
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRestraining the shuttle effect of polyiodides and modulating the deposition of zinc ions to enhance the cycle lifespan of aqueous Zn–I2 batteries†
Qu
Yue
*a,
Yu
Wan
a,
Xiaoqin
Li
a,
Qian
Zhao
a,
Taotao
Gao
a,
Guowei
Deng
 b,
Bing
Li
c and
Dan
Xiao
*ad
b,
Bing
Li
c and
Dan
Xiao
*ad
aInstitute for Advanced Study, School of Mechanical Engineering, Chengdu University, Chengdu, 610106, P. R. China. E-mail: yuequ@cdu.edu.cn; xiaodan@scu.edu.cn
bSichuan Provincial Key Laboratory for Structural Optimization and Application of Functional Molecules, College of Chemistry and Life Science, Chengdu Normal University, Chengdu, 611130, P. R. China
cHubei Key Laboratory of Wudang Local Chinese Medicine Research, Hubei University of Medicine, Shiyan, 442000, P. R. China
dCollege of Chemical Engineering, Sichuan University, No. 24 South Section 1, Yihuan Road, Chengdu 610064, PR China
First published on 18th March 2024
Abstract
The boom of aqueous Zn-based energy storage devices, such as zinc–iodine (Zn–I2) batteries, is quite suitable for safe and sustainable energy storage technologies. However, in rechargeable aqueous Zn–I2 batteries, the shuttle phenomenon of polyiodide ions usually leads to irreversible capacity loss resulting from both the iodine cathode and the zinc anode, and thus impinges on the cycle lifespan of the battery. Herein, a nontoxic, biocompatible, and economical polymer of polyvinyl alcohol (PVA) is exploited as an electrolyte additive. Based on comprehensive analysis and computational results, it is evident that the PVA additive, owing to its specific interaction with polyiodide ions and lower binding energy, can effectively suppress the migration of polyiodide ions towards the zinc anode surface, thereby mitigating adverse reactions between polyiodide ions and zinc. Simultaneously, the hydrogen bond network of water molecules is disrupted due to the abundant hydroxyl groups within the PVA additive, leading to a decrease in water activity and mitigating zinc corrosion. Further, because of the preferential adsorption of PVA on the zinc anode surface, the deposition environment for zinc ions is adjusted and its nucleation overpotential increases, which is favorable for the dense and uniform deposition of zinc ions, thus ensuring the improvement of the performance of the Zn–I2 battery. This investigation has inspired the development of a user-friendly and high-performance Zn–I2 battery.
1. Introduction
The necessity to formulate rechargeable energy storage apparatus with elevated safety, improved efficiency, and diminished operating expenditure has elicited significant interest. This is due to their capability to not only merely store surplus electricity for staggered utilization, but also assimilate electricity generated from renewable energy sources (e.g., photovoltaic and wind energy) into the grid.1–5 Amongst secondary batteries, aqueous zinc–iodine (Zn–I2) batteries exhibit considerable appeal because of the advantages of elemental Zn and I, including ample reserves (50–60 mg iodine per Locean), reasonable price point, significant theoretical capacity (Zn: 820 mA h g−1 and I2: 211 mA h g−1), low redox potential of Zn (−0.76 V vs. SHE) and high redox potential of I2 (approximately 0.53 V vs. SHE).6 Regrettably, crucial challenges that necessitate resolution for the deployment of aqueous Zn–I2 batteries include both the cathode and the anode. The former pertains to the low electronic conductivity of iodine monomers (10−6–10−9 S m−1), their solubilization in iodide ion solutions, and sluggish redox kinetics,6–8 whereas the latter refers to problems at the Zn interface (e.g., side reactions, Zn dendrites, etc.).9–14Strategies have been designed to resolve these issues, involving cathode framework construction, electrolyte modulation, separator modification, and Zn interfacial protection. For I2 cathodes, host materials such as carbonaceous materials, metal–organic frameworks (MOF), polymer–organic materials, etc. are usually utilized to confine the iodine within the cathode electrode and boost their charge transfer kinetics.15–20 Among them, carbon materials, which have contributed to the development of clean energy technologies,21–23 play a crucial role in energy storage.15,16,24–27 For instance, Pan et al. restrained I2 within the nanoscale pores of an activated carbon fiber cloth as a cathode material for an aqueous Zn–I2 battery.24 Owing to the robust adsorption of I3−, the battery demonstrated a long cycle life, with a decreased self-discharge capacity loss. Zhi et al. selected 2D Nb-based MXene as the I2 matrix for the development of cathode materials employed in the Zn–I2 battery, which exhibited exceptional performance and ultralong cycle life, as a result of the robust confinement of I2 and immediate charge replenishment from the MXene.16 Significant progress has been made in utilizing host materials to encapsulate iodine; however challenges remain in resolving the shuttle phenomenon of multiple iodide ions, boosting the charge transfer kinetic, and augmenting the durability of Zn–I2 batteries, particularly long-lasting ones.
The approach of fabricating cation-exchange membranes or specialized protective coatings, such as multifunctional MOF membranes,28 PEDOT:PSS nanopapers,29 ZnHCF films,30 silicon-based anticorrosion films,31 zeolite-based cation-exchange protecting layers,32 and tannin acid based anticorrosive films,33 aims to impede the arrival of I3− to the surface of the Zn anode and to mitigate side-reactions on the Zn anode. Membranes must demonstrate exemplary electrical insulation properties alongside a superior ionic conductivity. At the same time, their electrochemical stability coupled with robust mechanical properties under operating conditions is an imperative consideration. Moreover, they should focus on resolving dendrite growth and parasitic reactions by regulating the interfacial electric field, the migratory routes for Zn2+, and the entrance of polyiodide ions.30 Based on the structure and properties of these films or coatings, the charge/discharge efficiency and cycle lifespan of the Zn–I2 battery have been improved, but further efforts are needed.
Electrolyte modulation is an effective strategy to solve cathode and anode problems. Up to now, the common method has been to develop electrolytes with decreased water content and activity, which can prohibit the formation, dissolution, and diffusion of polyiodide species and protect the Zn anode from side reactions. For example, Hong et al. suggested a water-in-salt electrolyte containing 5 M KI and 15 M ZnCl2, wherein iodide resides in the [ZnIx(OH2)4−x]2−x complex, eliminating free iodide anions and avoiding their interaction with I2 to generate polyiodides.34 Likewise, Wu et al. suppressed free iodide anions via a 10 M ZnI2 electrolyte.35 Li et al. demonstrated a Zn–I2 aqueous battery with superior energy efficiency and longevity, where a solid electrolyte interphase film (SEI) was created on the Zn anode via the utilization of the electrolyte of Zn(CF3SO3)2.15 Nevertheless, the elevated salt concentration increases the expense of the battery and brings about additional environmental considerations, which are not conducive to practical applications.36 With regard to cost considerations, starch has been added into Zn–I2 batteries.20,37,38 Zhao et al. developed a starch gel electrolyte for Zn–I2 batteries to inhibit the migration effect of I3− attributable to the starch binding with I5− and I3−.37 Apart from natural polymers such as starch and cyclodextrin,39 certain synthetic polymers, with electron-rich main or branch chains, are also inclined to adsorb iodine. For example, polyvinylpyrrolidone was utilized as a substrate to synthesize povidone iodine that can bind with I3−, thus averting its shuttle effect.38 Inspired by these studies, the addition of PVA into the electrolyte of Zn–I2 batteries is under consideration, as I3− can bind with PVA,40–42 potentially preventing the migration effect of I3− thereby enhancing the performance of Zn–I2 batteries. In addition, PVA has a large number of hydroxyl groups, which as an additive may have a positive effect on the electrolyte or zinc anode.
Here, we illustrate an advanced performance of Zn–I2 batteries by adding PVA into the electrolyte. An array of material analysis and electrochemical assessments have determined that PVA is capable of adsorbing I3− in solution, while Density Functional Theory (DFT) calculation results demonstrate that PVA–I3− has the lowest binding energy among the models developed. These findings imply that PVA additives in the electrolyte can efficiently bind with I3−, thus confining their shuttle effect in the Zn–I2 battery. Moreover, due to the PVA additives, the hydrogen bond network of water molecules is disrupted, resulting in a decrease in water activity. Further, because of the preferential adsorption of PVA on the zinc anode surface, the nucleation overpotential for the deposition of zinc ions is increased, resulting in a dense and uniform deposition of zinc ions which ensures an improved performance of the Zn–I2 battery. Extensive battery performance studies underscore that Zn–I2 batteries containing PVA exhibit higher specific capacity, long cycling stability, and superior coulombic efficiency compared to Zn–I2 batteries without PVA. This exploration opens an avenue for improving the performance of Zn–I2 battery electrolytes subjected to the shuttle effect and the zinc interfaces.
2. Experimental section
2.1 Chemical reagents
PVA 1799 (A.R.) was purchased from Chengdu Kelong Chemical Co., Ltd. Zn(Ac)2 (A.R.) and KI (A.R.) were purchased from Shanghai Titan Scientific Co., Ltd. I2 (>99.9%) was purchased from Chengdu Jinshan Chemical Test. Porous carbon was obtained from Jiangsu Xianfeng Nanomaterials Technology Co. The Zn foil (50 μm in thickness) was provided by Qinghe Safeway Metal Materials Co. Polyvinylidene difluoride (PVDF) and N-methylpyrrolidone were provided by Shanghai Macklin Biochemical Co., Ltd. Ketjen black (KB) was purchased from Shandong Xiya Chemical Co., Ltd. Glass fibers were provided by GE Whatman.2.2 Preparation of the carbon/iodine material
Porous carbon was mixed with iodine monomers at a mass ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Then, the mixtures were heated at 90 °C for 4 h and the carbon/iodine material was obtained.
:2. Then, the mixtures were heated at 90 °C for 4 h and the carbon/iodine material was obtained.
2.3 Preparation of the Zn–I2 battery
The slurry of the cathode was prepared with a weight ratio of 8:1:1 (carbon/iodine material:acetylene black:PVDF). Then the mixture was coated onto carbon papers and dried at 90 °C in a vacuum for 4 h. The battery performance was investigated using CR2032 coin batteries. Porous carbon immobilized I2 was used as the cathodic electrode, while Zn foil served as the anode electrode. The mass loading of I2 was controlled to be about 0.6 mg cm−2. The electrolyte was composed of 1 M Zn(Ac)2 and PVA (2.5 wt%). Zn foil was used directly without polishing. Glass fiber was employed as the separator. For symmetric batteries and asymmetric batteries, the anode, cathode, and separator were cut into disks with a diameter of 14 mm, 14 mm, and 19 mm, respectively. The specific capacity was calculated from the mass of iodine.
2.4 Material characterization
Scanning electron microscopy (SEM) images were recorded on a Hitachi SU8100 instrument operating at 5 kV. X-ray diffraction (XRD) was performed on a Bruker D8 Advance (Bruker AXS GmbH, German) diffractometer with Cu Kα radiation (λ = 1.5406 Å) at a scan rate of 8° min−1. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha instrument and Al Kα monochromatized radiation was employed as an X-ray source. UV-vis spectra were collected on a UV-2700I instrument (Shimadzu, Japan). Fourier transform infrared (FTIR) mapping was performed on a Niolet iN10 spectroscopy instrument. Raman spectroscopy was carried out on a WiTech alpha300R with a laser wavelength of 532 nm.2.5 Electrochemistry
An electrochemical workstation (Autolab M204) was used to record cyclic voltammograms and electrochemical impedance spectra. The cyclic voltammetry (CV) curves of the Zn–I2 battery with and without PVA were recorded at 0.1, 0.2, 0.5, 1.0, and 2.0 mV s−1 in the potential range of 0.5–1.6 V. The CV curves of the Zn||Cu coin battery with and without PVA were recorded at 0.5 mV s−1. Electrochemical impedance spectroscopy was performed in the frequency range of 100 kHz to 0.1 Hz with an alternating current (AC) voltage amplitude of 5 mV at the open-circuit voltage. The deposition/stripping process of Zn was also conducted in the coin battery. Galvanostatic charge–discharge profiles were obtained on a Neware BTS-51 battery testing system. All batteries were charged to a cutoff potential of 1.6 V and then discharged to 0.5 V. The specific capacity values were referenced to the mass of iodine in the cathode electrode.2.6 DFT calculations
All calculations were performed using the DMol3 program package in Materials Studio 2018. The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional method was used to describe the exchange–correlation interaction. The global orbital cutoff was set as 5.2 Å and 0.005 Ha smearing was used for the orbital occupation. The thresholds of energy, force, and displacement are 10−5 hartree, 4 × 10−3 hartree per atom for the maximum force, and 5 × 10−3 Å for displacement. Self-consistent field (SCF) procedures were performed with a convergence criterion of 2 × 10−5 Ha on the total energy to achieve accurate electronic convergence. The binding energy (Ebind) between vinyl alcohol, PVA5, PVA10, H2O, and I3− was calculated using the formula:| Ebind = Etotal − (Ea + Eb) | (1) |
For the adsorption optimization, the Zn (002) slab was constructed with lattice constants of a = b = 15.9894 Å, c = 19.9468 Å, α = β = 90° and γ = 120°. In the Z direction, there is about a 20 Å vacuum layer to eliminate the interaction between the upper periodic Zn atoms. The adsorption energy (Eads) of S on the Zn (002) slab was calculated using the formula:
| Eads = EM+S − (EM + ES) | (2) |
3. Results and discussion
3.1 The interaction between PVA and iodine
In common Zn–I2 batteries, the interaction between I− and I2 at the cathode typically results in the formation of I3−,7 which is inevitable during the electrochemical redox procedure in Zn–I2 batteries. Such intermediate species exhibit high solubility and usually migrate to the surface of the metallic Zn anode. This phenomenon is commonly acknowledged as the “shuttle effect”. As a consequence of the shuttle effect, I3− reacts with Zn to generate I− (Zn + I3− ↔ Zn2+ + 3I−), which subsequently returns to the cathode for re-oxidation.6 This occurrence leads to a self-discharge and depletion of Zn activity, and it is one of the primary reasons for the precipitous decline in the cycle life of zinc iodine batteries, as illustrated in Fig. 1. Therefore, the development of electrolytes with the function of trapping polyiodide ions is necessary.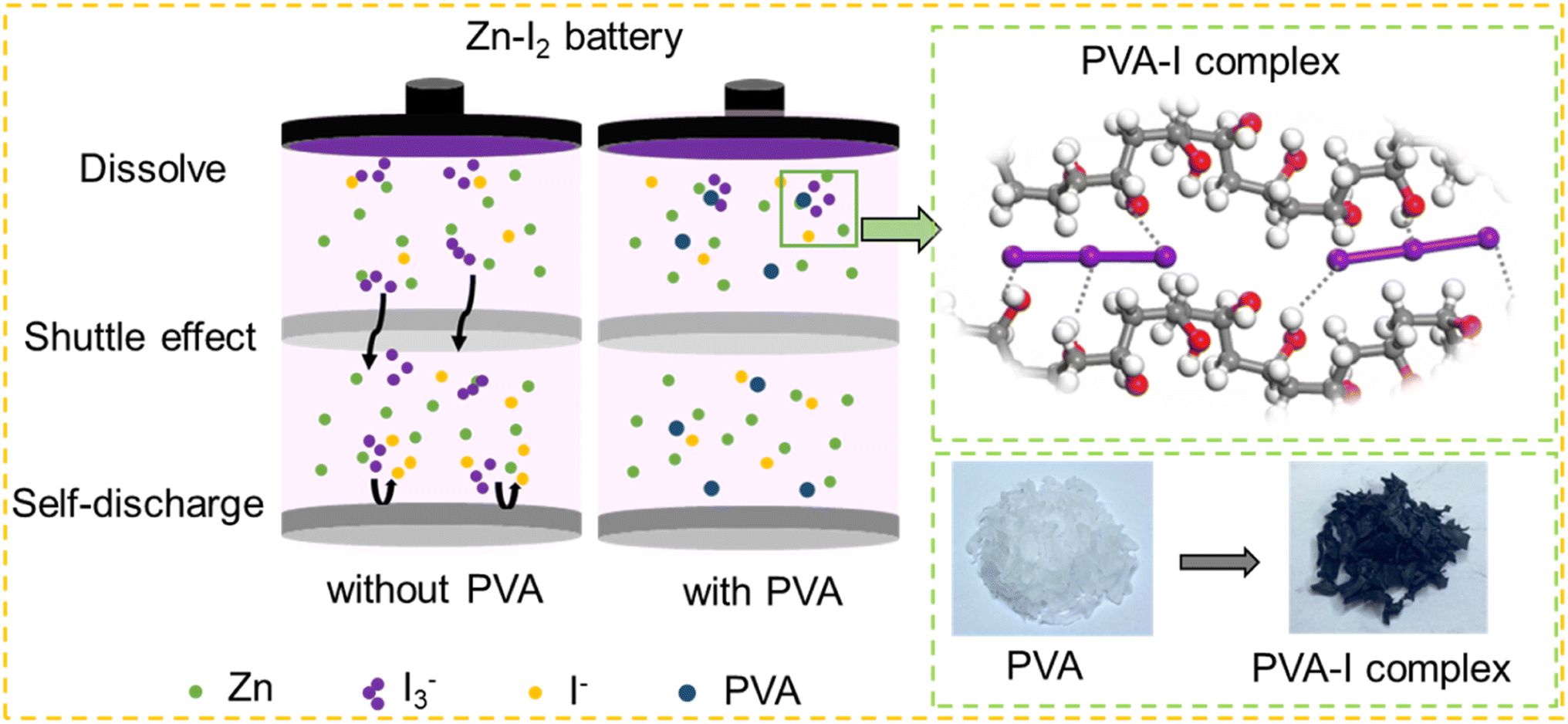 | ||
| Fig. 1 Illustration of the mechanisms for Zn–I2 batteries with and without PVA, the schematic diagram of iodine adsorption by PVA, and actual samples of PVA and PVA adsorbed iodine. | ||
Lewis base iodine has been reported to form complexes with the electron-rich binding of hydroxyl groups situated on the side chains of PVA, and the iodine sorbed PVA exhibits a distinctive coloration of deep blue.42–44 The adsorption process of I3− by PVA is schematically presented in Fig. S1.† Upon introduction of a polyiodide solution comprising 1 M KI and 0.01 M I2 into a bottle containing PVA, a deep blue mixture forms, while a colorless supernatant results post-resting and filtration. The complete sorption of polyiodides by PVA means that it may have the potential of alleviating the shuttle of polyiodides in Zn–I2 batteries.
To intuitively observe the shuttling polyiodides in Zn–I2 batteries, a visualization experiment in H-type cells is firstly applied to demonstrate the blocking capability. In the H-type Zn–I2 cells with or without PVA, the carbon/iodine material acts as the cathode, Zn as the anode, and a glass fiber as the separator (Fig. S2†). The electrolyte exhibited a light yellow color due to the mild precipitation of iodine upon exposure of its initial solution to atmospheric conditions. After 60 minutes of charging and discharging at a current density of 2 A g−1, the color of the PVA-free electrolyte changed to a dark yellow owing to the formation of brown polyiodides. With the addition of PVA, the colorlessness electrolyte after 60 minutes suggests an interaction between I3− and PVA, which can inhibit the diffusion of polyiodides. Ultraviolet-visible (UV) measurement was performed to investigate their interaction, as shown in Fig. 2a and b. When the electrolyte with and without PVA experienced several charge/discharge cycles, a distinctive peak attributed to I− emerges at 210 nm,20 meaning the dissolution of I2 from the cathode. For the PVA-free electrolyte, the peaks at 290 nm and 350 nm can be identified as the I3− characteristic peak,20 implying the presence of I3− species in the electrolyte. However, for the electrolyte with PVA, a broad peak from 270–400 nm was detected as a result of PVA–I3− complex formation.42,44 Raman spectroscopy was further applied to investigate the interaction between I3− and PVA, as displayed in Fig. 2c. With PVA-free electrolytes, the peak at 110 cm−1 in Raman spectra was associated with the I3− characteristic peak.45 Conversely, with PVA-containing electrolytes, the peak experienced a significant blue-shift from 110 cm−1 to 107 cm−1, implying an interaction between I3− and PVA. XRD measurements were employed on pure PVA and PVAI composites to substantiate their interaction, as shown in Fig. 2d. The PVA exhibits certain diffraction peaks at 19°, 29°, and 30°, while these peaks disappears in the PVAI sample, verifying the interaction between I3− and PVA.46 To examine the impact of the PVA additive on the I2 cathode, a H-type electrolytic cell was assembled, in which a carbon paper coated with the carbon/iodine material (I2: 20 wt%) was the cathode, Zn foil was the anode, Zn(Ac)2 or Zn(Ac)2 + PVA was the electrolyte. After several charge/discharge cycles, XPS spectroscopy of the cathode was performed. The full profile of the XPS spectrum confirms the existence of I, C and O (Fig. S3†). As demonstrated in high-resolution I 3d spectra (Fig. 2e), two characteristic peaks located at 630.6 and 619.2 eV can be assigned to the typical I 3d3/2 and I 3d5/2, respectively, indicating the presence of I2.16,37,47,48 A stronger I 3d peak at the cathode cycled in PVA electrolyte suggests a less dissolution of active I2, which can be verified by the iodine content in the cathode. Fig. S4 and S5† show that the iodine content (18.2 wt%) on the cathode after cycling in the PVA electrolyte exceeds the cathodic iodine content (17.4 wt%) in the PVA-free electrolyte. The elemental distribution mappings display that iodine element is uniformly distributed throughout the cathode. These findings suggest that adding PVA to the electrolyte of the Zn–I2 battery, due to the interaction between I3− and PVA, can relieve the dissolution of I2 on the cathode and restrain the shuttle effect of polyiodides in the electrolyte, which may probably prolong the cycling performance of the Zn–I2 battery.
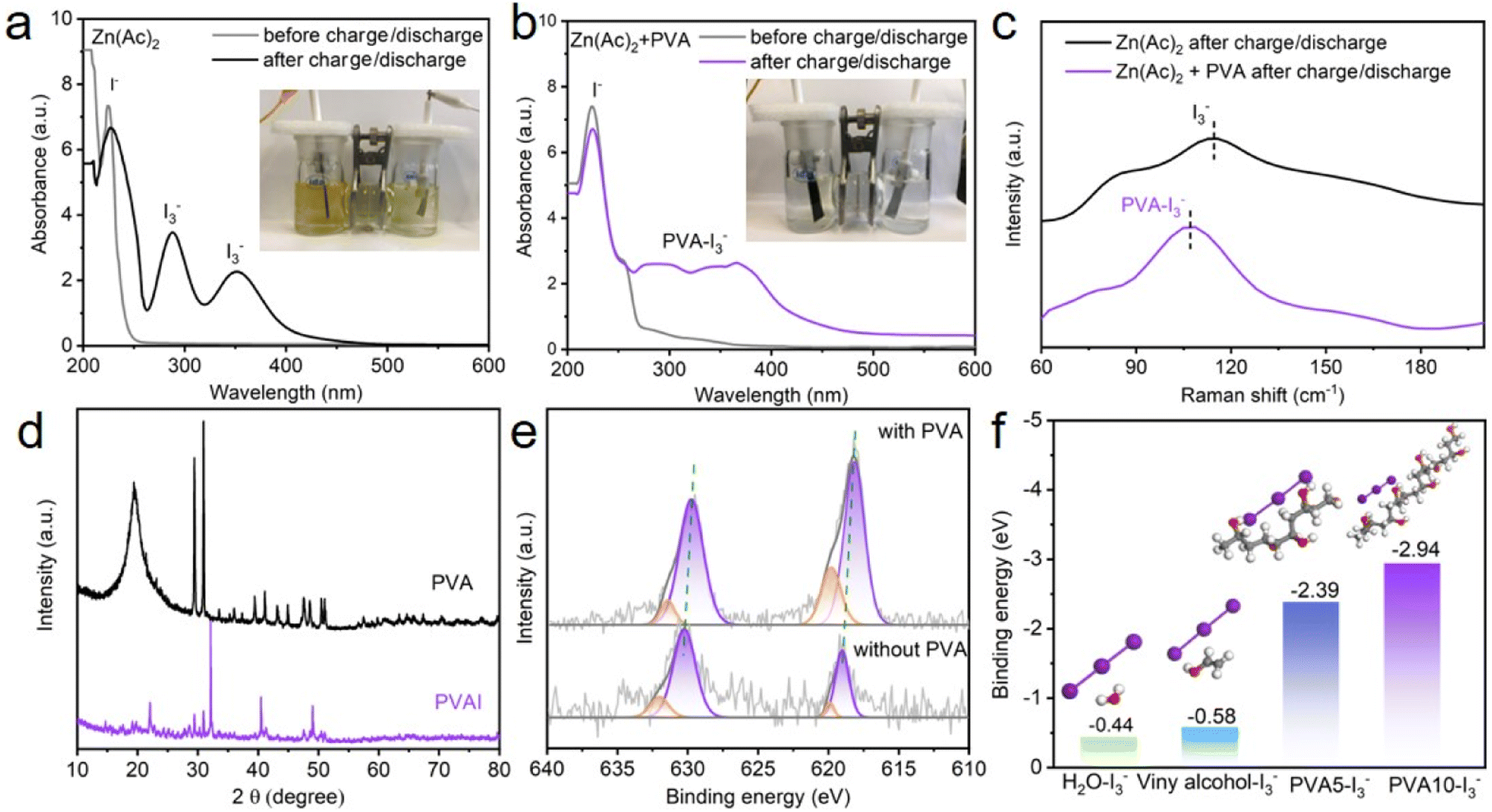 | ||
| Fig. 2 (a) and (b) The UV spectra and (c) the Raman spectra of the electrolyte without and with PVA before and after charge/discharge, respectively, (d) XRD patterns of PVA and PVAI compounds, (e) I 3d XPS spectra of the cathode after cycling in PVA and PVA-free electrolytes, and (f) DFT calculations of binding energy for H2O, vinyl alcohol–I3−, PVA5–I3−, and PVA10–I3− models. | ||
DFT calculations were applied to investigate the binding energy values between PVA and I3− (Fig. 2f). To simplify the calculation model, a single vinyl alcohol molecule, PVA with a polymerization degree of 5 (denoted as PVA5), and PVA with a polymerization degree of 10 (denoted as PVA10) were constructed to represent –OH groups in compounds, because of their key role in confining the shuttling and diffusion of iodine.37 Calculation results show that the binding energy of PVA10–I3−, PVA5–I3−, vinyl alcohol–I3−, and H2O–I3− is −2.94 eV, −2.39 eV, −0.58 eV, and −0.44 eV, respectively. The negative value of Ebind indicates that the process is an exothermic reaction and lower binding energy signifies easier interaction in thermodynamics. The binding energy of PVA–I3− decreases as the polymerization degree increases, implying that PVA as an additive can efficiently adsorb I3− in the electrolyte to form a PVA–I complex. It has been known that I3− orbitals have lone pair electrons and the OH bonds of PVA have anti-bonding character.41 Therefore, this complexation may probably relate to the hydrogen bond between iodine and the OH group of PVA. Once I3− ions are generated in the electrolyte, the intermolecular OH⋯O hydrogen bonds between the neighboring PVA chains are broken, releasing OH groups, which combine with iodide ions instantly through OH⋯I hydrogen bonds (Fig. 1). Accordingly, due to the interaction between PVA and I3−, the introduction of PVA into the electrolyte can effectively eliminate free I3−, existing as a PVA–I3− complex. It exhibits potential to mitigate the shuttle effect caused by I3− in Zn–I2 batteries.
3.2 The impact of PVA on Zn anodes
To check the impact of PVA on Zn anodes, the interaction between PVA and zinc metal was studied. DFT simulations were initially employed to reveal the adsorption energy of different molecules onto the Zn (002) surface. As presented in Fig. 3a, the adsorption energy values for H2O, Zn, viny alcohol, and PVA5 are −0.51 eV, −0.60 eV, −0.62 eV, and −2.70 eV, respectively. Although the adsorption energies of vinyl alcohol and Zn atom do not differ significantly, the adsorption energy decreases substantially with the increase in the degree of polymerization of vinyl alcohol molecules. Evidently, the adsorption energy of PVA5 on the Zn (002) surface is the least, signifying that the PVA additive in the electrolyte tends to be preferentially adsorbed on the surface of the Zn anode. To examine whether PVA additives can reduce the side reaction of the Zn anode, commercial Zn foil was soaked in the electrolyte of Zn(Ac)2 with and without PVA for 13 days. As displayed in Fig. S6,† the surface of zinc foil immersed in the PVA-free solution shows uneven and differently sized protrusions, whereas that in the PVA containing solution exhibits uniform and very tiny particles. This suggests that Zn foil corrosion can be alleviated by PVA additives. Through the XRD patterns, these products can be identified and estimated (Fig. 3b). After 13 days of immersion in PVA-free Zn(Ac)2 solution, characteristic peaks of hydrated zinc acetate (PDF#48-1917) at 13.1° and 19.9° (denoted by ) and alkaline zinc acetate (PDF# 19-1472) at 33.3° and 33.8° (denoted by
) and alkaline zinc acetate (PDF# 19-1472) at 33.3° and 33.8° (denoted by  ) appear on the surface of Zn foil. In contrast, after 13 days of immersion in Zn(Ac)2 solution containing PVA, no characteristic peaks of alkaline zinc acetate are detected on the surface of the zinc foil, while the intensity of the characteristic peaks of hydrated zinc acetate is relatively weak, signifying that the incorporation of PVA additives can weaken the side reactions of zinc. SEM results further verified this suggestion, as shown in Fig. 3c, d and S6.†
) appear on the surface of Zn foil. In contrast, after 13 days of immersion in Zn(Ac)2 solution containing PVA, no characteristic peaks of alkaline zinc acetate are detected on the surface of the zinc foil, while the intensity of the characteristic peaks of hydrated zinc acetate is relatively weak, signifying that the incorporation of PVA additives can weaken the side reactions of zinc. SEM results further verified this suggestion, as shown in Fig. 3c, d and S6.†
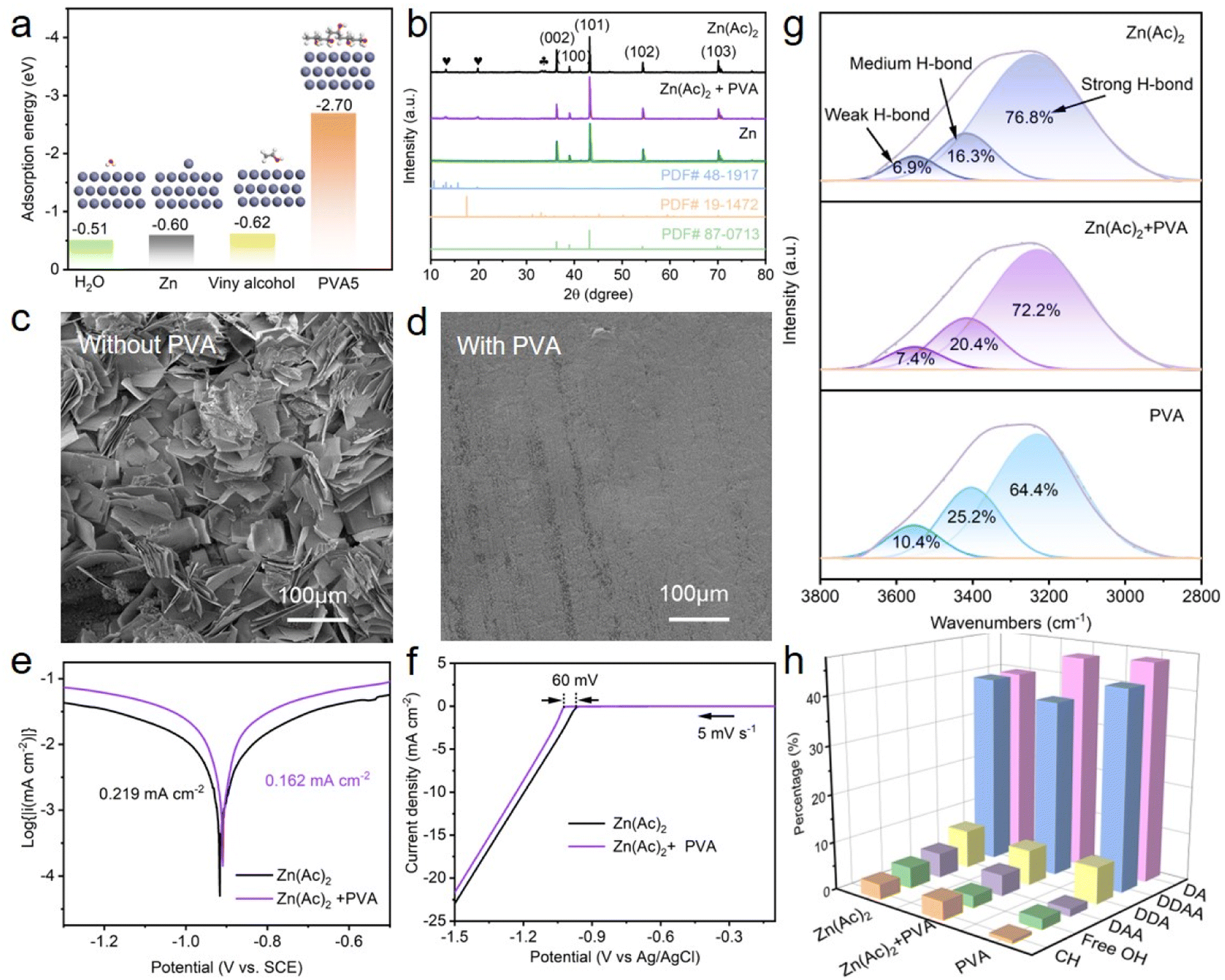 | ||
| Fig. 3 (a) Adsorption energy values of H2O, the Zn atom, viny alcohol, and the PVA5 molecule onto the Zn (002) surface, (b) XRD patterns and (c and d) SEM of Zn foil after immersing in the electrolyte with and without PVA for 13 days, (e) corrosion Tafel plots of the Zn electrode tested in the electrolyte with and without PVA, (f) LSV curves of the electrolyte with and without PVA at a scan rate of 5 mV s−1, (g) FTIR spectra of Zn(Ac)2, Zn(Ac)2 + PVA, and PVA electrolytes, respectively, and (h) the percentage of C–H, DAA, DDAA, DA, DDA, and free OH. | ||
The endurance of Zn to corrosion within the electrolyte with and without PVA was concurrently examined. As shown in Fig. 3e, the corrosion current of Zn in PVA-containing electrolyte (0.162 mA cm−2) is lower than that in PVA-free electrolyte (0.219 mA cm−2). Meanwhile, an enhanced corrosion voltage in the PVA-containing electrolyte is realized when compared to the PVA-free electrolyte, indicating a diminished tendency of Zn electrodes towards corrosion in both thermodynamic and kinetic terms.49 Linear scan voltammetry (LSV) analysis confirms that the overpotential of the hydrogen evolution reaction (HER) increases by 60 mV when the PVA additive is employed, signifying the suppression of the HER (Fig. 3f). FTIR and Raman characterization were performed to investigate the hydrogen bindings between H2O molecules in the electrolyte with and without PVA. As demonstrated in Fig. 3g, compared to pure Zn(Ac)2 electrolyte, the areas for strong hydrogen bindings (3229 cm−1) decrease with the adding of PVA, while the areas of medium (3418 cm−1) and weak hydrogen bindings (3553 cm−1) increase, suggesting that the hydrogen bond network between H2O molecules can be weakened by PVA additives.50 Raman results and detailed explanation further prove the above suggestion, as can be seen in Fig. S7.† With the addition of PVA, the proportion of DAA and free OH is decreased (Fig. 3h), implying that the activity of water molecules is weakened.51 These findings confirm that the hydrogen bond network and the activity of H2O molecule can be decreased in the electrolyte with PVA, alleviating side reactions caused by the H2O molecule.
The performance of the zinc anode is fundamentally related to the plating and stripping behavior of Zn2+ ions, which are predominantly influenced by the properties of the solution and interface. Therefore, a comprehensive series of tests and characterization are conducted on both the electrolyte and interface. Firstly, the influence of PVA on the deposition behavior of zinc was investigated in detail. As shown in Fig. 4a, a crossover characteristic of nucleation processes is observed while sweeping the potential from −0.2 V to 0.4 V versus Zn/Zn2+ in the positive direction. The potential located at the crossover point A is known as the crossover potential. Zn ions start to be reduced on substrates at the points B and C. The potential disparity between A and B/C is called the nucleation overpotential (NOP), which is regarded as an index to evaluate the extent of polarization.52,53 The larger the NOP, the smaller the radius of the nucleated particles.54 The correlation between the critical Zn nucleus radius (r) and NOP can be described as follows:55,56
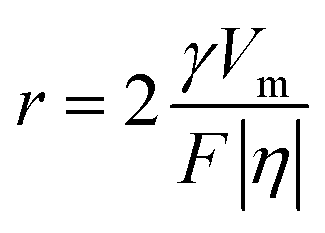 | (3) |
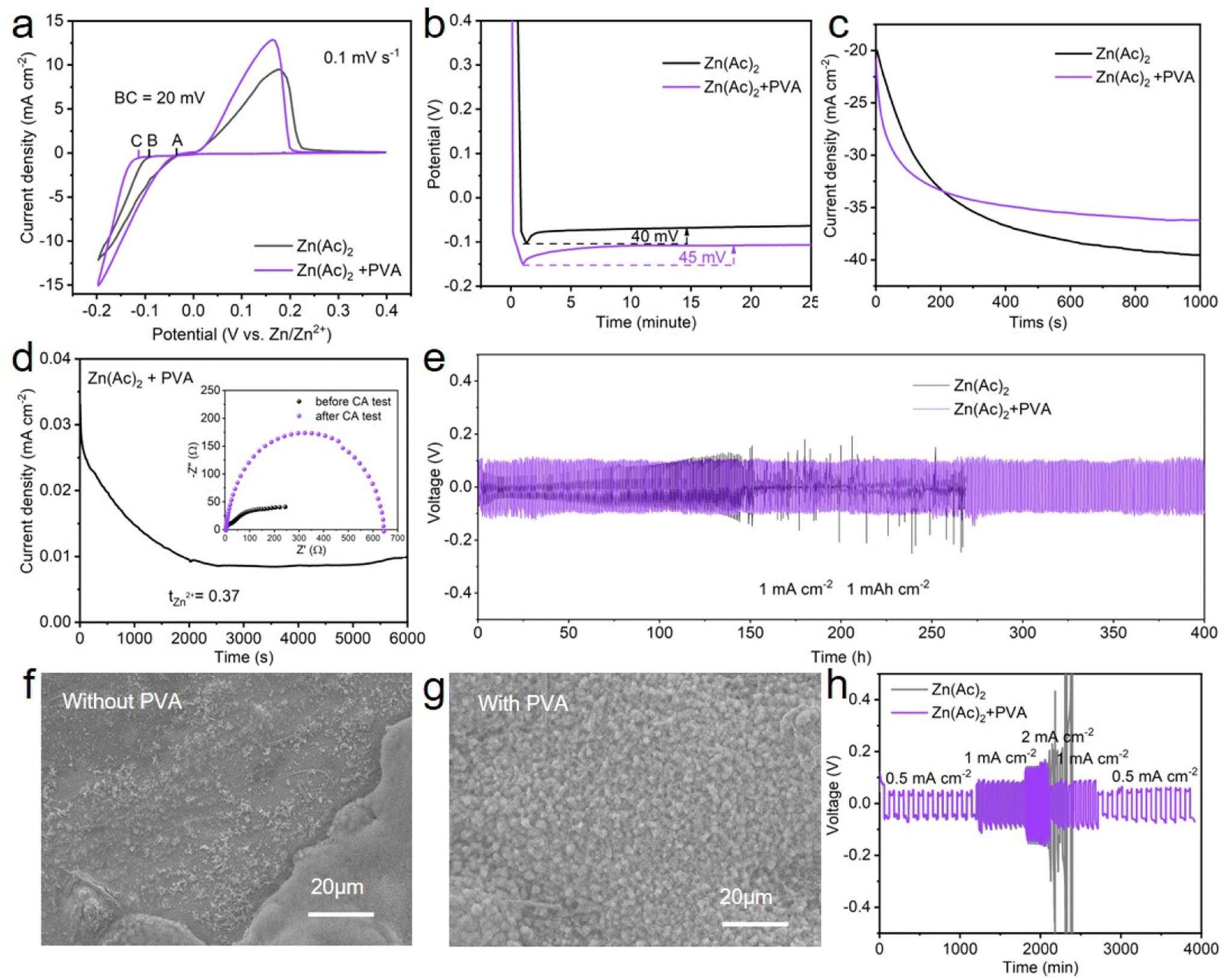 | ||
| Fig. 4 (a) Cyclic voltammograms for Zn nucleation on Zn||Cu batteries at a scan rate of 0.5 mV s−1, (b) voltage–time profiles of nucleation overpotential on Zn||Cu batteries at 1 mA cm−2, (c) current–time profiles of the Zn electrode and (d) the CA curve of the Zn||Zn battery with PVA (the inset show the corresponding Nyquist plots at the initial and steady states), (e) cycling performances of Zn||Zn batteries with and without PVA under 1 mA cm−2 and 1 mA h cm−2, (f) and (g) SEM of the Zn anode surface using Zn||Zn batteries without and with PVA after Zn plating and stripping for 20 cycles at a current density of 2.0 mA cm−2, and (h) the rate performance of Zn||Zn batteries with and without PVA at different current densities. | ||
Secondly, to characterize the Zn nucleation and growth, chronoamperometry (CA) measurements were performed at −150 mV, by observing the Zn2+ ion diffusion behavior on the surface of the Zn anode in the Zn||Zn batteries with and without PVA.52 From the current–time profile depicted in Fig. 4c, the current density of the Zn||Zn battery with PVA stabilizes at ∼6.1 mA cm−2 after a short two-dimensional (2D) diffusion period of approximately 150 s, whereas the current density of the Zn||Zn battery without PVA persistently increases for over 500 s indicating an extended and rampant 2D diffusion process of Zn2+ ions, which correlates with the uneven nucleation and dendritic growth.52,61 Contact angle tests were applied to assess the alteration of wettability of the electrolyte with PVA. As shown in Fig. S10,† the contact angle of Zn metal decreases from 98° to 86° in PVA-free and PVA-containing electrolytes, attributed to the unique characteristics of PVA, which has both a hydrophobic (CH2–CH2) backbone and hydrophilic (–OH) side-groups.62 Endowed with these –OH groups, PVA exhibits high affinity with the H2O molecule and Zn anode, potentially influencing the environment of the Zn electrode.61 This may facilitate the formation of molecular channels for 3D diffusion of Zn ions during the process of Zn deposition, while concurrently establishing a novel layer of water scarcity that impedes H2O corrosion.11,61,63 It is known that a lower value of tZn2+ in electrolyte can aggravate the concentration polarization of Zn2+ at the Zn anode/electrolyte interface, leading to harmful Zn dendrites and parasitic side reactions. The Zn2+ transference number (tZn2+) of the electrolyte with and without PVA was calculated, by performing electrochemical impedance spectroscopy (EIS) at the corresponding initial and steady states and CA of the Zn symmetric battery at a constant polarization potential of 10 mV. According to the curves (Fig. 4d and S11†) and eqn (4):
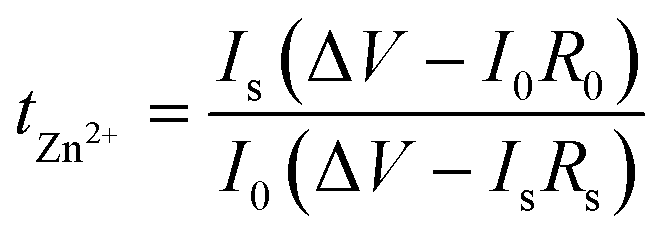 | (4) |
Subsequently, Zn||Zn batteries with and without PVA were constructed to evaluate the reversibility of Zn plating/stripping at a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2. Fig. S12† displays the results of the optimization of electrolyte composition. The Zn(Ac)2 + PVA (2.5 wt%) electrolyte has longer battery cycling stability, probably because this concentration of electrolyte not only regulates the deposition environment of zinc ions, but also does not impede the migration of zinc ions and introduce high voltage hysteresis during Zn plating/stripping processes. Under these conditions, the battery shows a prolonged lifespan exceeding 400 h, while the short circuit occurs rapidly for the battery without PVA after cycling about 140 h (Fig. 4e). These results suggest that homogeneous Zn deposition is achievable in the electrolyte with PVA. With such a speculation, the morphology of Zn anodes cycled in electrolyte with and without PVA was examined via SEM, as shown in Fig. 4f, g and S13. For PVA-free electrolyte, irregular protrusions and varying degrees of corrosion appear on the surface of Zn foil. In contrast, even nanosheets are observable on the surface of Zn foil in electrolyte with PVA, indicating a uniform Zn deposition performance brought about by the addition of PVA. Additionally, the Zn||Zn batteries with PVA show a good rate performance at the current densities of 0.5 mA cm−2, 1.0 mA cm−2, 2.0 mA cm−2 (Fig. 4h). To reveal the efficiency of Zn deposition, the CE of Zn||Cu batteries with and without PVA was studied (Fig. S14†). The results demonstrate that the Zn||Cu batteries with PVA can attain an average CE of 99.5% at a current density of 1 mA cm−2 with a capacity of 1 mA h cm−2 and keep stable for 250 cycles. Although the CE of the Zn||Cu batteries without PVA is 99.1%, it can only retain stability for 60 cycles. These results indicate the excellent performance of plating and stripping behavior of Zn2+ by the addition of PVA.
3.3 The impact of PVA on the electrochemical performance of the Zn–I2 full battery
Motivated by the above observations, PVA as an additive into Zn–I2 batteries may potentially alleviate the shuttle phenomenon of I3− ions, hence mitigating the decline in cycling life and severe self-discharge (Fig. 1). To evaluate this hypothesis, Zn–I2 batteries with and without PVA were assembled, followed by a series of experimental procedures.Fig. 5a demonstrates the redox behavior of I− and I2 in the Zn–I2 full batteries with and without PVA. The oxidative peak at 1.3 V is attributed to the oxidation of I−, while the reductive peak at 1.25 V is assigned to the reduction of I2. The shape of the two curves exhibits no discernible discrepancy, indicating that the addition of PVA does not influence the redox mechanism of the battery. In addition, it can be seen from the CV curves in the voltage range of 1.4–1.6 V that the current of water oxidation is significantly reduced by the addition of PVA, indicating an inhibition of water oxidation. Fig. 5b depicts that the charge transfer resistance of the Zn–I2 battery with PVA is just slightly larger than that of the battery without PVA. To investigate the chemical reaction kinetics of the Zn–I2 battery with PVA, extensive analysis was conducted on CV curves at different scan rates. Fig. S15† demonstrates that the potential polarizations between the anodic and cathodic peaks increase with the increased scan rate. Using the equation
| i = aνb | (5) |
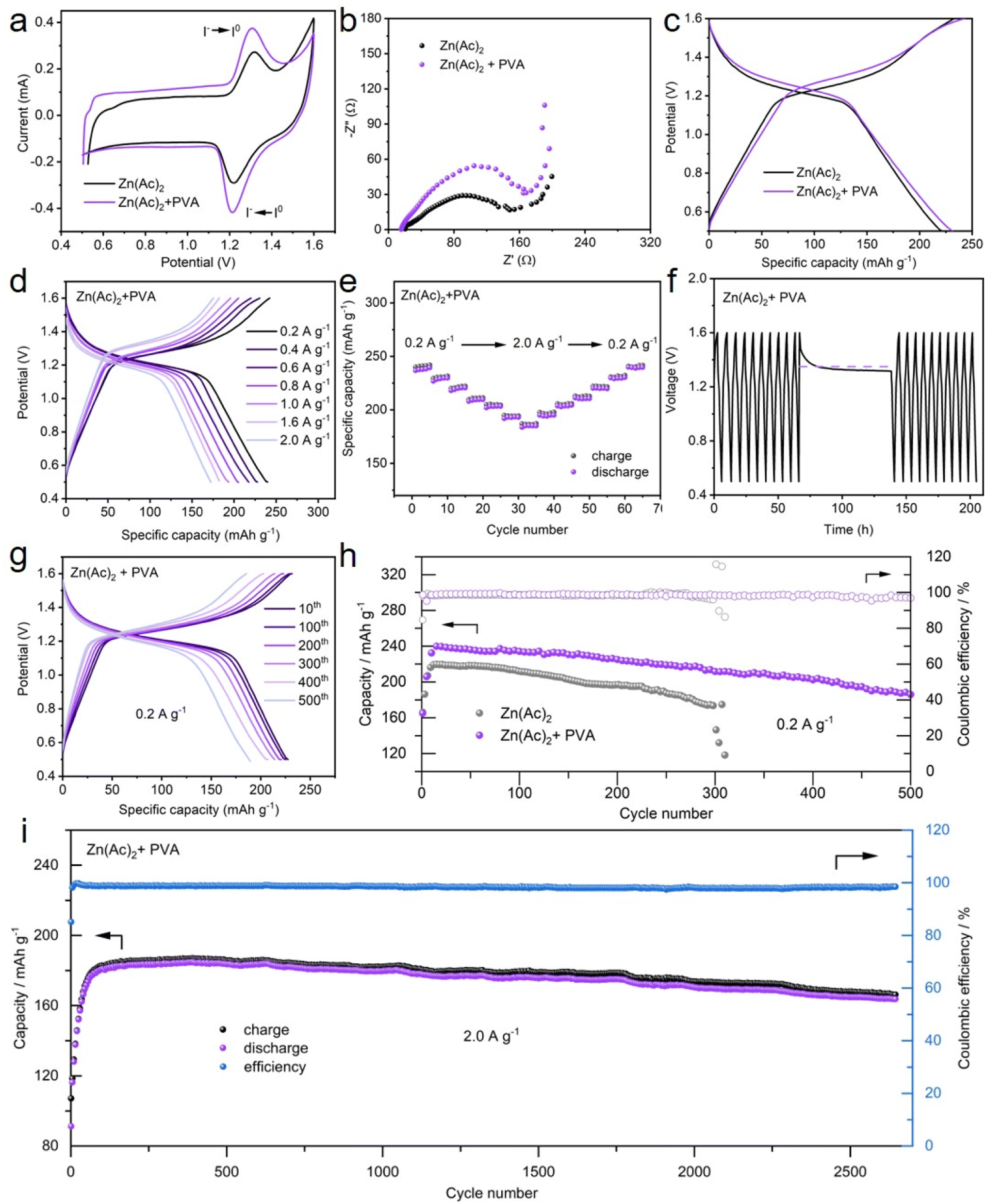 | ||
| Fig. 5 (a) CV profiles of the Zn–I2 batteries with and without PVA at 0.2 mV s−1, (b) the EIS spectra of the Zn–I2 battery with and without PVA, (c) charge/discharge profiles of the Zn–I2 batteries with and without PVA at 0.2 A g−1 for the 50th cycle, (d) galvanostatic charge/discharge profiles and (e) rate performance of the Zn–I2 battery with PVA at different current densities, (f) self-discharge characteristics for Zn–I2 batteries with PVA, (g) galvanostatic charge/discharge profiles of different cycles at 0.2 A g−1 in the Zn–I2 battery with PVA, (h) long-term cycling performance of the Zn–I2 battery with and without PVA at 0.2 A g−1, and (i) long-term cycling performance of the Zn–I2 battery with PVA at 2.0 A g−1. | ||
The typical galvanostatic charge/discharge profiles show that both Zn–I2 batteries exhibit a charge/discharge platform at approximately 1.3 V (Fig. 5c), in accordance with the forementioned CV measurements. Although the potential gap between the charge/discharge plateaus of the battery with PVA is 0.05 V, its specific capacity (240 mA h g−1) slightly surpasses that of the PVA-free battery (225 mA h g−1). The reason for this specific capacity exceeding the theoretical specific capacity of 211 mA h g−1 lies in the fact that the porous carbon also contributes a portion of reversible capacity (Fig. S17†).65,66 On the basis of the total current measured by CV, the capacitive contribution can be calculated through the following equation:
| i = k1ν + k2ν1/2 | (6) |
To investigate the specific capacity and rate performance of the Zn–I2 battery with and without PVA, various current densities encompassing 0.2 A g−1 to 2.0 A g−1 were implemented. As demonstrated in Fig. 5d and S19,† the batteries with PVA show a superior specific capacity (240 mA h g−1, 220 mA h g−1, 200 mA h g−1, and 185 mA h g−1, at 0.2 A g−1, 0.6 A g−1, 1.0 A g−1, and 2.0 A g−1, respectively) compared to the PVA-free batteries (225 mA h g−1, 206 mA h g−1, 192 mA h g−1, and 173 mA h g−1, at 0.2 A g−1, 0.6 A g−1, 1.0 A g−1, and 2.0 A g−1, respectively), indicating an improved performance of the electrolyte with PVA. Fig. 5e shows that the charge/discharge specific capacity of the battery decreases progressively as the current density increases from 0.2 A g−1 to 2.0 A g−1. When the current density returns from 2.0 A g−1 to 0.2 A g−1, its capacity is nearly completely recovered, manifesting good rate capability of the Zn–I2 battery with PVA. Conversely, the Zn–I2 battery without PVA presents a gradual decrease in capacity in rate performance (Fig. S20†), likely due to the shuttle effect of I3− generated at the cathode. SEM further examined the surface appearance of Zn anodes after charging and discharging 20 cycles of the Zn–I2 full battery with/without the PVA additive. As shown in Fig. S21,† on the surface of Zn foil used in PVA-free electrolyte, many large deposits and irregular corrosion areas appear, whereas on the surface of zinc foil used in the PVA-containing electrolyte, only smaller-sized deposits are observed and there are no obvious corrosion areas. The above results provide credible evidence that PVA additives can slow down capacity decay and prevent zinc corrosion.
Self-discharge tests were employed to estimate the self-discharge capacity loss in the Zn–I2 batteries with and without PVA. The batteries are charged/discharged for 10 cycles and then fully charged to 1.6 V and allowed to rest for 72 h. As displayed in Fig. 5f, the open-circuit potential can stabilize at 1.3 V for more than 72 h, implying the stability of the battery with PVA. Although the battery capacity decreased marginally after 72 h of rest, the capacity retention was 85% (from 240 to 206 mA h g−1). Furthermore, the battery capacity promptly returned to 235 mA h g−1 in the subsequent cycling, indicating a “self-healing” capability. In contrast, the open-circuit potential and the discharge capacity of the battery without PVA decreased to 1.2 V and 145 mA h g−1 after enduring 72 h of rest, with a capacity retention of 68% (Fig. S22†). This enhanced preservation of capacity retention after resting suggests that PVA in the electrolyte can mitigate the capacity decline by confining the shuttle effect of polyiodides.
Given the criticality of battery stability, the stable cycling performance of the Zn–I2 battery with and without PVA was evaluated at 0.2 A g−1, 1.0 A g−1, and 2.0 A g−1 (Fig. 5g–i and S23–S25†). At a current density of 0.2 A g−1, the constructed Zn–I2 batteries with PVA and without PVA yield an initial specific capacity of 160 mA h g−1 and 180 mA h g−1, respectively (Fig. 5h). However, the capacity of the Zn–I2 battery with PVA gradually climbs up to 240 mA h g−1, while that for the Zn–I2 battery without PVA is just 220 mA h g−1. Meanwhile, the battery with PVA shows a reversible specific capacity of 211 mA h g−1 after 300 cycles, alongside a capacity retention of 88% and a good CE of 98.5%, exceeding that of the battery without PVA (a specific capacity of 173 mA h g−1, a retention of 78% and CE of 96.1%) (Fig. 5h and S23†). After 500 cycles, the battery with PVA yet maintains a specific capacity of 185 mA h g−1. At a current density of 1.0 A g−1 and 2.0 A g−1, the Zn–I2 battery with PVA achieves a substantial reversible capacity of 175 mA h g−1 and 160 mA h g−1 after 1600 cycles and 2600 cycles, respectively, with an average CE value of 98%. Conversely, the discharge capacity of the Zn–I2 battery without PVA displayed a decline from 171 mA h g−1 to 75 mA h g−1 after 1000 cycles at a current density of 2.0 A g−1, indicating rapid capacity degradation (Fig. S25†). Highly capacity retention and long-term stability are attributed to the role of PVA, which can adsorb polyiodide species formed in the electrolyte, prohibiting polyiodide species dissolution and diffusion, as well as protecting the Zn anode from side reactions.
4. Conclusion
In summary, PVA possessing electron-rich hydroxyl groups has been demonstrated as an efficient and economic additive electrolyte in a Zn–I2 aqueous battery to confine the shuttle effect of polyiodides and modulate zinc deposition performance. On the basis of a series of experimental evaluation, characterization, and theoretical calculation, it is evident that PVA additives play the following important role: (1) PVA has a pronounced affinity for polyiodides, hindering their migration to the surface of the zinc electrode. (2) These hydroxyl groups can disrupt the hydrogen bonding network of water molecules, reducing water activity and thus alleviating zinc electrode corrosion. (3) The preferential adsorption of PVA on the zinc anode alters the zinc ion deposition environment, leading to an increase in the nucleation overpotential, which is favorable for the formation of dense and uniform zinc ion deposition. Consequently, at a current density of 1.0 A g−1 and 2.0 A g−1, the Zn–I2 full battery with PVA yet achieves a substantial reversible capacity of 175 mA h g−1 and 163 mA h g−1 after 1600 cycles and 2600 cycles, respectively, with an average CE value of 98%. This study demonstrates the influence of PVA in enhancing the lifespan of the Zn–I2 battery and provides insight into the design of user-friendly electrolytes for aqueous batteries.Data availability
Data will be made available on request.Author contributions
Qu Yue: conceptualization, investigation, writing – original draft, supervision. Yu Wan: investigation, formal analysis. Xiaoqin Li: investigation. Qian Zhao: investigation. Taotao Gao: investigation. Guowei Deng: investigation, funding acquisition. Bing Li: investigation, funding acquisition. Dan Xiao: software, writing – review & editing, supervision.Conflicts of interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Opening Project of Structural Optimization and Application of Functional Molecules Key Laboratory of Sichuan Province (2023GNFZ-04) and the Opening Project of Hubei Key Laboratory of Wudang Local Chinese Medicine Research (Hubei University of Medicine) (WDCM2023008). The authors would like to thank Xie Han from Shiyanjia Lab (http://www.shiyanjia.com) for the XPS tests.References
- T. M. Gür, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- W. Zhao, X. Ma, L. Gao, X. Wang, Y. Luo, Y. Wang, T. Li, B. Ying, D. Zheng, S. Sun, Q. Liu, Y. Zheng, X. Sun and W. Feng, Adv. Mater., 2024, 36, 2305190 CrossRef CAS PubMed.
- L. Yue, D. Wang, Z. Wu, W. Zhao, Y. Ren, L. Zhang, B. Zhong, N. Li, B. Tang, Q. Liu, Y. Luo, A. M. Asiri, X. Guo and X. Sun, Chem. Eng. J., 2022, 433, 134477 CrossRef CAS.
- L. Yue, C. Ma, S. Yan, Z. Wu, W. Zhao, Q. Liu, Y. Luo, B. Zhong, F. Zhang, Y. Liu, A. A. Alshehri, K. A. Alzahrani, X. Guo and X. Sun, Nano Res., 2022, 15, 186–194 CrossRef CAS.
- L. Yue, J. Liang, Z. Wu, B. Zhong, Y. Luo, Q. Liu, T. Li, Q. Kong, Y. Liu, A. M. Asiri, X. Guo and X. Sun, J. Mater. Chem. A, 2021, 9, 11879–11907 RSC.
- M. Han, D. Chen, Q. Lu and G. Fang, Small, 2023, 2310293 CrossRef PubMed.
- M. Xing, Z. Z. Zhao, Y. J. Zhang, J. W. Zhao, G. L. Cui and J. H. Dai, Mater. Today Energy, 2020, 18, 100534 CrossRef CAS.
- Z. Pei, Z. Zhu, D. Sun, J. Cai, A. Mosallanezhad, M. Chen and G. Wang, Mater. Res. Bull., 2021, 141, 111347 CrossRef CAS.
- C. Li, L. Wang, J. Zhang, D. Zhang, J. Du, Y. Yao and G. Hong, Energy Storage Mater., 2022, 44, 104–135 CrossRef.
- A. Naveed, T. Rasheed, B. Raza, J. Chen, J. Yang, N. Yanna and J. Wang, Energy Storage Mater., 2022, 44, 206–230 CrossRef.
- X. Li, J. Xiang, H. Liu, P. Wang, C. Chen, T. Gao, Y. Guo, D. Xiao and Z. Jin, J. Colloid Interface Sci., 2024, 654, 476–485 CrossRef CAS PubMed.
- M. Tang, Q. Zhu, P. Hu, L. Jiang, R. Liu, J. Wang, L. Cheng, X. Zhang, W. Chen and H. Wang, Adv. Funct. Mater., 2021, 31, 2102011 CrossRef CAS.
- Y. Li, Y. Wang, Y. Xu, W. Tian, J. Wang, L. Cheng, H. Yue, R. Ji, Q. Zhu, H. Yuan and H. Wang, Small, 2022, 18, 2202214 CrossRef CAS.
- J. Wang, Y. Yang, Y. Zhang, Y. Li, R. Sun, Z. Wang and H. Wang, Energy Storage Mater., 2021, 35, 19–46 CrossRef.
- W. Li, K. Wang and K. Jiang, J. Mater. Chem. A, 2020, 8, 3785–3794 RSC.
- X. Li, N. Li, Z. Huang, Z. Chen, G. Liang, Q. Yang, M. Li, Y. Zhao, L. Ma, B. Dong, Q. Huang, J. Fan and C. Zhi, Adv. Mater., 2021, 33, 2006897 CrossRef CAS PubMed.
- Q. Guo, H. Wang, X. Sun, Y. N. Yang, N. Chen and L. Qu, ACS Mater. Lett., 2022, 4, 1872–1881 CrossRef CAS.
- T. Liu, H. Wang, C. Lei, Y. Mao, H. Wang, X. He and X. Liang, Energy Storage Mater., 2022, 53, 544–551 CrossRef.
- Z. Wang, J. Huang, Z. Guo, X. Dong, Y. Liu, Y. Wang and Y. Xia, Joule, 2019, 3, 1289–1300 CrossRef CAS.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- T. Gao, S. Yu, Y. Chen, X. Li, X. Tang, S. Wu, B. He, H. Lan, S. Li, Q. Yue and D. Xiao, J. Colloid Interface Sci., 2023, 642, 120–128 CrossRef CAS PubMed.
- T. Gao, X. Tang, X. Li, H. Lan, S. Yu, S. Wu, Q. Yue and D. Xiao, Inorg. Chem. Front., 2023, 10, 1447–1456 RSC.
- Q. Yue, T. Gao, S. Wu, H. Yuan and D. Xiao, J. Colloid Interface Sci., 2022, 605, 906–915 CrossRef CAS PubMed.
- H. Pan, B. Li, D. Mei, Z. Nie, Y. Shao, G. Li, X. S. Li, K. S. Han, K. T. Mueller, V. Sprenkle and J. Liu, ACS Energy Lett., 2017, 2, 2674–2680 CrossRef CAS.
- J. Xu, W. Ma, L. Ge, M. Ren, X. Cai, W. Liu, J. Yao, C. Zhang and H. Zhao, J. Alloys Compd., 2022, 912, 165151 CrossRef CAS.
- Q. Zhao, Y. Meng, L. Su, W. Cen, Q. Wang and D. Xiao, J. Colloid Interface Sci., 2022, 608, 265–274 CrossRef CAS PubMed.
- Q. Zhao, Y. Meng, L. Yang, X. He, B. He, Y. Liu and D. Xiao, J. Colloid Interface Sci., 2019, 551, 61–71 CrossRef CAS PubMed.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, 2004240 CrossRef CAS PubMed.
- Y. Zhang, T. Zhao, S. Yang, Y. Zhang, Y. Ma and Z. Wang, J. Energy Chem., 2022, 75, 310–320 CrossRef CAS.
- Z. Huang, W. Zhang, H. Zhang, L. Fu, B. Song, W. Zhang, Q. Chen and K. Lu, J. Power Sources, 2022, 523, 231036 CrossRef CAS.
- T. T. Su, J. B. Le, K. Wang, K. N. Liu, C. Y. Shao, W. F. Ren and R. C. Sun, J. Power Sources, 2022, 550, 232136 CrossRef CAS.
- W. Shang, Q. Li, F. Jiang, B. Huang, J. Song, S. Yun, X. Liu, H. Kimura, J. Liu and L. Kang, Nano-Micro Lett., 2022, 14, 82 CrossRef CAS PubMed.
- P. F. Zhang, Z. Wu, S. J. Zhang, L. Y. Liu, Y. Tian, Y. Dou, Z. Lin and S. Zhang, Nano Energy, 2022, 102, 107721 CrossRef CAS.
- J. J. Hong, L. Zhu, C. Chen, L. Tang, H. Jiang, B. Jin, T. C. Gallagher, Q. Guo, C. Fang and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 15910–15915 CrossRef CAS PubMed.
- W. Wu, C. Li, Z. Wang, H. Y. Shi, Y. Song, X. X. Liu and X. Sun, Chem. Eng. J., 2022, 428, 131283 CrossRef CAS.
- A. Li, J. Li, Y. He and M. Wu, J. Energy Chem., 2023, 83, 209–228 CrossRef CAS.
- D. Zhao, Q. Zhu, Q. Zhou, W. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2022, e12522 Search PubMed.
- Y. Zhang, D. Tao, F. Xu and T. Li, Chem. Eng. J., 2022, 427, 131592 CrossRef CAS.
- F. S. Cai, Y. Q. Duan and Z. H. Yuan, J. Mater. Sci.: Mater. Electron., 2018, 29, 11540–11545 CrossRef CAS.
- K. Tashiro, H. Kitai, S. M. Saharin, A. Shimazu and T. Itou, Macromolecules, 2015, 48, 2138–2148 CrossRef CAS.
- T. Takahama, S. M. Saharin and K. Tashiro, Polymer, 2016, 99, 566–579 CrossRef CAS.
- S. M. Saharin, T. Takahama, S. Nonogaki, K. Saito and K. Tashiro, Macromolecules, 2015, 48, 8867–8876 CrossRef CAS.
- P. Chetri, N. N. Dass and N. S. Sarma, Polym. Bull., 2007, 58, 489–494 CrossRef CAS.
- S. Moulay, J. Polym. Eng., 2013, 33, 389–443 CAS.
- H. Ren, B. Gao and M. Wang, Polymer, 2022, 249, 124828 CrossRef CAS.
- U. K. Fatema, F. Okino and Y. Gotoh, J. Mater. Sci., 2014, 49, 1049–1057 CrossRef CAS.
- Q. Zhang, Z. Wu, F. Liu, S. Liu, J. Liu, Y. Wang and T. Yan, J. Mater. Chem. A, 2017, 5, 15235–15242 RSC.
- L. Zhang, M. Zhang, H. Guo, Z. Tian, L. Ge, G. He, J. Huang, J. Wang, T. Liu, I. P. Parkin and F. Lai, Adv. Sci., 2022, 9, 2105598 CrossRef CAS PubMed.
- Y. Lin, Z. Mai, H. Liang, Y. Li, G. Yang and C. Wang, Energy Environ. Sci., 2023, 16, 687–697 RSC.
- D. Feng, Y. Jiao and P. Wu, Angew. Chem., Int. Ed., 2023, 62, e202314456 CrossRef CAS PubMed.
- H. Yang, D. Chen, R. Zhao, G. Li, H. Xu, L. Li, X. Liu, G. Li, D. Chao and W. Han, Energy Environ. Sci., 2023, 16, 2910–2923 RSC.
- J. Zhu, W. Deng, N. Yang, X. Xu, C. Huang, Y. Zhou, M. Zhang, X. Yuan, J. Hu, C. Li and R. Li, Small, 2022, 18, 2202509 CrossRef CAS PubMed.
- F. Zhao, Z. Jing, X. Guo, J. Li, H. Dong, Y. Tan, L. Liu, Y. Zhou, R. Owen, P. R. Shearing, D. J. L. Brett, G. He and I. P. Parkin, Energy Storage Mater., 2022, 53, 638–645 CrossRef.
- H. Cao, X. Huang, Y. Liu, Q. Hu, Q. Zheng, Y. Huo, F. Xie, J. Zhao and D. Lin, J. Colloid Interface Sci., 2022, 627, 367–374 CrossRef CAS PubMed.
- C. Meng, W. He, L. Jiang, Y. Huang, J. Zhang, H. Liu and J. J. Wang, Adv. Funct. Mater., 2022, 32, 2207732 CrossRef CAS.
- H. Tian, J. L. Yang, Y. Deng, W. Tang, R. Liu, C. Xu, P. Han and H. J. Fan, Adv. Energy Mater., 2023, 13, 2202603 CrossRef CAS.
- Z. Hu, F. Zhang, A. Zhou, X. Hu, Q. Yan, Y. Liu, F. Arshad, Z. Li, R. Chen, F. Wu and L. Li, Nano-Micro Lett., 2023, 15, 171 CrossRef CAS PubMed.
- Y. Cao, X. Tang, L. Li, H. Tu, Y. Hu, Y. Yu, S. Cheng, H. Lin, L. Zhang, J. Di, Y. Zhang and M. Liu, Nano Res., 2023, 16, 3839–3846 CrossRef CAS.
- K. Wang, J. B. Le, S. J. Zhang, W. F. Ren, J. M. Yuan, T. T. Su, B. Y. Chi, C. Y. Shao and R. C. Sun, J. Mater. Chem. A, 2022, 10, 4845–4857 RSC.
- J. Xu, W. Lv, W. Yang, Y. Jin, Q. Jin, B. Sun, Z. Zhang, T. Wang, L. Zheng, X. Shi, B. Sun and G. Wang, ACS Nano, 2022, 16, 11392–11404 CrossRef CAS PubMed.
- Y. Quan, M. Yang, M. Chen, W. Zhou, X. Han, J. Chen, B. Liu, S. Shi and P. Zhang, Chem. Eng. J., 2023, 458, 141392 CrossRef CAS.
- S. Wu, M. Hua, Y. Alsaid, Y. Du, Y. Ma, Y. Zhao, C. Y. Lo, C. Wang, D. Wu, B. Yao, J. Strzalka, H. Zhou, X. Zhu and X. He, Adv. Mater., 2021, 33, 2007829 CrossRef CAS PubMed.
- T. Wei, X. Zhang, Y. Ren, Y. Wang, Z. Li, H. Zhang and L. Hu, Chem. Eng. J., 2023, 457, 141272 CrossRef CAS.
- X. Gong, H. Yang, J. Wang, G. Wang and J. Tian, ACS Appl. Mater. Interfaces, 2023, 15, 4152–4165 CrossRef CAS PubMed.
- D. Yu, A. Kumar, T. A. Nguyen, M. T. Nazir and G. Yasin, ACS Sustainable Chem. Eng., 2020, 8, 13769–13776 CrossRef CAS.
- S. Chai, J. Yao, Y. Wang, J. Zhu and J. Jiang, Chem. Eng. J., 2022, 439, 135676 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00792a |
| This journal is © The Royal Society of Chemistry 2024 |