
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pair of conjoined trinuclear sub-frameworks in a pentanuclear double-cavity discrete coordination cage†
Shruti
Sharma
,
Shobhana
Krishnaswamy
 ,
Soumyakanta
Prusty‡
and
Dillip Kumar
Chand
*
,
Soumyakanta
Prusty‡
and
Dillip Kumar
Chand
*
IoE Center of Molecular Architecture, Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: dillip@iitm.ac.in
First published on 12th June 2024
Abstract
Combination of Pd(II) with selected bis-monodentate ligands produces the familiar multinuclear PdmL2m type self-assembled “single-cavity discrete coordination cages” (SCDCC). If the ligand provides parallel coordination vectors, then it forms a binuclear Pd2L4 type cage, whereas utilization of ligands having appropriately divergent coordination vectors results in specific higher nuclear complexes. In contrast, preparation of emergent “multi-cavity discrete coordination cages” (MCDCC) using Pd(II) and designer ligands is quite captivating where the neighboring cavities of the framework are conjoined with each other through a common metal center. A pair of conjoined binuclear Pd2L4 type sub-frameworks are present in a trinuclear Pd3L4 type double-cavity cage prepared from Pd(II) and a tris-monodentate ligand having parallel coordination vectors. The present work envisioned a design to make double-cavity coordination cages having a pair of conjoined trinuclear Pd3L6 type sub-frameworks. To fulfill the objective we combined Pd(II) with a mixture of designer bis-monodentate ligand (L) and tris-monodentate ligand (L′) in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:4 ratio in one pot to afford the targeted pentanuclear
:4:4 ratio in one pot to afford the targeted pentanuclear 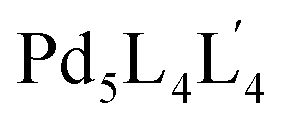 type cage. The choice of bis-monodentate ligand L is based on the divergent nature of the coordination vectors suitable to produce a Pd3L6 type SCDCC. The tris-monodentate ligand L′ having two arms is designed in such a manner that each of the arms reasonably resembles L. Study of the complexation behavior of Pd(II) with L′ provided additional guiding factors essential for the successful making of
type cage. The choice of bis-monodentate ligand L is based on the divergent nature of the coordination vectors suitable to produce a Pd3L6 type SCDCC. The tris-monodentate ligand L′ having two arms is designed in such a manner that each of the arms reasonably resembles L. Study of the complexation behavior of Pd(II) with L′ provided additional guiding factors essential for the successful making of 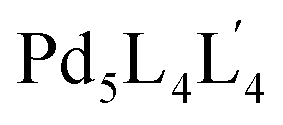 type MCDCC by integrative self-sorting. A few other MCDCC including lower symmetry versions were also prepared in the course of the work.
type MCDCC by integrative self-sorting. A few other MCDCC including lower symmetry versions were also prepared in the course of the work.
1 Introduction
Building of coordination architectures by a metal-driven self-assembly route using the participants from a library of metal and ligand components has strategic importance in supramolecular chemistry.1–9 Combination of Pd(II) with one type of symmetrical bis-monodentate ligand to prepare “single-cavity discrete coordination cages” (SCDCC) of PdmL2m formulations is well known.10–15Fig. 1A and B depict binuclear Pd2L4 and trinuclear Pd3L6 type cages (simplest but most studied PdmL2m). Diverging coordination vectors and appropriate bend angles of the ligands are important parameters that decide the nuclearity of the ensuing cages. Preparation of Pd(II)-based low-symmetry SCDCC has been achieved by either mixing of Pd(II) with a designer unsymmetrical ligand14,16–21 to afford orientational selectivity of ligand components, or mixing of Pd(II) with more than one type of ligand14,22–26 targeting mixed ligated integrative self-sorting. | ||
| Fig. 1 Cartoon representation depicting Pd(II)-based (A) and (B) single-cavity bi-/trinuclear regular cages, and (C)–(H) double-cavity tri-/tetra-/pentanuclear conjoined cages (the constituent ligands are also shown). The depicted conjoined cages have characteristic metal sharing where apparent conjoining of (C) a “binuclear” with a “binuclear” of equal size, (D) a “binuclear” with a “binuclear” of unequal size, (E)/(G) a “binuclear” with a “trinuclear”, and (F)/(H) a “trinuclear” with a “trinuclear” are presented. | ||
Making the rare “multi-cavity discrete coordination cages” (MCDCC) is quite captivating because multiple but defined number of cavities are present in their architectures.14,27–29 This is in contrast to the single-cavity and infinite cavities of the prevalent SCDCC and “metal organic frameworks” (MOF), respectively.30–33 We introduced34 a double-cavity trinuclear Pd3L4 type MCDCC (Fig. 1C(i)); such conjoined cages of Pd3L4 type have received considerable attention14,35–39 recently. In the framework of a Pd3L4 type cage there are two units of Pd2L4 type sub-framework that are apparently conjoined with each other using a common square planar coordination environment of a Pd(II) centre. Low-symmetry Pd3L4 cages possessing two unequal sized Pd2L4 sub-frameworks have been reported40–42 as shown in Fig. 1D(i). Another class of double-cavity tetranuclear conjoined cage where a Pd2L4 type sub-framework is conjoined with a Pd3L6 type sub-framework (see Fig. 1E) has been reported40 by us that is a sole example of its own type.
In the present work we have successfully lowered the symmetry of the above mentioned tetranuclear MCDCC (compare Fig. 1E with G). However, the major disclosure of this work is the achievement of an entirely new design that is a double-cavity pentanuclear conjoined cage where a triangular Pd3L6 type sub-framework is conjoined with another Pd3L6 type sub-framework as shown in Fig. 1F; further, the symmetry of the pentanuclear cage could be lowered as shown in Fig. 1H. One-pot complexation of Pd(II) with a bis-monodentate ligand and a tris-monodentate ligand in a 5:4:4 ratio is required for achieving the targeted pentanuclear complexes. A thorough analysis of the results obtained provides us with necessary guidelines for the making of double-cavity pentanuclear MCDCC.
2 Results and discussion
Design of the double-cavity pentanuclear conjoined cage
Making of the double-triangle type pentanuclear conjoined cages, depicted in Fig. 1F and H, via complexation of Pd(II) with tailored ligands is the primary objective of this work. Designing aspects of ligands to achieve the targeted cages is described hereafter. Complexation of Pd(II) with a bis-monodentate ligand such as Lc (or Ld) having appropriate divergence of the coordination vector, hence suitable bend angle, would form the well-known Pd3L6 type trinuclear framework, i.e. [Pd3(Lc)6]6+ (or [Pd3(Ld)6]6+), shown in Fig. 1B.43 It should be possible to prepare tris-monodentate ligands such as Lcc and Ldd by taking inspiration from Lc and Ld type ligands (see Fig. 1C(ii) for Lcc). Combination of Pd(II) with Lcc would not result in a discrete structure keeping the Pd3L6 type sub-framework intact. Thus, it might form a strained coordination cage like [Pd3(Lcc)4]6+ (Fig. 1C(ii)) and/or oligomerized complexes unless the ligand finds way to form an alternate but sufficiently stable cage framework; the same comments apply for complexation of Pd(II) with Ldd.We asked ourselves whether or not a one-pot mixture of Pd(II), Lc and Lcc would result in integrative self-sorting. The answer being: in order to avoid the imposition of strain/oligomerization mentioned above, narcissistic (or statistical) self-sorting would not occur, hence giving way to integrative self-sorting from a mixture of Pd(II), Lc and Lcc in a 5:4:4 ratio to afford a double-cavity conjoined cage like [Pd5(Lc)4(Lcc)4]10+ (Fig. 1F). Symmetry of this pentanuclear complex can be lowered locally at the trinuclear moieties by mixing Pd(II), Ld and Lcc to prepare a conjoined cage, i.e. [Pd5(Ld)4(Lcc)4]10+ (Fig. 1H), on the condition that the ligands Lc and Ld are of comparable lengths or averaged non-bonded distances between Pd(II) centres in [Pd3(Lc)6]6+ are comparable with that of [Pd3(Ld)6]6+. Similarly, cages like [Pd5(Ld)4(Ldd)4]10+ and [Pd5(Lc)4(Ldd)4]10+ are conceivable. However, the process of integrative self-sorting might get inhibited and hence narcissistic/statistical self-sorting might partially or completely take over if the ligand Lcc or Ldd finds alternate stable architectures without involving the ancillary ligand Lc or Ld.
The designs shown in Fig. 1F and H are practically realised in the present work with logical ligand designs where the orthogonal isomer-II is formed in preference to the planar isomer-I. Detailed investigation of the ligand systems of this work assisted in the mapping of Pd(II)-coordination behaviour of individual ligands (Lcc and Ldd) with the prospect of their participation in narcissistic or integrative self-sorting in the presence of an ancillary ligand like Lc or Ld. In the course of our research work, all cage varieties shown in Fig. 1 (except 1A, 1C(i) and 1D(i)) were prepared.
Synthesis of ligands L1 to L6
For a systematic study, we have prepared a series of pyridine-appended semi-flexible/-rigid organic ligands (L1–L6) as shown in Fig. 2. This series includes symmetrical bis-monodentate ligands (L1 and L2), unsymmetrical tris-monodentate ligands (L3 and L4), and symmetrical tris-monodentate ligands (L5 and L6). The conformations of ligands shown in Fig. 2 are selected on the basis of their probable conformation in the targeted complexes (as learned during the course of the work); some of the related conformers of L1, L2, L5 and L6 are shown in ESI Fig. S1 and S2.†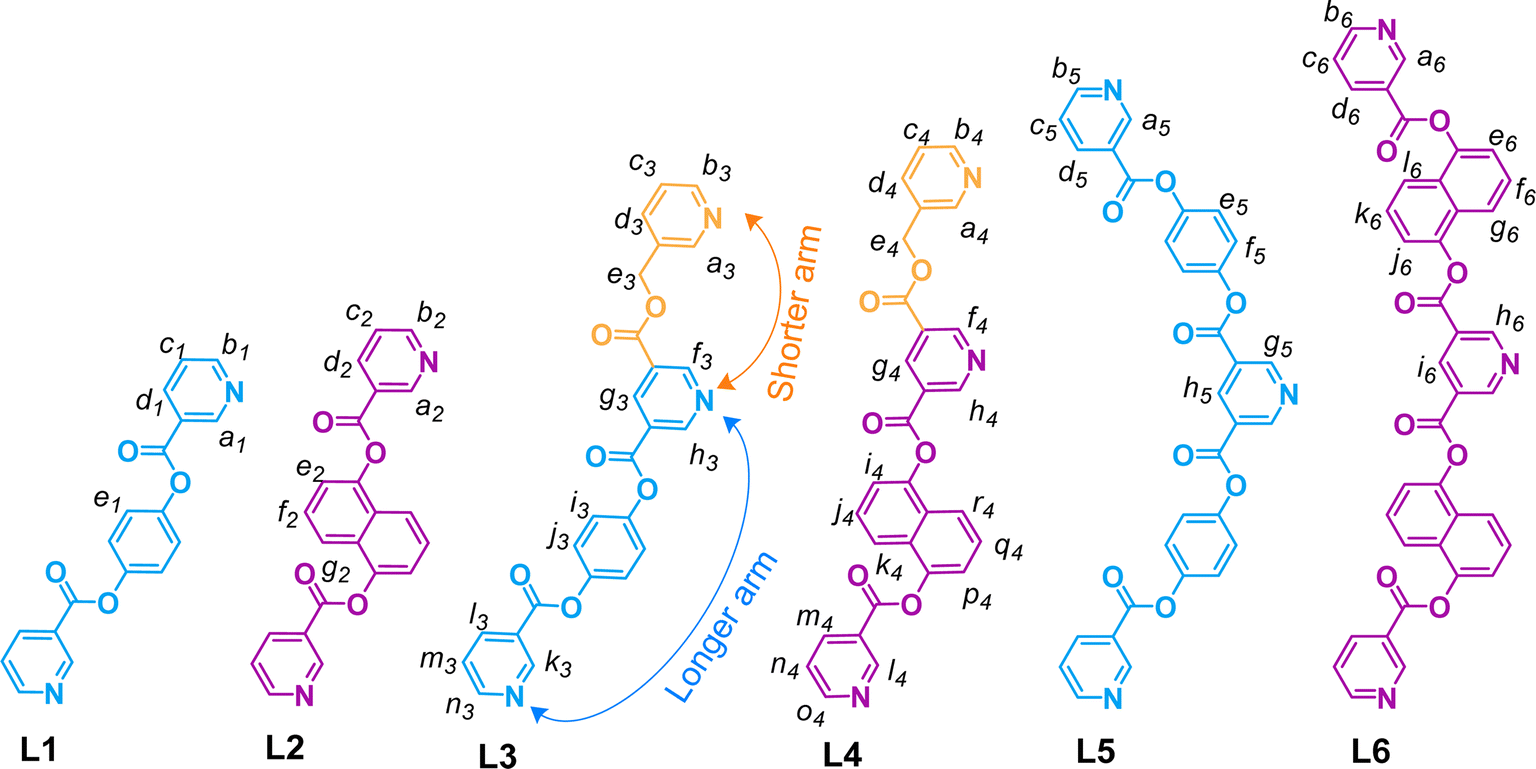 | ||
| Fig. 2 Chemical structures of the symmetrical bis-monodentate (L1, L2), unsymmetrical tris-monodentate (L3, L4) and symmetrical tris-monodentate (L5, L6) ligands. | ||
The ligands L1 and L2 were synthesized, by slight modification of known procedures,44,45 in one-step condensation reactions of two equivalents of nicotinic acid with one equivalent of 1,4-dihydroxybenzene and 1,5-dihydroxynaphthalene, respectively (ESI Schemes S1 and S2†). Synthesis of the ligands L3–L6 required esterification reactions using four possible combinations comprising of one of the mono-/dicarboxylic acid precursors (from (i) pyridine-3,5-dicarboxylic acid and (ii) 5-((pyridin-3-ylmethoxy)carbonyl)nicotinic acid, A) and one of the monohydroxy precursors (from (i) 4-hydroxyphenyl nicotinate, B, and (ii) 5-hydroxynapthalene-1-yl nicotinate, C). The precursor A was prepared following a method established in our laboratory40 whereas B and C were prepared by condensation of nicotinic acid with three equivalents of 1,4-dihydroxybenzene and 1,5-dihydroxynapthalene, respectively (ESI Schemes S3 and S4†). Then, the ligands L3 and L4 were synthesized in one-step condensation reactions of the monocarboxylic acid A with monohydroxy compounds B and C, respectively (ESI Schemes S5 and S6†). The ligands L5 and L6 were obtained in one-step condensation reactions of pyridine-3,5-dicarboxylic acid with B and C, respectively (ESI Schemes S7 and S8†). All the ligands except L5 are soluble in DMSO/DMSO-d6; ligand L5 is sparingly soluble in CHCl3/CDCl3. The precursors B and C and all the ligands were characterized by ESI-MS and various NMR techniques including 1H, 13C (not for L5), H–H COSY, C–H COSY (not for L5) and H–H NOESY (not for L1 and L2) (ESI Fig. S3–S46†). The ligands L1 and L2 are known44,45 whereas the ligands L3–L6 are new. However, L1 has not been used for complexation with any metal ions but it is ribosylated at pyridine nitrogen positions to prepare compounds of pharmaceutical values.44 The ligand L2 binds as a guest in the cleft of a specific bis-zinc porphyrin host.45
Complexation of Pd(II) separately with bis-monodentate ligands L1 and L2
Complexation of Pd(NO3)2 with the ligand L1 in a 1:2 ratio (Scheme 1(i), ESI Scheme S9†) was performed in DMSO-d6 at room temperature (27 °C) keeping 10 mM as the concentration with respect to Pd(II). Spontaneous formation (within 5 min) of a single discrete product was confirmed by monitoring the reaction mixture using 1H NMR spectroscopy. The spectrum (Fig. 3(ii)) showed a single set of downfield-shifted sharp signals with substantial shift for inward- and outward-pointing α-pyridine protons (Δδ = 0.85 and 0.87 ppm for Ha1 and Hb1, respectively) with respect to the free ligand (Fig. 3(i and ii)). ESI-MS study indicated the formation of a Pd3L6 type complex: [Pd3(L1)6](NO3)6, 1a (ESI Fig. S54†).
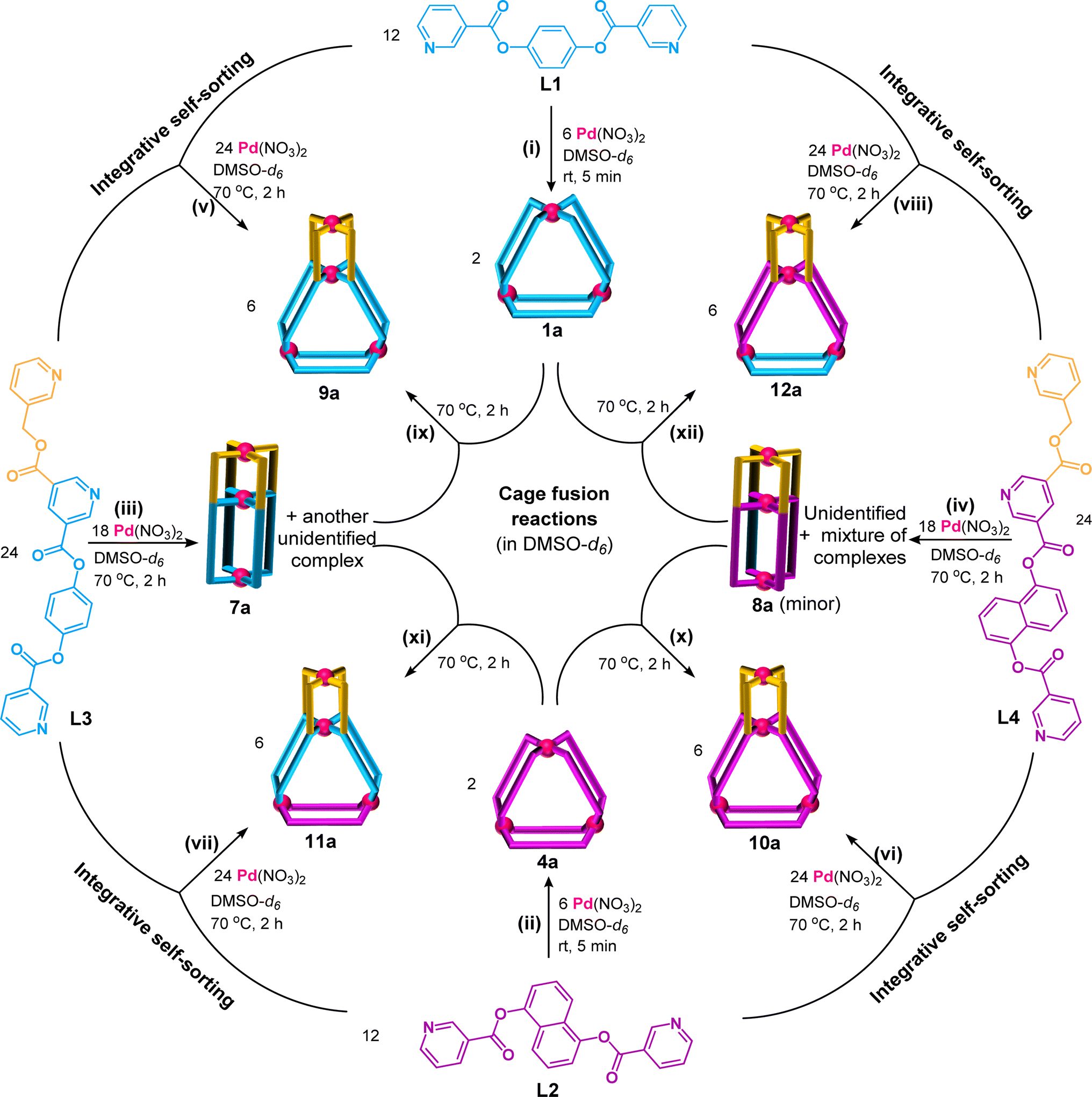 | ||
| Scheme 1 ((i) to (iv)) Complexation of Pd(NO3)2 with one type of ligand, ((v) to (viii)) complexation of Pd(NO3)2 with two types of ligands, and ((ix) to (xii)) cage fusion reactions, to prepare (i) [Pd3(L1)6](NO3)6, 1a; (ii) [Pd3(L2)6](NO3)6, 4a; (iii) [Pd3(L3)4](NO3)6, 7a; (iv) [Pd3(L4)4](NO3)6, 8a and unidentified complexes; (v)/(ix) [Pd4(L1)2(L3)4](NO3)8, 9a; (vi)/(x) [Pd4(L2)2(L4)4](NO3)8, 10a; (vii)/(xi) [Pd4(L2)2(L3)4](NO3)8, 11a; and (viii)/(xii) [Pd4(L1)2(L4)4](NO3)10, 12a. (Stoichiometry of reactants are factored and mapped in all three schemes to get integral number for all products.) | ||
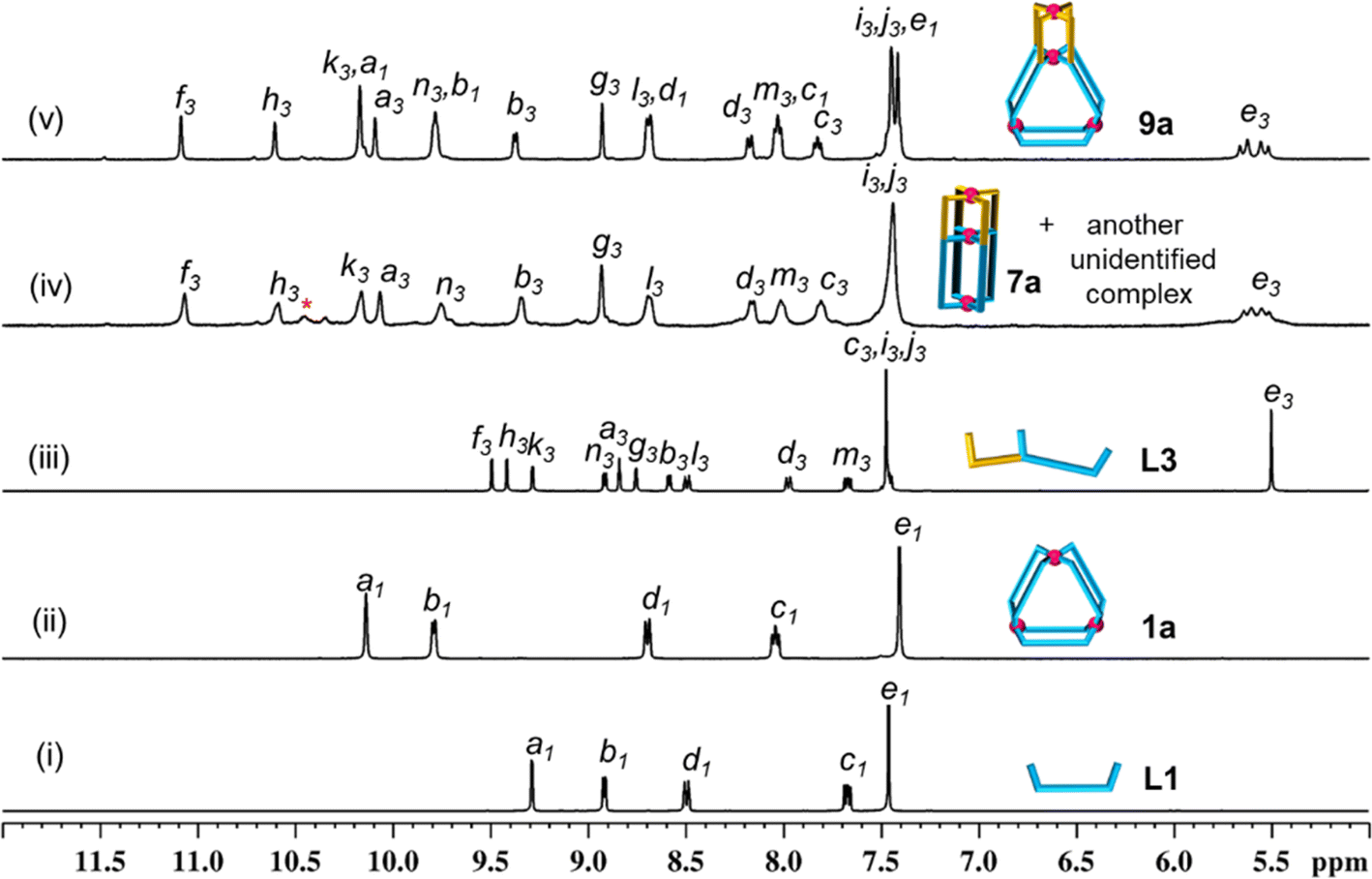 | ||
| Fig. 3 Partial 1H NMR spectra (400 MHz, DMSO-d6, 300 K) of (i) ligand L1; (ii) [Pd3(L1)6](NO3)6, 1a; (iii) ligand L3; (iv) [Pd3(L3)4](NO3)6, 7a (and another unidentified complex); and (v) [Pd4(L1)2(L3)4](NO3)8, 9a. Note: complex 9a can be easily prepared from one-pot combination of Pd(NO3)2, L1 and L3 in a 4:2:4 ratio or cage fusion of 1a with 7a (and another unidentified complex) in a 1:3 ratio. | ||
It is reported that complexation of Pd(II) with L1′ (a positional isomer of L1) containing a meta-phenylene spacer also formed a Pd3L6 type complex: [Pd3(L1′)6](NO3)6, 1a′. However, the complexation reaction at lower and higher concentrations (1 mM and 30 mM of metal) (ESI Schemes S25–S27†) resulted in detectable amounts of Pd2L4 and Pd4L8 type complexes [Pd2(L1′)4](NO3)4, 2a′, and [Pd4(L1′)8](NO3)8, 3a′, respectively, along with 1a′ as studied by 1H NMR spectroscopy and then confirmed by ESI-MS.40 However, during the complexation of Pd(NO3)2 with L1, no new complexes other than 1a were observed at either lower or higher concentrations (1 mM or 30 mM of metal) in their 1H NMR spectra (ESI Fig. S53†). Incidentally, Pd2L4 type [Pd2(L1)4](NO3)4, 2a, could be detected by ESI-MS (ESI Fig. S54†) but not Pd4L8 type [Pd4(L1)8](NO3)8, 3a. Thus, the influence of concentration on nuclearity is neglected (as other complexes were not detected by NMR) for the complexation of Pd(II) with L1. However, the presence of an undetectable amount of 3a cannot be ruled out in the samples.
Complexation of Pd(NO3)2 with the ligand L2 also gave a Pd3L6 type trinuclear complex [Pd3(L2)6](NO3)6, 4a, as shown in Scheme 1(ii) and ESI Scheme S10.†1H NMR spectral study (Fig. 4(i) and (ii)) revealed that the magnitude of downfield shift for inward- and outward-pointing α-pyridine protons of 4a, as compared to L2 (Δδ = 0.84 and 0.78 ppm for Ha2 and Hb2, respectively), is similar to that of 1a as compared to L1. This is also true for β-/γ-pyridine protons. Concentration variation study (1 to 30 mM range) showed additional peaks at higher concentration in the 1H NMR spectra of the generated samples (ESI Fig. S60†). Since these peaks are negligible, we considered 4a as an almost exclusive product in a range of concentrations.
 | ||
| Fig. 4 Partial 1H NMR spectra (400 MHz, DMSO-d6, 300 K) of (i) ligand L2; (ii) [Pd3(L2)6](NO3)6, 4a; (iii) ligand L3; (iv) [Pd3(L3)4](NO3)6, 7a (and another unidentified complex); and (v) low-symmetry [Pd4(L2)2(L3)4](NO3)8, 11a. (Note: complex 11a can be easily prepared from one-pot combination of Pd(NO3)2, L2 and L3 in a 4:2:4 ratio or mixing 4a and 7a (and another unidentified complex) in a 1:3 ratio.). | ||
As mentioned above, binuclear 2a could be detected whereas tetranuclear 3a could not be detected by ESI-MS during complexation study of L1. Thus for complexation of L2 at lower concentration, we could not rule out the coexistence of 4a along with undetectable amount of [Pd2(L2)4](NO3)4, 5a; but [Pd4(L2)8](NO3)8, 6a, might not coexist even at higher concentration. While ESI-MS study detected the presence of the usual Pd3L6 type complex [Pd3(L2)6](BF4)6, 4b, only (ESI Fig. S61†), markedly binuclear 5a (analogue of 2a) and tetranuclear 6a (analogue of 3a) were not detected. Thus, the influence of concentration on the nuclearity is also ruled out for complexation of Pd(II) with L2.
The complexes 1a and 4a were isolated in the solid state by a precipitation method using excess of EtOAC from their solutions in DMSO or DMSO-d6. 1H NMR spectra of the isolated and in situ prepared complexes are found to be identical. The complexes 1a and 4a were characterized by ESI-MS and various NMR techniques including 1H, 13C, H–H COSY, C–H COSY and H–H NOESY (ESI Fig. S47–S51 and S55–S59†). Gas-phase energy-minimized structures of the cationic part of the complexes 1a and 4a are shown in ESI Fig. S161.† The crystal structure of 1a is also discussed in a later section. The metal centres in the complexes are located at the corners of an imaginary triangle where the calculated average non-bonded distances between two Pd centres in 1a and 4a are comparable (approximately 15 Å).
Complexation of Pd(II) separately with unsymmetrical tris-monodentate ligands L3 and L4
The ligand L3 has a 3,5-disubstituted internal/central pyridine where the substituents are pyridine-appended arms of unequal lengths. The fragment spanning the length of the ligand from the internal/central pyridine to the pyridine of the shorter arm is known to form a Pd2L4 type binuclear complex as reported earlier,40 whereas the fragment comprising the internal/central pyridine which extends up to the pyridine unit of the longer arm (i.e.L1) forms a Pd3L6 type complex as established in the present work. Self-assembly of Pd(NO3)2 with the unsymmetrical tris-monodentate ligand L3 was examined by combining them in a 3:4 ratio in DMSO-d6 at 70 °C (Scheme 1(iii), ESI Scheme S11†). The 1H NMR spectrum of the solution appeared like a single set of complexation-induced downfield shifted peaks, but slightly broad, when compared with free L3 (Fig. 3(iii and iv)). Careful analysis indicated another set of overlapping signals (one of the peaks marked with an asterisk is not overlapped). Formation of double-cavity Pd3L4 type [Pd3(L3)4](NO3)6, 7a, is identified by ESI-MS study (ESI Fig. S69†) as one of the products. While DOSY NMR analysis showed the presence of two close-by bands where the major species is possibly 7a (ESI Fig. S67†) and we could not identify the other compound. In addition the possibility of Pd6L8 type complex [Pd6(L3)8](NO3)12 cannot be ruled out (ESI Scheme S11†). The 1H NMR peak pattern remained unchanged when the complexation reactions were performed at variable concentrations ranging from 2 to 40 mM with respect to metal (ESI Fig. S68†). Incidentally, mixing of Pd(NO3)2 with L3′ (isomeric to L3 having meta-phenylene spacer) is known to produce a mixture of double-cavity Pd3L4 and triple-cavity Pd6L8 type complexes [Pd3(L3′)4](NO3)6, 7a′, and [Pd6(L3′)8](NO3)12 (ESI Scheme S28†). The mixture of 7a and unidentified complex was characterized by recording 1H, 13C NMR, H–H COSY, C–H COSY and H–H NOESY spectra (ESI Fig. S62–S66†). Two unequal sized Pd2L4 type cavities, conjoined with a common metal centre,40 are present in the structure of the cage 7a (as in 7a′). The longer arm entity (see Fig. 2) of L3 in its native form (i.e.L1) is ideal for making Pd3L6 type arrangement (i.e.1a), but for L3 as such this longer arm is not able to participate in Pd3L6 formation due to the presence of a connected, binuclear cage forming, shorter arm entity. Thus, the longer arm in L3 has two choices: to form (i) entropically unfavourable oligomerized products or (ii) entropically favourable but probably thermodynamically unfavourable discrete structures. Experimentally, the latter took place and 7a was formed, but along with another unidentified compound (possibly Pd6L8).
Similarly, complexation of Pd(II) with L4 resulted in a mixture of products (Scheme 1(iv), ESI Scheme S12†) as suggested by the broad peaks and noisy baseline in the 1H NMR spectrum of the sample (ESI Fig. S70 and S71†). The mixture might contain Pd3L4 type complex [Pd3(L4)4](NO3)6, 8a, and other unidentified complexes probably including Pd6L8 type cage. The DOSY NMR spectrum was collected for the reaction mixture which showed two nearby bands where major species could be 8a (ESI Fig. S76†). However, the presence of 8a is indirectly inferred from the ESI-MS data that showed the existence of [Cl⊂Pd3(L4)4](NO3)5, 8ab (ESI Fig. S77†). The difference between the two related ligands L3 and L4 is evident in their spacer units that are 1,4-phenylene and naphthalene-1,5-diyl, respectively. This structural difference must be among the possible reasons for the variance in their complexation behaviours. Such a difference was not observed in the case of complexation of L1 and L2; thus, the shorter arms of L3 and L4 also played some roles remotely.
One-pot complexation of Pd(II) with mixture of L1 and L3 (or mixture of L2 and L4)
One-pot combination of Pd(NO3)2 with a mixture of bis-monodentate ligand L1 and tris-monodentate ligand L3 was performed in a 4:2:4 ratio in anticipation of the tetranuclear mixed ligated cage [Pd4(L1)2(L3)4](NO3)8, 9a (Scheme 1(v), ESI Scheme S13†) through integrative self-sorting.40 The reaction was performed in DMSO-d6 at 70 °C and monitored by 1H NMR spectroscopy (ESI Fig. S78†) to obtain a single set of sharp peaks within 2 h. Downfield shift of all the α-pyridyl protons was observed in the 1H NMR spectrum of 9a (Fig. 3(v)) as compared to the free ligands (six signals from L3 and two from L1) indicating coordination of all pyridine moieties with Pd(II). For bound L3 units: Δδ = 1.58, 1.18, 0.88, 1.25, 0.79 and 0.88 ppm for Hf3, Hh3, Hk3, Ha3, Hb3 and Hn3, respectively; and for bound L1 units: Δδ = 0.88 and 0.87 for Hk1 and Hn1, respectively. ESI-MS data of 9a (ESI Fig. S85†) confirmed the proposed molecular formula. The success of integrative self-sorting is probably due to the unfavourable strain in one of the products (i.e.7a) if narcissistic self-sorting had occurred to produce 7a and 1a. The strain could be evaded when integratively self-sorted product 9a was formed. In a similar fashion, complexation of Pd(NO3)2 with a mixture of L2 and L4 produced integratively self-sorted product [Pd4(L2)2(L4)4](NO3)8, 10a (Scheme 1(vi), and ESI Scheme S14 and Fig. S86–S87†). The complexes 9a and 10a were obtained as single species confirmed by DOSY NMR (ESI Fig. S84 and S92†) and also further characterized by ESI-MS and various NMR techniques including 1H, 13C, H–H COSY, C–H COSY and H–H NOESY (ESI Fig. S79–S83, S85 and S87–S91†). The complex 9a was also characterized by the single-crystal XRD method as discussed in a later section.
One-pot complexation of Pd(II) with mixture of L2 and L3 (or mixture of L1 and L4)
The calculated non-bonded distances between metal centres in the trinuclear complexes [Pd3(L1)6](NO3)6, 1a, and [Pd3(L2)6](NO3)6, 4a, are comparable (approximately 15 Å), as discussed above. Thus, it was proposed to prepare complexes like [Pd4(L2)2(L3)4](NO3)8, 11a (by one-pot combination of Pd(II), L2 and L3 in a 4:2:4 ratio) and [Pd4(L1)2(L4)4](NO3)8, 12a (by one-pot combination of Pd(II), L1 and L4 in a 4:2:4 ratio) (ESI Schemes S15 and S16†). In fact, the proposed complexes 11a and 12a could be easily prepared as shown in Scheme 1(xi) and (xii), by performing the reactions at 70 °C in DMSO-d6. The presence of a single band in DOSY NMR spectra revealed the formation of single discrete products 11a and 12a (ESI Fig. S100 and S108†). Further characterization was completed by various NMR techniques, namely 1H, 13C NMR, H–H COSY, C–H COSY and H–H NOESY spectra (ESI Fig. S94–S99 and S102–S107†). ESI-MS data of both the complexes are given in ESI Fig. S101 and S109.†
The complexes 9a and 10a belong to a rare variety of tetranuclear complex of which only one example is known so far in the literature,40i.e. [Pd4(L1′)2(L3′)4](NO3)8, 9a′ (ESI Scheme S29†). The complexes 11a and 12a are unusual as the longer arms in the trinuclear entity of 11a (or 12a) are unequal thus lowering the local symmetry at triangular builds, unlike the symmetrical triangular builds 9a′, 9a, and 10a.
Complexation of Pd(II) separately with symmetrical tris-monodentate ligands L5 and L6
The ligands L5 and L6 have a 3,5-disubstituted internal/central pyridine where the substituents are pyridine-appended arms of equal lengths for a given ligand. These arms of L5 and L6 along with the internal/central pyridine are essentially the ligands L3 and L4, respectively. Thus, a question arises as to whether complexation of Pd(II) separately with the ligands L5 and L6 in a 3:4 ratio would result in double-cavity Pd3L4 type complexes [Pd3(L5)4](NO3)6, 13a, and [Pd3(L6)4](NO3)6, 14a. In the case where the strain is considerably high in the structure then the complexation of Pd(II) with L5 (or L6) should possibly yield only oligomerized products, at the cost of discrete products.
Keeping the above in mind, Pd(NO3)2 was treated with ligand L5 in a 3:4 ratio in DMSO-d6 (Scheme 2A(i), ESI Scheme S17†) and stirred at room temperature (27 °C) whereupon a turbid reaction mixture was formed. The mixture was heated at 70 °C for 10 min to get a clear solution and it was monitored by recording 1H NMR spectra as a function of time. At 10 min it showed broad unassignable signals (Fig. 5(ii)) and the peak pattern remained unchanged even after 48 h; thus, formation of a mixture of products was suspected as can be seen in the DOSY NMR spectrum (ESI Fig. S114†). ESI-MS study of the sample indicated the existence of 13a as one of the products in the mixture though its proportion could not be ascertained from NMR study (ESI Fig. S110 and S115†).
 | ||
| Scheme 2 (A) (i)/(ii) Complexation of Pd(NO3)2 with L5 in 3:4 and 2:4 ratios showing formation of 13a and 15a, respectively; (iii)/(iv) combination of 15a with one equivalent of Pd(NO3)2 per cage or simply heating at 70 °C, resulting in 13a. (B) (i)/(ii) Complexation of Pd(NO3)2 with L6 in 3:4 and 2:4 ratios showing formation of 16a in both cases; (iii)/(iv) combination of 16a with one equivalent of Pd(II) per cage or simply heating at 70 °C resulting in no changes. | ||
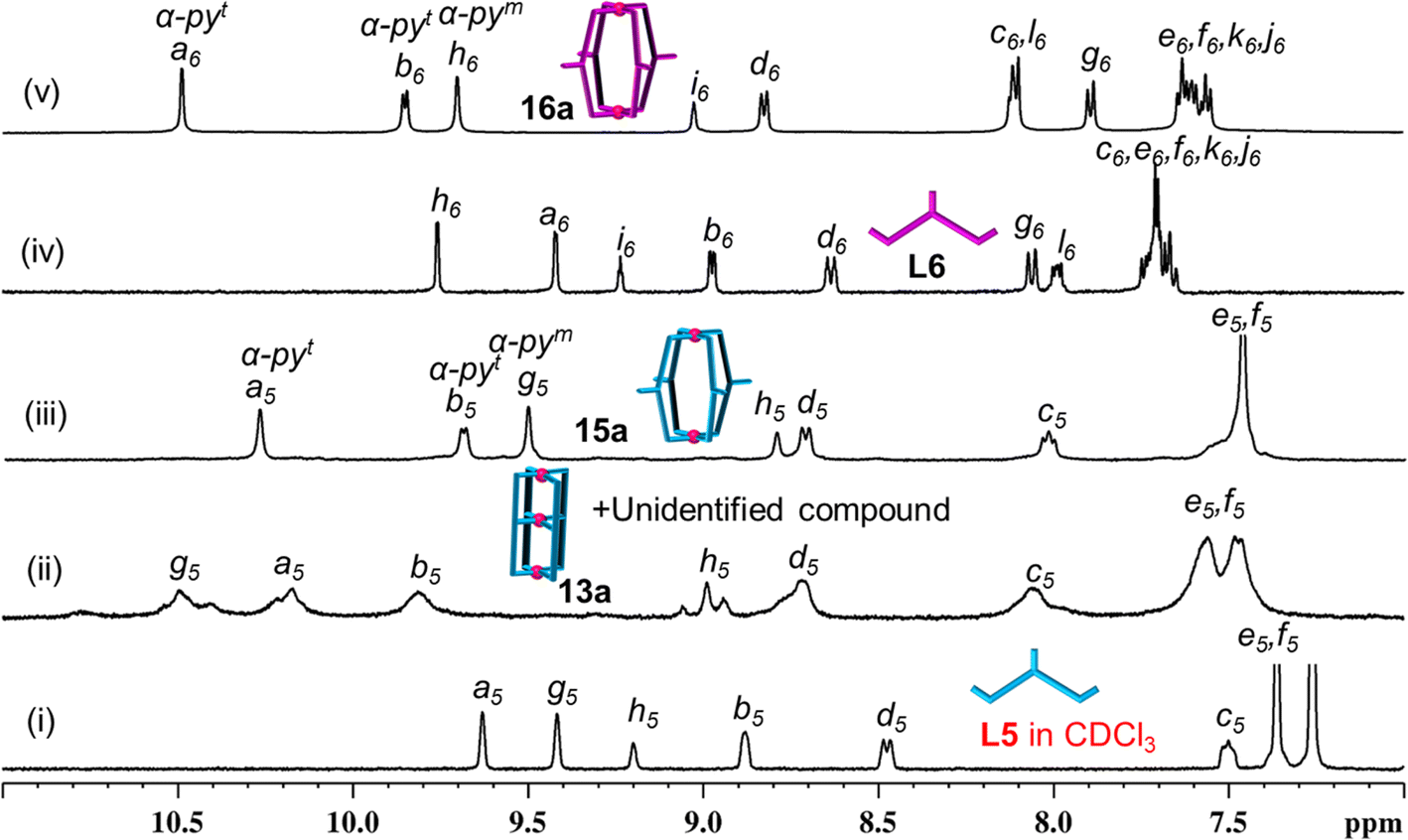 | ||
| Fig. 5 Partial 1H NMR spectra (400 MHz, DMSO-d6, 300 K) of (i) ligand L5 (in CDCl3); (ii) [Pd3(L5)4](NO3)6, 13a, and unidentified complexes; (iii) [Pd2(L5)4](NO3)4, 15a; (iv) ligand L6; and (v) [Pd2(L6)4](NO3)4, 16a. (Note: spectrum (ii) corresponds to complexation reaction of Pd(II) with ligand L5 in 3:4 ratio.). | ||
Complexation of Pd(NO3)2 with L6 in DMSO-d6 was also performed in a 3:4 ratio (Scheme 2A(i), ESI Scheme S18†) whereupon a clear solution was obtained even at room temperature (27 °C). Surprisingly, the 1H NMR spectrum recorded after 5 min of reaction time showed a single set of downfield-shifted and reasonably sharp signals (Fig. 5(v)). The simple nature of the NMR spectrum was a surprise to us as we did not expect to get the complex 14a so easily (due to the inevitable strain expected from the naphthalene spacer). Careful analysis of the spectrum in comparison with that of the free ligand (Fig. 5(iv) and (v)) indicated that the terminal pyridine moieties of L6 are coordinated with Pd(II) whereas the internal/central pyridine is uncoordinated; the protons of terminal pyridines are downfield shifted (Δδ = 1.07 and 0.88 ppm for Ha6 and Hb6, respectively) and those of internal/central pyridine showed negligible but upfield shift. DOSY NMR verified the presence of single species (ESI Fig. S128†). This finding led us to believe that 14a is not formed but that a binuclear Pd2L4 type complex [Pd2(L6)4](NO3)4, 16a, is formed and one equivalent of Pd(II) per cage remained unutilized. This proposal of binuclear complex formation was confirmed by ESI-MS data of [Pd2(L6)4](BF4)4, 16b (ESI Fig. S129†). When the reaction was continued for a longer time, or even after heating at (70 °C), no further changes were observed in the 1H NMR spectral peak pattern. The complex 16a was characterized by 1H, 13C, NMR, H–H COSY, C–H COSY and H–H NOESY (ESI Fig. S123–S127†).
Looking back at the already performed complexation of Pd(II) with L5 in a 3:4 ratio that resulted in a mixture of products including trinuclear 13a, it was decided to perform the reaction of Pd(II) with L5 in a 2:4 ratio in DMSO-d6 (Scheme 2A(ii), ESI Scheme S19†). Interestingly, a clear solution was obtained within 2 h of reaction when carried out at room temperature. The 1H NMR spectrum of the solution showed a single set of sharp peaks (Fig. 5(iii)), the DOSY NMR spectrum showed a single band (ESI Fig. S121†) and ESI-MS confirmed the formation of Pd2L4 type complex [Pd2(L5)4](NO3)4, 15a (ESI Fig. S122†). The complex 15a remained stable in solution for at least 4 days, after which new broad peaks started appearing in the 1H NMR spectrum but 15a still remained as major product (ESI Fig. S116†). The appearance of such new peaks was expediated upon heating the reaction solution at 70 °C; the complex 15a became a minor product within 10 min of heating (ESI Fig. S117†). The pattern of the new peaks resembled the pattern shown in Fig. 5(ii) corresponding to the sample obtained from one-step combination of Pd(II) with L5 in a 3:4 ratio; the solution was turbid indicating dissociation of some amount of the ligands from the complex 15a (Scheme 2A(iv)). Upon addition of another equivalent of Pd(II) per cage of 15a (i.e. Pd(II) to L5 ratio was 3:4) (Scheme 2A(iii)), with or without heating, the peaks of binuclear 15a completely disappeared and the spectrum resembled that shown in Fig. 5(ii). Thus, the unidentified products along with 13a are anything but 15a. The complex 15a was characterized by 1H NMR, H–H COSY, and NOESY (ESI Fig. S118–S120†).
Subsequently, the complexation of Pd(II) with L6 was performed in a 2:4 ratio (Scheme 2B(ii), ESI Scheme S20†) whereupon 16a was formed as expected. Addition of another equivalent of Pd(II) to 16a did not bring about any changes (Scheme 2B(iii)). The complex 16a is considered to be quite stable as it remained unchanged after heating or after addition of another equivalent of Pd(II). However, the complex 15a was found to be unstable as it rearranged giving a mixture, simply upon standing for few days at room temperature without requiring another equivalent of Pd(II). While the complex [Pd3(L5)4](NO3)6, 13a, was detected by ESI-MS, the complex [Pd3(L6)4](NO3)6, 14a, was not detected from complexation reactions of Pd(II) with ligands at 3:4 ratios.
One-pot complexation of Pd(II) with mixture of L1 and L5 (or mixture of L2 and L5)
A mixture of Pd(NO3)2, L1 and L5 in a 5:4:4 ratio taken in DMSO-d6 (Scheme 3(v), ESI Scheme S21†) formed a suspension (as L5 is insoluble in DMSO medium). The suspension was stirred at room temperature (27 °C) and monitored by recording 1H NMR spectra as a function of time (ESI Fig. S130†). The spectrum recorded after 10 min showed only one set of sharp peaks corresponding to the formation of 1a (due to interaction of Pd(II) with L1) while L5 was still insoluble. After 1 h of stirring, the peaks were slightly broadened and small humps also appeared around 10.5, 9.0 and 7.6 ppm due to partial dissolution of L5 and thereby its participation in self-assembly processes, while the sample was still turbid. The turbidity disappeared gradually and humps turned to prominent peaks giving a single set of peaks (Fig. 6A(i)) after 4 days of stirring, indicating formation of a discrete product, thereafter no further changes were observed, suggesting completion of the reaction. When the reaction was carried out at 70 °C a clear solution was obtained within 4 h and the reaction was completed as confirmed by monitoring the reaction by the 1H NMR method (ESI Fig. S131†). We proposed the formation of our targeted single discrete product [Pd5(L1)4(L5)4](NO3)10, 17a, through integrative self-sorting which required 4 days at room temperature or 4 h at 70 °C. In the event of narcissistic self-sorting of Pd(II), L1 and L5 the formation of a mixture of products including 1a, 13a and unidentified complexes is conceivable, and broad signals in the 1H NMR spectrum (like Fig. 5(ii)), but that was not the case. Formation of a single species, i.e.17a, was further supported by diffusion-ordered spectroscopy (DOSY), where the spectrum clearly showed a single band (Fig. 6(B), ESI Fig. S137†) with diffusion coefficient (D) of 6.45 × 10−11 m2 s−1. Further, the molecular composition of 17a was confirmed from ESI-MS data (ESI Fig. S138†).
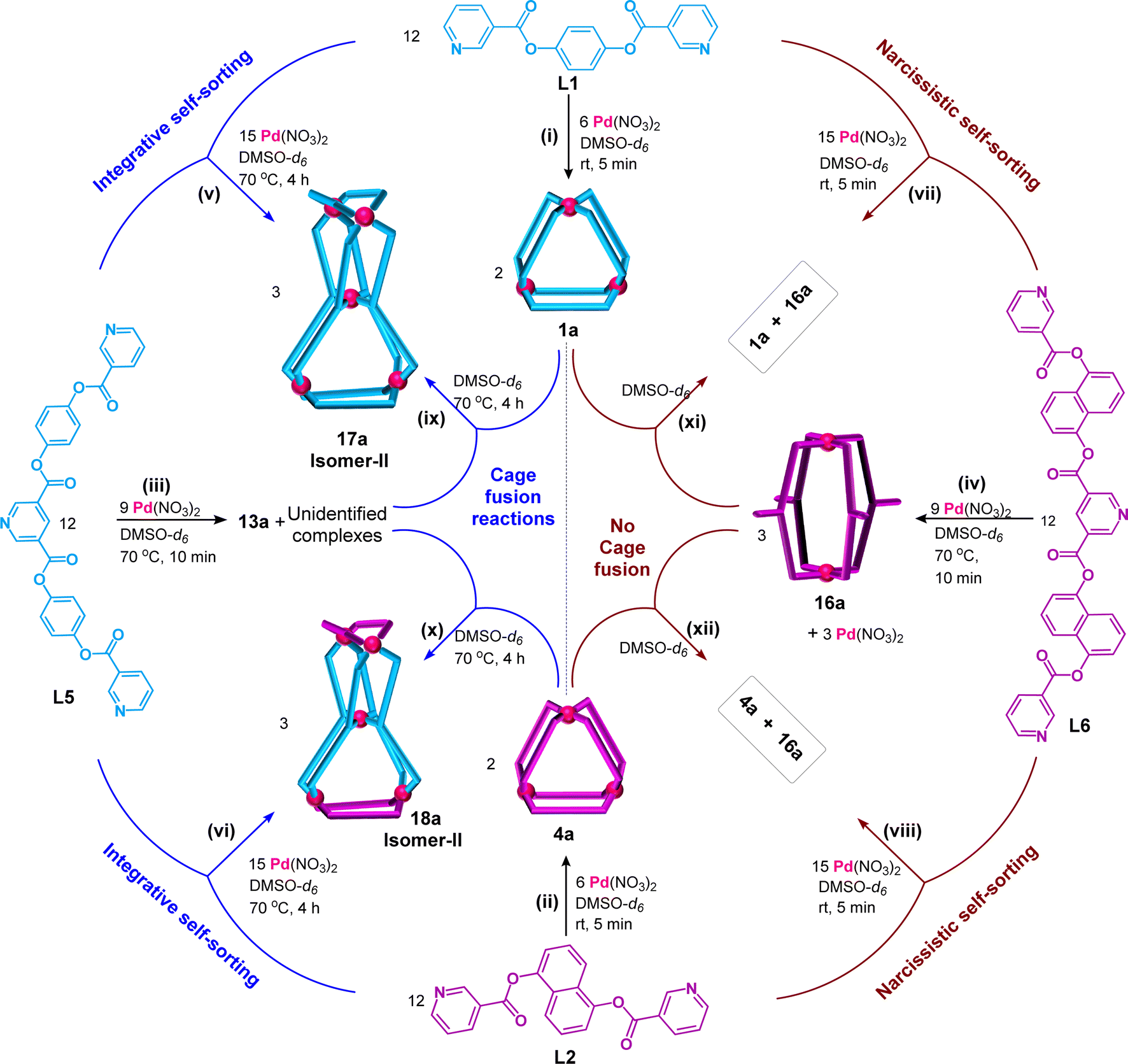 | ||
| Scheme 3 ((i)–(iv)) Complexation of Pd(NO3)2 separately with ligands L1, L2, L5 and L6 giving 1a, 4a, 13a, and 16a, respectively; ((v) and (vi)) integrative self-sorting due to one-pot combination of Pd(NO3)2, L5 and L1 (or L2) resulting in isomer-II of [Pd5(L1)4(L5)4](NO3)10, 17a (or [Pd5(L2)4(L5)4](NO3)10, 18a); (vii) and (viii) narcissistic self-sorting due to one-pot combination of Pd(NO3)2, L6 and L1 (or L2) resulting in 1a and 16a (or 4a and 16a); (ix) and (x) cage fusion reactions of 13a with 1a (or 4a) giving isomer-II of 17a (or 18a); (xi) and (xii) combination of 16a with 1a (or 4a) in absence/presence of extra Pd(NO3)2 resulting in no changes. | ||
 | ||
| Fig. 6 (A) Partial 1H NMR spectra (400 MHz, DMSO-d6, 300 K) of (i) [Pd5(L1)4(L5)4](NO3)10, 17a; (ii) [Pd5(L2)4(L5)4](NO3)10, 18a; (iii) mixture of 1a and 16a; (iv) mixture of 4a and 16a. (Note: the complexes are prepared from Pd(II) and the relevant ligands taken in appropriate stoichiometries through (i)/(ii) integrative self-sorting and (iii)/(iv) narcissistic self-sorting.) (B) 1H DOSY NMR spectrum of complex 17a. | ||
For the complex 17a, there should be two sets of terminal pyridine signals due to Ha1, Hb1, Hc1 and Hd1 (originated from bound L1) and Ha5, Hb5, Hc5 and Hd5 (originated from bound L5); four signals of one set are found to be overlapped with the signals of the other set. As expected, the positions of these signals are closely comparable with corresponding pyridine signals of 1a as well as that of 13a. Notably, for 17a the chemical shift values of internal/central pyridine protons Hg5 and Hh5 (originated from bound L5) having sharp signals are also closely comparable to the corresponding signals of protons of 13a, being broad signals. This is indicative of coordination of all pyridine units with Pd(II) and the existence of a single product is already proposed above through DOSY. The use of the 1H NMR technique for calculation of complexation induced Δδ values for 17a as compared to L5 moiety is not possible (due to solubility issues of L5), but Δδ values with respect to L1 moiety can be calculated easily and are found to be 0.88 and 0.89 ppm for Ha1 and Hb1, respectively. The Hg5 protons of 17a are downfield shifted by 0.97 ppm as compared to the Hg5 protons of 15a (in 15a the central pyridine of L5 is not coordinated with a metal centre). This comparison is an additional support for claiming the coordination of internal/central pyridine in 17a. Characterization of 17a was also carried out by NMR techniques including 1H, 13C, H–H COSY, C–H COSY and H–H NOESY (ESI Fig. S132–S136†).
Preparation of the tetranuclear [Pd4(L1)2(L3)4](NO3)8, 9a, and a lower symmetry tetranuclear [Pd4(L2)2(L3)4](NO3)8, 11a, by integrative self-sorting is described in the previous sections. In a similar line and taking inspiration from the synthesis of pentanuclear 17a we targeted the lower symmetry pentanuclear complex [Pd5(L2)4(L5)4](NO3)10, 18a. The complex 18a was prepared through integrative self-sorting of Pd(NO3)2, L2 and L5 in a 5:4:4 ratio taken in DMSO-d6 (Scheme 3(vi), ESI Scheme S22†) which required stirring for 4 days at room temperature or 4 h at 70 °C. A single set of sharp peaks (Fig. 6A(ii)) with well separated and assignable signals in the 1H NMR spectrum, a single band with a diffusion coefficient of 6.3 × 10−11 m2 s−1 (ESI Fig. S144†) and convincing ESI-MS data provided enough support for the formation of single discrete structure 18a (ESI Fig. S145†). The complete characterization of complex 18a was done by NMR techniques including 1H, 13C, H–H COSY, C–H COSY and H–H NOESY (ESI Fig. S139–S143†).
Two isomeric arrangements, i.e. planar isomer-I and orthogonal isomer-II, are conceivable for the three-dimensional structures of 17a and 18a. The structure determined using single-crystal X-ray diffraction data corresponded with isomer-II for 18a (discussed in a later section), whereas we were not successful in growing single crystals of 17a after several trials. DFT calculation (discussed in a later section) supported energetic preference for isomer-II for both the complexes. Hence, formation of isomer-II is also proposed for the complex 17a. Isomers I and II of a given cage have different bonding patterns with respect to metal-to-ligand bonds; these are not conformers of each other. In isomer-I it is possible to locate a pair of tris-monodentate ligands completely describing an edge of one triangle and an edge of another triangle, whereas in isomer-II this is not so. A pair of tris-monodentate ligands in isomer-II completely describe the edge of only one of the triangles and partially describe two edges of another triangle. In the planar isomer-I, both of the triangular moieties are portrayed to be located almost on the same plane, whereas in the orthogonal isomer-II the triangles are orthogonally placed with respect to each other.
One-pot complexation of Pd(II) with mixture of L2 and L6 (or mixture of L1 and L6)
Detailed investigation indicated that the internal/central pyridine of L6 is unsuitable for coordination with Pd(II). Therefore it is not expected to afford pentanuclear complexes like [Pd5(L2)4(L6)4](NO3)10, 19a, and [Pd5(L1)4(L6)4](NO3)10, 20a. Nevertheless, complexation reactions were tried by combining Pd(NO3)2, L2, and L6 in a 5:4:4 ratio in DMSO-d6 (Scheme 3(viii), ESI Scheme S23†). Unsurprisingly, narcissistic self-sorting was observed spontaneously at room temperature (27 °C) resulting in a mixture of [Pd3(L2)6](NO3)6, 4a, and [Pd2(L6)4](NO3)4, 16a, as confirmed by monitoring the reaction by 1H NMR spectroscopy (Fig. 6A(iv)). Longer reaction time and heating at 70 °C did not bring about any change to the product profile. In a similar line combination of Pd(NO3)2, L1, and L6 in a 5:4:4 ratio in DMSO-d6 (Scheme 3(vii), Fig. 6A(iii), ESI Scheme S24†) also exhibited narcissistic self-sorting to produce [Pd3(L1)6](NO3)6, 1a, and [Pd2(L6)4](NO3)4, 16a. It is assumed that unfavourable strain hindered the formation of the imaginary molecules 19a and 20a. 1H NMR spectra of relevant ligands and complexes are plotted together for a quick reference and comparison (ESI Fig. S146 and S147†).
Cage fusion reactions
The tetranuclear complexes (9a, 10a, 11a, and 12a) and pentanuclear complexes (17a and 18a) prepared in the present work were exclusively formed by integrative self-sorting of Pd(II) and a pair of appropriate ligands (L1 to L6). We considered a library of complexes (1a, 4a, 7a, 8a, 13a, and 16a) prepared from Pd(II) and individual ligands. Understandably, combination of two selected complexes/cages from the library would essentially undergo cage fusion reactions leading to the integratively self-sorted tetra- and pentanuclear complexes cited above. For instance, complexation of Pd(II) with L1 and L3 separately results in cages [Pd3(L1)6](NO3)6, 1a, and [Pd3(L3)4](NO3)6, 7a, respectively, whereas one-pot mixing of Pd(II), L1 and L3 produces cage [Pd4(L1)2(L3)4](NO3)8, 9a. For the cage fusion reaction, cages 1a and 7a were combined in a 1:3 ratio whereupon three equivalents of 9a were generated exclusively (Scheme 1(ix)). Cage fusion reactions were successfully performed for all other complexes that are formed by integrative self-sorting processes (Schemes 1(x)–(xii) and 3(ix) and (x)). The progress of the cage fusion reactions generating 9a was monitored by 1H NMR spectroscopy for a better evaluation (ESI Fig. S148†). Similarly, the progress of the reaction for other cage fusion reactions was also monitored (ESI Fig. S149–S151 and S152–S155†). It is well known that thermodynamic stability drives the formation of integratively self-sorted products in contrast to narcissistically sorted products. In the present work, we believe that thermodynamic stability is the reason for the formation of pentanuclear mixed-ligated double-cavity cages 17a and 18a.
A mixture of complexes 1a and 16a was formed by narcissistic self-sorting using Pd(II), L1 and L6. Obviously, combination of isolated 1a and 16a would not undergo cage fusion and that was the case (Scheme 3(vii), ESI Fig. S156†). Similarly, a mixture of 4a and 16a also remained intact and there was no cage fusion (Scheme 3(viii), ESI Fig. S157†).
Single-crystal X-ray diffraction analysis
Structures of [Pd3(L1)6](NO3)6, 1a, [Pd4(L1)2(L3)4](NO3)8, 9a, and [Pd5(L2)4(L5)4](NO3)10, 18a, are unequivocally confirmed by single-crystal X-ray diffraction study. The structures are shown in Fig. 7 and a brief description is given below. Further description about the crystallographic data, structural refinement and ORTEP diagrams are provided in the ESI (Table S1 and Fig. S158–S160†). | ||
| Fig. 7 Crystal structures showing cationic frameworks and encapsulated solvents/counter-anions corresponding to the cages (A) 1a, (B) 9a, and (C) 18a. | ||
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule of the tetranuclear complex, eight nitrate ions and six DMSO molecules in the asymmetric unit. The electron density corresponding to other disordered solvent molecules was squeezed. The small cavity houses a single nitrate anion which is involved in C–H⋯O contacts with the pyridyl protons of the ligand. Three DMSO solvent molecules are present in the larger cavity. A second nitrate ion is also present in the central portion of the triangular cavity and interacts with the DMSO molecules via noncovalent interactions.
with one molecule of the tetranuclear complex, eight nitrate ions and six DMSO molecules in the asymmetric unit. The electron density corresponding to other disordered solvent molecules was squeezed. The small cavity houses a single nitrate anion which is involved in C–H⋯O contacts with the pyridyl protons of the ligand. Three DMSO solvent molecules are present in the larger cavity. A second nitrate ion is also present in the central portion of the triangular cavity and interacts with the DMSO molecules via noncovalent interactions.
DFT study to comprehend bend angle of free and bound ligands
Non-chelating bis-monodentate ligands are known to form PdnL2n types of complexes where the value of “n” is relatable to the bend angle (θ) of the coordination vectors of bound-ligand units. For binuclear Pd2L4 and hexanuclear Pd6L12 type complexes having the metal ions arranged at the end of a line and the vertices of an octahedron, the ideal values of θ are 0° and 90°, respectively.12,46 In such cases, the square planar coordination geometry around the metal centre is either maintained or shows a small deviation. For ring-like trinuclear Pd3L6, tetranuclear Pd4L8 and pentanuclear Pd5L10 type complexes the values of θ are below 90°.12 The values of θ calculated from the crystal structures of a few reported systems are approximately 0° for Pd2L4 type complexes and in the range of 40° to 50° for Pd3L6 type complexes. If the bend angle of free ligand (in one of the stable conformers) is comparable/matching with the required bend angle of the bound ligand in a PdnL2n type complex then we can expect the formation of such a complex if Pd(II) is combined with the ligand. The strain imposed in the overall structure of the coordination architecture should be within the limit of tolerance whenever the bend angles match.Keeping this in mind, energy-minimized structures of the free ligands (L1–L6) and the cationic frameworks of a range of the Pd(II) complexes of the ligands, including mixed ligated complexes, were obtained by the density functional theory (DFT) method (ESI Fig. S161–S163†). The bend angles of the free ligands and bound ligands are estimated in order to qualitatively understand whether or not considerable strain is imposed on the ligand backbone of some of the coordination architectures (ESI Fig. S161–S163†). The calculated bend angles of ligands L1 and L2 are found to be suitable for supporting Pd3L6 architecture (as in 1a and 4a); the longer arms of the ligands L3 and L4 are also suitable for Pd3L6 sub-framework but participation of either L1 or L2 as ancillary ligand is required (as in 9a to 12a) for making a discrete structure.
The arms of ligand L5 are well suited for the making of Pd3L6 fragments but by including either L1 or L2 as ancillary ligand (as in 17a and 18a). In the absence of ancillary ligand, the ligand L5 is forced to support Pd2L4 sub-framework (as in 13a) of a Pd3L4 cage. The ligand L6 could not support Pd3L4 (as in imaginary 14a); also, even in the presence of ancillary ligand it did not support Pd3L6 sub-frameworks (as in imaginary 19a and 20a) due to formation of alternate stable structure, i.e.16a where bend angle is not an issue.
DFT study on planarly/orthogonally conjoined triangles
DFT calculations were performed to find out whether the structure of isomer-II obtained from single-crystal X-ray diffraction data is energetically preferred over isomer-I for the cationic framework [Pd5(L2)4(L5)4]10+ of the pentanuclear complex 18a. The gas-phase calculations (ESI Tables S8 and S9†) showed that the isomer-II, i.e.18a-II, is considerably preferred (by 67 kJ mol−1) as compared to 18a-I (Fig. 8(B)). Similar calculations for the two isomers of the cationic framework [Pd5(L1)4(L5)4]10+ of the pentanuclear complex 17a in the gas phase revealed that 17a-II is preferred (by 7.8 kJ mol−1) over 17a-I (Fig. 8(A)). Various views of both the isomers of the complexes 17a and 18a are shown in ESI Fig. S164–S167,† for better three-dimensional visualization. However, energy optimization of the complexes 17a and 18a in implicit DMSO was not successful. In isomer-I, both of the triangular moieties appear to be located on the same plane, whereas in the orthogonal isomer-II the triangles are orthogonally placed with respect to each other. The planar isomer-I was found to be not completely planar in the DFT structure. A triangle is slightly twisted from the plane of the other and the square planar nature of Pd(II) centres is also disturbed making isomer-I less preferred.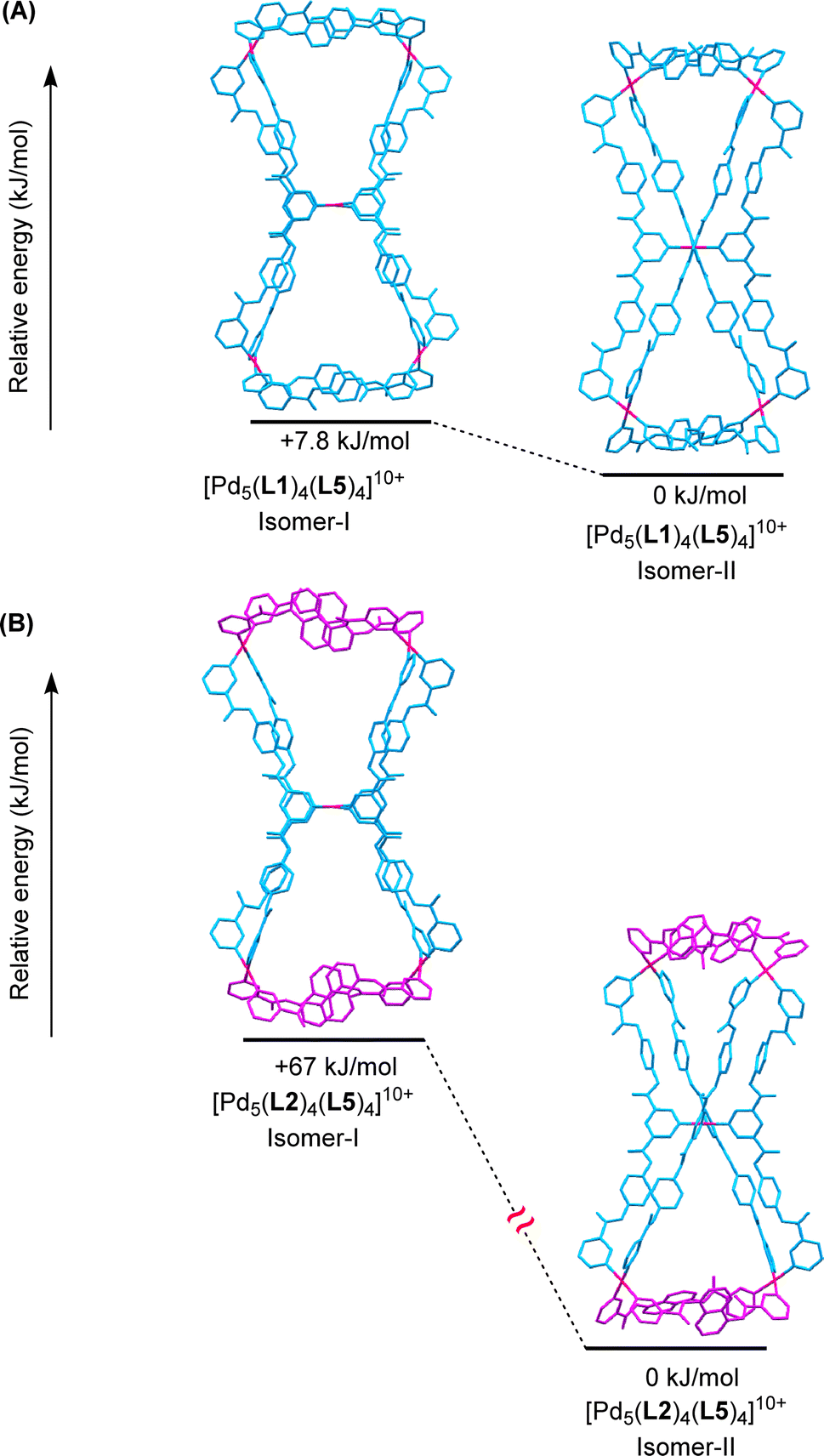 | ||
| Fig. 8 Energy-minimized structures (B3LYP-D3/SDD,6-31G(d)) showing energetic preference between planarly conjoined (isomer-I) and orthogonally conjoined (isomer-II) triangles for (A) [Pd5(L1)4(L5)4]10+, 17a, and (B) [Pd5(L2)4(L5)4]10+, 18a, in the gas phase. (Hydrogen atoms are omitted for clarity; counter-anions/solvents were not included in the calculations.) | ||
3 Conclusions
In summary, we have carefully designed a set of symmetrical bis-monodentate (L1), unsymmetrical tris-monodentate (L3) and symmetrical tris-monodentate (L5) ligands. Complexation of Pd(II) with the individual ligands L1, L3 and L5 resulted in relaxed [Pd3(L1)6]6+ (exclusively), [Pd3(L3)4]6+ (almost exclusively), and [Pd3(L5)4]6+ (in a mixture). This complexation behaviour provided a basis for the targeted mixed-ligated MCDCC. Complexation of Pd(II) with a mixture of L1 and L3 in a 4:2:4 ratio resulted in integrative self-sorting to form a relaxed double-cavity [Pd4(L1)2(L3)4]8+ framework containing binuclear and trinuclear sub-frameworks that are conjoined at a common metal centre. Integrative self-sorting of Pd(II), L1 and L5 in a 5:4:4 ratio resulted in the relaxed double-cavity [Pd5(L1)4(L5)4]10+ framework where two trinuclear sub-frameworks are conjoined. The complexation behaviour of another set of ligands L2, L4 and L6 that are comparable to L1, L3 and L5 was also studied. The tris-monodentate ligand L6 behaved very differently from L5 in that complexation of Pd(II) with L6 resulted in a stable binuclear single-cavity complex [Pd2(L6)4]4+. Integrative self-sorting was observed for the combination of Pd(II), L2 and L4 in a 4:2:4 ratio resulting in [Pd4(L2)2(L4)4]8+ whereas narcissistic self-sorting was observed for the combination of Pd(II), L2 and L6 resulting in the individual complexes [Pd3(L2)6]6+ and [Pd2(L6)4]4+. The success of the observed integrative self-sorting is ascribed to two factors, i.e. (i) size complementarity of the two ligands in the targeted design and (ii) strained/oligomeric complexes formed by at least one of the ligands. Lowering the symmetry of multi-cavity complexes was achieved by integrative self-sorting leading to exclusive formation of [Pd4(L2)2(L3)4]8+, [Pd4(L1)2(L4)4]8+ and [Pd5(L2)4(L5)4]10+. However, we did not expect the formation of [Pd5(L1)4(L6)4]10+, and it did not form.
Devising methods for lowering the symmetry of SCDCC is a relatively new research area; here we have gone a step further and demonstrated a strategy to lower the symmetry in MCDCC. Our work provided design principles for the construction of a new and novel pentanuclear double-cavity framework composed of two trinuclear sub-frameworks. We recommend the following two points for selection of bis-monodentate and tris-monodentate ligands for preparation of new pentanuclear MCDCC. (1) It is crucial to select a bis-monodentate ligand that is capable of forming a relaxed trinuclear complex upon complexation with Pd(II). (2) It is necessary to prepare a tris-monodentate ligand that resembles two units of the bis-monodentate ligand having a common donor site and then evaluate the complexation behaviour of Pd(II) with the tris-monodentate ligand; select those ligands that yield strained complexes or oligomers and reject those that form stable complexes. The pentanuclear MCDCC design introduced by us should inspire the making of many more such complexes by variation of spacer in the ligands to modulate the shapes, sizes and ultimately the behaviour/reactivity aspects of MCDCC.
Data availability
All relevant data have been included in the paper and ESI.†Author contributions
D. K. C. and S. S. carried out the research, analyzed the data and wrote the manuscript. S. P. synthesized and characterized the complexes 1a and 4a; S. S. took over the work from S. P. to proceed further with the synthesis of ligands, complexation study, growing of all crystals, and computational study. S. P. helped in scientific discussion. S. K. carried out X-ray diffraction experiments and refined two crystal structures and contributed to the manuscript preparation. D. K. C. is the principal investigator and managed the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. K. C. thanks the Science and Engineering Research Board (SERB), Department of Science and Technology, Government of India (project no. CRG/2022/004413), for financial support. D. K. C. thanks IIT Madras for financial support through a Mid-Career Institute Research and Development Award (IRDA-2019) and through a Center under Institute of Eminence program (IoE Center of Molecular Architecture). We thank SAIF, IIT Madras for single-crystal XRD facility (especially Dr P. K. S. Antharjanam for refining crystal structure of 9a). We thank the Department of Chemistry, IIT Madras, for NMR facility (especially Dr R. Baskar for helping in DOSY NMR study), and DST-FIST funded ESI-MS facility. We thank the P. G. Senapathy Centre for Computing Resources, IIT Madras, for providing access to the Gaussian16 package. S. S. thanks IIT Madras for a fellowship, S. P. thanks CSIR, New Delhi, India for a fellowship. This Edge Article is dedicated to Prof. Ashok K. Mishra on the occasion of his superannuation.References
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed.
- M. Yoshizawa, Chem. Lett., 2017, 46, 163–167 CrossRef CAS.
- N. B. Debata, D. Tripathy and H. S. Sahoo, Coord. Chem. Rev., 2019, 387, 273–298 CrossRef CAS.
- E. Raee, Y. Q. Yang and T. B. Liu, Giant, 2021, 5, 100050 CrossRef CAS.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- A. Tarzia and K. E. Jelfs, Chem. Commun., 2022, 58, 3717–3730 RSC.
- Y. Domoto and M. Fujita, Coord. Chem. Rev., 2022, 466, 214605 CrossRef CAS.
- T. Tateishi, M. Yoshimura, S. Tokuda, F. Matsuda, D. Fujita and S. Furukawa, Coord. Chem. Rev., 2022, 467, 214612 CrossRef CAS.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef CAS.
- J. E. M. Lewis, Chem. Commun., 2022, 58, 13873–13886 RSC.
- S. Sharma, M. Sarkar and D. K. Chand, Chem. Commun., 2023, 59, 535–554 RSC.
- A. Schmidt, A. Casini and F. E. Kuhn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed.
- D. Tripathy, N. B. Debata, K. C. Naik and H. S. Sahoo, Coord. Chem. Rev., 2022, 456, 214396 CrossRef CAS.
- S. S. Mishra and D. K. Chand, Dalton Trans., 2022, 51, 11650–11657 RSC.
- R.-J. Li, A. Tarzia, V. Posligua, K. E. Jelfs, N. Sanchez, A. Marcus, A. Baksi, G. H. Clever, F. F. Tirani and K. Severin, Chem. Sci., 2022, 13, 11912–11917 RSC.
- P. Molinska, A. Tarzia, L. Male, K. E. Jelfs and J. E. M. Lewis, Angew. Chem., Int. Ed., 2023, 62, e202315451 CrossRef CAS PubMed.
- J. D. Montmollin, A. B. Solea, D. W. Chen, F. F. Tirani and K. Severin, Inorg. Chem., 2024, 63, 4583–4588 CrossRef PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- D. Bardhan and D. K. Chand, Chem.–Eur. J., 2019, 25, 12241–12269 CrossRef CAS PubMed.
- J. L. Algar and D. Preston, Chem. Commun., 2022, 58, 11637–11648 RSC.
- C.-B. Tian and Q.-F. Sun, Chem.–Eur. J., 2023, 29, e202300195 CrossRef CAS PubMed.
- R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Asian J., 2017, 12, 2513–2523 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- C. T. McTernan, J. A. Davies and J. R. Nitschke, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef CAS PubMed.
- Q. Sun, L. Qin, C. Lai, S. Liu, W. Chen, F. Xu, D. Ma, Y. Li, S. Qian, Z. Chen, W. Chen and H. Ye, J. Hazard. Mater., 2023, 447, 130848 CrossRef CAS PubMed.
- S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem.–Eur. J., 2014, 20, 13122–13126 CrossRef CAS PubMed.
- M. D. Johnstone, E. K. Schwarze and G. H. Clever, Chem.–Eur. J., 2015, 21, 3948–3955 CrossRef CAS PubMed.
- K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- G. Sarada, A. Kim, D. Kim and O.-S. Jung, Dalton Trans., 2020, 49, 6183–6190 RSC.
- H. Dasary, M. Sarkar and D. K. Chand, Chem. Commun., 2022, 58, 8480–8483 RSC.
- S. Samantray, S. Krishnaswamy and D. K. Chand, Nat. Commun., 2020, 11, 880 CrossRef CAS PubMed.
- S. S. Mishra, S. Krishnaswamy and D. K. Chand, J. Am. Chem. Soc., 2024, 146, 4473–4488 CrossRef CAS PubMed.
- X. Huang and Q. Zhang, Acta Chim. Sin., 2023, 81, 217–221 CrossRef CAS.
- D. K. Chand, K. Biradha, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Chem.–Asian J., 2006, 1, 82–90 CrossRef CAS PubMed.
- J. N. Kremsky, B. Szczepankiewicz, H. Toop and J. Morris, Nicotinamide riboside analogs, pharmaceutical compositions, and use thereof, US Pat., 11180521B2, 2021 Search PubMed.
- S. P. Gaynor, M. J. Gunter, M. R. Johnston and R. N. Warrener, Org. Biomol. Chem., 2006, 4, 2253–2266 RSC.
- S. M. Jansze, G. Cecot, M. D. Wise, K. O. Zhurov, T. K. Ronson, A. M. Castilla, A. Finelli, P. Pattison, E. Solari, R. Scopelliti, G. E. Zelinskii, A. V. Vologzhanina, Y. Z. Voloshin, J. R. Nitschke and K. Severin, J. Am. Chem. Soc., 2016, 138, 2046–2054 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2326106, 2326107 and 2326108. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01078g |
| ‡ Current address: Department of Chemistry, VIT-AP University, Amaravati 522237, India. |
| This journal is © The Royal Society of Chemistry 2024 |