
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCsp2–H functionalization of phenols: an effective access route to valuable materials via Csp2–C bond formation
Giulia
Brufani†
a,
Benedetta
Di Erasmo†
ab,
Chao-Jun
Li
 b and
Luigi
Vaccaro
*a
b and
Luigi
Vaccaro
*a
aLaboratory of Green S.O.C., Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it
bDepartment of Chemistry, FRQNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A0B8, Canada
First published on 21st February 2024
Abstract
In the vast majority of top-selling pharmaceutical and industrial products, phenolic structural motifs are highly prevalent. Non-functionalized simple phenols serve as building blocks in the synthesis of value-added chemicals. It is worth mentioning that lignin, being the largest renewable biomass source of aromatic building blocks in nature, mainly consists of phenolic units, which enable the production of structurally diverse phenols. Given their remarkable applicability in the chemical value chain, many efforts have been devoted to increasing the molecular complexity of the phenolic scaffold. Among the key techniques, direct functionalization of Csp2–H is a powerful tool, enabling the construction of new Csp2–C bonds in an economical and atomic manner. Herein we present and summarize the large plethora of direct Csp2–H functionalization methods that enables scaffold diversification of simple, unprotected phenols, leading to the formation of new Csp2–C bonds. In this review article, we intend to summarize the contributions that appeared in the literature mainly in the last 5 years dealing with the functionalization of unprotected phenols, both catalytic and non-catalytic. Our goal is to highlight the key findings and the ongoing challenges in the stimulating and growing research dedicated to the development of new protocols for the valorization of phenols.
1 Introduction
Phenols are simple aromatic building-blocks widely used to synthesize value-added chemicals such as natural products (NPs), active pharmaceutical ingredients (APIs), agrochemicals, dyes, flavours, fragrance components and resins.1 Phenolic compounds are also widely distributed in nature and in the human body.2 Examples are the amino acid tyrosine, the neurotransmitter serotonin, and the thyroid hormone levothyroxine. Phenols and their derivatives are highly prevalent structural motifs in the vast majority of top-selling pharmaceutical and industrial products.3 FDA-approved drug products in the United States have reported 138 compounds in which phenolic structural motifs appear.4Most of the large volumes of simple phenols are found downstream of the cumene peroxidation pathway (Hock process) via the oxidation of cumene to cumene-hydroperoxide which decomposes to form acetone and phenols, accounting for more than 106 tons per year of production of phenols,5 or via Vilsmeier–Haack formylation followed by Dakin oxidation.6 In any case, cumene is obtained primarily from crude oil or from the alkylation of benzene with propene over an acid catalyst, making the Hock process the last part of a very energy-intensive and waste-producing strategy.
Interestingly, phenolic units are the main constituents of lignin, the largest renewable biomass source of aromatic building blocks in nature.7 Structurally diverse phenols, generally containing methoxy groups, are obtainable via depolymerization processes from this natural and renewable raw material, increasing the interest in the development of efficient synthetic strategies for the transformation of lignin-derived phenols (i.e. guaiacols) (Fig. 1).8,9
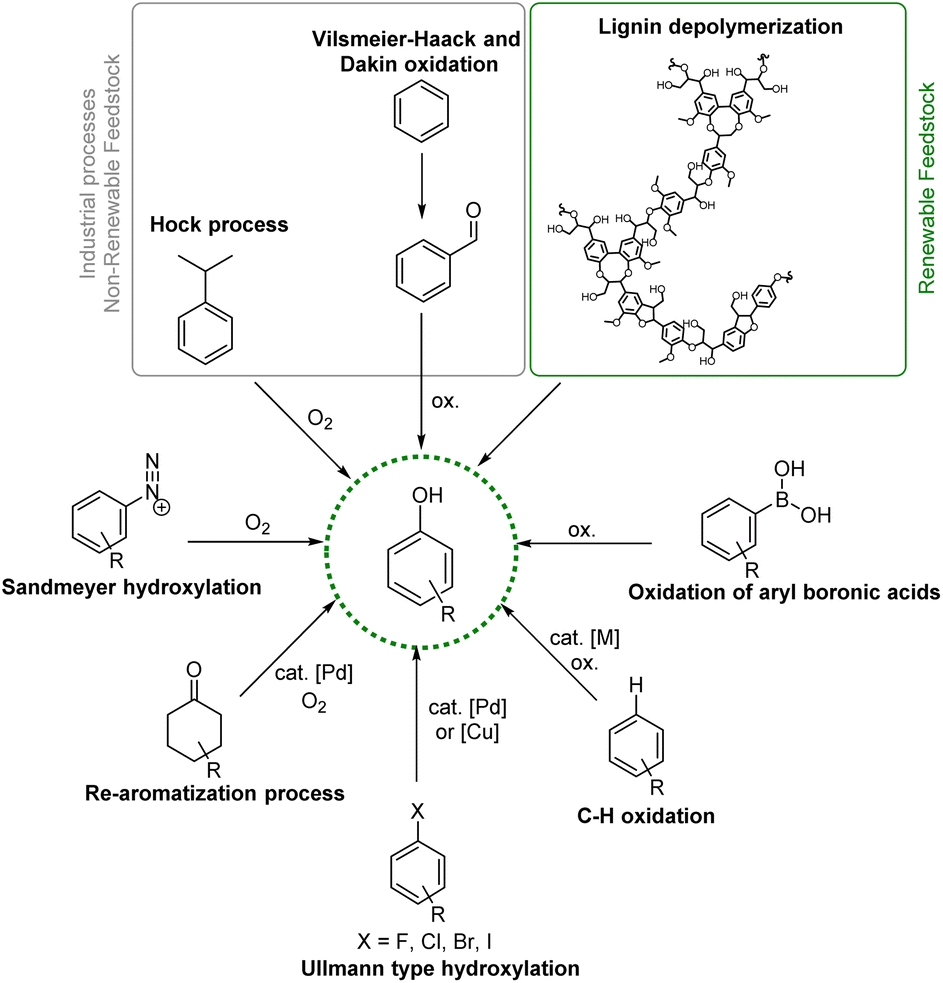 | ||
| Fig. 1 Synthetic pathway for phenol production. | ||
Given the wide range of potential applications of phenols in the chemical value chain, the development of new and useful synthetic methodologies aimed at increasing the molecular complexity of phenolic structural moieties is an interesting and useful research arena (Fig. 1). Highly functionalized phenols required for the synthesis of fine chemicals are commonly prepared via transition metal-promoted Csp2–O bond formation from a wide assortment of starting materials, such as aryl halides via Pd- or Cu-catalysed Ullmann type hydroxylation;10,11 arene diazonium salts and derivatives via Sandmeyer hydroxylation;12,13 and aryl boronic acids via direct oxidation or the Csp2–H activation/borylation/oxidation of arenes.14–16 While these methodologies are well-known and robust, their applicability to complex molecular architecture is constrained by the generally harsh reaction conditions needed. Moreover, installing the halide or boryl group with high site-selectivity is challenging or not currently possible on complex molecules therefore requiring specific substituents or substitution patterns, making more complicated and less atom-economical the overall approach.
Siegel and Ritter independently reported a challenging strategy for the synthesis of phenols via direct Csp2–H oxidation of arenes.17–19 Furthermore, phenols can also be synthesized from non-aromatic precursors such as cyclohexanone via an oxidative aromatization strategy.20
Compared to the use of aryl halides, boron compounds, or arene diazonium salts, starting from simple unprotected phenols is highly advantageous considering their lower price and their origin from more sustainable and renewable aromatic feedstocks.
Among the key techniques for obtaining highly functionalized phenols, direct Csp2–H functionalization is certainly one of the most interesting and powerful as it enables the efficient construction of novel Csp2–C bonds, in a step- and atom-economical manner. This method is even more interesting if applied to simple unfunctionalized phenols, offering the advantage of preserving the phenolic functional group and avoiding additional protection/deprotection/derivatization steps. However, the control of chemo- and regioselectivity of phenol C–H functionalization is still a challenge for three main reasons:
(1) The reactivity can be determined by the nucleophilicity of the OH– functionality instead of Csp2–H, favouring the functionalization of the hydroxy group, via substitution or cross-coupling reactions.21,22
(2) The site difference in the nucleophilic reactivity among the ortho- and para-positions of phenol leads to the formation of a mixture of two products when reacting with various electrophiles.23–26 Generally, a slight preference is observed for the substitution at the ortho-position thanks to the capability of the hydroxy group to participate in the formation of hydrogen bonds and/or metal complexes.
(3) Phenols are sensitive to oxidants: they can undergo oxidative decomposition.27
For these reasons, it's often necessary to employ directing groups (DGs) and generally, the installation of acyl, silyl, and pyridyl groups on the hydroxy functionality is an efficient method to realize highly ortho-selective Csp2–H bond functionalizations.28,29 However the need for additional steps to both install and remove a DG limits the practical application of this approach, undermining the atom- and step-economy of the overall strategy. For these reasons, it is desirable to develop methodologies relying on the use of free phenols exploiting the OH- group as DG itself.30,31
In this review article, we present and summarize the wide plethora of methodologies reported in the period 2019–2023 on the Csp2–H functionalization of simple unprotected OH-phenols that allow the direct diversification of this valuable molecular scaffold with the formation of novel Csp2–C. We leave the reader to refer to the previous excellent report published by Lumb et al.32
In this review, each section has been organized according to the type of Csp2–C bonds formed during the transformation. Accordingly, we anticipate that in the timeframe selected, all the contributions available in the literature mostly deal with the following reactions: alkylation, aminomethylation, allylic alkylation, alkenylation, arylation, carboxylation, and spiroannulation (Fig. 2). The alkynylation reaction does not appear in the list as in recent years this reaction has not been used for simple functionalization of phenols but for the one-pot synthesis of O-heterocyclic compounds. Similarly, alkenylation protocols utilized for one-pot approaches towards heterocycles have not been included in the present review.
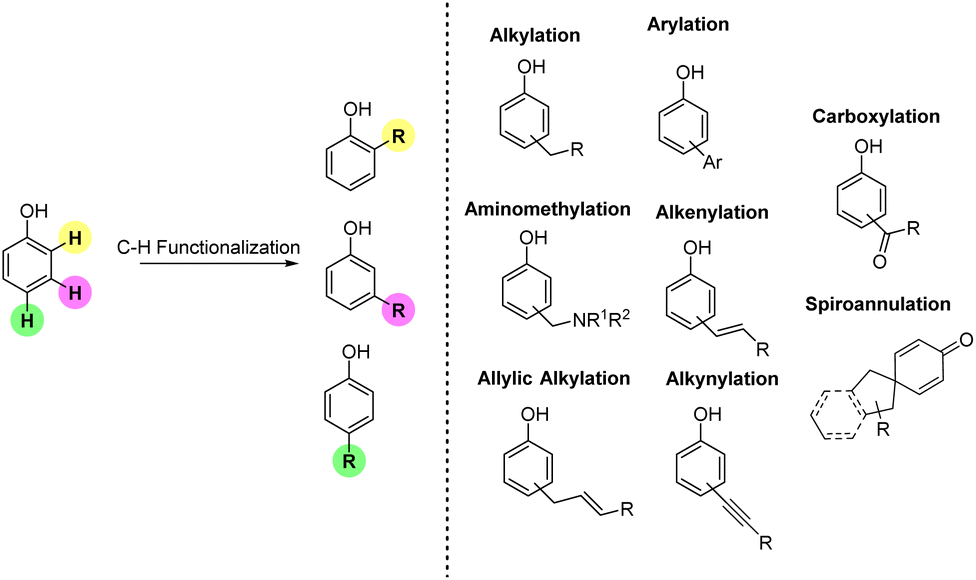 | ||
| Fig. 2 The potential of Csp2–H functionalization of phenols. | ||
Among all the methodologies reported in the literature, a central role is certainly played by Csp2–H functionalization catalysed using transition-metals, acids/bases, and light. Also several examples deal with non-catalytic protocols utilizing acid/base, metal-free, photo-promoted and electro-assisted protocols.
Synthetic methodologies starting from derivatized phenols such as aryl halides, arene diazonium salts, benzyne intermediates, cyclohexanone, and aryl boronic compounds to obtain highly functionalized phenols have not been considered in this review.
1.1. Alkylation
Alkylated phenol is an important industrial target, as many polymers and polymer additives are based on this simple molecular scaffold. Furthermore, the alkylated phenolic structural motif is commonly found in molecules endowed with several intriguing properties such as the active pharmaceutical ingredients (APIs) propofol, olodaterol, cannabidiol and mesoprocol; agrochemicals such as dinoterb, dinoseb and DNOC; and the plethora of catecholamines such as dopamine, tyrosine, epinephrine, noradrenaline and levodopa (Fig. 3).4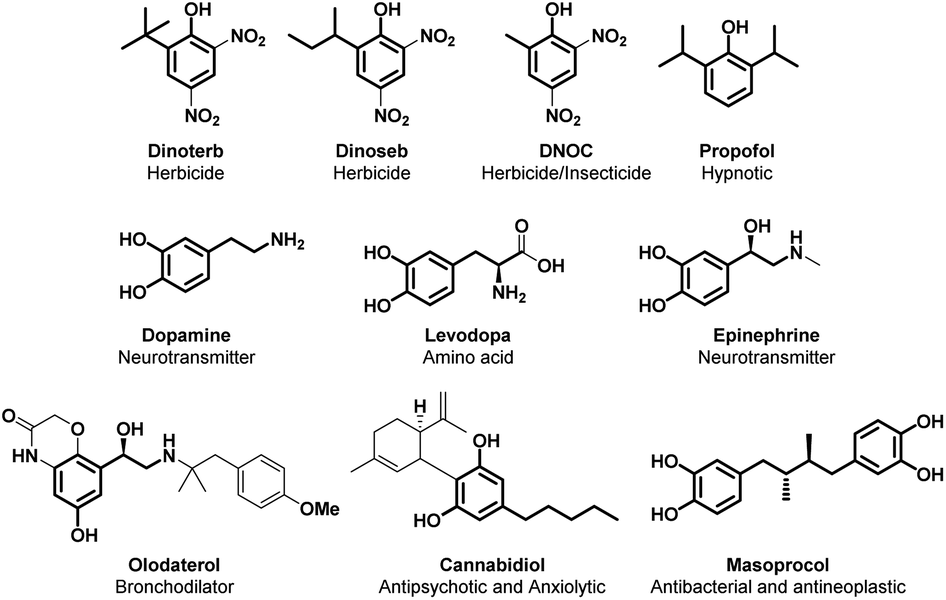 | ||
| Fig. 3 Alkylated phenol structural moieties in API and agrochemical compounds. | ||
The alkylation of phenolic compounds is mainly referred to as Friedel–Crafts electrophilic substitution.33,34 Plenty of examples that describe this transformation are reported using several electrophiles (e.g. alkenes, ketones, acetals, unsaturated aldehydes, imines, α-aryl-α-diazoesters, N-tosylaldimines, etc.) with different catalysts (mostly Lewis acids).35–37 This strategy, however, yields a mixture of ortho- and para-substituted compounds and it often leads to over-alkylated products.
To better promote the regioselectivity, a DG on the phenolic hydroxy group is often installed, at the expense of atom- and step-economy.38–41 For this reason, the alkylation of the OH– group followed by a Claisen rearrangement has been considered as an alternative to the direct electrophilic substitution.42,43 Another variant to synthesize alkylated phenols is oxidative coupling reactions via the single-electron-transfer (SET) mechanism.44,45
Nowadays, transition-metal-catalysed Csp2–H alkylation reactions are gaining a great deal of attention and several researchers have contributed in this direction.46,47
In this section, we describe the most recent strategies for obtaining alkylated phenolic compounds, classifying them into catalysed or non-catalysed protocols. Regarding catalysed protocols, we have organized them into three different classes depending on the activation species: transition metals, acids/bases, and light.
1.1.1.1 Metal catalysis. The use of a transition-metal catalyst to promote Csp2–H alkylation is a generally efficient and chemo-selective approach for the late-stage functionalization of organic molecules.48 Representatively, in 2012 Yi et al.49 reported the use of a Ru-based catalyst to access alkylated and alkenylated phenols. They also accessed oxacyclic derivatives via dehydrogenative coupling. Moreover, in 2009, the Mo/o-chloranil catalyst system was used by Yamamoto and Itonaga to access chromans.50 Takai et al. developed a regioselective Re-catalysed alkylation using terminal alkenes as the alkylating agents, that proceeded with the sole formation of the branched product.51–53
A relevant procedure for the alkylation of phenols using Pd-catalysis was reported by Li, Zeng and coworkers in 2021. They investigated a process in which a primary alcohol is the alkylating agent and Pd/C is the heterogeneous catalyst. The protocol features a large substrate scope with various phenols and primary alcohols. In general, an electron-withdrawing group (EWG) on the phenol, leads to poor or no conversion, while the presence of an electron-donating group (EDG) allows the corresponding alkylated phenols in high yields. The use of primary linear alcohols gave the highest yields. This process is highly chemo-selective: in a competitive experiment using a substrate with both a primary and a secondary hydroxyl functionality, only the primary hydroxyl group reacts. The mechanism of this process is peculiar since it leads to an excellent selectivity towards the ortho-alkylated product. Validated by mechanistic studies, a de-aromatization/re-aromatization pathway has been proposed. They assumed that the primary alcohol is converted to the corresponding aldehyde thanks to the action of the Pd-catalyst while the originated HPd(II)H species can selectively hydrogenate the phenol derivative to the corresponding cyclohexanone. At this stage, the role of tBuOLi is to catalyse the aldol-type reaction followed by dehydration that produces the α,β-unsaturated ketone. The latter undergoes oxidative aromatization to produce the alkylated phenol while regenerating HPd(II)H (Scheme 1).54
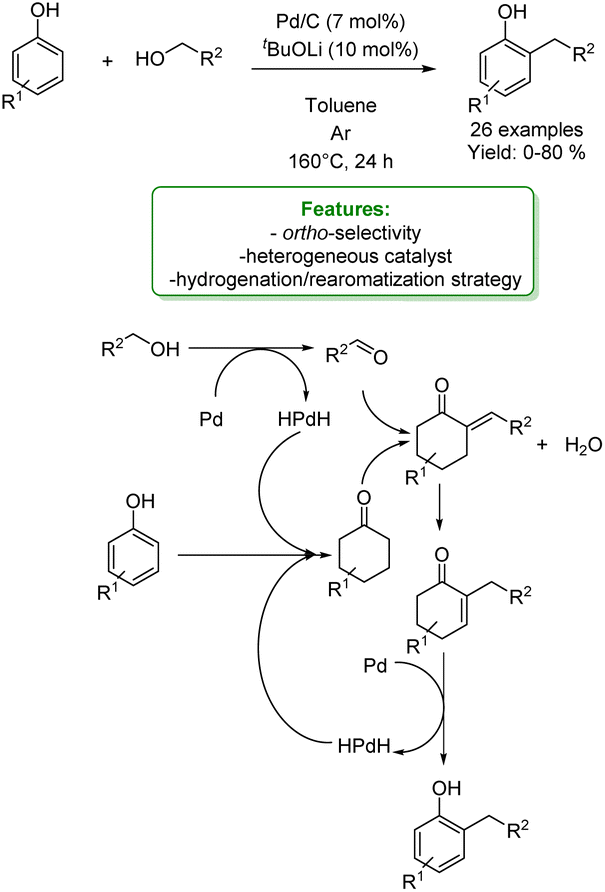 | ||
| Scheme 1 Pd-catalysed alkylation of phenols with alcohols.54 | ||
R. G. Iafe et al. developed a gold-catalyzed Friedel–Crafts-like benzylation of non-activated benzylic alcohols, resulting in the production of 1,1-diarylalkanes up to excellent yields (98%) under mild reaction conditions. Isolated yields using non-substituted phenol indicated a preference for the para-position. Additionally, reactions with disubstituted phenols favoured the ortho-position of the more electron-releasing substituents (Scheme 2).55
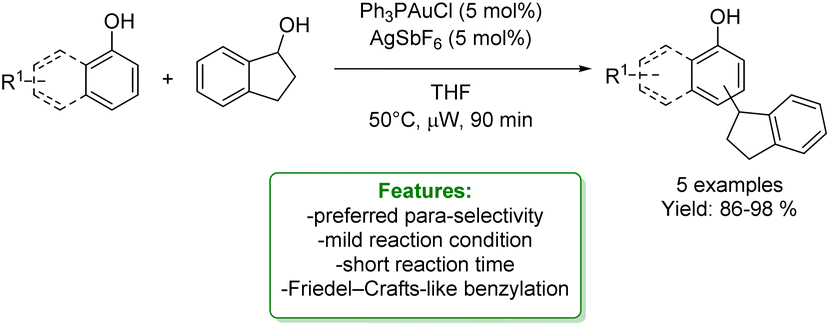 | ||
| Scheme 2 Au-catalysed alkylation of phenols with benzylic alcohols.55 | ||
A. Kumar et al. developed an ortho-alkylation of phenols catalyzed by a pincer–ruthenium complex. The reaction of 1-phenyl ethanol with phenol, in the presence of molecular oxygen, leads to both cross-etherification and ortho-alkylation. The corresponding reactions of either β-naphthol with 1-phenyl ethanol derivatives or phenol with diphenylmethanol were selective toward the formation of ortho-alkylated products. The reaction involves a radical pathway with the formation of a hydroxyl radical from 1-phenyl ethanol, followed by the formation of the secondary radical. Evidence for the formation of the secondary radical was obtained by trapping it with radicals generated from phenols, exploiting their selective ortho-alkylation. In this context, resonance-stabilized phenoxy radicals show significant potential as coupling counterparts, potentially leading to the creation of cross-etherification products (Scheme 3).56
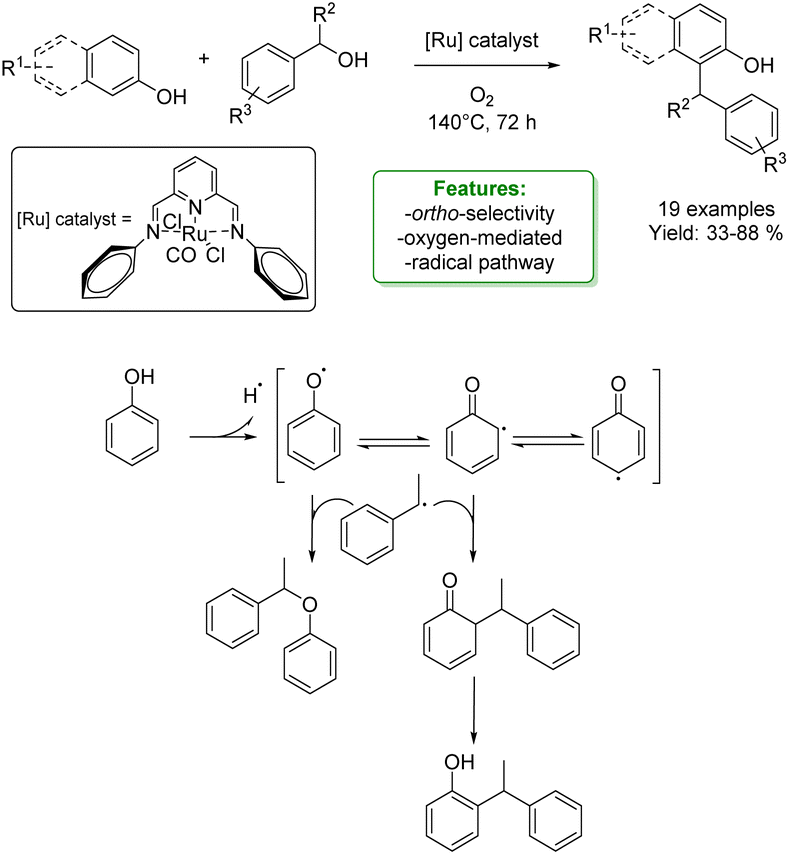 | ||
| Scheme 3 Ru-catalysed alkylation of phenols with benzylic alcohols.56 | ||
Yi, Baik et al. reported a cationic Ru hydride complex [(C6H6)-(PCy3)(CO)RuH]+BF4− to perform a dehydrative C–H coupling between phenols and aldehydes. Changing the type of aldehyde used, different phenol-derived products were obtained. Ortho-alkylated phenols were synthesized with non-branched alkylating agents, in conjunction with an H-source (2-propanol), resulting in isolated yields of up to 90%.
The utilization of α-substituted aldehydes, in combination with CO addition, led to the synthesis of benzofurans. Additionally, the coupling of phenols with salicylaldehyde and the use of 1,4-dioxane as the solvent facilitated the formation of xanthanes. By combining experimental studies (deuterium labelling study, Hammett study, deuterium and carbon kinetic isotope effects – KIE, and spectroscopic detection of catalytically relevant species) with DFT computational results, a mechanism was elucidated.
After an arene exchange between the Ru-catalyst and the phenol, the Ru–phenoxo complex is formed, followed by the subsequent substitution of the aldehyde. The C–C bond formation occurs in a two-step process: the first step involves an electrophilic addition of the aldehyde to the ortho-arene carbon of the coordinated phenoxo ligand. The second step promotes a stepwise proton and hydride transfer with the phenoxydienone tautomerization, resulting in the formation of the hydroxy-coordinated Ru-aryloxo species (Scheme 4).57
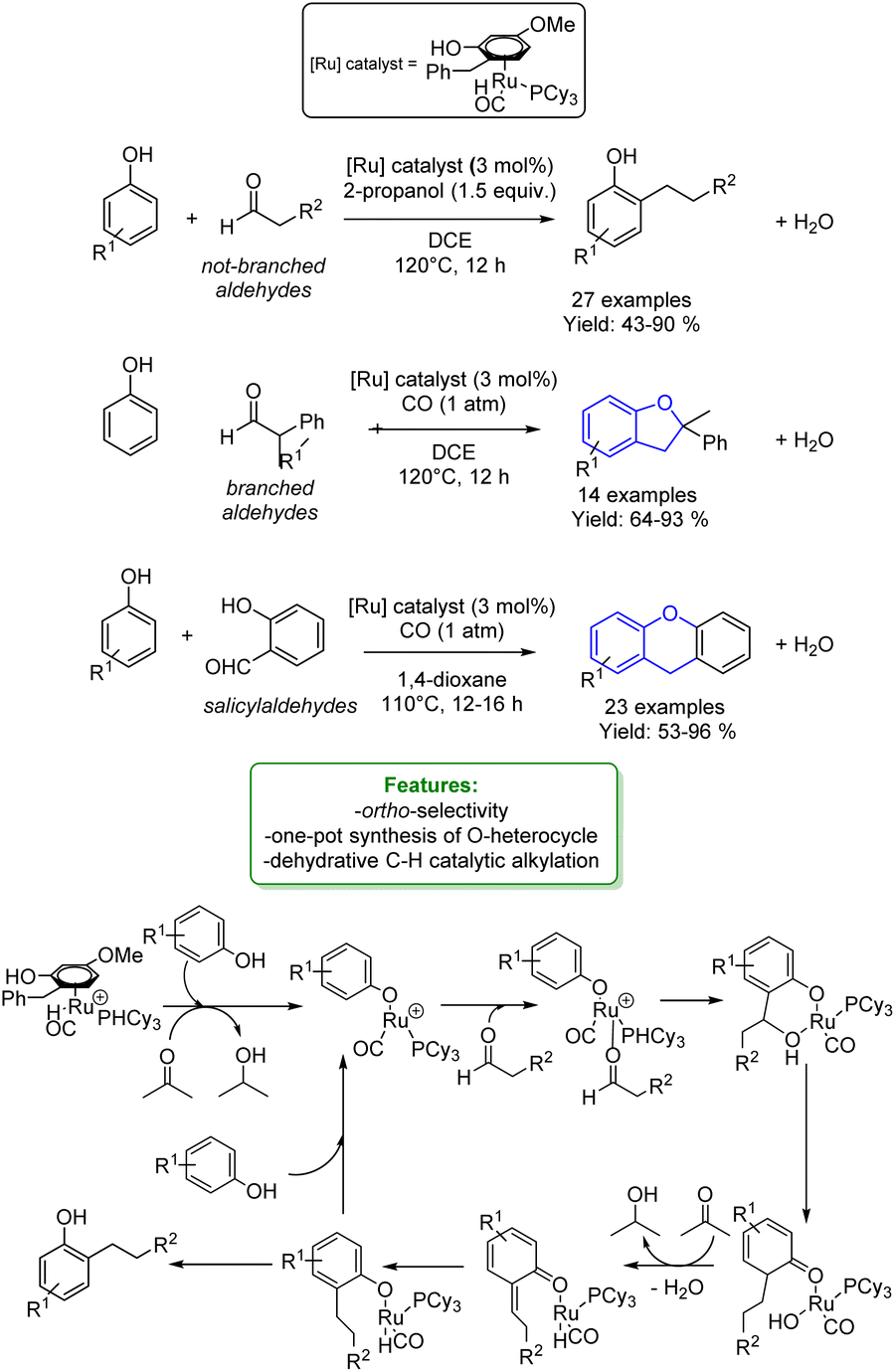 | ||
| Scheme 4 Ru-catalysed alkylation of phenols with aldehydes.57 | ||
L. Gong et al. developed a strategy utilizing iron(III) halides under visible light conditions for the direct and selective coupling of phenols with benzylic and allylic derivatives. The reaction was conducted under mild conditions with air serving as the oxidant, and it proved to be effective for a diverse array of substituents. A wide range of cross-coupling products with high yields and notable chemo- and site-selectivity could be prepared. The authors reported the synthesis of various bioactive molecules; notably, nafenopin, a potent hypolipidemic drug, and a beclorbrate derivative were synthesized starting from phenol. Mechanistic studies revealed the multifunctional role of the iron halide, which responds to visible light, initiates C-centered radicals, induces single-electron oxidation to carbocations, and engages in a subsequent Friedel–Crafts-type process (Scheme 5).58
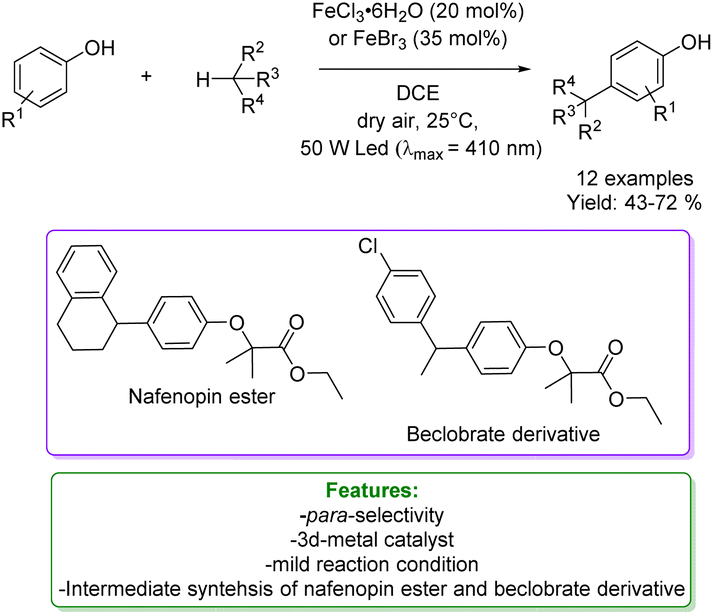 | ||
| Scheme 5 Fe-mediated visible light coupling of phenols with aliphatic hydrocarbons.58 | ||
Diazo-compounds have emerged as effective phenolic alkylating agents through transition-metal catalysed carbene transfer reactions. They have become a powerful synthetic tool for accessing regio- and chemo-selective aromatic compounds via Csp2–H bond functionalization, ensuring step- and atom-economy.59,60
In 2014, Shi and Zhang independently developed a site-selective para-C–H bond functionalization of phenols with α-aryl-α-diazoesters via Au-catalysis.61,62 In a subsequent contribution in 2016, Zhang and coworkers identified the conditions to change the regio-selectivity for obtaining a highly selective ortho-Csp2–H process catalysed by (C6F5)3B.36 In 2019, the same group developed a new alkylation of naphthol with α-aryl-α-diazoesters to access bioactive molecules, and ligands via a regio-divergent process depending on the Au-catalyst's ligands. Using the JohnPhos ligand along with AgOTf creates crowded chloride-bridged di-gold complexes. This inhibits the coordination of the directing group with the metal, preventing the intermolecular attacks of the carbene and improving the preference for para-positions. On the other hand, to obtain the ortho-alkylated product, a carbene ligand that promotes coordination between the hydroxyl group and the metal is employed (NHC–gold complex). The method exhibits a wide substrate scope, both for EWG and EDG substituted naphthols (an overall of 43 examples with 52–95% yields) and propranolol analogues among them. Finally, late-stage functionalization of the alkylated product has been conducted (Scheme 6).63
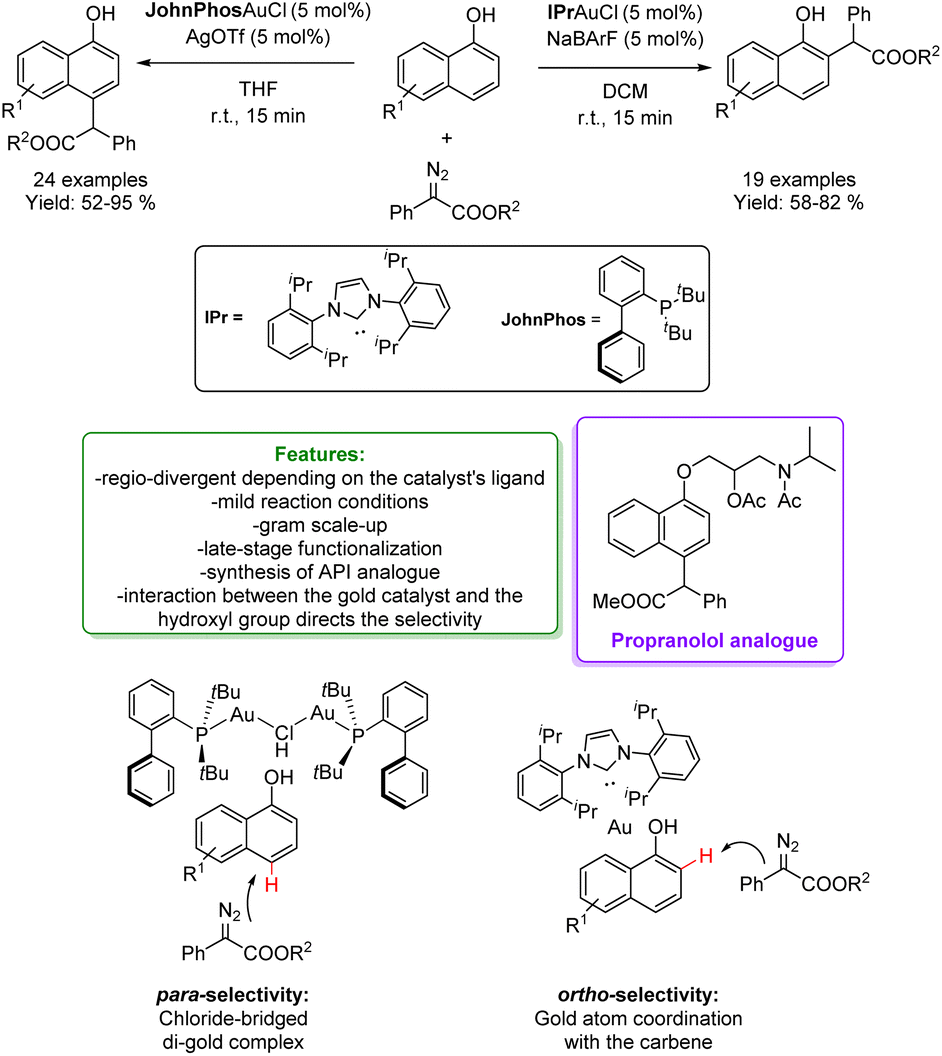 | ||
| Scheme 6 Regio-divergent alkylation of phenols and naphthol with α-aryl-α-diazoesters catalysed by a Au-catalyst.63 | ||
In 2020, Zhang and Liu reported a Cu-catalysed reaction between naphthols and α-aryl-α-diazoesters, under very mild reaction conditions. To elucidate the transformation, two distinct mechanisms have been proposed: one involving a Cu-carbene process and the other one a Lewis acid (CuCl2) mediated pathway. The former mechanism is based on the diazo decomposition to form the Cu-carbene, which subsequently undergoes electrophilic addition at the ortho-position of the phenol. On the other hand, when CuCl2 acts as a Lewis acid, a carbocation is generated from the α-aryl-α-diazoesters. This carbocation then coordinates with the phenol's hydroxyl group, facilitating the electrophilic addition.64
In 2023, to clarify the reaction mechanism, the authors reported a study combining DFT calculations and experiments. They revealed that the ortho-selective products are obtained from the C–H insertion of naphthols using monometallic carbenes. It is proposed that the H-bond interactions between the Cu carbenes and the substrates play an essential role in stabilizing the site-selectivity-determining TSs in all cases, resulting in a lower energy barrier, and generating the experimentally observed ortho-selective products (Scheme 7).65
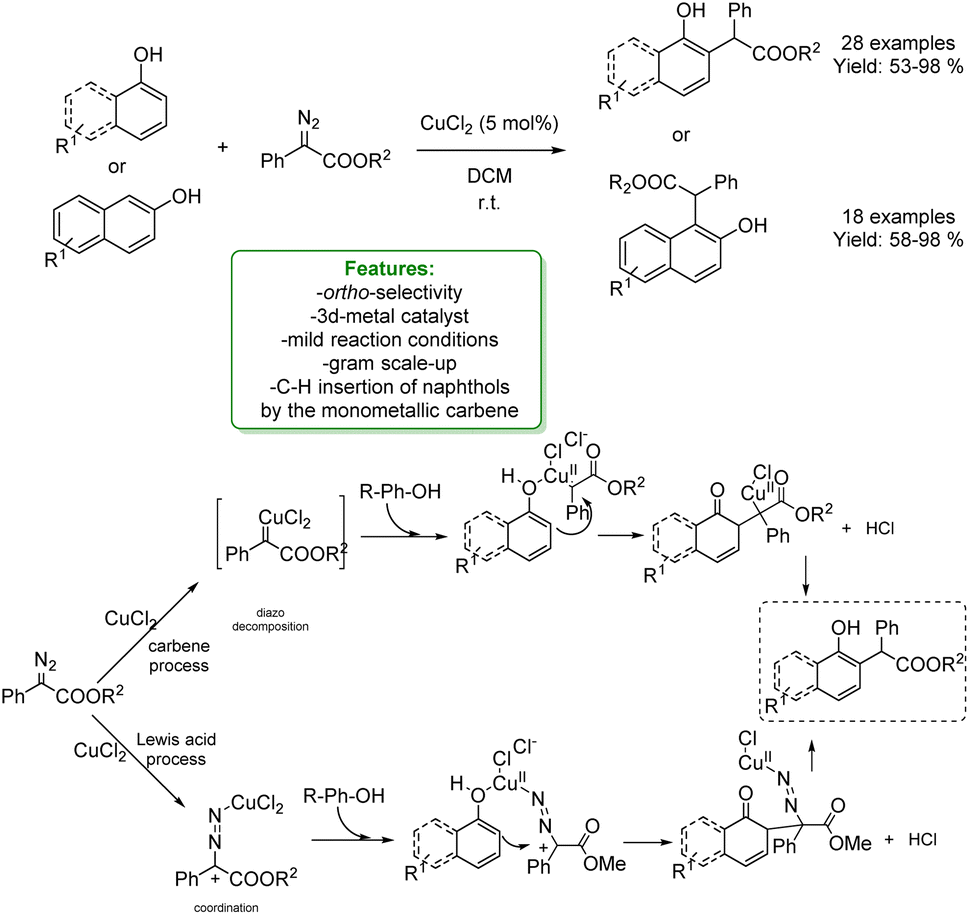 | ||
| Scheme 7 Ortho-alkylation of phenols, 1-naphthols and 2-naphthols with α-aryl-α-diazoesters catalysed by a Cu-catalyst.64 | ||
In 2021 the same authors reported the ortho-alkylation of 1-naphthol with different α-aryl-α-diazoesters catalysed by an Fe-catalyst. A porphyrin ring has been employed as the Fe-ligand to enhance the control over regioselectivity. A broad array of ortho-alkylated 1-naphthols, featuring both the EDG and EWG on the diazo compound, were synthesized in moderate to good yields ranging from 43% to 78%. The authors proposed a Lewis acid pathway, although the Fe-carbene pathway could not be ruled out completely.
The proposed mechanism starts from the coordination between the Fe-porphyrin and the phenolic OH– which leads to the formation of Fe-phenolate. Fe-species can coordinate with the diazo-compound through the nitrogen atom acting as a Lewis acid. Then, the electrophilic substitution at the ortho-position occurs. In the end, the final product is obtained by tandem aromatization, denitrogenation and protonation (Scheme 8).66
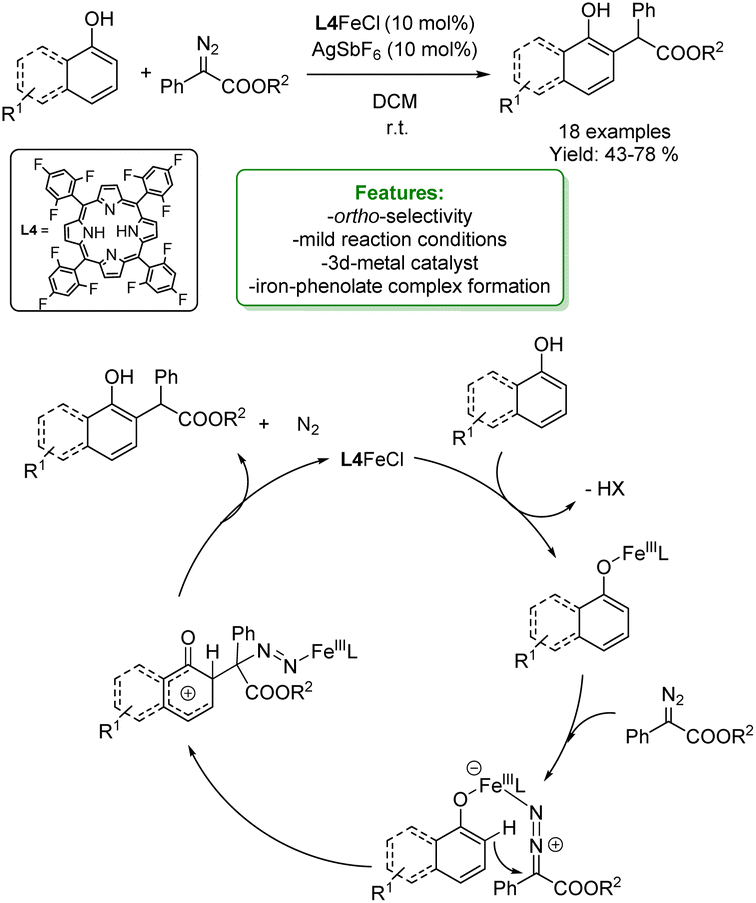 | ||
| Scheme 8 Ortho-alkylation of 1-naphthol and phenols with α-aryl-α-diazoesters catalysed by an Fe-catalyst.66 | ||
Another interesting example of alkylation of phenols with diazo compounds is reported by A. Vidal-Ferran et al. They developed a phosphite-based Au(I)catalytic system selective for the para-alkylation, avoiding the O–H functionalization. The catalytic performance and selectivity of the supramolecularly regulated catalysts have been rationalised using DFT calculations. Regulation of the steric congestion around the catalytic Au(I) centre via ion–dipole interactions with an external regulation agent (i.e., CsBF4 and RbBF4) led to an enhancement of both the activity and selectivity of the reaction favouring the insertion of the carbene into the para-Csp2–H bond. This new approach enabled the derivatisation of an array of substituted phenols and naphthols, and the preparation of an advanced synthetic intermediate of the anticancer agent tamoxifen under mild reaction conditions and short reaction time (Scheme 9).67
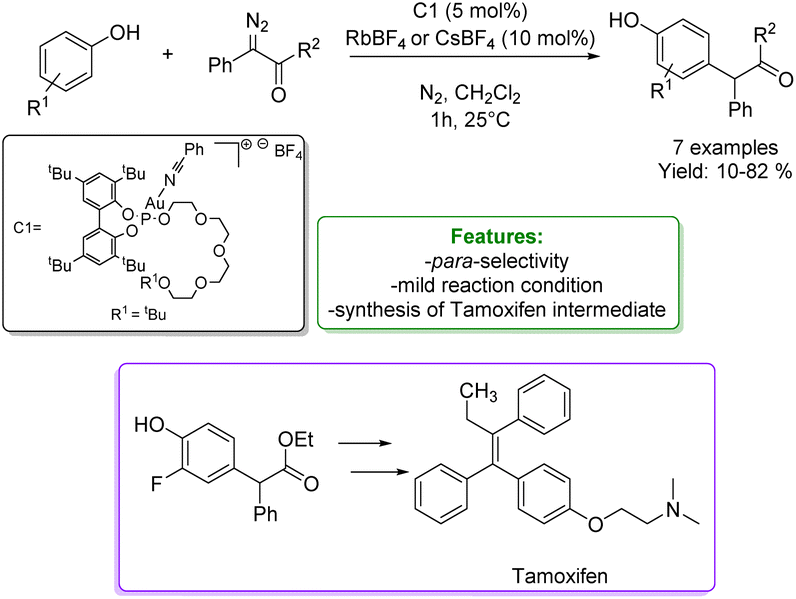 | ||
| Scheme 9 Phosphite-based Au(I)catalytic systems applied in the para-alkylation of phenols with diazo compounds.67 | ||
M. C. Kozlowski reported a Cu(OTf)2 catalysed oxidative arylation of a tertiary carbon-containing substrate. The optimized reaction conditions were applicable for a wide range of arenes, including phenols obtaining moderate to good isolated yield. The aryl malononitriles regioselectively coupled at the para-position of the phenols. When the para-position of the phenol was blocked, coupling occurred at the ortho-position and was accompanied by tandem coupling/cyclization to form benzofuran-2-one derivatives. Mechanistic studies support the formation of a cationic intermediate via a two-electron oxidation (Scheme 10).68
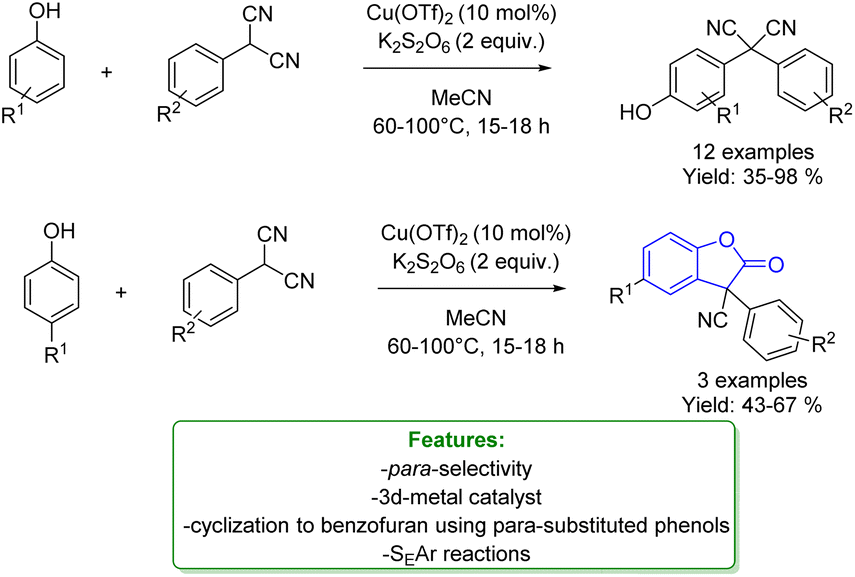 | ||
| Scheme 10 Cu-catalysed alkylation of phenols with aryl malononitriles.68 | ||
Z. Ke et al. presented the concept of hypervalent chalcogenonium⋯π bonding catalysis utilizing 1,2-oxaselenolium salts. The catalyst was employed in the intermolecular hydro-functionalization of styrenes and phenols. In reactions involving p-cresol as the substrate, both electron-rich and electron-deficient styrenes exhibited notable reactivity, yielding their respective products with favorable yields. Electron-donating substituents on phenols facilitated successful reactions with styrene. With electron-deficient substituents the reaction did not work, indicative of a catalytic effect rooted in the Lewis acidity. Notably, 2-naphthols yielded an ortho-selective product, and 2,4-disubstituted phenols proved to be excellent substrates, furnishing the desired products smoothly.
To elucidate the mechanism of the selenium ion's activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in styrene and the cationic chalcogenonium⋯π bonding interaction mode involving the selenium ion, density functional theory (DFT) calculations were performed. Then, the authors proposed a mechanistic pathway for the formation of the final products. The trisubstituted selenonium catalyst reacts with the olefin to form the seleniranium ion-like intermediate. This intermediate is subject to attack by the OH–, leading to the formation of an intermediate that readily releases a proton. Subsequent proton transfer events culminate in the production of the final product and the restoration of the selenonium species (Scheme 11).69
C double bond in styrene and the cationic chalcogenonium⋯π bonding interaction mode involving the selenium ion, density functional theory (DFT) calculations were performed. Then, the authors proposed a mechanistic pathway for the formation of the final products. The trisubstituted selenonium catalyst reacts with the olefin to form the seleniranium ion-like intermediate. This intermediate is subject to attack by the OH–, leading to the formation of an intermediate that readily releases a proton. Subsequent proton transfer events culminate in the production of the final product and the restoration of the selenonium species (Scheme 11).69
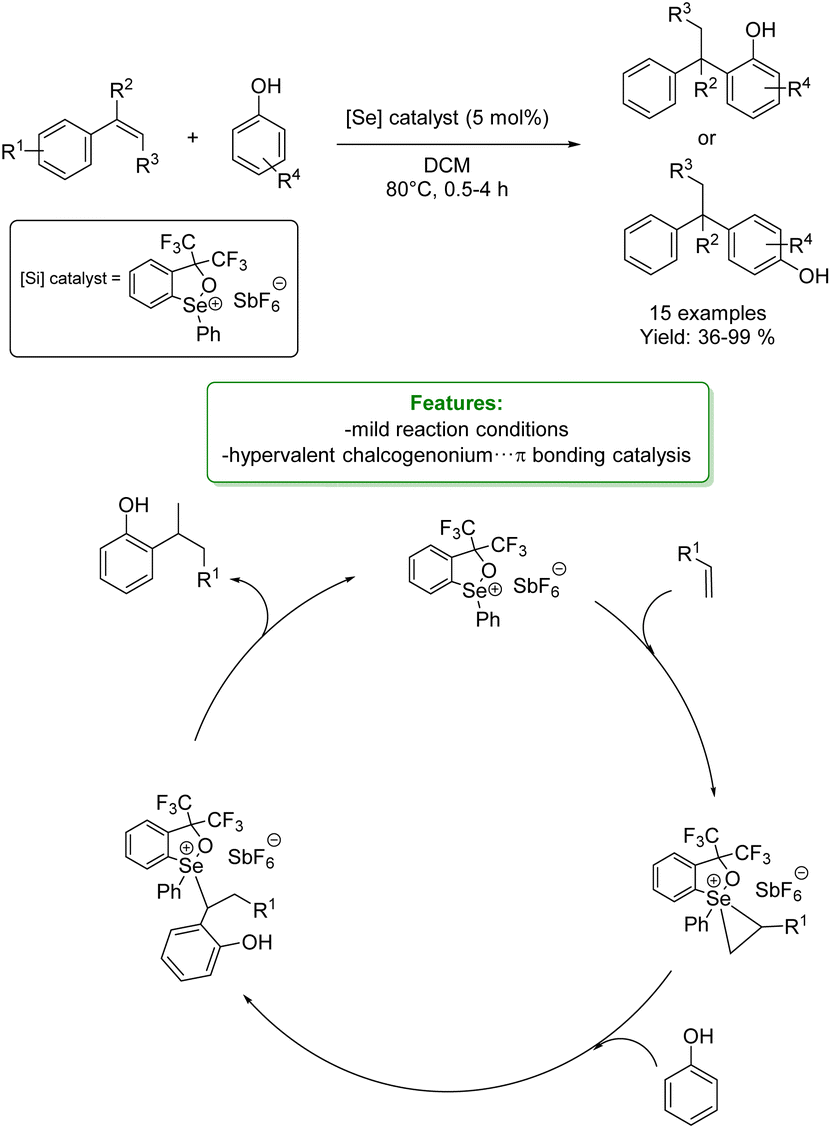 | ||
| Scheme 11 Se-catalysed alkylation of phenols with styrenes.69 | ||
H. Lu et al. reported a recyclable bismuth complex-catalysed addition of various nucleophiles, including naphthols, to para-quinone methides. The bismuth complex was efficiently recycled multiple times. The catalyst was applied in the reaction of p-QMs bearing both electron-donating and electron-deficient aromatic moieties with naphthalen-2-ol, yielding triarylmethane derivatives in good to excellent yields (up to 98%). To demonstrate the robustness of the protocol, a gram-scale reaction was performed (Scheme 12).70
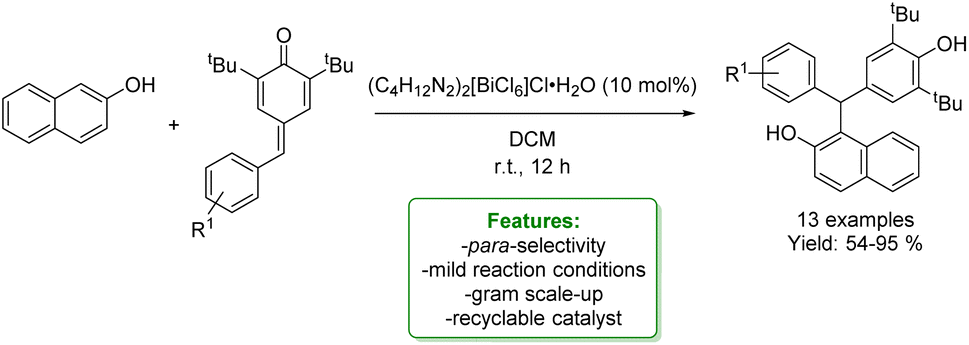 | ||
| Scheme 12 Recyclable Bi-catalysed addition to para-quinone methides.70 | ||
Regarding the previous example of reactivity, more recently, W.-Y. Wong et al. developed a silver-catalysed regioselective 1,6-hydroarylation of para-quinone methides (p-QMs) with phenols and naphthols. The mild reaction conditions were applied to a broad range of phenols and para-quinone methides, yielding the corresponding 1,6-hydroarylation products with moderate to good yields. The reaction demonstrated good regioselectivity at the para-position of the phenols. A large-scale reaction with phenol was performed under the optimized conditions. Furthermore, the optimized protocols were applied in the late-stage modification of ethinylestradiol and 1,3,5(10)-estratrien-3-ol-17-one. The authors proposed a plausible mechanism in which AgBF4 initially attacks the carbonyl group of p-QMs. In the presence of a nucleophile, the previously formed intermediate could act as an electrophile, and the target product is generated through hydrogen-atom transfer and intramolecular aromatization processes. The silver source is then regenerated and reused in the next catalytic cycle (Scheme 13).71
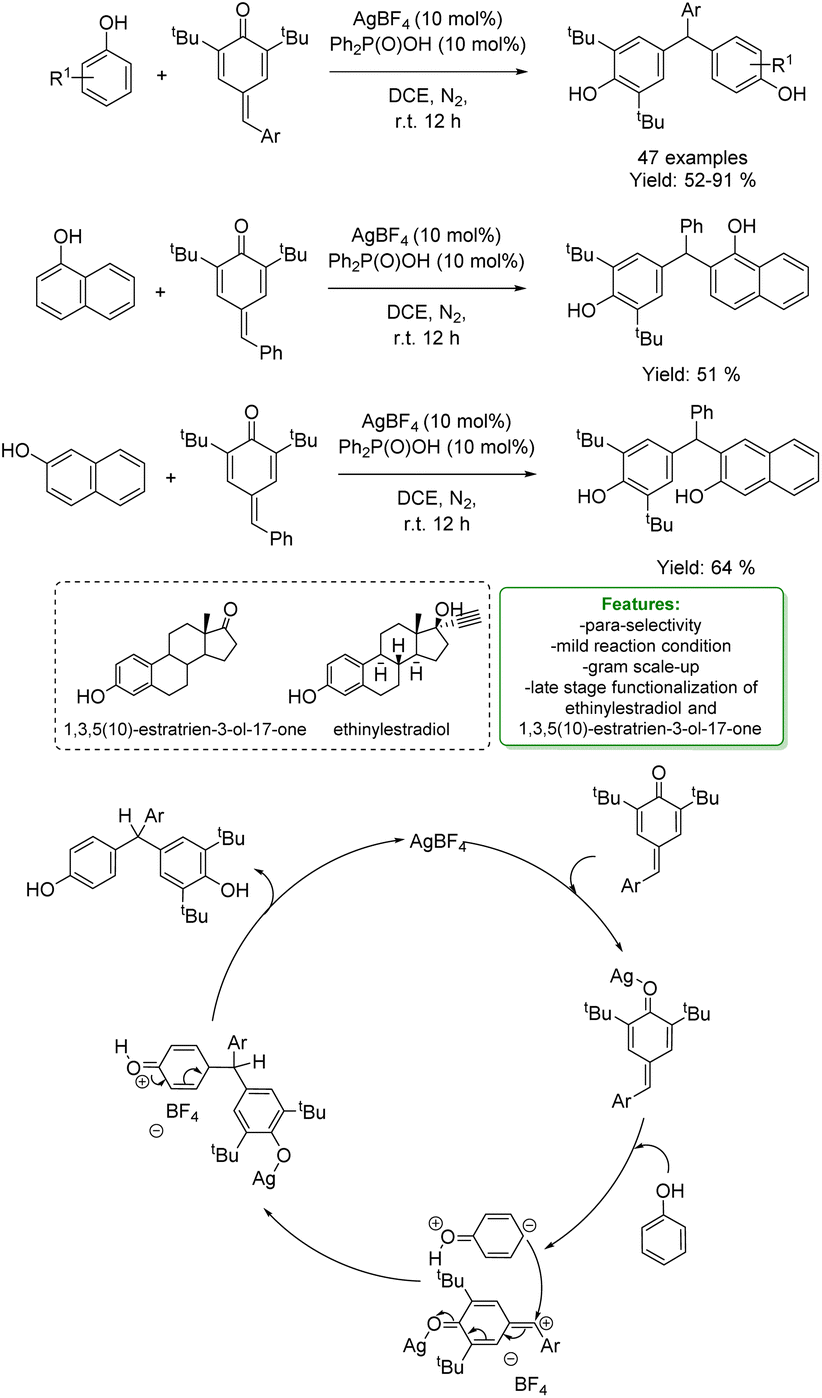 | ||
| Scheme 13 Ag-catalysed para-selective 1,6-hydroarylation of para-quinone methides with phenols and naphthols.71 | ||
J. A. Bull et al. reported the synthesis of 3,3-diarylazetidines by calcium(II)-catalysed Friedel–Crafts alkylation with readily available azetidinols. The reaction condition was compatible with a wide range of phenols. A mixture of para- and ortho-substituted products was afforded. The reaction using o-cresol as a nucleophile could be scaled to 4.0 mmol preserving a high yield (Scheme 14).72
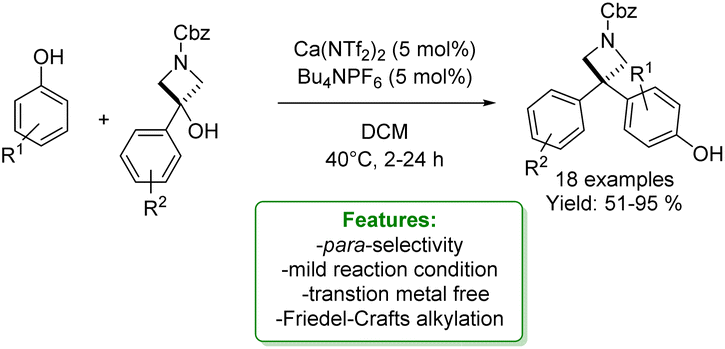 | ||
| Scheme 14 Calcium catalysed synthesis of 3,3-diarylazetidines with azetidinols.72 | ||
J.-J. Feng et al. reported a silver-catalyzed cyclobutylation of phenols and naphthols with bicyclo[1.1.0]butanes, yielding the desired product in moderate to excellent yields. Mechanistic experiments and DFT calculations were performed to gain insights into the reaction mechanism, revealing that the silver catalyst functions as a carbophilic π-acid rather than an oxygenophilic Lewis acid (Scheme 15).73
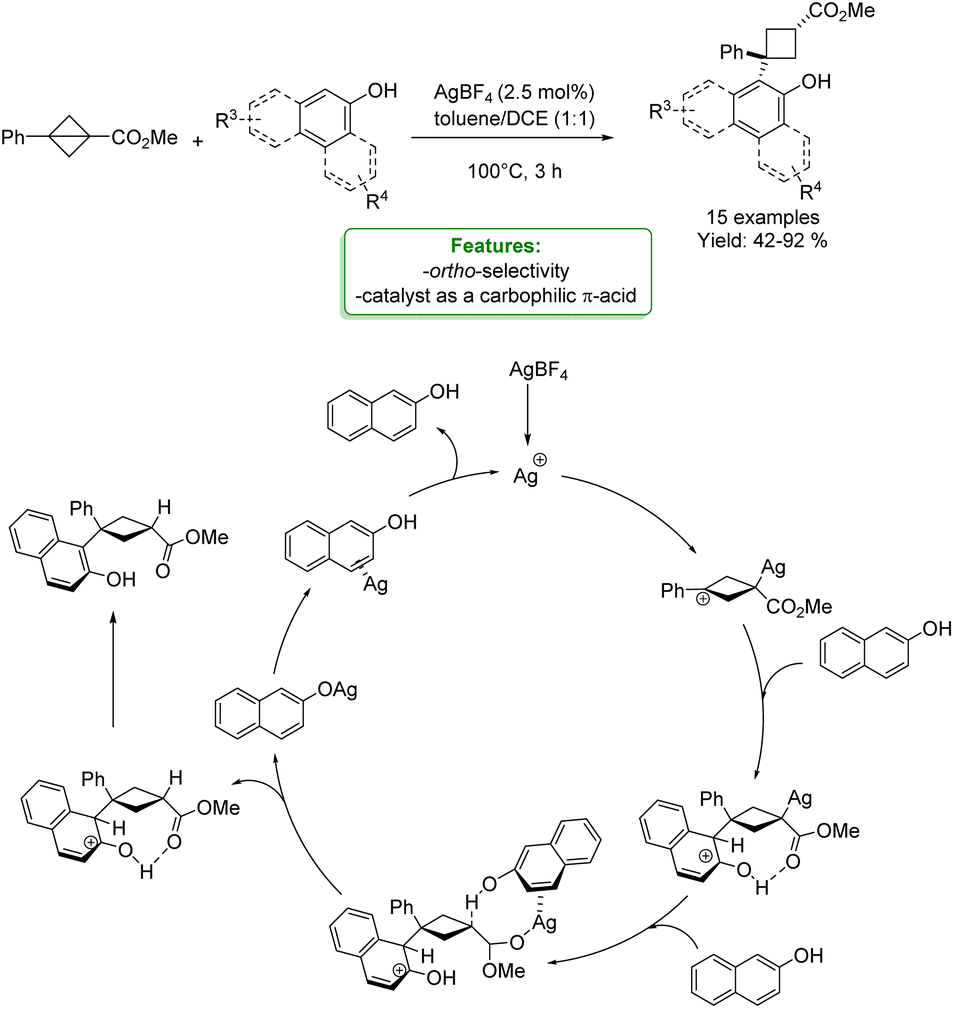 | ||
| Scheme 15 Ag-catalyzed cyclobutylation of phenols and naphthols with bicyclo[1.1.0]butanes.72 | ||
1.1.1.2 Acid/base catalysis. Electrophilic aromatic substitution (Friedel–Crafts alkylation) is the traditional method used to access alkylated phenols using Lewis acids as catalysts.74 However, this method suffers from different drawbacks: beyond the low chemo- and/or regioselectivities described before, it needs harsh reaction conditions. Great efforts have been made to overcome these issues and recently some elegant hydroarylation protocols have been developed expressing excellent Markovnikov or anti-Markovnikov regioselectivity towards the alkene or the alkyne and large functional groups tolerance.75,76
As highlighted in the preceding section, diazo-compounds have emerged as versatile alkylating agents. Here, we provide a series of examples related to the use of diazo-compounds with acid or base catalysts instead of transition metal catalysts.
A. C. B. Burtoloso et al. reported a Friedel–Crafts alkylation of phenols, as nucleophile species, with diazocarbonyl with silica-supported Brønsted acid, H2SO4–SiO2 as a catalyst under mild reaction conditions and short reaction time. The developed protocols enabled to afford a wide range of products in good to excellent yield. The authors provided a plausible reaction mechanism in which the first step is likely to be protonation of diazo ester, followed by N2 expulsion leading to a quinone type intermediate. This intermediate then undergoes the Friedel–Crafts-type reaction with a phenolic-based nucleophile. The reaction could be driven by the re-aromatization of the aryl ring system. Control experiments were conducted to clarify the mechanism (Scheme 16).77
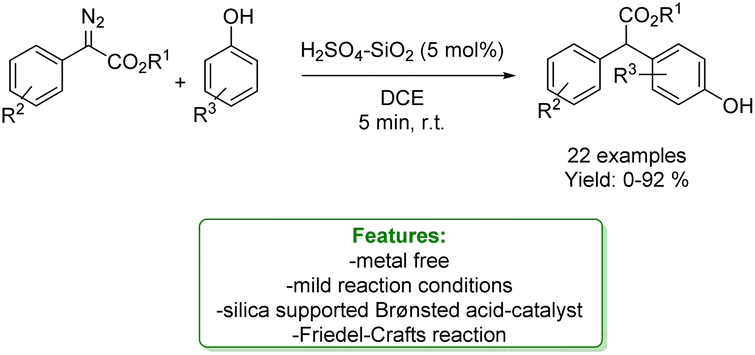 | ||
| Scheme 16 Friedel–Crafts reaction of phenols catalysed by a supported Brønsted acid.77 | ||
An interesting example of phenol alkylation with alcohols under metal-free conditions is reported by C. K. Hazra et al. The authors described a Brookhart acid-catalysed dehydrative coupling, utilizing cyclopropyl carbinol as an electrophile and a wide range of nucleophiles, including phenols. The para-alkylated phenols were obtained in good to excellent yield under mild reaction conditions. Control experiments suggested that the method was a Brønsted acid-catalysed process (Scheme 17).78
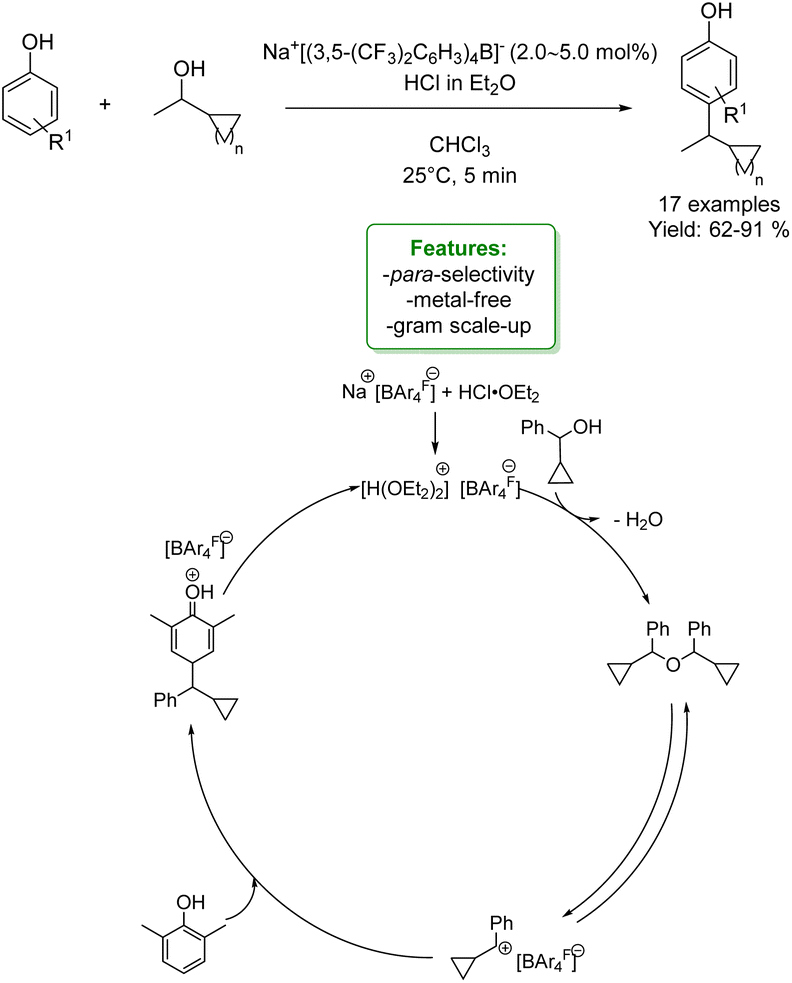 | ||
| Scheme 17 Brookhart acid-catalysed dehydrative coupling of cyclopropyl carbinol and phenols.78 | ||
Patureau et al. performed a hydroarylation of styrene with various phenols catalysed by AgBF4 or HBF4·Et2O, obtaining the branched (Markovnikov) ortho-alkylated product as a major product. Mechanistic studies revealed that the reaction could proceed via three possible transition states depending on the reaction conditions: Beller's (Brønsted acid catalysis), Lewis acid or electron-hole catalysis (if the catalysis plays an oxidant role). The authors concluded that the OH– functional group is essential to address the reactivity and the ortho-selectivity via a concerted protonation/C–C bond-formation pathway (Scheme 18).79
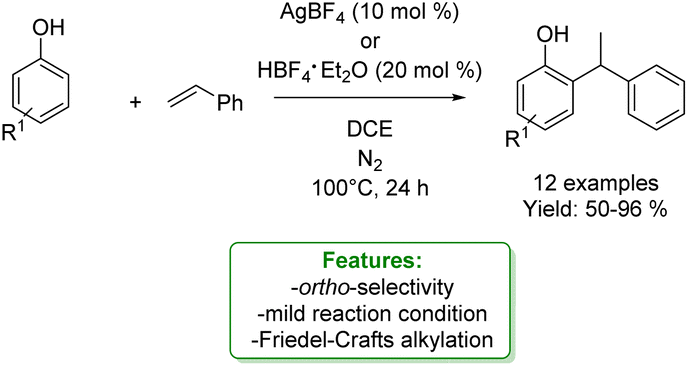 | ||
| Scheme 18 Ortho-selective Friedel–Crafts alkylation of phenols with tertiary styrene.79 | ||
Phosphorus-based acids are common acids used in organic synthesis, and the organic derivatives are commonly applied as an organocatalytic system.80–84 Inorganic acids, such as H3PO3 and H3PO4, have been less used: however, they turned out to be extremely efficient in the alkylation of phenols and naphthols with alkenes in Friedel–Crafts-type alkylation (Scheme 26 and 27). Zhou's research group reported the use of phosphorus in the synthesis of 39 examples of alkylated phenols with isolated yields ranging from 31% to 96%. The reaction of the simplest phenol under the optimized conditions gave a mixture of para- and ortho-alkylated products with an isolated yield of 52% and 32%, respectively. They demonstrated the broad functional group tolerance by applying the process to late-stage functionalization of estrone and raspberry ketone, resulting in yields of 59% and 72%, respectively (Scheme 19).85
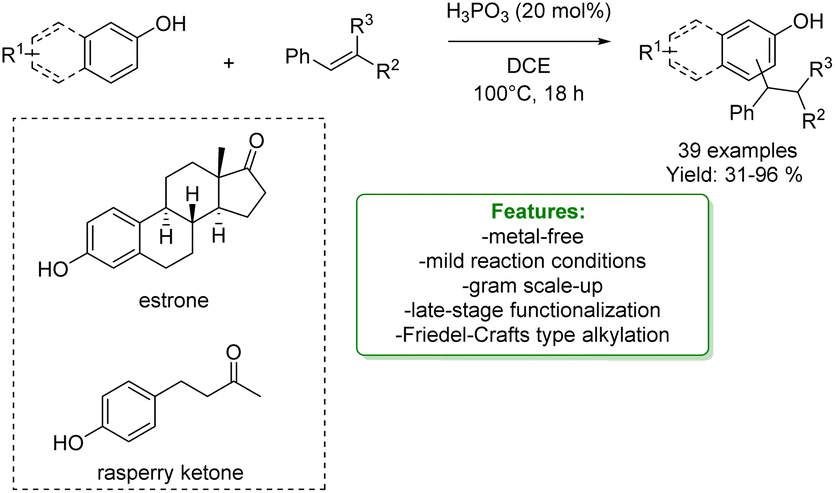 | ||
| Scheme 19 H3PO3-catalysed alkylation of phenols with alkenes.85 | ||
D. Lebœuf et al. reported interesting examples of the hydroarylation of α-(trifluoromethyl)styrenes to access a trifluoromethylated quaternary center, wherein phenols were efficiently utilized as arylating agents. The process was catalysed by the Brønsted acid triflic acid in hexafluoroisopropanol (HFIP). This reaction provided the products in good yields. When simple phenol was applied, a mixture of ortho- and para-products was obtained; however, the para-position was favoured in the process. DFT computations were provided to elucidate the reaction mechanism (Scheme 20).86
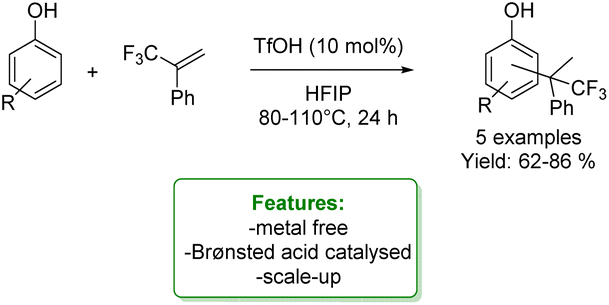 | ||
| Scheme 20 Triflic acid catalysed hydroarylation of α-(trifluoromethyl)styrenes with phenols.86 | ||
C. K. Hazra et al. developed a one-pot Brønsted-acid-catalyzed reaction involving benzaldehydes, silyl enolates, and phenols as nucleophiles. Triflic acid catalyzed a Mukaiyama aldol reaction followed by benzylic arylation with phenols. β,β-Diarylesters were obtained in high yields. The protocols were shown to be selective for the para-position. Moreover, the optimized protocols offer the opportunity to synthesize indanone cores in one-pot procedures. A plausible reaction mechanism was proposed: the benzaldehyde is protonated by TfOH and subsequently undergoes nucleophilic attack. Elimination of trimethylsilanol results in the generation of the carbocationic species, which readily reacts with the external arene nucleophile (the phenols). Finally, aromatization furnishes the β,β-diarylester product and regenerates the proton that further catalyses the benzylic arylation. To perform Friedel–Crafts acylation, the carbonyl functionality of the ester formed in situ undergoes HFIP-assisted protonation, followed by intramolecular cyclization through the nucleophilic attack of an arene, and subsequent removal of a proton, resulting in the synthesis of the 3-aryl-1-indanone scaffold (Scheme 21).87
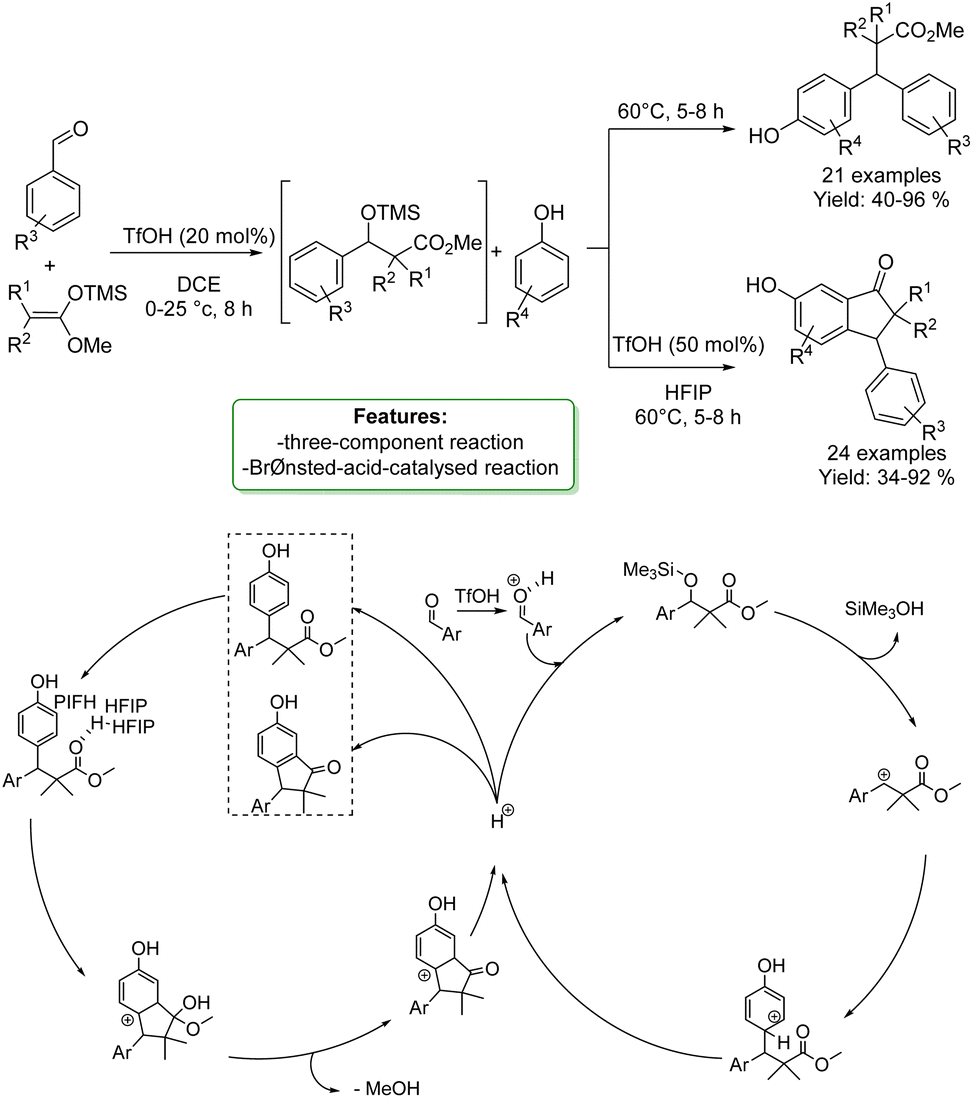 | ||
| Scheme 21 One-pot Brønsted-acid-catalyzed reaction involving benzaldehydes, silyl enolates, and phenols.87 | ||
G. Song et al. developed a novel Brønsted acid-catalyzed cyclization of o-alkynylbenzoic acids followed by an ortho-regioselective electrophilic alkylation of phenols. Using triflic acid as the catalyst, the corresponding regioselective products at the ortho-position of the phenolic ring were obtained in moderate to excellent yields. A plausible mechanism was proposed: the cyclization can occur by hydrogen bonding between the triple bond and the TfOH, which activates the alkyne toward an intramolecular nucleophilic addition. Then, the intermediate is protonated, and it is electrophilically attacked by electron-rich aromatic rings to give the final products (Scheme 22).88
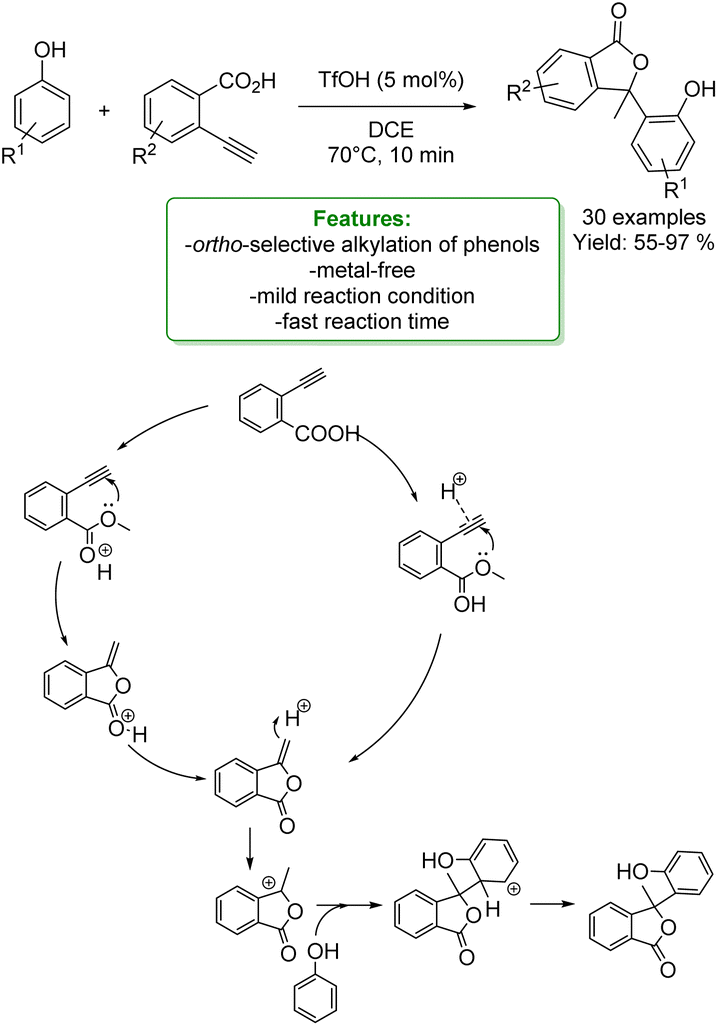 | ||
| Scheme 22 Brønsted acid-catalyzed cyclization of o-alkynylbenzoic acids followed by an ortho-regioselective electrophilic alkylation of phenols.88 | ||
R. Qiu et al. developed a transition-metal-free protocol for the efficient cross-dehydrogenative coupling of 3-aryl benzofuran-2(3H)-ones with phenols using DTBP as an oxidant. The ortho-functionalization of phenols delivered triaryl all-carbon quaternary centres in good yields. A gram-scale experiment was performed, yielding favourable results. A reaction mechanism involving radical participation is proposed. 3-Aryl benzofuran-2(3H)-ones and phenols respectively generate free carbon radicals with DTBP, and these radicals then undergo cross-coupling. It is likely that the 3-aryl benzofuran-2(3H)-ones could also generate relative radicals by reacting with benzyl radical species, which can be easily trapped by another benzyl radical to form the target products (Scheme 23).89
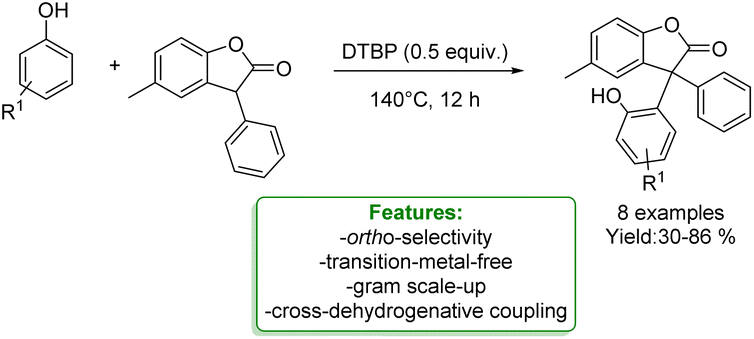 | ||
| Scheme 23 Transition-metal-free protocol for the cross-dehydrogenative of phenols with benzofuran-2(3H)-ones.89 | ||
Xiong, Wang et al. also used a phosphorus-based acid catalyst, H3PO4, for the regioselective synthesis of α-diarylmethyl-substituted phenols in water as a safe solvent and under mild reaction conditions. They developed an ortho-selective reaction between mostly naphthols and para-quinone methides: 65 products have been formed in 59–99% yield. Phenol gave the correspondent para-alkylated product in 67%. Enantioselective investigations with a chiral Brønsted acid resulted in an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 enantiomeric ratio (Scheme 24).90
:20 enantiomeric ratio (Scheme 24).90
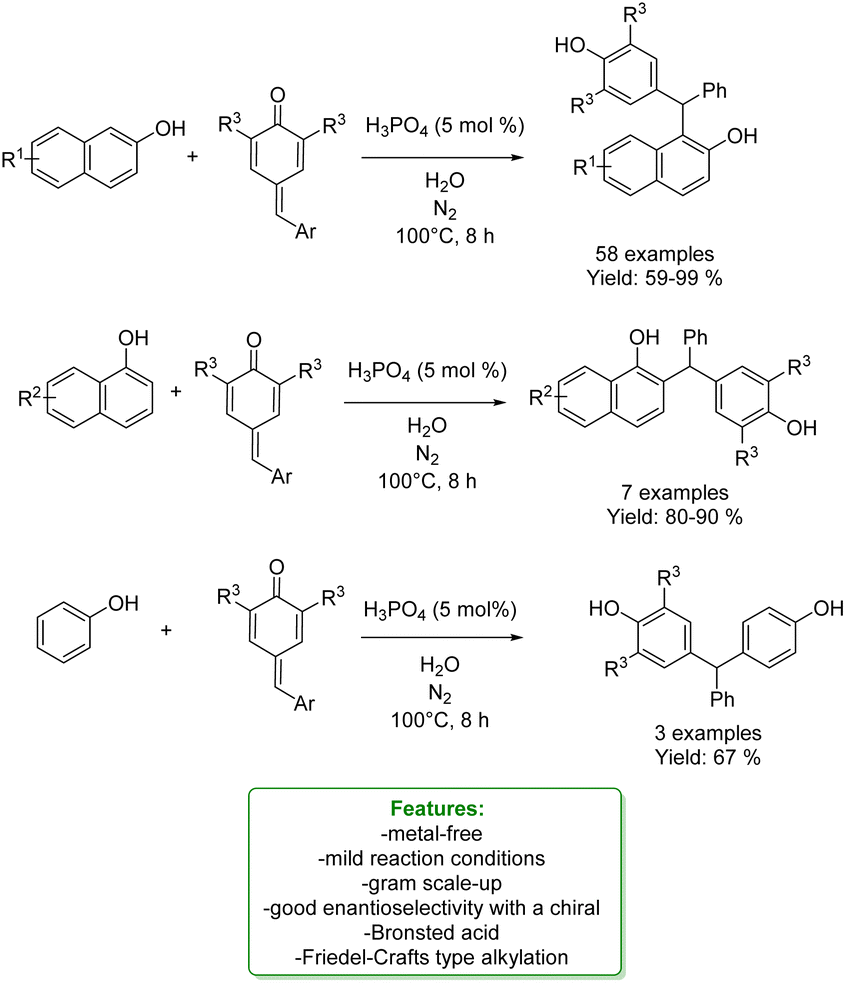 | ||
| Scheme 24 H3PO4-catalysed para-alkylation phenols with para-quinomethides.90 | ||
W. Li, P. Li et al. developed an organocatalytic enantioselective conjugate addition of 2-naphthols to ortho-hydroxyphenyl substituted para-quinone methides. A series of enantioenriched triarylmethanes were obtained in good to excellent yields under mild reaction conditions (Scheme 25).91
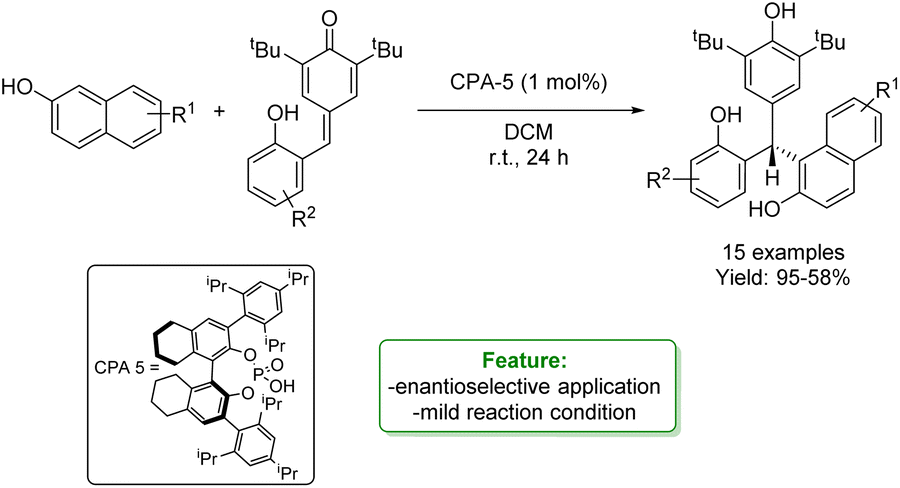 | ||
| Scheme 25 Organocatalytic enantioselective conjugate addition of 2-naphthols to ortho-hydroxyphenyl substituted para-quinone methides.91 | ||
M. Rubin et al. have devised a straightforward method for constructing 4-aryl-5-alkynylpyrimidines through a chemo- and regio-selective acid-promoted electrophilic alkylation of phenols with 5-bromopyrimidine. Notably, when employing simple phenols, the para-product emerges as the predominant outcome. To showcase the versatility of the reaction protocols, a subsequent oxidative aromatization of the resulting dihydropyrimidine moieties, followed by a Sonogashira cross-coupling reaction, was successfully executed. The mechanism involves a SEAr pathway. Specifically, the protonation of one of the nitrogen atoms in pyrimidine in the presence of a strong Brønsted acid is expected to generate a highly electrophilic pyrimidinium species. This species can serve as an electrophile in a Friedel–Crafts-like reaction with electron-rich arenes (Scheme 26).92
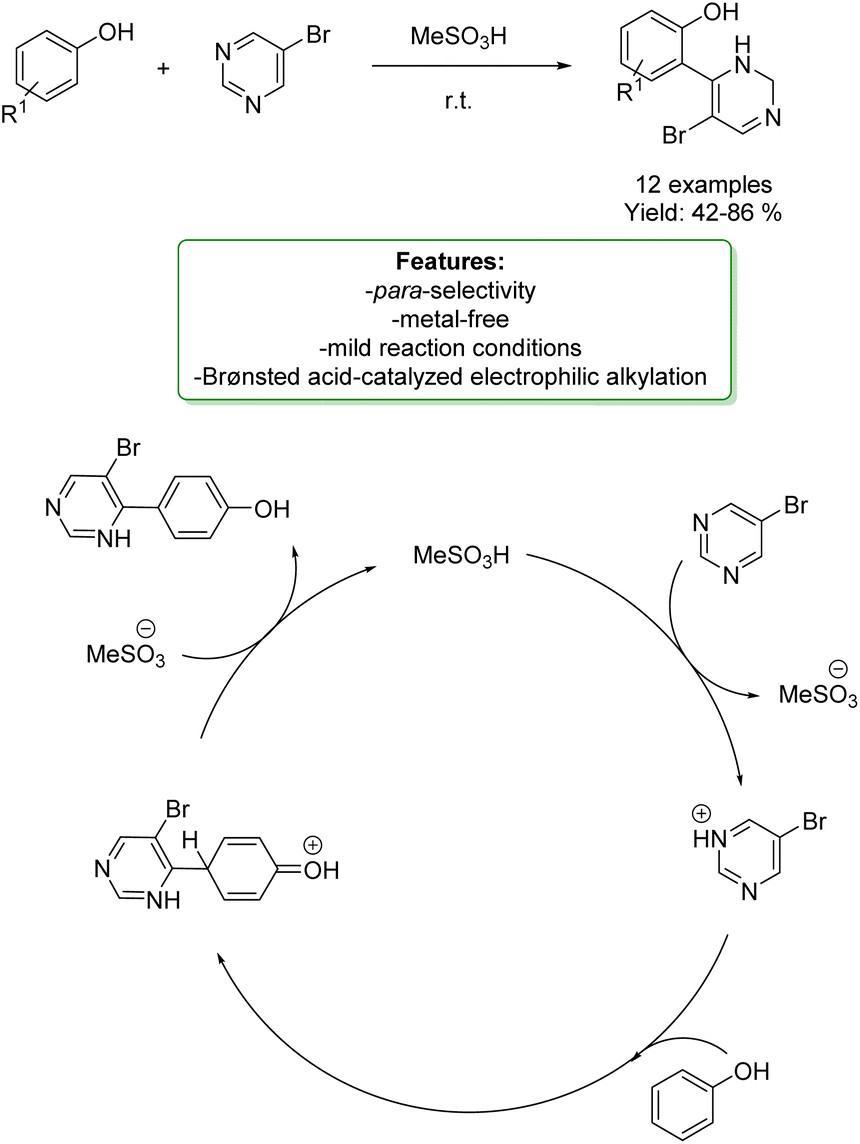 | ||
| Scheme 26 4-Aryl-5-alkynylpyrimidines through a chemo- and regioselective acid-promoted electrophilic alkylation of phenols.92 | ||
Several solid acids were developed for the alkylation of phenol with smaller, non-branched alcohols, enabling the synthesis of products with significant commercial applications (Scheme 27).104 These products are widely used in the production of antioxidants, phenolic resins, agrochemicals, rubber chemicals, printing ink, varnish, surface coatings, fungicides, ultraviolet absorbers, petroleum additives, and heat stabilizers for polymeric materials as feedstock.93
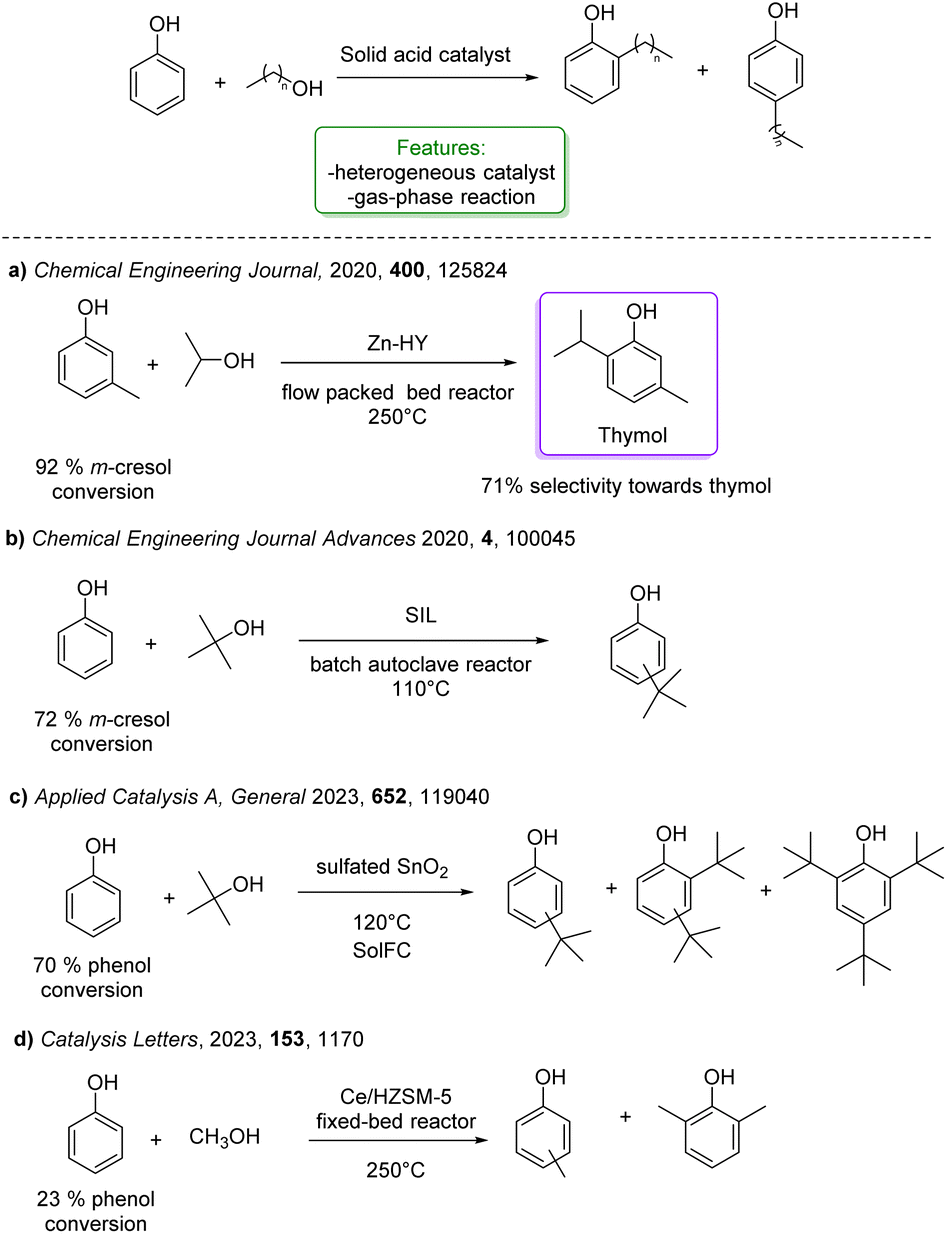 | ||
| Scheme 27 Solid acid catalysed ortho-alkylation of phenols with simple and short alcohols. (a) Alkylation of phenol with iPrOH Zn-HY modified zeolite catalysts.94 (b) Alkylation of phenol with tBuOH supported ionic liquid catalysts.95 (c) Alkylation of phenol with tBuOH metal oxide catalysts (SnO2, TiO2 and ZrO2).96 (d) Alkylation of phenol with methanol catalysed by Ce/HZSM-5.97 | ||
Upadhyayula and coworkers reported the gas phase alkylation of m-cresol with iso-propanol to thymol over a solid acidic catalyst.94 The best results were achieved using a zinc-modified HY zeolite in a packed-bed reactor at 250 °C, yielding a 92% conversion of m-cresol, with 71% selectivity toward thymol. Mechanistic and kinetic investigations were conducted confirming that the mechanism for this reaction consists first in the dehydration of iso-propanol to propylene and water, either directly or through di-isopropyl ether. Then, both O- and C-alkylation occur via isomerization processes that lead to thymol (Scheme 27a).105 The same authors investigated the gas-phase t-butylation of phenol over a mesoporous silica material (SIL), covalently bonded to sulphonic acid functional ionic liquid.95 They obtained 72% conversion of phenol and a product distribution of 24% ortho-butylated phenol, 46% para-butylated phenol, 46% 2,4-dibutylated phenol, 4% 2,6-dialkylated phenol and 2% etherified phenol (Scheme 27b).
More recently, Tsilomelekis and Zuber reported the application of several sulphated solid superacid metal oxide catalysts (SnO2, TiO2 and ZrO2) in the solvent-free alkylation of phenol with tBuOH. They improved the acidity properties by treating the supports with various solutions of H2SO4. The solid acid catalysts were employed for mono-alkylation under industrially relevant conditions. Among the catalysts studied, SnO2 treated with 1.0 M H2SO4 yielded the best results (70% phenol conversion with 10% of the mono-alkylated product) (Scheme 27c).96
In the same year, Wang et al. reported the vapor-phase alkylation of phenol with methanol using an HZSM-5-supported Ce-catalyst (Ce/HZSM-5). The authors disclosed that the preparation method significantly affects the physicochemical properties of Ce/HZSM-5 resulting in different catalytic performances with variously functionalized phenols. Among the three different preparation methods, ball milling, impregnation and solid-state ion-exchange (SSIE), the last one turned out to be the best, achieving 23% of phenol conversion (Scheme 27d).97
The alkylation of phenols is often used to provide a more economical and sustainable method for synthesizing high-density biofuel precursors, with the aim of alleviating the energy crisis and environmental pollution, typically starting from a potential lignin-derived substrate. Cyclic ketones or cyclic alcohols serve as alkylating agents in the acid-catalyzed alkylation of phenol, followed by hydrodeoxygenation (HDO) to obtain bicyclic alkanes or polycyclic alkanes, which are classified as high-density biofuels (Scheme 28). In this sense, an interesting application concerns the synthesis of perhydrofluorene, a component of high-density jet fuel where renewable lignin-derived phenols and benzyl acetate were used as starting materials. The first step is the alkylation reaction of the starting materials catalysed by solid acid montmorillonite (MMT), followed by the HDO of alkylated products catalysed by Pd/C (Scheme 28a).98
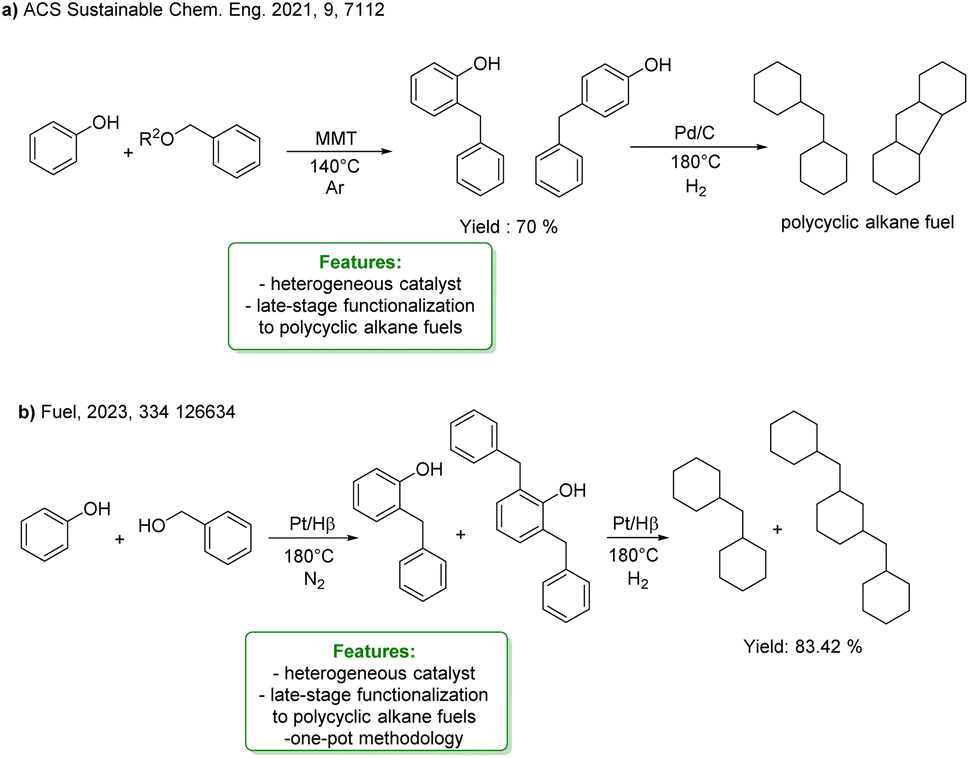 | ||
| Scheme 28 Acid-catalysed alkylation of phenol, and subsequent HDO to obtain bi- or poly-cycloalkanes or high-density biofuels (a) two-step synthesis of perhydrofluorene, from phenols and benzyl acetate catalysed by solid acid montmorillonite (MMT), and Pd/C.98 (b) One-pot strategy for preparing polycyclic alkane from phenol and benzyl alcohol over a bifunctional Pt/Hβ catalyst.99 | ||
More recently, Zou et al. reported an interesting example of application of alkylation of phenols for high-density fuel synthesis via a one-pot strategy for preparing polycyclic alkane from phenol and benzyl alcohol over a bifunctional Pt/Hβ catalyst. The activity of different zeolites for catalysing the alkylation of phenol was explored: the pore size, acidity and specific surface area of zeolites were found to affect the reaction results. The selectivity of alkylphenol can reach more than 97%, and almost all products can be hydrodeoxygenated to bi-cycloalkane under H2 conditions. The yield of bicyclic and tricyclic in the final product can reach 83.42% (Scheme 28b).99
1.1.1.3 Photocatalysis. Phenolates are the corresponding anions of phenols, formed upon their deprotonation. They are strongly electron-rich and have physiochemical properties. For instance, compared to the neutral phenol, their UV-Vis absorption is red-shifted, due to the increased conjugation between the negative oxygen and the benzene ring. This makes them photochemically active electron-rich intermediates that can be used to generate reactive open-shell species.100,101
In 2021, Dell’Amico et al. exploited the photochemical activity of a halogen-bonded complex between electron-rich phenolate anions and α-iodosulfones, with which they generated a scope of 27 ortho-alkylated products with yields up to 97%. They developed a flow strategy that consists of a micro-fluidic photoreactor, furnished with a modular light source (370 nm or 456 nm). The alkylated products were obtained in a short reaction time (10 min), under mild reaction conditions (r.t.), with a high productivity rate (0.6 mmol h−1) and very good ortho-selectivity (>20:1). DFT calculations highlighted the formation of the halogen-bonded complex and furnished radical mechanism insights with quantum yields measurements. Direct light excitation promotes the SET within the halogen-bonded complex forming the sulfonyl and the phenolate radicals where the first one is the electrophilic species that is added to the ortho-position. Late-stage alkylation of biorelevant phenols, such as paracetamol, resulted in good to excellent yields (50–95%) after the removal of the sulfonyl functional group through reductive cleavage (Scheme 29).102
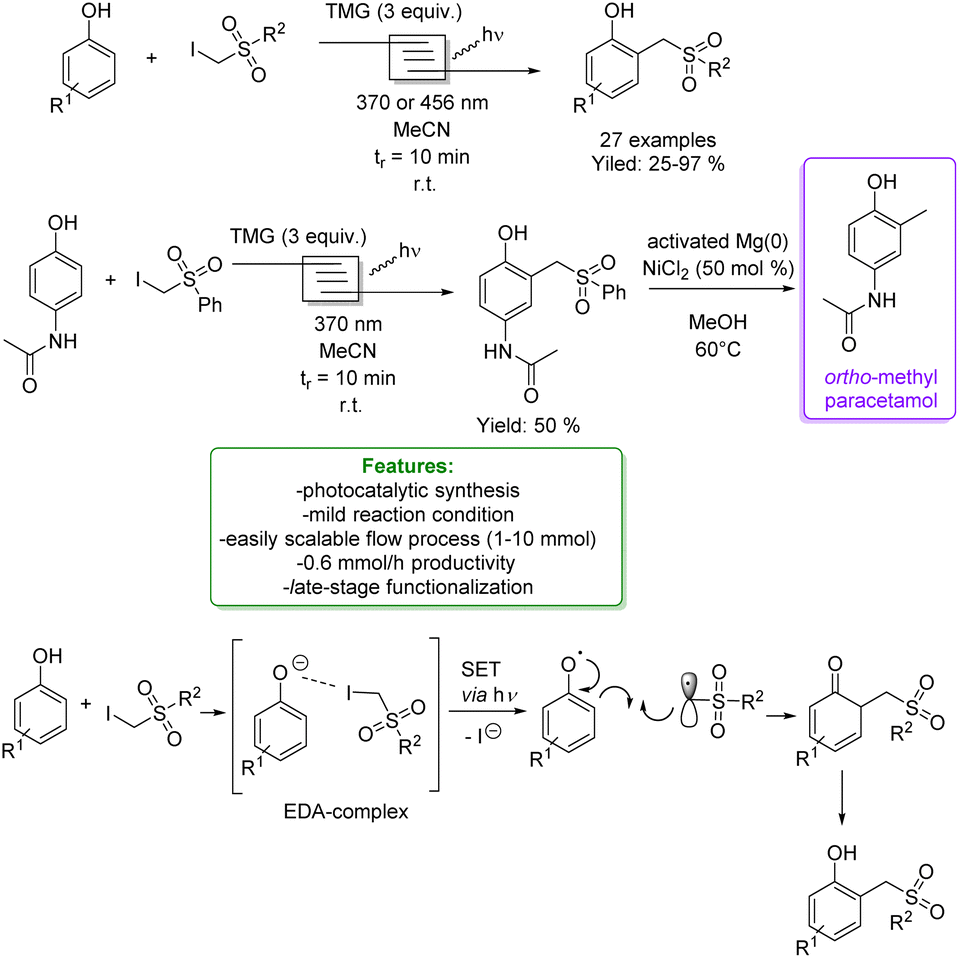 | ||
| Scheme 29 Alkylation of phenol with α-iodosulfones via a photocatalytic flow strategy.102 | ||
1.1.2.1 Metal-free. Lewis acid-assisted Friedel–Crafts alkylation of phenols provided a straightforward way for the Csp2–H functionalization of phenols. However, it suffers from poor site selectivity, reducing its applications. Recently, Zhao et al. reported an interesting example of highly para-selective Friedel–Crafts alkylation of phenols with tertiary alkyl bromide promoted by hexafluoroisopropanol (HFIP). This process leads to a broad substrate scope of para-alkylated products in moderate to excellent yields under mild reaction conditions. Mechanistic studies showed that HFIP acts as a mild Lewis acid towards the haloalkane, helping the generation of the carbocation intermediate. Subsequently, HFIP establishes a hydrogen bond with the phenolic OH– group increasing its steric hindrance and determining the functionalization of the less hindered para-position (Scheme 30).103
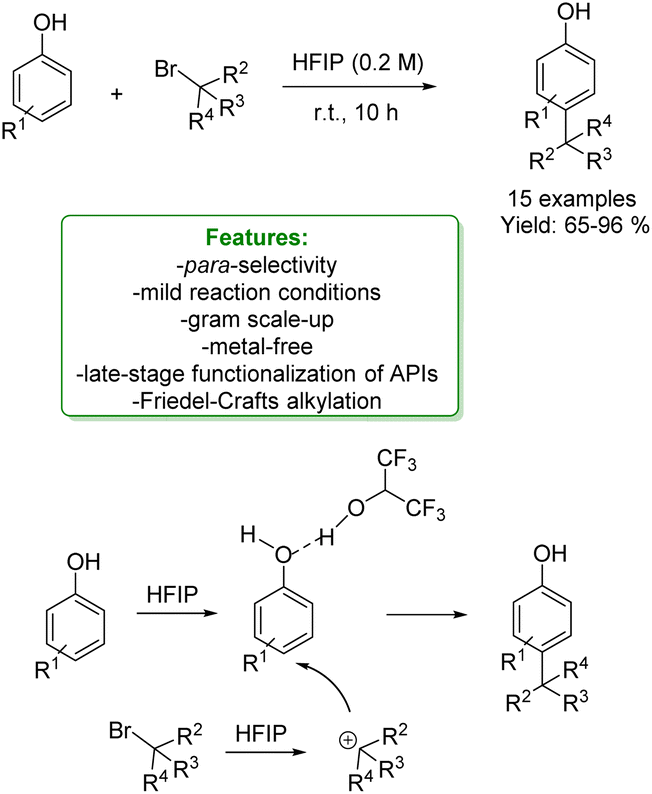 | ||
| Scheme 30 Para-selective Friedel–Crafts alkylation of phenols with tertiary alkyl bromide.103 | ||
1.1.2.2 Electrosynthesis. Ackermann et al. developed a sequential selective electrochemical Csp3–H fluorination, avoiding the use of expensive electrophilic fluorine reagents by utilizing readily available NEt3·3HF. Fluorides were employed as strategic intermediates for the subsequent metal-free benzylation of electron-rich arenes, such as phenols. The optimized protocols were applied in the late-stage functionalization of tolterodine precursors, hexylresorcinol, and estrone. The authors proposed a plausible reaction mechanism in which the arene is oxidized to a radical cation, stabilized by the HFIP co-solvent. The heterolytic C–H cleavage furnishes the radical intermediate. In a second anodic oxidation, the corresponding benzyl cation is formed and then trapped by a fluoride ion to give the desired product (Scheme 31).104
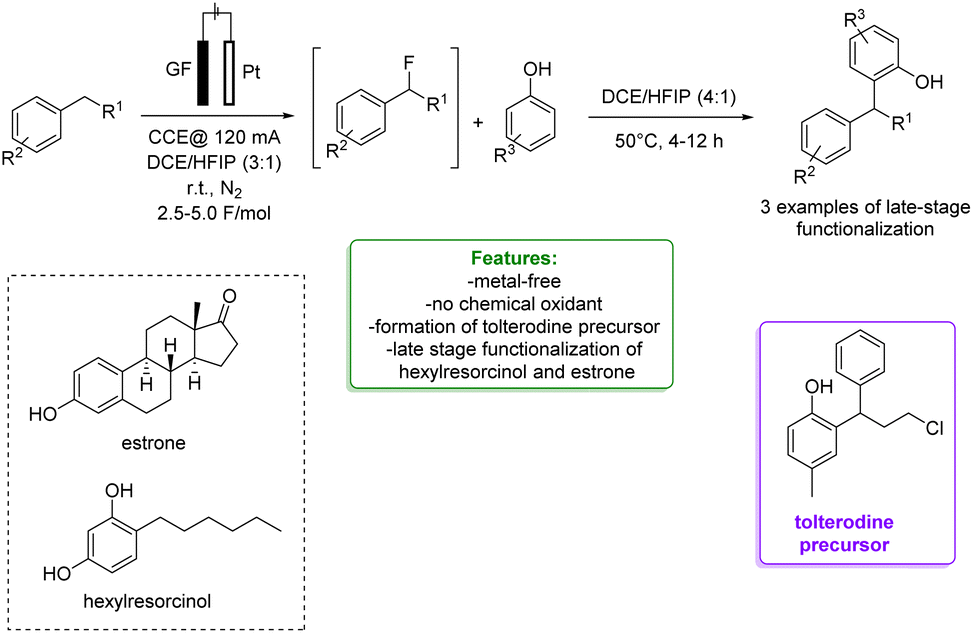 | ||
| Scheme 31 Sequential electrochemical Csp3–H fluorination followed by metal-free benzylation of phenols.104 | ||
1.2. Aminomethylation
Ortho-aminomethylation of phenols is a useful retrosynthetic tool for accessing the synthesis of N-containing scaffolds found in interesting natural products (such as capsaicin), APIs (including PKS13 inhibitors, PARP1 inhibitors, amodiaquine, lavendustin A, and topotecan), as well as ligands for transition metals (Fig. 4).4,105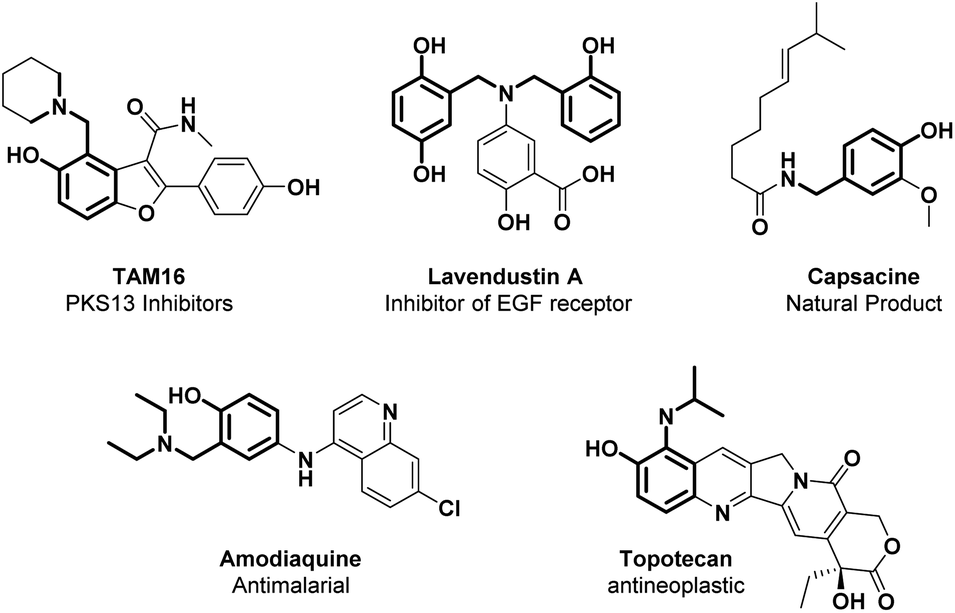 | ||
| Fig. 4 Aminomethylated phenol structural moieties in API and agrochemical compounds. | ||
Given the interesting applications of this class of compounds, the development of synthetic strategies for the ortho-aminomethylation of phenols is a significant challenge. Traditionally, this structural moiety is obtained by utilizing Eschenmoser's salt, but stoichiometric amounts of reactive species such as pre-generated salt itself, N-oxide, or BrCCl3 are needed.106–109 Thus, different synthetic methodologies have been devolved to access this strategical structural unit, such as the Mannich reaction and cross-dehydrogenative-coupling (CDC). The Mannich reaction is a multicomponent condensation involving a primary or secondary amine, formaldehyde, and phenol. One of the earliest examples was reported in 1994 by B. L. Feringa et al.121 On the other hand, the CDC reaction can provide an atom-economic strategy avoiding a multistep synthetic procedure and pre-functionalization of the amine coupling partner.110,111
In the following section, we explore the latest strategies developed to obtain amino methylated phenols dividing them into metal catalysis, and acid/base catalysis.
1.2.1.1 Metal catalysis. Transition-metal catalysed Csp2–H CDC of phenols is a widely and well-established method to access ortho-aminomethylation. In 2017, Hong et al. developed a Ru-catalysed protocol with methanol, amine and phenol. The metal catalyst dehydrogenates MeOH to form formaldehyde that couples with the amine to the corresponding imine cation. This intermediate is then attacked by the phenolate anion, and the amino-methylated product is achieved.112 In the same year, Wang et al. reported the first two-component Cu(II)-catalysed aerobic ortho-aminomethylation of free phenols with potassium aminomethyltrifluoroborates.113 In 2018, F. W. Patureau reported a Cu(II)-catalysed ortho-selective aminomethylation of phenols with N,N-dimethylanilines by a direct intermolecular CDC reaction.114 In the same year, N. Jain et al. reported the use of GO–Cu7S4 NPs as a reusable heterogeneous catalyst for ortho-selective aminomethylation of phenols and naphthols under solvent-free conditions.115 Among the most recent examples, in 2021, N. Liu et al. reported the use of a Cr-catalyst for the ortho-aminomethylation of phenols with N,N-dimethylanilines via a radical-cation mechanism, where the Cr(III) intermediate is the active species. The approach shows excellent site selectivity at the ortho-position of phenols, a broad substrate scope and functional group compatibility for both N,N-dimethylanilines and phenols (Scheme 32).116
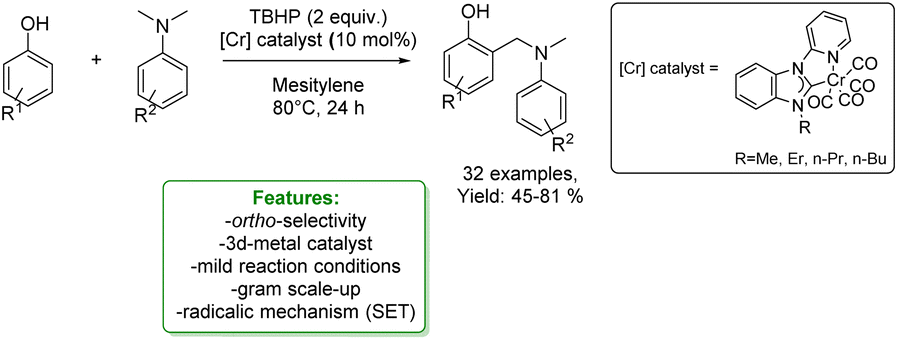 | ||
| Scheme 32 Cr-catalysed ortho-aminomethylation of phenols with N,N-dimethylaniline.116 | ||
In the same year, Y. Liu et al. reported the use of the heterogeneous Cu(II)-modified nitrogen-rich covalent organic polymer (Cu/N-COP-1) catalyst employed in the CDC ortho-aminomethylation of phenols with N,N-dimethylanilines. The Cu/N-COP-1 catalyst displayed excellent catalytic activity under mild conditions without any ligands. Various substituted phenols and N,N-dimethylanilines were successfully converted to the corresponding products. Moreover, the catalyst was readily reused for five consecutive runs (Scheme 33).117
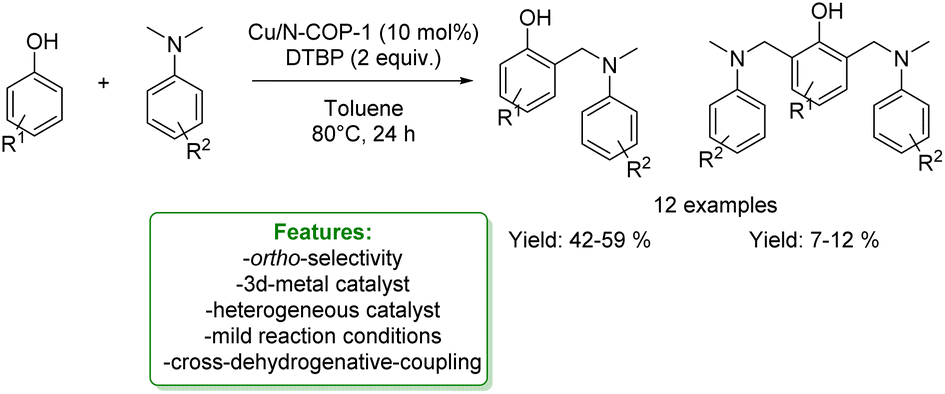 | ||
| Scheme 33 Heterogeneous Cu-catalysed ortho-aminomethylation of phenols with N,N-dimethylaniline.117 | ||
N. Noroozi Pesyan et al. designed a Zn transition metallic complex on functionalized mesoporesilica MCM-41. The catalyst was applied in the one-pot three-component synthesis of amino benzyl quinolinols and naphthols (6 examples), obtaining excellent yields in a short reaction time. The heterogeneous catalyst was efficiently recovered and reused with no loss in selectivity (Scheme 34).118
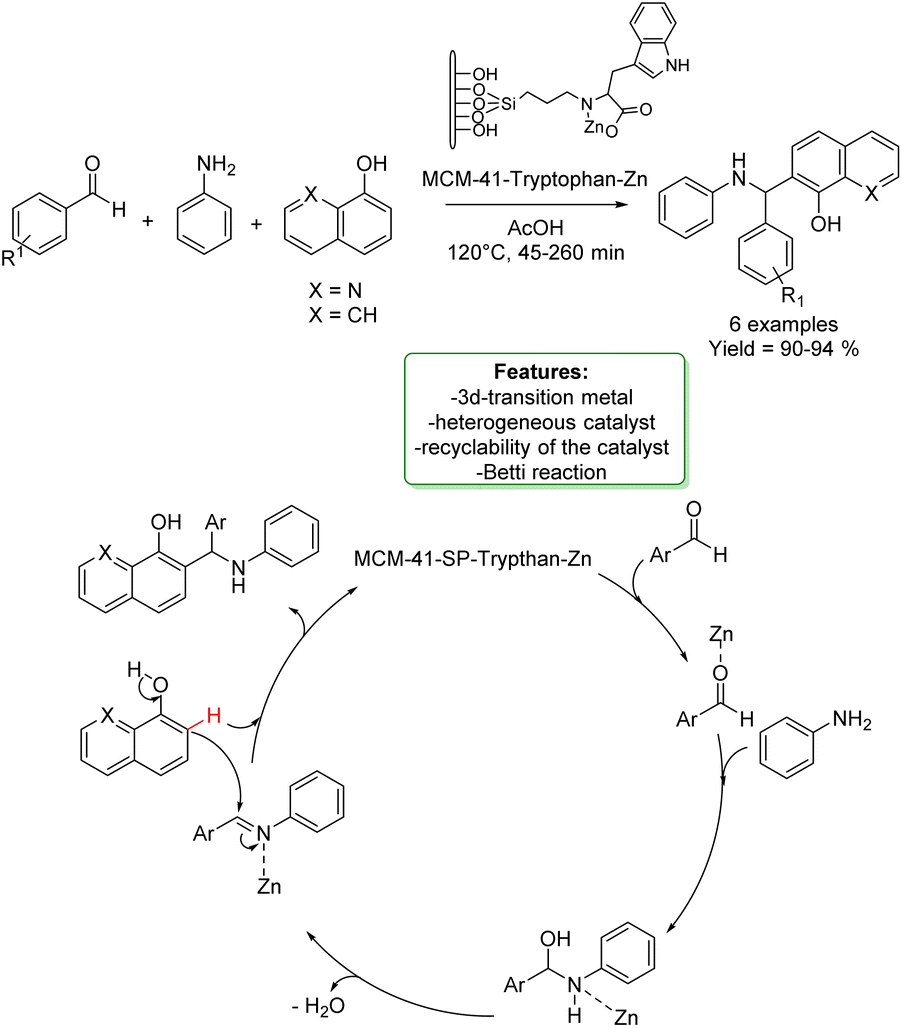 | ||
| Scheme 34 Zn transition metallic complex on functionalized mesoporesilica MCM-41 catalysed the one-pot three-component synthesis of amino benzyl quinolinols and naphthols. | ||
An asymmetric version of amination of phenols was reported by T. Arai et al. They developed an asymmetric para-selective acid-catalyzed aza-Friedel–Crafts reaction of phenols with sulfonylaldimines. Chiral bis(imidazolidine)pyridine (PyBidine)–Ni(OAc)2 catalysts allowed the formation of a wide range of products in good to excellent yields with excellent enantioselectivity. To demonstrate the synthetic utility of the optimized asymmetric protocols, an intermediate in the synthesis of an orexin antagonist was performed. DFT calculations suggest that cooperative HOMO–LUMO activation, involving nickel phenoxide formation (HOMO activation) and hydrogen bonding activation of the sulfonylaldimine (LUMO activation), is the key to switching regioselectivity (Scheme 35) 119 Lots of efforts were spent to access functionalized phenols with α-substituted cyclic amines, which are ubiquitous scaffold in natural alkaloids, bioactive molecules, drugs, agrochemicals, organocatalysts, and functional materials. In addition, cyclic amines serve as bulk chemicals since they are extensively used as organic solvents, bases, and synthetic building blocks.120,121 In 2020, Zhang’s research group developed a synthetic strategy based on the Hydrogen Evolution Cross-Coupling (HECC) between a variety of phenolic monomers and N-alkyl cyclic amines, catalysed by Ru.122 This mechanism was firstly reported for N-heteroarenes and indoles by Zhang et al.123 It consists in a first Ru-catalysed hydride transfer from amine forming Ru-hydride species and iminium. Then, the α-deprotonation to form the correspondent azomethine ylide occurs thanks to Ru-hydride that, at the same time, returns in its Ru(0) form generating H2. In the end, p-TSA helps the formation of the iminium having an endocyclic double bond in which the nucleophilic addition of phenol happens. They reported a large substrate scope, made of 47 examples with isolated yields up to 85 % (Scheme 36).
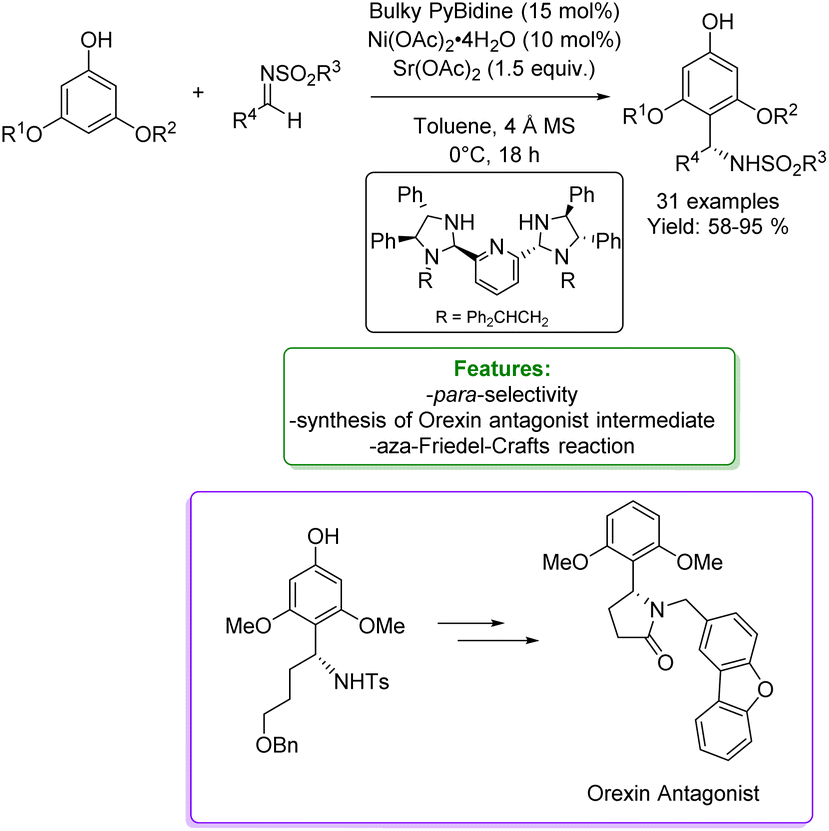 | ||
| Scheme 35 Asymmetric para-selective acid-catalyzed aza Friedel–Crafts reaction of phenols with sulfonylaldimines.119 | ||
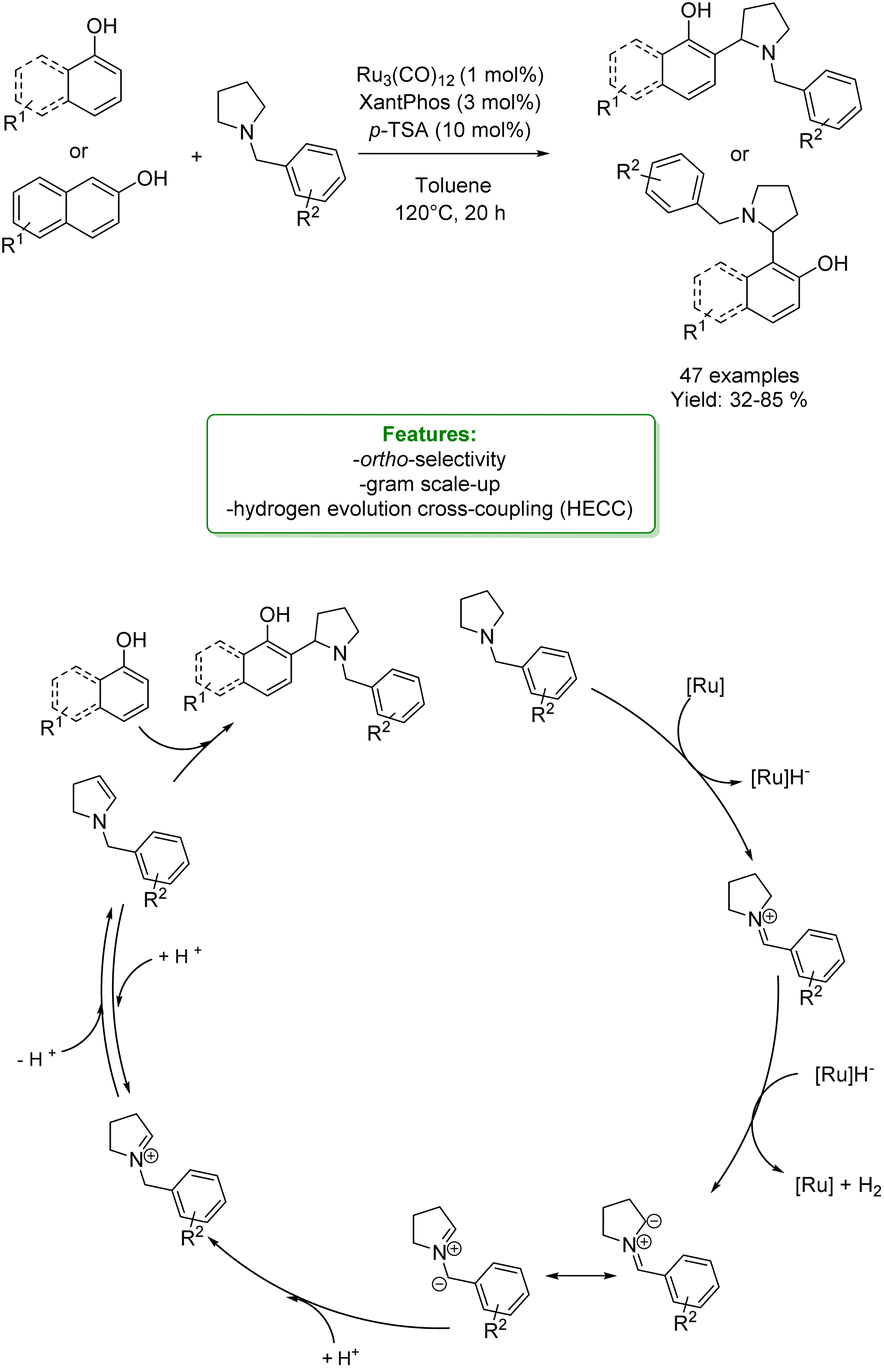 | ||
| Scheme 36 Ru-catalysed aminomethylation of phenols with N-alkyl cyclic amines.122 | ||
1.2.1.2 Acid/Base Catalysis. The Mannich reaction is a step-economical strategy for aminomethylation of phenols under metal-free conditions. However, this strategy suffers from drawbacks such as high temperature, narrow substrate scope and low ortho-selectivity. Therefore, some pre-activated reagents can be used to guarantee the site-selectivity and milder conditions. N,O-Acetal is a stable and readily available reagent for aminomethylation, which can react with various nucleophiles via the in situ formation of iminium intermediates. In 2021, L. Liu et al. reported a metal-free Friedel–Crafts type Mannich ortho-aminomethylation of phenols with N,O-acetals catalysed by trifluoro acetic acid (TFA) under mild reaction conditions, yielding various aminoethyl substituted phenol derivatives in good to excellent yields (Scheme 37).124
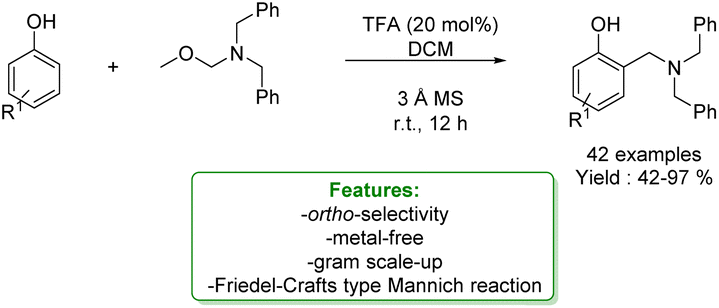 | ||
| Scheme 37 Brønsted acid catalysed aminomethylation of phenols with N,O-acetals.124 | ||
With the same goal, Tian et al. developed a transition-metal-free ortho-aminomethylation of phenols with trifluoroborate reagents catalysed by an iodine–sodium percarbonate complex in aqueous media. This method exhibited wide substrate scope and excellent tolerance to functional groups, as exemplified by the late-stage ortho-Csp2–H aminomethylation of ezetimibe, estrone, O-desmethylvenlafaxine, L-tyrosine and Tyr-Val dipeptide. The mechanistic findings indicated that a radical process may be involved (Scheme 38).125
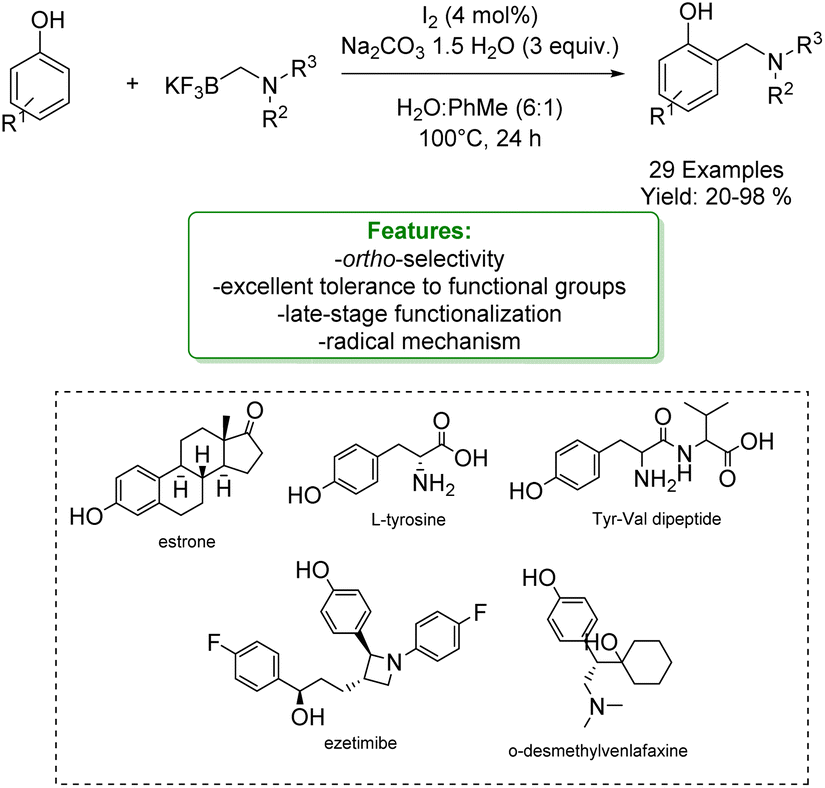 | ||
| Scheme 38 Iodine–sodium percarbonate catalysed aminomethylation of phenols with trifluoroborate reagents.125 | ||
P. S. Nagarajan et al. developed a one-pot three-component strategy for the synthesis of various amidoalkyl-2-naphthol dihydropyrimidin-2(1H)-ones using 2,6-pyridine dicarboxylic acid (PDCA), as an organocatalyst. The optimized reaction conditions allowed amido-2-naphthols to be obtained in excellent yields (up to 96%). At the end of the reaction, the catalyst was recovered and reused (Scheme 39).126
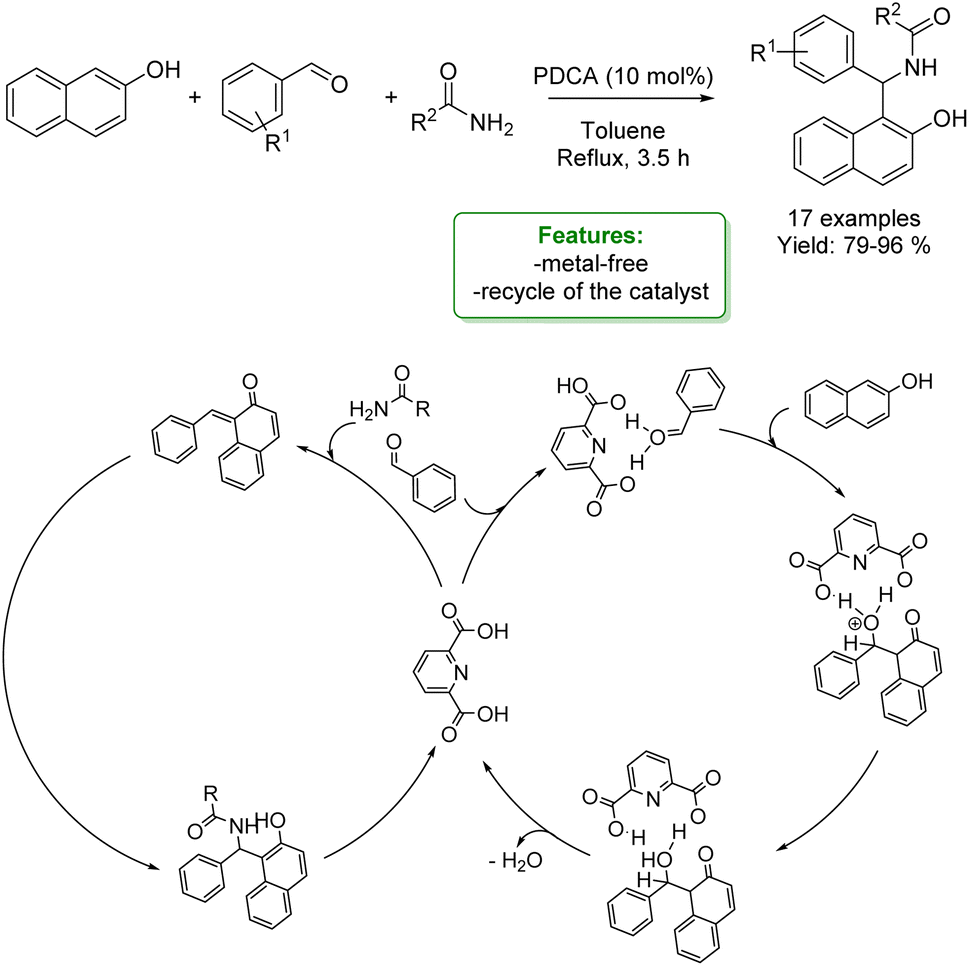 | ||
| Scheme 39 One-pot three-component strategy for the synthesis of amidoalkyl-2-naphthol dihydropyrimidin-2(1H)-ones.126 | ||
H. Tran et al. developed a three-component synthesis of 1-amidoalkyl naphthols and polyhydroquinolines catalysed by a deep eutectic solvent, [CholineCl][ZnCl2]3. The reaction proceeds smoothly at low temperatures for a short reaction time under solvent-free conditions affording moderate to excellent yields. The authors proposed a plausible reaction mechanism: first, benzaldehyde forms a complex with the catalyst so the benzamide can attack the carbonyl group of the activated aldehyde. Then, 2-naphthol is attacked and the so-formed intermediate undergoes a deprotonation generating the 1-amidoalkyl-2-naphthol (Scheme 40).127
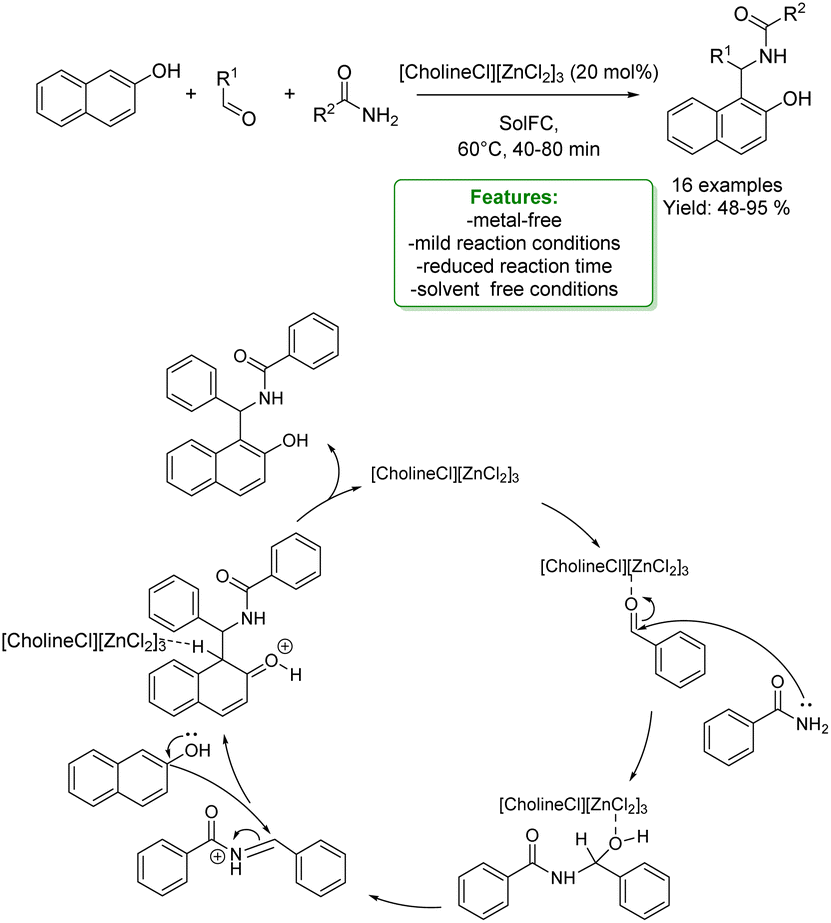 | ||
| Scheme 40 Deep eutectic solvent catalysed three-component synthesis of 1-amidoalkyl naphthols.127 | ||
The enantioselective aza-Friedel–Crafts reaction is one of the most straightforward and efficient strategies for the introduction of a new Csp2–C bond-bearing quaternary stereocenter.128,129 Li et al. reported an aza-Friedel–Crafts reaction with isatin-derived ketamines using Bi(OH)3/CPA as the catalytic system to obtain para-alkylated phenols. The process consists in the acid-catalysed addition of electron-rich aromatic compounds (in this case electron-rich phenols) to imines (isatin-derived ketamines). The authors obtained 20 examples of para-substituted phenols with 68–99% yields and excellent enantioselectivity (91–99% ee) (Scheme 41).81
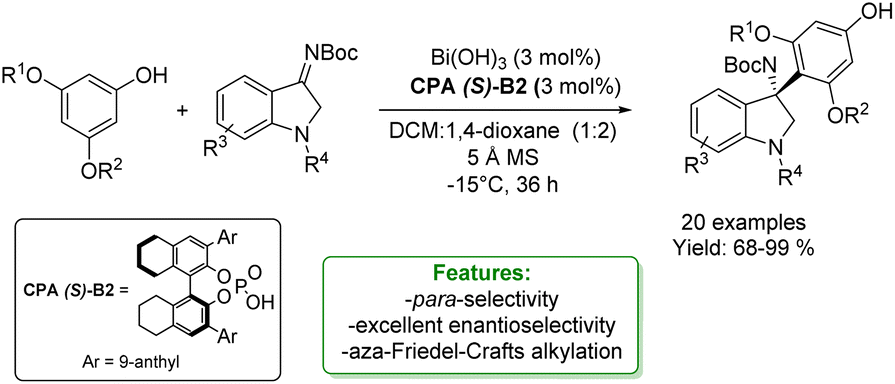 | ||
| Scheme 41 Para-selective aza-Friedel–Crafts reaction of phenols with isatin-derived ketamines.81 | ||
B. Fan et al. developed an enantioselective aza-Friedel–Crafts addition of naphthols with isatin-derived ketimines catalyzed by H8-BINOL-derived chiral biaryl phosphoric acid. A wide range of isatin-derived ketimines, as well as naphthols, were successfully employed, yielding products with sufficient to excellent yields and high enantioselectivities (Scheme 42).130
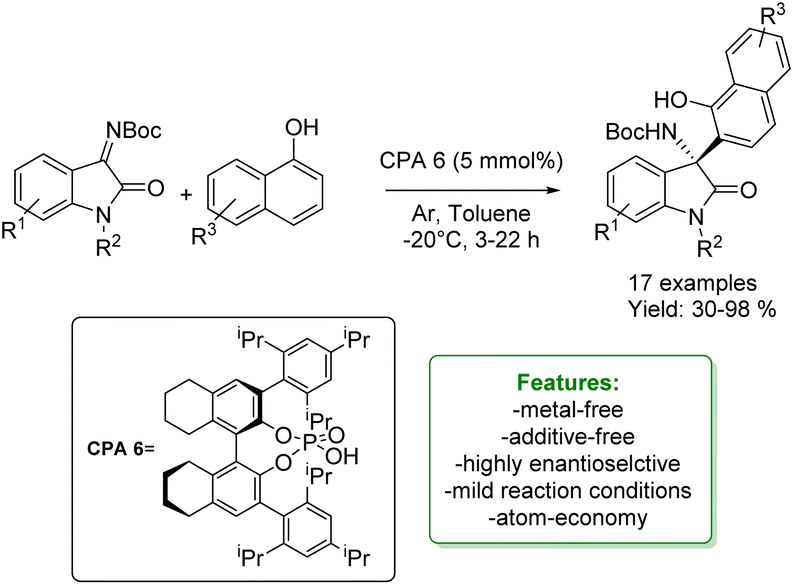 | ||
| Scheme 42 H8-BINOL-derived chiral biaryl phosphoric acid catalysed aza-Friedel–Crafts addition of naphthols with isatin-derived ketimines.130 | ||
The enantioselective aza-Friedel–Crafts functionalization of phenols and naphthols with isatin-derived ketimines has been extensively investigated using various metal-free catalysts to achieve high enantioselectivity. Y. Jin et al. reported the application of a Takemoto-type organocatalyst. This catalyst smoothly promotes the reaction to provide the desired product in excellent yields and enantioselectivities (Scheme 43).131
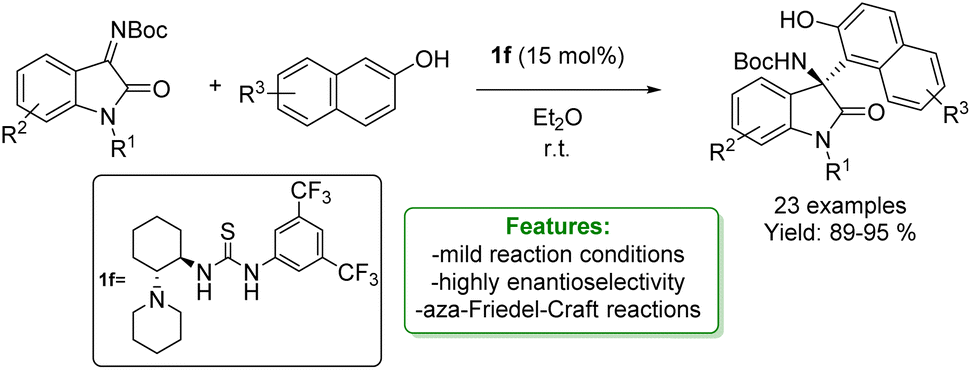 | ||
| Scheme 43 Takemoto-type organocatalyst catalysed aza-Friedel–Crafts addition of naphthols with isatin-derived ketimines.131 | ||
J. M. Andrés, R. Pedrosa et al. developed the application of a supported bifunctional chiral thiourea-based catalyst in the synthesis of 3-amino-2-oxindoles through an enantioselective aza-Friedel–Crafts reaction. The desired products were obtained with good to excellent yields and high enantioselectivities. The supported organocatalyst was recycled without loss of activity and was employed as a catalyst under continuous flow conditions (Scheme 44).132
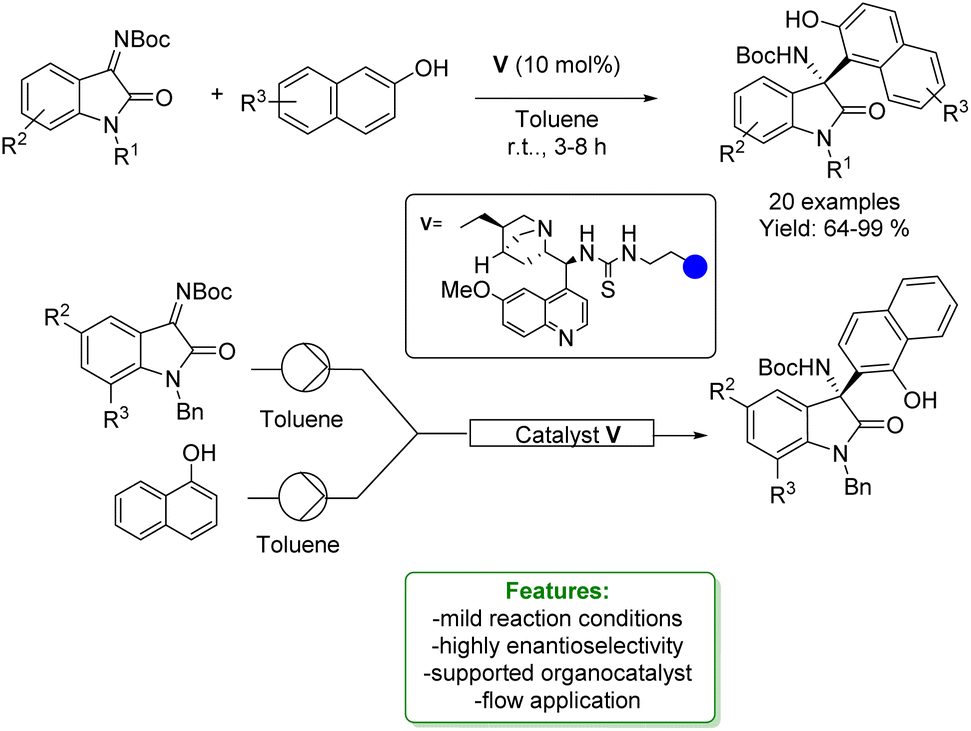 | ||
| Scheme 44 Supported bifunctional chiral thiourea-based catalyst catalysed aza-Friedel–Crafts addition of naphthols with isatin-derived ketimines.132 | ||
M. Gredičak et al. developed a formal Betti reaction between phenols and isoindolinone alcohol. The Betti reaction, a Mannich-type multicomponent reaction involving aldehydes, primary amines, and naphthols, is employed for synthesizing α-secondary amines known as Betti bases. In the reported protocols, the authors demonstrated the use of phenols as nucleophiles. The desired Betti bases were obtained in low to excellent yields under mild reaction conditions. When the unsubstituted phenol was used, only the para-regioisomer was observed (Scheme 45).133
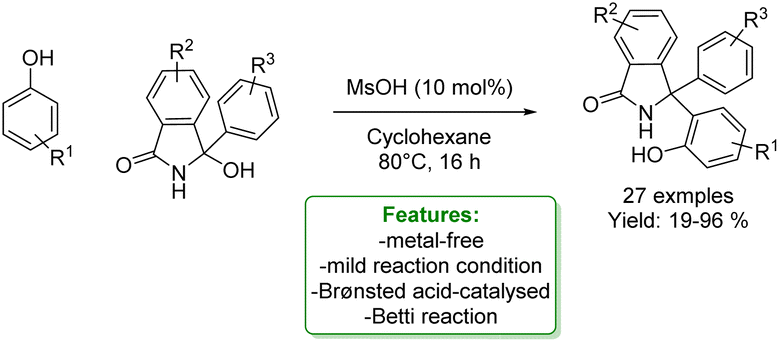 | ||
| Scheme 45 Brønsted acid-catalyzed Betti reaction of phenols with isoindolinone alcohol.133 | ||
More recently, the same authors reported a stereoselective variant of the Betti reaction using phenols as nucleophiles. They developed a chiral phosphoric acid-catalysed reaction for accessing chiral Betti base precursors featuring a congested, triaryl-substituted stereogenic center. The transformation proceeds smoothly with a broad range of phenols and isoindolinone alcohol, yielding products in good to excellent yields and moderate enantioselectivities. Notably, the reaction operates under mild conditions, albeit with a considerably long reaction time (Scheme 46).134
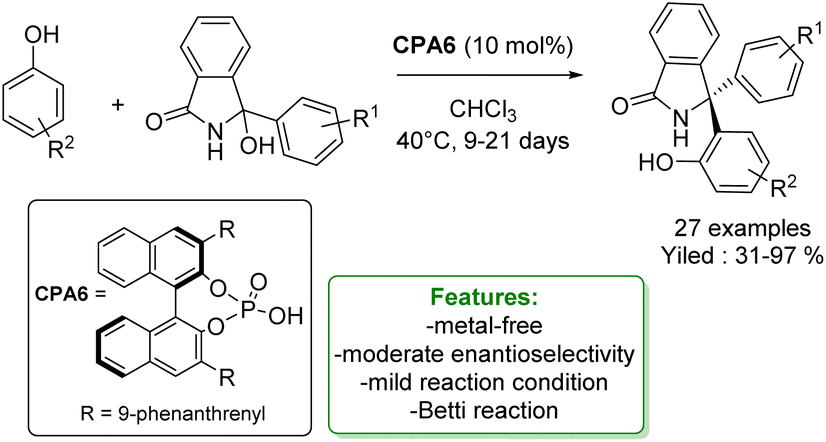 | ||
| Scheme 46 Chiral phosphoric acid-catalyzed Betti reaction of phenols with isoindolinone alcohol.134 | ||
H. Song, Q. Li, and S. Ban et al. developed an asymmetric Michael-type Friedel–Crafts alkylation of naphthol and unsaturated pyrazolones through a chiral squaramide-base catalyst. A wide range of substrates is tolerated, obtaining excellent yields (up to 99%) with a moderate to excellent level of enantioselectivity. The scalability was also demonstrated by a gram-scale reaction. The authors proposed a plausible mechanism for asymmetric transformations catalyzed by bifunctional squaramides. The basic nitrogen atom of the catalyst deprotonates the naphthol and the unsaturated pyrazolone is activated by the squaramide moiety via the formation of two hydrogen bonds. The deprotonated naphthol then attacks the unsaturated pyrazolone from the Re-face to afford the S-configured stereocenter, followed by tautomerization to give the pyrazolone (Scheme 47).135
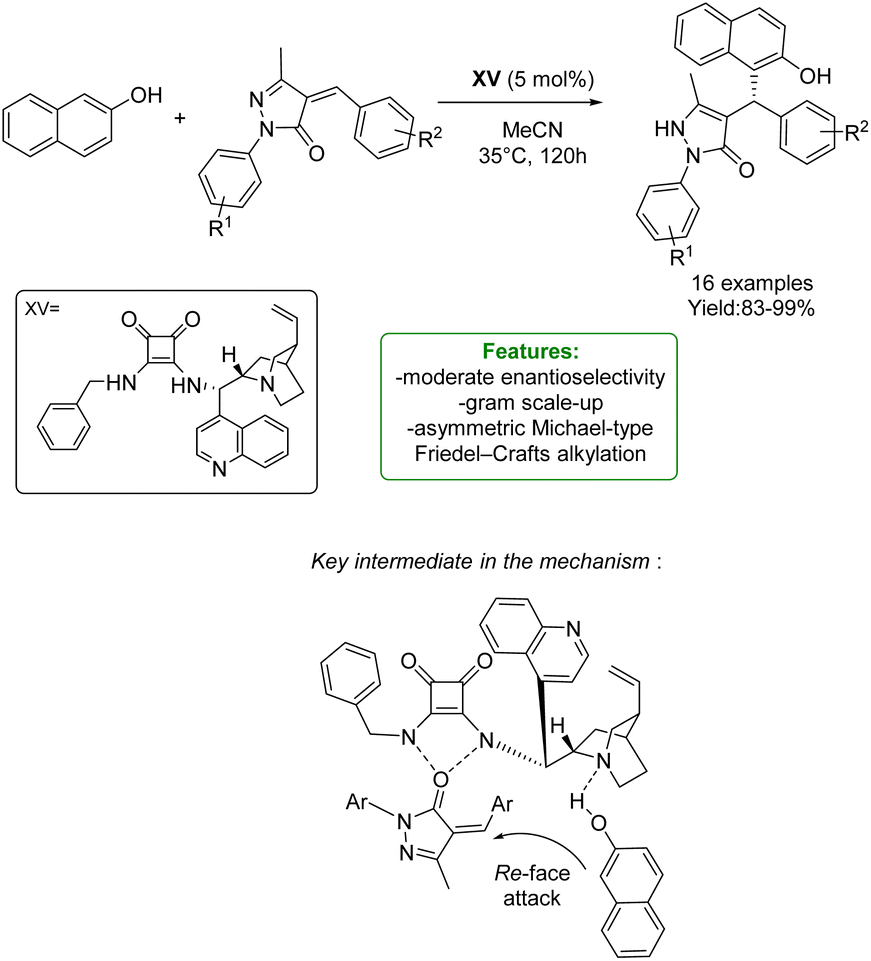 | ||
| Scheme 47 Chiral squaramide-catalyzed Michael-type Friedel–Crafts alkylation of naphthol and unsaturated pyrazolones.135 | ||
X.-J. Zhao et al. reported a highly enantioselective aza-Friedel–Crafts reaction of phenols and naphthols with cyclic-ketimines catalysed by a chiral phosphoric acid system (C3). The optimized reaction conditions provide a broad range of chiral indolin-3-one derivatives bearing a quaternary stereocenter at the C2 position in high yields and excellent enantioselectivities, up to 97%. These advantages can be attributed to two H-bonds and π–π interactions enabled by the chiral phosphoric acid (Scheme 48).82
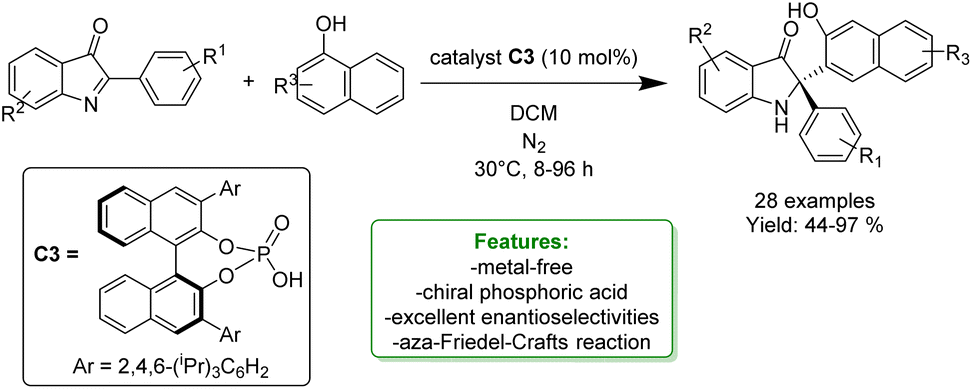 | ||
| Scheme 48 Ortho-selective aza-Friedel–Crafts reaction of naphthols catalysed by a chiral phosphoric acid system.82 | ||
L.-X. Wang et al. developed an organocatalytic enantioselective aza-Friedel–Crafts reaction between benzothiazolimines and 2-naphthols. The squaramide organocatalyst allowed the obtainment of a series of chiral 2′-aminobenzothiazolomethyl naphthols in excellent yields (up to 99% yield) with excellent enantioselectivities (up to >99% ee) under mild conditions. The synthetic potential of the developed protocol was evaluated in a scale-up preparation (Scheme 49).136
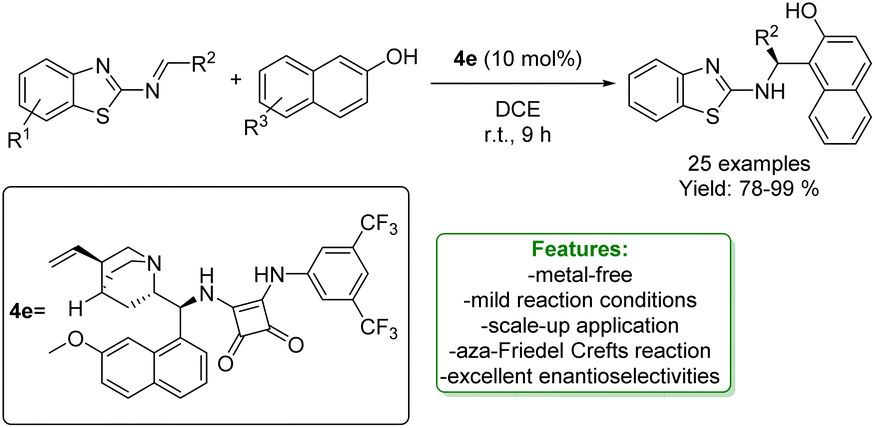 | ||
| Scheme 49 Aza-Friedel–Crafts reaction of benzothiazolimines and 2-naphthols. | ||
1.2.2.1 Metal-free. L. Liao, X. Zhao et al. developed a Lewis acid-mediated three-component electrophilic thiolative functionalization of enamides. A series of phenols were successfully incorporated into the substrates with high regio- and stereo-selectivities under mild conditions. β-Aminosulfides were obtained in moderate to very good yields (up to 90%). To demonstrate the practicability of the method, a scale-up experiment was conducted. The authors proposed a Fries-like rearrangement pathway for this reaction, in which the Lewis acid, TMSOTf, activates the sulphur reagent (Scheme 50).137
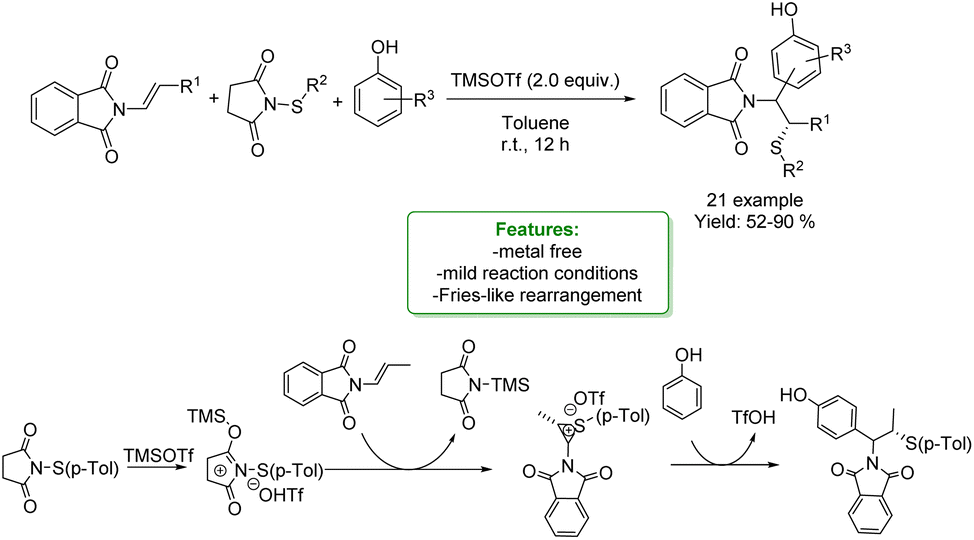 | ||
| Scheme 50 Lewis acid-mediated three-component electrophilic thiolative functionalization of enamides.137 | ||
1.3. Allylic alkylation
The cinnamyl phenol core displays a broad range of biological activities such as anti-bacterial (novobiocin and obtusastyrene), anti-inflammatory, anti-allergic, anti-thrombotic, antioxidant, anti-cancer and immunosuppressant (mycophenolic acid) properties.138,139 Furthermore, many plant metabolites, such as eugenol and chavicol, are typical flavour/fragrance components of essential oils from herbs and spices (Fig. 5).140,141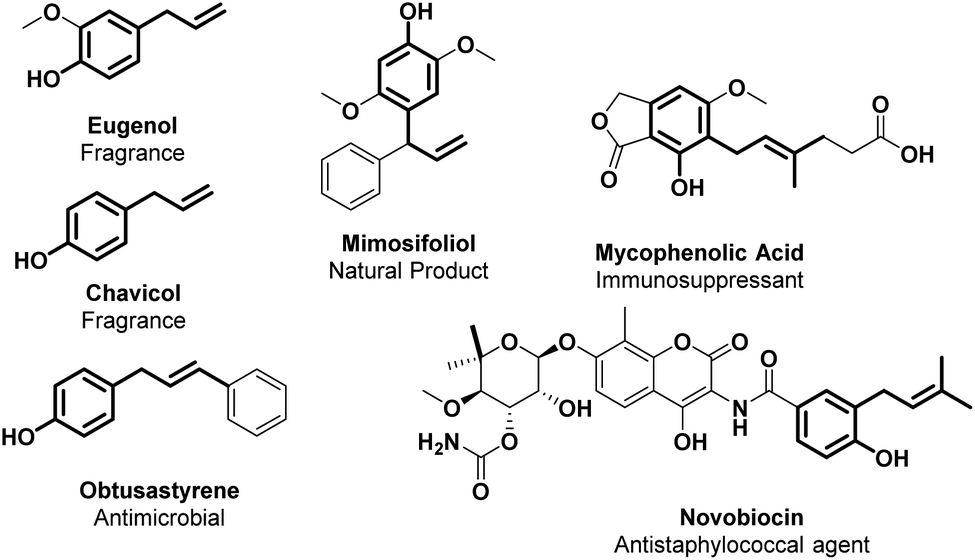 | ||
| Fig. 5 Allylic alkylated phenol structural moieties in API and agrochemical compounds. | ||
Considering the importance of this structural moiety, it is both essential and challenging to develop straightforward and regioselective synthetic approaches. Traditionally, Csp2–H allylic alkylation is accounted via a first O-alkylation followed by an aromatic Claisen rearrangement.42 Indeed, there are some issues to face to obtain a direct Csp2–H allylic alkylation, such as the poor regioselectivity (a mixture of regioisomers is often observed); the facility for ortho-allyl phenols to undergo intramolecular cyclization; and the common necessity of a pre-activated reaction partner bearing a leaving group.
Herein we report the latest strategies developed to address these challenges by employing simple-unprotected phenols.
1.3.1.1 Metal catalysis. Many efforts were made before 2019 to overcome limitations of allylic alkylation. For instance, metal-catalysts such as Mo,142,143 Ru,49,144 Pd,145,146 Rh147 and Ir148 were applied in the allylic alkylation of phenols. Generally, this reactivity requires the activation of the allylic alcohol moiety either as carbonates, acetates, or aryl boronic acids. Despite this, often the desired products are obtained with poor regioselectivity. Among the most recent examples, in 2021, Gedu's research group developed a synthetic strategy for Pd-catalysed highly para-selective allylation of phenols with inactivated allylic alcohols as reaction partners. The protocol was successful under an open atmosphere and mild reaction conditions. A good number of 1,3-diarylpropene derivatives were achieved in good to excellent yields. Among them, they obtained obtusastyrene and obtustyrene. The proposed mechanism is based on the Tsuji–Trost reaction, where an η3π–allylic complex is formed between the Pd-catalyst and the alcohol to activate the hydroxyl group for the coupling with the phenol (Scheme 51).149
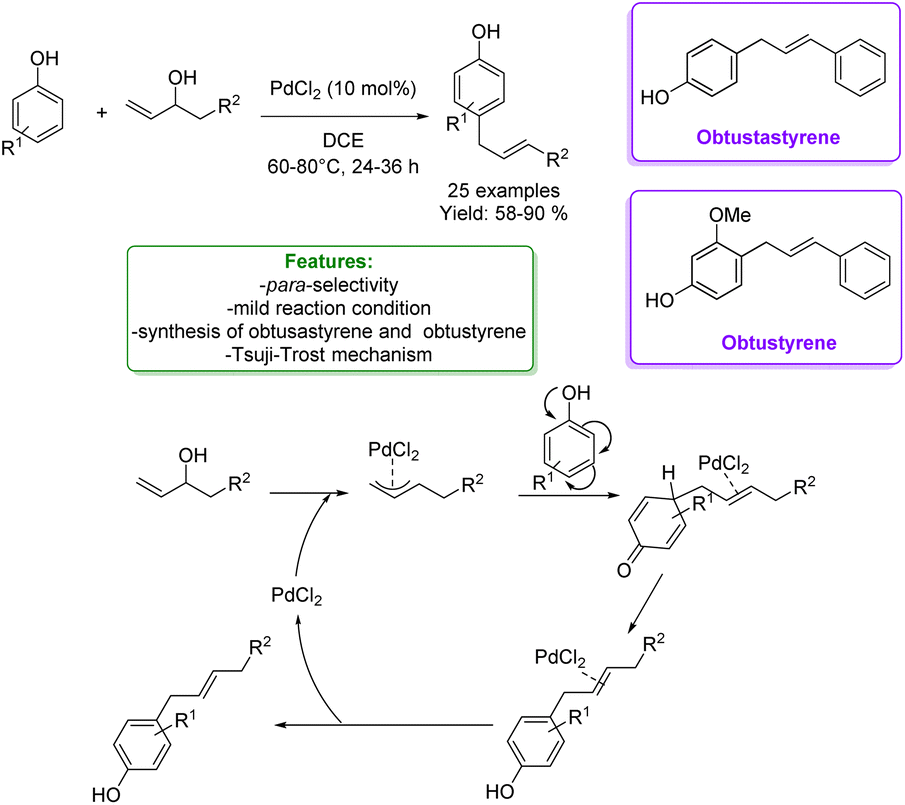 | ||
| Scheme 51 Pd-catalysed para-allylic alkylation of phenols.149 | ||
Shifting from para to ortho selectivity, in 2023, Yin et al. reported an ortho-Csp2–H allylic alkylation with 1,3-dienes. They implemented a Pd-catalysed allylic alkylation of electron-rich phenols and 2-naphthols with a diphosphine ligand to activate the catalyst, yielding a broad substrate scope with good chemo- and regioselectivity. Resorting to the use of bulky and electron-rich chiral phosphine ligand it was possible to perform the enantiomeric version reaching up to 55% ee (Scheme 52).150
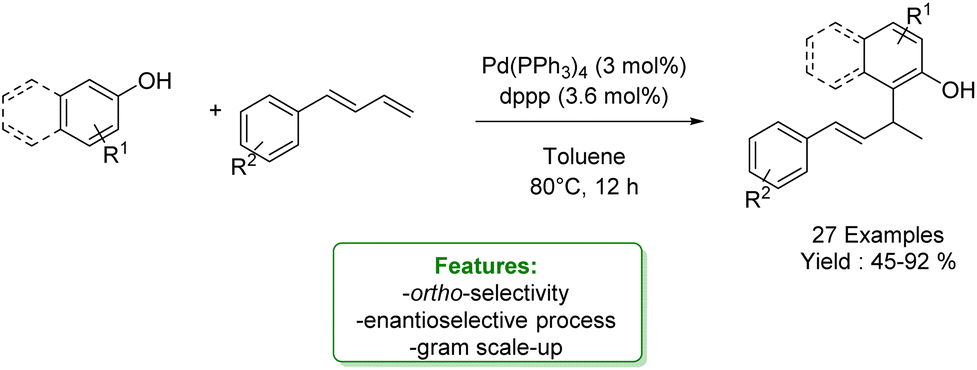 | ||
| Scheme 52 Pd-catalysed ortho-allylic alkylation of phenols.150 | ||
Several methods for transition-metal catalysed de-aromative allylic alkylation of naphthols have been developed in the last years151,152 in order to generate naphthalenones, which are interesting components of the pharmaceutical and agrochemical industries.153,154 You and coworkers proposed a highly enantioselective Rh-catalysed allylic de-aromatization of β-naphthols with racemic aryl–vinyl carbinol, in the presence of a chiral-ligand with TFA as an additive. The functionalized β-naphthalenone compounds were obtained in good yields with excellent enantioselectivity, up to 97% ee. Two further transformations on the β-naphthalenone product were carried out: a selective reduction of the ketone functionality to the corresponding alcohol; and the reaction with CH3Li, obtaining good yields with 72% ee (Scheme 53).155
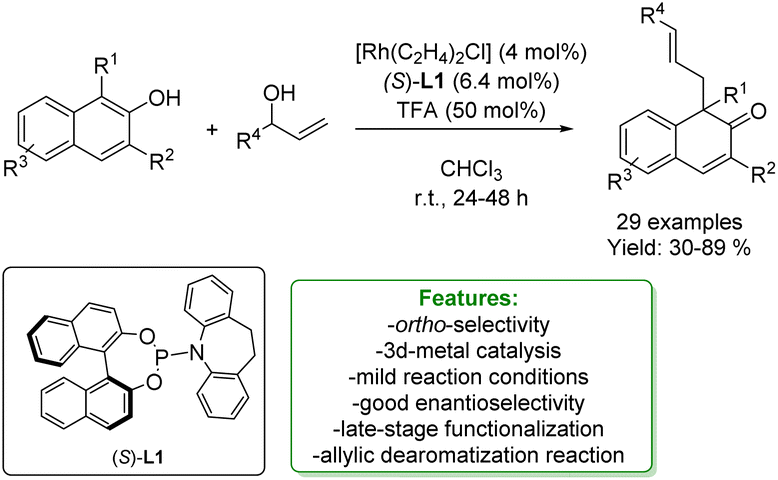 | ||
| Scheme 53 Rh-catalysed dearomatize allylic alkylation of 2-naphthols.155 | ||
With the same purpose, You et al. in 2020 explored the de-aromative allylic alkylation of 2-napthols using a Ni-catalyst, cheaper and more abundant compared to other 4d and 5d transition-metals. By utilizing Ni(cod)2 as a catalyst precursor, DPEPhos as a ligand and 4 Å molecular sieves as additives, the de-aromatization reaction of β-naphthols with aryl allylic alcohols proceeded smoothly under mild conditions. The desired β-naphthalenone products bearing a quaternary carbon centre were synthesized in moderate to good yields with excellent linear selectivity. Acidic molecular sieves played a crucial role in the activation of the reaction pathway since they could generate the π-allyl nickel species from allylic alcohol and Ni(0). The direct allylic alkylation to the naphthol is the most viable process, however, an alternative pathway involving O-allylation with a subsequent α-C alkylation cannot be excluded. The authors also performed a gram scale-up to validate the synthetic utility of this reaction (Scheme 54).156
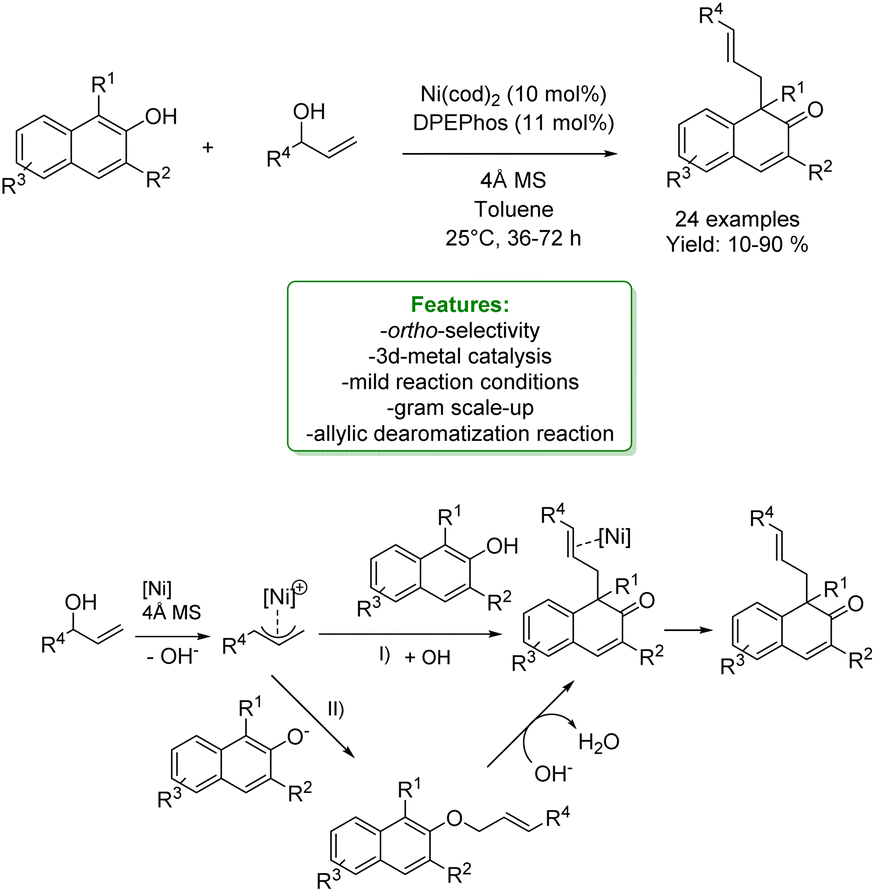 | ||
| Scheme 54 Ni-catalysed allylic alkylation of 2-naphthols.156 | ||
X. Wang et al. reported a Rh(II)-catalyzed reaction to afford the para-selective Csp2–H functionalization of free phenols in a multicomponent reaction with diazoesters and allylic carbonates. Mechanistic studies suggested that the reaction occurred through a tandem process of carbene-induced para-selective Csp2–H functionalization, followed by Rh(II)/XantPhos-catalysed allylation. Moreover, it was found that the base additive plays a crucial role in guaranteeing the para-selectivity functionalization. The optimized protocol was applied for the late-stage modification of different pharmaceuticals such as L-menthol and S-(−)-β-citronellol (Scheme 55).157
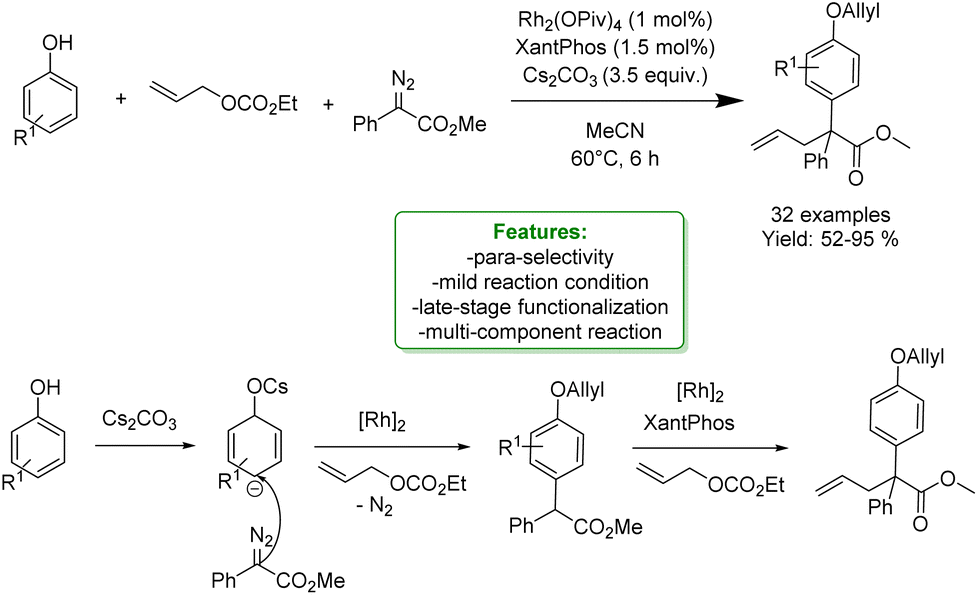 | ||
| Scheme 55 Rh(II)/Xantphos-catalysed allylic alkylation of phenols.157 | ||
Y. Xia et al.158 reported a rhodium-catalysed allylation of phenols and naphthols with gem-difluorinated cyclopropanes. These reactants were employed as highly reactive allyl surrogates, providing allyl arene derivatives in good yields with high regioselectivity under mild conditions (Scheme 56).
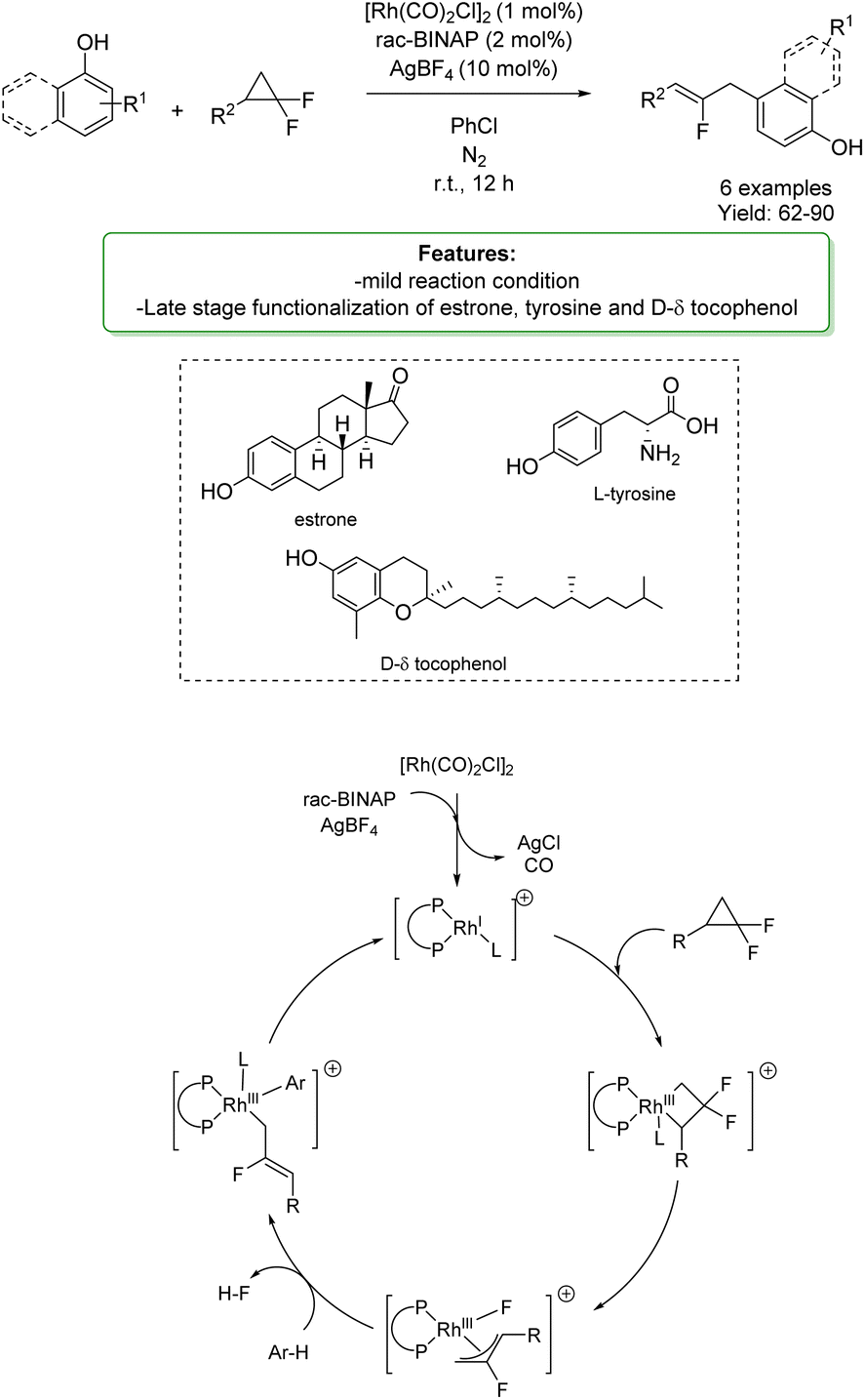 | ||
| Scheme 56 Rh-catalysed allylation of phenols and naphthols with gem-difluorinated cyclopropanes.158 | ||
Allylation occurred preferentially at the para-position; however, when the para-position was blocked, the ortho-position was favored. The developed protocol was applied in the late-stage functionalization of biologically active compounds: estrone, protected tyrosine, and D-δ-tocopherol. The authors proposed a plausible mechanism: initially, the pre-catalyst [Rh(CO)2Cl]2 de-dimerizes to form a highly active cationic rhodium(I) species in the presence of rac-BINAP and AgBF4. A C–C bond oxidative addition of the gem-difluorinated cyclopropane occurs to form a rhodium complex, which then undergoes a β-fluoride elimination to generate the key intermediate. As proposed, the fluorinated allylrhodium species would be highly electrophilic, triggering a C–H bond activation of the arene through an electrophilic metalation pathway. Finally, a reductive elimination process occurs to afford the desired allylation product, regenerating the active catalyst (Scheme 56).158
1.3.1.2 Acid/base catalysis. Allylic alkylation of phenols using 1,3-dienes under acidic conditions has always been quite challenging. Dienes are extremely reactive, often leading to the formation of dimerization or polymerization products. Moreover, the resulting substituted phenols are significantly more reactive than the starting materials. This increased reactivity can lead to the formation of multi-alkylated products or to intramolecular cyclization, resulting in indane or dihydrobenzopyran products. In 2019 Li and coworkers performed a B(C6F5)3-catalysed hydroarylation process where the borane enhances the acidity of the hydroxy group triggering the protonation of 1,3-dienes for subsequent electrophilic alkylation reactions. They explored a large variety of products obtaining good to excellent yields (40–91%). Estrone and ethinylestradiol were also used as substrates in a late-stage functionalization process gaining 70% and 83% of isolated yields, respectively. The authors provided both theoretical and experimental mechanistic insights that shed light on the key-role played by the Lewis acid adduct in accessing the ortho-allylated phenol via Friedel–Crafts type reaction. Product transformations to access heterocycles such as benzofurans and benzopyrans were performed to test the synthetic relevance of this protocol, obtaining very good yields (75% and 79% respectively) (Scheme 57).159
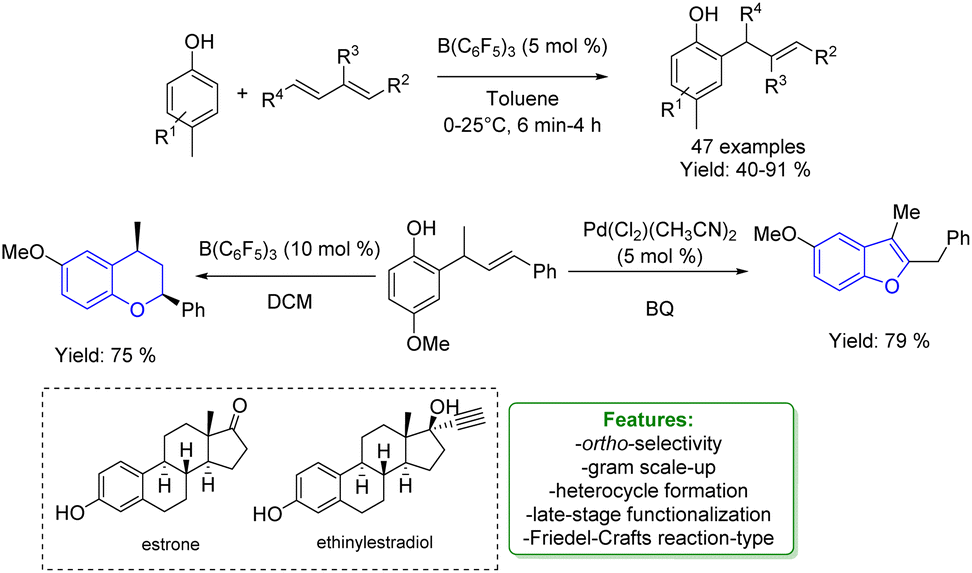 | ||
| Scheme 57 Borane-catalysed ortho-allylic alkylation of phenols.159 | ||
Another example of allylic allylation of phenols with 1,3-dienes via the acid Friedel–Crafts reaction-type was proposed by Zhang, Liu and coworkers. Triflic acid was used as a catalyst to access para-allylic phenols under mild reaction conditions, yielding sufficient to very good yields. An ortho-allylic product was obtained in a 40% yield when a para-substituted phenol was used as the substrate. Further transformations of the product such as the debromination on the double bond, the Suzuki coupling with a phenyl boronic acid and the hydroxy group transformation were developed (Scheme 58).160
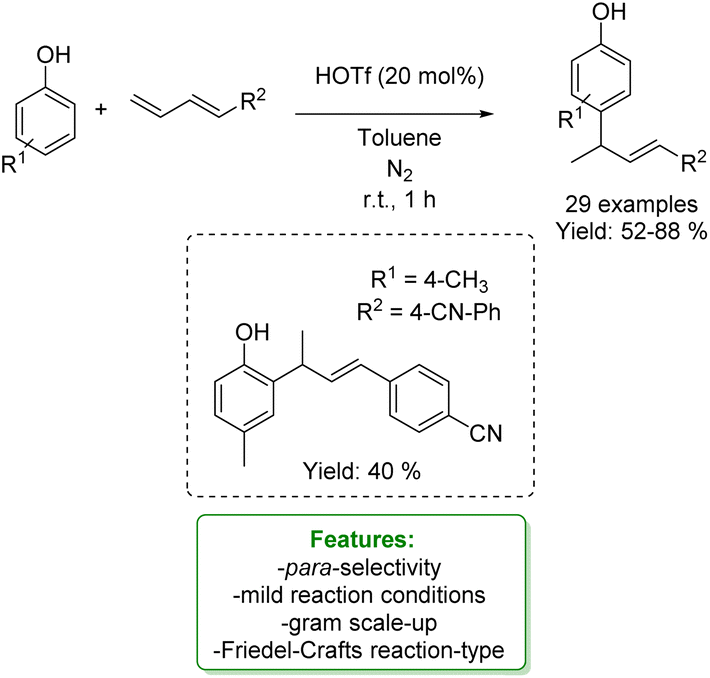 | ||
| Scheme 58 Triflic acid-catalysed para-allylic alkylation of phenols.160 | ||
In 2021, Liu and coworkers proposed a Lewis-acid-promoted para-allylic alkylation of phenol. Diazo compounds were employed as exceptionally reactive and versatile reagents capable of generating carbenes in order to synthesize para-allylated phenols through an insertion reaction. A substrate scope of 25 examples was demonstrated with isolated yields ranging from 50% to 99%. Other transformations occurred on the formed products, including ortho-bromination, selective reduction of ester groups to carboxylic groups, and various conversions of hydroxy groups to acetate, methoxy, and triflate groups (Scheme 59).161
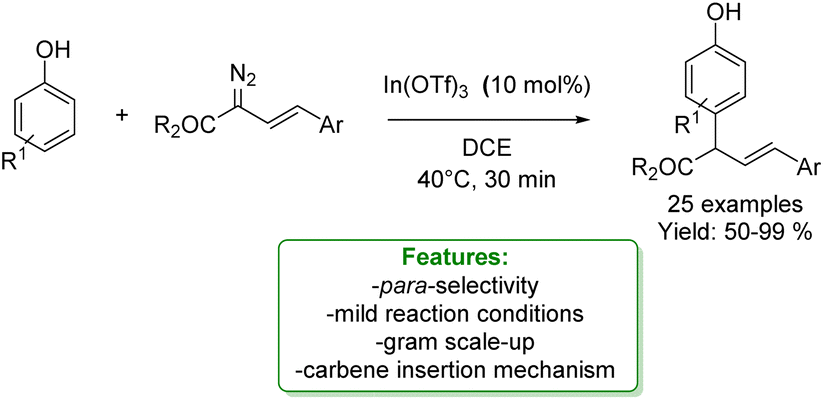 | ||
| Scheme 59 Lewis acid-catalysed para-allylic alkylation of phenols.161 | ||
N. Shankaraiah et al. reported an example of para-propargylation of phenol via the electrophilic Friedel–Crafts reaction conducted on 3-hydroxy-3-phenylethynyloxindoles under microwave irradiation (MW) assisted heating. Lewis acid BF3·OEt2 was used to catalyse the Friedel–Crafts arylation of 3-hydroxy-3-phenylethynyloxindoles with 2,6-disubstituted phenols to access various substituted 3-substituted-3-propargyl oxindole scaffolds under mild reaction condition. These new scaffolds can be utilized in the drug discovery process for their potential therapeutic interest (Scheme 60).162
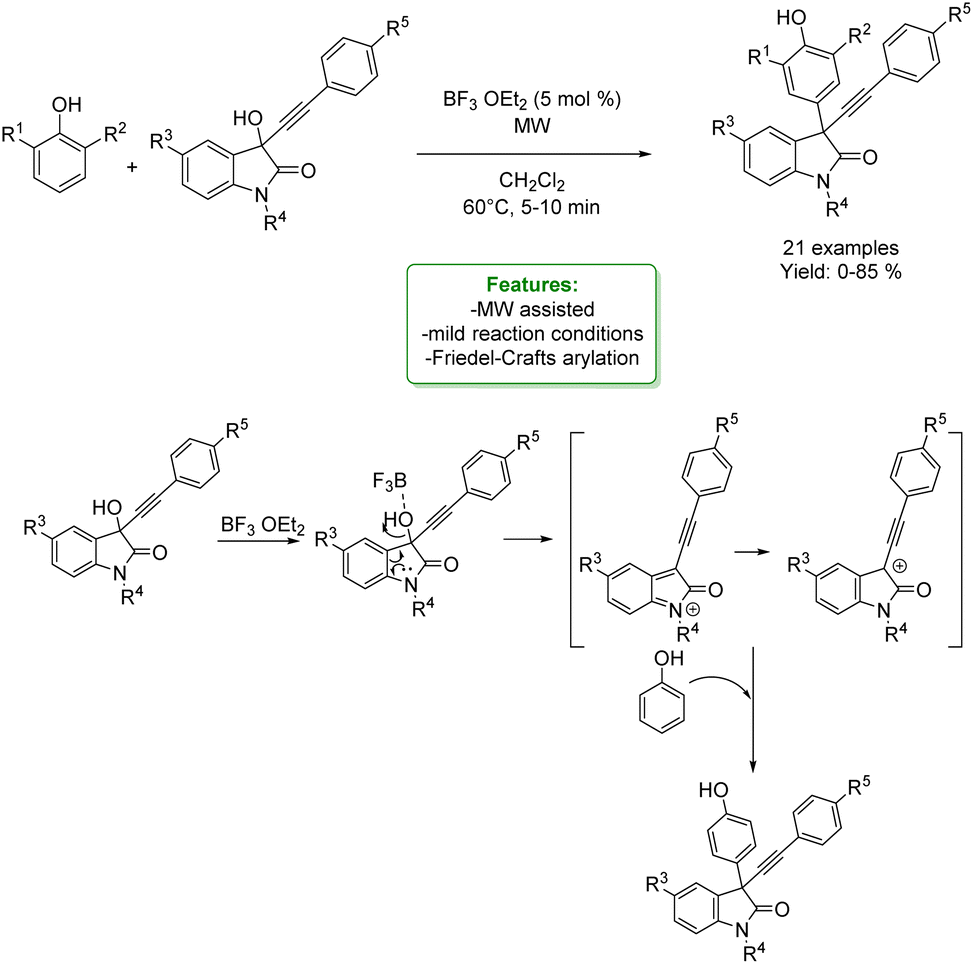 | ||
| Scheme 60 Microwave-assisted Friedel–Crafts propargylation of phenols to 3-substituted-3-propargyl oxindole scaffolds.162 | ||
S. Li et al. reported another interesting metal-free example of propargylation of phenol. The authors developed a borane-catalyzed, B(C6F5)3, sequential addition of terminal alkynes to para-substituted phenols, affording a wide range of ortho-propargylic alkylated phenols bearing congested quaternary carbons. The methodology involved a sequential phenol alkenylation/hydroalkynylation to afford the ortho-propargyl phenols. The propargylation products could undergo a wide range of transformations to afford heterocycles and other phenol analogues. The authors reported DFT calculations and control experiments for a better understanding of the reaction mechanism. The reaction starts with the proton transfer from the OH group to the boron catalyst, forming a tight ion-pair with the alkyne species. This consists of a highly electrophilic vinyl cation and a borate-phenol anion as the counter-anion, which could readily undergo electrophilic addition reactions to afford the Wheland intermediate. The subsequent re-aromatization followed by dissociation of B(C6F5)3 allows the formation of alkylated phenol by restoring the catalyst. Competing with catalyst regeneration, the complex may also undergo intramolecular protonation to generate a borane-stabilized tertiary carbenium ion that undergoes electrophilic addition reactions with another alkyne molecule to form a zwitterionic intermediate. This is then deprotonated to provide the neutral complex. Finally, the dissociation of Lewis acid regenerates B(C6F5)3 and releases the ortho-propargylation product (Scheme 61).163
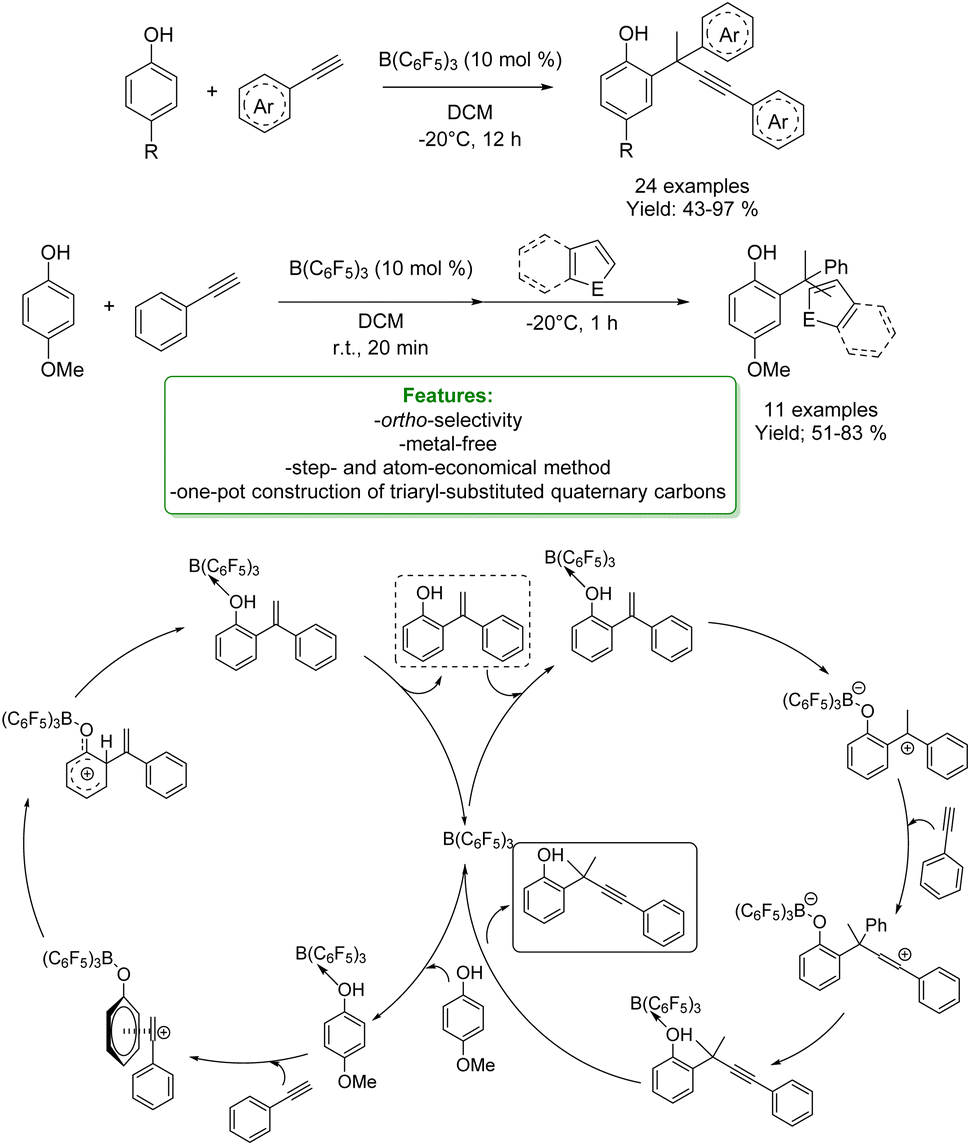 | ||
| Scheme 61 Borane-catalysed ortho-propargylation of para-substituted phenols with alkynes.163 | ||
1.4. Alkenylation and alkynylation
Alkenylated phenols are important structural motifs in natural products (such as resveratrol, dienestrol, caffeic acid, and curcumin) and pharmaceuticals (such as entacapone) (Fig. 6).4 These scaffolds are crucial intermediates in synthetic organic chemistry making accessible the cyclization to O-heterocycle compounds.46,164,165 For this reason the direct Csp2–H addition of olefins to phenols is an attractive way to access those structures in an atom-efficient manner.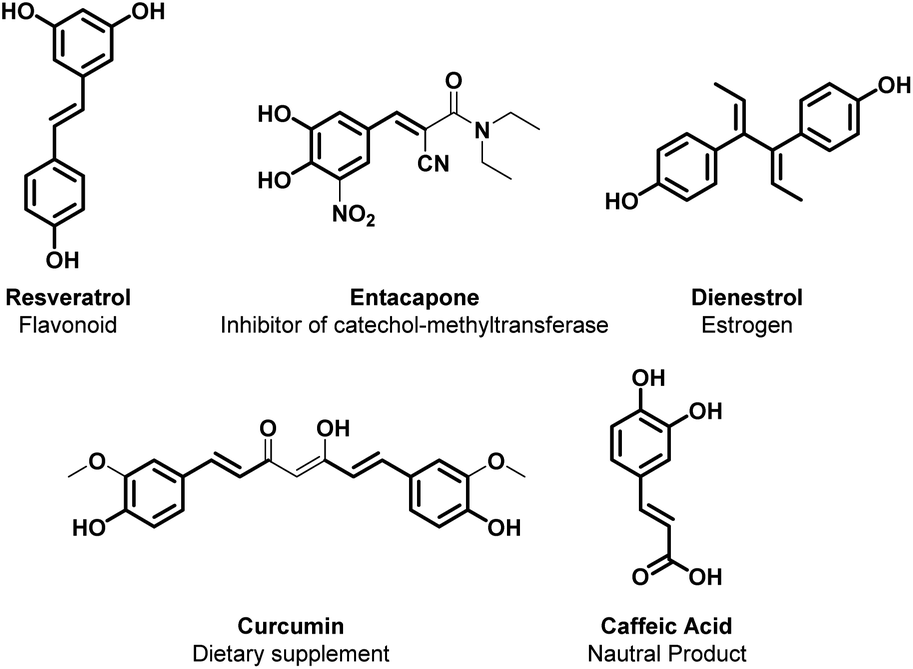 | ||
| Fig. 6 Alkenylated phenol structural moieties in API and agrochemical compounds. | ||
Traditionally, ortho-alkenylation of phenols is achieved through consecutive ortho-halogenation/Mizoroki–Heck cross-coupling reactions with alkenes.166,167 However, these methods suffer from limited functional group (FG) compatibility, poor atom-economy, and harsh reaction conditions. Thus, the development of a new approach to obtain vinylated phenols is highly desirable. The pioneering work in the direct Csp2–H alkenylation was reported by Yamaguchi using a stoichiometric amount of SnCl4 or GaCl3.168,169 Catalytic protocols remain challenging due to the preferred functionalization of the unprotected OH group, which typically acts as an oxygen-based nucleophile, resulting in preferential C–O bond formation. Furthermore, alkenylation leads to the formation of regioisomers (ortho- and para-products). Moreover, some phenol derivatives are sensitive to oxidants and acids, requiring mild conditions for their transformations.
Analogue considerations can be translated also to the alkynylation of phenols. Yamaguchi et al. reported the first examples of ortho-ethynylation of phenols with chloroethynyl-triethylsilane in the presence of GaCl3 as the catalytic system. It first reacts with phenol to form the active species, liberating hydrogen chloride. The phenoxy gallium species formed attack the silylated-ethyne to form a vinyl gallium intermediate. The β-elimination then leads to the ortho-ethynylatedphenol and to the regeneration of GaCl3.170 A. M. Echavarren et al. reported a Ru-catalysed peri-alkynylation of naphthol with bromoalkynes via insertion and elimination.171 The introduction of the alkynyl functional group opens many other transformations leading to O-heterocycle compounds via intramolecular cyclization. Examples of transitionmetal-catalysed synthesis of O-heterocycles are reported for the synthesis of benzochromenes.172 A related Rh(III)-catalysed reaction of 1-naphthylaminederivatives gives 1H-benzo[de]quinolines.173–176 H. Jiang and co-workers reported a one-pot procedure for the construction of highly functionalized benzo[b]furans via Pd-catalysed multicomponent reaction of haloalkynes, o-aminophenol and isocyanides.177 Inspired by this work H. Jiang et al. reported a Pd-catalysed sequential C–H alkynylation/annulation of 2-phenylphenols with haloalkynes.178 Interesting examples are reported for the synthesis of coumarins via ortho-substitution of phenols with alkynoates initiated by an ortho-palladation. Indeed, treatment of alkynoates with electron-rich phenols in the presence of a Pd-catalyst and an acid does generate coumarins.164
In the following sections, we report the latest transition metal catalysed Csp2–H alkenylation.
1.4.1.1 Metal catalysis. Transition-metal catalysis is the privileged method to access the alkenylation of unprotected phenols. The Fujiwara–Moritani reaction is a highly explored strategy, and many efforts have been devoted to realizing regioselective vinylation of phenol.179,180
Among the more recent examples, K. Takai et al. reported a Re2(CO)10 catalysed ortho-alkenylation of phenol with 1-phenyl-1-propyne obtaining a mixture of two stereoisomers with the Z-configuration being the predominant one (E/Z = 90:10 to 82:18). Sterically hindered phenols with a methyl group at the ortho-position prevented the alkenylation at any position, indicating that the interaction of the phenoxy oxygen atom with the Re centre is the key for the transformation. By increasing the reaction time and alkene amount they reported a selective formation of 2H-chromene derivatives via [3 + 2 + 1] cycloaddition (Scheme 62a).181 The same authors reported the use of Re2(CO)10 as a catalyst in the ortho-alkenylation with 1-aryl-1-propyne for late-stage modification of biologically active molecules containing phenolic structural moieties. Alkenylation was also performed with allenes yielding good results. The regioselective C-alkenylation de-hydrogenative cyclization was performed to access the benzofuran skeleton promoted by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). A mechanistic study was conducted to understand the reactivity of Re2(CO)10. Single-crystal XRD analysis revealed the formation of a (m-aryloxo) Re complex. Several control experiments were conducted, concluding that the OH group assisted the electrophilic alkenylation, instead of classic Friedel–Crafts type electrophilic functionalization (Scheme 62b).182
 | ||
| Scheme 62 Re-catalysed ortho-alkenylation of phenols with alkynes and cyclization to 2H-chromene.181,182 | ||
A. S. K. Hashmi et al. reported an Au-catalysed alkenylation of phenols. Thanks to the carbophilic π-acidic nature, Au demonstrated to be a promising metal for site-selective alkenylation. Using IPr as the ligand and sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF) as the co-catalyst, good to excellent yields were obtained at room temperature. Products displayed good selectivity for the formation of Z-isomers. When employing ortho-substituted phenols, the process exclusively delivered para-addition products. In the case of only meta-substituted phenols, mixtures of ortho- and para-addition products were observed, and yields were lower. A series of phenols with blocked para-position yielded ortho-addition at the sterically less hindered position. The kinetic study and kinetic isotope experiment revealed that the Csp2–H bond cleavage of phenol is not involved in the rate-determining step. Therefore, the Csp2–H activation of the phenol should proceed via the nucleophilic addition of the phenol onto the π-activated alkyne. The authors proposed that Au directly coordinates with the chloro-substituted alkyne carbon, and only then phenol attacks the highly electrophilic alkyne. The observed para-selectivity should result from both steric and electrostatic repulsion between the polarized alkyne and the OH moiety. The obtained products are of high interest as intermediates in the synthesis of benzofurans. For industrial purposes, they performed a gram-scale synthesis and a late-stage modification of estrone (Scheme 63).183
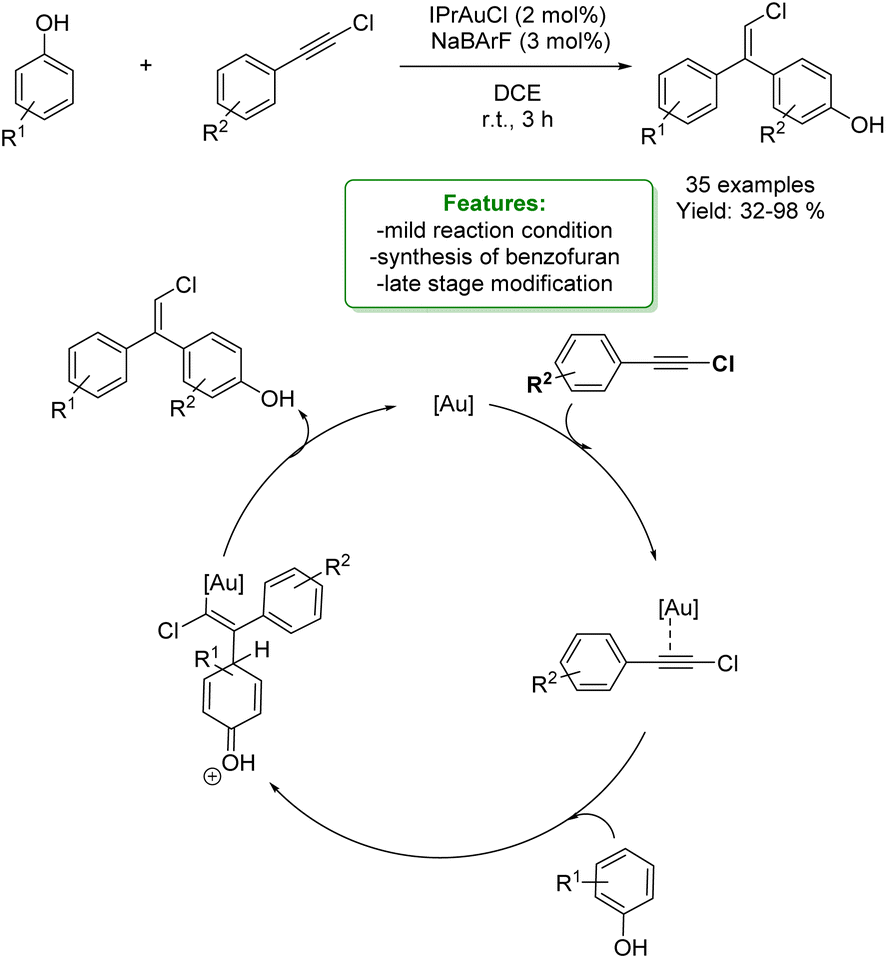 | ||
| Scheme 63 Au-catalysed alkenylation of phenols with chloroalkyne.183 | ||
L. Zhang et al. reported another interesting protocol of Au-catalysed alkenylation of phenols, with phenylacetylene derivatives. The remote amide group of the bifunctional ligand WangPhos plays a crucial role in the transformation, enabling the Au-catalysed hydroarylation of terminal alkynes under mild reaction conditions. This transformation featured a broad substrate scope and exhibited moderate to excellent yields. The synthetic utility of this chemistry was also demonstrated by the gram-scale synthesis and the one-step conversion of the product to versatile naphthol[2,1-b]furan derivatives (Scheme 64).184
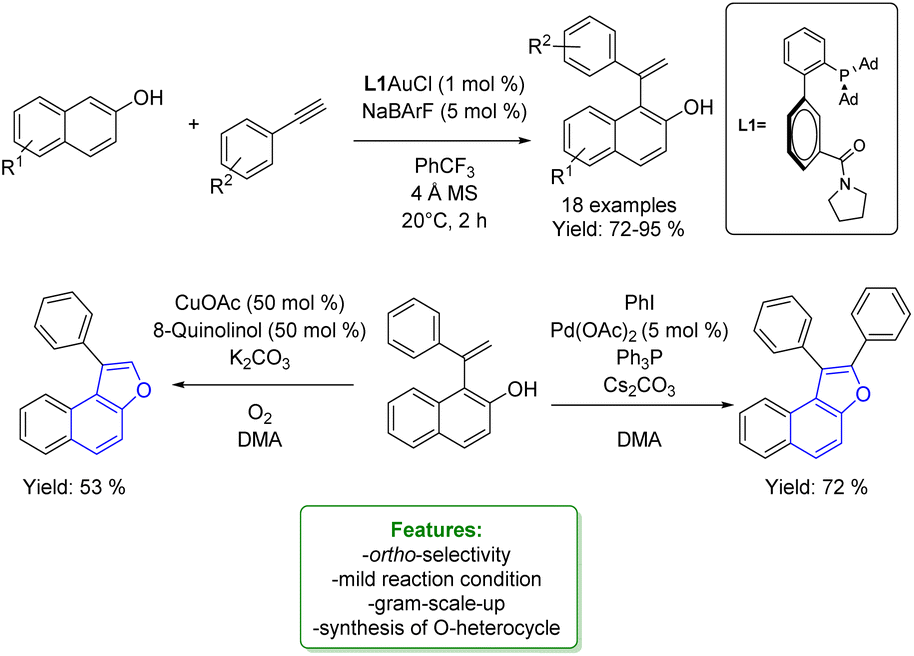 | ||
| Scheme 64 Au-catalysed alkenylation of phenols with terminal alkynes.184 | ||
Q. Zhu et al. reported an efficient and highly selective Pd-catalysed Csp2–H alkenylation of phenols with ethyl acrylates under mild reaction conditions. They accessed 32 ortho-substituted products in good yields, up to 95%. The required oxidative reaction condition was provided by K2S2O8. To further show the wide applicability of this method, the alkynylated phenols were converted into benzofurans and coumarins in good yields. The intramolecular cyclization of olefinic phenol to coumarin was further carried out under the same conditions. Late-stage modification of biologically active molecules with structural diversity, typical phenol-containing bioactive compounds, such as estrones, oestradiol, and ethinyl-oestradiol, was performed. The authors proposed a reaction mechanism in which phenol coordinates with the Pd-catalyst in the presence of the acetate counteranion. Then, a 1,3-migration of palladium happened, generating a C–H bond metalated intermediate. The insertion of alkene, followed by the β-H elimination, releases the alkenylated product and the Pd-species, which is oxidized to the Pd(II) species to complete the catalytic cycle (Scheme 65).185
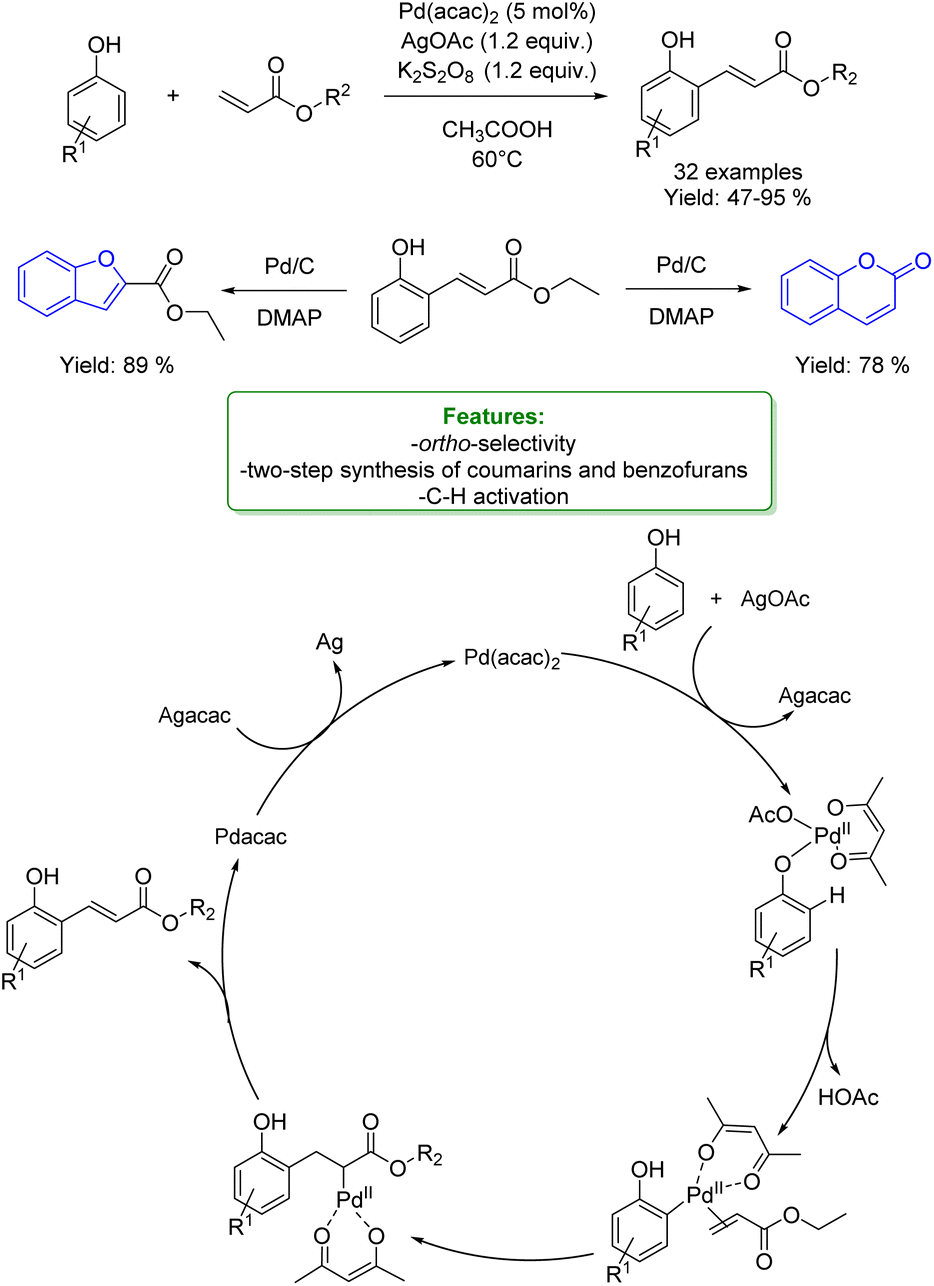 | ||
| Scheme 65 Pd-catalysed alkenylation of phenols with alkynes and cyclization to benzofuran and coumarins.185 | ||
1.4.1.2 Acid/base catalysis. L. Huang et al. reported a borane-catalysed (B(C6F5)3) hydroarylation of terminal alkynes with phenols, leading to the synthesis of 2-gem-vinyl-phenols with good regio-selectivity. When the reaction was performed with simple phenol, only ortho-alkenylated products were obtained. (B(C6F5)3) acted as a Lewis acid, activating the hydroxyl group and increasing the acidity of phenol, which was beneficial to the protonation of the terminal alkyne, followed by the ortho Friedel–Crafts-type addition of phenol. Once the Lewis adduct is formed, the proton transfer from the phenolic OH group to the alkyne occurs forming an ion-pair intermediate. Then an electrophilic attack of the carbocation to the phenol anion affords the de-aromatized intermediate. Re-aromatization, followed by the dissociation of (B(C6F5)3), provides the desired product and regenerates the catalyst. The robustness of the (B(C6F5)3)-catalyzed process was demonstrated by the gram-scale application (Scheme 66).186
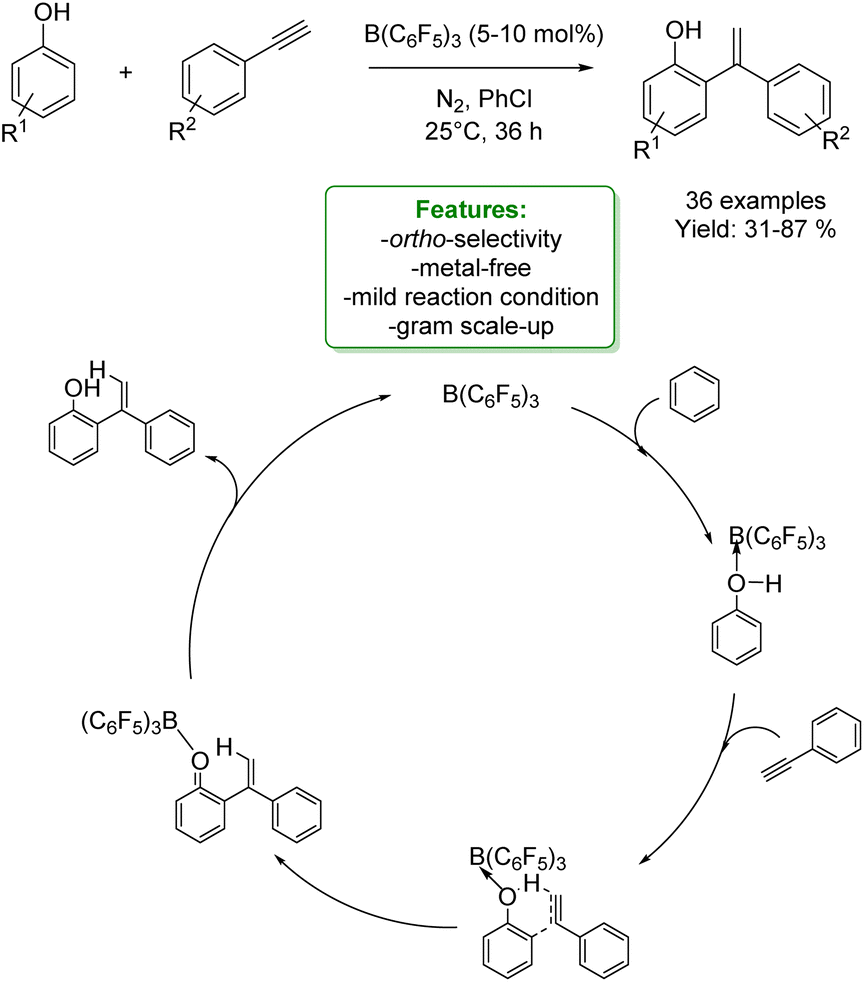 | ||
| Scheme 66 Borane-catalysed hydroarylation of terminal alkynes with phenols.186 | ||
G. Wang, S. Li et al. reported another interesting example of borane-catalyzed (B(C6F5)3) hydroarylation of aryl alkynes with phenols to obtain Z-alkenylated phenols under mild reaction conditions and reduced reaction time. The optimized protocols showed selectivity in providing ortho-alkenylation products. The origin of Z-selectivity was investigated by DFT calculations, suggesting that the E-alkenyl phenol was less stable than its Z-configuration. The isomerization of the E-product proceeds through a borane-assisted protonation/deprotonation sequence. The mechanism proposed by the authors confirms what was previously reported for the same catalyst (Scheme 67).187
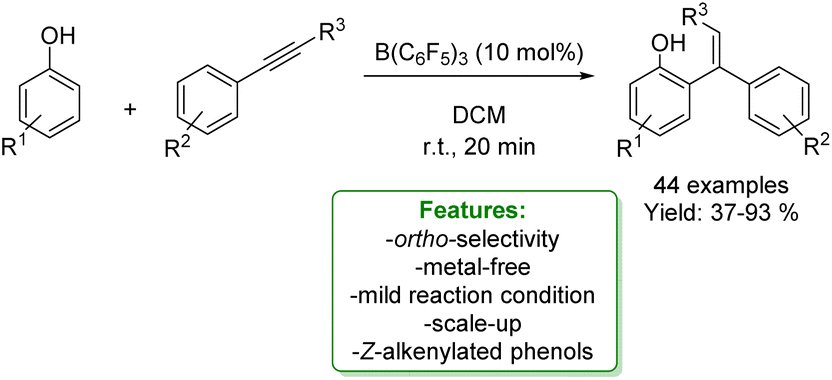 | ||
| Scheme 67 Borane-catalysed hydroarylation of terminal alkynes with phenols.187 | ||
K. N. Houk, B. Tan et al. reported the enantioselective synthesis of EBINOL scaffolds from naphthols. To construct this axially chiral motif asymmetrically, a chiral phosphoric acid-catalyzed asymmetric hydroarylation of alkynes was employed. This approach features complete E/Z-selectivity, with good to excellent yields (up to 99%), and excellent enantioselectivities (up to 99% ee). DFT calculations revealed the reaction mechanism and the origins of the enantioselectivity and E/Z-selectivities in this chiral phosphoric acid-catalyzed hydroarylation of alkynes. The potential applications of EBINOL-derived products were demonstrated by a series of asymmetric catalytic reactions (Scheme 68).188
 | ||
| Scheme 68 Chiral phosphoric acid-catalysed asymmetric hydroarylation of alkynes with naphthols to obtain the EBINOL scaffold.187 | ||
X. Lin et al. reported an asymmetric dearomatization reaction for the synthesis of axial chiral allene-derived naphthalenones from 2-naphthols through γ-addition and β,γ-alkynyl-α-iminoesters with complete atom economy. The chiral phosphoric acid catalyst (R)-A1 provided facile and efficient access to the asymmetric construction of a broad range of products in moderate to excellent yields (up to 98%) with high diastereoselectivities and enantioselectivities. Control experiments suggested that the stereoselectivity arises from dual hydrogen bonding interactions with chiral phosphoric acid, and the substituent at position 1 of β-naphthol plays a crucial role in controlling the regioselectivity (Scheme 69).189
 | ||
| Scheme 69 Chiral phosphoric acid symmetric dearomatization reaction for the synthesis of axial chiral allene-derived naphthalenones.189 | ||
S. Bhar et al. have devised a highly regioselective, transition metal-free protocol utilizing alumina–sulfuric acid as a recyclable heterogeneous solid acid catalyst for the hydroarylation of arylacetylenes and styrene with differently substituted phenols and naphthols. The protocol yielded a range of 1,1-diarylalkenes and 1,1-diarylalkanes in good to excellent yields within a reasonable reaction time. The reaction selectively occurred at the ortho-position. Presumably, the reaction is initiated by the initial polarization of the carbon–carbon multiple bonds by the protic hydrogen of alumina–sulfuric acid, leading to the development of incipient electron deficiency at the α-carbon. Subsequent intermolecular reactions with phenolic compounds might occur through a six-membered cyclic transition state involving the phenolic-OH moiety, resulting in an intermediate with exclusive ortho-substitution, followed by aromatization through tautomerization (Scheme 70).190
 | ||
| Scheme 70 Alumina–sulfuric heterogeneous solid acid catalyst for the hydroarylation of arylacetylenes and styrene with phenols and naphthols. | ||
1.4.2.1 Metal-free. T. Li and X. Qin et al. reported a metal-free tandem annulation reaction of 2-alkynylbenzaldehydes with 2,6-disubstituted phenols, yielding 2,3-diarylated indenones in good to excellent yields under mild reaction conditions. The authors disclosed two complementary modes of reaction, furnishing either ‘non-rearranged’ indenones or ‘rearranged’ indenones with high selectivity based on electronic effects and reaction temperature. A plausible mechanism was proposed for both pathways. In the ‘non-rearranged’ mechanism, dearomatizing addition of phenolate to the alkyne produces an intermediate, which undergoes intramolecular nucleophilic addition to the aldehyde. Re-aromatization generates inden-1-ol, which is oxidized to the ‘non-rearranged’ indenone product by TEMPO. In the ‘rearranged’ mechanism, dearomatizing addition of phenolate to the aldehyde generates an intermediate, followed by 5-exo-dig cyclization and rearomatization to phthalan. Fragmentation of the latter to para-quinone methide and intramolecular 1,6-conjugate addition furnishes a dihydroindenone intermediate. This intermediate is oxidized to the ‘rearranged’ indenone product by TEMPO or oxygen (Scheme 71).191
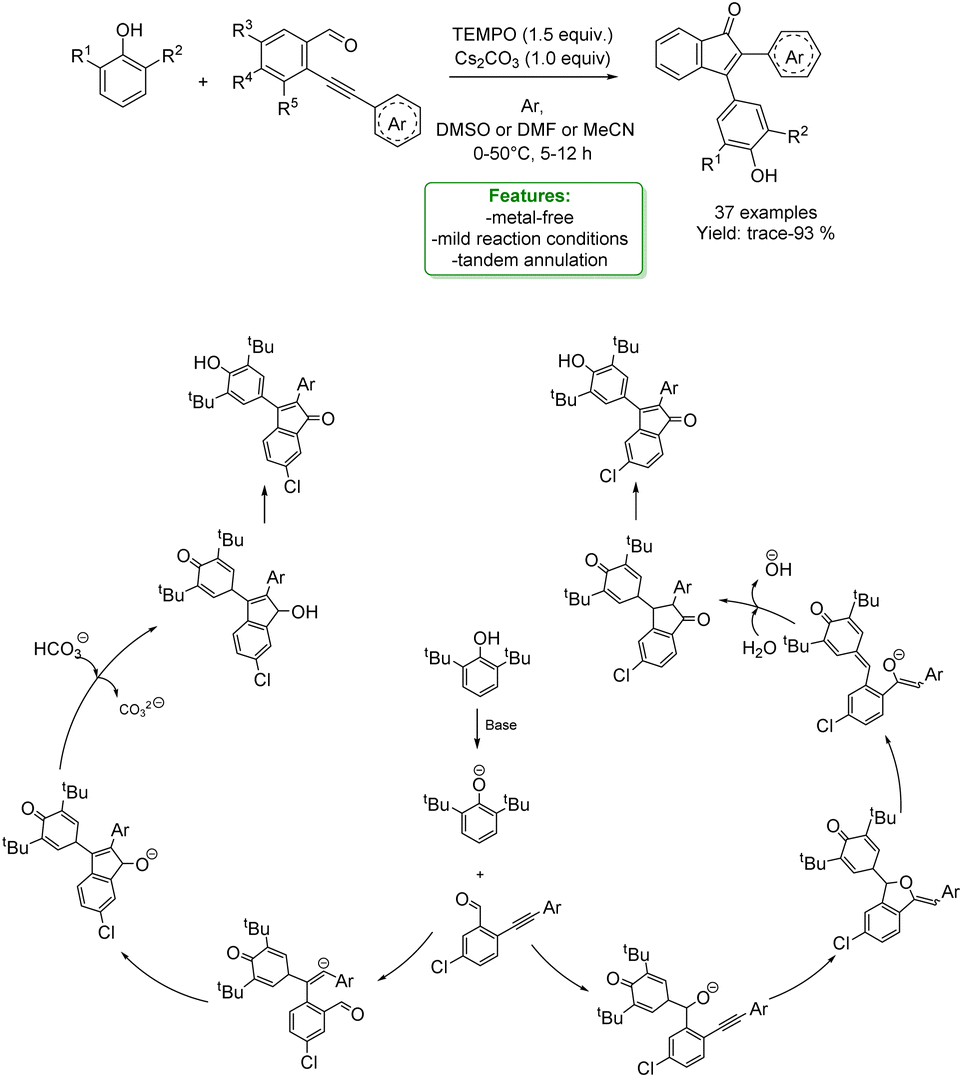 | ||
| Scheme 71 Tandem annulation reaction of 2-alkynylbenzaldehydes with 2,6-disubstituted phenols.191 | ||
R. Chegondi et al. reported a transition-metal-free Csp2–H arylation of cyclopentene-1,3-diones with phenols. The developed oxidative protocols with 2-naphthols proceed through a sequential base-mediated C-Michael addition, followed by oxygen insertion and subsequent α-hydroperoxy elimination. This operationally simple transformation is highly scalable and does not require any pre-functionalization, providing α-substituted β-naphthols bearing all-carbon quaternary centers in good to excellent yields. The potential applicability of this arylation reaction was also highlighted with a gram-scale reaction. A plausible mechanism has been proposed. Initially, the base-mediated C-Michael addition of β-naphthol to the α-position of pentene-1,3-dione occurs. Later, aromatization/protonation of the formed intermediate, followed by the interaction of O2 with hydrogen, gives the hydroperoxy intermediate. This oxidation step likely proceeds through the formation of hydroperoxide radicals via hydrogen transfer to oxygen, followed by C–O bond formation with the resulting carbon radicals. Furthermore, base-mediated elimination and subsequent aromatization afford the formal Csp2–H arylation product (Scheme 72).192
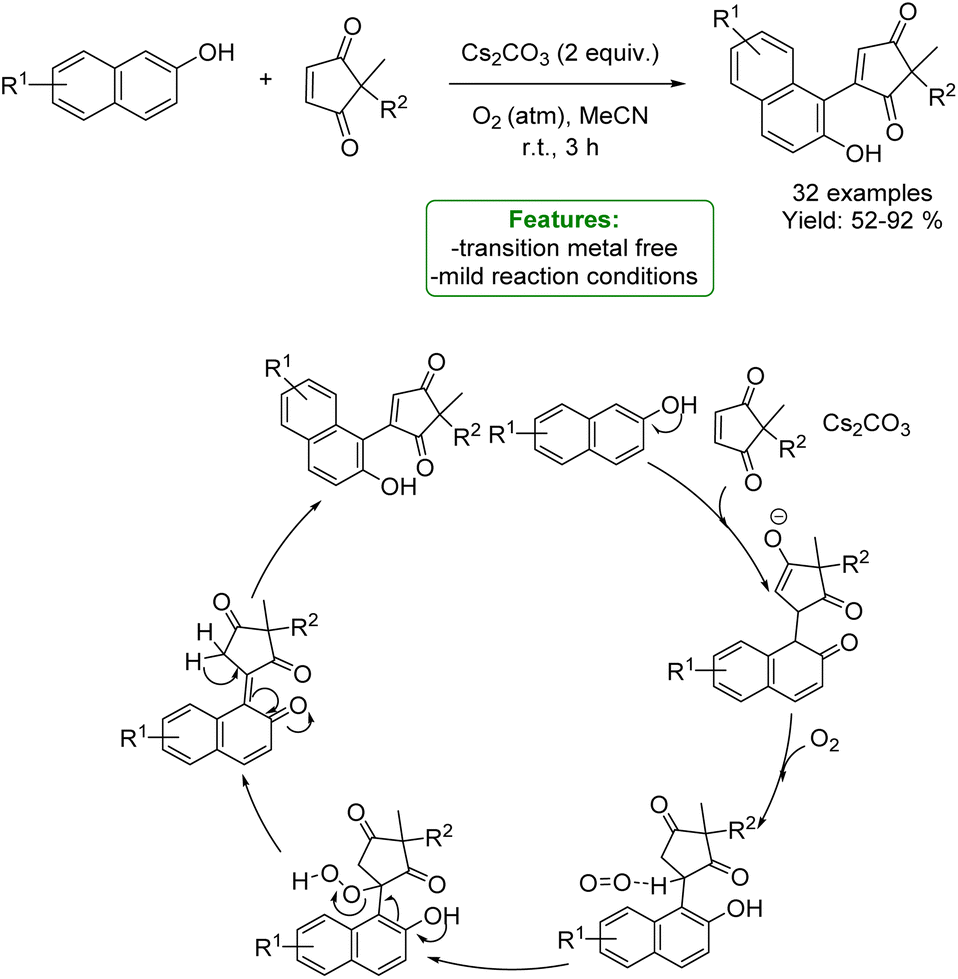 | ||
| Scheme 72 Transition-metal-free Csp2–H arylation of cyclopentene-1,3-diones with phenols.192 | ||
1.5. Arylation
Aryl-substituted phenols and naphthols, are privileged structural motifs in biologically active molecules including natural products (such as honokiol and piperitylmagnolol) and commercial APIs (such as bazedoxifene, reloxifene and eltrombopag). Several examples show antimalarial, anti(retro)viral, anti-inflammatory (diflunisal) or cytotoxic properties.25,26,193,194 These scaffolds are extensively applied as versatile reactants,195,196 building blocks for sensors197 and intermediates in the synthesis of catalysts and ligands (Fig. 7).198–201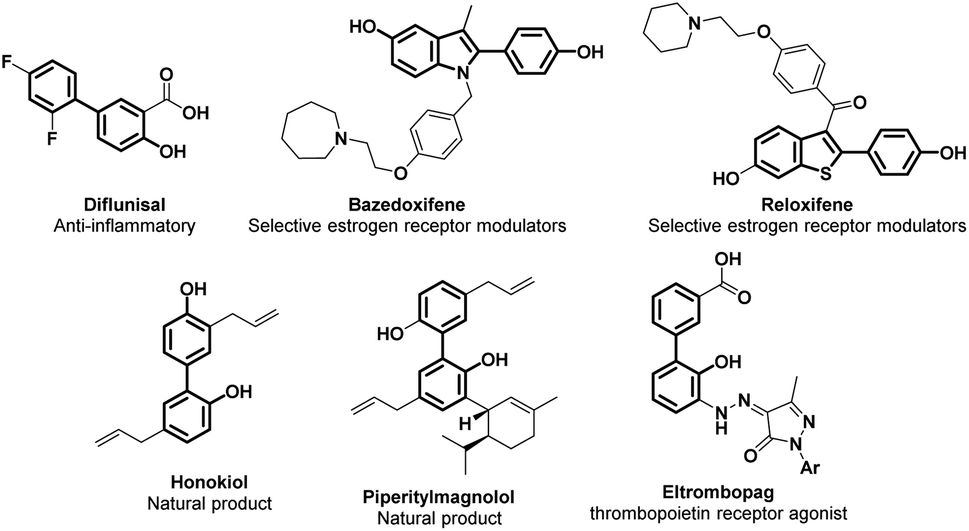 | ||
| Fig. 7 Arylation of phenol structural moieties in API and agrochemical compounds. | ||
Due to the importance of this structural motif, significant research efforts have been dedicated to the development of novel, effective methodologies for obtaining it. The most common synthetic methodology to access such an interesting structural motif involves transition-metal catalysed cross-coupling, which typically requires challenging ortho-selective halogenation or borylation of the phenols.202,203 The required pre-functionalization steps are necessary to achieve the control of the regio- and chemo-selectivity, betraying the atom- and step-economy of the overall protocol. A more desirable, but also particularly challenging, route involves the direct Csp2–H functionalization of the desired position. However this reactivity demands installation and subsequent removal of DG.204–206 In 2003, R. B. Bedford et al. reported a pioneering approach that entirely avoids the use of DG to selectively obtain ortho-aryl-phenols.207,208 On the heels of this work, great efforts have been made to achieve regioselective arylation of phenols via Csp2–H functionalization over the years.
In the upcoming sections, we report the latest transition-metal catalysed, acid/base catalysed and photocatalyzed protocols. We also focus on the non-catalytic processes: metal-free, photo-promoted and electrosynthesis examples of Csp2–H arylation of phenols.
1.5.1.1 Metal catalysis. Transition-metal catalysed Csp2–H arylation of phenols is a privileged route to access selective functionalization. Many attempts were performed before 2019 resorting to the use of different metal-catalysts (Rh and Pd), to perform para-functionalization, meta-functionalization and ortho-functionalization.204,208-210
Focusing attention on the earliest reported examples, in 2019, M. C. Kozlowski et al. reported a combination of experimental and computational studies of a Cr-Salen phenol–phenol cross-coupling with oxygen as the oxidant. The reaction between 2,6-substituted phenols and phenols with only unsubstituted ortho-positions available resulted in the selective formation of ortho–para cross-coupling products. Cross-coupling between 2,6-substituted phenols and phenols that had both ortho- and para-positions available resulted in the selective formation of para–para cross-coupled products. Reactions with 2-naphthol and its derivatives, with the EWG and EDG, lead to cross-coupling products in good yields, up to 88%. To probe the reaction mechanism, kinetic studies of the Cr-Salen catalysed cross-coupling were performed. The results showed that the rate-determining step of the reaction is the oxidation of the Cr(III) to the active Cr(V). DFT calculations and kinetic isotope experiments were performed to understand the origin of the selectivity which is determined during the C–C bond formation (Scheme 73).211
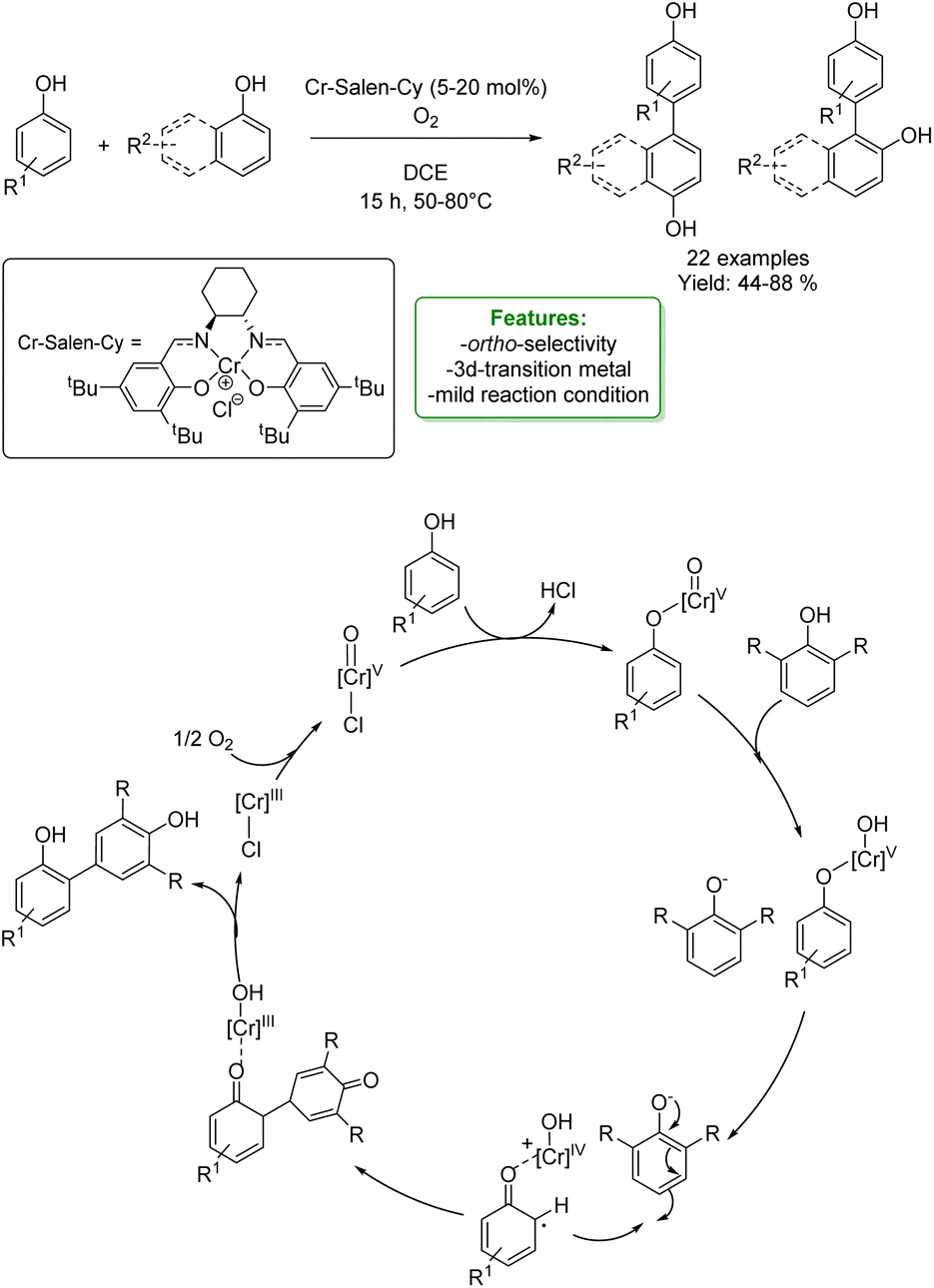 | ||
| Scheme 73 Cr-catalysed phenol–phenol cross-coupling.211 | ||
With the same goal, D. Pappo et al. reported a Co(II)[salen]-catalysed aerobic oxidative phenol–phenol cross-coupling to non-symmetric bi-phenols under mild reaction conditions. Aerobic conditions were guaranteed by conducting the reaction under air. The latter advantage in the greenness of the process was conferred by the possible recyclability of the 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) solvent. Under the optimized reaction conditions, different unsymmetrical bi-phenols were isolated in moderate to very good yields. In general, readily oxidized phenols with either ortho- or para-methoxy groups and 2-naphthols substituted with an EDG or weak EWG were suitable coupling partners. The authors postulated that the mechanism involves the coupling between a liberated phenoxyl radical and a ligated naphthoxyl radical, whereas the selectivity was attributed to the difference in the binding ability of the two phenols to the catalyst in HFIP (Scheme 74).212
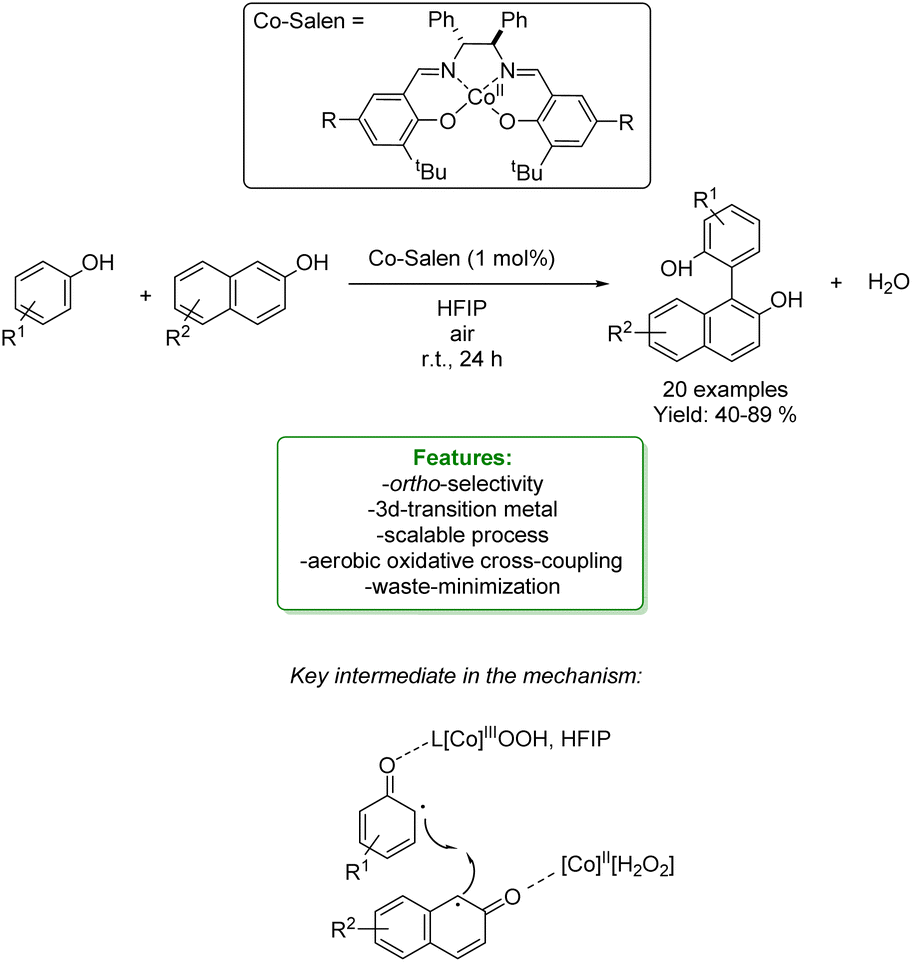 | ||
| Scheme 74 Co-catalysed phenol–phenol cross-coupling.212 | ||
Another example of the synthesis of bi-phenols was reported by B. Tan et al. in 2021. However, this case is not a phenol–phenol cross-coupling, since the authors reported a Pd-catalysed arylation of phenols with diazoquinones (1-DNQ) as arylating reagents for the construction of privileged bi-aryl frameworks, BINOL and NOBIN. The Pd-catalytic system plays a pivotal role in the excellent selectivity since it is the most effective to inhibit the undesired X–H insertion reactions for both 2-naphthols and N-Boc-2-naphthamines. This approach demonstrates good functional group compatibility and a broad range of structurally diversified BINOLs and BONIN derivatives were obtained in up to 88% yield (Scheme 75).213
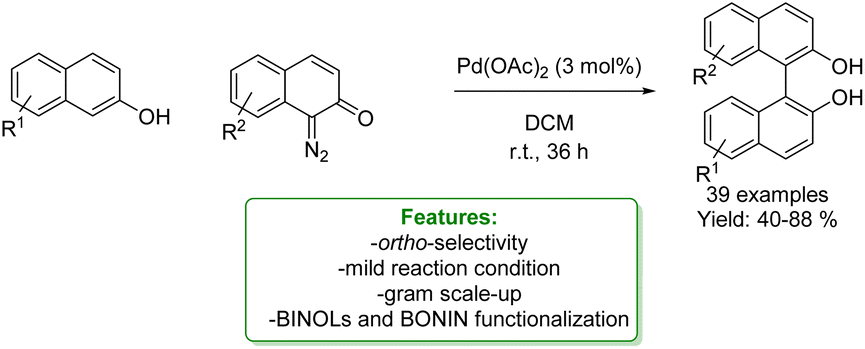 | ||
| Scheme 75 Pd-catalysed synthesis of bisphenols.213 | ||
In 2020, D. Pappo et al. reported a Fe-catalysed para-selective oxidative amination of phenols with primary and secondary anilines, resorting to the use of a peroxide (tBuOOH) and HFIP as privileged solvent under mild reaction conditions. This process provides a direct entry to benzoquinone anils via C–N coupling, or to N,O-biaryl compounds via C–C coupling, depending on the nature of the R1 substituent of the phenolic ring. When R = H or OMe, benzoquinone anils were obtained via sequential oxidative amination/de-hydrogenation (R = H) or oxidative amination/elimination (R = OMe) pathways. On the other hand, N,O-biaryl compounds are formed when R = alkyl through an oxidative amination/[3,3]-sigmatropic rearrangement (quinamine rearrangement) process. The postulated mechanism involves the coupling of a liberated anilino radical and an iron-ligated (4-R)phenoxyl radical (Scheme 76).214
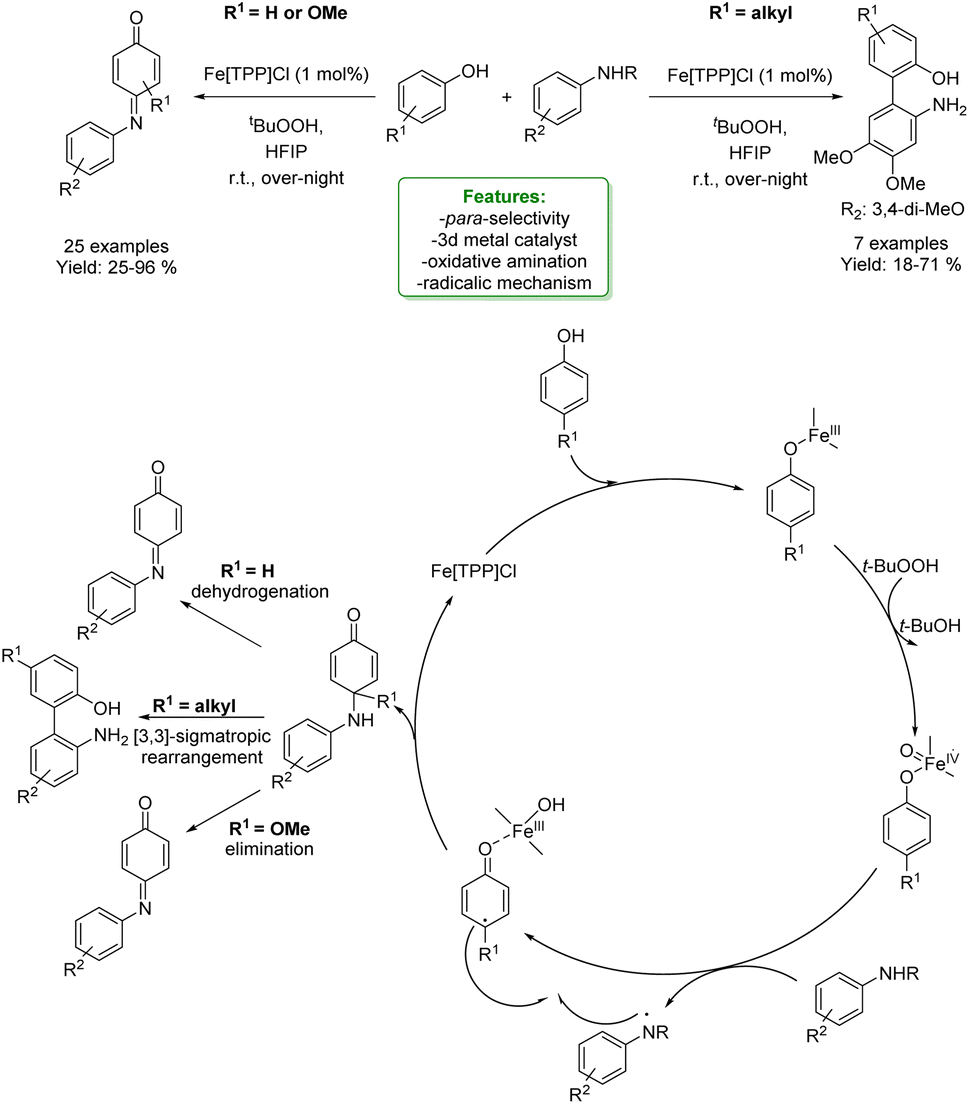 | ||
| Scheme 76 Fe-catalysed oxidative cross-coupling of phenol with anilines.214 | ||
Later in 2021, the same authors reported another Fe-catalysed oxidative cross-coupling of phenol with primary, secondary, and tertiary 2-aminonaphthalene resorting to the use of peroxide (tBuOOtBu) as the oxidant and HFIP as privileged solvent under mild reaction condition as well. The generality of this scalable method provides a sustainable alternative to obtain a wide range of variously functionalized N,O-biaryl compounds from moderate to excellent yields. A comprehensive kinetic investigation showed how the ternary complex that interacts with both the coupling partners and the oxidant is the crucial oxidative coupling stage. Furthermore, the studies showed that the reaction is regulated by off-cycle acid–base and ligand exchange processes215 (Scheme 77).
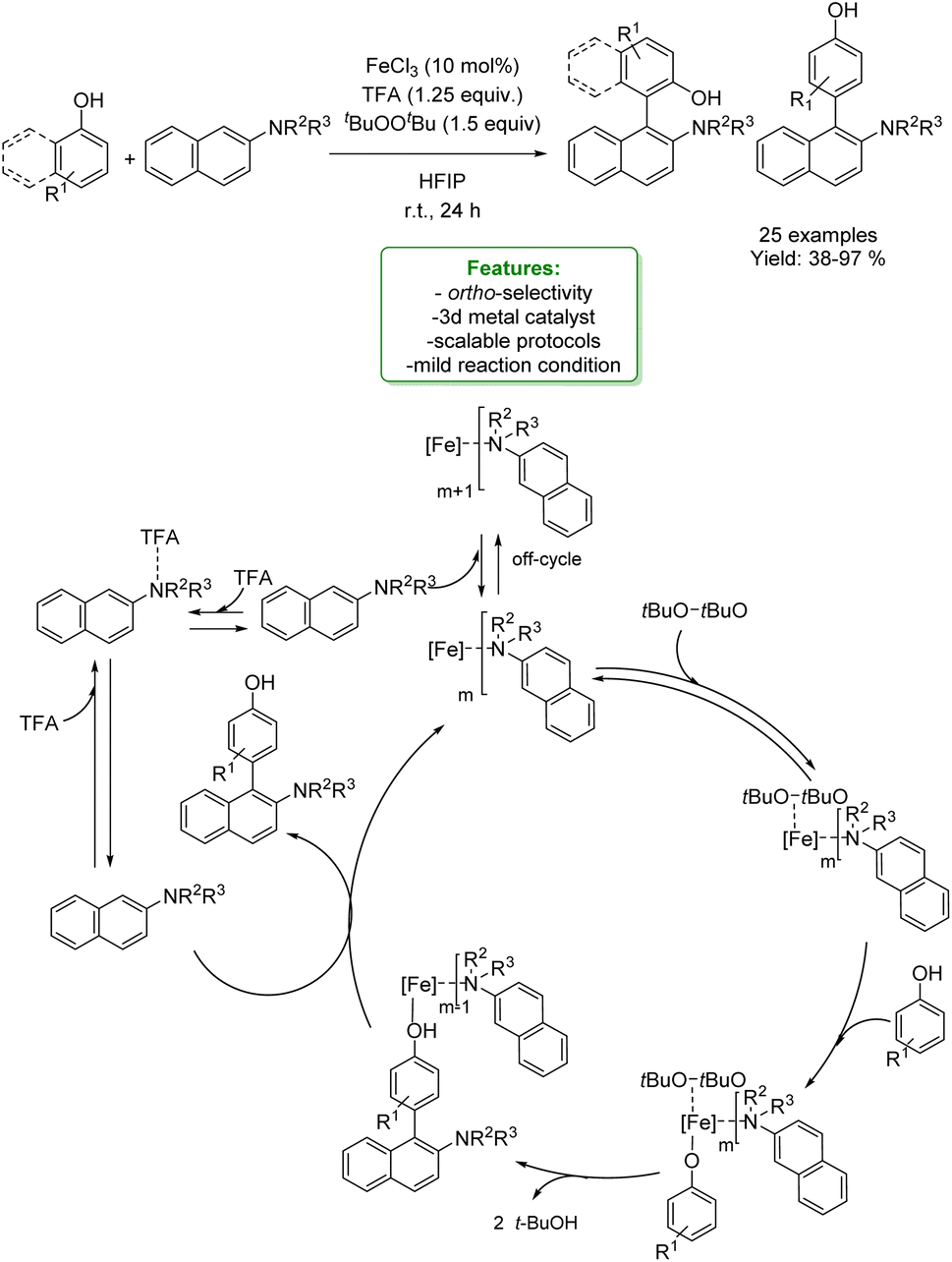 | ||
| Scheme 77 Fe-catalysed oxidative cross-coupling of phenol with aminonaphthalenes.215 | ||
1.5.1.2 Acid/base catalysis. Shin et al. reported an ortho-indolylation of phenols, with exceptional chemo- and regioselectivity, using two catalytic systems, HNTf2 or [Cu(OTf)]2 C6H6, under mild reaction conditions. Although HNTf2 provided an efficient metal-free catalytic system, the Cu-catalyst was applied to a broader range of substrates, obtaining no less than 49% isolated yield. The authors proposed that the reaction proceeded through an SN2 substitution for Brønsted acid catalysis or through oxidative addition for Cu(I)-catalysis. Interestingly, an EDG at the m-position of phenols, accelerated the coupling reaction, compared to the p-position. This is presumably because the m-substituent was placed para- to the reacting center, increasing the electron-density in the proposed [3,3]-sigmatropic rearrangement. Likewise, other electron-rich (hetero) arenes, such as indoles, pyrroles, and benzofurans, failed to provide the corresponding coupled product, under both conditions (Scheme 78).216
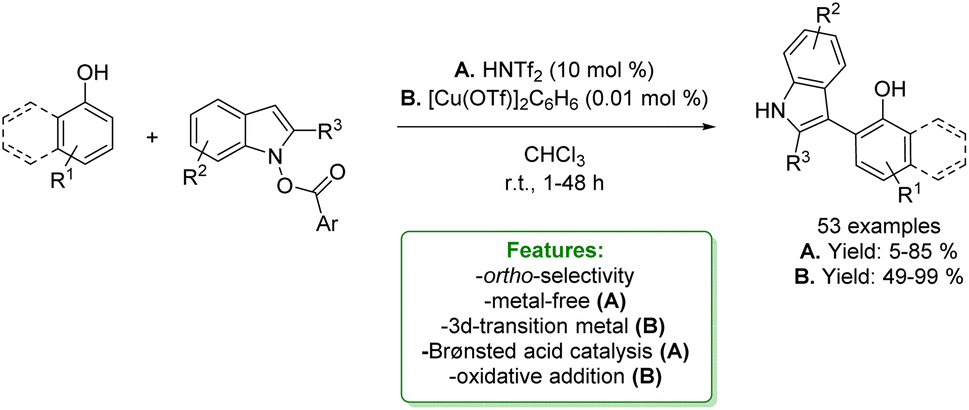 | ||
| Scheme 78 Brønsted (A) and Lewis (B) acid catalysed arylation of phenols with N-carboxyindoles.216 | ||
1.5.1.3 Photocatalysis. In the field of bi-phenol synthesis via phenol–phenol homo- and cross-coupling, a novel oxidative photocatalytic protocol was reported by M. C. Kozlowski et al. using MesAcr+BF4− as the photocatalyst. Air was used to guarantee the oxidative reaction conditions and HFIP as the reaction medium. Mechanistic studies suggested that an oxidation of the phenol followed by the nucleophilic addition of another phenol is a plausible pathway. The developed process afforded a range of new and known compounds in high yields via phenols homo- and cross-coupling. Poor selectivity is generally observed with homocoupling. For this reason, homocoupling reaction requires specific substitution patterns, which include an alkyl group at the para-position. On the other hand, cross-coupling resulted in being more effective with monosubstituted phenols thanks to the high oxidation potential (Scheme 79).217
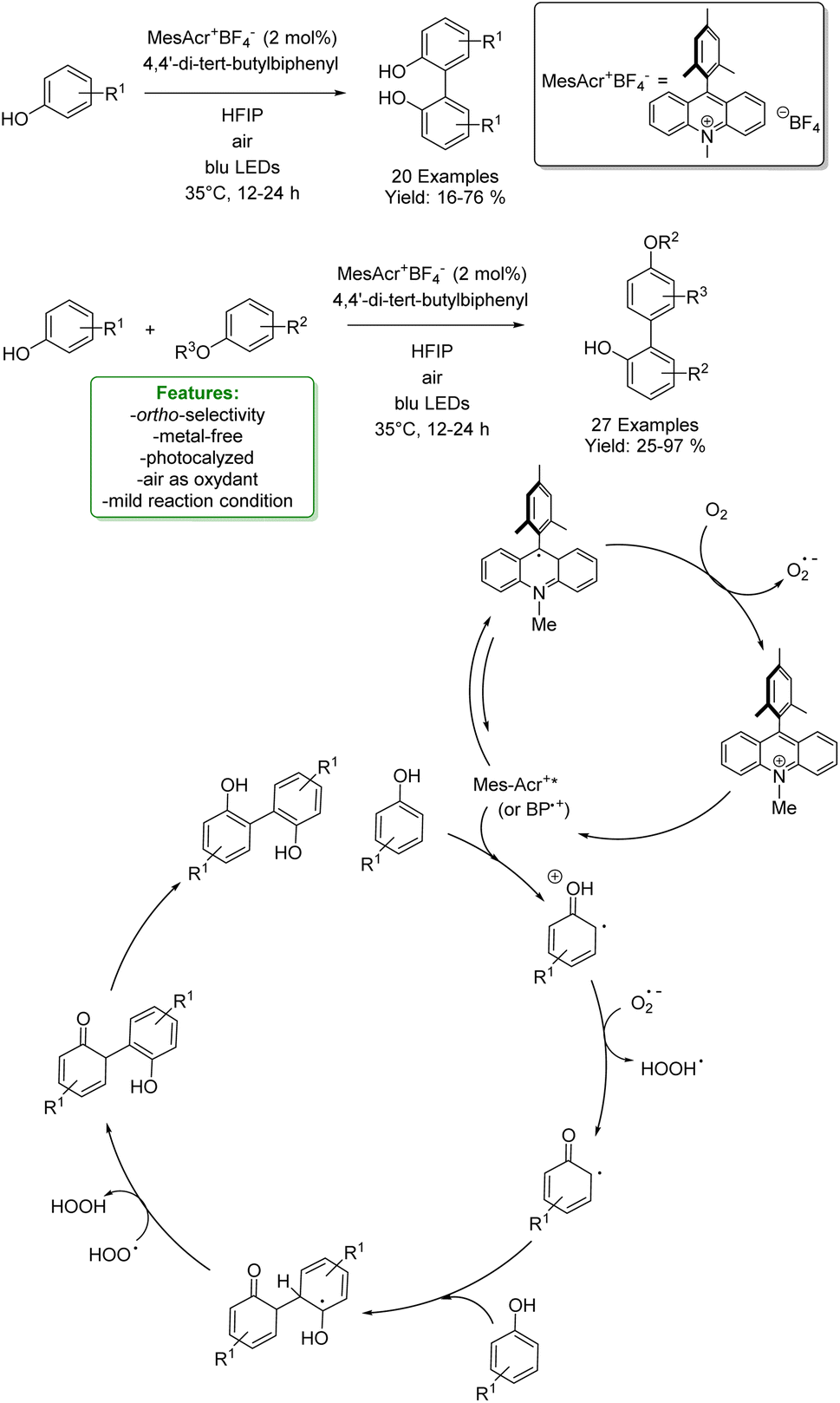 | ||
| Scheme 79 Photocatalytic phenol–phenol and phenol–ether coupling.217 | ||
An intriguing novel methodology reported the development of modular bismacycles as general solution for this challenge. L. T. Ball et al. in 2020 reported a telescoped Bi(V)-mediated ortho-arylation of phenols and naphthols exhibiting a broad substrate scope and tolerating synthetically useful functionalities. The reaction proceeded under mild conditions without the need to exclude air or moisture (Scheme 80a).218 In 2022, they reported the method for the meta-selective Csp2–H arylation of sterically congested phenols via Bi(V)-mediated electrophilic arylation and subsequent 1,2-migration. This protocol employed readily available reagents exhibited a broad substrate scope and tolerated synthetically useful functionalities (Scheme 80b).219
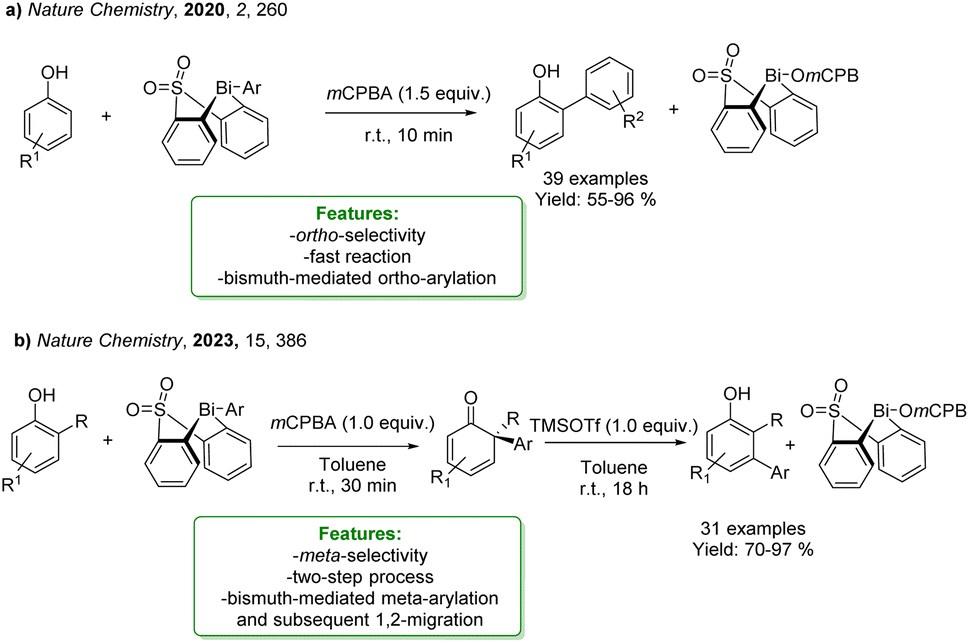 | ||
| Scheme 80 Arylation of phenols with modular bismacycles. (a) Ortho-arylation of phenols with modular bismacycles.218 (b) Meta-arylation of phenols with modular bismacycles.219 | ||
1.5.2.1 Metal-free. As far as transition metal-free methods are concerned, distinct approaches that facilitate the efficient incorporation of an aryl substituent into the structure of a phenol are gaining attention. Examples include Friedel–Crafts–type reactions and Brønsted acid-promoted reactions. However, these methods have inherent limitations because they are only capable of incorporating electron-rich aryl groups.
To overcome these constraints in a transition-metal-free context, M. Kalek et al. in 2019 developed a Csp2–H arylation of 2-naphthols using diaryliodonium salts as aryl-donors. The developed method enabled the introduction of electron-poor aryl rings, diaryliodonium salts. The reaction proceeded in good to excellent yields for a variety of 2-naphthol substrates and an array of substituents in positions 6 and 7 was tolerated. This protocol was particularly suited for the preparation of fluorinated products. Further transformations, for example, upon intramolecular cyclization under basic conditions allowed p-extended dibenzofuran derivatives to be obtained. The mechanistic computational investigation demonstrates that the overall Csp2–Csp2 coupling occurred by a de-aromatization/re-aromatization sequence (Scheme 81).220
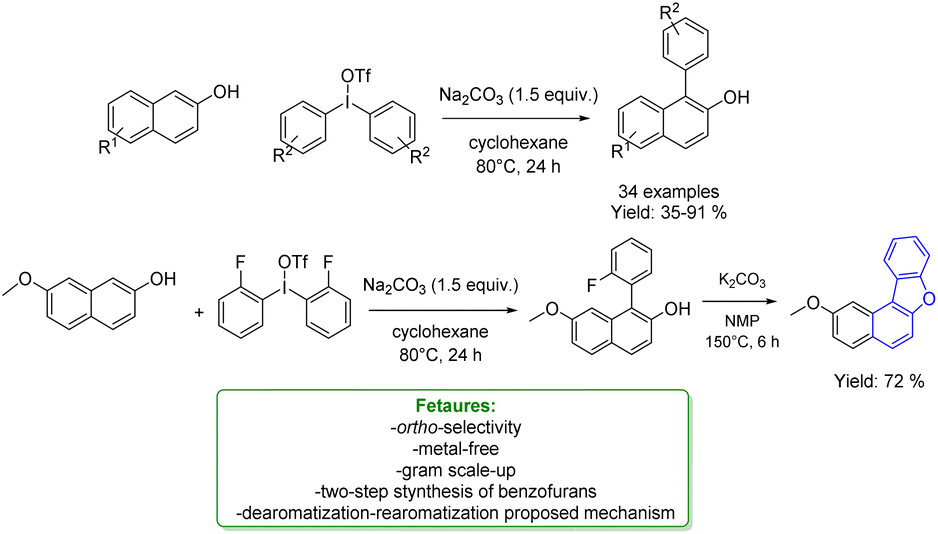 | ||
| Scheme 81 Transition metal-free arylation of 2-naphthols with diaryliodonium salts.220 | ||
Another interesting example of transition metal-free arylation of phenols was reported by D. J. Procter et al. with variously functionalized benzothiophenes activated as their S-oxides. This para-selective process needed trifluoroacetic anhydride and an electron-withdrawing group at C3 of the benzothiophene for taking place. Computational studies suggested a mechanism involving heterolytic cleavage of an aryloxy-sulfur species to give a benzothiophene and a phenoxonium cation. The synthetic utility of the metal-free, direct arylation process was underlined by the selective manipulation of the versatile benzothiophene products (Scheme 82).221
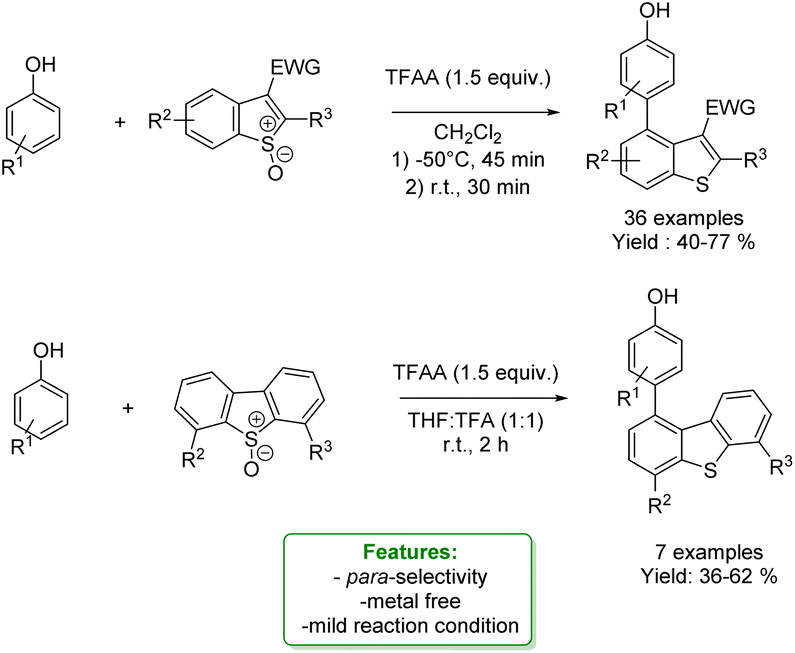 | ||
| Scheme 82 Metal-free arylation of phenols with variously functionalized benzothiophenes.221 | ||
The same research group reported a sulfoxide-mediated oxidative cross-coupling of phenols with various nucleophiles, including arenes, 1,3-diketones and other phenols. The metal-free protocol was mediated by a sulfoxide which allowed the selective ortho-arylation cross-coupling versus homo-coupling. The sulfoxide 4a was activated using trifluoroacetic acid (TFA) before the addition of the arylating agent, obtaining a wide plethora of desired products from good to excellent yields. A wide range of biaryls was achieved. Benzofurans were obtained via an iterative coupling of three nucleophiles (Scheme 83).222
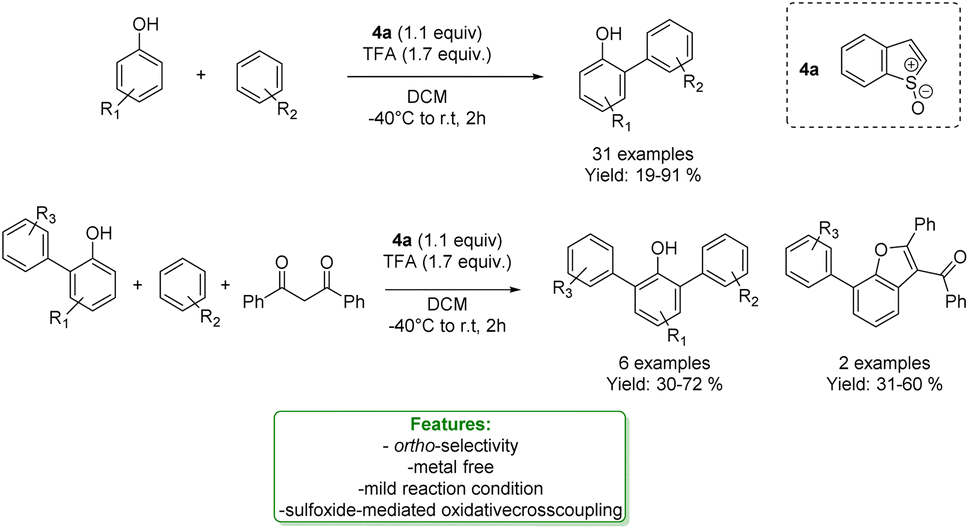 | ||
| Scheme 83 Metal-free sulfoxide-mediated oxidative cross-coupling of phenols with various nucleophiles.222 | ||
B. Tan et al. presented a metal-free oxidative cross-coupling reaction between isoquinolines and 2-naphthols, furnishing structurally diversified QUINOLs.223 This protocol boasts high yields and exclusive chemoselectivity. To underscore the robustness of the developed protocols, a gram-scale experiment was conducted. A plausible mechanistic pathway was elucidated. The reaction is initiated as Tf2O activates the isoquinoline, forming acyl isoquinolinium.
The nucleophilic 2-naphthol selectively adds to the iminium carbon of the activated species from the C1 position, facilitated by proton abstraction from the hydroxyl group by the proximal triflate anion. Subsequent tautomerization of the naphthol ring then induces dearomatization. Such intermolecular synergistic activation-addition events likely play a crucial role in overcoming the otherwise challenging chemoselectivity issues posed by the competitive nucleophilic and electrophilic sites on these reactants. Finally, an oxidative aromatization process yields the target product QUINOL, accompanied by the removal of the Tf group (Scheme 84).223
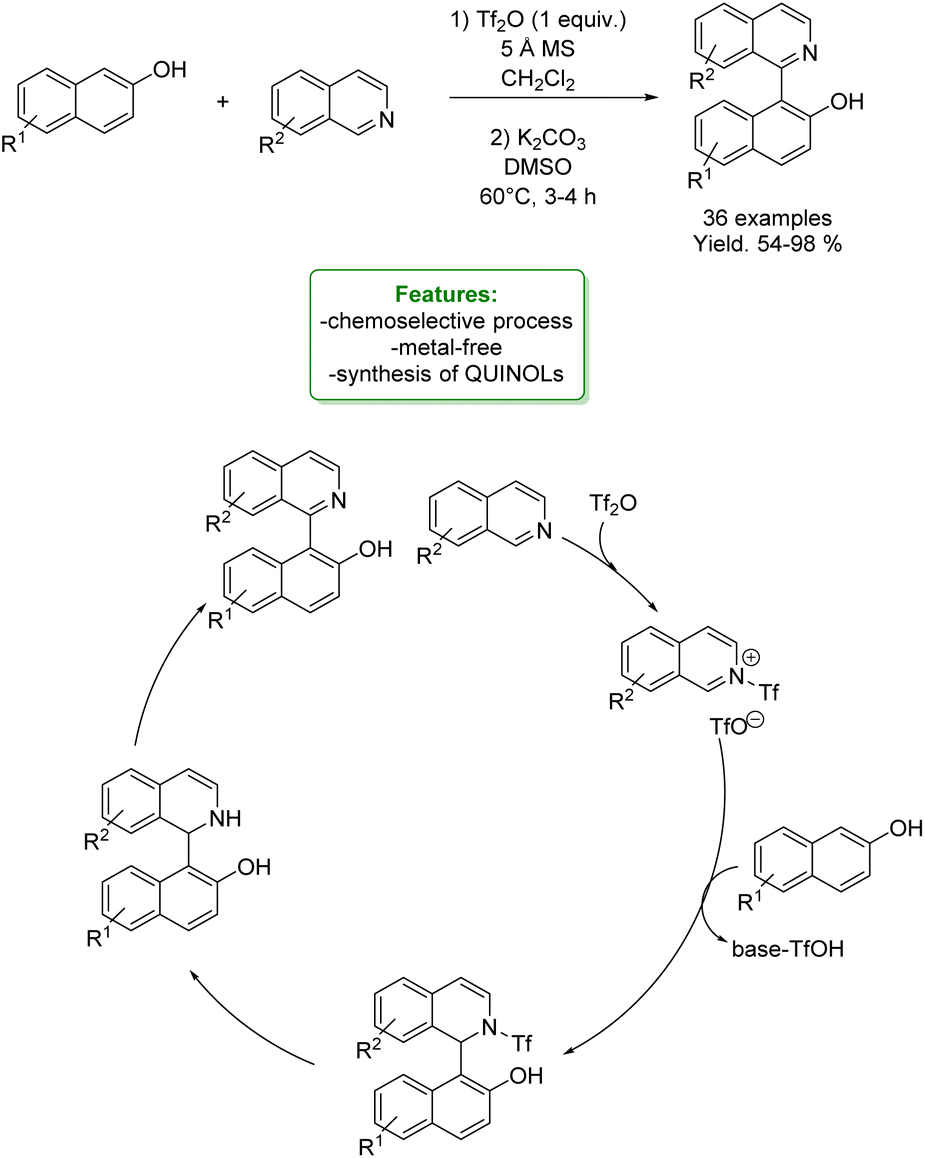 | ||
| Scheme 84 Metal-free oxidative cross-coupling reaction between isoquinoline and 2-naphthol.223 | ||
I. A. Utepova et al. disclosed a one-pot oxidative coupling of 1,3-diazines, 1,2,4-triazines, and 2-quinoxalone with 2-naphthols, facilitated by Brønsted acid. The reaction selectively occurs at the C1 position of 2-hydroxynaphthalene, yielding products ranging from low to excellent yields under mild reaction conditions. The pivotal role of K3[Fe(CN)6] as an oxidant is evident, enabling the re-aromatization of the dihydroderivative intermediate formed after the acid-promoted addition of 1,3-diazines, 1,2,4-triazines, and 2-quinoxalone to 2-naphthols (Scheme 85).224
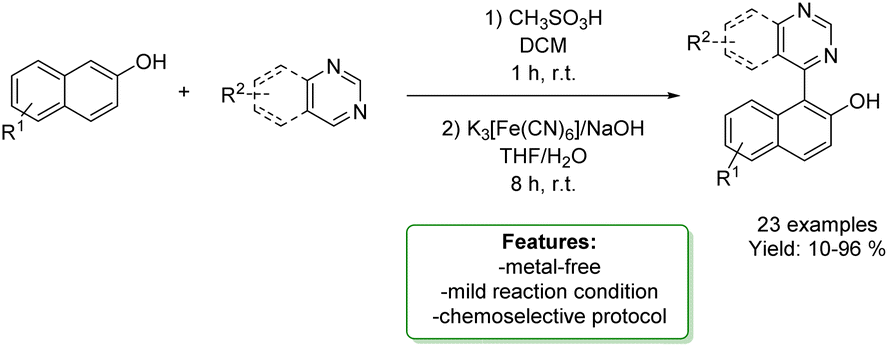 | ||
| Scheme 85 One-pot oxidative coupling of 1,3-diazines, 1,2,4-triazines, and 2-quinoxalone with 2-naphthols.223 | ||
1.5.2.2 Photosynthetic protocols. Given the concerns in the development of transition-metal-free arylation of phenols, photosynthetic processes were developed to overcome the main challenges involving the use of an electron donor–acceptor (EDA) complex formed by electron-rich phenolates with aryl halides. In 2022, H.-X. Li et al. developed a transition-metal-free visible-light-driven arylation of phenols with arylbromides at room temperature under irradiation of a blue LED or natural sunlight. Phenols bearing an EWG at the para-position were converted into ortho-arylated phenols. This reaction proceeded through two possible pathways. The first one involved the visible light photoexcitation of an EDA complex to trigger SET from phenolate to aryl bromide, resulting in the formation of an aryl radical and phenoxyl radical. Then, the latter isomerizes into a phenoxonium radical, and finally, the radical–radical cross-combination gives the desired product after aromatization. Alternatively, the addition of the aryl radical to phenoxyl radical, reduction by ArBr, and aromatization afforded the product (Scheme 86).225
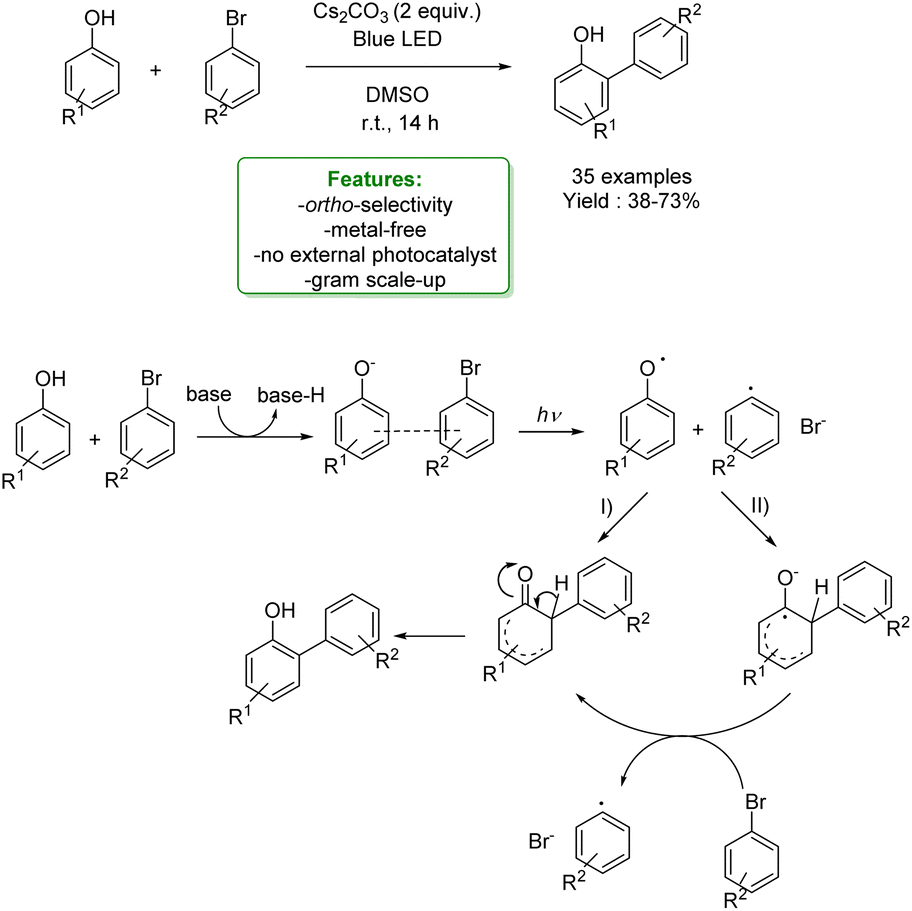 | ||
| Scheme 86 Transition-metal-free photoinduced arylation of phenols with aryl halides.225,226 | ||
Yu and collaborators reported a light-driven arylation of phenols to biphenols by using di-t-butyl peroxide (DTBP) under blue LEDs, without an external photocatalyst. They proposed a radical mechanism starting from the generation of tBuO· that abstracts a hydrogen from the phenol. Then, through a cascade process, products were formed (Scheme 87).226
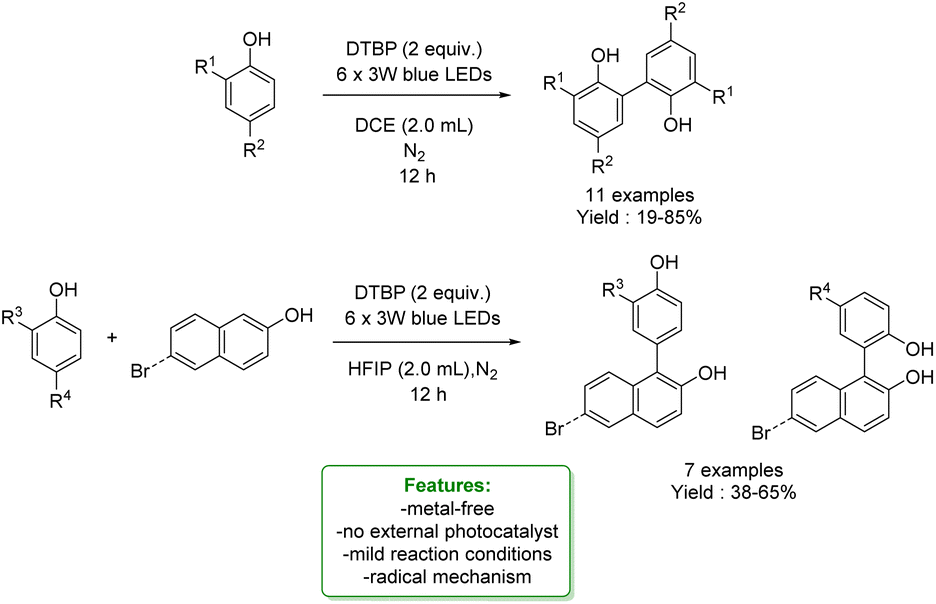 | ||
| Scheme 87 Transition-metal-free photoinduced arylation of phenols with aryl halides.225,226 | ||
1.5.2.3 Electrosynthesis. Another interesting approach to overcome the principal limitation of the transition-metal-free arylation of phenols is furnished by electrosynthesis which represents a remarkably simple, inherently safe, and sustainable method to implement the desired phenol arylation.
S. R. Waldvogel et al. in 2019 reported a highly para-selective electrochemical protocol for the anodic dehydrogenative 2,5- and 2,6-substituted phenol–phenol cross-coupling and homocoupling. This synthetic strategy gave access to a broad range of non-symmetric and symmetric 4,4′-biphenols under mild reaction conditions. To successfully prevent over-oxidation and therefore side reactions, the use of HFIP and 2,5-substituted phenols was a suitable and efficient strategy, resulting in yields up to 77%. Concerning the dehydrogenative homo-coupling protocol, the HFIP/H2O mixture was the key to access the desired symmetrical products in yields up to 60% (Scheme 88).227
 | ||
| Scheme 88 Electrosynthesis of arylated phenols with arenes.227–229 | ||
In 2020, the same authors reported a supporting-electrolyte-free and scalable flow process for the electrochemical dehydrogenative homocoupling of phenols to afford 3,3′,5,5′-tetramethyl-2,2′-biphenol. The key to this process was the use of pyridine as an additive in HFIP, which can simply be removed by distillation and recovery. This facilitates the downstream process, making it simple, economical, and technically viable (Scheme 89).228
 | ||
| Scheme 89 Electrosynthesis of arylated phenols with arenes.227–229 | ||
More recently, in 2021, they developed a dehydrogenative phenol-arene cross-coupling by direct anodic oxidation of phenols to bi-aryls resorting to the use of boron-doped diamond (BDD) electrodes in combination with 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) (Scheme 90).229
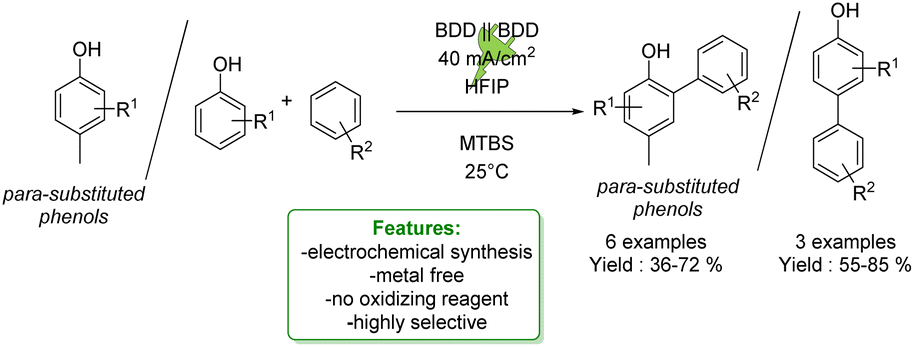 | ||
| Scheme 90 Electrosynthesis of arylated phenols with arenes.227–229 | ||
Formation of benzothiophene derivatives is a cutting-edge process to access pharmaceuticals and organic electronic materials.230,231 Liu, Yue and others developed a Csp2–H coupling of phenols with thiophene to obtain 3-phenylbenzothiophenes. They used an undivided cell with Pt as the cathode and RVC as the anode with HFIP and CH2Cl2 as solvents. After a large screening of different electrolytes, nBu4NPF6 was selected as the best one. They first studied a single C–H coupling, obtaining 16 products in moderate to very good yields (33–89%). Changing the current from 10 mA to 5 mA, they obtained 12 double-functionalized products in moderate to very good yields (33–80%). They also proposed a mechanism based on the SET oxidation of both the reaction partners: at the anode, phenol generated the phenol oxygen radical while the 3-phenylbenzothiophene was the cation radical. Then, a radical coupling between these two intermediates occurred and, after deprotonation, benzothiophene derivative products were obtained (Scheme 91).232
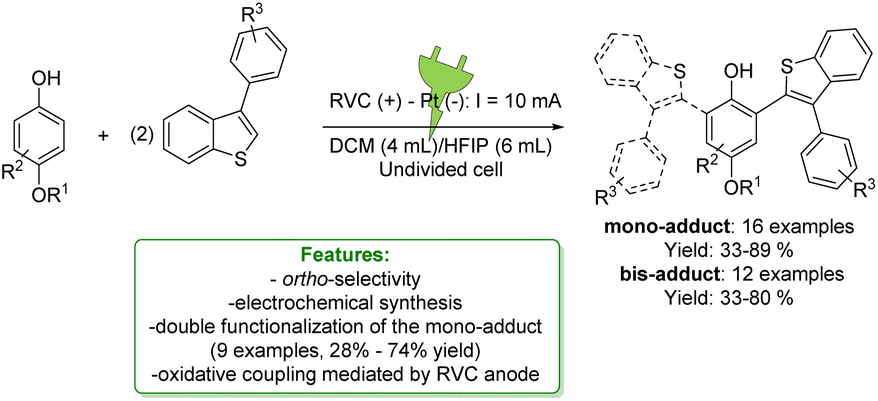 | ||
| Scheme 91 Electro-oxidative coupling of phenols and 3-phenyl-benzothiophenes.232 | ||
1.6. Acylation
Acylation reactions of phenolic compounds are widely used in the synthesis of fine chemicals such as flavours (i.e. vanillin) and a wide range of anti-inflammatory compounds (mesalamine, diflusinal, salicylic Acid, sulfasalazine) and antibiotics (novobiocin) (Fig. 8).4 | ||
| Fig. 8 Acylated phenol structural moieties in API and flavor compounds. | ||
The acylation of phenolic compounds may proceed through two different pathways: O-acylation or direct C-acylation. The first one yields the corresponding ester, which can be further transformed into an aromatic ketone via Fries rearrangement. The latter one is the direct Friedel–Crafts acylation of the benzene ring. If one or the other mechanism is followed depends on different features: the acylating agent used, if the reaction is in vapor or liquid phase, the presence of water, the solvent used and the acid/base properties of the catalyst.233,234 Padró and others studied the correlation between the two different mechanisms and the acid catalyst used to promote the reaction: they explained that phenol esters are mainly formed by Brønsted acids, while strong Lewis acid sites generally promoted direct C-acylation.235,236
1.6.1.1 Metal catalysis. Zhao and Ji's research group developed a Ni-catalysed free-radical carboxylation strategy using 1,10-phenanthroline as the ligand to gain steric hindrance directing the regioselectivity towards the para-product. They obtained para-acylated phenols in sufficient to very good yields (42–86%). They proposed a one-step synthesis of three natural products (methyl syringate, methyl gallate and dimethyl gallate) in 43–75% isolated yields. Late-stage functionalization of seven bioactive molecules was achieved in 34–72% yields. They performed mechanistic studies in which they proved the free radical mechanism and the Ni–OH interaction to activate the phenolic ring.
The proposed mechanism consists of the first reduction of Ni(II) to Ni(0) which reacted with a CBr4 molecule to form BrNi(I)Ln species and a carbon tribromide radical. Since the Ni(I) complex is highly active, it continues to react with other carbon tetrabromide molecules to access Br2Ni(II)Ln intermediate. This coordinated the phenol reducing its aromaticity so the carbon tribromide radical can attack the para-position. Then, through a SET process, BrNi(I)Ln was formed again, and phenol aromaticity was restored. In the end, through a double methanolysis process, the para-acylated phenol was obtained (Scheme 92).237
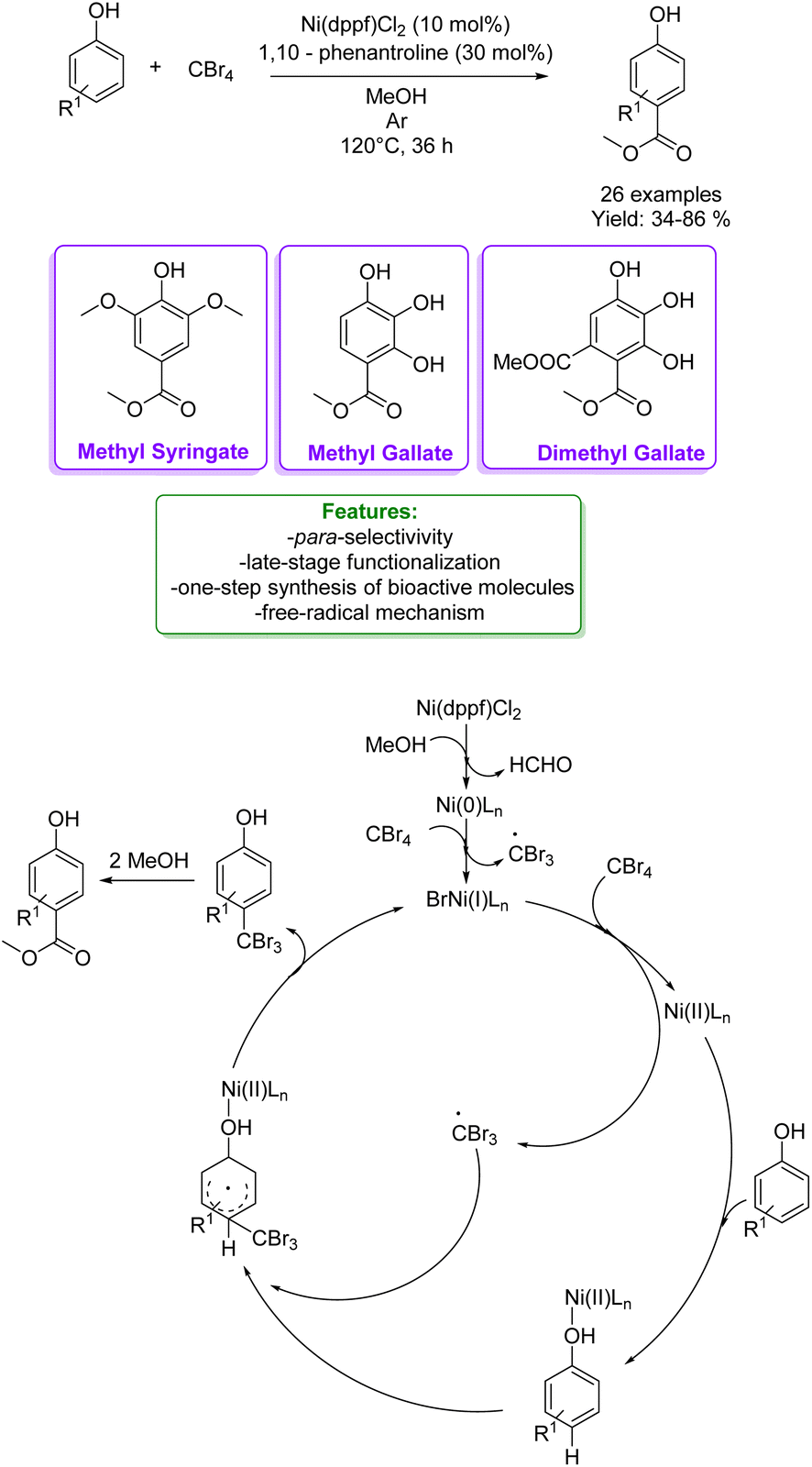 | ||
| Scheme 92 Ni-catalysed acylation of phenols.237 | ||
1.6.1.2 Acid/base catalysis. Cresols, guaiacols, catechols, and resorcinols are key starting materials to be functionalized since they are directly formed from lignin depolymerization processes. Inspired by Yadav's work on Friedel–Crafts acylation of guaiacol with various acylating agents over solid acid catalysts,238 Sad et al. reported a vapor phase flow reaction between guaiacol and acetic acid over different zeolites.239 They obtained very low conversions (16.0% to 24.0%, depending on the catalyst used) and poor selectivity (O-acylated product is mainly formed). However, they proved catalyst reusability obtaining 24% of guaiacol conversion. Later in 2021 Moreno, Coronado and others studied vapor phase acylation of guaiacol with acetic acid over microcrystalline, nanocrystalline and hierarchical zeolites (ZSM-5 and Beta). After the catalysts' characterization with different techniques, they investigated the formation of hydroxymethoxyacetophenones (HMPAs). They noted that the better accessibility of the pores of hierarchical and nanocrystalline zeolites gave better conversions (40–60% for ZSM; ∼30% for Beta) than the microcrystalline one (<20%). The selectivity towards the HMAPs was better when a zeolite with Lewis/Brønsted acid site ratio of 0.5 (nano-crystalline and hierarchical zeolites) was used (Scheme 93).240
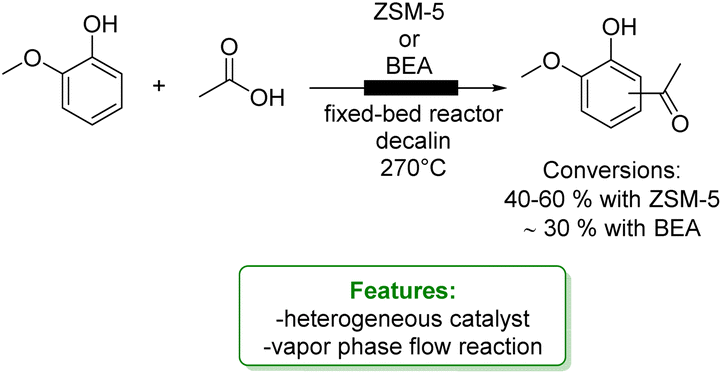 | ||
| Scheme 93 Acylation of phenolic compounds with acetic acid.240–242 | ||
Crossley et al. developed acylation of m-cresol with acetic acid over different zeolites establishing a vapor phase flow strategy. After the pretreatment of the catalyst to remove the physiosorbed water, starting materials m-cresol and acetic acid were introduced in the inlet of the reactor that is heated at 250 °C to vaporize the reagents. Upon varying respectively cresol and acetic acid pressures the initial turn-over frequency (TOF0) was affected. The rate of 2-hydroxy-4-methylacetophenone formation resulted to be zero-order with respect to phenol and acetic acid. The reaction rate equation was derived based on the proposed acylation pathway via active intermediate ester, m-tolylacetate. This intermediate was formed at the MFI channel intersections; it was subsequently decomposed in the Brønsted acid sites to form acylium ion that acylates m-cresol. The deactivation of the zeolite catalysts was due to the subsequent esterification of the already-acylated product forming higher molecular mass species (Scheme 94).241
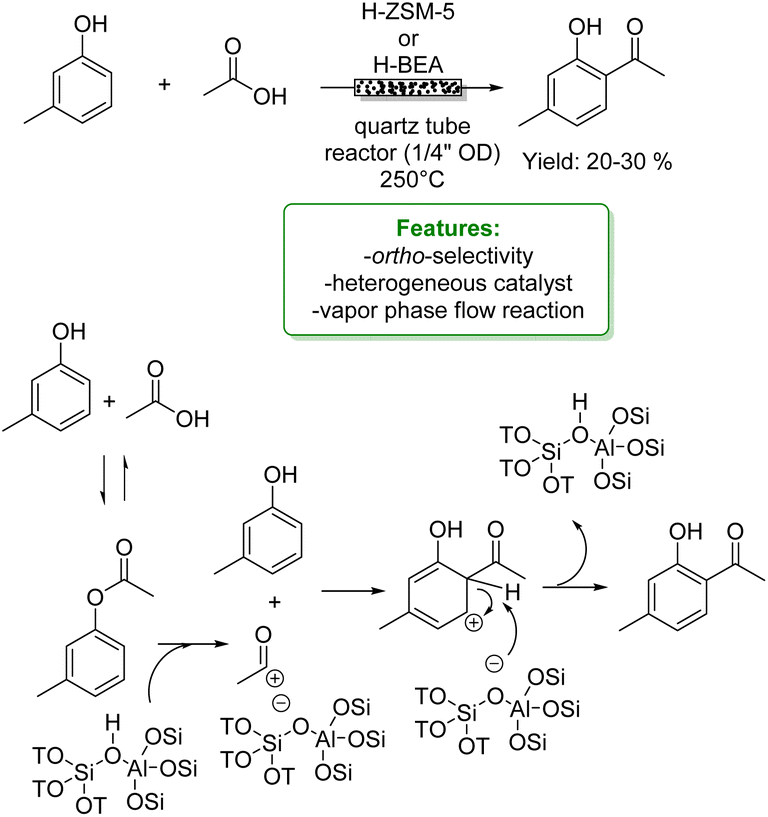 | ||
| Scheme 94 Acylation of phenolic compounds with acetic acid.240–242 | ||
A. B. Charette's group proposed the acylation of phenol derivatives in the presence of the Lewis acid BF3·Et2O in a PFA reactor. Running the reaction on a 20 mmol scale with a flow rate of 10 mL min−1 and residence time of 1 min they obtained a productivity of 98/99 g h−1 of the acylated product (Scheme 95).242
 | ||
| Scheme 95 Acylation of phenolic compounds with acetic acid.240–242 | ||
1.7. Spiroannulation
 | ||
| Fig. 9 Bioactive and drugs spirocycle-containing compounds. | ||
Transition-metal catalysed spiroannulation of phenols and naphthols is an attractive tool combining Csp2–H bond functionalization and sequential de-aromatization of arenes to access spirocycles in a step- and atom-economical approach.245–254 The dearomatizing spiroannulation of phenol derivatives is particularly intriguing since it enables the incorporation of a carbonyl group into the resulting spirocyclic compounds. It is noteworthy that this functionality is usually employed as an acceptor unit in organic optoelectronic molecules.255 However, the spiroannulation suffers from several limitations, for example, the only ortho-position functionalization, and the harsh reaction conditions, which limits the applicability to a restricted substrate scope. Thus, the development of novel and most effective approaches for phenols and naphthols functionalization to spirocycles is highly desirable.
1.7.1.1 Metal catalysis. L. Liu et al. developed an Au-catalyzed protocol to de-aromatize phenols and naphthols to access spirocyclic compounds via Csp2–H bond functionalization with o-alkynylaryl-α-diazoesters. The reaction delivered alkynyl phenol derivatives, which were able to further undergo a carbocyclization de-aromatization reaction. This protocol provided diverse highly spirocyclic molecules in good to excellent yields with high chemo- and regio-selectivity and good diastero-selectivity (Scheme 96).256
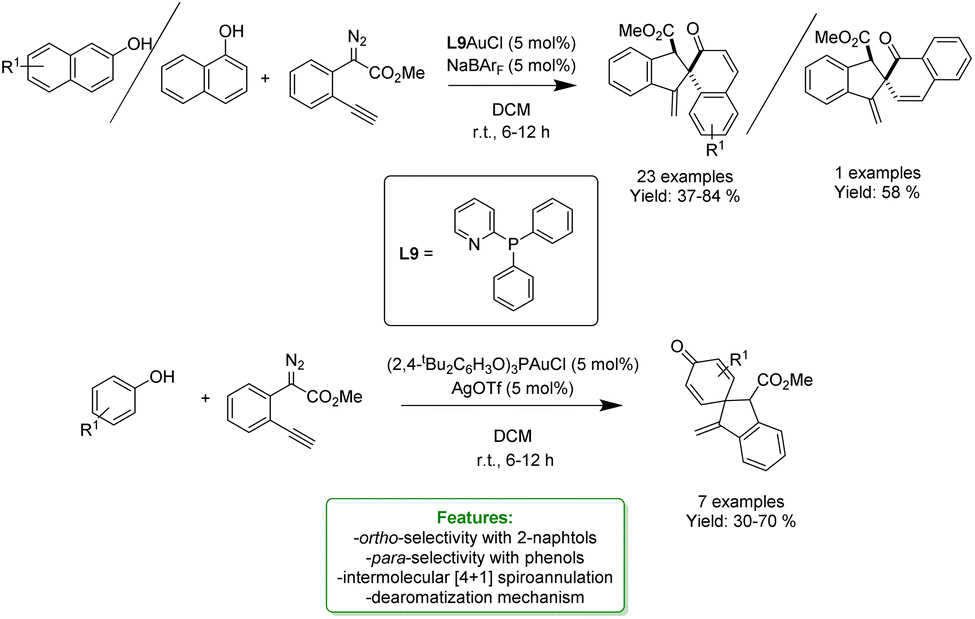 | ||
| Scheme 96 Au-catalysed spiroannulation of phenols and naphthols.256 | ||
Z. Bin, J. You et al. disclosed a Pd-catalysed direct [4 + 1] spiroannulation of ortho-Csp2–H bonds of naphthols with cyclic-diaryliodonium salts. They synthesized a wide range of spirofluorenyl naphthalenones (SFNP) under mild reaction conditions. The so-formed spirocycle products were further used as acceptors to construct structurally new thermally activated delayed fluorescent (TADF) materials (Scheme 97).257
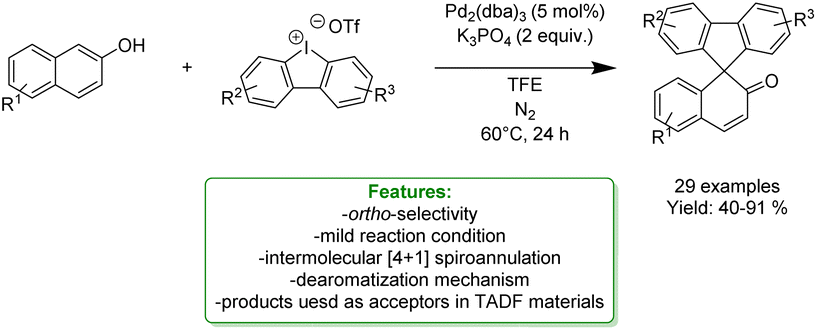 | ||
| Scheme 97 Pd-catalysed spiroannulation of naphthols.257 | ||
The X. Luan research group reported a Pd(0)-catalysed chemo- and regio-selective three-component dearomative [2 + 2 + 1] spiroannulation of 2-naphthols with 1,2-dihaloarene and alkynes. The authors conducted mechanistic studies revealing that the domino reaction proceeded through a cascade of oxidative addition to Pd(0), alkyne migratory insertion, and 2-naphthol facilitated dearomatizing [4 + 1] spiroannulation. Various substituents on the 1,2-dihaloarene ring and 2-naphthols were well tolerated, including electron-donating and electron-withdrawing groups. A broad range of symmetrical alkynes, bearing various aromatic, heterocyclic and aliphatic groups were used as well, yielding good results (Scheme 98).258
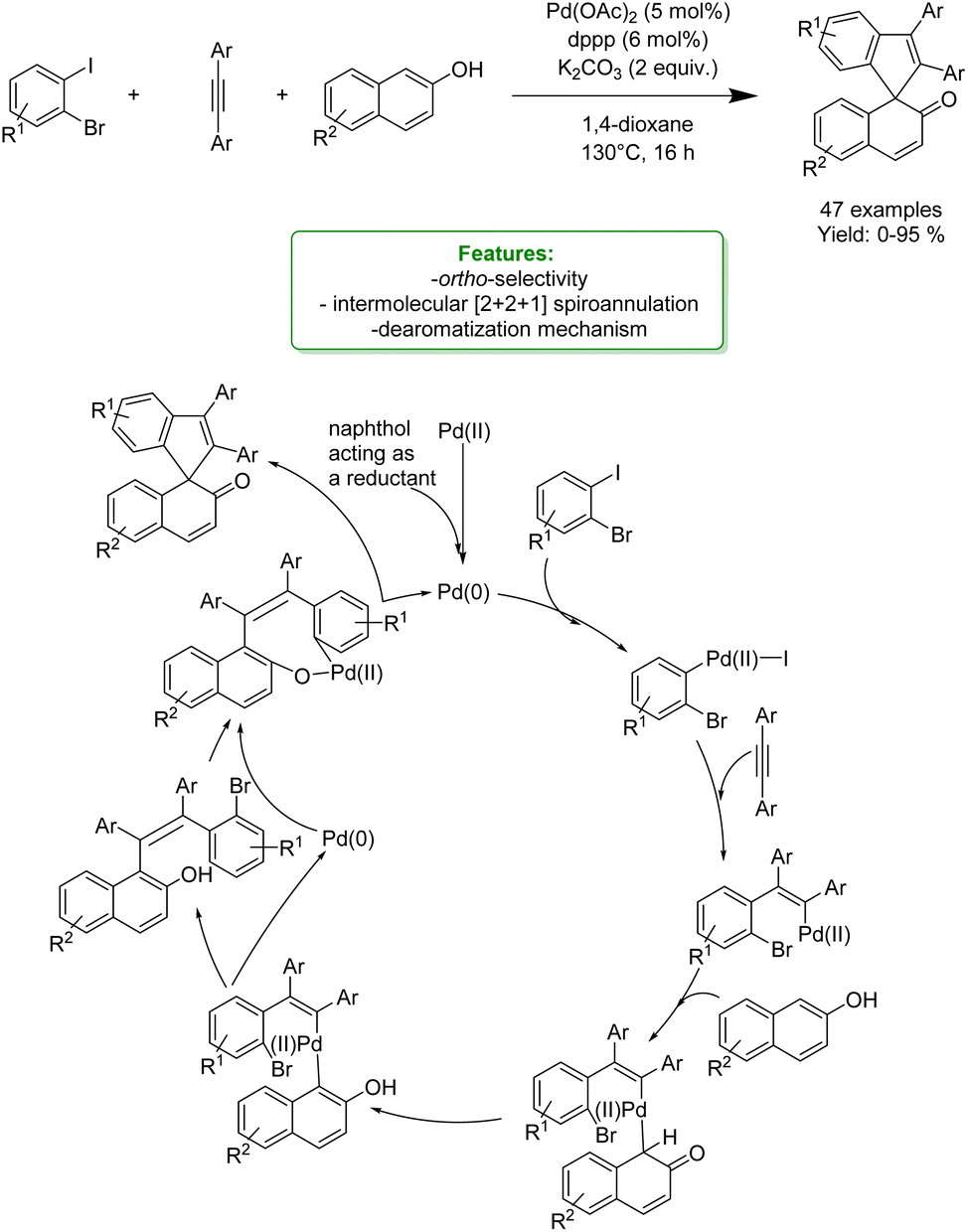 | ||
| Scheme 98 Pd-catalysed spiroannulation of naphthol via three-component reactions.258 | ||
2 Conclusions
In this review, we have highlighted the current state of the art on the Csp2–H functionalization of simple, unprotected phenols. By surveying the literature of the past five years, the growing interest in such methodologies is certainly evident. In fact, this versatile tool enables access to a wide plethora of functionalized phenols, that often constitute essential structural moieties for target materials with a diverse range of properties and activities. Csp2–H functionalization facilitates the efficient synthesis, in a step- and atom-economical manner, of versatile intermediates, NPs and APIs, as well as the late-stage functionalization of biologically active products. Csp2–H alkylation and arylation constitute reliable alternatives to traditional cross-coupling methods. Csp2–H alkenylation is a well-established method to access the synthesis of O-containing heterocyclic compounds.Transition-metal catalysed Csp2–H functionalization remains the preferred method to ensure high selectivity and substrate versatility. Additionally, metal-free procedures are also of great interest to achieve functionalization while reducing the environmental footprint. These strategies include photocatalysis and electrosynthesis.
The ortho-functionalization of unprotected phenols is observed to be favoured over other positions. Selective functionalization at the para-position has also been explored and it seems to be in agreement with classical steric considerations. However, selective meta-functionalization of unprotected phenols is still a challenge. To achieve this selectivity the installation of a directing group on the hydroxy group is necessary.
The efforts to valorise lignin have resulted in the generation of well-defined streams of phenolic building blocks, which can be integrated into the chemical value chain.
This review has also tried to draw attention to the development of efficient and inherently environmentally friendly Csp2–H functionalization methods for simple, unprotected phenols, with the goal of promoting their utilization as lignin-derived feedstock. Indeed, in the pursuit of innovative synthetic methodologies, both the utilization of renewable feedstock and the development of environmentally sustainable approaches play pivotal roles. The fusion of these two concepts represents a significant advancement in the design of new synthetic production processes for phenols' valorisation and future investigations will certainly contribute in this direction.
Author contributions
G. B. and B. D. started the literature survey proposed by L. V. in agreement with C.-J. L. The manuscript was drafted by G. B. and B. D. with revisions by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. The University of Perugia is acknowledged for financial support to the university project “Fondo Ricerca di Ateneo, edizione 2021”. G. B. and L. V. wish also to thank INPS and Sterling SpA for the PhD grant and training offered to G. B. C.-J. L. thanks NSERC, CFI, Canada Research Chair Foundation, FQRNT and CCVC for the financial support.Notes and references
- M. A. Rahim, S. L. Kristufek, S. Pan, J. J. Richardson and F. Caruso, Angew. Chem., Int. Ed., 2019, 58, 1904–1927 CrossRef CAS PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouységu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- K. A. Scott, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2022, 65, 7044–7072 CrossRef CAS PubMed.
- R. J. Schmidt, Appl. Catal., A, 2005, 280, 89–103 CrossRef CAS.
- A. Corma, J. Catal., 2004, 221, 67–76 CrossRef CAS.
- R. Sun, ChemSusChem, 2020, 13, 4385–4393 CrossRef CAS PubMed.
- C. Zhang and F. Wang, Acc. Chem. Res., 2020, 53, 470–484 CrossRef CAS PubMed.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS PubMed.
- Q. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Angew. Chem., 2021, 133, 20554–20562 CrossRef.
- P. Ni, L. Yang, Y. Shen, L. Zhang, Y. Ma, M. Sun, R. Cheng and J. Ye, J. Org. Chem., 2022, 87, 12677–12687 CrossRef CAS PubMed.
- Z. Yu, X. Ye, Q. Xu, X. Xie, H. Dong and W. Su, Org. Process Res. Dev., 2017, 21, 1644–1652 CrossRef CAS.
- H. Yi and A. Lei, Chem.–Eur. J., 2017, 23, 10023–10027 CrossRef CAS PubMed.
- L. Hao, G. Ding, D. A. Deming and Q. Zhang, Eur. J. Org Chem., 2019, 2019, 7307–7321 CrossRef CAS.
- Y.-T. Xu, C.-Y. Li, X.-B. Huang, W.-X. Gao, Y.-B. Zhou, M.-C. Liu and H.-Y. Wu, Green Chem., 2019, 21, 4971–4975 RSC.
- C. Yuan, A. M. Eliasen, A. M. Camelio and D. Siegel, Nat. Protoc., 2014, 9, 2624–2629 CrossRef CAS PubMed.
- D. Saha, P. Das, P. Biswas and J. Guin, Chem.–Asian J., 2019, 14, 4534–4548 CrossRef CAS PubMed.
- R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed.
- D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213–8221 CrossRef CAS PubMed.
- Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed.
- M. Lanzi, T. Rogge, T. S. Truong, K. N. Houk and J. Wencel-Delord, J. Am. Chem. Soc., 2023, 145, 345–358 CrossRef CAS PubMed.
- M. Weber, M. Weber and M. Kleine-Boymann, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wiley, 1st edn, 2004 Search PubMed.
- Comprehensive organic synthesis, ed. P. Knochel and G. A. Molander, Elsevier, Amsterdam, Netherlands; Waltham, MA, 2nd edn, 2014 Search PubMed.
- J. H. P. Tyman and J. H. P. Tyman, Synthetic and natural phenols, Elsevier, Amsterdam [The Netherlands] New York, 1996 Search PubMed.
- Z. Rappoport, The Chemistry of Phenols, John Wiley & Sons, Chichester, 2004 Search PubMed.
- J. Sun, Q. Mu, H. Kimura, V. Murugadoss, M. He, W. Du and C. Hou, Adv. Compos. Hybrid Mater., 2022, 5, 627–640 CrossRef.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS.
- J. Xu, J. Chen, F. Gao, S. Xie, X. Xu, Z. Jin and J.-Q. Yu, J. Am. Chem. Soc., 2019, 141, 1903–1907 CrossRef CAS PubMed.
- S. W. Youn and C.-G. Cho, Org. Biomol. Chem., 2021, 19, 5028–5047 RSC.
- S. Sasmal, U. Dutta, G. K. Lahiri and D. Maiti, Chem. Lett., 2020, 49, 1406–1420 CrossRef CAS.
- Z. Huang and J.-P. Lumb, ACS Catal., 2019, 9, 521–555 CrossRef CAS.
- N. O. Calloway, Chem. Rev., 1935, 17, 327–392 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, M. Heydari and B. Masoumi, Chem. Rec., 2019, 19, 2236–2340 CrossRef CAS.
- M. Montesinos-Magraner, C. Vila, G. Blay and J. Pedro, Synthesis, 2016, 48, 2151–2164 CrossRef CAS.
- Z. Yu, Y. Li, J. Shi, B. Ma, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2016, 55, 14807–14811 CrossRef CAS PubMed.
- L. Yu, X. Xie, S. Wu, R. Wang, W. He, D. Qin, Q. Liu and L. Jing, Tetrahedron Lett., 2013, 54, 3675–3678 CrossRef CAS.
- C. Huang, B. Chattopadhyay and V. Gevorgyan, J. Am. Chem. Soc., 2011, 133, 12406–12409 CrossRef CAS PubMed.
- T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed.
- B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468–469 CrossRef CAS PubMed.
- A. John and K. M. Nicholas, J. Org. Chem., 2012, 77, 5600–5605 CrossRef CAS PubMed.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 1998, 120, 815–816 CrossRef CAS.
- T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582–14585 CrossRef CAS PubMed.
- X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387–17393 CrossRef CAS PubMed.
- A. Regev, H. Shalit and D. Pappo, Synthesis, 2015, 47, 1716–1725 CrossRef CAS.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS.
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., 2013, 125, 12901–12905 CrossRef.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- H. Lee, M. V. Mane, H. Ryu, D. Sahu, M.-H. Baik and C. S. Yi, J. Am. Chem. Soc., 2018, 140, 10289–10296 CrossRef CAS PubMed.
- Y. Yamamoto and K. Itonaga, Org. Lett., 2009, 11, 717–720 CrossRef CAS PubMed.
- Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914–9915 CrossRef CAS PubMed.
- Y. Kuninobu, M. Yamamoto, M. Nishi, T. Yamamoto, T. Matsuki, M. Murai and K. Takai, in Organic Syntheses, ed. A. S. Kende and J. P. Freeman, Wiley, 1st edn, 2018, pp. 280–291 Search PubMed.
- D. Lehnherr, X. Wang, F. Peng, M. Reibarkh, M. Weisel and K. M. Maloney, Organometallics, 2019, 38, 103–118 CrossRef CAS.
- J. Yu, C. Li and H. Zeng, Angew. Chem., Int. Ed., 2021, 60, 4043–4048 CrossRef CAS PubMed.
- J. V. Oakley, T. J. Stanley, K. A. Jesse, A. K. Melanese, A. A. Alvarez, A. L. Prince, S. E. Cain, A. G. Wenzel and R. G. Iafe, Eur. J. Org Chem., 2019, 2019, 7063–7066 CrossRef CAS.
- K. Das, E. Yasmin and A. Kumar, Adv. Synth. Catal., 2022, 364, 3895–3909 CrossRef CAS.
- N. Pannilawithana, B. Pudasaini, M.-H. Baik and C. S. Yi, J. Am. Chem. Soc., 2021, 143, 13428–13440 CrossRef CAS PubMed.
- Q.-Y. Li, S. Cheng, Z. Ye, T. Huang, F. Yang, Y.-M. Lin and L. Gong, Nat. Commun., 2023, 14, 6366 CrossRef CAS PubMed.
- H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed.
- H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733–738 CrossRef CAS PubMed.
- Y. Xi, Y. Su, Z. Yu, B. Dong, E. J. McClain, Y. Lan and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 9817–9821 CrossRef CAS PubMed.
- Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904–6907 CrossRef CAS PubMed.
- Z. Yu, Y. Li, P. Zhang, L. Liu and J. Zhang, Chem. Sci., 2019, 10, 6553–6559 RSC.
- B. Ma, Z. Tang, J. Zhang and L. Liu, Chem. Commun., 2020, 56, 9485–9488 RSC.
- X. Zhu, X. Liu, F. Xia and L. Liu, Molecules, 2023, 28, 1767 CrossRef CAS PubMed.
- Z. Yu, G. Li, J. Zhang and L. Liu, Org. Chem. Front., 2021, 8, 3770–3775 RSC.
- L. Carreras, A. Franconetti, A. Grabulosa, A. Frontera and A. Vidal-Ferran, Org. Chem. Front., 2020, 7, 1626–1634 RSC.
- P. Basnet, M. B. Sebold, C. E. Hendrick and M. C. Kozlowski, Org. Lett., 2020, 22, 9524–9528 CrossRef CAS.
- Q. Zhang, Y. Chan, M. Zhang, Y. Yeung and Z. Ke, Angew. Chem., Int. Ed., 2022, 61, e202208009 CrossRef CAS.
- X. Liang, H. Xu, H. Li, L. Chen and H. Lu, Eur. J. Org Chem., 2020, 2020, 217–226 CrossRef CAS.
- B. Xiong, S. Xu, W. Xu, Y. Liu, L. Zhang, K.-W. Tang, S.-F. Yin and W.-Y. Wong, Org. Chem. Front., 2022, 9, 3807–3817 RSC.
- C. Denis, M. A. J. Dubois, A. S. Voisin-Chiret, R. Bureau, C. Choi, J. J. Mousseau and J. A. Bull, Org. Lett., 2019, 21, 300–304 CrossRef CAS PubMed.
- L. Tang, Q.-N. Huang, F. Wu, Y. Xiao, J.-L. Zhou, T.-T. Xu, W.-B. Wu, S. Qu and J.-J. Feng, Chem. Sci., 2023, 14, 9696–9703 RSC.
- G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7558–7598 CrossRef CAS PubMed.
- M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6 DOI:10.3762/bjoc.6.6.
- Y. He, Y. Cai and S. Zhu, J. Am. Chem. Soc., 2017, 139, 1061–1064 CrossRef CAS.
- R. D. C. Gallo, P. B. Momo, D. P. Day and A. C. B. Burtoloso, Org. Lett., 2020, 22, 2339–2343 CrossRef CAS PubMed.
- N. Yadav, J. Khan, A. Tyagi, S. Singh and C. K. Hazra, J. Org. Chem., 2022, 87, 6886–6901 CrossRef CAS PubMed.
- C. K. Rank, B. Özkaya and F. W. Patureau, Org. Lett., 2019, 21, 6830–6834 CrossRef CAS PubMed.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed.
- L. Cai, X. Liu, J. Wang, L. Chen, X. Li and J.-P. Cheng, Chem. Commun., 2020, 56, 10361–10364 RSC.
- T. Ma, Y. He, X.-X. Qiao, C.-P. Zou, X.-X. Wu, G. Li and X.-J. Zhao, Org. Biomol. Chem., 2023, 21, 489–493 RSC.
- M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539 RSC.
- B. Han, X.-H. He, Y.-Q. Liu, G. He, C. Peng and J.-L. Li, Chem. Soc. Rev., 2021, 50, 1522–1586 RSC.
- S. Wu, J. Dong, D. Zhou, W. Wang, L. Liu and Y. Zhou, J. Org. Chem., 2020, 85, 14307–14314 CrossRef CAS PubMed.
- M. Vayer, R. J. Mayer, J. Moran and D. Lebœuf, ACS Catal., 2022, 12, 10995–11001 CrossRef CAS.
- P. Sharma, N. Taneja, S. Singh and C. K. Hazra, Chem.–Eur. J., 2023, 29, e202202956 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, Y. Xia, J. Wang, Y. Peng and G. Song, J. Org. Chem., 2023, 88, 12924–12934 CrossRef CAS PubMed.
- Z. Tong, X. Peng, Z. Tang, W. Yang, W. Deng, S.-F. Yin, N. Kambe and R. Qiu, RSC Adv., 2022, 12, 35215–35220 RSC.
- B. Xiong, W. Shang, W. Xu, Y. Liu, K. Tang and W. Wong, Asian J. Org. Chem., 2022, 11, e202200295 CrossRef CAS.
- Y. Cheng, Z. Fang, Y. Jia, Z. Lu, W. Li and P. Li, RSC Adv., 2019, 9, 24212–24217 RSC.
- S. S. Shcherbakov, A. Yu. Magometov, V. Yu. Shcherbakova, A. V. Aksenov, D. A. Domenyuk, V. A. Zelensky and M. Rubin, RSC Adv., 2020, 10, 10315–10321 RSC.
- Phenolic resins: a century of progress, ed. L. Pilato, Springer, Heidelberg: New York, 2010 Search PubMed.
- G. Afreen, S. Pathak and S. Upadhyayula, Chem. Eng. J., 2020, 400, 125824 CrossRef CAS.
- P. Elavarasan, S. Rengadurai and S. Upadhyayula, Chem. Eng. J. Adv., 2020, 4, 100045 CrossRef CAS.
- A. Zuber and G. Tsilomelekis, Appl. Catal., A, 2023, 652, 119040 CrossRef CAS.
- M. Luo, H. Li, H. Song, C. Shao and A. Wang, Catal. Lett., 2023, 153, 1170–1179 CrossRef CAS.
- J. Bai, Y. Zhang, X. Zhang, C. Wang and L. Ma, ACS Sustainable Chem. Eng., 2021, 9, 7112–7119 CrossRef CAS.
- Z. Shen, G. Zhang, C. Shi, J. Qu, L. Pan, Z. Huang, X. Zhang and J.-J. Zou, Fuel, 2023, 334, 126634 CrossRef CAS.
- B. Bartolomei, G. Gentile, C. Rosso, G. Filippini and M. Prato, Chem.–Eur. J., 2021, 27, 16062–16070 CrossRef CAS PubMed.
- G. Filippini, M. Nappi and P. Melchiorre, Tetrahedron, 2015, 71, 4535–4542 CrossRef CAS.
- S. Cuadros, C. Rosso, G. Barison, P. Costa, M. Kurbasic, M. Bonchio, M. Prato, G. Filippini and L. Dell'Amico, Org. Lett., 2022, 24, 2961–2966 CrossRef CAS PubMed.
- P. Huang, X. Jiang, D. Gao, C. Wang, D.-Q. Shi and Y. Zhao, Org. Chem. Front., 2023, 10, 2476–2481 RSC.
- M. Stangier, A. Scheremetjew and L. Ackermann, Chem.–Eur. J., 2022, 28, e202201654 CrossRef CAS PubMed.
- W. Zhao, B. Wang, Y. Liu, L. Fu, L. Sheng, H. Zhao, Y. Lu and D. Zhang, Eur. J. Med. Chem., 2020, 189, 112075 CrossRef PubMed.
- G. F. Grillot and W. T. Gormley, J. Am. Chem. Soc., 1945, 67, 1968–1969 CrossRef CAS.
- D.-R. Hwang and B.-J. Uang, Org. Lett., 2002, 4, 463–466 CrossRef CAS.
- W. Sun, H. Lin, W. Zhou and Z. Li, RSC Adv., 2014, 4, 7491 RSC.
- H. F. Anwar, L. Skattebøl and T. V. Hansen, Tetrahedron, 2007, 63, 9997–10002 CrossRef CAS.
- C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed.
- S. Kim and S. H. Hong, Adv. Synth. Catal., 2017, 359, 798–810 CrossRef CAS.
- J.-L. Dai, N.-Q. Shao, J. Zhang, R.-P. Jia and D.-H. Wang, J. Am. Chem. Soc., 2017, 139, 12390–12393 CrossRef CAS PubMed.
- C. Yu and F. W. Patureau, Angew. Chem., Int. Ed., 2018, 57, 11807–11811 CrossRef CAS PubMed.
- S. Gupta, N. Chandna, P. Dubey, A. K. Singh and N. Jain, Chem. Commun., 2018, 54, 7511–7514 RSC.
- J. Shi, Y. Wang, Q. Bu, B. Liu, B. Dai and N. Liu, J. Org. Chem., 2021, 86, 17567–17580 CrossRef CAS PubMed.
- J. Xie, M. Chen, L.-L. Peng, J.-Q. Wu, Q. Zhou, C.-S. Zhou, B.-Q. Xiong and Y. Liu, Catal. Commun., 2021, 159, 106348 CrossRef CAS.
- S. Hamedimehr, K. Ojaghi Aghbash and N. Noroozi Pesyan, ACS Omega, 2023, 8, 8227–8236 CrossRef CAS PubMed.
- R. Tajima, T. Saito and T. Arai, ACS Catal., 2023, 13, 9495–9501 CrossRef CAS.
- I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057–5191 CrossRef CAS PubMed.
- M. Lubben and B. L. Feringa, J. Org. Chem., 1994, 59, 2227–2233 CrossRef CAS.
- L. Cao, H. Zhao, Z. Tan, R. Guan, H. Jiang and M. Zhang, Org. Lett., 2020, 22, 4781–4785 CrossRef CAS PubMed.
- C. Chen, X. Chen, H. Zhao, H. Jiang and M. Zhang, Org. Lett., 2017, 19, 3390–3393 CrossRef CAS PubMed.
- Z. Tang, D. Li, Y. Yue, D. Peng and L. Liu, Org. Biomol. Chem., 2021, 19, 5777–5781 RSC.
- Z.-H. Zhou, B. Wang, Y. Ding, T.-P. Loh and J.-S. Tian, Chem. Commun., 2023, 59, 223–226 RSC.
- C. Govindhan and P. S. Nagarajan, ChemistrySelect, 2021, 6, 8716–8726 CrossRef CAS.
- V. T. Nguyen, H. T. Nguyen and P. H. Tran, New J. Chem., 2021, 45, 2053–2059 RSC.
- (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) P. D. MacLeod, Z. Li and C.-J. Li, Tetrahedron, 2010, 66, 1045–1050 CrossRef CAS; (c) P. D. MacLeod, Z. Li, J. Feng and C.-J. Li, Tetrahedron Letters, 2006, 47, 6791–6794 CrossRef CAS; (d) T. Huang and C.-J. Li, Tetrahedron Lett., 2000, 41, 6715 CrossRef CAS.
- Z. Turgut, E. Pelit and A. Köycü, Molecules, 2007, 12, 345–352 CrossRef CAS PubMed.
- M. Duan, J. Chen, T. Wang, S. Luo, M. Wang and B. Fan, J. Org. Chem., 2022, 87, 15152–15158 CrossRef CAS PubMed.
- Z. Chen, T. Zhang, Y. Sun, L. Wang and Y. Jin, New J. Chem., 2021, 45, 10481–10487 RSC.
- M. Rodríguez-Rodríguez, A. Maestro, J. M. Andrés and R. Pedrosa, Adv. Synth. Catal., 2020, 362, 2744–2754 CrossRef.
- D. Glavač, N. Topolovčan and M. Gredičak, J. Org. Chem., 2020, 85, 14253–14261 CrossRef PubMed.
- D. Glavač and M. Gredičak, New J. Chem., 2022, 46, 8760–8764 RSC.
- R. Wei, L. Gao, G. Li, L. Tang, G. Zhang, F. Zheng, H. Song, Q. Li and S. Ban, Org. Biomol. Chem., 2021, 19, 3370–3373 RSC.
- C.-Y. Li, M. Xiang, J. Zhang, W.-S. Li, Y. Zou, F. Tian and L.-X. Wang, Org. Biomol. Chem., 2021, 19, 7690–7694 RSC.
- Y. Zhang, J. Wu, W. Qiu, L. Liao, B. Wang and X. Zhao, Org. Lett., 2023, 25, 5173–5178 CrossRef CAS PubMed.
- D. G. Lynn, J. C. Steffens, V. S. Kamut, D. W. Graden, J. Shabanowitz and J. L. Riopel, J. Am. Chem. Soc., 1981, 103, 1868–1870 CrossRef CAS.
- R. E. Carlson and D. H. Dolphin, Phytochemistry, 1982, 21, 1733–1736 CrossRef CAS.
- F. Shahidi and P. Ambigaipalan, J. Funct. Foods, 2015, 18, 820–897 CrossRef CAS.
- L. B. Davin, M. Jourdes, A. M. Patten, K.-W. Kim, D. G. Vassão and N. G. Lewis, Nat. Prod. Rep., 2008, 25, 1015 RSC.
- A. V. Malkov, S. L. Davis, I. R. Baxendale, W. L. Mitchell and P. Kočovský, J. Org. Chem., 1999, 64, 2751–2764 CrossRef CAS PubMed.
- A. V. Malkov, P. Spoor, V. Vinader and P. Kočovský, J. Org. Chem., 1999, 64, 5308–5311 CrossRef CAS PubMed.
- I. Fernández, R. Hermatschweiler, F. Breher, P. S. Pregosin, L. F. Veiros and M. J. Calhorda, Angew. Chem., Int. Ed., 2006, 45, 6386–6391 CrossRef PubMed.
- T. Mino, T. Kogure, T. Abe, T. Koizumi, T. Fujita and M. Sakamoto, Eur. J. Org Chem., 2013, 2013, 1501–1505 CrossRef CAS.
- C. A. Discolo, A. G. Graves and D. R. Deardorff, J. Org. Chem., 2017, 82, 1034–1045 CrossRef CAS.
- C. Li and B. Breit, Chem.–Eur. J., 2016, 22, 14655–14663 CrossRef CAS PubMed.
- Q.-L. Xu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 2579–2581 CrossRef CAS PubMed.
- S. Chinnabattigalla, A. Choudhury and S. Gedu, Org. Biomol. Chem., 2021, 19, 8259–8263 RSC.
- K.-Q. Wu, H. Li, A. Zhou, W.-R. Yang and Q. Yin, J. Org. Chem., 2023, 88, 2599–2604 CrossRef CAS PubMed.
- C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2013, 52, 10056–10059 CrossRef CAS PubMed.
- D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Angew. Chem., 2017, 129, 3290–3294 CrossRef.
- J. T. Randolph, C. A. Flentge, P. P. Huang, D. K. Hutchinson, L. L. Klein, H. B. Lim, R. Mondal, T. Reisch, D. A. Montgomery, W. W. Jiang, S. V. Masse, L. E. Hernandez, R. F. Henry, Y. Liu, G. Koev, W. M. Kati, K. D. Stewart, D. W. A. Beno, A. Molla and D. J. Kempf, J. Med. Chem., 2009, 52, 3174–3183 CrossRef CAS PubMed.
- S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS PubMed.
- S.-B. Tang, H.-F. Tu, X. Zhang and S.-L. You, Org. Lett., 2019, 21, 6130–6134 CrossRef CAS PubMed.
- H.-J. Zhang, Q. Gu and S.-L. You, Org. Lett., 2020, 22, 3297–3301 CrossRef CAS PubMed.
- Y. Yang, B. Lu, G. Xu and X. Wang, ACS Cent. Sci., 2022, 8, 581–589 CrossRef CAS PubMed.
- Z. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626–10631 CrossRef CAS PubMed.
- G. Wang, L. Gao, H. Chen, X. Liu, J. Cao, S. Chen, X. Cheng and S. Li, Angew. Chem., Int. Ed., 2019, 58, 1694–1699 CrossRef CAS PubMed.
- Z. Liu, G. Li, T. Yao, J. Zhang and L. Liu, Adv. Synth. Catal., 2021, 363, 2740–2745 CrossRef CAS.
- D. Zhao, J. Luo, L. Liu and Y. Liu, Org. Chem. Front., 2021, 8, 6252–6258 RSC.
- K. Laxmikeshav, A. P. Sakla, S. Rasane, S. E. John and N. Shankaraiah, ChemistrySelect, 2020, 5, 7004–7012 CrossRef CAS.
- H. Chen, M. Yang, G. Wang, L. Gao, Z. Ni, J. Zou and S. Li, Org. Lett., 2021, 23, 5533–5538 CrossRef CAS PubMed.
- B. M. Trost, F. D. Toste and K. Greenman, J. Am. Chem. Soc., 2003, 125, 4518–4526 CrossRef CAS PubMed.
- G. Brufani, F. Valentini, F. Sabatelli, B. Di Erasmo, A. M. Afanasenko, C.-J. Li and L. Vaccaro, Green Chem., 2022, 24, 9094–9100 RSC.
- R. Martinez, F. Voica, J.-P. Genet and S. Darses, Org. Lett., 2007, 9, 3213–3216 CrossRef CAS PubMed.
- J. K. Belardi and G. C. Micalizio, J. Am. Chem. Soc., 2008, 130, 16870–16872 CrossRef CAS PubMed.
- M. Yamaguchi, A. Hayashi and M. Hirama, J. Am. Chem. Soc., 1995, 117, 1151–1152 CrossRef CAS.
- M. Yamaguchi, Pure Appl. Chem., 1998, 70, 1091–1096 CrossRef CAS.
- K. Kobayashi, M. Arisawa and M. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 8528–8529 CrossRef CAS.
- E. Tan, A. I. Konovalov, G. A. Fernández, R. Dorel and A. M. Echavarren, Org. Lett., 2017, 19, 5561–5564 CrossRef CAS.
- S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847–851 CrossRef CAS PubMed.
- X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4830–4833 CrossRef CAS PubMed.
- V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Org. Lett., 2012, 14, 3416–3419 CrossRef CAS PubMed.
- L. Hu, M. C. Dietl, C. Han, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2021, 60, 10637–10642 CrossRef CAS PubMed.
- A. Kobayashi, T. Matsuzawa, T. Hosoya and S. Yoshida, RSC Adv., 2023, 13, 839–843 RSC.
- B. Liu, Y. Li, M. Yin, W. Wu and H. Jiang, Chem. Commun., 2012, 48, 11446 RSC.
- G. Jiang, S. Fang, W. Hu, J. Li, C. Zhu, W. Wu and H. Jiang, Adv. Synth. Catal., 2018, 360, 2297–2302 CrossRef CAS.
- X. Chen, K. M. Engle, D. Wang and J. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- K. Naksomboon, C. Valderas, M. Gómez-Martínez, Y. Álvarez-Casao and M. Á. Fernández-Ibáñez, ACS Catal., 2017, 7, 6342–6346 CrossRef CAS.
- M. Murai, M. Yamamoto and K. Takai, Org. Lett., 2019, 21, 3441–3445 CrossRef CAS PubMed.
- M. Murai, M. Yamamoto and K. Takai, Chem.–Eur. J., 2019, 25, 15189–15197 CrossRef CAS PubMed.
- T. Adak, J. Schulmeister, M. C. Dietl, M. Rudolph, F. Rominger and A. S. K. Hashmi, Eur. J. Org Chem., 2019, 2019, 3867–3876 CrossRef CAS.
- T. Li, Y. Yang, B. Luo, B. Li, L. Zong, W. Kong, H. Yang, X. Cheng and L. Zhang, Org. Lett., 2020, 22, 6045–6049 CrossRef CAS PubMed.
- Y. Dou, Kenry, J. Liu, J. Jiang and Q. Zhu, Chem.–Eur. J., 2019, 25, 6896–6901 CrossRef CAS PubMed.
- J. Zhou, J. Huang, C. Lu, H. Jiang and L. Huang, Adv. Synth. Catal., 2021, 363, 3962–3967 CrossRef CAS.
- H. Chen, L. Gao, X. Liu, G. Wang and S. Li, Eur. J. Org Chem., 2021, 2021, 5238–5242 CrossRef CAS.
- Y.-B. Wang, P. Yu, Z.-P. Zhou, J. Zhang, J. Wang, S.-H. Luo, Q.-S. Gu, K. N. Houk and B. Tan, Nat. Catal., 2019, 2, 504–513 CrossRef CAS.
- A. G. Woldegiorgis, Z. Han and X. Lin, Org. Lett., 2021, 23, 6606–6611 CrossRef CAS PubMed.
- A. Pramanik, A. Ghatak, S. Khan and S. Bhar, Tetrahedron Lett., 2019, 60, 1091–1095 CrossRef CAS.
- T. Yao, S. Zhao, T. Liu, Y. Wu, Y. Ma, T. Li and X. Qin, Chem. Commun., 2022, 58, 4592–4595 RSC.
- S. Maurya, S. B. Jadhav and R. Chegondi, Org. Biomol. Chem., 2022, 20, 2059–2063 RSC.
- D. Zofou, F. Ntie-Kang, W. Sippl and S. M. N. Efange, Nat. Prod. Rep., 2013, 30, 1098 RSC.
- H. Aldemir, R. Richarz and T. A. M. Gulder, Angew. Chem., Int. Ed., 2014, 53, 8286–8293 CrossRef CAS PubMed.
- W. Sun, G. Li, L. Hong and R. Wang, Org. Biomol. Chem., 2016, 14, 2164–2176 RSC.
- L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235–2261 CrossRef.
- Ullmann's Encyclopedia of Industrial Chemistry, ed. M. Bohnet, Wiley-VCH, Weinheim, 6th edn, 2003 Search PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef CAS PubMed.
- Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed.
- P. Wawrzyniak and J. Heinicke, Tetrahedron Lett., 2006, 47, 8921–8924 CrossRef CAS.
- C. Huang and V. Gevorgyan, J. Am. Chem. Soc., 2009, 131, 10844–10845 CrossRef CAS PubMed.
- R. B. Bedford, M. Betham, A. J. M. Caffyn, J. P. H. Charmant, L. C. Lewis-Alleyne, P. D. Long, D. Polo-Cerón and S. Prashar, Chem. Commun., 2008, 990 RSC.
- G. B. Bajracharya and O. Daugulis, Org. Lett., 2008, 10, 4625–4628 CrossRef CAS PubMed.
- T. Truong and O. Daugulis, Chem. Sci., 2013, 4, 531–535 RSC.
- R. B. Bedford, S. J. Coles, M. B. Hursthouse and M. E. Limmert, Angew. Chem., Int. Ed., 2003, 42, 112–114 CrossRef CAS PubMed.
- R. B. Bedford and M. E. Limmert, J. Org. Chem., 2003, 68, 8669–8682 CrossRef CAS PubMed.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed.
- R. B. Bedford, R. L. Webster and C. J. Mitchell, Org. Biomol. Chem., 2009, 7, 4853 RSC.
- Y. Nieves-Quinones, T. J. Paniak, Y. E. Lee, S. M. Kim, S. Tcyrulnikov and M. C. Kozlowski, J. Am. Chem. Soc., 2019, 141, 10016–10032 CrossRef CAS PubMed.
- H. Reiss, H. Shalit, V. Vershinin, N. Y. More, H. Forckosh and D. Pappo, J. Org. Chem., 2019, 84, 7950–7960 CrossRef CAS PubMed.
- J.-W. Zhang, F. Jiang, Y.-H. Chen, S.-H. Xiang and B. Tan, Sci. China: Chem., 2021, 64, 1515–1521 CrossRef CAS.
- V. Vershinin and D. Pappo, Org. Lett., 2020, 22, 1941–1946 CrossRef CAS PubMed.
- V. Vershinin, H. Forkosh, M. Ben-Lulu, A. Libman and D. Pappo, J. Org. Chem., 2021, 86, 79–90 CrossRef CAS PubMed.
- N. H. Nguyen, S. M. Oh, C.-M. Park and S. Shin, Chem. Sci., 2022, 13, 1169–1176 RSC.
- K. A. Niederer, P. H. Gilmartin and M. C. Kozlowski, ACS Catal., 2020, 10, 14615–14623 CrossRef CAS PubMed.
- M. Jurrat, L. Maggi, W. Lewis and L. T. Ball, Nat. Chem., 2020, 12, 260–269 CrossRef CAS PubMed.
- A. Senior, K. Ruffell and L. T. Ball, Nat. Chem., 2023, 15, 386–394 CrossRef CAS PubMed.
- M. K. Ghosh, J. Rzymkowski and M. Kalek, Chem.–Eur. J., 2019, 25, 9619–9623 CrossRef CAS PubMed.
- R. Bisht, M. V. Popescu, Z. He, A. M. Ibrahim, G. E. M. Crisenza, R. S. Paton and D. J. Procter, Angew. Chem., Int. Ed., 2023, 62, e202302418 CrossRef CAS PubMed.
- Z. He, G. J. P. Perry and D. J. Procter, Chem. Sci., 2020, 11, 2001–2005 RSC.
- P.-Y. Jiang, K.-F. Fan, S. Li, S.-H. Xiang and B. Tan, Nat. Commun., 2021, 12, 2384 CrossRef CAS PubMed.
- I. A. Utepova, A. I. Nemytov, V. A. Ishkhanian, O. N. Chupakhin and V. N. Charushin, Tetrahedron, 2020, 76, 131391 CrossRef CAS.
- D.-L. Zhu, S. Jiang, D. J. Young, Q. Wu, H.-Y. Li and H.-X. Li, Chem. Commun., 2022, 58, 3637–3640 RSC.
- H. Jia, M. He, S. Yang, X. Yu and M. Bao, Eur. J. Org Chem., 2022, 2022, e202101469 CrossRef CAS.
- B. Dahms, P. J. Kohlpaintner, A. Wiebe, R. Breinbauer, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2019, 25, 2713–2716 CrossRef CAS PubMed.
- R. A. Green, R. C. D. Brown, D. Pletcher and B. Harji, Org. Process Res. Dev., 2015, 19, 1424–1427 CrossRef CAS.
- M. Hielscher, E. K. Oehl, B. Gleede, J. Buchholz and S. R. Waldvogel, ChemElectroChem, 2021, 8, 3904–3910 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- Y. Yue, J. Chao, Z. Wang, Y. Yang, Y. Ye, C. Sun, X. Guo and J. Liu, Org. Biomol. Chem., 2021, 19, 7156–7160 RSC.
- I. Neves, F. Jayat, P. Magnoux, G. Pérot, F. R. Ribeiro, M. Gubelmann and M. Guisnet, J. Mol. Catal., 1994, 93, 169–179 CrossRef CAS.
- M. Ghiaci, R. J. Kalbasi, M. Mollahasani and H. Aghaei, Appl. Catal., A, 2007, 320, 35–42 CrossRef CAS.
- C. L. Padró, M. E. Sad and C. R. Apesteguía, Catal. Today, 2006, 116, 184–190 CrossRef.
- C. Padro, J. Catal., 2004, 226, 308–320 CrossRef CAS.
- G. Tu, G. Ju, Z. Huang, S.-J. Ji and Y. Zhao, Org. Chem. Front., 2022, 9, 3876–3881 RSC.
- G. D. Yadav and A. R. Yadav, Ind. Eng. Chem. Res., 2013, 52, 10627–10636 CrossRef CAS.
- M. K. Montañez Valencia, C. L. Padró and M. E. Sad, Appl. Catal., B, 2020, 278, 119317 CrossRef.
- S. Gutiérrez-Rubio, M. Shamzhy, J. Čejka, D. P. Serrano, I. Moreno and J. M. Coronado, Appl. Catal., B, 2021, 285, 119826 CrossRef.
- H. K. Chau, D. E. Resasco, P. Do and S. P. Crossley, J. Catal., 2022, 406, 48–55 CrossRef CAS.
- K. Saint-Jacques and A. B. Charette, J. Flow Chem., 2023, 13, 193–199 CrossRef CAS.
- N. W. Gaikwad, G.-S. Hwang and I. H. Goldberg, Org. Lett., 2004, 6, 4833–4836 CrossRef CAS PubMed.
- C. S. Kim, B. Shin, O. W. Kwon, S. Y. Kim, S. U. Choi, D.-C. Oh, K. H. Kim and K. R. Lee, Tetrahedron Lett., 2014, 55, 6504–6507 CrossRef CAS.
- J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880–4883 CrossRef CAS PubMed.
- C. Zheng, J. Zheng and S.-L. You, ACS Catal., 2016, 6, 262–271 CrossRef CAS.
- G. Duarah, P. P. Kaishap, B. Sarma and S. Gogoi, Chem.–Eur. J., 2018, 24, 10196–10200 CrossRef CAS PubMed.
- I. Khan, S. R. Chidipudi and H. W. Lam, Chem. Commun., 2015, 51, 2613–2616 RSC.
- J. Nan, Z. Zuo, L. Luo, L. Bai, H. Zheng, Y. Yuan, J. Liu, X. Luan and Y. Wang, J. Am. Chem. Soc., 2013, 135, 17306–17309 CrossRef CAS PubMed.
- L. Han, H. Wang and X. Luan, Org. Chem. Front., 2018, 5, 2453–2457 RSC.
- A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 7607–7610 CrossRef CAS PubMed.
- S. Kujawa, D. Best, D. J. Burns and H. W. Lam, Chem.–Eur. J., 2014, 20, 8599–8602 CrossRef CAS PubMed.
- S. Gu, L. Luo, J. Liu, L. Bai, H. Zheng, Y. Wang and X. Luan, Org. Lett., 2014, 16, 6132–6135 CrossRef CAS PubMed.
- V. A. D’yakonov, O. A. Trapeznikova, A. De Meijere and U. M. Dzhemilev, Chem. Rev., 2014, 114, 5775–5814 CrossRef PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- Y. Li, Z. Tang, J. Zhang and L. Liu, Chem. Commun., 2020, 56, 8202–8205 RSC.
- W. Liang, Y. Yang, M. Yang, M. Zhang, C. Li, Y. Ran, J. Lan, Z. Bin and J. You, Angew. Chem., Int. Ed., 2021, 60, 3493–3497 CrossRef CAS PubMed.
- J. Wu, L. Bai, L. Han, J. Liu and X. Luan, Chem. Commun., 2021, 57, 1117–1120 RSC.
Footnote |
| † These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2024 |