
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcitation generated preferential binding sites for ethane on porous carbon–copper porphyrin sorbents: ethane/ethylene adsorptive separation improved by light†
Shi-Chao
Qi
 ,
Yun-Jie
Zhao
,
Xiao-Jie
Lu
,
Yong-Lan
Liu
,
Zhen
Sun
,
Xiao-Qin
Liu
and
Lin-Bing
Sun
*
,
Yun-Jie
Zhao
,
Xiao-Jie
Lu
,
Yong-Lan
Liu
,
Zhen
Sun
,
Xiao-Qin
Liu
and
Lin-Bing
Sun
*
State Key Laboratory of Materials-Oriented Chemical Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), College of Chemical Engineering, Nanjing Tech University, 211816, Nanjing, China. E-mail: lbsun@njtech.edu.cn
First published on 16th April 2024
Abstract
Energy-efficient separation of C2H6/C2H4 is a great challenge, for which adsorptive separation is very promising. C2H6-selective adsorption has big implications, while the design of C2H6-sorbents with ideal adsorption capability, particularly with the C2H6/C2H4-selectivity exceeded 2.0, is still challenging. Instead of the current strategies such as chemical modification or pore space modulation, we propose a new methodology for the design of C2H6-sorbents. With a Cu-TCPP [TCPP = 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin] framework dispersed onto a microporous carbon and a hierarchical-pore carbon, two composite sorbents are fabricated. The composite sorbents exhibit enhanced C2H6-selective adsorption capabilities with visible light, particularly the composite sorbent based on the hierarchical-pore carbon, whose C2H6 and C2H4 adsorption capacities (0 °C, 1 bar) are targetedly increased by 27% and only 1.8% with visible light, and therefore, an C2H6-selectivity (C2H6/C2H4 = 10/90, v/v) of 4.8 can be realized. With visible light, the adsorption force of the C2H6 molecule can be asymmetrically enhanced by the excitation enriched electron density over the adsorption sites formed via the close interaction between the Cu-TCPP and the carbon layer, whereas that of the C2H4 molecule is symmetrically altered and the forces cancelled each other out. This strategy may open up a new route for energy-efficient adsorptive separation of C2H6/C2H4 with light.
Introduction
As a vital feedstock for the petrochemical industry, C2H4 is usually produced via C2H6 cracking. Owing to the small relative volatility (only 1.2), the separation of C2H6/C2H4 to produce polymer-grade C2H4 with a purity of 99.99% is one of the most challenging assignments, so its traditional cryogenic distillation usually operates under extreme conditions of −90 to −15 °C and 0.7–2.8 MPa along with >100 tray columns, which is highly energy-intensive.1–3 As an energy-efficient process, adsorptive separation of C2H6/C2H4 has drawn much attention, and various sorbents are being explored.4–8 There are two categories for the sorbents, C2H4-selective sorbents and C2H6-selective ones. The active C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of C2H4 and its smaller molecular size than C2H6 facilitate the design of C2H4-selective sorbents, and so a series of C2H4 sorbents with very high C2H4/C2H6 selectivity have been successfully prepared, based on the mechanisms of π-complexation,9 molecular sieving,10 kinetic separation,11etc. For example, the C2H4/C2H6 selectivity over a porous polymer named Cu(OPTz) can reach 75.12 However, the impurity component in cracking gas is C2H6, so the C2H6-selective sorbent possesses broader application prospects.
C bond of C2H4 and its smaller molecular size than C2H6 facilitate the design of C2H4-selective sorbents, and so a series of C2H4 sorbents with very high C2H4/C2H6 selectivity have been successfully prepared, based on the mechanisms of π-complexation,9 molecular sieving,10 kinetic separation,11etc. For example, the C2H4/C2H6 selectivity over a porous polymer named Cu(OPTz) can reach 75.12 However, the impurity component in cracking gas is C2H6, so the C2H6-selective sorbent possesses broader application prospects.
Preparation of C2H6 sorbents with both high adsorption capacity and high selectivity is challenging. Most of the C2H6-selective sorbents have still not exceeded 2.0 in ideal adsorption solution theory (IAST) selectivity,13,14 even if there have been some unconventional strategies to design C2H6-selective sorbents with unconventional materials such as post-synthetic shielding,15 pore modulation,16 and pore space partition (PSP).17 Li et al. oxidized the original Fe(II) sites in the metal–organic framework (MOF) of Fe2(dobdc) which is inclined to capture C2H4via π-complexation, and then the Fe(II) sites turned to be C2H6-affinitive, which led to an impressive C2H6/C2H4 selectivity of 4.4.18 Zhang et al. employed the PSP strategy to split the hexagonal channel of the MIL-88D MOF into two small rectangular channels to fabricate a NbU-12 MOF, which exhibited a C2H6/C2H4 selectivity of 1.5.19
The studies on C2H6-selective sorbents are concentrated on MOF-based materials in recent years,20–23 so the hydrothermal stability and the costs of sorbents are additional obstacles along with further improving and balancing the C2H6 capacity and selectivity. Meanwhile, the above-mentioned strategies to construct the selective adsorption sites are limited by the mechanistic theory, being specific to the base materials employed. Note that the underlying cause of an adsorption site working should be its specific charge density distribution contributed by the molecular skeleton and the only corresponding electron density distribution (EDD). This means that the adsorption ability and selectivity of an adsorption site can be modulated via electronic excited states, provided that the excited states are stable enough to meet the timescale of microscopic adsorption equilibrium.24–26
In this study, we propose and verify a new strategy to realize C2H6-selective adsorptive separation. Metalloporphyrins are widely found in nature and living organisms, such as cytochrome, heme, and chlorophyll, and are easily excited by visible light, and then exhibit the photoelectric conversion efficiently.27–29 Carbonaceous materials have many conjugated electrons, whose EDD would be remarkably changed once excited. Therefore, using a Cu-TCPP [TCPP = 5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin] framework (CuT for short) as the photosensitizer, we disperse CuT onto a microporous carbon (MC) and onto a hierarchical-pore carbon (HC) to fabricate two composite materials of CuT/MC and CuT/HC, respectively (Fig. 1). The pristine adsorption capacities of CuT/HC for C2H6 and C2H4 are 2.06 and 1.67 mmol g−1 (0 °C, 1 bar), respectively, and the calculated IAST selectivity is only 1.5 (C2H6/C2H4 = 10/90, v/v; 1 bar). Once exposed to visible radiation (Vis) at 420 nm, the adsorption capacity (0 °C, 1 bar) of C2H6 over CuT/HC can increase by 27%, and in contrast, that of C2H4 is increased only 1.8%. Therefore, a competitive IAST selectivity of 4.8 (C2H6/C2H4 = 10/90, v/v; 1 bar) can be obtained. Furthermore, we verify that for this strategy, the adsorption force of the C2H6 molecule can be asymmetrically enhanced by the excitation modulated EED over the adsorption sites formed via the close interaction between Cu-TCPP and the carbon layer, whereas that of the C2H4 molecule is symmetrically altered and the forces cancelled each other out. This is why the preferential binding sites for C2H6 are targetedly generated.
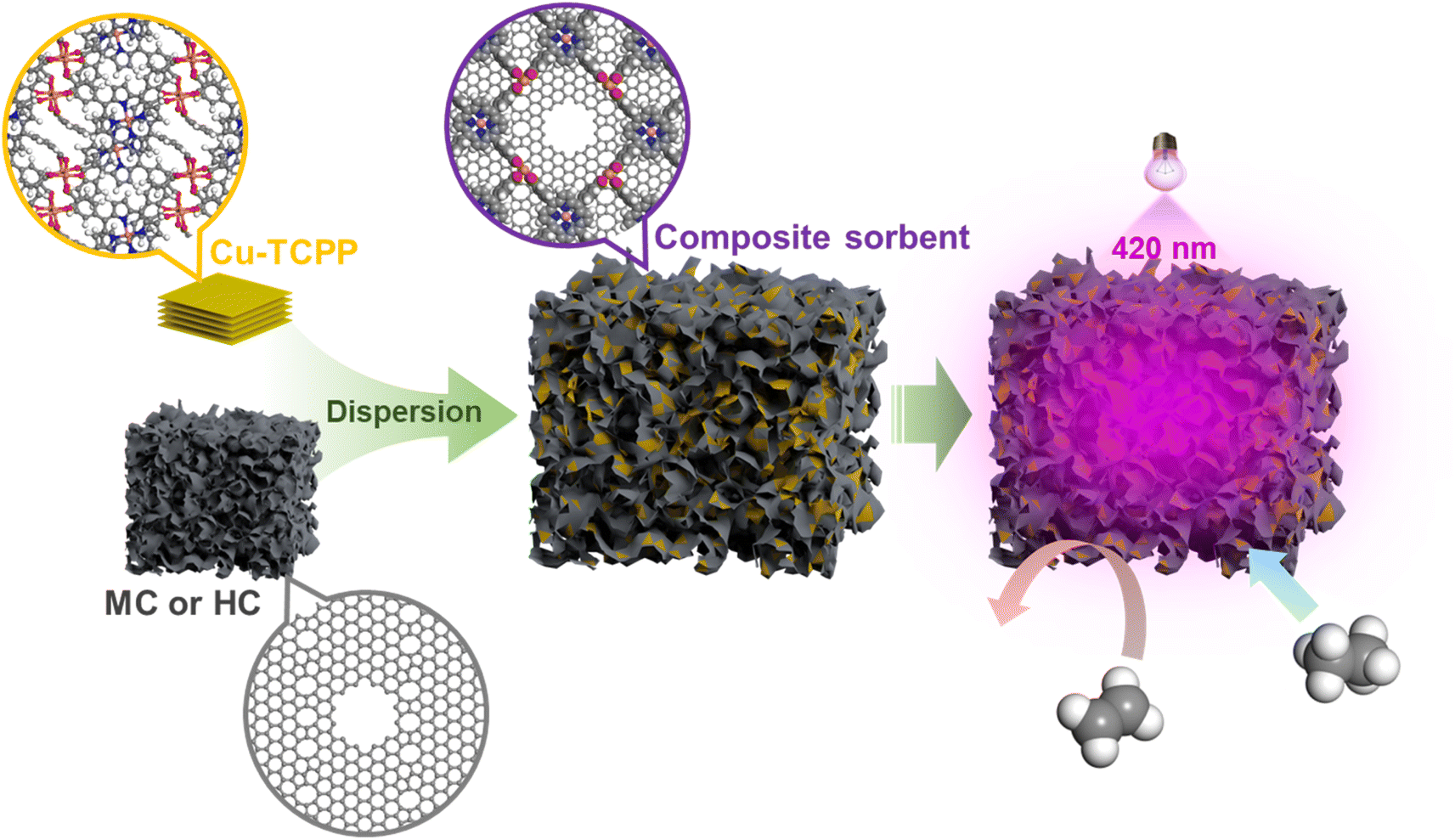 | ||
| Fig. 1 The scheme for composite sorbent construction and the adsorptive separation of C2H6/C2H4 enhanced by visible radiation. | ||
Results and discussion
Materials characterization
The morphology of pristine CuT can be seen in the scanning electron microscope (SEM) image (Fig. S1†), which is a mixture of CuT crystal particles with different grain sizes, but with the solvent treatment for the composite sorbent preparation, CuT can be reassembled and then well dispersed onto the carbonaceous materials to closely interact with the supports.25 According to the thermogravimetric (TG) profiles (Fig. S2†), the final mass losses for CuT/MC and CuT/HC are well consistent with the theoretical value of the CuT load (0.8/1.0, w/w) adopted in this study. This CuT load is verified to be optimal, which would result in more obvious light-responsiveness and better C2H6/C2H4 separation than other reference CuT loads as discussed in the following section. According to their SEM images, the morphologies of composite sorbents are similar to those of the MC and HC, owing to the well dispersed CuT (Fig. S3†). The presence of CuT on the host materials can be verified with energy dispersive spectrometry (EDS), and CuT is spread all over the host materials. Moreover, compared with the X-ray powder diffraction (XRPD) pattern of CuT, some peaks of high-order crystal planes for CuT/MC and CuT/HC weaken, e.g. [320], or even disappear, e.g. [002] and [200], indicating the increased dispersion of the supported CuT (Fig. S4†). In view of the Fourier transform infrared spectroscopy (FTIR) patterns (Fig. S5†), all the characteristic peaks of CuT can be observed in the patterns of CuT/MC and CuT/HC, especially those located at 1002 (N–Cu), 1407, and 1608 cm−1 (–COOCu), which indicate the presence of Cu-porphyrin and of secondary building units (SBUs) among the Cu-porphyrin rings.30,31 This means that the 2D-frameworks of the dispersed CuT can be preserved, or the original CuT chips disperse as much smaller chips on the MC and HC. In the high-resolution electron microscopy (HREM) images, the preserved CuT frameworks on the MC and HC can be distinguished from the wormhole-like structures of the amorphous carbonaceous supports, and the elemental mappings further demonstrate the widespread CuT on the supports (Fig. 2A and S6†). | ||
| Fig. 2 Textural and optoelectronic properties. (A) The HRTEM image of the representative CuT/HC sample. (B) The XPS deconvolution for the Cu 2p3/2 of CuT, CuT/MC, and CuT/HC. (C) The N2 adsorption–desorption isotherms of the pristine and composite materials. (D) The pore diameter distributions of the pristine and composite materials. | ||
The dispersed CuT 2D-framework interacts with the support closely, so the X-ray photoelectron spectroscopy (XPS) of Cu 2p3/2 is markedly influenced. As shown in Fig. 2B, the two main deconvoluted peaks of Cu 2p3/2 for CuT are located at 931.7 and 931.2 eV, respectively, indicating the presence of the SBUs and Cu-porphyrin in CuT.32,33 The porphyrin ring with large conjugate orbitals donate electrons so intensely that the coordinated Cu(II) exhibits a tendency to be reduced. Therefore, the porphyrin-coordinated Cu(II) gives rise to not only a peak at 931.2 eV, but also a small satellite peak at 929.4 eV, which should be attributed to further electron donation among different CuT framework layers.34 Loaded onto the MC or HC, the CuT chips exhibit wider Cu 2p3/2 peaks owing to the interaction with the host materials. The deconvoluted peak at 932.2 eV for CuT/MC can be attributed to the interaction between partial SBUs and the O-containing groups on the MC surface, while the redshifted peaks of CuT/MC located at 931.4, 930.8, and 929.3 eV compared with those of the CuT (931.7, 931.2, and 929.4 eV) demonstrate that the conjugate graphite surface of the MC likewise strongly donates electrons to the loaded CuT. Compared with CuT/MC, the peaks of CuT/HC further shift towards lower binding energy. A noticeable deconvoluted peak located at 929.3 eV, much stronger than that of CuT (929.4 eV) or CuT/MC (929.3 eV), and a small satellite peak at an even lower binding energy of 927.8 eV can be observed for CuT/HC, clearly indicating that CuT on the HC ought to interact with the host material better than that on the MC. As seen in Fig. 2C, the commercial MC and HC we used exhibit typical type-I and type-IV N2 adsorption–desorption isotherms, respectively. Both of them exhibit rapid N2 uptakes at the low P/P0 stage, indicating the presence of abundant micropores. A hysteresis loop of type-H4 can be observed in the HC isotherm, demonstrating the presence of mesopores and hierarchical structures. The Brunauer–Emmett–Teller specific surface area (SBET) values of the MC and HC are 1020 and 640 m2 g−1, respectively, but the SBET value of pristine CuT is only 30 m2 g−1. Therefore, the total N2 uptakes of CuT/MC and CuT/HC are lower than that of their host materials, and the calculated SBET values are reduced to 790 and 400 m2 g−1, respectively (Table S1†). The mesoporous structure of the HC can be clearly seen in the inset of Fig. 2D, which is the most obvious among all the materials. Anyway, the pore structure features of the host materials are preserved after CuT loading, and only the pore diameter distributions are somewhat altered because of the CuT chips covering up, especially for the mesopore structures of the host HC (Fig. 2D).
Adsorption experiments
The IAST selectivity of C2H6/C2H4 for the HC is already better than that for the MC (1.6 vs. 1.0; Table S1†), whereas the HC exhibits lower adsorption capacities for both C2H6 and C2H4 than the MC (Fig. S7 and Table S1†). This implies that both C2H6 and C2H4 tend to be indiscriminately captured by micropores, and the mesopore structures provide better C2H6/C2H4 selectivity. Xiang et al. theoretically proved that the pore structures with larger diameters facilitate the adsorption of larger molecules, which was dominated by Knudsen diffusion, such that the kinetic separation of C2H6/C2H4 can be realized over sorbents with hierarchical structures.35 The C2H6 and C2H4 adsorption capacities of CuT/MC and CuT/HC are lower than those of their host materials because of the reduced SBET values, and in fact, the C2H6 and C2H4 adsorption capacities of pristine CuT are lower than those of all the other sorbents (Fig. S8 and Table S1†), so without visible radiation, simply loading CuT onto the MC and HC does not essentially help the selective adsorption of C2H6. As for CuT/MC, there are some new hierarchical structures generated owing to CuT loading, as shown in the inset of Fig. 2D, which is beneficial to the adsorption selectivity of C2H6.Once exposed to the vis at 420 nm, the adsorption performances of Cu/MC and CuT/HC would be substantially changed. As metalloporphyrin derivatives, the ultraviolet-visible (UV-vis) absorption spectra of CuT present two characteristic bands, i.e. Soret and Q bands, which are also present in those of CuT/MC and CuT/HC (Fig. S9†). Therefore, the vis at 420 nm is chosen to excite the sorbents. Furthermore, it is demonstrated that once the metalloporphyrin derivatives and the conjugated carbon systems with metalloporphyrin are excited, the excited states would be stable enough to meet the molecular adsorption equilibrium, in view of the effective lifetimes of their excited states persisting for more than 35 μs according to their phosphor decay profiles (Fig. S10†). With visible radiation, the C2H6 adsorption capacities at 0 °C and 1 bar over CuT/MC and CuT/HC are increased to 2.21 and 2.62 mmol g−1, respectively, in contrast to that in the dark, i.e. 1.80 mmol g−1 for CuT/MC and 2.06 mmol g−1 for CuT/HC (Fig. 3A and Table S1†). The change rates reach 23% for CuT/MC and 27% for CuT/HC, respectively (Fig. 3B). Meanwhile, the photo-responsiveness of C2H4 adsorption over CuT/MC is much weaker, and its C2H4 capacity at 0 °C and 1 bar only increases from 1.75 to 1.91 mmol g−1, increased by 9%. Moreover, the C2H4 adsorption over CuT/HC is almost unresponsive to visible radiation, and its C2H4 capacity at 0 °C and 1 bar only increases from 1.67 to 1.70 mmol g−1, increased by only 1.8% (Fig. 3B).
 | ||
| Fig. 3 The static adsorption and dynamic breakthrough results of C2H6 and C2H4 tested with visible radiation and in the dark over the composite sorbents at 0 °C. (A) The static adsorption isotherms, in which the blue arrow indicates the variation trend of the C2H6 adsorption isotherm under visible radiation with respect to that in the dark, and the red arrow shows the variation trend of the C2H4 adsorption isotherm. (B) The rates of change for the light-responsive adsorption capacities of C2H6 and C2H4 at 1 bar. (C) The breakthrough profiles of C2H4 and C2H6 over CuT/MC and CuT/HC with visible radiation and in the dark, in which the arrows indicate the variation trends of the breakthrough profiles under visible radiation with respect to that in the dark. (D) The calculated IAST selectivity at 0 °C and 1 bar on the variable of the molar fraction of C2H6. | ||
Owing to the remarkably photo-gained C2H6 adsorption and the photo-irresponsiveness of C2H4 adsorption, the enhanced selectivity for C2H6/C2H4 can be achieved over the two composite sorbents with visible radiation, particularly over CuT/HC. As seen in Fig. 3D, the calculated IAST selectivity of C2H6/C2H4 at 0 °C and 1 bar is negatively associated with the molar fraction of C2H6. This means the sorbents are suitable for the actual separation of C2H6/C2H4 during which C2H6 is the impurity to be separated. An IAST selectivity of 4.8 over CuT/HC with visible radiation can be seen at a C2H6/C2H4 molar ratio of 10/90, and with this molar ratio fixed, the calculated IAST selectivity remains high in the entire pressure range from 0 to 1 bar (Fig. S11†). The visible radiation influence on the selectivity of C2H6/C2H4 over the two composite sorbents can be further confirmed by the dynamic breakthrough experiments with a gas mixture of C2H6/C2H4 (10/90, v/v), and competitive adsorption between C2H6 and C2H4 would occur. As Fig. 3C exhibits, neither CuT/MC nor CuT/HC presents an efficient separation of C2H6 over C2H4 in the dark. However, once there is visible radiation, the efficiency of C2H6/C2H4 separation for both CuT/MC and CuT/HC increases. As for CuT/HC with visible radiation, C2H4 was first eluted through a fixed bed at 8 min g−1, and the breakthrough of C2H6 occurs at 24 min g−1, so during this period the high-purity C2H4 product could be directly collected. Such impressive selectivity of C2H6/C2H4 is competitive with that of other representative benchmark C2H6-selective sorbents, especially when we consider that most of the current C2H6-selective sorbents are completely based on well-designed MOFs (Table S2†). In addition, both CuT/MC and CuT/HC exhibit ideal reversibility, whose C2H6 adsorption capacity both in the dark and with visible radiation can be well maintained even after five cycles, showing the stability of the composite sorbents so prepared (Fig. S12†).
Note that none of the pristine CuT, MC, and HC can separate C2H6 and C2H4 effectively, either in the dark or with visible radiation. As for pristine CuT, the adsorption capacities for C2H6 and C2H4 are both limited owing to its low SBET and poor pore structure (Fig. S8 and Table S1†). With visible radiation, the adsorption capabilities for both C2H6 and C2H4 can be increased slightly and indistinguishably. Therefore, both the adsorption capacity and the selectivity of C2H6/C2H4 over CuT are lackluster. As carbonaceous materials, the MC and HC only show a negative response to light, such that the adsorption capacities of both C2H6 and C2H4 over them drop with visible radiation, along with the further reduced C2H6/C2H4 selectivity (Fig. 3B, S7 and Table S1†). In addition, it is worth noting that both CuT/MC and CuT/HC are based on carbonaceous materials, with the same CuT load and the same preparation method, but CuT/HC performs much better than CuT/MC in terms of adsorption capacity and selectivity for C2H6, with visible radiation. In order to confirm the optimal CuT load of CuT/MC and CuT/HC (0.8/1.0, w/w) we focus on, reference composite sorbents with other mass ratios between the CuT and the support (0.1/1.0, 0.2/1.0, or 0.4/1.0; w/w) are prepared, which are then tested via the static adsorption of C2H6/C2H4. As seen in Fig. S13 and S14,† with different CuT loads, the reference sorbents exhibit light-responsiveness at different levels, as well as different adsorption capacities and selectivities for C2H6. With the optimal CuT load, CuT/MC and CuT/HC balance the light-responsiveness and the selective adsorption capability for C2H6, and then the highest C2H6/C2H4 selectivity of 4.8 can be obtained over CuT/HC with visible radiation. All the experimental facts demonstrate that it is required to fabricate the composite materials between CuT and the carbonaceous host material. The co-excitation of the CuT photosensitizer-carbon layer and the hierarchical pore structure of the host HC must jointly contribute to C2H6-selective adsorption over CuT/HC with visible radiation.
Mechanism investigation
To investigate the underlying mechanism of the C2H6-selective adsorption in this study, a series of simulation models are built for the calculation with density functional theory (DFT). As shown in Fig. S15,† we employ two graphitic carbon layers overlapped with a c-axial distance of 1 nm to simulate the carbonaceous micropore structure (tagged by C-M), and use a single graphitic carbon layer to simulate the carbonaceous mesopore larger pore structures (tagged by C-L), because the distances between the carbonaceous walls of the mesopores (2–50 nm) and larger pores (>50 nm) are too far to be considered in quantum computing. As Fig. 4 shows, based on the C-M model, we then place two CuT 2D-framework layers near the outside of the two carbon layers to mimic the micropore structure of CuT/MC and CuT/HC (tagged by CP-M), and locate a CuT 2D-framework layer near the C-L carbon layer to simulate the mesopore and large pore structure of CuT/MC and CuT/HC (tagged by CP-L). The mesoscopic mass transfer in the hierarchical structures is impossible to be simulated with quantum computing, but the static adsorption state of C2H6/C2H4 over the hierarchical structure can be simulated as mentioned above. | ||
| Fig. 4 The DCD images of the excited composite sites adsorbing C2H4 and C2H6 with respect to their ground states. DCD = CD (excited state) − CD (ground state); DCD: differential charge density; CD: charge density. (A) and (B) The DCD images of the excited CP-M site respectively adsorbing C2H4 and C2H6, in which the CP-M simulates the micropore structures in the composite sorbent, constructed via placing two CuT 2D-framework layers near the outside of the two carbon layers with a c-axial distance of 1 nm. (C) and (D) The DCD images of the excited CP-L site respectively adsorbing C2H4 and C2H6, in which the CP-L simulates the mesopore and large pore structures in the composite sorbent, constructed via locating a CuT 2D-framework layer near a carbon layer. | ||
The calculated adsorption binding energy (ABE) values of C2H6 and C2H4 over the C-M and C-L sites at ground states are the same as those at the low-order excited states (Table S3†). This means that the excited states of the carbonaceous pore structure, if any, cannot improve the affinity either to C2H6 or to C2H4. In fact, pristine carbonaceous materials are difficult to be excited with visible radiation, and even their low-order excited states are unstable. In contrast, the C2H6 ABE over CP-M is increased from −49 kJ mol−1 at the ground state to −63 kJ mol−1 at the excited state, whereas the C2H4 ABE over the site is somewhat weakened with excitation (from −47 to −46 kJ mol−1). As for the CP-L site, the C2H4 ABE is not changed by the excitation therein, while the C2H6 ABE is increased from −20 to −25 kJ mol−1 with excitation. The enhanced C2H6 ABE values and the weakened or unchanged C2H4 ABE values mean the formation of the sites for preferential binding of C2H6, and correspondingly, higher C2H6 adsorption capacity with invariant C2H4 capacity, and an improved C2H6/C2H4 selectivity can be obtained. In addition, we calculate the ABE values of C2H4 and C2H6 over monolayer or bilayer CuT frameworks, to explore the adsorption ability of CuT with different dispersions (Table S4†). It can be seen that the dispersion of CuT does not determine its adsorption ability. Compared with the bilayer CuT, the monolayer one provides somewhat increased adsorption ability for C2H4 (from −21 to −29 kJ mol−1), but the adsorption ability for C2H6 largely decreases owing to the increased dispersion of CuT (from −40 to −12 kJ mol−1). This implies that the increased dispersion of CuT would not necessarily result in better adsorption ability herein.
With the differential charge density (DCD) analysis, the different adsorption abilities of the excited model sites for C2H4 and C2H6 can be further explained. As Fig. 4A shows, the electron density modulated by excitation is enriched on both sides of the CP-M structure when CP-M adsorbs a C2H4 molecule, because the π-electrons of C2H4 link the conjugated electrons on both sides of the structure. The enriched electron density indeed enhances the van der Waals' force towards the C2H4 molecule, but the opposite enhanced forces cancel each other out.36–40 In contrast, the electron density altered by excitation is enriched only on one side of the CP-M structure when confining an C2H6 molecule, such that the enriched electron density would effectively enhance the van der Waals' force towards the C2H6 molecule (Fig. 4B). As for the excited CP-L site (Fig. 4C and D), the electron excitation mainly occurs on the SBUs of the CuT 2D-framework when adsorbing a C2H4 molecule, whereas the conjugated electrons of the carbon layer also participate in the excitation once a C2H6 molecule is adsorbed. The adsorption of C2H4 ought to have reduced the energy levels of the conjugated electrons of the carbon layer of CP-L, so the frontier electrons of the SBUs are excited instead. Therefore, the adsorption force of the C2H6 molecule is effectively enhanced by the excitation-enriched electron density of the CP-L site. In addition, such adsorption selectivity for C2H6 enhanced by the excited electrons would disappear if the CuT 2D-framework is not present near the carbon layers. Regardless of whether it is C2H4 or C2H6, the electron density altered by excitation would be enriched on both sides of the C-M structure and on the carbon layer of the C-L site (Fig. S15†).
CuT is chosen as a photosensitizer rather than as the adsorption site, and harvests light and make it easier to excite the composite sorbents and to stabilize the excited states. The carbonaceous MC and HC cannot generate stable excited states, so they only exhibit reduced adsorption capacities under visible radiation. Only after loading CuT and with visible radiation, the MC and HC are able to exhibit markedly enhanced adsorption capabilities for C2H6. In addition, it is worth noting that the SBET value and the resultant adsorption capacity of the HC are obviously lower than those of the MC, which are the opposite of their C2H6 adsorption selectivities (Table S1†). This means the mesopores or the hierarchical structures of the HC play an important role in the C2H6/C2H4 adsorptive separation.41–44 Owing to Knudsen diffusion,45–47 the micropore structures are inclined to adsorb the C2H4 molecules with a smaller molecular size, and the mesopore structures preferentially adsorb the C2H6 ones, when C2H6 and C2H4 coexist. Such kinetic separation of C2H6/C2H4 is still important for the adsorption of the composite sorbents with visible radiation. With an identical preparation method and the same CuT load, CuT/HC performs much better than CuT/MC for C2H6-selective adsorption. Although the SBET value of CuT/HC is obviously lower than that of CuT/MC, the calculated total pore volume of Cu/HC and its mesopore volume are larger than that of Cu/MC. More mesopore structures provide not only more locations for better dispersion of the CuT 2D-frameworks, but also more appropriate spaces for the Knudsen diffusion of C2H6 molecules. Based on the above-mentioned mechanism, excitation-modulated adsorption sites for preferential binding of C2H6 can be generated at both the micropores and the mesopores of the composite sorbents with visible radiation. The hierarchical structures of CuT/HC inherited from its host HC not only provide more preferential binding sites at the mesopore structures for C2H6, but also provide the new sites at the micropore structures to C2H6via facilitating the selective adsorption of C2H6.
Conclusions
In summary, instead of the current strategies to design C2H6-selective sorbents, we propose a new strategy that utilizes the excited states of the sorbents to generate new adsorption sites for preferential binding of C2H6, or in other words, employs visible radiation to facilitate C2H6/C2H4 adsorptive separation, and prove its feasibility. With CuT as the photosensitizer to be dispersed onto the carbonaceous materials of the MC and HC, CuT interacts with the host materials closely, which offers the possibility of a positive response to visible radiation for generating excited states, and that of the excitation models targeted for capturing the C2H6 molecule. In contrast, without the CuT dispersion, the host materials of the MC and HC only respond negatively to visible radiation, such that both the adsorption capacity and the selectivity for C2H6 are reduced over the pristine carbonaceous sorbents because of the visible radiation. Compared with CuT/MC, CuT/HC with visible radiation exhibits much better C2H6 adsorption capability, particularly C2H6/C2H4 selectivity. Along with the excitation which generated preferential binding sites for C2H6, CuT/HC inherits the hierarchical structure from its host HC, which facilitates the selective adsorption of C2H6. This strategy may open up a new route for energy-efficient adsorptive separation of C2H6/C2H4, for which sorbents are designed focusing on light-responsiveness instead of the current mechanistic theory.Data availability
All experimental and computational procedures and associated data are provided in the ESI.†Author contributions
Conceptualization, S. C. Q. and L. B. S.; methodology, S. C. Q., X. J. L., and L. B. S.; investigation, S. C. Q., Y. J. Z., X. J. L., Y. L. L., Z. S., X. Q. L., and L. B. S.; writing – original draft, S. C. Q.; writing – review & editing, S. C. Q., L. B. S., and X. Q. L.; funding acquisition, L. B. S., X. Q. L., and S. C. Q.; supervision, L. B. S., S. C. Q., and X. Q. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support of this work by the National Key Research and Development Program of China (2022YFB3806800), and the National Natural Science Foundation of China (22178163, 22125804, and 22078155). We are also grateful to the High-Performance Computing Center of Nanjing Tech University for supporting the computational resources.Notes and references
- K. J. Chen, D. G. Madden, S. Mukherjee, T. Pham, K. A. Forrest, A. Kumar, B. Space, J. Kong, Q. Y. Zhang and M. J. Zaworotko, Science, 2019, 366, 241–246 CrossRef CAS PubMed.
- V. F. D. Martins, A. M. Ribeiro, J. C. Santos, J. M. Loureiro, K. Gleichmann, A. Ferreira and A. E. Rodrigues, AIChE J., 2016, 62, 2490–2500 CrossRef CAS.
- D. E. Jaramillo, A. Jaffe, B. E. R. Snyder, A. Smith, E. Taw, R. C. Rohde, M. N. Dods, W. DeSnoo, K. R. Meihaus, T. D. Harris, J. B. Neaton and J. R. Long, Chem. Sci., 2022, 13, 10216–10237 RSC.
- J. Q. Liu, H. Wang and J. Li, Chem. Sci., 2023, 14, 5912–5917 RSC.
- X. J. Xie, Y. Wang, Q. Y. Cao, R. Krishna, H. Zeng, W. G. Lu and D. Li, Chem. Sci., 2023, 14, 11890–11895 RSC.
- F. Anwar, M. Khaleel, K. Wang and G. N. Karanikolos, Ind. Eng. Chem. Res., 2022, 61, 12269–12293 CrossRef CAS.
- D. F. Lv, P. J. Zhou, J. H. Xu, S. Tu, F. Xu, J. Yan, H. X. Xi, W. B. Yuan, Q. Fu, X. Chen and Q. B. Xia, Chem. Eng. J., 2022, 431, 133208 CrossRef CAS.
- J. X. Wang, C. C. Liang, X. W. Gu, H. M. Wen, C. H. Jiang, B. Li, G. D. Qian and B. L. Chen, J. Mater. Chem. A, 2022, 10, 17878–17916 RSC.
- N. A. Khan and S. H. Jhung, J. Hazard. Mater., 2017, 325, 198–213 CrossRef CAS PubMed.
- K. H. Cho, J. W. Yoon, J. H. Lee, J. C. Kim, K. Kim, U. H. Lee, S. K. Kwak and J. S. Chang, Microporous Mesoporous Mater., 2020, 307, 110473 CrossRef CAS.
- Q. Ding, Z. Zhang, C. Yu, P. Zhang, J. Wang, X. Cui, C. H. He, S. Deng and H. Xing, Sci. Adv., 2020, 6, 10596–10602 Search PubMed.
- C. Gu, N. Hosono, J. J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363, 387–391 CrossRef CAS PubMed.
- X. Lin, Y. S. Yang, X. Wang, S. Lin, Z. B. Bao, Z. J. Zhang and S. C. Xiang, Sep. Purif. Technol., 2014, 330, 125252 CrossRef.
- Y. Q. Wu and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2021, 60, 18930–18949 CrossRef CAS.
- L. Yang, W. Zhou, H. Li, A. Alsalme, L. T. Jia, J. F. Yang, J. P. Li, L. B. Li and B. L. Chen, Chin. J. Chem. Eng., 2020, 28, 593–597 CrossRef CAS PubMed.
- J. Y. Pei, J. X. Wang, K. Shao, Y. Yang, Y. J. Cui, H. Wu, W. Zhou, B. Li and G. D. Qian, J. Mater. Chem. A, 2020, 8, 3613–3620 RSC.
- S. T. Zheng, J. T. Bu, Y. F. Li, T. Wu, F. Zuo, P. Y. Feng and X. H. Bu, J. Am. Chem. Soc., 2010, 132, 17062–17064 CrossRef CAS PubMed.
- L. B. Li, R. B. Lin, R. Krishna, H. Li, S. C. Xiang, H. Wu, J. P. Li, W. Zhou and B. L. Chen, Science, 2018, 362, 443–446 CrossRef CAS.
- Q. L. Zhang, J. Chen, X. C. Zhu, J. Li and D. P. Wu, Inorg. Chem., 2020, 59, 16829–16832 CrossRef CAS.
- W. W. Liang, F. Xu, X. Zhou, J. Xiao, Q. B. Xia, Y. W. Li and Z. Li, Chem. Eng. Sci., 2016, 148, 275–281 CrossRef CAS.
- Y. X. Ye, Y. Xie, Y. S. Shi, L. S. Gong, J. Phipps, A. M. Al-Enizi, A. Nafady, B. L. Chen and S. Q. Ma, Angew. Chem., Int. Ed., 2023, 62, e202302564 CrossRef CAS PubMed.
- S. Q. Yang, F. Z. Sun, P. X. Liu, L. B. Li, R. Krishna, Y. H. Zhang, Q. W. Li, L. Zhou and T. L. Hu, ACS Appl. Mater. Interfaces, 2021, 13, 962–969 CrossRef CAS.
- H. Zeng, X. J. Xie, M. Xie, Y. L. Huang, D. Luo, T. Wang, Y. F. Zhao, W. G. Lu and D. Li, J. Am. Chem. Soc., 2019, 141, 20390–20396 CrossRef CAS.
- S. C. Qi, Y. F. Yu, Z. H. Yang, X. Y. Liu, X. J. Lu, X. Q. Liu and L. B. Sun, AIChE J., 2023, 69, e17994 CrossRef CAS.
- S. C. Qi, Y. L. Liu, X. J. Lu, Y. J. Zhao, J. X. Li, X. Q. Liu and L. B. Sun, Angew. Chem., Int. Ed., 2023, 62, e202304367 CrossRef CAS PubMed.
- S. C. Qi, Z. Sun, Z. H. Yang, Y. J. Zhao, J. X. Li, X. Q. Liu and L. B. Sun, Research, 2023, 6, 0261 CrossRef CAS PubMed.
- X. R. Yang, P. Zhang, H. J. Hou, J. P. Hu, L. Liu, L. S. Wu, S. J. Chen, K. L. Pan, S. Liang, S. S. Yuan and J. K. Yang, Chem. Eng. J., 2023, 451, 138688 CrossRef CAS.
- M. W. Liu, J. Wen, R. S. Xiao, R. Tan, Y. Qin, J. L. Li, Y. X. Bai, M. Z. Xi, W. H. Yang, Q. Fang, L. Y. Hu, W. L. Gu and C. Z. Zhu, Nano Lett., 2023, 23, 5358–5366 CrossRef CAS.
- D. J. Li, Y. B. Tian, Q. Lin, J. Zhang and Z. G. Gu, ACS Appl. Mater. Interfaces, 2022, 14, 33548–33554 CrossRef CAS.
- F. M. F. Ayman, M. Taha, A. A. Farghali and R. M. Abdelhameed, CrystEngComm, 2023, 25, 6697–6709 RSC.
- S. C. Qi, Z. H. Yang, R. R. Zhu, X. J. Lu, D. M. Xue, X. Q. Liu and L. B. Sun, J. Mater. Chem. A, 2021, 9, 24510–24516 RSC.
- P. Brand and H. Freiser, Anal. Chem., 1974, 46, 1147 CrossRef CAS.
- B. Folkesson, P. Sundberg, L. Johansson and R. Larsson, J. Electron Spectrosc. Relat. Phenom., 1983, 32, 245–256 CrossRef CAS.
- K. Zhu, M. Q. Zhang, X. Y. Feng, L. X. Qin, S. Z. Kang and X. Q. Li, Appl. Catal., B, 2020, 268, 118434 CrossRef CAS.
- H. Xiang, X. L. Fan and F. R. Siperstein, Sep. Purif. Technol., 2020, 241, 116635 CrossRef CAS.
- X. D. Zhu, J. B. He, R. J. Zhang, C. X. Cong, Y. X. Zheng, H. Zhang, S. Y. Wang, H. B. Zhao, M. P. Zhu, S. W. Zhang, S. J. Li and L. Y. Chen, Nano Res., 2022, 15, 2674–2681 CrossRef CAS.
- C. Y. Son and Z. G. Wang, Proc. Natl. Acad. Sci. U.S.A., 2021, 118, e2020615118 CrossRef CAS PubMed.
- X. W. Sheng, L. Mentel, O. V. Gritsenko and E. J. Baerends, J. Comput. Chem., 2011, 32, 2896–2901 CrossRef CAS PubMed.
- P. Tarakeshwar, K. S. Kim, E. Kraka and D. Cremer, J. Chem. Phys., 2001, 115, 6018–6029 CrossRef CAS.
- B. G. Rao and M. A. Murcko, J. Comput. Chem., 1994, 15, 1241–1253 CrossRef CAS.
- V. Daoo and J. K. Singh, ACS Appl. Mater. Interfaces, 2024, 16, 6971–6987 CrossRef CAS PubMed.
- J. Yan, Y. Tan, L. Wei, Z. Liu, Q. Wang, H. Sun, Z. Wang, D. Li, Y. Qian and S. Guo, Ind. Eng. Chem. Res., 2022, 61, 13453–13460 CrossRef CAS.
- X. Ma, M. Fang, B. Liu, R. Chen, R. Shi, Q. Wu, Z. Zeng and L. Li, Chem. Eng. J., 2022, 428, 130985 CrossRef CAS.
- S. Laha, N. Dwarkanath, A. Sharma, D. Rambabu, S. Balasubramanian and T. K. Maji, Chem. Sci., 2022, 13, 7172–7180 RSC.
- P. J. Donaubauer and O. Hinrichsen, Chem. Eng. Sci., 2018, 185, 282–291 CrossRef CAS.
- H. Nagasawa, T. Niimi, M. Kanezashi, T. Yoshioka and T. Tsuru, AIChE J., 2014, 60, 4199–4210 CrossRef CAS.
- R. Krishna and J. M. van Baten, J. Membr. Sci., 2011, 369, 545–549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00898g |
| This journal is © The Royal Society of Chemistry 2024 |