
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating substrate binding in the light-dependent protochlorophyllide oxidoreductase†
Penelope
Pesara
a,
Katarzyna
Szafran
b,
Henry C.
Nguyen‡
c,
Abhishek
Sirohiwal
d,
Dimitrios A.
Pantazis
 *a and
Michal
Gabruk
*b
*a and
Michal
Gabruk
*b
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470, Mülheim an der Ruhr, Germany. E-mail: dimitrios.pantazis@kofo.mpg.de
bDepartment of Plant Physiology and Biochemistry, Faculty of Biochemistry, Biophysics and Biotechnology, Jagiellonian University, Gronostajowa 7, 30-387, Cracow, Poland. E-mail: michal.gabruk@uj.edu.pl
cDepartment of Biochemistry & Biophysics, Quantitative Biosciences Institute, University of California, San Francisco, CA, USA
dDepartment of Biochemistry and Biophysics, Arrhenius Laboratory, Stockholm University, 10691, Stockholm, Sweden
First published on 30th April 2024
Abstract
The Light-Dependent Protochlorophyllide Oxidoreductase (LPOR) catalyzes a crucial step in chlorophyll biosynthesis: the rare biological photocatalytic reduction of the double C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in the precursor, protochlorophyllide (Pchlide). Despite its fundamental significance, limited structural insights into the active complex have hindered understanding of its reaction mechanism. Recently, a high-resolution cryo-EM structure of LPOR in its active conformation challenged our view of pigment binding, residue interactions, and the catalytic process. Surprisingly, this structure contrasts markedly with previous assumptions, particularly regarding the orientation of the bound Pchlide. To gain insights into the substrate binding puzzle, we conducted molecular dynamics simulations, quantum-mechanics/molecular-mechanics (QM/MM) calculations, and site-directed mutagenesis. Two Pchlide binding modes were considered, one aligning with historical proposals (mode A) and another consistent with the recent experimental data (mode B). Binding energy calculations revealed that in contrast to the non-specific interactions found for mode A, mode B exhibits distinct stabilizing interactions that support more thermodynamically favorable binding. A comprehensive analysis incorporating QM/MM-based local energy decomposition unraveled a complex interaction network involving Y177, H319, and the C131 carboxy group, influencing the pigment's excited state energy and potentially contributing to substrate specificity. Importantly, our results uniformly favor mode B, challenging established interpretations and emphasizing the need for a comprehensive re-evaluation of the LPOR reaction mechanism in a way that incorporates accurate structural information on pigment interactions and substrate-cofactor positioning in the binding pocket. The results shed light on the intricacies of LPOR's catalytic mechanism and provide a solid foundation for further elucidating the secrets of chlorophyll biosynthesis.
C bond in the precursor, protochlorophyllide (Pchlide). Despite its fundamental significance, limited structural insights into the active complex have hindered understanding of its reaction mechanism. Recently, a high-resolution cryo-EM structure of LPOR in its active conformation challenged our view of pigment binding, residue interactions, and the catalytic process. Surprisingly, this structure contrasts markedly with previous assumptions, particularly regarding the orientation of the bound Pchlide. To gain insights into the substrate binding puzzle, we conducted molecular dynamics simulations, quantum-mechanics/molecular-mechanics (QM/MM) calculations, and site-directed mutagenesis. Two Pchlide binding modes were considered, one aligning with historical proposals (mode A) and another consistent with the recent experimental data (mode B). Binding energy calculations revealed that in contrast to the non-specific interactions found for mode A, mode B exhibits distinct stabilizing interactions that support more thermodynamically favorable binding. A comprehensive analysis incorporating QM/MM-based local energy decomposition unraveled a complex interaction network involving Y177, H319, and the C131 carboxy group, influencing the pigment's excited state energy and potentially contributing to substrate specificity. Importantly, our results uniformly favor mode B, challenging established interpretations and emphasizing the need for a comprehensive re-evaluation of the LPOR reaction mechanism in a way that incorporates accurate structural information on pigment interactions and substrate-cofactor positioning in the binding pocket. The results shed light on the intricacies of LPOR's catalytic mechanism and provide a solid foundation for further elucidating the secrets of chlorophyll biosynthesis.
Introduction
Chlorophyll (Chl) is the most essential natural pigment in organisms that carry out photosynthesis – the process of capturing sunlight and transforming it into chemical energy, powering Earth's biosphere.1 A crucial step in chlorophyll biosynthesis is the conversion of protochlorophyllide (Pchlide) to chlorophyllide (Chlide) by reduction of the C17C18 double bond of the former2 (Fig. 1). This reaction, a rare example of light-driven biosynthetic transformation, is catalyzed by the light-dependent protochlorophyllide oxidoreductase (LPOR) that employs NADPH as a cofactor. Owing to the photocatalytic nature of the reaction, LPOR has also a regulatory role in Chl biosynthesis,3,4 but the mechanism of action of the enzyme remains poorly understood.2,5,6 The main challenges so far have been related to the involvement of several intermediates, and the lack of accurate structural information regarding the active form of the enzyme and the precise positioning of the substrate and the cofactor. Nevertheless, some insights into the reaction have been accumulated over the years.
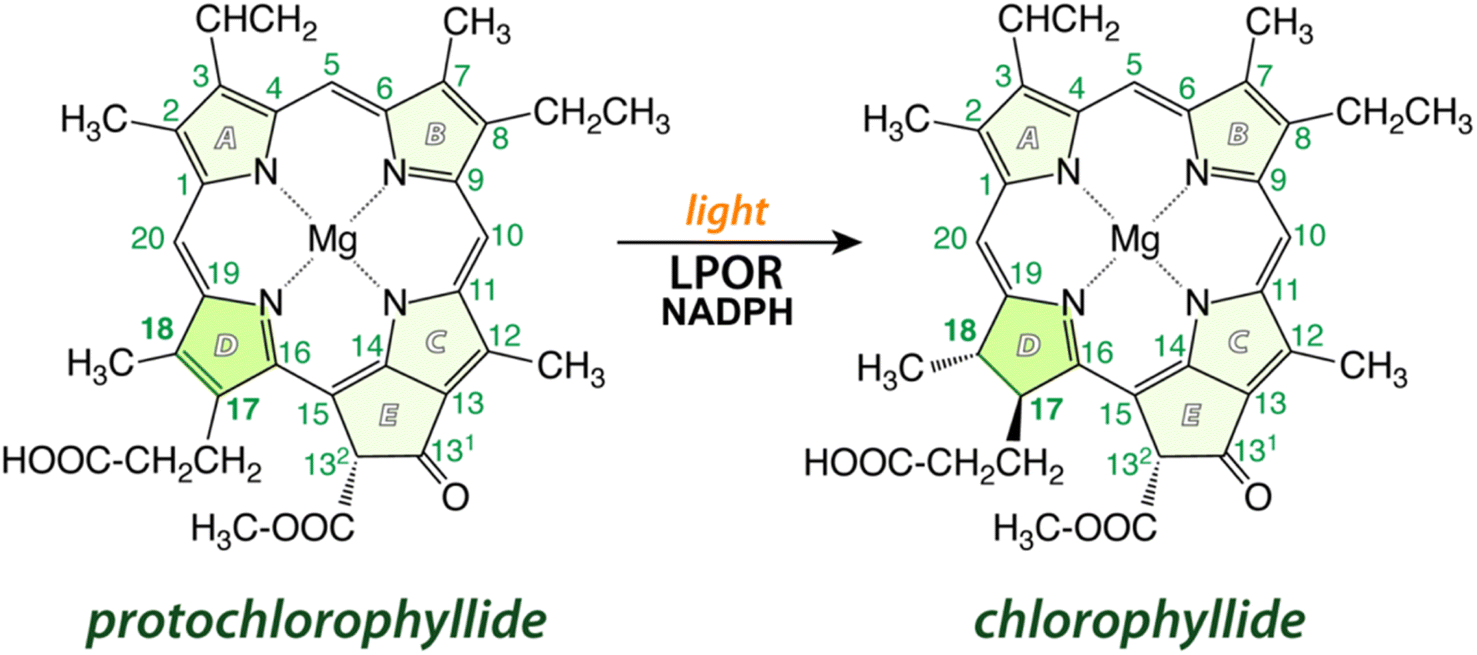 | ||
| Fig. 1 A schematic of the reaction catalyzed by LPOR: in the presence of light and NADPH, the enzyme reduces the C17C18 double bond in ring D of protochlorophyllide. | ||
An early NMR study by Begley and Young suggested that the pro-S face of NADPH delivers a hydride to C17 of Pchlide, while an amino acid residue was assumed to subsequently protonate C18 to complete the reaction.7 A suitable candidate for such a residue was proposed based on the sequence homology between LPOR and short-chain dehydrogenase reductase (SDR) family.8 Most enzymes of the family share a conservative catalytic motif that consists of Y276 and K280 residues (the numeration for PORB isoform of A. thaliana).9 The classical SDR mechanism considers that the interaction between the tyrosine and the lysine residues lowers the pKa of the hydroxyl group of the former so it can serve as a proton donor. Numerous site-directed mutagenesis studies confirmed the importance of Y276 for the activity of LPOR.10,11 However, in all published experiments the mutation of Y276 only lowered and did not abolish LPOR activity as would be expected for the putative catalytic residue. Based on this finding it was suggested that the solvent may be the most likely candidate for a proton donor.10 Moreover, it was shown that the deuterated NADPH (NADP2H) influenced the kinetic of hydride transfer but not proton transfer, while the use of heavy water affected the rate of proton transfer but not hydride transfer.12 Additionally, the rate of hydride transfer was orders of magnitude faster than the rate of proton transfer, what clearly suggests a two-step mechanism of the reaction.12 The accumulated experimental data allowed the proposal of a plausible picture of the active site of the enzyme (mode A, Fig. 2B), which served as a starting point for several attempts at computational modelling of mechanistic scenarios. In these studies, either a homologous model of the enzyme or some experimentally derived structures were employed.6,13,14 Crucially, in all these studies the conformation of the enzyme and the binding mode of the substrate were obtained with molecular docking techniques or were presumed to accommodate past assumptions regarding the stereospecificity of the reaction, the involvement of Y276, and the orientation of NADPH. However, even though the assumed binding mode was the same in two recent computational studies on the mechanism of LPOR, i.e., mode A, the actual conclusions in these studies diverge drastically on the nature and sequence of events.6,14
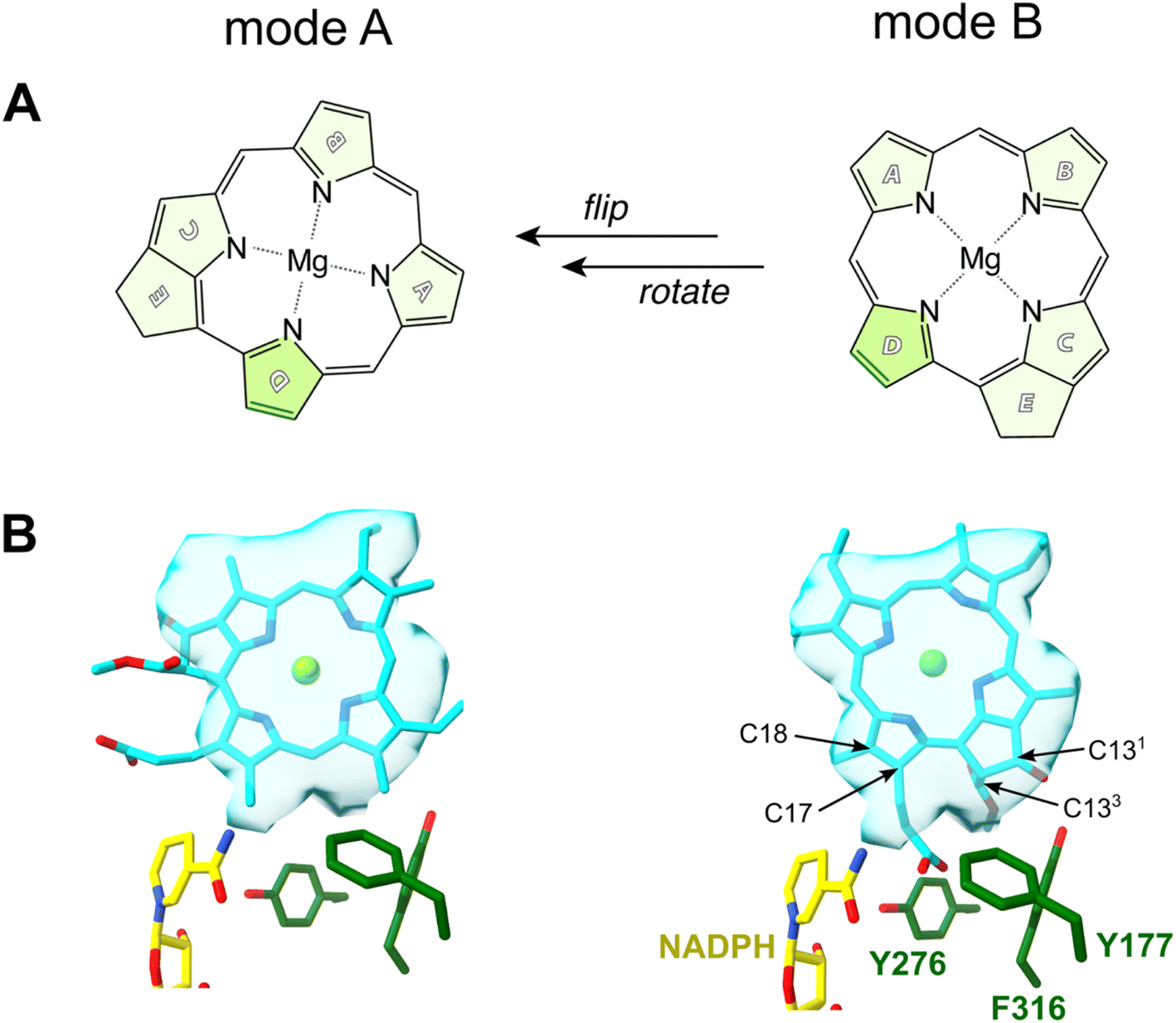 | ||
| Fig. 2 Two possible modes of Pchlide binding to LPOR: mode A, based on previous proposals, mode B, based on structural data. (A) A schematically visualized orientation difference between the two modes. (B) The fit of the Pchlide structure to the density found in the cryoEM structure of LPOR (PDB: 7JK9) for two possible modes of binding. The orientations of the residues are shown as in the published structure (7JK9). | ||
Johannissen et al. proposed a hydride transfer (HYT) from NADPH to C17 of Pchlide followed by proton transfer (PT) from a tyrosine residue to C18.6 In contrast, Silva and Cheng supposed an initial electron transfer (ET) followed by PT from a tyrosine residue to C18, after which the HYT from NADPH to C17 would take place.14 The involvement of a cysteine residue (C309) was also suggested.
In 2021 the atomic structure of helical assembly of LPOR on a lipid membrane was reported that for the first time revealed the architecture of the active site of the protein with bound Pchlide.15 This reconstruction showed that Pchlide is embedded within a helical lattice partially in the outer leaflet of the membrane sandwiched between the helix α10 and a region of the LPOR-specific loop (“the Pchlide loop”). Surprisingly, a head of one of the lipids was found next to the pigment, namely the galactosyl of monogalactosyldiacylglycerol (MGDG). The resolution of the protein within the structure allowed to determine the positions of all amino acid residues, including these responsible for Pchlide binding and those suggested to be involved in the reaction. Intriguingly, the conformation of the protein in the active state is nearly identical to the other LPOR structures obtained for the protein lacking the pigment, except for parts interacting with Pchlide. The two parts of the enzyme sandwiching Pchlide show two distinct conformations depending on the presence of the pigment. These conformations may represent two states of the enzyme: the apo and the holo forms, of which the latter is active. Crucially, even though the resolution of the electron density found in the pigment binding pocket of the active form of the enzyme was not sufficient to propose the orientation of Pchlide with complete confidence,15 the best fit of the pigment to the electron density clearly disagrees with the conventional assumptions on the binding of Pchlide, and hence on the most likely mechanism of the reaction (Fig. 2). Specifically, the nicotinamide ring of NADPH and the Y276 are found in neither the proximity nor the orientation previously assumed with respect to C17 and C18 (mode B, Fig. 2B). Moreover, the pigment is flipped and rotated compared to the previous assumptions (Fig. 2). This suggests a different mechanistic pathway than those discussed in the past due to the fundamentally distinct positioning of Pchlide (mode B), which even challenges the conventional interpretation of the Begley–Young experiment7 where NADPH is supposed to reduce C17. Nguyen et al. therefore hypothesized the involvement of other alternative proton donors or water molecules, but nevertheless, the question on the mechanism of action of LPOR remains wide open.
Given the central importance of the binding mode of Pchlide in addressing mechanistic details, here we evaluate the validity of both competing binding modes A and B taking advantage of the new structure of the enzyme that depicts LPOR in its active form and combining a range of new computational and experimental data.
Materials and methods
Experimental
LPOR WT and the mutants were expressed in E. coli and purified according to the previously described protocol.16 The AtPORB C309S, AtPORB C309A and AtPORB Y276F mutants have been obtained from a previous study.15 The AtPORB F327A_F330A, Q331A and Q331E mutants were obtained using PCR with previously described protocol.16,17 The primers and temperature conditions are described in ESI (Table S8†).For spectra measurements, reaction mixtures were prepared according to previously described protocol16,18 with protochlorophyllide purified from etiolated wheat seedlings.19 The reaction mixtures were prepared under dim, green light, that was previously shown not to trigger the reaction catalyzed by LPOR, and consisted of: 200 μM NADPH, 5 μM Pchlide, 15 μM LPOR, and the lipids: MGDG:DGDG:PG (50 mol%, 35 mol%, 15 mol%, Avanti Polar Lipids) in a phosphate buffer (37.5 mM phosphate, 225 mM NaCl, 150 mM imidazole, 7 mM 2-mercaptoethanol, 25% w/w glycerol). Samples were incubated for 30 minutes in darkness at room temperature, before being transferred into glass capillaries and frozen in liquid nitrogen. The spectra were measured at 77 K with PerkinElmer LS50b spectrofluorometer set to the following parameters: excitation 440 nm, emission 600–750 nm, slits: 9 nm/9 nm, 400 nm min−1. To verify enzymatic activity, samples were thawed in darkness, illuminated with white light (80 μmol photons per m2 per s) for 20 seconds, then frozen in liquid nitrogen, and measured again. For data analysis, the spectra were normalized at maximum intensity. To calculate the photoactive Pchlide as compared to WT, the spectra of free Pchlide, Pchlide in a complex with WT enzyme and the mutants were used. For each, the ratio R between the intensities at 655 nm (emission maximum of oligomeric complex) and at 632 nm (emission maximum of free Pchlide) were calculated. For each experimental condition at least two independent reaction mixtures were analyzed. The photoactive Pchlide was calculated according to the formula: (Rmut − RPch)/(RWT − RPch), where Rx is 655/632 intensity ratio of a given mutant (mut), WT enzyme or free Pchlide in a buffer (Pch).
Samples for electron microscopy were negatively stained according to the previously published protocol15 and visualized with JEOL JEM2100 HT CRYO LaB6 electron microscope. At least two independent samples were analyzed and representative micrographs are presented.
Computational
The molecular mechanics (MM) based set-up for multiscale simulations was constructed using the cryo-EM structure of LPOR (PDB ID: 7JK9).15 A monomer was extracted from the larger oligomeric structure, including the protein and three protein-bound co-factors (Pchlide, NADPH, and MGDG) to build the WT and mutants models. To account for missing water molecules in the cryo-EM structure, a systematic hydration protocol was applied, followed by equilibration and 400 ns of cumulative production simulations. Binding free energy computations were performed using the MM-PBSA protocol.20 Quantum Mechanics/Molecular Mechanics (QM/MM) multiscale calculations were performed with the additive scheme along with electrostatic embedding as implemented in ORCA 5.0.21 For geometry optimizations, density functional theory (DFT) computations were performed using the PBE22 functional with the def2-SVP basis set.23 Further details can be found in the ESI.† The QM region contains Pchlide, NADPH, MGDG, and amino acid side chains within 9 Å to the central magnesium atom of Pchlide (Fig. 3). The active MM region contains residues within a radius of 5 Å from any atom of the QM region. The regions were manually adapted such that they are chemically meaningful, for example adjusting the backbone from the QM region. QM/MM system consists of 72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 270 atoms, where the QM subsystem contains 401 atoms (link atoms included). The regions are visualized in Fig. S1.† Long-range electrostatic interactions are essential for modeling biological processes and in our work we used the electrostatic embedding scheme, where the QM electron density interacts explicitly with the MM charges. In the additive QM/MM multiscale approach, link atoms are created and the charges are corrected with a charge shifting scheme. Force switching was used for the Lennard-Jones (LJ) interaction and force shifting for the electrostatic interaction. A smooth switching function was used to truncate the LJ potential energy starting after a cut-off of 10 Å, going to 0 after 12 Å. The electrostatic potential was shifted to zero after 12 Å from the QM region (more details can be found in ESI†).
270 atoms, where the QM subsystem contains 401 atoms (link atoms included). The regions are visualized in Fig. S1.† Long-range electrostatic interactions are essential for modeling biological processes and in our work we used the electrostatic embedding scheme, where the QM electron density interacts explicitly with the MM charges. In the additive QM/MM multiscale approach, link atoms are created and the charges are corrected with a charge shifting scheme. Force switching was used for the Lennard-Jones (LJ) interaction and force shifting for the electrostatic interaction. A smooth switching function was used to truncate the LJ potential energy starting after a cut-off of 10 Å, going to 0 after 12 Å. The electrostatic potential was shifted to zero after 12 Å from the QM region (more details can be found in ESI†).
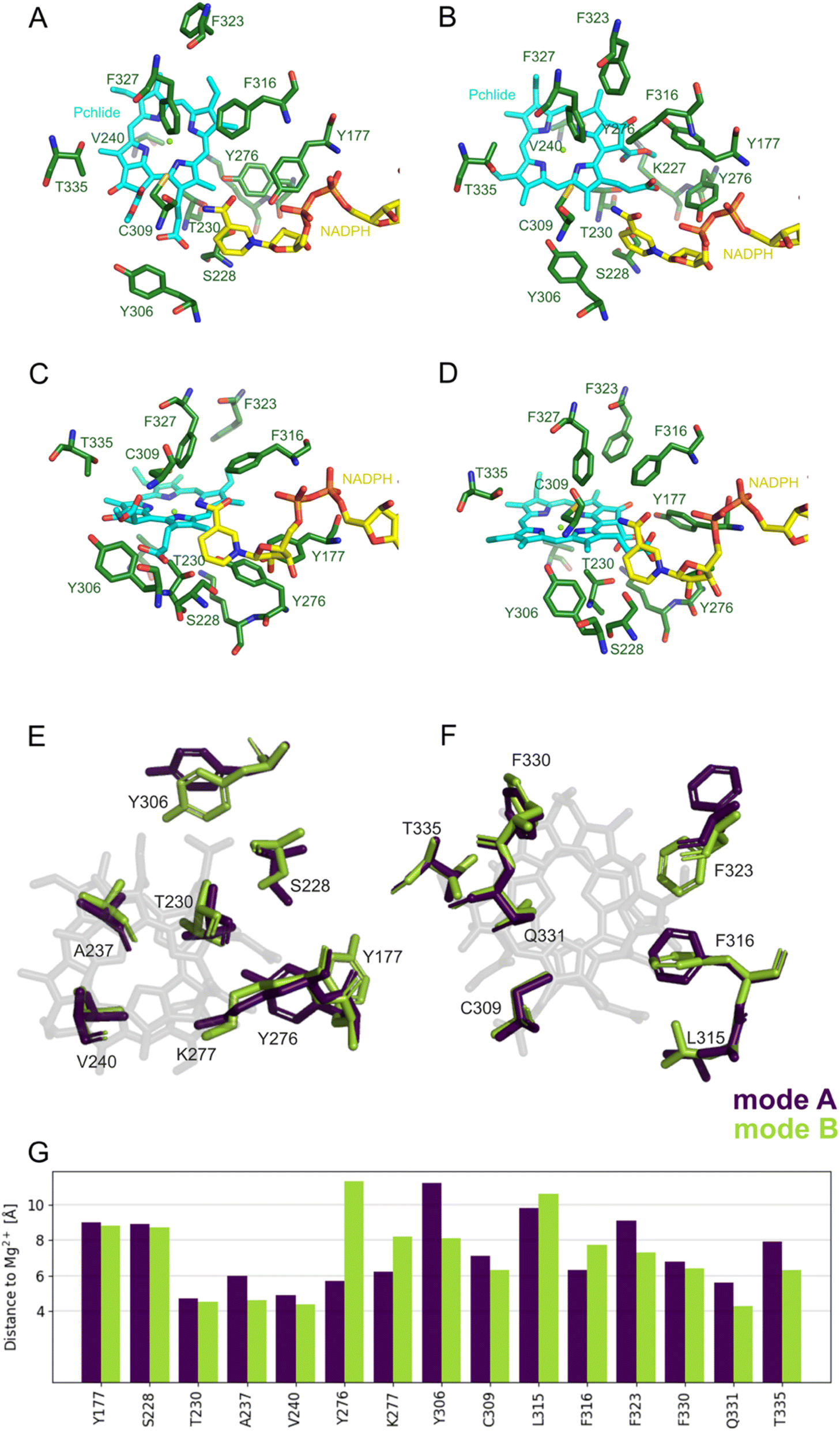 | ||
| Fig. 3 Selected residues of the pigment binding pocket in two binding modes within the optimized QM/MM model. (A) Top view of the active site for mode A. (B) Top view for mode B. (C) Lateral view for mode A. (D) Lateral view for mode B. (E and F) The positioning of selected residues located at separate sites on the tetrapyrrole ring in two distinct binding modes. (G) Distances between selected residues and central magnesium ion of Pchlide in two binding modes. | ||
Local Energy Decomposition (LED) analysis24,25 of the binding energy was performed using the DLPNO-CCSD(T) approach (domain-based local pair natural orbital approach to coupled cluster theory with singles, doubles, and perturbative triples excitations26). The DLPNO-CCSD(T)/def2-TZVP energy was decomposed into a series of additive contributions corresponding to the interaction of pairs of the defined fragments. The fragmentation includes Pchlide as one fragment, as well as NADPH and the lipid MGDG, and a series of amino acid side chains within 9 Å to the central Mg atom representing other fragments. Additional detailed information on the computational protocols is provided in the ESI.†
Results
Structural comparison of binding modes
To investigate the two potential binding modes A and B of Pchlide within the substrate pocket of LPOR, we conducted a comprehensive analysis using two distinct computational methodologies encompassing classical atomistic molecular dynamics simulations and hybrid quantum mechanics/molecular mechanics (QM/MM). For Pchlide in mode B, we used as starting point the cryo-EM structure (PDB ID: 7KJ9), while mode A was created following cryo-EM structure refinement using a “flipped” orientation of the Pchlide and subsequent MM-based minimization to adapt the pigment in novel orientation to the binding pocket. Both models were eventually fully optimized within a Quantum Mechanics/Molecular Mechanics (QM/MM) approach. The selected QM region and resulting models are demonstrated in Fig. S1.† To give an understanding of the active site architecture for both binding modes, selected residues, as well as the pigment, are shown in Fig. 3. Fig. 3E and F depicts essential amino acids residues and the pigment within both modes, highlighting the manner in which Pchlide is positioned within the protein pocket for both binding modes with respect to the central Mg2+ ion of Pchlide. The orientation of most residues is nearly identical in both modes (Fig. 3G, Tables S1 and S2†), except for F323, Y276, Y306, and T335, which exhibit the most distinct positioning.Additionally, we analyzed the distances evolution in the MD simulations between C17/C18 carbon atoms of Pchlide and the residues investigated in the previous study,15 namely S228, Y276 and C309 (Fig. S2 and S3†). We found that for mode A, the distance between Y276 and C17 is around 4 Å, but for mode B, the distance is over 7 Å between both crucial carbon atoms of Pchlide and the residue. In contrary, for mode B, the distance between C17 and S228 is around 4.57 Å, while C18 and C309 are about 4 Å apart (Fig. 4, S2 and S3†).
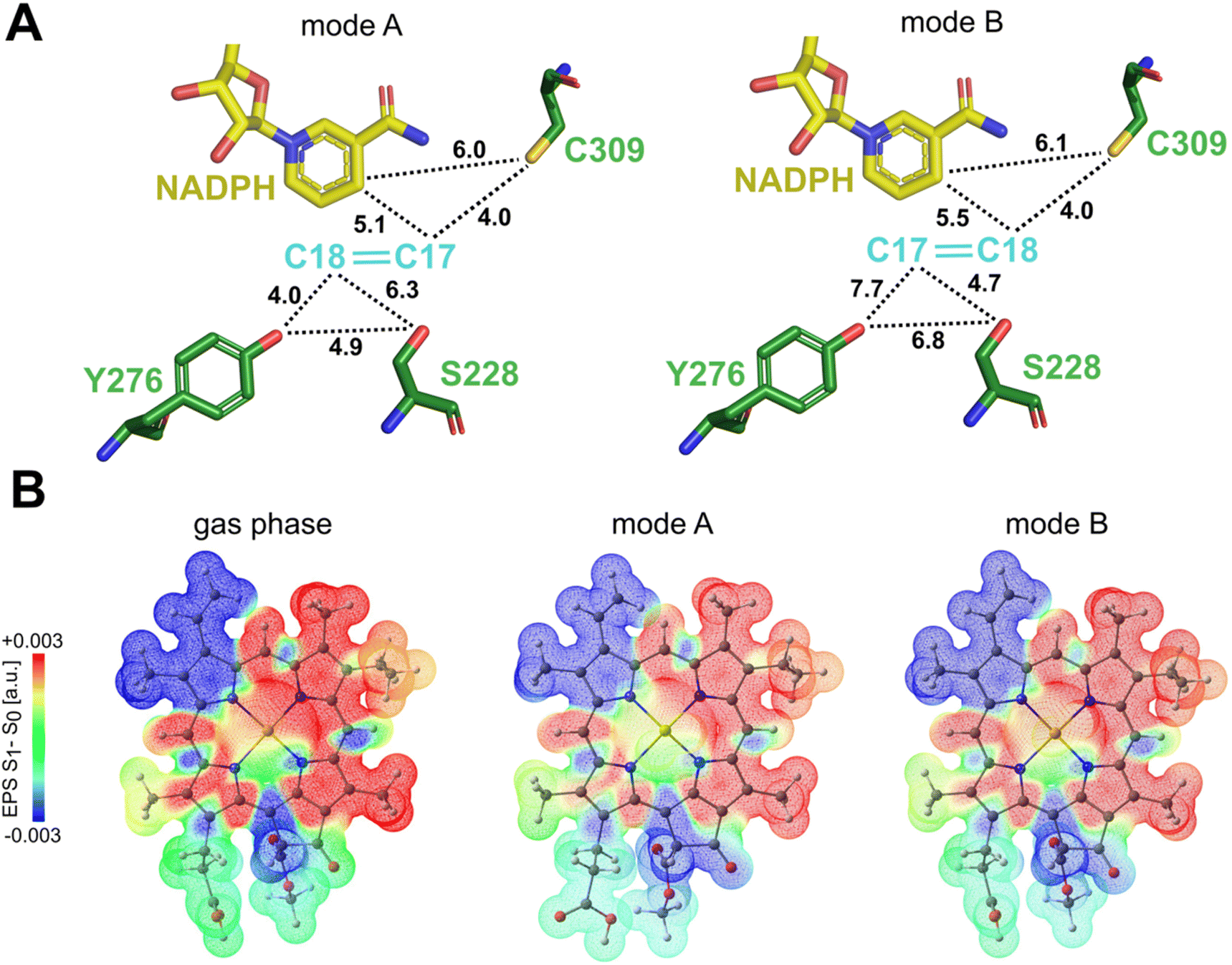 | ||
| Fig. 4 Mapping molecular proximity and electrostatic potentials in Pchlide binding modes (QM/MM optimized structures). (A) A schematic representation illustrating the average distances between C17C18 atoms of Pchlide, NADPH and the crucial residues for both mode of binding. (B) Difference electrostatic potential map from TD-DFT calculations (S1 minus S0) associated with the S0 → S1 (Qy) excitation for Pchlide in gas phase, binding mode A and binding mode B. | ||
Excited states of the two binding modes
Polarization along the C17–C18 bond in the excited state is a key driving force for the proton-coupled electron transfer process. Therefore, we assessed the possible impact of the different Pchlide modes on this bond. Our excited state TD-DFT computations with the ωB97X-D3BJ functional27,28 reveal that the polarization of the C17–C18 bond (judged by the S1–S0 ESP difference, see Fig. 4B) shows similar behavior in mode A and mode B. The computed S1 energies are also practically the same for the two binding modes, 1.986 eV (mode A) and 1.984 eV (mode B). Since computations reveal similar bond polarization, this implies that the excited-state polarization of this bond is intrinsic to the electronic structure of Pchlide. Therefore, the different binding modes do not alter the intrinsic properties of the pigment and the fundamental features of light-driven excitation as judged by the S1–S0 ESP difference. However, the different binding does have implications for the nature of subsequent mechanistic events, namely for the residues involved and their role in reactivity.Computational analysis of protochlorophyllide binding
Our subsequent aim was to determine the Pchlide binding affinity in both modes and further gain atomic-level insights into the functional role of the protein in substrate stabilization. The first approach involved a comprehensive MM-PBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) based characterization, enabling the computation of the Pchlide binding energy along the dynamic progression while explicitly considering protein flexibility. The conformational flexibility of the protein and co-factors was achieved through large-scale classical molecular dynamics simulations for 200 ns. We applied the MM-PBSA approach systematically over the complete simulated time-scales to account for the local and global functional motion of the protein. In the case of the MM-PBSA approach, the binding energies were averaged over a large ensemble of snapshots (see ESI† for details).The second approach employed a QM/MM technique using the DLPNO-CCSD(T) method as the basis for the Local Energy Decomposition (LED) scheme, which enables decomposition of the DLPNO-CCSD(T) interaction energy into fragment-pairwise additive contributions. This approach assessed the interaction energies between the substrate and the protein by explicitly considering the quantum interactions among electron orbitals localized on distinct fragments.
Our MM-PBSA revealed a stronger binding affinity of Pchlide for mode B than mode A (Fig. 5A). The binding energy of Pchlide in mode B was found to be more exergonic (−9.79 ± 0.219 kcal mol−1) compared to that of mode A (−1.71 ± 0.247 kcal mol−1). To gain detailed insights, the binding free energy was decomposed into per-residue contributions to identify which residues are important for the binding process within each mode. For mode A, the most significant contributors were T230, A237, V240, Y306, C309, F316, F327, F330, and T335. For mode B, the key residues were Y177, T230, A237, V240, A273, C309, L315, F316, F323, F327, F330, Q331, and T335 (Fig. 5B). Most of these residues exhibited stronger interactions for mode B, except for Y306, C309, F316 and F327, where the interactions were predicted to be stronger for mode A. Overall, the amino acid residues in the substrate binding pocket significantly contribute to the overall binding energy (see Fig. 5) and exhibit differential stabilization between the two modes of Pchlide binding, particularly favoring mode B.
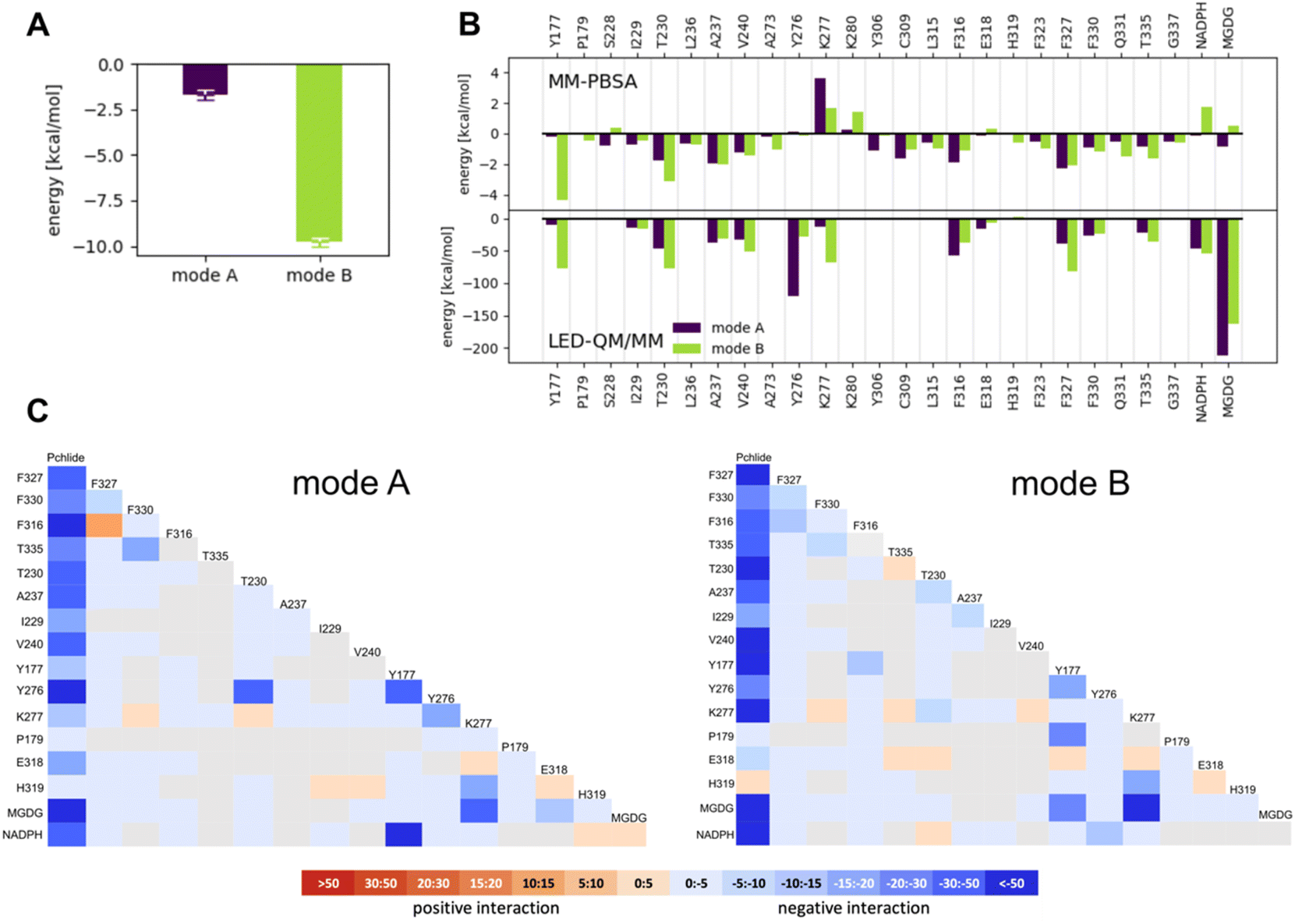 | ||
| Fig. 5 Computational analysis of Pchlide binding. (A) Total Pchlide binding energy calculated with MM-PBSA approach. (B) The contribution of selected residues to the Pchlide binding energy as calculated with MM-PBSA and LED QM/MM approaches for modes A and B. (C) LED interaction map for mode A and B. All energies shown in the LED maps are given in kcal mol−1. | ||
To go beyond the classical mechanics picture and further elucidate the energetics of Pchlide binding at the quantum chemical level, we employed a QM/MM-based LED analysis (Fig. 5B and C). We found that for mode A the binding arises primarily from interaction terms with T230, A237, Y276, F316 and F327, as well as with NADPH and MGDG (Fig. 5B, C and S1†). In contrast, for mode B, LED analysis reveals a complex network of interactions that effectively stabilize the pigment in this specific conformation (Fig. 5C). On one side of the pigment, we observed notable electrostatic and exchange interactions between the tetrapyrrole ring and three phenylalanine residues (316, 327, and 330) as well as T335 (Fig. 6B). On the other side of the pigment, a chain of hydrogen bonds involving T230, K277, and MGDG facilitates the connection between the two carbonyl oxygens of the Pchlide molecule at carbon atoms C131 and C133 (Fig. 6A). The LED analysis suggests that these interactions primarily arise from electrostatic and exchange forces, but electrostatic interactions dominate the MGDG–Pchlide interaction.
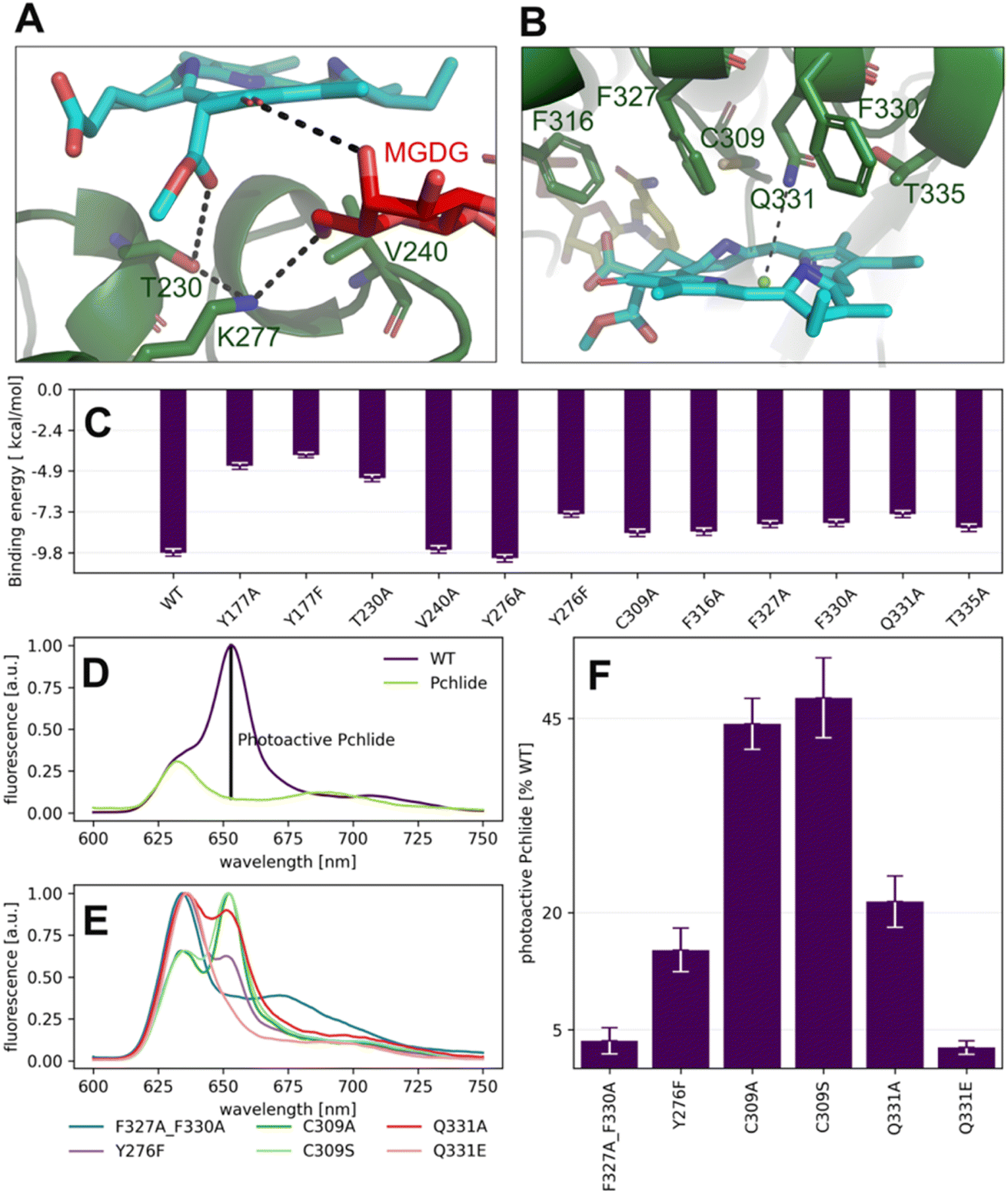 | ||
| Fig. 6 Pchlide binding in mode B. (A and B) Major interactions between Pchlide, amino acid residues and MGDG in mode B. (C) Impact of point mutations on binding energy assessed via the MM-PBSA methodology. (D and E) The low-temperature fluorescence spectra of free Pchlide in a buffer (D: Pchlide), Pchlide in the reaction mixture with LPOR-WT, NADPH and lipids (D: WT), and Pchlide in the same conditions but with different mutants (E). The fluorescence intensity assigned to photoactive Pchlide in marked (D). (F) The amount of photoactive Pchlide compared to WT-LPOR calculated for different mutants. Site-directed mutagenesis and validation of the Pchlide binding mode. | ||
Overall, despite the fundamentally distinct theoretical approaches of molecular mechanics and the correlated wave function approaches employed here, all computational results favor binding of Pchlide in mode B, which is consistent with the recent cryo-EM data, rather than in the previously hypothesized mode A. Moreover, both approaches identify a similar set of residues as contributing to the favorable binding in mode B.
To further evaluate the influence of specific residues on the binding affinity of Pchlide in mode B, we conducted a computational (using MM-PSBA) and experimental mutational screening. We selected residues with significant contributions to the binding energy and performed selective substitutions (Fig. 6C) in order to examine the corresponding changes in the binding free energy (ΔΔG). Among the chosen mutations, the system with Y177A, Y177F and T230A led to a significant decrease of Pchlide binding energies by about half compared to the WT (Fig. 6C). The substitutions of Y276F, C309A, Q331A, T335A and the three phenylalanine residues: F316A, F327A, and F330A led to a reduction in binding energies by about a quarter (Fig. 6C). Conversely, the mutations of V240A and Y276A exhibited minimal to negligible effects on the binding energies. Overall, these findings underscore the importance of specific residues in modulating the binding affinity of Pchlide in mode B and provide valuable insights into the molecular mechanisms governing this process.
To experimentally validate the computational predictions regarding the contributions of these residues to Pchlide binding, we performed site-directed mutagenesis on selected residues. Based on the available data and structural information, we chose to study the following mutants: Y177F, Y276F, C309A, C309S, Q331E, Q331A, and the double mutant F327A_F330A. To evaluate the impact of these mutations on Pchlide binding, we compared the low-temperature fluorescence emission spectra of Pchlide in a complex with lipids, NADPH and AtPORB: the WT and the respective mutants (Fig. 6D–F). The peaks corresponding to free non-photoactive Pchlide, and the pigment bound to LPOR exhibit distinct differences, with maximum emission peaks separated by 23 nm (Fig. 6D). These spectral variations have been extensively employed to estimate the quantity of photoactive Pchlide (i.e., the pigment bound to the enzyme in the active holo conformation, Fig. 6D) in etiolated seedlings.29,30 In our study, we employed the same approach to estimate the amount of bound Pchlide in the mutant variants (Fig. 6E and F). Our findings revealed that the double mutant F327A_F330A and the Q331E mutant were barely able to bind the pigment, while the Y276F and Q331A mutants exhibited binding at about 20% compared to the wild type. Additionally, the C309A and C309S mutants showed binding at around 45% compared to the wild type. The close-up analysis of the emission maxima of the peaks originating from remaining non-photoactive Pchlide revealed that they resemble the emission maximum of Pchlide:LPOR complex except for F327A_F330A and C309A, for which the maxima where blue-shifted towards the emission maximum characteristic of free Pchlide (Fig. S4†).
Our calculations predicted that the interaction between Y177 and Pchlide is one of the strongest in mode B (Fig. 5B). LED analysis predominantly attributes this interaction to electrostatic and exchange energies, which align with the cryo-EM structure. The structure indicates a potential hydrogen bond between the hydroxyl group of tyrosine and a C131 carboxyl of the pigment (Fig. 7A). This interaction is only possible in binding mode B because, in the flipped orientation (mode A), the carboxyl oxygen points towards the solvent while Y177 points towards ring A of Pchlide (Fig. 2).
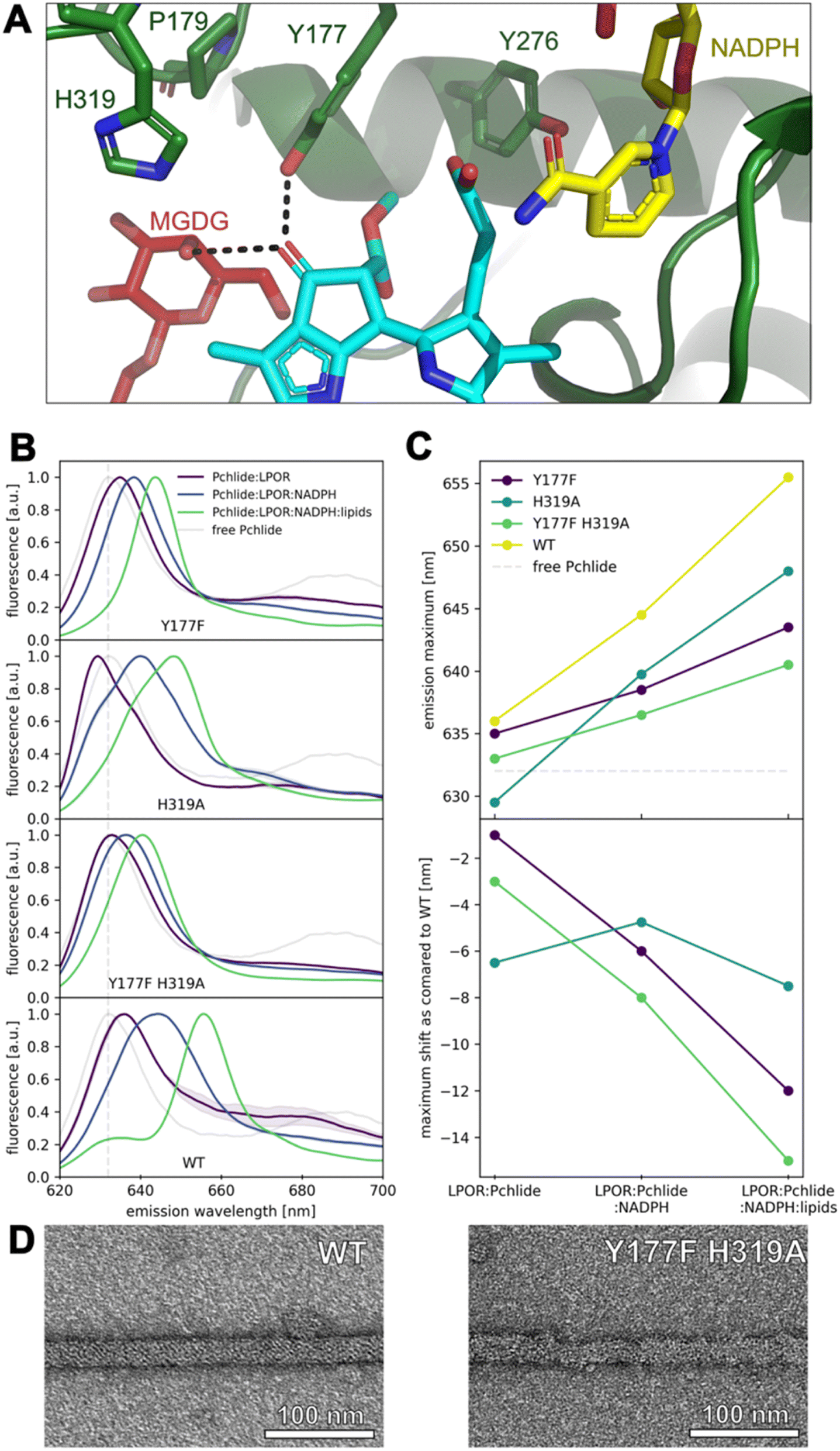 | ||
| Fig. 7 Hydrogen-bonding interactions with the keto group at C131 of Pchlide affect the emission maximum of the pigment. (A) Residues in proximity to keto group at C131. (B) Fluorescence emission spectra of free Pchlide in buffer and in complexes with LPOR WT and mutants. Reagents concentrations: 15 μM LPOR, 5 μM Pchlide, 200 μM NADPH and 100 μM lipids (50 mol% MGDG, 35 mol% DGDG, 15 mol% PG). Spectra represent averages from at least two independent experimental replicates, with standard deviations depicted as bands. (C) Fluorescence emission maxima of Pchlide in different complexes with WT LPOR and mutants and shifts of the emission maxima relative to WT enzyme. (D) Negative-stain electron microscopy micrographs of LPOR:Pchlide:NADPH:lipids oligomers of WT enzyme and Y177F_H319A mutant. | ||
We further performed mutation studies on Y177 to assess its role in the pigment binding. The Y177F mutant showed the ability to bind Pchlide and form complexes with NADPH and lipids, all of which exhibited enzymatic activity in the presence of NADPH and Pchlide (Fig. 7B and S5†). Interestingly, the fluorescence emission maxima of Pchlide bound by the Y177F mutant were blue-shifted compared to the wild-type enzyme (Fig. 7B and C). To explore further, we also mutated the adjacent residue H319 (Fig. 7A), even though it was predicted to interact weakly with Pchlide (Fig. 5B). The H319A mutant was also enzymatically active in the presence of NADHP and Pchlide, and capable of complex formation with NADPH and lipids (Fig. 7B and S5†). Both mutations, Y177F and H319A, influenced the emission maxima of Pchlide in the complexes with LPOR, LPOR and NADPH, as well as LPOR:NADPH:lipids, but the strength of the observed effects was distinct for each mutant.
In the case of H319A, the emission maximum of the LPOR:Pchlide complex was blue-shifted compared to free Pchlide, while for Y177F, it was red-shifted, albeit not to the extent of the WT enzyme (Fig. 7B and D). The addition of NADPH to the reaction mixture caused a red-shift of the maxima for both mutants, but it was less pronounced compared to the WT enzyme, with Y177F showing a milder effect than H319A (Fig. 7B and D). As for LPOR WT, the addition of lipids containing MGDG to the reaction mixture, along with NADPH and Pchlide, further red-shifted the emission maximum of the pigment to the characteristic value of 655 nm and led to the formation of filamentous oligomers (Fig. 7D). The addition of the lipids to the reaction mixtures of the mutants also resulted in the further red-shift of emission, but not as profound as for the WT enzyme. The observed red-shift was less profound for Y177F than for H319A (Fig. 7B and D).
Finally, we also characterized a double mutant, Y177F_H319A, which was found to be enzymatically active in the presence of NADPH and Pchlide, and capable of forming the complexes with Pchlide, NADPH, and lipids (Fig. 7B and S5†). In terms of emission maxima of the complexes, the double mutant behaved similarly to the Y177F mutant, but with even less pronounced red-shifts, approximately by 2–3 nm compared to Y177F (Fig. 7C). We confirmed that the Y177F_H319A double mutant could form filamentous oligomers in the presence of Pchlide, NADPH, and lipids (Fig. 7D), although they were less organized than those formed by the WT and were not as abundant on the grid.
Discussion and conclusions
The light-dependent catalysis carried out by LPOR has been a subject of great interest for many years. However, due to the limited availability of data on the structure of the active complex, investigations into the reaction mechanism have been built on uncertain foundations. Fortunately, with the recent determination of a high-resolution structure of the enzyme in its active conformation,15 we are now able to evaluate various hypotheses regarding pigment binding, the involvement of specific residues in this process, and ultimately, the reaction mechanism. This breakthrough provides a solid basis for advancing our understanding of the intricate workings of LPOR and its role in light-dependent catalysis. In this paper, we focused our efforts to compare two binding modes of Pchlide.Protochlorophyllide binding
We employed classical molecular dynamics and hybrid quantum-mechanics/molecular-mechanics (QM/MM) simulations to produce the “best-case” structural solutions for the two binding modes and we evaluated them using a range of criteria, including binding free energies determined using the MM-PBSA,31 and combine such simulations with experimental mutation data.The MM-PBSA calculations performed for the previously assumed mode A indicate that the binding interactions are much weaker compared to the cryo-EM structure-based mode B, however it is thermodynamically stable. A limited number of residues can contribute to the binding energy in this mode, but overall, the observed interactions are nonspecific in nature. Previous simulations exploring this binding mode also demonstrated its thermodynamic stability but similarly did not reveal any specific interactions with the pigment, except for interactions with the propionate group. These interactions were attributed to either S228 (T145 in T. elongatus LPOR)32 or I229 (V142 in Synechocystis LPOR).14 Our analysis assigned only mild interactions between the pigment and these residues (Fig. 5). In one of these studies, which provided relevant data, the remaining essential elements of the pigment were observed to either interact with water molecules (the magnesium ion, the C131 keto group) or demonstrate a lack of interactions entirely (the C133 keto group).32
Notably, any modifications to these pigment components have been shown to significantly impair or eliminate the affinity between modified Pchlide derivatives and LPOR, highlighting the enzyme's sensitivity towards these parts of the pigment.32,33 The enzyme's exceptional substrate specificity is expected, considering that Pchlide is among several chlorophyll intermediates present in plastids. To uphold such high substrate specificity, LPOR must possess specific residues that interact exclusively with Pchlide, enabling the enzyme to discriminate it from other similar intermediates.
This is indeed realized in binding mode B, for which we have discovered a distinct ensemble of interactions between the residues of LPOR and the pigment. Collectively, these interactions result in nearly six times higher (more negative) binding energy compared to mode A, indicating significantly enhanced thermodynamic stability. Notably, a subset of the identified interactions involves vital components of Pchlide, namely the keto groups at C131 and C133, suggesting their potential contribution to the substrate specificity of LPOR. Both carboxyl groups are formed by the Mg-protoporphyrin IX monomethyl ester cyclase, which catalyses a preceding reaction in the chlorophyll biosynthetic pathway, namely the formation of divinyl-Pchlide. Our computational analysis of the two modes of Pchlide binding reveals different orientations of the crucial carboxyl group at C131. In mode A, this group points towards the solvent, while in mode B, this group is involved in one of the strongest binding interactions we see between the pigment and the enzyme in both analyzed modes.
Previous computational studies have demonstrated that a strong hydrogen bond with the carboxyl group at C131 is responsible for the red shift observed in the fluorescence emission spectrum of the pigment during Pchlide binding to LPOR.13 Our site-directed mutagenesis experiments clearly show that the Y177F mutation allows the enzyme to bind Pchlide, but the resulting complexes exhibit a blue-shifted emission maximum compared to the wild type (Fig. 7B and C). Additionally, one of the hydroxyl groups of MGDG is predicted to have a strong interaction with the carboxyl group at C131. Our previous experiments have shown that the addition of MGDG causes a red shift in the emission maximum of the complex.16 These findings suggest that both Y177 and MGDG red shift the emission of the pigment, by forming strong bonds with carboxyl group at C131.
A spectral blue shift in emission, similar to that observed in the Y177F mutant, is also seen in the H319A mutant. However, in the fully assembled complex as determined by cryoEM, the interaction between the residue and the pigment is predicted to be weak and repulsive (Fig. 5B and C). Interestingly, the H319A mutant is the only mutation discovered thus far that induces a blue shift in the emission maximum of the pigment in the LPOR:Pchlide complex compared to free Pchlide in a buffer solution (Fig. 7C). Previous studies in model systems have demonstrated that the emission maximum of the pigment is influenced by the solvent's dielectric constant.34 These observations suggest that the binding pocket of the H319A mutant may be more hydrophobic than the water-based buffer. If this is indeed the case, it suggests that in the absence of NADPH, H319 may interact with the pigment, specifically with the carboxyl group at C131. Such an interaction would likely necessitate the enzyme to adopt a different conformation in this complex compared to the structure determined by cryoEM, most likely the apo conformation (Fig. 8). However, to validate this hypothesis, it is crucial to obtain a detailed structure of the LPOR:Pchlide complex lacking NADPH.
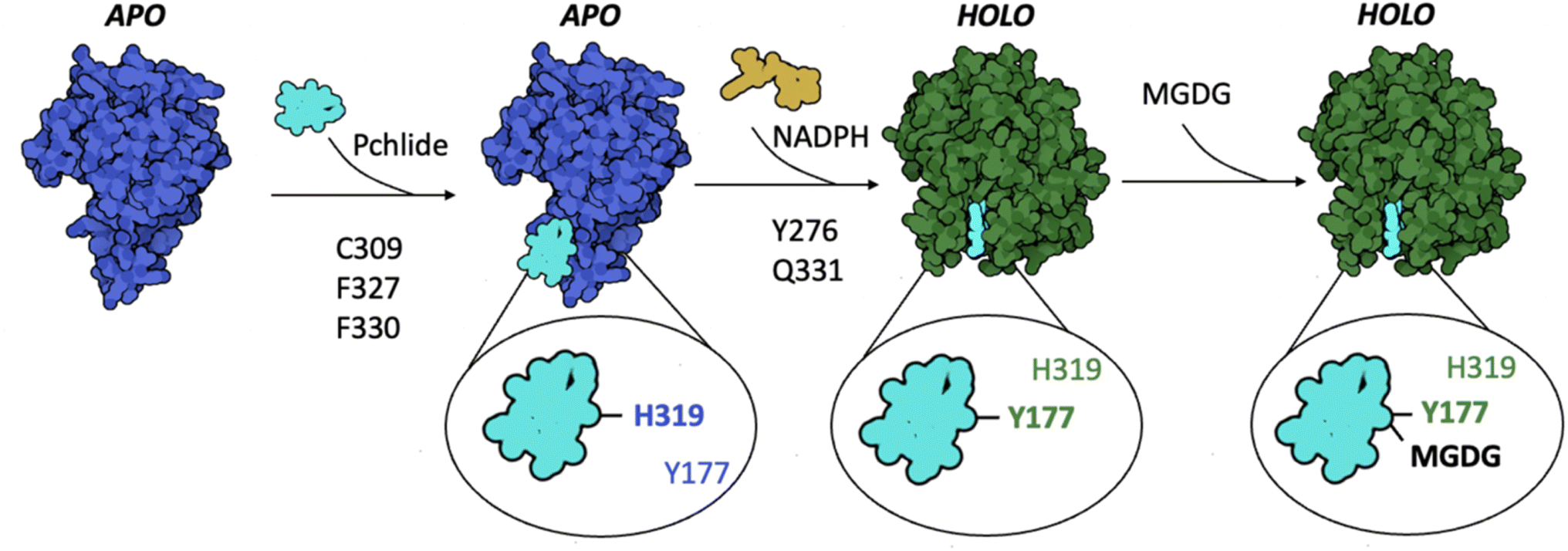 | ||
| Fig. 8 A model of Pchlide binding to LPOR. Pchlide attaches to the enzyme's apo conformation, with C309, F327, and F330 playing crucial roles in the process, along with H319 which interacts with carboxyl at C131 of Pchlide, red-shifting fluorescence emission maximum of the complex. The subsequent binding of NADPH changes the conformation of LPOR to active holo state, in which Y177 starts to interact with the pigment, causing an additional red-shift of the emission maximum. The interactions between the pigment and the residues Y276 and Q331 are important for the conformational change. On the membrane, the enzyme binds MGDG, which interacts with carboxyl at C131 of Pchlide, red-shifting the emission maximum of the complex. | ||
An additional interaction specific to mode B, with potential implications for substrate specificity, involves a strong bond between T230 and the C133 keto group. This interaction is stabilized by the presence of K277, as evidenced by LED analysis (Fig. 5B, C and 6A). Notably, in a bacterial LPOR variant, mutation of the residue corresponding to T230 (T. elongatus T147) exhibited a major impact on steady-state activity and NADPH affinity, while only mildly affecting Pchlide binding.35 Supplementary spectral data revealed, however, a blue-shifted absorbance maximum for the mutant complex compared to the WT, indicating a potential variation in the conformation of the binding pocket between the mutant and WT enzyme.
The other identified interactions are not specific to a particular binding mode. For instance, the interaction between the pigment and Q331, which is most likely a coordination bond, can occur irrespective of the ring's orientation. Similarly, the hydrophobic interactions involving F316, F327, and F330 are also independent of the binding mode. Mutations of these residues resulted in a significant decrease in pigment binding, confirming their involvement in the process.
Upon comparing the calculated binding energies of mutants with experimental data, we observed agreement for mutations of Y276, F237, F330, C309, and Q331, that is decrease in Pchlide binding. The decrease in the photoactive Pchlide is, however, greater for the mutants than what was expected from the calculation. The emission maxima determined for the remaining non-photoactive Pchlide show some variation between analyzed mutants: for some of them it resembles the one of Pchlide:LPOR complex, while for the others it is blue-shifted towards the free Pchlide in a buffer. This suggest that some of them, namely F327A_F330A and C309A, the mutations affected the binding of Pchlide already to the apo conformation, while for the rest, the decrease of the photoactive Pchlide may be associated with impaired transition between apo and holo state (Fig. 8). Both the emission maxima and the shapes of the peaks corresponding to the non-photoactive Pchlide suggest a good binding of Pchlide to the apo state of Y276F, C309S, Q331A and Q331E, but a different technique should be applied to assess these interactions with better precision. In previous studies the mutations of the residues corresponding to Y276 and C309 in T. elongatus (Y193 and C227, respectively) were associated with decrease in Pchlide binding.10,36 For these calculations, however, the absorbance at 642 nm was used to monitor the binding, which reflects the formation of the active holo state and therefore these analyses did not discriminate between the impaired conformational transition and the affected binding to the apo state.10,36
Additionally, some noticeable discrepancies between calculations and experiments were found for Y177. The Y177F mutant exhibited an effective Pchlide binding, resulting in no visible band originating for a free pigment in the spectrum (Fig. 7B). Computational predictions, on the other hand, indicate that the bond between Y177 and Pchlide in the active, holo state contributes significantly to the pigment binding, which may be interpreted as finding that supports mode A, in which the interaction with the residue is predicted to be insignificant. However, the emission spectrum of Pchlide:LPOR complex for the WT enzyme and the Y177F mutant are highly similar, which suggests that Y177 may not interact with the pigment in the apo state, unlike H319 (Fig. 8). The effect of the mutation can be seen only when NADPH is added to the reaction mixture, which induces the conformation change to the active holo state (Fig. 7C). The unexpectedly good binding of Pchlide to Y177F mutant may therefore be explained by the fact that the interaction between the pigment and the residue forms after the Pchlide binds to the apo conformation of the enzyme. Moreover, it is worth noting that the calculations do not account for the experimental conditions, particularly the presence of lipids, which induce an oligomeric state of the enzyme, causing the pigment to be sequestered in the outer leaflet of the membrane and effectively locked in the complex. An artistic visualization of the process of Pchlide binding in mode B based on the structure of PDB 9JK7 is presented in a movie provided as ESI material.†
Implications for the reaction mechanism
The overall findings of the present paper, based on both computational and experimental data, conclusively support mode B over mode A for Pchlide binding to LPOR. The electrostatic potential maps revealed that the binding mode does not affect the polarization of the C17–C18 in the excited state (Fig. 4B), but it has crucial implications for the residues involved in the photocatalysis owing to the stereospecificity of the reduction. Specifically, our conclusion regarding the binding mode appears – at least superficially – to contradict the conventional interpretation of the NMR study by Begley and Young, which aimed to identify the carbon atom reduced by NADPH. The atom proposed in that paper, C17 of Pchlide, appears too distant from the pro-S face of NADPH in mode B to accommodate direct hydride transfer. Moreover, the direct reduction of C17 atom by NADPH in mode B would lead to the incorrect stereoisomer of the pigment. To align the stereochemistry of the reaction with the binding mode B and with the Begley–Young interpretation, the indirect hydride transfer from NADPH to C17 is a possible solution. This process could involve the carboxyl group of Pchlide and/or S228, however there is no evidence for such a process. Alternatively, the Begley–Young observations may need to receive an alternative interpretation, or the NMR experiment will have to be revisited in light of the new structural knowledge. If the Begley–Young interpretation is not assumed to be an actual constraint, then a structure-based proposal for the mechanism would be that C18 can be directly reduced by NADPH with the possible involvement of C309, while C17 could be reduced by S228 with the involvement of Y276 and F316 (Fig. 9C). The involvement of both the solvent and multiple residues in proton transfer has been previously suggested.10 Interestingly, differences have been observed between cyanobacterial and plant LPOR homologs in both the solvent isotope effect and viscosity dependence.37,38 This suggests that plant variants rely less on long-range solvent-coupled protein motions for proton transfer, which may be an effect of evolutionary optimization.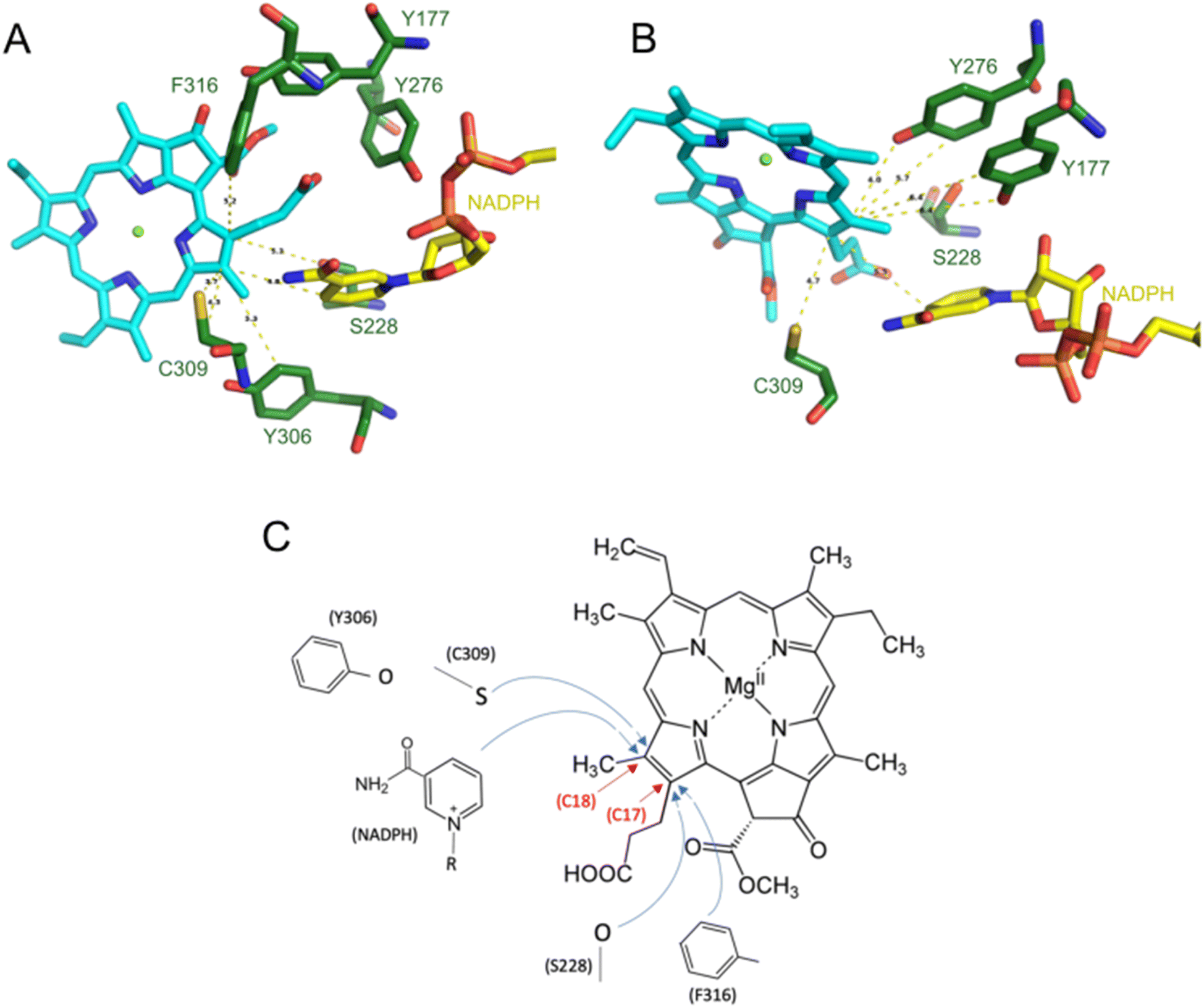 | ||
| Fig. 9 Implications of the present study for a possible reaction mechanism of Pchlide reduction by LPOR. (A) Residues potentially involved in the reaction for binding mode B. (B) Residues potentially involved for mode A. (C) A reaction mechanism that can be proposed in light of the favorable mode B binding of Pchlide. | ||
The scenarios of the reaction discussed above are speculative; therefore, the mechanism of the light-dependent reaction catalyzed by the enzyme remains an open question. A comprehensive analysis of both suggested reaction pathways incorporating detailed QM/MM simulations at the excited state of the pigment using the structurally-consistent binding mode of the substrate will be necessary. Such analysis is underway and will explicitly take into account the critical interactions between the pigment and the LPOR residues highlighted in this work.
Data availability
The data that support the findings of this study are available from the corresponding author.Author contributions
P. P.: investigation, analysis, writing – original draft; K. S.: investigation, analysis; H. C.·N.: analysis; A. S.: investigation, analysis, writing – original draft; D. A. P.: methodology, supervision, writing – review and editing; M. G.: conceptualization, methodology, investigation, analysis, supervision, writing – original draft; writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jerzy Kruk for his help with Pchlide purification. This work (presented in Fig. 6) was supported by SONATA project (2019/35/D/NZ1/00295) granted by NCN to MG. This research was funded by the Priority Research Area BioS under the program Excellence Initiative – Research University at the Jagiellonian University in Krakow (B.1.7.2021.16) granted to MG. PP and DAP acknowledge support by the Max Planck Society. Open Access funding provided by the Max Planck Society.References
- P. Wang, S. Ji and B. Grimm, J. Exp. Bot., 2022, 73, 4624–4636 CrossRef CAS PubMed.
- M. Gabruk and B. J. Mysliwa-Kurdziel, Biochemistry, 2015, 54, 5255–5262 CrossRef CAS PubMed.
- P. Brzezowski, A. S. Richter and B. Grimm, Biochim. Biophys. Acta, Bioenerg., 2015, 1847, 968–985 CrossRef CAS PubMed.
- T. Masuda and Y. Fujita, Photochem. Photobiol. Sci., 2008, 7, 1131–1149 CrossRef CAS PubMed.
- O. A. Sytina, D. J. Heyes, C. N. Hunter and M. L. Groot, Biochem. Soc. Trans., 2009, 37, 387–391 CrossRef CAS PubMed.
- L. O. Johannissen, A. Taylor, S. J. O. Hardman, D. J. Heyes, N. S. Scrutton and S. Hay, ACS Catal., 2022, 12, 4141–4148 CrossRef CAS PubMed.
- T. P. Begley and H. Young, J. Am. Chem. Soc., 1989, 111, 3095–3096 CrossRef CAS.
- K. L. Kavanagh, H. Jörnvall, B. Persson and U. Oppermann, Cell. Mol. Life Sci., 2008, 65, 3895–3906 CrossRef CAS PubMed.
- U. Oppermann, C. Filling, M. Hult, N. Shafqat, X. Wu, M. Lindh, J. Shafqat, E. Nordling, Y. Kallberg, B. Persson and H. Jörnvall, Chem.-Biol. Interact., 2003, 143–144, 247–253 CrossRef CAS PubMed.
- B. R. K. Menon, J. P. Waltho, N. S. Scrutton and D. J. Heyes, J. Biol. Chem., 2009, 284, 18160–18166 CrossRef CAS PubMed.
- N. Lebedev, O. Karginova, W. McIvor and M. P. Timko, Biochemistry, 2001, 40, 12562–12574 CrossRef CAS PubMed.
- D. J. Heyes, M. Sakuma, S. P. de Visser and N. S. Scrutton, J. Biol. Chem., 2009, 284, 3762–3767 CrossRef CAS PubMed.
- S. Gholami, A. Nenov, I. Rivalta, M. Bocola, A. K. Bordbar, U. Schwaneberg, M. D. Davari and M. Garavelli, J. Phys. Chem. B, 2018, 122, 7668–7681 CrossRef CAS PubMed.
- P. J. Silva and Q. Cheng, ACS Catal., 2022, 12, 2589–2605 CrossRef CAS PubMed.
- H. C. Nguyen, A. A. Melo, J. Kruk, A. Frost and M. Gabruk, Nat. Plants, 2021, 7, 437–444 CrossRef CAS PubMed.
- M. Gabruk, B. Myśliwa-Kurdziel and J. Kruk, Biochem. J., 2017, 474(7), 1307–1320 CrossRef CAS PubMed.
- M. Gabruk, Z. Nowakowska, B. Skupien-Rabian, S. Kędracka-Krok, B. Mysliwa-Kurdziel and J. Kruk, Biochim. Biophys. Acta, Proteins Proteomics, 2016, 1864(12), 1757–1764 CrossRef CAS PubMed.
- M. Gabruk and B. Mysliwa-Kurdziel, Biochem. J., 2020, 477, 2221–2236 CrossRef CAS PubMed.
- J. Kruk and B. Myśliwa-Kurdziel, Chromatographia, 2004, 60(1), 117–123 CAS.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case and T. E. Cheatham, Acc. Chem. Res., 2000, 33, 889–897 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- W. B. Schneider, G. Bistoni, M. Sparta, M. Saitow, C. Riplinger, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2016, 12, 4778–4792 CrossRef CAS PubMed.
- A. Altun, M. Saitow, F. Neese and G. Bistoni, J. Chem. Theory Comput., 2019, 15, 1616–1632 CrossRef CAS PubMed.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. S. Lin, G. De Li, S. P. Mao and J. Da Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- B. Schoefs and F. Franck, Photosynth. Res., 2008, 96, 15–26 CrossRef CAS PubMed.
- O. B. Belyaeva and F. F. Litvin, Biochemistry, 2007, 72, 1458–1477 CAS.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- S. Zhang, D. J. Heyes, L. Feng, W. Sun, L. O. Johannissen, H. Liu, C. W. Levy, X. Li, J. Yang, X. Yu, M. Lin, S. J. O. Hardman, R. Hoeven, M. Sakuma, S. Hay, D. Leys, Z. Rao, A. Zhou, Q. Cheng and N. S. Scrutton, Nature, 2019, 574, 722–725 CrossRef CAS PubMed.
- H. Klement, M. Helfrich, U. Oster, S. Schoch and W. Rüdiger, Eur. J. Biochem., 1999, 265, 862–874 CrossRef CAS PubMed.
- B. Myśliwa-Kurdziel, M. R. M. R. Amirjani, K. Strzałka and C. Sundqvist, Photochem. Photobiol., 2003, 78, 205–212 CrossRef.
- B. R. K. Menon, S. J. O. Hardman, N. S. Scrutton and D. J. Heyes, J. Photochem. Photobiol., B, 2016, 161, 236–243 CrossRef CAS PubMed.
- B. R. K. Menon, P. A. Davison, C. N. Hunter, N. S. Scrutton and D. J. Heyes, J. Biol. Chem., 2010, 285, 2113–2119 CrossRef CAS PubMed.
- D. J. Heyes, C. Levy, M. Sakuma, D. L. Robertson and N. S. Scrutton, J. Biol. Chem., 2011, 286, 11849–11854 CrossRef CAS PubMed.
- R. Hoeven, S. J. O. Hardman, D. J. Heyes and N. S. Scrutton, Biochemistry, 2016, 55, 903–913 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional methodological details, additional data, figures, tables and Cartesian coordinates of computational models. See DOI: https://doi.org/10.1039/d4sc00923a |
| ‡ Present address: Asher Biotherapeutics, South San Francisco, CA, USA. |
| This journal is © The Royal Society of Chemistry 2024 |