
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePadlocking dihydrofurannulation for the control of small degree of helicity built on a fused-tetracyclic core†
Arthur
Gaucherand
 a,
Expédite
Yen-Pon
a,
Diego
García-López
a,
Jean-Valère
Naubron
b,
Sara
Chentouf
b,
Michel
Giorgi
b,
Stéphane
Humbel
a,
Marion
Jean
a,
Jean
Rodriguez
a and
Damien
Bonne
*a
a,
Expédite
Yen-Pon
a,
Diego
García-López
a,
Jean-Valère
Naubron
b,
Sara
Chentouf
b,
Michel
Giorgi
b,
Stéphane
Humbel
a,
Marion
Jean
a,
Jean
Rodriguez
a and
Damien
Bonne
*a
aAix Marseille Université, CNRS, Centrale Marseille, iSm2, Marseille, France
bAix Marseille Université, CNRS, Centrale Marseille, FSCM, Spectropole, Marseille, France
First published on 22nd April 2024
Abstract
Enantioselective construction of small molecules displaying a configurationally stable helical shape built on a fused-tetracyclic core is a daunting synthetic challenge even more pronounced when five-membered rings are incorporated in the structure. The resulting higher configurational lability strongly hampers their access, and therefore the development of new efficient methodologies is timely and highly desirable. In this context, we describe a padlocking approach via the enantioselective organocatalytic domino furannulation of appropriately designed achiral fused-tricyclic precursors resulting in the synthesis of configurationally locked helically chiral tetracyclic scaffolds featuring one or two five-membered rings with the simultaneous control of central and helical chiralities.
Introduction
Stereochemically complex molecules are particularly important for the development of new bioactive substances,1 functional materials,2 catalysts3 and valuable supramolecular structures.4 In this context, synthetically challenging molecules displaying a small helical shape built on a fused-tetracyclic core are particularly attractive given their unique structural and chiroptical features. However, their enantioselective assembly still constitutes a daunting challenge in organic synthesis because of their strong configurational lability at room temperature.The archetypal example is the all-carbon [4]helicene series featuring four ortho-fused phenyl rings pioneered by Newman in the 1950s, which can be stabilised only by introduction of substituents at one or both 1,12-termini of the helix fjord region. Known as the remote steric effect it has been exploited by several other groups more recently,5 including in their diastereoselective6 and enantioselective synthesis (Fig. 1a).7 However, the incorporation of five-membered ring heterocycles in the helix severely decreases their configurational stability and seriously hampers their enantioselective elaboration. For example, we have shown that dioxa[4]helicene 1 has a very low barrier to enantiomerisation of only 57 kJ mol−1 meaning a half-life time for racemisation of seconds at room temperature despite a double remote steric effect (Fig. 1b).8 In 2018, the synthesis and resolution of a photoresponsive molecular switch featuring three different stereogenic elements was reported by Feringa and co-workers (Fig. 1c).9 Regarding enantioselective access to these stereochemically complex molecules, the group of Yan reported the synthesis of stereochemically complex oxygen- or nitrogen-containing molecules featuring either a four-cycle helical shape10 or a saddle-shaped conformation11 (Fig. 1c).
 | ||
| Fig. 1 Synthetic challenge and our design in the synthesis of molecules bearing multiple stereogenic elements. | ||
In 2023, Yang and Li's groups independently reported an enantioselective Povarov reaction leading to centrally and helically chiral scaffolds, precursors of azahelicene after oxidation.12 Contemporaneously, the group of Wang managed the control of both central and helical stereogenic elements via enantioselective phospha-Michael addition under phosphonium salt organocatalysis.13 Therefore, the development of efficient methodologies to address the control of new small helically shaped chiral scaffolds built on a fused-tetracyclic core is timely and highly desirable.14
Results and discussion
For this endeavor, it was envisioned to forge the helical chirality through enantioselective organocatalytic domino dihydrofurannulation15 of appropriately designed achiral fused-tricyclic precursors 3, resulting in the synthesis of tetracyclic derivatives 4 with simultaneous control of two different stereogenic elements (Fig. 1b).16 The required configurational stability would be guaranteed by a remote steric effect ensuring kinetically affordable but thermodynamically impossible diastereomerisation. Essentially, the central chirality would act like a padlock that imposes the configuration of the helix, which cannot be inverted. This scenario enables the option to introduce an additional stereogenic axis by adding an aryl group (R1 = Ar), delivering a product with up to three different stereogenic elements (center, axis and helix) in a close environment.Preliminary investigations were initiated using hydroxynaphthofuran 3a and chloronitroalkene 2a as model substrates (Table 1). Various types of hydrogen-bonding organocatalysts (A–F) were used in order to promote the centrally and helically selective domino Michael/C–O alkylation,17 in the presence of a weak base (K2HPO4) in chloroform. With catalyst A, a very slow reaction afforded the desired product in moderate yield and good enantiomeric excess (entry 1). Quinine-based squaramide organocatalysts B–E showed increased efficiency both in terms of enantioselectivity and reaction time (entries 2–5), the best being catalyst E which delivered 4aa in 97% ee (entry 5). Crystallisation of 4aa enabled assignment of its absolute configuration by single crystal X-ray diffraction analysis (SCXRD),18 and this confirmed its helical shape (vide infra).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | t (d) | Yield of 4aab[%] | ee of 4aac |
| a Reaction conditions: 2a (1.0 equiv.), 3a (1.0 equiv.) and base (1.2 equiv.) were combined with solvent [0.1 M] at 25 °C until the starting materials were consumed. b Yield of the analytically pure product after purification by flash chromatography. c Determined by HPLC on a chiral stationary phase. d 1.2 equiv. of 3a was employed. | ||||||
| 1 | A | K2HPO4 | CHCl3 | 12 | 30 | 86 |
| 2 | B | K2HPO4 | CHCl3 | 7 | 57 | 82 |
| 3 | C | K2HPO4 | CHCl3 | 7 | 76 | 94 |
| 4 | D | K2HPO4 | CHCl3 | 7 | 62 | 90 |
| 5 | E | K2HPO4 | CHCl3 | 4 | 57 | 97 |
| 6 | F | K2HPO4 | CHCl3 | 13 | 26 | 90 |
| 7 | E | K2CO3 | CHCl3 | 3 | 76 | 45 |
| 8 | E | Na2CO3 | CHCl3 | 4 | 67 | 97 |
| 9 | E | Na2CO3 | CH2Cl2 | 3 | 55 | 97 |
| 10 | E | Na2CO3 | Toluene | 7 | 50 | 78 |
| 11 | E | Na2CO3 | (ClCH2)2 | 3 | 71 | 95 |
| 12 | E | Na2CO3 | CHCl3 | 7 | 80 | 96 |
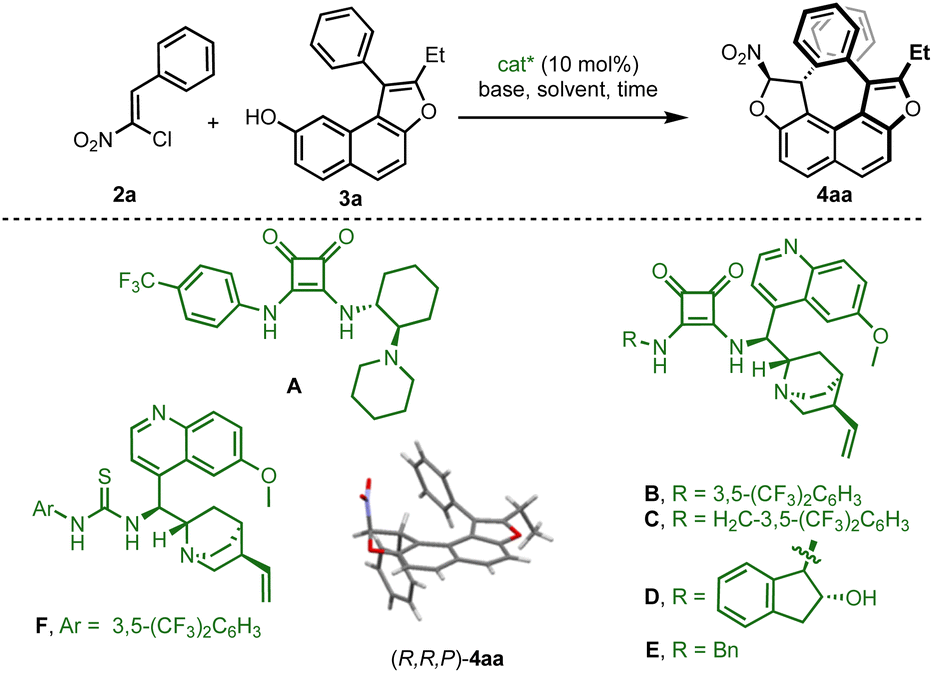
The use of thiourea catalyst F (entry 6) resulted in lower yield demonstrating the superiority of the squaramide subunit. Different bases were then screened with catalyst E (see the ESI†), and the use of K2CO3 (entries 7) resulted in an important decrease in the enantiomeric excess probably due to the ability of this base to promote the dihydrofurannulation reaction. However, the use of Na2CO3 allowed the isolation of the desired product in good yield and excellent enantioselectivity (entry 8). Other solvents were tested (entries 9–11) but none were revealed to be superior to chloroform both in terms of yield and enantioselectivity. Finally, the yield was maximised to 80% when the naphthol derivative was used in excess (entry 12), and these reactions conditions were selected to carry on the study on the scope.
With the optimised reaction conditions in hand, the generality of this new centrally/helically selective domino reaction was subsequently explored. The influence of the chloronitroalkene structure was initially studied, revealing that both electronic and steric parameters govern the reactivity, while the enantioselectivity remains consistently high (Scheme 1). Noticeably, the presence of an electron withdrawing substituent generally results in good to excellent yields (4ab, 4ac, 4ag, 4ah and 4ai), while a methoxy substitution gives 4aj in only 41% yield. The steric hindrance also has a negative impact on the yield as shown by the poor reactivity of ortho-substituted aryl nitroalkenes (4ad, 38% yield and 4ae, 23%), which required higher temperature resulting in lower enantiomeric excess (85% ee). Incorporation of other aryl substituents, such as 2-naphtyl (4al, 68%) proceeded smoothly compared with that of heteroaryl groups (4am, 27% and 4an, 35%), while maintaining a high level of enantioselectivity (95–97% ee).
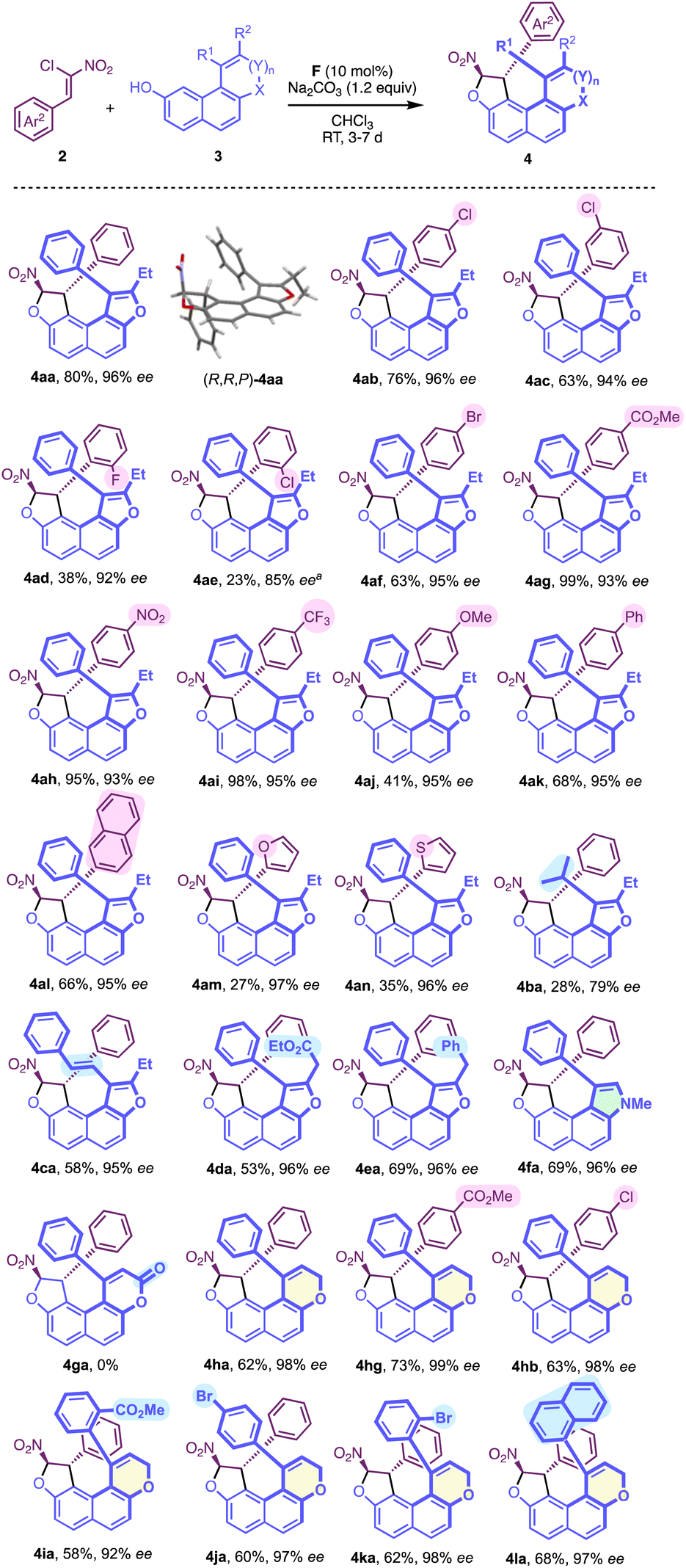 | ||
| Scheme 1 Reaction scope. [a] The reaction was run at 50 °C. | ||
We then looked at the structure of naphthol with the variation of R1, which can be either an alkyl or an alkenyl group (4ba and 4ca) with an overall lower efficiency for 4ba (R1 = iPr). Interestingly, an ester moiety could be introduced on the furan ring of 3d (R1 = Ph, R2 =CH2CO2Et, X = O, n = 0) delivering the desired product 4da in good yield and excellent enantioselectivity.
Gratifyingly, the corresponding N-methyl pyrrole-containing derivative 4fa was obtained with good efficiency. With a larger fused-heterocyclic system the situation became more complicated probably due to increased steric hindrance. Indeed, the corresponding δ-lactone 3g (R1 = Ph, R2 = H, X = O, n = 1, Y = C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), failed to react to give 4ga under the optimised conditions with 2a. However, and to our delight, the reactivity could be restored with the more flexible dihydropyran analog DHP-3h (R1 = Ph, R2 = H, X = O, n = 1, Y = CH2), allowing the efficient formation of the desired configurationally locked helically chiral bis-heterocycle 4ha in good yield and high enantioselectivity. Moreover, the efficiency of the transformation is not significantly affected by the substitution pattern in both aryl groups of the structure. Interestingly, the incorporation of a substituent in the ortho position of the aryl group of the DHP nucleus (4ia, 4ka and 4la) is possible, but this was not conducive to the formation of stable atropisomers due to the too low barriers to rotation in these cases. For 4ka, the diastereomerisation barrier was measured by dynamic NMR to be only 63 kJ.mol−1 (see the ESI†). We then aimed at increasing the complexity in the final structure by the incorporation of an additional stereogenic element. This was made possible starting with axially chiral and racemic naphthofuran 3m displaying a barrier to enantiomerisation ΔG≠exp = 113.7 kJ mol−1 (ΔG≠calc = 116.2 kJ mol−1) (Scheme 2). The organocatalyzed domino reaction with 2a led to the clean formation of a 1
O), failed to react to give 4ga under the optimised conditions with 2a. However, and to our delight, the reactivity could be restored with the more flexible dihydropyran analog DHP-3h (R1 = Ph, R2 = H, X = O, n = 1, Y = CH2), allowing the efficient formation of the desired configurationally locked helically chiral bis-heterocycle 4ha in good yield and high enantioselectivity. Moreover, the efficiency of the transformation is not significantly affected by the substitution pattern in both aryl groups of the structure. Interestingly, the incorporation of a substituent in the ortho position of the aryl group of the DHP nucleus (4ia, 4ka and 4la) is possible, but this was not conducive to the formation of stable atropisomers due to the too low barriers to rotation in these cases. For 4ka, the diastereomerisation barrier was measured by dynamic NMR to be only 63 kJ.mol−1 (see the ESI†). We then aimed at increasing the complexity in the final structure by the incorporation of an additional stereogenic element. This was made possible starting with axially chiral and racemic naphthofuran 3m displaying a barrier to enantiomerisation ΔG≠exp = 113.7 kJ mol−1 (ΔG≠calc = 116.2 kJ mol−1) (Scheme 2). The organocatalyzed domino reaction with 2a led to the clean formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (NMR, see the ESI†) of two diastereomers (R,R,P,aS)-4ma and (R,R,P,aR)-4ma in good yield. Significantly, both diastereomers were separated by column chromatography and isolated in high enantiomeric excesses. These observations indicate that this organocatalyzed domino reaction is totally under catalyst control, and the stereogenicity of the starting material 3m does not influence the activity of the catalyst. Analogously, nonchiral naphthofuran 3n (ΔGcalc = 55.7 kJ mol−1) was also employed in the dihydrofurannulation reaction, and we were pleased to observe the clean formation of product 4na in good yield, excellent enantioselectivity and in a 5:1 diastereomeric ratio, relative to the configuration of the stereogenic C–C bond. Interestingly, the barrier to diastereomerisation of the stereogenic axis was found to be quite high (ΔG≠calc = 92.7 kJ mol−1), probably due to intramolecular C–H⋯p interactions, that can clearly be seen in the crystal structure (vide infra and see the ESI†). Noteworthily, generation of this still rare family of enantioenriched compounds featuring axial, central and helicoidal stereogenic elements was achieved through a one-step synthesis.
:1 ratio (NMR, see the ESI†) of two diastereomers (R,R,P,aS)-4ma and (R,R,P,aR)-4ma in good yield. Significantly, both diastereomers were separated by column chromatography and isolated in high enantiomeric excesses. These observations indicate that this organocatalyzed domino reaction is totally under catalyst control, and the stereogenicity of the starting material 3m does not influence the activity of the catalyst. Analogously, nonchiral naphthofuran 3n (ΔGcalc = 55.7 kJ mol−1) was also employed in the dihydrofurannulation reaction, and we were pleased to observe the clean formation of product 4na in good yield, excellent enantioselectivity and in a 5:1 diastereomeric ratio, relative to the configuration of the stereogenic C–C bond. Interestingly, the barrier to diastereomerisation of the stereogenic axis was found to be quite high (ΔG≠calc = 92.7 kJ mol−1), probably due to intramolecular C–H⋯p interactions, that can clearly be seen in the crystal structure (vide infra and see the ESI†). Noteworthily, generation of this still rare family of enantioenriched compounds featuring axial, central and helicoidal stereogenic elements was achieved through a one-step synthesis.
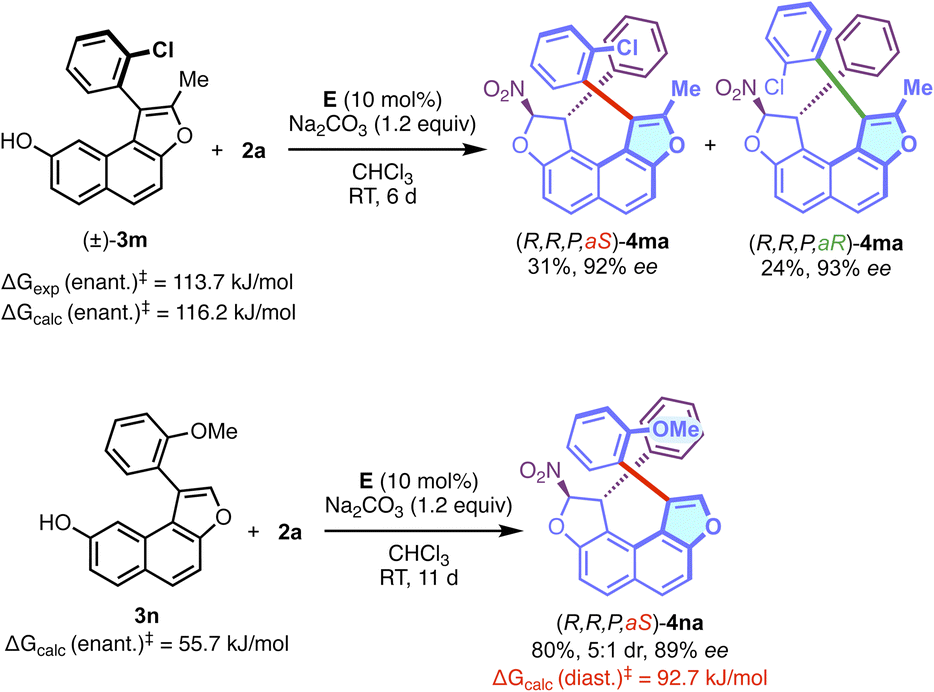 | ||
| Scheme 2 Access to enantioenriched molecules with three different stereogenic elements. | ||
The high configurational stability of our centrally/helically chiral molecules was exploited and post-functionalisations were carried out (Scheme 3). Hence, a Suzuki-type coupling at 110 °C allowed the conversion of 4af to 4ak in 48% yield while preserving high enantiomeric purity. Compound 4ha could also be functionalised by chemoselective chlorination with NCS delivering the helically chiral chlorinated dihydropyran 4ha-Cl with very good efficiency.
 | ||
| Scheme 3 Post-functionalisations. | ||
Complementarily, to circumvent the absence of reactivity of δ-lactone 3g, a complementary strategy was designed starting with dihydrofuran 5a (see the ESI†). From here, esterification of the phenol function of 5a with 3-phenylpropiolic acid allowed the formation of 6, which was then engaged in a gold-catalyzed intramolecular hydroarylation,19 affording the desired δ-lactone 4ga in good yield, also as a single diastereomer, via a highly efficient central-to-helical chirality transfer (Scheme 4). Then, DBU-promoted elimination of HNO2 was carried out to examine the configurational stability of dioxa[4]helicene-type 7. At 110 °C, the elimination afforded the desired compound 7 in 30% yield with slight erosion of the enantiomeric excess, whose structure and configuration were confirmed by X-ray diffraction analysis. The barrier to enantiomerisation was experimentally determined to be 123.4 kJ mol−1, corresponding to a half-life of racemisation of 14 hours at 90 °C. Alternatively, the lactone moiety of 4ga could be selectively reduced using DIBAL-H to afford the desired centrally and axially chiral styrene 8 as a single diastereomer, via a helical-to-axial chirality conversion. Having established an enantioselective organocatalyzed synthesis of molecules bearing multiple stereogenic elements, we pursued the study of the structural parameters of a series of tetracyclic fused compounds 4aa, 4na, 4ma, 4fa, 4ha and 4ga. In this series, SCXRD analysis allowed us to determine the absolute configurations as well as measure the torsion of these molecules. Hence, we selected three parameters to describe and measure the helicity (Fig. 2a): (i) the sum of the torsional angles ψ (in black) which characterizes the degree of twist of the helix; (ii) the interplanar angle φ (in orange) between both terminal ring planes, which indicates the compacity of the helical structure and (iii) the wedge angle θ (in red), which directly depends on the nature of the heteroatom and the size of the ring (5-membered vs. 6-membered). Conveniently, all three parameters establish directly proportional relationships with the degree of helicity of our compounds. Incorporation of a substituent at the ortho position of the phenyl ring in C5 has a strong influence on the compacity of the helical molecule going from an interplanar angle φ of 3.5° for (R,R,P)-4aa to 7.9° for (R,R,P,aS)-4na bearing an o-methoxyphenyl substituent (Fig. 2c). This effect increases with the size of this substituent, φ = 14.1° for (R,R,P,aS)-4ma bearing an o-chlorophenyl substituent.
 | ||
| Scheme 4 Alternative strategies and the X-ray structure of (P)-7. | ||
 | ||
| Fig. 2 Structural features of a series of helically chiral molecules and the computed diastereomerisation profile. | ||
There is also an important increase in the torsion when going from an oxygen atom in (R,R,P)-4aa to a nitrogen atom in (R,R,P)-4fa which follows the van der Waals radius order O < N. This behavior has been rationalised in related di-heterobridged [4]helicenium ions.20 The size of the heterocycle involved in the helical structure is even more crucial and constitutes an essential parameter to modulate the helical shape of the final molecule. Hence, when going from (R,R,P)-4aa to (R,R,P)-4ha, the sum of torsional angles ψ went from 12.0° to 36.0° and the interplanar angle φ increased from 3.5° to 29.5°. The strong influence of the ring size on the helicity can be explained by the large difference in term of wedge angles θ for these two heterocycles: 30.6° for furan versus 52.6° for dihydropyran. Consequently, the molecule is forced to adopt a more twisted configuration to avoid steric clash. Moreover, the presence of a methylene subunit in the dihydropyran ring of (R,R,P)-4ha renders the molecule quite flexible, which allows its strong twisting. In term of reactivity, it is interesting to note that although 3h (X = O, Y = CH2, R1 = Ph, R2 = H) is more hindered than 3a (X = O, n = 0, R1 = Ph, R2 = Et), it is still more reactive (lower reaction times are observed) because it is more flexible. However, the replacement of the methylene subunit in the DHP ring by a carbonyl function in (R,R,P)-4ga, makes the molecule much more rigid, which partially explains the absence of reactivity of 3g in the dihydrofurannulation reaction. This phenomenon of increasing rigidity is accompanied by a flattening of the molecule, which is clearly visible by comparing the interplanar angle φ between (R,R,P)-4ha and (R,R,P)-4ga, going from 29.5° to 24°, respectively. Finally, aromatisation of the dihydrofuran ring to the corresponding furan resulted in a strong increase in the torsion of the molecule. Hence, when going from (R,R,P)-4ga to (P)-7, the sum of torsional angles ψ went from 37.0° to 44.7° and the interplanar angle φ increased from 24.0° to 32.3°. This shows that helically chiral dihydrofurans 4 display rather small degrees of torsion in comparison to more classical “true” heterohelicenes. Importantly, these small degrees of helicity are configurationally stable thanks to the presence of the stereogenic carbon atoms of dihydrofuran.
To elucidate the fundamental aspects and characteristics of the studied compounds, the diastereomerisation of 4aa was studied by means of DFT calculations (Fig. 2b). The experimentally observed diastereomer, (R,R,P)-4aa, exhibited a diastereomerisation process that was found to be kinetically accessible but thermodynamically unfavorable. A transition state free energy of 93.4 kJ mol−1 feasibly interconverts (R,R,P)-4aa into the analogous (R,R,M)-4aa with an inverted helix. Nevertheless, the inverted diastereomer (R,R,M)-4aa is 84.1 kJ mol−1 higher in energy than its (R,R,P)-4aa counterpart, which makes the reverse free energy barrier from (R,R,M)-4a to (R,R,P)-4aa 9.3 kJ mol−1. Hence, the free energy profile of compound 4aa (Fig. 2b) accounts for the totally helicoselective formation (R,R,P)-4aa upon heteroannulation. As confirmed by our DFT studies, the central chirality serves as a constraining element, imposing and stabilizing the helical configuration, thereby precluding any possibility of inversion.
Finally, the chiroptical properties of (R,R,P)-4aa and (R,R,P)-4ha were subjected to a preliminary evaluation and ECD and VCD spectra have been measured and compared with their calculated ones (Fig. 3). Excellent agreement between measured and calculated spectra unambiguously establishes their absolute configuration. For compound (R,R,P)-4aa, the VCD spectra show six outstanding bands numbered 1 to 6 in Fig. 3. Analysis of the vibrational modes calculated for these bands enables their association with the chiral elements of which they are characteristic: band 1, positive and intense, located at 1560 cm−1, is characteristic of the central chirality of the carbon atom bearing the NO2 moiety. Bands 2 to 4, all negatives and of moderate intensities, located at 1540, 1440 and 1370 cm−1, can be linked respectively to a helix alone, to both central and helical stereogenic elements, and to stereogenic carbon atoms. Finally, bands 5 and 6, respectively positive and negative and both of moderate intensity, located at 1280 and 1265 cm−1 are related to both stereogenic centers, like band 4. For compound (R,R,P)-4ha, it is interesting to note additional negative (1620 cm−1 and 1230 cm−1) and positive (1220 cm−1) bands tagged A, B and C, respectively (Fig. 3), which distinguish the dihydropyran moiety. The ECD spectra show non-standard characteristics for a helical derivative. Contrary to what is generally observed, enantiomers (R,R,P)-4aa and (R,R,P)-4ha with P helicity are characterised by an intense positive band centered at around 250 nm followed by a weak negative band between 280 and 300 nm. Calculated UV and ECD spectra reproduce the shape of the measured ones very satisfactorily (see Fig. 3 and the ESI†). For the dominant transitions in the spectral range of 220–350 nm, analysis of the involved molecular orbital (MO) pair contributions reveals a strong p–p* character for the positive–negative ECD bands. The MO-pairs involved in these excitations are the delocalised MOs p and p* of the helix and phenyl moieties. In addition, weaker transitions with a noteworthy charge transfer signature also contribute to the positive–negative ECD bands. With low to medium intensities, these excitations involve mainly the MOs p of the helix and phenyl moieties (HOMO, HOMO-1, HOMO-2, and HOMO-3), with the LUMO located almost exclusively on the p system of the nitro moiety (see the ESI†).
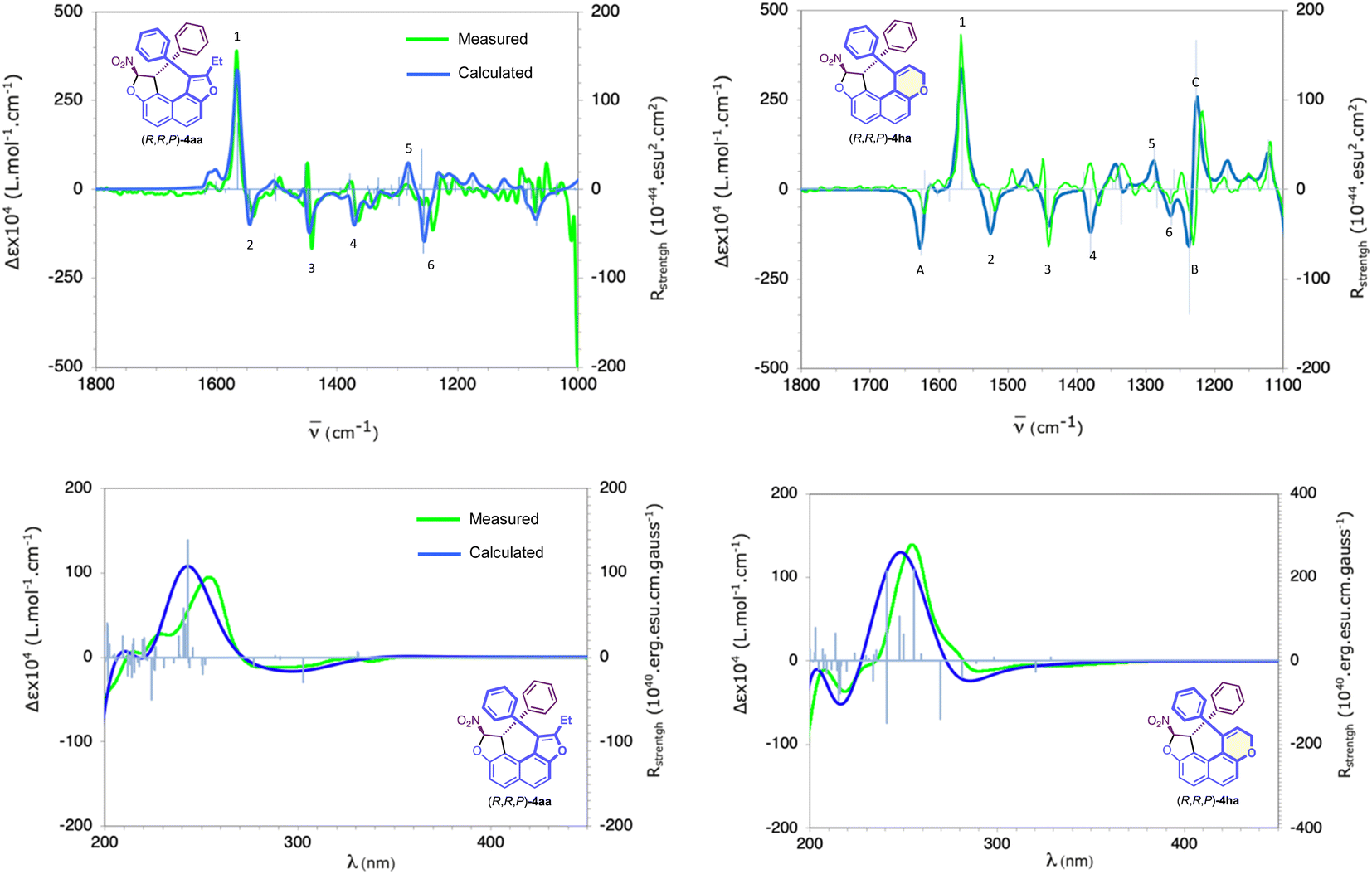 | ||
| Fig. 3 VCD (top)/ECD (bottom) spectra measured in CH3CN for (R,R,P)-4aa and (R,R,P)-4ha, and calculated using TD(60)-SMD(ACN)/CAM-B3LYP/6–31++G(d,p)//SMD(ACN)/B3LYP/TZVP (Gaussian shape in dark blue, dipole and rotational strength in clear blue). | ||
Conclusions
In conclusion, we have developed an organocatalyzed domino approach for the enantioselective construction of configurationally locked helically chiral tetracyclic scaffolds displaying up to three different stereogenic elements (point/axe/helix) from easily accessible starting materials. Importantly, the configurations of both stereogenic centers and the helix are simultaneously controlled, and the configuration of the stereogenic carbon atoms dictates the configuration of the helix rendering the diastereomerisation impossible. We show here that this concept can be applied in highly challenging small helically chiral heterocyclic systems otherwise impossible to access. In this methodology, we have also showed that the helicity can be easily modulated by subtle modification of the structure of the final molecule, providing a simple platform for further synthetic developments.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
D. B. and J. R. conceived and supervised the project. D. B. wrote the manuscript. A. G. and E. Y.-P. performed condition optimisation, scope study and product derivatisation. D. G.-L. and S. H. performed DFT studies. J.-V. N. and S. C. conducted chiroptical studies. M. G. performed SCXRD analyses. M. J. performed chiral HLPC analyses and experimental enantiomerisation barrier determination. All the authors proofread and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agence Nationale pour la Recherche (ANR-21-CE07-0036), Aix-Marseille Université, the Centre National de la Recherche Scientifique (CNRS), and Centrale Marseille is gratefully acknowledged. This work was supported by the computing facilities of the CRCMM, ‘Centre Régional de Compétences en Modélisation Moléculaire de Marseille’.Notes and references
- (a) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, The Challenge of Atropisomerism in Drug Discovery, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed; (b) A. Zask, J. Murphy and G. A. Ellestad, Biological Stereoselectivity of Atropisomeric Natural Products and Drugs, Chirality, 2013, 25, 265–274 CrossRef CAS PubMed.
- (a) J. Brandt, F. Salerno and M. J. Fuchter, The added value of small-molecule chirality in technological applications, Nat. Rev. Chem., 2017, 1, 45 CrossRef CAS; (b) F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros and T. Sierra, Orthogonal Action of Noncovalent Interactions for Photoresponsive Chiral Columnar Assemblies, Angew. Chem., Int. Ed., 2010, 49, 4910–4914 CrossRef CAS PubMed.
- (a) F. Lauterwasser, M. Nieger, H. Mansikkamäki, K. Nättinen and S. Bräse, Structurally Diverse Second-Generation [2.2]Paracyclophane Ketimines with Planar and Central Chirality: Syntheses, Structural Determination, and Evaluation for Asymmetric Catalysis, Chem.–Eur. J., 2005, 11, 4509–4525 CrossRef CAS PubMed; (b) H. Wölfle, H. Kopacka, K. Wurst, K.-H. Ongania, H.-H. Görtz, P. Preishuber-Pflügl and B. Bildstein, Planar chiral ferrocene salen-type ligands featuring additional central and axial chirality, J. Organomet. Chem., 2006, 691, 1197–1215 CrossRef.
- (a) D. Zhang, J.-C. Mulatier, J. R. Cochrane, L. Guy, G. Gao, J.-P. Dutasta and A. Martinez, Helical, Axial, and Central Chirality Combined in a Single Cage: Synthesis, Absolute Configuration, and Recognition Properties, Chem.–Eur. J., 2016, 22, 8038–8042 CrossRef CAS PubMed; (b) J. Buendía, E. E. Greciano and L. Sánchez, Influence of Axial and Point Chirality in the Chiral Self-Assembly of Twin N-Annulated Perylenecarboxamides, J. Org. Chem., 2015, 80, 12444–12452 CrossRef PubMed.
- (a) M. K. Lakshman, P. L. Kole, S. Chaturvedi, J. H. Saugier, H. J. C. Yeh, J. P. Glusker, H. L. Carrell, A. K. Katz, C. E. Afshar, W.-M. Dashwood, G. Kenniston and W. M. Baird, Methyl Group-Induced Helicity in 1,4-Dimethylbenzo[c]phenanthrene and Its Metabolites: Synthesis, Physical, and Biological Properties, J. Am. Chem. Soc., 2000, 122, 12629–12636 CrossRef CAS; (b) H. Okubo, M. Yamaguchi and C. Kabuto, Macrocyclic Amides Consisting of Helical Chiral 1,12-Dimethylbenzo[c]phenanthrene-5,8-dicarboxylate, J. Org. Chem., 1998, 63, 9500–9509 CrossRef CAS; (c) C. Herse, D. Bas, F. C. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. W. Laursen and J. A. Lacour, Highly Configurationally Stable [4]Heterohelicenium Cation, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef CAS PubMed; (d) R. Bam, W. Yang, G. Longhi, S. Abbate, A. Lucotti, M. Tommasini, R. Franzini, C. Villani, V. J. Catalano, M. M. Olmstead and W. A. Chalifoux, Four-Fold Alkyne Benzannulation: Synthesis, Properties, and Structure of Pyreno[a]pyrene-Based Helicene Hybrids, Org. Lett., 2019, 21, 8652–8656 CrossRef CAS PubMed.
- M. C. Carreño, Á. Enríquez, S. García-Cerrada, J. Sanz-Cuesta, A. Urbano, F. Maseras and A. Nonell-Canalsowards, Configurationally Stable [4]Helicenes: Enantioselective Synthesis of 12-Substituted 7,8-Dihydro[4]helicene Quinones, Chem.–Eur. J., 2008, 14, 603–620 CrossRef PubMed.
- (a) T. Hartung, R. Machleid, M. Simon, C. Golz and M. Alcarazo, Enantioselective Synthesis of 1,12-Disubstituted [4]Helicenes, Angew. Chem., Int. Ed., 2020, 59, 5660–5664 CrossRef CAS PubMed; (b) K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, Enantioselective Synthesis and Enhanced Circularly Polarised Luminescence of S-Shaped Double Azahelicenes, J. Am. Chem. Soc., 2014, 136, 5555–5558 CrossRef CAS PubMed; (c) P. Ravat, R. Hinkelmann, D. Steinebrunner, A. Prescimone, I. Bodoky and M. Juríček, Configurational Stability of [5]Helicenes, Org. Lett., 2017, 19, 3707–3710 CrossRef CAS PubMed; (d) U. Dhawa, C. Tian, T. Wdowik, J. C. A. Oliveira, J. Hao and L. Ackermann, Enantioselective Pallada-Electrocatalyzed C−H Activation by Transient Directing Groups: Expedient Access to Helicenes, Angew. Chem., Int. Ed., 2020, 59, 13451–13457 CrossRef CAS PubMed.
- See the ESI†.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen, E. Otten and B. L. Feringa, Central-to-Helical-to-Axial-to-Central Transfer of Chirality with a Photoresponsive Catalyst, J. Am. Chem. Soc., 2018, 140, 17278–17289 CrossRef CAS PubMed.
- K. Li, S. Huang, T. Liu, S. Jia and H. Yan, Organocatalytic Asymmetric Dearomatizing Hetero-Diels−Alder Reaction of Nonactivated Arenes, J. Am. Chem. Soc., 2022, 144, 7374–7381 CrossRef CAS PubMed.
- S. Huang, H. Wen, Y. Tian, P. Wang, W. Qin and H. Yan, Organocatalytic Enantioselective Construction of Chiral Azepine Skeleton Bearing Multiple-Stereogenic Elements, Angew. Chem., Int. Ed., 2021, 60, 21486–21493 CrossRef CAS PubMed.
- (a) W. Liu, T. Qin, W. Xie, J. Zhou, Z. Ye and X. Yang, Enantioselective Synthesis of Azahelicenes through Organocatalyzed Multicomponent Reactions, Angew. Chem., Int. Ed., 2023, 62, e202303430 CrossRef CAS PubMed; (b) C. Li, Y.-B. Shao, X. Gao, Z. Ren, C. Guo, M. Li and X. Li, Enantioselective synthesis of chiral quinohelicenes through sequential organocatalyzed Povarov reaction and oxidative aromatisation, Nat. Commun., 2023, 14, 3380 CrossRef CAS PubMed.
- J.-H. Wu, S. Fang, X. Zheng, J. He, Y. Ma, Z. Su and T. Wang, Organocatalytic Dynamic Kinetic Resolution Enabled Asymmetric Synthesis of Phosphorus-Containing Chiral Helicenes, Angew. Chem., Int. Ed., 2023, 62, e202309515 CrossRef CAS PubMed.
- For reviews on enantioselective synthesis of helically chiral molecules, see: (a) W. Liu, T. Qin and X. Yang, Chem.–Eur. J., 2022, 28, e202202369 CrossRef CAS PubMed; (b) Y. Wang, Z.-H. Wu and F. Shi, Chem Catal., 2022, 2, 3077 CrossRef CAS.
- P. Liu, X. Bao, J.-V. Naubron, S. Chentouf, S. Humbel, N. Vanthuyne, M. Jean, L. Giordano, J. Rodriguez and D. Bonne, Simultaneous Control of Central and Helical Chiralities: Expedient Helicoselective Synthesis of Dioxa[6]helicenes, J. Am. Chem. Soc., 2020, 142, 16199–16204 CrossRef CAS PubMed.
- For a recent review, see: H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Catalytic Asymmetric Synthesis of Atropisomers Bearing Multiple Chiral Elements: An Emerging Field, Angew. Chem., Int. Ed., 2023, 62, e202311053 Search PubMed.
- (a) C. Jarava-Barrera, F. Esteban, C. Navarro-Ranninger, A. Parra and J. Alemán, Chem. Commun., 2013, 49, 2001–2003 RSC; (b) J.-Y. Pan, X.-S. Li, D.-C. Xu and J.-W. Xie, Aust. J. Chem., 2013, 66, 1415–1421 CrossRef CAS; (c) D. Becerra, W. Raimondi, D. Dauzonne, T. Constantieux, D. Bonne and J. Rodriguez, Synthesis, 2017, 49, 195–201 CAS; (d) Z. D. Susam, B. D. Özcan, E. Kurtkaya, E. Yildirim and C. Tanyeli, Org. Biomol. Chem., 2022, 20, 8725 RSC.
- CCDC 2263627 (4aa) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- K. Nakamura, S. Furumi, M. Takeuchi, T. Shibuya and K. Tanaka, J. Am. Chem. Soc., 2014, 136, 5555–5558 CrossRef CAS PubMed.
- J. Elm, J. Lykkebo, T. J. Sørensen, B. W. Laursen and K. V. Mikkelsen, Racemisation Mechanisms and Electronic Circular Dichroism of [4]Heterohelicenium Dyes: A Theoretical Study, J. Phys. Chem. A, 2011, 115, 12025–12033 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation for all new compounds. CCDC 2263627, 2263636–2263640 and 2306282. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00745j |
| This journal is © The Royal Society of Chemistry 2024 |