiff-stilbene cores, i.e. (E)-S1 and (E)-S3, had been engineered for responsiveness to visible-light. Additionally, in our design, the two phenolic groups anchored to the scaffolds that are essential for Rieche formylation facilitate late-stage modifications, e.g., O-alkylation, which is a desirable feature for design of the synthetic functional materials and photo-triggered dynamic systems.41 Given the distinct advantages of this design, we anticipate that this new type of aldehyde-functionalized stiff-stilbene photoswitches will serve as prominent candidates to construct all-visible-light-driven functional materials and dynamic systems.Data availability
The experimental and computational data associated with this article have been provided in the document of ESI.†Author contributions
B. L. F., J. S. and F. X. conceived this project. J. S. and F. X. designed synthetic routes. F. X. synthesized the stiff-stilbenes and performed the UV-vis and NMR studies. J. S. synthesized indanones, assisted with UV-vis and NMR measurements and prepared the crystals. C. N. Stindt resynthesized S6 and performed X-ray diffraction measurements. C. N. S., M. F. H. and W. J. B. performed transient absorption measurements. S. C. performed computational studies. J. S. conducted UV-vis experiments for the QY determination and W. D. processed the data for QY determination. All authors discussed the results and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Netherlands Organization for Scientific Research (NWO-CW), the European Research Council (ERC; advanced grant No.694345 to B. L. sF.), the Dutch Ministry of Education, Culture and Science (Gravitation program No.024.001.035), the China Scholarship Council (CSC; No.201707040064 to F. X. and CSC PhD Fellowship No. 201808330459 to J. S.), and the Marie Skłodowska-Curie Actions (Individual Fellowship No. 838280 to S. C. and 101027639 to W. D.) is gratefully acknowledged.References
- B. L. Feringa and W. R. Browne, Molecular Switches, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Adv. Mater., 2020, 32, 1905966 CrossRef CAS PubMed.
- L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- Molecular Photoswitches : Chemistry, Properties, and Applications, ed. Z. L. Pianowski, Wiley-VCH, 2022 Search PubMed.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, e1905227 CrossRef PubMed.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052–1079 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- H.-B. Cheng, S. Zhang, E. Bai, X. Cao, J. Wang, J. Qi, J. Liu, J. Zhao, L. Zhang and J. Yoon, Adv. Mater., 2022, 34, 2108289 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS PubMed.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- Z. Zhang, W. Wang, M. O'Hagan, J. Dai, J. Zhang and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202205758 CrossRef CAS PubMed.
- A. Leistner and Z. L. Pianowski, Eur. J. Org. Chem., 2022, 2022, e202101271 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem, 2019, 3, 133–146 CrossRef CAS.
- E. Ishow, A. Brosseau, G. Clavier, K. Nakatani, R. B. Pansu, J.-J. Vachon, P. Tauc, D. Chauvat, C. R. Mendonça and E. Piovesan, J. Am. Chem. Soc., 2007, 129, 8970–8971 CrossRef CAS PubMed.
- L. Pfeifer, M. Scherübl, M. Fellert, W. Danowski, J. Cheng, J. Pol and B. L. Feringa, Chem. Sci., 2019, 10, 8768–8773 RSC.
- D. Villarón, N. Duindam and S. J. Wezenberg, Chem.–Eur. J., 2021, 27, 17346–17350 CrossRef PubMed.
- T. Fukaminato, T. Hirose, T. Doi, M. Hazama, K. Matsuda and M. Irie, J. Am. Chem. Soc., 2014, 136, 17145–17154 CrossRef CAS PubMed.
- X. Guo, J. Zhou, M. A. Siegler, A. E. Bragg and H. E. Katz, Angew. Chem., Int. Ed., 2015, 54, 4782–4786 CrossRef CAS PubMed.
- J. Li, H. K. Bisoyi, S. Lin, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2019, 58, 16052–16056 CrossRef CAS PubMed.
- S. Fredrich, R. Göstl, M. Herder, L. Grubert and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1208–1212 CrossRef CAS PubMed.
- H. Xi, Z. Zhang, W. Zhang, M. Li, C. Lian, Q. Luo, H. Tian and W.-H. Zhu, J. Am. Chem. Soc., 2019, 141, 18467–18474 CrossRef CAS PubMed.
- A. Cnossen, L. Hou, M. M. Pollard, P. V. Wesenhagen, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2012, 134, 17613–17619 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed.
- A.-L. L. Leistner, S. Kirchner, J. Karcher, T. Bantle, M. L. Schulte, P. Gödtel, C. Fengler and Z. L. Pianowski, Chem.–Eur. J., 2021, 27, 8094–8099 CrossRef CAS PubMed.
- L. N. Lameijer, S. Budzak, N. A. Simeth, M. J. Hansen, B. L. Feringa, D. Jacquemin and W. Szymanski, Angew. Chem., Int. Ed., 2020, 59, 21663–21670 CrossRef CAS PubMed.
- V. W. W. Yam, C. C. Ko and N. Zhu, J. Am. Chem. Soc., 2004, 126, 12734–12735 CrossRef CAS PubMed.
- J. Sheng, W. Danowski, S. Crespi, A. Guinart, X. Chen, C. Stähler and B. L. Feringa, Chem. Sci., 2023, 14, 4328–4336 RSC.
- L. Köttner, E. Ciekalski and H. Dube, Angew. Chem., Int. Ed., 2023, 62, e202312955 CrossRef PubMed.
- C. Petermayer, S. Thumser, F. Kink, P. Mayer and H. Dube, J. Am. Chem. Soc., 2017, 139, 15060–15067 CrossRef CAS PubMed.
- S. Thumser, L. Köttner, N. Hoffmann, P. Mayer and H. Dube, J. Am. Chem. Soc., 2021, 143, 18251–18260 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- V. Josef, F. Hampel and H. Dube, Angew. Chem., Int. Ed., 2022, 61, e202210855 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth, M. Di Donato, S. Doria, C. N. Stindt, M. F. Hilbers, F. L. Kiss, R. Toyoda, S. Wesseling, W. J. Buma, B. L. Feringa and W. Szymański, Angew. Chem., Int. Ed., 2021, 60, 25290–25295 CrossRef CAS PubMed.
- M. Nishijima, K. Mutoh, R. Shimada, A. Sakamoto and J. Abe, J. Am. Chem. Soc., 2022, 144, 17186–17197 CrossRef CAS PubMed.
- N. Moriyama and J. Abe, J. Am. Chem. Soc., 2023, 145, 3318–3322 CrossRef CAS PubMed.
- H. Ito, K. Mutoh and J. Abe, J. Am. Chem. Soc., 2023, 145, 6498–6506 CrossRef CAS PubMed.
- D. Villarón and S. J. Wezenberg, Angew. Chem., Int. Ed., 2020, 59, 13192–13202 CrossRef PubMed.
- J. F. Xu, Y. Z. Chen, D. Wu, L. Z. Wu, C. H. Tung and Q. Z. Yang, Angew. Chem., Int. Ed., 2013, 52, 9738–9742 CrossRef CAS PubMed.
- C.-L. Sun, C.-H. Tung, L.-Z. Wu, Y. Wang, Q.-Z. Yang, L.-Y. Niu and Y.-Z. Chen, Polym. Chem., 2017, 8, 3596–3602 RSC.
- X. Yan, J. F. Xu, T. R. Cook, F. Huang, Q. Z. Yang, C. H. Tung and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8717–8722 CrossRef CAS PubMed.
- F. Xu, L. Pfeifer, S. Crespi, F. K. C. Leung, M. C. A. Stuart, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 5990–5997 CrossRef CAS PubMed.
- D. Y. Alene, R. Arumugaperumal, M. Shellaiah, K. W. Sun and W. S. Chung, Org. Lett., 2021, 23, 2772–2776 CrossRef CAS PubMed.
- F. Xu, S. Crespi, L. Pfeifer, M. C. A. Stuart and B. L. Feringa, CCS Chem., 2022, 4, 2212–2220 CrossRef CAS.
- Q. Z. Yang, Z. Huang, T. J. Kucharski, D. Khvostichenko, J. Chen and R. Boulatov, Nat. Nanotechnol., 2009, 4, 302–306 CrossRef CAS PubMed.
- T. J. Kucharski, Z. Huang, Q. Z. Yang, Y. Tian, N. C. Rubin, C. D. Concepcion and R. Boulatov, Angew. Chem., Int. Ed., 2009, 48, 7040–7043 CrossRef CAS PubMed.
- K. Imato, A. Ishii, N. Kaneda, T. Hidaka, A. Sasaki, I. Imae and Y. Ooyama, JACS Au, 2023, 3, 2458–2466 CrossRef CAS PubMed.
- S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 324–327 CrossRef CAS PubMed.
- S. J. Wezenberg and B. L. Feringa, Nat. Commun., 2018, 9, 1984 CrossRef PubMed.
- D. Villarón, M. A. Siegler and S. J. Wezenberg, Chem. Sci., 2021, 12, 3188–3193 RSC.
- J. Sheng, S. Crespi, B. L. Feringa and S. J. Wezenberg, Org. Chem. Front., 2020, 7, 3874–3879 RSC.
- M. P. O’Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Chem.–Eur. J., 2020, 26, 6224–6233 CrossRef PubMed.
- Y. H. Wu, K. Huang, S. F. Chen, Y. Z. Chen, C. H. Tung and L. Z. Wu, Sci. China: Chem., 2019, 62, 1194–1197 CrossRef CAS.
- T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Nat. Rev. Chem, 2017, 1, 0096 CrossRef CAS.
- D. R. S. Pooler, A. S. Lubbe, S. Crespi and B. L. Feringa, Chem. Sci., 2021, 12, 14964–14986 RSC.
- N. Koumura, R. W. J. Zijistra, R. A. Van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423–9431 CrossRef CAS PubMed.
- J. Sheng, D. R. S. Pooler and B. L. Feringa, Chem. Soc. Rev., 2023, 52, 5875–5891 RSC.
- J. Sheng, W. Danowski, A. Sardjan, S. Crespi, J. Hou, M. P. Domínguez, A. Ryabchun, W. J. Buma, W. Browne and B. L. Feringa, Nat. Chem. Search PubMed , in press.
- M. Quick, F. Berndt, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, C. Knie, R. Mahrwald, D. Lenoir, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 1389–1402 CrossRef CAS PubMed.
- T. Shimasaki, S. Kato and T. Shinmyozu, J. Org. Chem., 2007, 72, 6251–6254 CrossRef CAS PubMed.
- K. Imato, A. Sasaki, A. Ishii, T. Hino, N. Kaneda, K. Ohira, I. Imae and Y. Ooyama, J. Org. Chem., 2022, 87, 15762–15770 CrossRef CAS PubMed.
- F. E. Doany, E. J. Heilweil, R. Moore and R. M. Hochstrasser, J. Chem. Phys., 1984, 80, 201–206 CrossRef CAS.
- W. Fuß, C. Kosmidis, W. E. Schmid and S. A. Trushin, Angew. Chem., Int. Ed., 2004, 43, 4178–4182 CrossRef PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J. M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Caldeweyher, J.-M. Mewes, S. Ehlert and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 8499–8512 RSC.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2319368, 2319369 and 2319370. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00983e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |