DOI:
10.1039/D4SC01182A
(Edge Article)
Chem. Sci., 2024,
15, 12928-12938
An intramolecular cobalt-peptoid complex as an efficient electrocatalyst for water oxidation at low overpotential†
Received
19th February 2024
, Accepted 5th July 2024
First published on 17th July 2024
Abstract
Water electrolysis is the simplest way to produce hydrogen, as a clean renewable fuel. However, the high overpotential and slow kinetics hamper its applicability. Designing efficient and stable electrocatalysts for water oxidation (WO), which is the first and limiting step of the water splitting process, can overcome this limitation. However, the development of such catalysts based on non-precious metal ions is still challenging. Herein we describe a bio-inspired Co(III)-based complex i.e., a stable and efficient molecular electrocatalyst for WO, constructed from a peptidomimetic oligomer called peptoid – N-substituted glycine oligomer – bearing two binding ligands, terpyridine and bipyridine, and one ethanolic group as a proton shuttler. Upon binding of a cobalt ion, this peptoid forms an intramolecular Co(III) complex, that acts as an efficient electrocatalyst for homogeneous WO in aqueous phosphate buffer at pH 7 with a high faradaic efficiency of up to 92% at an overpotential of about 430 mV, which is the lowest reported for Co-based homogeneous WO electrocatalysts to date. We demonstrated the high stability of the complex during electrocatalytic WO and that the ethanolic side chain plays a key role in the stability and activity of the complex and also in facilitating water binding, thus mimicking an enzymatic second coordination sphere.
Introduction
Mimicking the natural water splitting process is an attractive strategy for the production of oxygen and hydrogen, the latter having the potential to be used as an alternative renewable fuel.1 The first step in this process is the four-electron oxidation of water (2H2O → O2 + 4H+ + 4e−; ΔE° = 1.23 V at pH 0),2 which is challenging both kinetically and thermodynamically.3 Therefore, developing effective electrocatalysts for water oxidation, which are also highly stable and perform with low overpotential, i.e. operate near neutral pH with minimum applied potential, is a long standing goal towards the production of hydrogen as a renewable energy source.4 Indeed, significant research efforts have been made to design WO catalysts based on non-precious earth-abundant first-row transition metal ions (mainly Mn,5 Fe,6 Co,7 Ni8 and Cu9). Among them, Co-based WO catalysts show good catalytic activity under ambient pH conditions with relatively high turnover frequency (TOF). However, it is a challenging task to design and generate soluble Co-based WO electrocatalysts that are stable over time during electrocatalysis thus enabling homogeneous electrocatalytic WO to proceed for long periods of time towards high TON, as some cobalt complexes tend to get irreversibly oxidized to form Co-based oxides or hydroxides during WO, leading to heterogeneous catalytic systems.10 An example representing this challenge is a dinuclear Co(III) complex initially reported as an active homogeneous WO catalyst, but in later studies, it was found to generate heterogeneous CoOx which acts as the actual catalyst.10c A reason for this might be an intermolecular coordination of Co by the ligands, which is typically weaker than an intramolecular coordination,11 and thus can lead to ligand dissociation during catalysis,12 facilitating destabilization of the Co ion and its irreversible oxidation to Co oxides.10c Therefore, it is important to choose a chelating system that: (i) enables intramolecular coordination via strong electron-donating ligands that can stabilize Co in its high oxidation states, (ii) enables the formation of an intramolecular Co-complex that has at least one free coordination site for water binding, and (iii) can stabilize the catalytic center during WO electrolysis, even in the case of ligand dissociation. One way to achieve these goals is to design chelating systems that not only comprise specific ligands for Co coordination, but also include a second coordination sphere mimic about the Co center, akin to enzymes. This second coordination sphere mimic can provide additional weak interactions, such as hydrogen bonding, to facilitate the stabilization of Co in different oxidation states and/or of formed intermediates obtained during WO,13 even if the coordinating ligand is dissociated during the catalytic process.
To construct chelating systems that include both a direct coordination sphere and a second coordination sphere mimic, our lab is using synthetic peptide mimics called peptoids14 – N-substituted glycine oligomers – that are constructed from primary amines rather than from amino acids. Peptoids can be synthesized efficiently on a solid support using the “sub-monomer” method,15 which enables the incorporation of different side chains in a specific manner, including metal-binding ligands for direct metal coordination as well as structure directing elements and nucleophiles that can mimic second coordination sphere effects.16 In addition, peptoids are chemically inert towards many catalytic transformations, are highly stable under various pH and oxidative conditions,17a and can stabilize metal ions in their high oxidation state.17b Hence, peptoids are excellent candidates for developing efficient bio-inspired catalysts.17c,d Indeed, our group has reported catalytic metallopeptoids containing non-catalytic sites, which together with the peptoid backbone, aim to mimic a second coordination sphere that facilitates efficient oxidative transformations, including electrocatalytic WO.18–21 The activity of metallopeptoid-based electrocatalysts for WO, however, was only reported at pH > 9, limiting the requirement for low overpotential. Moreover, so far, only one Co-based peptoid complex has been reported as an electrocatalyst for WO – an intermolecular complex where Co3+ is coordinated to two terpyridine-based peptoid trimers.20 As a consequence of this intermolecular binding, the stability of this complex over time, similar to other Co-based WO electrocatalysts (Table A1, ESI†), was limited, hampering its activity.10c,22
Aiming to obtain a stable Co-peptoid electrocatalyst for WO that is active over time and performs at neutral pH with low overpotential, we have designed a peptoid trimer (TBE) bearing two metal-binding ligands, namely 2,2′;6′,2′′-terpyridine (Terpy) and 2,2′-bipyridine (Bipy), aiming to form an intramolecular penta-coordinated Co-peptoid complex with a free coordination site for water binding and an ethanolic side chain that, together with the peptoid backbone, will serve as a second coordination sphere mimic. We anticipated that the –OH group from the ethanolic side chain would either assist in stabilizing the metal center during WO,19b facilitate proton shuttling5e and/or stabilize Terpy or Bipy in case one of them dissociates from the Co center.20 As control peptoids for intramolecular binding of Co, the two trimers TTE and BBE having two Terpy ligands or two Bipy ligands respectively, in addition to the ethanolic side chain were also designed, aiming to investigate whether a vacant site for water binding is crucial for catalytic activity and which peptoid out of the three forms the most stable WO electrocatalyst.
Results and discussion
Synthesis and characterization of TBE and its corresponding Co complex
The peptoid TBE (Fig. 1a) was synthesized by the “sub-monomer” solid-phase method15 (see the ESI† for details), cleaved from the solid support and purified by high-performance liquid chromatography (HPLC, >95% purity) (Fig. S1†). The formation of the peptoid was further supported by high-resolution mass spectrometry (HR-MS), as the measured mass corresponded to the expected mass for its sequence (Fig. S2 and S3†). The corresponding peptoid Co complex was obtained by treating the peptoid with one molar equiv. of Co(OAc)2·4H2O in methanol and after 2 hours of stirring, the reddish-brown product was precipitated out by using excess sodium perchlorate.19,20 The precipitate was isolated, washed, and dried. The UV-vis spectra of TBE and its isolated complex, both in 0.1 M phosphate buffer solution (PBS) at pH 7, showed that the absorbance bands near 236 and 285 nm (assigned to the π–π* and n–π* transition of the Terpy and Bipy ligands present in the peptoid sequence) shift to 240 and 282 nm respectively, and a new absorption band near λ = 314 nm was produced (attributed to the metal to ligand charge transfer (MLCT) transition of CoTBE species, Fig. S4†). These observations support the formation of a cobalt–peptoid complex.16d,f,j,k Further characterization using UV-vis spectra in PBS pH 7 revealed that (i) the absorption band linearly varies with the complex concentration following the Beer–Lambert law, which indicates that the complex exists as a single species in PBS without dimerization (Fig. S5†)23 and (ii) the spectrum of the complex did not show any significant change after allowing it to stand for 24 hours in air, demonstrating that the complex is stable under these conditions (Fig. S6†). The HR-MS of the isolated complex in acetonitrile showed a peak at m/z = 763.27 corresponding to the mass of CoTBE as well as a peak at 863.23, which matches the calculated mass of CoTBE + ClO4− (Fig. S7 and S8†). Notably, this chromatogram did not show any other masses corresponding to the full or half-masses of species with a ratio Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBE 1:2 or 2:2, indicating the formation of the 1:1 Co:TBE complex, a.k.a. the intramolecular Co-peptoid complex CoTBE as the only product (Fig. 1b). To further support this composition, a metal-to-peptoid ratio plot was constructed from UV-Vis titration of TBE with Co ions where a plateau was obtained at a molar ratio of 1 (Fig. 2a). This also supports the formation of an intramolecular complex with a ratio of 1:1 Co:TBE. In addition, by keeping the total molar concentration of a mixture solution, which contains both Co and TBE constant at 33 μM and varying their mole fraction, a Job plot was also constructed (Fig. 2b). The absorbance proportional to complex formation was plotted against the mole fraction and from the intersection point at χ = 0.47, a stoichiometric ratio was determined to be 0.90,16f,j supporting the 1:1 metal-to-peptoid ratio obtained from the UV titrations. Altogether, the results from both UV-Vis titration, the Job plot and HR-MS confirmed the formation of the intramolecular complex CoTBE. Furthermore, the isolated CoTBE complex was characterized using solid phase FT-IR spectrum.
:TBE 1:2 or 2:2, indicating the formation of the 1:1 Co:TBE complex, a.k.a. the intramolecular Co-peptoid complex CoTBE as the only product (Fig. 1b). To further support this composition, a metal-to-peptoid ratio plot was constructed from UV-Vis titration of TBE with Co ions where a plateau was obtained at a molar ratio of 1 (Fig. 2a). This also supports the formation of an intramolecular complex with a ratio of 1:1 Co:TBE. In addition, by keeping the total molar concentration of a mixture solution, which contains both Co and TBE constant at 33 μM and varying their mole fraction, a Job plot was also constructed (Fig. 2b). The absorbance proportional to complex formation was plotted against the mole fraction and from the intersection point at χ = 0.47, a stoichiometric ratio was determined to be 0.90,16f,j supporting the 1:1 metal-to-peptoid ratio obtained from the UV titrations. Altogether, the results from both UV-Vis titration, the Job plot and HR-MS confirmed the formation of the intramolecular complex CoTBE. Furthermore, the isolated CoTBE complex was characterized using solid phase FT-IR spectrum.
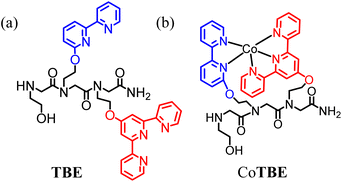 |
| | Fig. 1 (a) Peptoid ligand TBE and (b) the Co-peptoid complex CoTBE. | |
 |
| | Fig. 2 (a) UV-Vis spectra for the titration of peptoid TBE with Co ions (20 μM) in 0.1 M PBS at pH 7.0 and a metal-to-peptoid ratio plot for Co binding with TBE (inset). (b) Job-plot of TBE with Co measured in methanol (33 μM total concentration). | |
A comparison between the FT-IR spectra of TBE and its Co complex showed shifts from 1121 to 1080 cm−1 and 1533 to 1571 cm−1 corresponding to the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond stretching respectively, within the terpyridine and bipyridine center.24 In contrast, the FTIR spectra of TBE and its Co complex showed no shift at 1663 cm−1 and 3206 cm−1 associated with the CO of the amide bond and terminal N–H group, respectively. This indicates that neither the terminal N–H group nor the CO of the amide bond participates in Co coordination (Fig. S9†).16f,j,l,19b Collectively, all these results indicate that both Terpy and Bipy bind to one cobalt ion forming the intramolecular complex CoTBE. Importantly, the 1H-NMR spectrum of CoTBE in DMSO-d6 showed sharp well-resolved peaks (Fig. S10 and S11†), and the electron paramagnetic resonance (EPR) spectra of CoTBE was silent (Fig. S12†). Both results indicate that CoTBE is a diamagnetic complex and that the Co center is stable in the Co(III) oxidation state.20,25 Interestingly, the ESI-MS spectrum of the CoTBE, measured in water (Fig. S13†), showed an additional peak at 881.99 compared to the ESI-MS spectrum in acetonitrile. This peak is consistent with [CoTBE (H2O)] + ClO4−, suggesting that CoTBE coordinates H2O via the vacant binding site in solution to form a stable hexa-coordinated geometry.
N bond stretching respectively, within the terpyridine and bipyridine center.24 In contrast, the FTIR spectra of TBE and its Co complex showed no shift at 1663 cm−1 and 3206 cm−1 associated with the CO of the amide bond and terminal N–H group, respectively. This indicates that neither the terminal N–H group nor the CO of the amide bond participates in Co coordination (Fig. S9†).16f,j,l,19b Collectively, all these results indicate that both Terpy and Bipy bind to one cobalt ion forming the intramolecular complex CoTBE. Importantly, the 1H-NMR spectrum of CoTBE in DMSO-d6 showed sharp well-resolved peaks (Fig. S10 and S11†), and the electron paramagnetic resonance (EPR) spectra of CoTBE was silent (Fig. S12†). Both results indicate that CoTBE is a diamagnetic complex and that the Co center is stable in the Co(III) oxidation state.20,25 Interestingly, the ESI-MS spectrum of the CoTBE, measured in water (Fig. S13†), showed an additional peak at 881.99 compared to the ESI-MS spectrum in acetonitrile. This peak is consistent with [CoTBE (H2O)] + ClO4−, suggesting that CoTBE coordinates H2O via the vacant binding site in solution to form a stable hexa-coordinated geometry.
Electrochemical characterization and testing of the control peptoids
The electrochemical and electrocatalytic properties of CoTBE were evaluated in 0.1 M phosphate buffer at pH 7. Cyclic voltammetry (CV) was performed using glassy carbon (GC) as the working electrode, Ag/AgCl as the reference electrode and a Pt wire as the counter electrode. All the potentials are reported versus the normal hydrogen electrode (NHE) by adding 0.197 V to the measured potential. As shown in Fig. 3, the CV of CoTBE showed a redox wave at E1/2 = + 0.26 V vs. NHE assigned to CoII/III and at high oxidative potential, a sharp anodic wave was observed at E = +1.53 V, which is assigned to the catalytic event. However, when the CV was recorded in the same potential range at a slower scan rate (50 mV s−1), an additional pre-peak was observed at an onset potential of +1.21 V. The pre-peak is associated with CoIII/IV redox species.20 This peak, however, is not well observed at higher scan rates because the very small current of CoIII/IV conversion overlaps with the high anodic current obtained from the catalytic process. To further confirm that the anodic wave at E = +1.53 V is related to the catalytic process, we performed CV measurements at different scan rates within the range of 5–100 mV s−1. The normalized current at 1.53 V has a reverse dependency on the scan rate i.e., it increases with decreasing scan rates (Fig. S14†), consistent with a catalytic behavior.26 Notably, Co(ClO4)2·6H2O is unstable in PBS at pH 7, rapidly forming a purple precipitate, as demonstrated in the CV solution (Fig. S15†).This observation highlights the crucial role of TBE in stabilizing the Co-center and preventing the formation of un-soluble species under neutral phosphate buffer conditions.19b,20
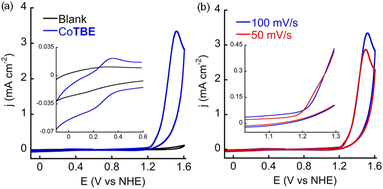 |
| | Fig. 3 (a) CVs of 0.5 mM CoTBE and blank at scan rate 100 mV s−1 (b) CVs of 0.5 mM CoTBE at scan rates 100 and 50 mV s−1; all the CVs are obtained in 0.1 M phosphate buffer at pH 7, using Ag/AgCl as the reference electrode with glassy carbon as the working electrode (0.07 cm2) and a Pt wire as the counter electrode. | |
The control peptoids TTE and BBE and their corresponding cobalt complexes (see ESI, Scheme S1†) were synthesized and characterized following the same procedure as that for the peptoid TBE and its corresponding Co complex CoTBE (Fig. S16–S23†). Notably, CoTTE was unstable in PBS at pH 7 as indicated by its UV-Vis spectrum, which changed with time (Fig. S24†).20 The CoBBE complex, on the other hand, was stable in PBS at pH 7 (Fig. S25†); however, it was not stable during electrocatalytic water oxidation as seen from a continuous CV scan experiment27 performed in PBS at pH 7 (Fig. S26†). These results indicate that among all the three complexes, CoTBE is the only one that remains stable before and during electrocatalytic WO, suggesting that using both Terpy and Bipy on one peptoid scaffold is required for the stability of its Co complex in PBS at pH 7.
Electrochemical water oxidation
Evolution of molecular oxygen was confirmed by a controlled potential electrolysis (CPE) experiment which was performed at an applied potential (Eapp) of 1.25 V vs. NHE, the onset potential of the reaction was catalyzed by CoTBE, with the 0.5 mM CoTBE complex in 0.1 M phosphate buffer solution at pH = 7 using porous glassy carbon as the working electrode (Fig. 4). CPE was carried out for 10 hours both in the presence and absence of CoTBE. During the CPE experiment with CoTBE, a charge of 22.4 C was accumulated and about 45 μmol of oxygen were evolved. In contrast, only 4.6 C and 2.2 μmol of oxygen were detected during an analogous experiment without the catalyst. Considering a 4e− process and the initial amount of the catalyst in solution, the faradaic efficiency (FE%) of the CPE experiment conducted in the presence of CoTBE was calculated to be approximately 78% before background correction (calculated from the CPE experiment performed without CoTBE) and 92% after this correction. The catalytic turnover number (TON) is 17.1 in 10 hours at an overpotential of about 433 mV (according to an applied potential of 1.25 V). This overpotential is significantly lower compared to that of the intermolecular Co-peptoid catalyst as well as to those of all the reported Co-based complexes that act as molecular electrocatalysts for homogeneous WO in the pH range 6–11 (see ESI, Table A1†). Notably, the current measured during the CPE rapidly increased in the first 6 minutes of the reaction from an initial value of 1.10 mA up to a value of about 1.28 mA and then slowly decreased over the next 4 hours. Importantly, after 4 hours, the catalyst remained active with a steady current of 0.5 mA for 6 more hours (Fig. S27†), representing that in addition to the significantly low overpotential, it has long durability compared with other Co-based homogeneous WO electrocatalysts (see ESI, Table A1†). However, the initial increase in current implies that at the beginning of the reaction some active species might be formed via rapid conformational changes.7g,8c,19b This active species could be either an insoluble nanoparticle or a soluble molecular species formed in the solution. To probe the possibility of nanoparticle formation we performed dynamic light scattering (DLS) measurements on the CPE solution before and after electrolysis. The DLS spectra obtained before and after the CPE experiment were found to be identical, ruling out the formation of insoluble nanoparticles (Fig. S28†),5f,7e and supporting the homogeneity of the CPE experiment. Considering the structural change for the cobalt peptoid complex during the catalysis,19b,20 we propose that CoTBE may act as a precatalyst, forming an active catalyst upon applying potential (this will be further discussed later in the paper).
 |
| | Fig. 4 (a) The evolution of O2 and (b) total accumulated charge during CPE in 0.1 M phosphate buffer at pH 7.0 containing 0.5 mM catalyst CoTBE and the buffer only using porous glassy carbon as the working electrode at +1.25 V vs. NHE for 10 hours. Evolution of O2 was measured with a fluorescent probe. | |
Homogeneity studies
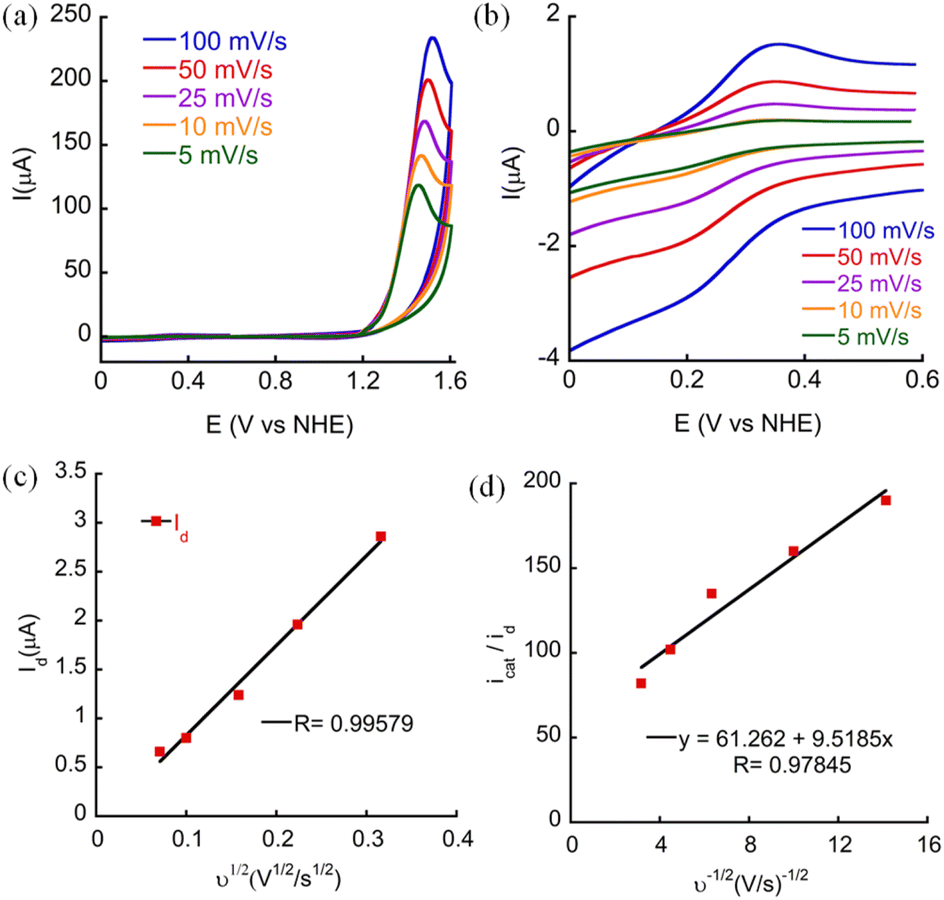
To assure that CoTBE is a soluble molecular catalyst and that the catalytic process is homogeneous, a CV scan was carried out in PBS at pH 7 in the absence of the catalyst (blank) and 20 continuous CV scans were performed in the presence of the catalyst using a glassy carbon (GC) working electrode (Fig. S29 and S30†).27 This experiment revealed that there is no increase in catalytic current suggesting that no film deposition on the electrode surface takes place during the catalytic process.10c In fact, it was observed that the catalytic current decreases after subsequent CV scans, possibly due to the formation of oxygen bubbles on the surface of the working electrode, decreasing its active surface area. To confirm that no cobalt oxide film was deposited on the electrode during the catalytic process, the working electrode was removed from the solution of CoTBE in 0.1 M PBS (pH 7.0) after 20 successive CV scans and it was rinsed with deionized water but not polished, and a CV scan was performed in a fresh buffer solution. This scan was identical to the blank CV scan, indicating that no active particles were absorbed on the working electrode. Moreover, scanning electron microscope (SEM) images of the GC electrode surface taken before and after 20 continuous scans showed no material deposition on the surface (Fig. S31†). Moreover, the CVs at different scan rates revealed that the peak current (id, corresponding to the reduction peak current of CoIII/II) varies linearly with the square root of the scan rate ν1/2 (Fig. 5a–c), which is consistent with the Randles–Sevcik equation displayed below:7e,27b,28,29| | | id = 0.446 ndFA[Co](ndFνDCo/RT)1/2 | (1) |
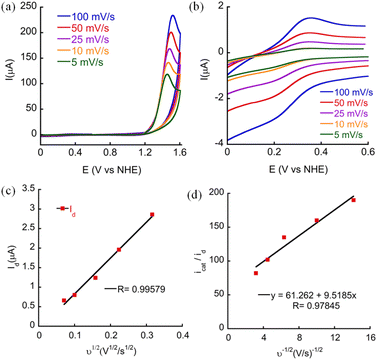 |
| | Fig. 5 CVs of 0.5 mM CoTBE at different scan rates in 0.1 M phosphate buffer pH 7 in a (a) broad scanning range and (b) narrow scanning range. (c) Linear regression of idversus ν1/2. (d) Linear regression of icat/idversus ν−1/2. | |
This implies that the process is diffusion controlled and thus, homogeneous.19a,28 Taken together, these outcomes indicate that the catalytic process is truly homogeneous.
Kinetic studies
CV scans of different concentrations of CoTBE were examined to gain some insight regarding its kinetics. The linear dependence of the catalytic peak current with the catalyst concentration (Fig. S32†) points out that WO is performed by a single site molecular catalyst with first order kinetics.30 Therefore, the catalytic process obeys the relationship displayed in eqn (2) as follows:28,31| | | icat = ncatFA[Co](kcatDCo)1/2 | (2) |
The correlation between icat/id and ν−1/2 can be obtained by dividing eqn (1) with eqn (2) and the value of the rate constant for water oxidation can be calculated using the linear slope of icat/id and ν−1/2 (Fig. 5d), as shown in eqn (3):7e,27b
| | | icat/id = 0.359ncat/nd3/2kcat1/2ν−1/2 | (3) |
From these equations, kcat, also defined as the turnover frequency (TOF) of the catalyst was calculated to be 44 s−1. This value is the highest reported compared with the other Co-based molecular electrocatalysts for WO and their catalytic activity was reported at pH 7 (see ESI, Table A1†). Further FOWA was performed from the CV data collected at different scan rates and the average Kobs was found to be 5.76 × 10 4 s−1 (Fig. S33†).32 The value calculated by this method is comparable to those of other reported Co-based molecular catalysts.7e,26
Structural stability of the catalyst
To gain some insight into the catalytic mechanism of CoTBE during WO, we wished to investigate its structural stability during the process. For this aim, we performed CV, UV-Vis, FTIR and ESI-MS analysis before and after 10 hours of CPE at an Eapp of +1.25 V. The CV recorded after the CPE showed that the intensity of the Co(II)/(III) oxidation wave was unaffected but the catalytic wave of CoTBE was completely suppressed (Fig. S34†). In addition, significant changes were observed in the UV-Vis spectrum of CoTBE after electrolysis: the intensity of the absorption band at 282 nm (associated with the n–π* transition of both Terpy and Bipy) increased, the absorption band at 314 nm (attributed to the MLCT transition of CoTBE) disappeared, and two new shoulder bands at 328 and 342 nm appeared (Fig. S35†). These observations suggest the dissociation of either Terpy or Bipy from the Co center, leading to the formation of new Co-peptoid complexes replacing CoTBE. As Terpy is a tridentate ligand that can stabilize the Co center more effectively than Bipy,33 we propose that it is the Bipy group that dissociates from the Co center,16j leading to the formation of a new dissociated Co-peptoid complex (see, Scheme 1).
 |
| | Scheme 1 Plausible mechanistic cycle of CoTBE for water oxidation, where X is H2O, H2PO4, ClO4 or their combination. | |
To support this hypothesis, we synthesized two control peptoids TE and BE with Terpy or Bipy as the only binding site, respectively, aiming to test the ability of each ligand-bound peptoid to coordinate Co. In both the peptoids, the 2nd binding site has been substituted by a napthyl group (Fig. 6). Both the peptoids were synthesized on a solid support via the submonomer method, cleaved from the resin, purified to >99% purity and analyzed by HPLC and MS (Fig. S36–S39†). To determine the binding properties of the peptoids TE and BE, we conducted UV-vis titration experiments with three different metal ions (Co, Ni and Zn) in PBS at pH 7. We observed that TE could bind all the three metal ions, whereas BE could only bind to Ni (Fig. S40 and S41†). To investigate which ligand dissociates from CoTBE during CPE, we conducted a CPE experiment with CoTBE and at the end of the reaction we collected the solution, dried it, re-dissolved the dried sample in PBS at pH 7 and performed a UV-vis titration experiment with all the three metal ions using the dried isolated solution from the CPE experiment. The resulting spectra were compared with the spectra of UV-vis titration experiments performed with TE and BE. Interestingly, when the dried CPE sample was titrated with Ni, we observed that two new bands appeared near 315 nm and 329 nm. These bands correspond to the binding of Ni (Table S1, ESI†) to the free binding site created during the CPE experiment (Fig. 7a). However, when the dried CPE sample solution was titrated with Co and Zn, no change in the UV-Vis spectrum was observed (Fig. S42†), indicating no binding. Based on the observation that both Co and Zn can be bound to TE but not to BE, the results from the post-CPE experiments imply that it is the Bipy ligand that dissociates during CPE. To gain more insights into the structure of the dissociated Co-peptoid complex (see, Scheme 1), FTIR and ESI-MS measurements were carried out before and after the CPE experiment. In the FTIR spectrum, a broad O–H stretching frequency was observed initially before electrolysis, indicating the presence of an ethanolic O–H group in the peptoid sequence. However, after electrolysis, additional broadening of O–H stretching frequency was observed in the region between 3000 and 3400 cm−1. The further broadening might be attributed to the stretching of the O–H bond of water molecules, indicating that a water molecule coordinates to Co during electrolysis, supporting the formation of the dissociated cobalt complex (see, Scheme 1).7h,20,22 In addition, a significant change in the CN bond stretching region (1500–1700 cm−1) was observed after electrolysis, possibly due to the dissociation of the Bipy center from Co during CPE (Fig. S43†). The ESI-MS analysis of the dried CPE sample solution showed masses of 938.7, 960.5 and 982.4, corresponding to the masses of [CoTBE(H2PO4)K2–H+], [CoTBE(H2PO4) (ClO4)] and [CoTBE(H2PO4) (ClO4) + Na–H+], respectively. These results support the formation of the dissociated Co-peptoid complex (see, Scheme 1), where X is H2O, H2PO4, ClO4 or their combination (Fig. S44 and S45†). Interestingly, when the isolated dried CPE solution was dissolved back in PBS at pH 7, the CV experiment showed that the activity of the complex was partially restored (Fig. S46†). We therefore suggest that some of the catalyst irreversibly decomposes during electrolysis to form the inactive complex VI, while some forms the active intermediate V, which continues the catalytic water oxidation cycle (see, Scheme 1). Altogether, these results demonstrate that CoTBE remains soluble during electrolysis, but its structure is modified during CPE.34 Together with this structural change we also observed that the pH of the solution decreased during the 10 hours of CPE to about 6 in the first 2 hours and to 5.4 in the following 2 hours and reached 4.5 at the end of the reaction. Accordingly, we wished to understand (i) whether the structural changes are a consequence of the drop in the pH, the oxidation process or both and (ii) whether the decrease in the catalytic activity after 10 hours of CPE is attributed to the drop in pH (according to the Le-Chatelier principle, WO is favorable at high pH and not favourable at low pH), to the structural changes of CoTBE or to both. To address the first question, we performed two experiments: UV-Vis titration of CoTBE with the chemical oxidant H2O2 and a CV experiment at pH 6 and 4.5. When we titrated CoTBE with sequential additions of 2.5% H2O2, a significant increase in the band near 282 nm, a decrease in the absorbance band at 314 nm and the appearance of two new bands near 328 and 342 nm were obtained, resulting in a spectrum which is identical to the spectrum obtained after electrolysis (Fig. 7b). This suggested that the structural change is a consequence of the oxidation process. On the other hand, CV performed at pH 6 already suggested a significant decrease in the catalytic activity compared to CV performed at pH 7, whereas CV performed at pH 4.5 suggested no catalytic activity. Notably, the latter CV was very similar to the CV obtained after the CPE experiment (Fig. S47†). To detect any structural changes that might occur at low pH, we also measured the UV-Vis spectrum of CoTBE in PBS at pH 6 and 4.5 for 24 hours. While there was almost no change in the spectrum of CoTBE at pH 6, the spectrum of CoTBE at pH 4.5 showed a significant increase in the intensity of the band at 282 nm and only a negligible change in the intensity of the band near 314 nm, indicating that this overall spectrum was very similar to the spectrum obtained after electrolysis (Fig. S48–S49†). All together, these results indicated that the structural change is a consequence of both the oxidation process and the drop in pH.
 |
| | Fig. 6 (a) Peptoid ligand TE and (b) peptoid ligand BE. | |
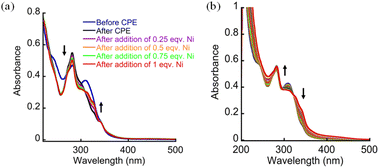 |
| | Fig. 7 (a) UV-Vis titration spectra of the dried sample after CPE with Ni and (b) titration of the 25 μM complex CoTBE with 1 μL aliquots of 2.5% H2O2 followed by UV-Vis spectroscopy. All the experiments were performed in 0.1 M PBS pH 7 at room temperature. | |
To address the second question, we re-adjusted the pH of the solution after 10 hours of CPE back to 7, analyzed it by UV-vis spectroscopy and performed a CV experiment on this solution. Interestingly, although the UV-Vis spectrum was identical to the one measured after CPE when the pH was 4.5 (indicating that CoTBE was not recovered and that the structural change is not reversible), the current intensity of the catalytic peak was about half of the catalytic peak current observed in the CV measured before CPE (Fig. S34†). This suggests that the decrease in activity depends on both the pH and the structural changes: it is partially related directly to the decrease in pH (which can be adjusted, thus making part of the decrease in activity to some reversible) and partly to some irreversible structural changes that occurred during CPE, resulting from both the oxidation process and the pH drop.
The role of Bipy and the ethanolic side chain as a second coordination sphere mimic that facilitates the electrocatalytic activity of CoTBE
Although CoTBE is structurally changing during WO, it is quite a remarkable electrocatalyst for WO; compared with all the Co-based molecular electrocatalysts reported, that operate in a pH range of 6–11, it is active at pH 7 with high TOF and the lowest overpotential compared to other electrocatalysts, including the ones operating at pH 6 for at least 10 hours, while remaining homogeneous in solution. The low overpotential and long durability of the catalyst might be related to the relatively slow decrease in the pH of the solution during CPE; we observed that during CPE the pH of the solution decreased from 7 to 6 in the first two hours, from 6 to 5.4 in the next two hours, and reached 4.5 at the end of the CPE experiment after 6 additional hours. This pH drop is significantly lower compared with other reported WO electrocatalytic systems, where the pH drop was about 2 units in the first two hours of CPE,19a,20 suggesting that the pH in our system is regulated. One option that can explain this regulation is that the Bipy ligand acts as a proton acceptor helping to control the pH drop of the solution upon rapid proton evolution during electrolysis. We previously showed that when there is one ethanolic side chain incorporated into a metallopeptoid WO electrocatalyst, the –OH group within this side chain participates in the stabilization of the complex during WO via H-bonding interactions.19 We therefore suggest that in this current system, protonation of Bipy is facilitated via H-bonding interactions with the ethanolic side chain, which stabilizes the protonated Bipy during WO and enables its participation in the proton transfer step. Hence, the ethanolic side chain acts as a proton shuttler within the second coordination sphere, enhancing the catalyst's overall stability and activity.35 To probe the significance of the ethanolic side chain in the electrocatalytic WO, we generated a set of control Co-peptoid complexes by modifying the sequence of TBE such that the ethanolic side chain was replaced by other groups. Thus, four additional Co-based peptoid complexes having a methoxy group (CoTB-OCH3), a methyl group (CoTB-CH3), or a benzyl group (CoTB-BZ) side chains instead of the ethanolic side chain, as well as CoTB having no additional side chain at the N-terminal (see ESI, Scheme S2†) were synthesized and characterized following the same procedure as that for CoTBE (Fig. S50–S65†). As shown in Fig. 8, the catalytic activity of all the control Co-peptoid complexes is much lower compared to that of CoTBE, indicating that the ethanolic group side chain plays a key role in facilitating the electrocatalytic activity of CoTBE towards WO. We suggest that the role of the –OH group is to act as a H-bonding donor that stabilizes the dissociated Bipy ligand in its protonated state, increasing its ability to serve as a proton acceptor at pH 7 (ref. 20 and 36) and thus facilitating the WO process. Based on these results, we propose that the peptoid, with its sidechains, acts as a second coordination sphere mimic, leading to the unique properties of CoTBE as a WO electrocatalyst, including its low overpotential and long durability.
 |
| | Fig. 8 CV scans of 0.1 M phosphate buffer solution at pH 7 in the absence of a catalyst and in the presence and of 0.5 mM CoTBE, CoTB-OCH3, CoTB-CH3, CoTB-BZ and CoTB (scan rates = 100 mV s−1). | |
Mechanistic studies and the proposed reaction mechanism
As we mentioned earlier, in solution CoTBE spontaneously coordinates to a water molecule, leading to the formation of [CoTBE(H2O)] (complex I, Scheme 1). The formation of I can be considered the first step in the WO process. We also saw that during electrocatalysis a structural change occurs, leading to the formation of a dissociated CoTBE complex coupled with the oxidation of CoIII to CoIV during electrolysis (Scheme 1, step 2). To quantify the number of proton and electron transfer(s) involved in the subsequent reaction in step 2, we performed differential pulse voltammetry (DPV) scans ranging from pH 6.5 to 9. The obtained results were then plotted versus the catalytic potentials to obtain a Pourbaix diagram. Based on the Pourbaix diagram, we suggest that in step 2 of the reaction mechanism, the oxidation from [CoIII–H2O] to [CoIVO] occurs via a 2H+/1e− transfer process (Scheme 1, step 2). This was supported by the obtained slope value of 0.126 (close to 0.12, Fig. 9a and S66†), which indicates the transfer of two protons and one electron. These transfers led to the formation of species II, in which Bipy is protonated by the protons evolved in this oxidation step. Subsequently, the kinetic isotope effect (KIE) of CoTBE was calculated based on different catalytic currents acquired in H2O and D2O (Fig. S67†). The lower catalytic current observed in D2O indicates that a proton from H2O is involved in this step. The KIE value was calculated to be 2.5 according to the equation kcat, H2O/kcat, D2O = (icat, H2O/icat, D2O)2.37 This obtained value suggests that the O–O bond formation step occurred via a water nucleophilic attack (WNA) process.26 To explore the possibility of the buffer solution (specifically HPO42−) acting as a proton acceptor during the O–O bond formation step, we obtained CVs of CoTBE at different buffer concentrations (0.025 to 0.1 M) and found that the catalytic peak current of WO remained constant irrespective of the buffer concentration (Fig. S68†). Based on this result, combined with the obtained KIE value we propose a WNA mechanism operating via cleavage of the O–H bond of water during the rate-limiting step – the O–O bond formation step-with a zero-order dependence of the catalytic current on the buffer concentration that rules out the possible role of HPO42− as a proton acceptor.26 These results support the potential role of the dissociated Bipy as an intramolecular proton acceptor stabilized via H-bonding interaction with the ethanolic group that assists in the proton transfer step during WO, thus enhancing the overall stability and activity of the catalyst.19,20,35,36 Consequently, in step 3 (Scheme 1), the high-valence cobalt-oxo intermediate undergoes nucleophilic attack by a water molecule leading to the formation of cobalt hydroperoxide intermediate (III).7d,f,g To confirm the formation of cobalt hydroperoxide species (III), CV experiments of buffer solution containing CoTBE in the presence of H2O2 were performed. These showed that both CoIII/IV peak and the catalytic peak current densities increased with the H2O2 concentration. This result suggests that CoTBE can catalyze the oxidation of the peroxide intermediate (Fig. S69†).38 Moreover, we have performed spectroelectrochemistry IR measurements with 1 mM of CoTBE at an applied potential of 1.25 V vs. NHE in 0.1 M PBS at pH 7. The IR spectrum showed the generation of a distinctive broad band between 870 and 895 cm−1 during electrolysis (Fig. 9b), which can be attributed to the ν(O–O) stretch.39
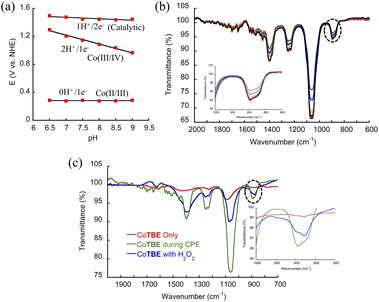 |
| | Fig. 9 (a) Pourbaix diagram of CoTBE in 0.1 M PBS in a pH range between 6.5 and 9. (b) FTIR spectra of the CoTBE (1 mM, 5 mL) complex during electrolysis at 1.25 V vs. NHE in 0.1 M PBS. At pH 7, (c) FTIR spectra of the CoTBE (1 mM, 1 mL) complex after addition of few drops of 2% H2O2 in 0.1 M PBS at pH 7. | |
To further support these findings, we performed a similar IR measurement after H2O2 was directly added to the catalyst solution. In this case also, a new broad band within the same spectral range emerged (Fig. 9c), which was initially absent in CoTBE solution.19a The IR results provide another strong indication that the peroxo species is electrochemically generated as an active species during catalysis. Overall, these findings indicate the formation of species (III) (Scheme 1). This is followed by 1H+/2e− transfer as evident from the Pourbaix diagram (Fig. 9a) to form species (IV) (Scheme 1). Finally, one electron transfer taking part in the last stage of the reaction occurs,40 leading to the release of O2 and subsequent coordination of H2O molecule to form the active intermediate V, which restarts the catalytic cycle while some of the catalyst irreversibly decomposes during electrolysis to form the inactive complex VI.
Conclusions
The challenge of stabilizing a Co-based WO electrocatalyst to perform in a homogeneous catalytic system at neutral pH, with low overpotential and high Faradaic efficiency, is addressed here by applying an intramolecular Co-peptoid complex. This complex, CoTBE, can catalyze O2 evolution for at least 10 hours at pH 7, with a high faradaic efficiency of 92% and a TOF of 44 s−1 (the highest reported at pH 7), and these, with an overpotential of only about 430 mV, are the lowest reported to date for Co-based homogeneous WO electrocatalysts. Based on the experimental characterization and control experiments we found that during electrolysis, the Bipy ligand dissociates from the Co-center and hence can act as a proton acceptor for the protons evolved during the reaction. Furthermore, the protonated Bipy ligand is stabilized by the –OH group of the ethanolic side chain, located near the catalytic center, via H-bonding. Thus, both Bipy and ethanol side chains form a second coordination sphere mimic around the Co center, which, akin to enzymatic systems and specifically to the tyrosine and histidine amino acid residues near the OEC of photosystem II, enables the formation of the active WO electrocatalyst and its unique properties including low overpotential and long durability. Altogether we showed that by rational design of peptoid scaffolds that provide both the first and second coordination spheres arround a Co center, it is possible to improve the stability and activity of the resulting WO electrocatalyst. Thus, our study represents a step forward towards developing stable and efficient biomimetic Co-based complexes for homogeneous electrocatalytic WO towards renewable energy production.
Data availability
All experimental data are available in the manuscript and ESI.†
Author contributions
S. P. conducted all the experiments and analyzed the data. G. M. conceptualized and directed the project, analyzed the data and drafted the manuscript together with S. P.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The research was funded by the Israel Ministry of Energy, grant number 221-11-090. The authors thank Mrs Larisa Panz for ESI-MS measurements and Dr Boris Tumansky from the Schulich Faculty of Chemistry for EPR measurements. The authors thank Dr Guilin Ruan for the fruitful discussions.
Notes and references
-
(a) S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC;
(b) J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CAS.
-
(a) H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS;
(b) A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS;
(c) M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446 CAS.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338 RSC.
-
(a) M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886 CrossRef CAS PubMed;
(b) K. J. Young, B. J. Brennan, R. Tagore and G. W. Brudvig, Acc. Chem. Res., 2015, 48, 567 CrossRef CAS PubMed;
(c) G. Maayan, N. Gluz and G. Christou, Nat. Catal., 2018, 1, 48–54 CrossRef CAS;
(d) T. Ghosh, G. Christou and G. Maayan, Angew. Chem., Int. Ed., 2019, 58, 2785–2790 CrossRef CAS PubMed;
(e) N. Gluz, G. Christou and G. Maayan, Chem.–Eur. J., 2021, 27, 6034–6043 CrossRef CAS PubMed;
(f) S. Biswas, S. N. Chowdhury, P. Lepcha, S. Sutradhar, A. Das, T. K. Paine, S. Paul and A. N. Biswas, Dalton Trans., 2023, 52, 16616–16630 RSC.
-
(a) W. C. Ellis, N. D. Mcdaniel, S. Bernhard and J. T. Collins, J. Am. Chem. Soc., 2010, 132, 10990–10991 CrossRef CAS;
(b) C. Panda, J. Debgupta, D. D. Díaz, K. K. Singh, S. S. Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273–12282 CrossRef CAS;
(c) M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465–468 CrossRef CAS PubMed;
(d) S. Karim, A. Chakraborty, D. Samanta, E. Zangrando, T. Ghoshd and D. Das, Catal. Sci. Technol., 2020, 10, 2830 RSC.
-
(a) T. Nakazono and T. Wada, Inorg. Chem., 2021, 60, 1284–1288 CrossRef CAS;
(b) D. K. Dogutan, R. McGuire and D. G. Nocera, J. Am. Chem. Soc., 2011, 133, 9178–9180 CrossRef CAS;
(c) D. Das, S. Pattanayak, K. K. Singh, B. Garai and S. Sen Gupta, Chem. Commun., 2016, 52, 11787 RSC;
(d) H.-Y. Du, Si-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS;
(e) P. Lepcha, S. Biswas, S. N. Chowdhury, S. Bose, J. Debgupta, S. Paul and A. N. Biswas, Eur. J. Inorg. Chem., 2023, 26, e202200611 CrossRef CAS;
(f) Y. Chen, X. Meng, X. Chen, X. Li, H. Ye, S. Liu, Z. Ruan, X. Liang and J. Lin, Sustainable Energy Fuels, 2023, 7, 242–247 RSC;
(g) H. Zheng, H. Ye, T. Xu, K. Zheng, X. Xie, B. Zhu, X. Wang, J. Lin and Z. Ruan, New J. Chem., 2022, 46, 7522–7527 RSC;
(h) S. Pahar, K. Majee and G. Maayan, Eur. J. Inorg. Chem., 2024, 27, e2023005 CrossRef;
(i) J. F. Khosrowabadi Kotyk, C. M. Hanna, R. L. Combs, J. W. Ziller and J. Y. Yang, Chem. Sci., 2018, 9, 2750 RSC;
(j) N. D. McMillion, A. W. Wilson, M. K. Goetz, M.-C. Chang, C.-C. Lin, W.-J. Feng, C. C. L. McCrory and J. S. Anderson, Inorg. Chem., 2019, 58, 1391 CrossRef CAS;
(k) A. Das, A. Ali, G. Gupta, A. Santra, P. Jain, P. P. Ingole, S. Paul and S. Paria, ACS Catal., 2023, 13, 5285–5297 CrossRef CAS.
-
(a) J. Shen, M. Wang, T. He, J. Jiang and M. Hu, Chem. Commun., 2018, 54, 9019–9022 RSC;
(b) J. W. Wang, W. J. Liu, D. C Zhong and T. B. Lu, Coord. Chem. Rev., 2019, 378, 237–261 CrossRef CAS;
(c) M. Zhang, M.-T. Zhang, C. Hou, Z.-F. Ke and T.-B. Lu, Angew. Chem., Int. Ed., 2014, 53, 13042–13048 CrossRef CAS;
(d) L.-H. Zhang, F. Yu, Y. Shi, F. Li and H. Li, Chem. Commun., 2019, 55, 6122–6125 RSC.
-
(a) S. M. Barnett, K. I. Goldberg and J. M. Mayer, Nat. Chem., 2012, 4, 498–502 CrossRef CAS;
(b) M. K. Coggins, M.-T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226 CrossRef CAS PubMed;
(c) K. J. Fisher, K. L. Materna, B. Q. Mercado, R. H. Crabtree and G. W. Brudvig, ACS Catal., 2017, 7, 3384 CrossRef CAS;
(d) S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586 CrossRef CAS;
(e) Q.-Q. Hu, X.-J. Su and M.-T. Zhang, Inorg. Chem., 2018, 57, 10481 CrossRef CAS;
(f) Y. Liu, Y. Han, Z. Zhang, W. Zhang, W. Lai, Y. Wang and R. Cao, Chem. Sci., 2019, 10, 2613–2622 RSC;
(g) M. Bera, K. Keshari, A. Bhardwaj, G. Gupta, B. Mondal and S. Paria, Inorg. Chem., 2022, 61, 3152–3165 CrossRef CAS PubMed.
-
(a) V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238 CAS;
(b) H.-Y. Wang, E. Mijangos, S. Ott and A. Thapper, Angew. Chem., Int. Ed., 2014, 53, 14499 CrossRef CAS PubMed;
(c) J.-W. Wang, P. Sahoo and T.-B. Lu, ACS Catal., 2016, 6, 5062 CrossRef CAS.
-
P. Flowers, K. Theopold, R. Langley and W. R. Robinson, Chemistry 2e, OpenStax, Houston, Texas, 2019 Search PubMed.
-
(a) J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehneh and E. Relsner, Nat. Catal., 2019, 2, 354 CrossRef CAS;
(b) E. C. Constable, K. Harris, C. E. Housecroft, M. Neuburger and J. A. Zampese, Dalton Trans., 2011, 40, 11441 RSC;
(c) K. Majee and S. K. Padhi, New J. Chem., 2019, 43, 3856 RSC.
- T. Zhang, C. Wang, S. Liu, J. L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
-
(a)
J. Seo, B.-C. Lee and R. N. Zuckermann in Comprehensive Biomaterials, ed. P. Ducheyne, K. E. Healy, D. W. Hutmacher, D. W. Grainger, C. J. Kirkpatrick, Elsevier, Amsterdam, 2011, vol. 2, pp. 53–76 Search PubMed;
(b) G. Maayan, Eur. J. Org Chem., 2009, 5699–5710 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646 CrossRef CAS.
-
(a) G. Maayan, M. D. Ward and K. Kirshenbaum, Chem. Commun., 2009, 1, 56–58 RSC;
(b) G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(33), 13679–13684 CrossRef CAS;
(c) L. Zborovsky, A. Smolyakova, M. Baskin and G. Maayan, Chem.–Eur. J., 2018, 24(5), 1159–1167 CrossRef CAS PubMed;
(d) M. Baskin, H. Zhu, Z. W. Qu, J. H. Chill, S. Grimme and G. Maayan, Chem. Sci., 2019, 10, 620–632 RSC;
(e) L. Zborovsky, H. Tigger-Zaborov and G. Maayan, Chem.–Eur. J., 2019, 25(38), 9098–9107 CrossRef CAS PubMed;
(f) M. Baskin and G. Maayan, Dalton Trans., 2018, 47, 10767–10774 RSC;
(g) G. Maayan and L.-K. Liu, Biopolymers, 2011, 96, 679–687 CrossRef CAS PubMed;
(h) T. Ghosh, N. Fridman, M. Kosa and G. Maayan, Angew. Chem., Int. Ed., 2018, 57, 7703–7708 CrossRef CAS;
(i) M. Baskin, L. Panz and G. Maayan, Chem. Commun., 2016, 52, 10350 RSC;
(j) A. E. Behar and G. Maayan, Chem.–Eur. J., 2023, 29, e202301118 CrossRef CAS PubMed;
(k) P. Ghosh, I. Rozenberg and G. Maayan, J. Inorg. Biochem., 2021, 217, 111388 CrossRef CAS;
(l) P. Ghosh and G. Maayan, Chem. Sci., 2020, 11, 10127–10134 RSC.
-
(a) H. R. Reese, C. C. Shanahan, C. Proulx and S. Menegatti, Acta Biomater., 2020, 102, 35–74 CrossRef CAS;
(b) A. D'Amato, P. Ghosh, C. Costabile, G. Della Sala, I. Izzo, G. Maayan and F. De Riccardis, Dalton Trans., 2020, 49, 6020–6029 RSC;
(c) D. Chandra Mohan, P. Ghosh, T. Ghosh and G. Maayan, Chem.–Eur. J., 2020, 26, 9573–9579 CrossRef;
(d) R. Schettini, B. Nardone, F. D. Riccardis, G. D. Sala and I. Izzo, Eur. J. Org Chem., 2014, 7793–7797 CrossRef CAS.
-
(a) K. J. Prathap and G. Maayan, Chem. Commun., 2015, 51, 11096 RSC;
(b) D. C. Mohan, A. Sadhukha and G. Maayan, J. Catal., 2017, 355, 139 CrossRef;
(c) Y. Stamatin and G. Maayan, Eur. J. Org Chem., 2020, 3147 CAS.
-
(a) T. Ghosh, P. Ghosh and G. Maayan, ACS Catal., 2018, 8, 10631 CrossRef CAS;
(b) G. Ruan, P. Ghosh, N. Fridman and G. Maayan, J. Am. Chem. Soc., 2021, 143, 10614–10623 CAS.
- G. Ruan, L. Engelberg, P. Ghosh and G. Maayan, Chem. Commun., 2021, 57, 939–942 CAS.
-
(a) G. Maayan, B. Yoo and K. Kirshenbaum, Tetrahedron Lett., 2009, 50, 4297 CrossRef CAS;
(b) M. Baskin and G. Maayan, Chem. Sci., 2016, 7, 2809 RSC;
(c) P. Ghosh and G. Maayan, Chem.–Eur. J., 2021, 27, 1383–1389 CAS.
- H. A. Younus, N. Ahmad, A. H. Chughtai, M. Vandichel, M. Busch, K. V. Hecke, M. Yusubov, S. Song and F. Verpoort, ChemSusChem, 2017, 10, 862–875 CAS.
- J. Shen, X. Zhang, M. Cheng, J. Jiang and M. Wang, ChemCatChem, 2020, 12, 1302–1306 CrossRef CAS.
- S. Aroua, T. K. Todorova, P. Hommes, L. M. Charnoreau, H. U. Reissig, V. Mougel and M. Fontecave, Inorg. Chem., 2017, 56, 5930 CrossRef CAS.
- H. M. Shahadat, H. A. Younus, N. Ahmad, S. G. Zhang, S. Zhuiykov and F. Verpoort, Chem. Commun., 2020, 56, 1968–1971 RSC.
- S. Biswas, S. Bose, J. Debgupta, P. Das and A. N. Biswas, Dalton Trans., 2020, 49, 7155–7165 RSC.
-
(a) D. J. Wasylenko, R. D. Palmer, E. Schott and C. P. Berlinguette, Chem. Commun., 2012, 48, 2107–2109 RSC;
(b) P. K. Das, S. Bhunia, P. Chakraborty, S. Chatterjee, A. Rana, K. Peramaiah, M. M. Alsabban, I. Dutta, A. Dey and K.-W. Huang, Inorg. Chem., 2021, 60, 614–622 CrossRef CAS.
-
A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
-
R is the ideal gas constant (8.314 J mol−1 K−1), T is the absolute temperature, 298 K; A is surface area of working electrode, (0.07 cm2), nd is the number of electron transferred in the corresponding process, nd =1; and [Co] is the bulk concentration of the CoTBE in solution.
- S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and T. M. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139(25), 8586–8600 CrossRef CAS PubMed.
-
A is the electrode surface area, F is the Faraday constant, icat is the peak current of catalytic wave, [Co] is the bulk concentration of the CoTBE in solution, DCo is diffusion coefficient of the catalyst, and ncat is the number of electrons transferred in each catalytic cycle which is 4 for water oxidation.
-
(a) C. Costentin, S. Drouet, M. Robert and J. M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS;
(b) C. Costentin and J. M. Savéant, ChemElectroChem, 2014, 1, 1226–1236 CrossRef CAS.
- D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49(5), 2202–2209 CrossRef CAS.
- D. D. Boer, Q. Siberie, M. A. Siegler, T. H. Ferber, D. C. Moritz, J. P. Hofmann and D. G. H. Hetterscheid, ACS Catal., 2022, 12, 4597–4607 CrossRef CAS.
-
(a) G. Smolentsev, M. A. Soldatov, B. Probst, C. Bachmann, N. Azzaroli, R. Alberto, M. Nachtegaal and J. A. van Bokhoven, ChemSusChem, 2018, 11, 3087–3091 CrossRef CAS;
(b) J. Xie, Q. Zhou, C. Li, W. Wang, Y. Hou, B. Zhanga and X. Wang, Chem. Commun., 2014, 50, 6520–6522 RSC;
(c) P. Wang, G. Liang, C. E. Webster and X. Zhao, Eur. J. Inorg. Chem., 2020, 3534–3547 CrossRef CAS;
(d) Z. Han, L. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659–14669 CrossRef CAS;
(e) F. H. Westheimer and O. T. Benfey, J. Am. Chem. Soc., 1956, 78(20), 5309–5311 CrossRef CAS;
(f) K. Nakamoto, J. Phys. Chem., 1960, 64, 1421 CrossRef.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS.
- Z. Chen, J. J. Concepcion, X. Hu, W. Yang, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(16), 7225–7229 CrossRef CAS.
- J. Shen, M. Wang, T. He, J. Jiang and M. Hu, Chem. Commun., 2018, 54, 9019–9022 RSC.
-
(a) D. D. Malik, A. Chandra, M. S. Seo, Y.-M. Lee, E. R. Farquhar, S. Mebs, H. Dau, K. Ray and W. Nam, Dalton Trans., 2021, 50, 11889 RSC;
(b) J. N. Campbell, A. C. Dengel and W. P. Griffith, Polyhedron, 1989, 8, 1379–1386 CrossRef.
- G. Ruan, N. Fridman and G. Maayan, Chem.–Eur. J., 2022, 28, e2022024 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*