
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA C–H functionalization approach to diverse nitrogenous scaffolds through conjugate addition of catalytic allyliron nucleophiles†
Sarah G. Scrivener and
Yi-Ming Wang *
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: ym.wang@pitt.edu
First published on 1st May 2024
Abstract
Cyclopentadienyliron(II) dicarbonyl complexes capable of coordinating to and enhancing the acidity of a range of unsaturated substrates have emerged as a new class of base-metal derived catalysts for C–H functionalization. In this manuscript, the iron-catalyzed C–H functionalization of allylic C(sp3)–H bonds using nitrogen containing α,β-unsaturated carbonyl compounds as coupling partners is reported. Employing a cationic cyclopentadienyliron dicarbonyl complex, this redox neutral process converts simple alkenes into allylic anion equivalents for 1,4-addition into maleimides, acyclic α,β-unsaturated imides, and vinylogous amides. The judicious pairing of pyridine and alkylamine bases with Lewis acid additives allowed each of these classes of substrates to be successfully employed, allowing for the formation of a diverse collection of cyclic and acyclic nitrogen-containing compounds featuring C–C unsaturation. The resulting Michael adducts can be further transformed into a variety of useful scaffolds such as allylated pyrroles, pyrrolidines, and carbocyclic acids.
Introduction
Nitrogen-containing building blocks, including heterocyclic scaffolds, are highly sought after targets of chemical synthesis, owing to their applicability as precursors toward the synthesis of pharmaceutical candidates and other bioactive compounds.1,2 Thus, the identification and investigation of general and modular approaches to access nitrogen-containing compounds endowed with commonly applicable synthetic handles represent important goals for the development of new synthetic methods. Approaches that provide access to nitrogenous compounds containing rapidly diversifiable olefin and carbonyl groups are especially valuable in this context.Succinimide derivatives in particular not only have independent bioactivity1–4 but also serve as versatile intermediates toward the synthesis of other five-membered nitrogen-containing heterocycles, including γ-lactams, pyrrolidines, and pyrroles, that feature prominently in drug candidates.5–7 In addition to providing a useful functional group that allows for further derivatization, the installation of allylic groups on to these azacycles can lead to bioactive compounds of interest in their own right (Scheme 1).1,3 Conjugate addition of carbon nucleophiles (Michael addition) to the readily available maleimide scaffold constitutes a straightforward and attractive route for quickly accessing 3-alkylated succinimides.8–11 However, there is a distinct lack of suitable nucleophiles for the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond allylation of maleimide derivatives. Common organometallic allyl nucleophiles such as magnesium,12 silicon,13 boron,14 indium,15 and tin16 reagents all selectively provide the 1,2-adduct of direct carbonyl addition, rather than 3-allylated succinimide derivative resulting from 1,4-addition.17 The installation of an allylic substructure at the 3-position by conjugate addition has only been successful in the context of additions by extended enolate species (Scheme 2).18
C bond allylation of maleimide derivatives. Common organometallic allyl nucleophiles such as magnesium,12 silicon,13 boron,14 indium,15 and tin16 reagents all selectively provide the 1,2-adduct of direct carbonyl addition, rather than 3-allylated succinimide derivative resulting from 1,4-addition.17 The installation of an allylic substructure at the 3-position by conjugate addition has only been successful in the context of additions by extended enolate species (Scheme 2).18
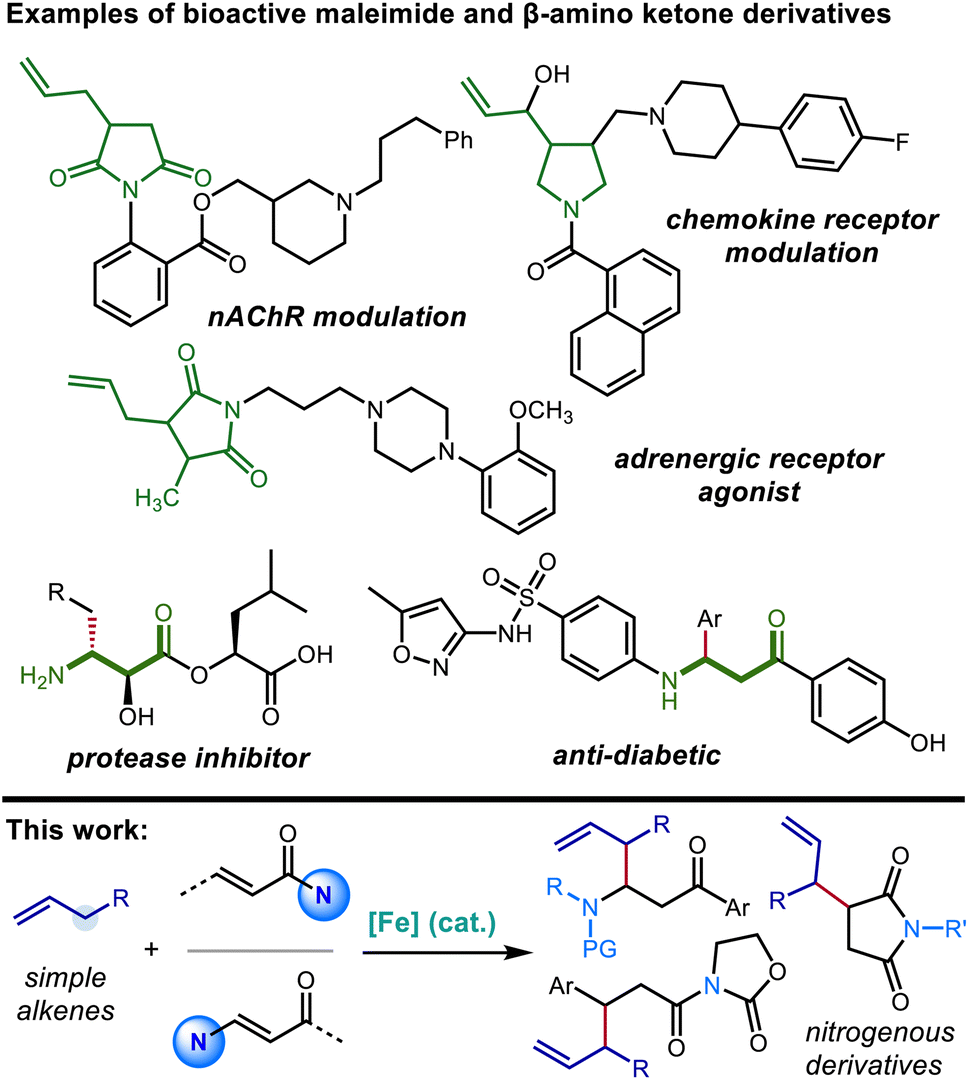 | ||
| Scheme 1 Examples of biologically active 3-allylated succinimides, β-substituted β-amino ketones (top) and overview of the transformations reported in this manuscript (bottom). | ||
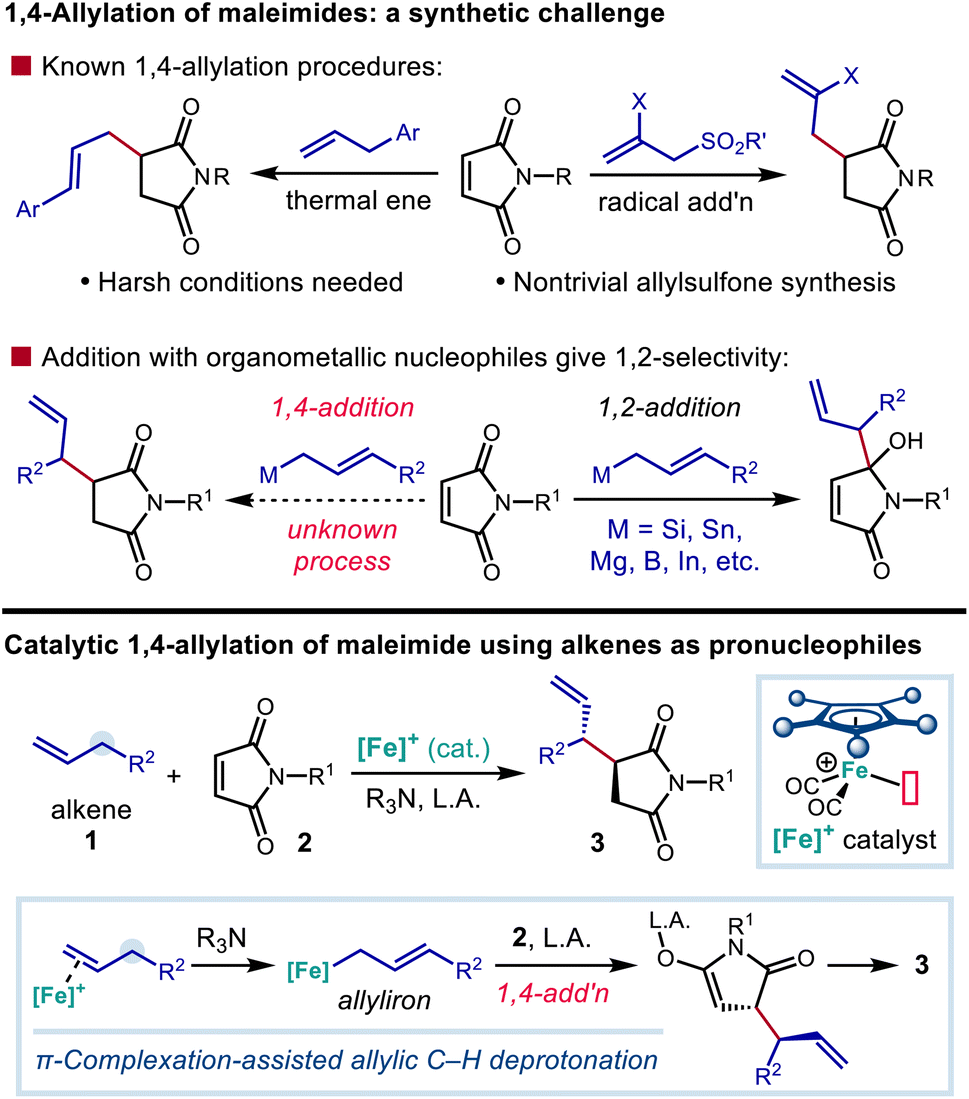 | ||
| Scheme 2 Allylation of maleimide derivatives as an unmet synthetic challenge and an approach to the synthesis of 3-allylated five-membered azacycles. | ||
Although 1,4-addition of allylic radicals to maleimides has been demonstrated using allylic sulfones as radical sources, the synthesis of the allylic sulfone precursors is often a nontrivial endeavour, and only the installation of the parent allyl group and a narrow range of 2-substituted derivatives have been demonstrated.19–21 Maleimides are also known to react with allylbenzene derivatives through the Alder-ene reaction22 to give 3-cinnamylated succinimides at very high reaction temperatures. Practically speaking, the current state-of-the-art for accessing allylated succinimide derivatives still relies on the reaction of succinimide-derived enolates with allylic bromides.23–25 However, highly reactive allylic bromides may be difficult to purify or handle and can give side products, including bis-allylated or regioisomeric adducts.23 In addition, they require additional steps to synthesize, creating hurdles when a functional group-rich allylic fragment is needed. A protocol for the regioselective addition of a nucleophilic allylic fragment to maleimide under catalytic conditions, particularly one employing simple hydrocarbon-based precursors (i.e., by C–H functionalization), would be a valuable tool for the synthesis of 3-allylated succinimides and other functionalized five-membered nitrogen-containing heterocycles.
Our group and others have shown that coordination of a cationic late transition metal complex to a C–C π-bond can greatly enhance the C–H acidity of the neighboring allylic, propargylic, or allenic protons.26,27 In particular, through the use of cyclopentadienyliron(II) dicarbonyl derivatives as catalysts,28 this acidity enhancement enables the use of weak, functional group-tolerant amine bases to effect the allylic deprotonation of olefins to generate nucleophilic allyliron intermediates that react with electrophiles with SE2′ selectivity.29–35 We further discovered that these nucleophilic organoiron species will undergo electrophilic functionalization with chalcone derivatives through a 1,4-addition process.36
In this article we report the development of a series of C–H functionalization methods for the coupling of alkenes with maleimides and other synthetically versatile nitrogen-containing α,β-unsaturated carbonyl compounds. These processes allow for the synthesis of high value nitrogen-containing building blocks using simple, readily available precursors. Moreover, these procedures can be used for the elaboration of complex scaffolds derived from known pharmaceuticals and bioactive compounds.
Results and discussion
Maleimide electrophiles
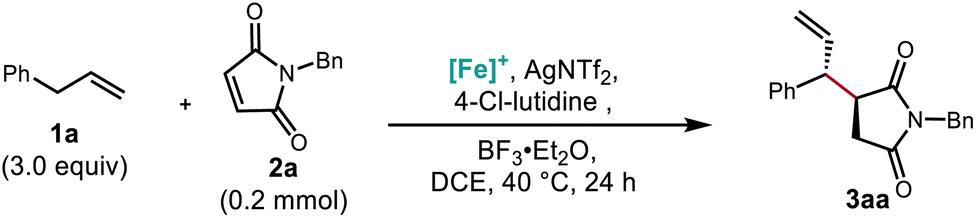
We hypothesized that this π-coordination-assisted deprotonation approach to allylic C–H functionalization could allow for conjugate addition into maleimides, delivering functionalized succinimide products in a highly regioselective manner. Exploring these electrophiles, we immediately found that silyl Lewis acids such as TIPSOTf (Table 1, entry 1) and TMSOTf (entry 2) provided no desired product. However, the boron Lewis acid BF3·Et2O (entry 3) did provide the desired coupling product, though with poor mass balance in part due to product decomposition. An increase in yield was observed upon inclusion of lithium bistriflimide as a co-Lewis acid additive (entry 4), as well as by decreasing base strength (entries 5–7). Stronger amine bases such as 2,2,6,6-tetramethylpiperidine (TMPH) appeared to be incompatible with the highly electrophilic starting material and the readily enolizable product, while weak and hindered bases like 4-chloro-2,6-lutidine (4-Cl-lutidine) were found to be optimal (entry 7). Reflecting the sensitivity of the starting material and product, lowering the temperature to 40 °C and reducing the amount of Lewis acid used likewise improved yield (entries 8 and 9). Gratifyingly, extensive screening of additives demonstrated that employing substoichiometric AgNTf2 as the co-Lewis acid and toluene as a solvent resulted in an 89% isolated yield while increasing the diastereomeric ratio (d.r.) from 3.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 7.6:1 (entry 11). The addition of Mg(NTf2)2 was nearly as effective for this purpose and may be a more economical alternative on larger scale (entry 10). The major regioisomer obtained was ascertained by X-ray crystallographic analysis and is consistent with an open transition state model (see the ESI†). Under all conditions evaluated, 1H NMR analysis of the crude material indicated that the branched regioisomer was formed exclusively (>20:1 r. r.).
:1 to 7.6:1 (entry 11). The addition of Mg(NTf2)2 was nearly as effective for this purpose and may be a more economical alternative on larger scale (entry 10). The major regioisomer obtained was ascertained by X-ray crystallographic analysis and is consistent with an open transition state model (see the ESI†). Under all conditions evaluated, 1H NMR analysis of the crude material indicated that the branched regioisomer was formed exclusively (>20:1 r. r.).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L.A. + additivea | Base | Temp. | Ratio (LA/base) | Yieldb (%) | dr |
| a Bistriflimide additive (0.35 equiv.) used for entries 4–11. b Yields determined by 1H NMR using 2,4-dinitrotoluene as the internal standard. c Isolated yield. Conditions: 1a (3.0 equiv.), 2a (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]−(20 mol%), BF3·Et2O (1.5 equiv.), 4-chlorolutidine (4.0 equiv.), AgNTf2 (0.35 equiv.), toluene [1.5 M], 40 °C, 24 h. | ||||||
| 1 | TIPSOTf | Collidine | 60 | 3/4 | 0 | — |
| 2 | TMSOTf | Collidine | 60 | 3/4 | 0 | — |
| 3 | BF3·Et2O | Collidine | 60 | 3/4 | 30 | — |
| 4 | BF3·Et2O + LiNTf2 | Collidine | 60 | 3/4 | 37 | — |
| 5 | BF3·Et2O + LiNTf2 | TMPH | 60 | 3/4 | 0 | — |
| 6 | BF3·Et2O + LiNTf2 | Lutidine | 60 | 3/4 | 40 | — |
| 7 | BF3·Et2O + LiNTf2 | 4-Cl-lutidine | 60 | 3/4 | 47 | — |
| 8 | BF3·Et2O + LiNTf2 | 4-Cl-lutidine | 40 | 3/4 | 64 | — |
| 9 | BF3·Et2O + LiNTf2 | 4-Cl-lutidine | 40 | 1.5/4 | 83 | 3.7:1 |
| 10 | BF3·Et2O + Mg(NTf2)2 | 4-Cl-lutidine | 40 | 1.5/4 | 75 | 6.4:1 |
| 11 | BF3·Et2O + AgNTf2 | 4-Cl-lutidine | 40 | 1.5/4 | 89c | 7.6:1 |
| 12 | BF3·Et2O | 4-Cl-lutidine | 40 | 1.5/4 | 9 | 6.3:1 |
With these reaction conditions in hand, we explored a variety of nitrogen substituents and alkene coupling partners. The scope of well tolerated N-substituents was broad: alkyl groups spanning a range of steric profiles (Scheme 3, 3ba and 3ca) were tolerated, as well as cycloalkyl groups (3da, 3ea), as was a maleimide derived from a pharmaceutical fragment (3ga). The reaction performed well with N-benzylic substituents (3aa and 3ha), giving good yields and good diastereoselectivity. Although N-aryl maleimides furnished products with somewhat reduced diastereoselectivity, succinimide products were nonetheless obtained in good to excellent yields for both electron-donating (3ka) and electron-withdrawing (3ia) aryl substituents. This reaction provided good to excellent yields for a range of alkene substrates, including electron-rich (3ab, 3ae) and electron-poor (3ad) allylbenzene derivatives. Unfortunately, unactivated olefins (without an additional aryl group at the α-position) were unsuccessful (<5% yield), presumably because the weakly basic 4-chlorolutidine (pKaH = 5.4) and lower reaction temperatures (40 °C) needed in this protocol are insufficient for deprotonation of these substrates.30
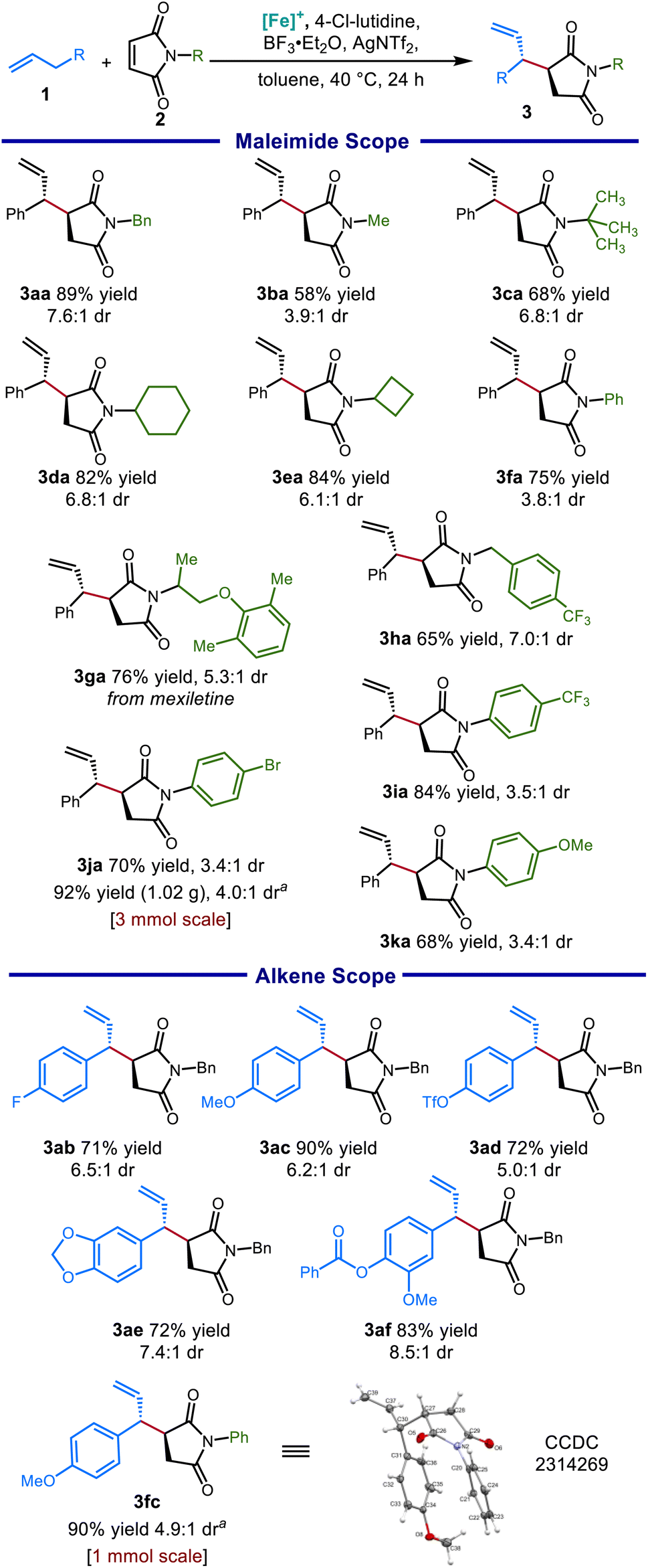 | ||
| Scheme 3 Scope of 3-allylic pyrrolidine-2,5-dione products obtained from catalytic C–H allylic functionalization. Conditions: 1 (3.0 equiv.), 2 (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]− (20 mol%), BF3·Et2O (1.5 equiv.), 4-chlorolutidine (4.0 equiv.), AgNTf2 (0.35 equiv.), toluene [1.5 M], 40 °C, 24 h. Isolated yields reported. aConditions: Mg(NTf2)2 (0.35 equiv.). | ||
A slightly modified protocol could be performed on gram scale while maintaining synthetic efficiency, giving the aryl bromide 3ja in excellent yield. Notably, both aromatic and saturated five-membered azacycles can be obtained as derivatives of the succinimide products. For example, reduction of 3fc using LiAlH4 selectively provided either the 3-allylic pyrrole or the 3-allylic pyrrolidine depending on the level of vigor of the reaction conditions (Scheme 4).
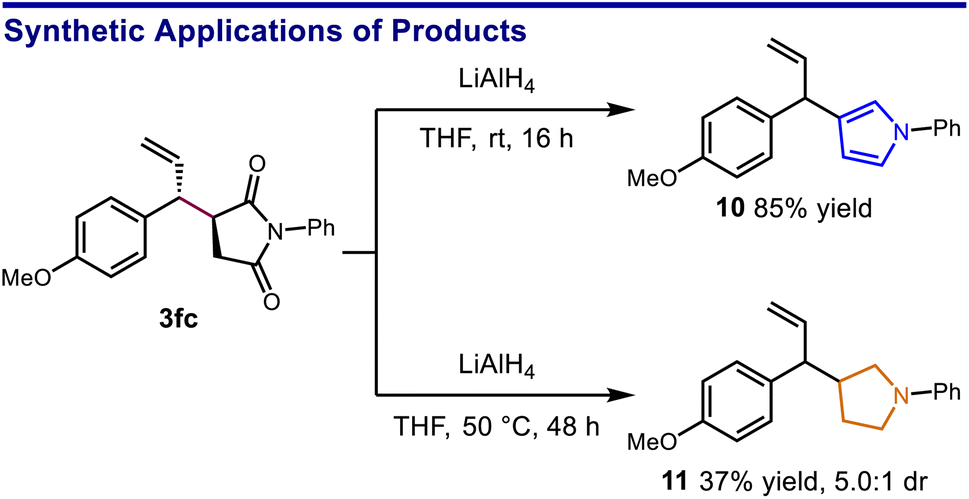 | ||
| Scheme 4 Diversification of five-membered nitrogen heterocycles. | ||
N-Acyl oxazolidinone electrophiles
Having unlocked a strategy for conjugate addition of alkenes into maleimides, we further hypothesized that alkene addition to acyclic α,β-unsaturated imides would also be possible. α,β-Unsaturated N-acyl oxazolidinones were chosen due to their ease of synthesis and synthetic utility as precursors to a variety of functional groups, including alcohols, aldehydes, carboxylic acids,37 hydroxamic acids,38 esters, and amides.39This reaction was found to require elevated temperatures relative to the maleimide coupling partners, in addition to a larger amount of the Lewis acids BF3·Et2O and LiNTf2. These reaction conditions reflect the decreased reactivity of the acyclic system, as well as the presence of additional Lewis basic sites on the oxazolidinone substrate. Although diastereoselectivities remained modest after optimization, the identity and stoichiometry of the co-Lewis acid additive nonetheless proved important for this reaction (Table 2).
|
|
|||
|---|---|---|---|
| Entry | Ratio (LA/base/add.) | Additive | Yielda (%) |
| a Yields determined by 1H NMR using 2,4-dinitrotoluene as the internal standard. b Isolated yield. Conditions: 1a (3.0 equiv.), 4a (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]− (20 mol%), BF3·Et2O (5.0 equiv.), 4-chlorolutidine (3.0 equiv.), LiNTf2 (1.0 equiv.), DCE [1.5 M], 80 °C, 24 h. | |||
| 1 | 1.5/4/0.2 | AgNTf2 | Trace |
| 2 | 5/3/0.2 | AgNTf2 | 19 |
| 3 | 5/3/0.2 | Cu(NTf2)2 | 42 |
| 4 | 5/3/0.2 | Yb(OTf)3 | 54 |
| 5 | 5/3/0.2 | Zn(OTf)2 | 57 |
| 6 | 5/3/0.2 | LiNTf2 | 67 |
| 7 | 5/3/1.0 | LiNTf2 | 85 (88)b |
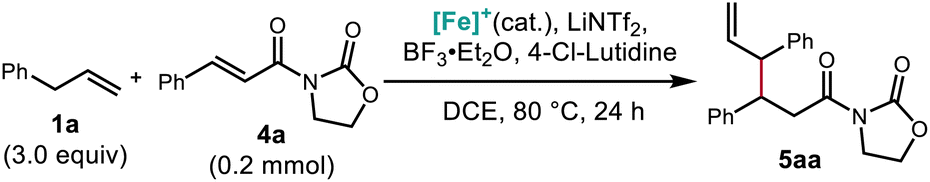
Optimized reaction conditions allowed for the coupling of α,β-unsaturated N-acyl oxazolidinone electrophiles with alkenes bearing synthetic handles such as aryl halides (5bg), and an alkene derived from a pharmaceutical fragment (5ah). Hydrolysis of the oxazolidinone with lithium hydroperoxide provided the δ,ε-unsaturated carboxylic acid 12 in excellent yield (Scheme 5). Moreover, ytterbium-catalyzed alcoholysis using propargyl alcohol afforded ester 13 bearing an additional terminal alkyne function in excellent yields.40
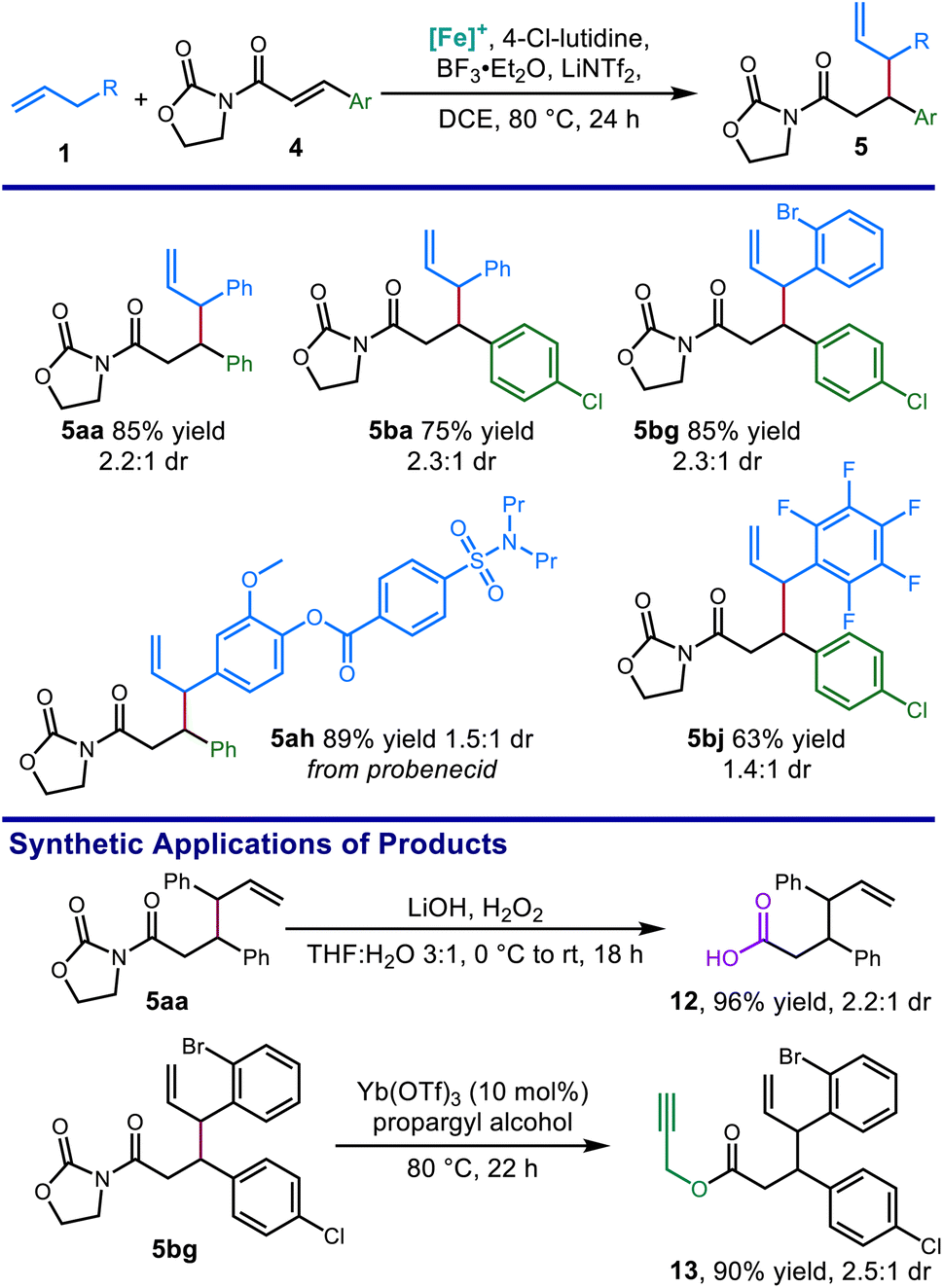 | ||
| Scheme 5 Scope of allylic N-acyl oxazolidinone products obtained from catalytic C–H allylic functionalization and their synthetic applications. Conditions: 1 (3.0 equiv.), 4 (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]− (20 mol%), BF3·Et2O (5.0 equiv.), 4-Cl-lutidine (3.0 equiv.), LiNTf2 (1.0 equiv.), DCE [1.5 M], 80 °C, 24 h. Isolated yields reported. | ||
Phthalimide-protected vinylogous amide electrophiles
Encouraged by the results from α,β-unsaturated imides, we hypothesized that our system could also provide allylation products by nucleophilic addition to vinylogous amides (β-enaminones), another challenging class of Michael acceptor susbtrates. Conjugate addition to the β-position of these substrates represents a straightforward and attractive route for quickly accessing β-substituted β-amino carbonyl compounds. However, this process faces two significant hurdles. First, the nitrogen substituent confers potentially undesirable nucleophilic character to the α-position. Moreover, electrophilicity of the β-position is attenuated by its unfavorable steric and electronic influence. Vinylogous amides thus tend to be sluggish Michael acceptors.41–46 Additionally, when addition does occur at the β-position, E1cb elimination of the nitrogen substituent readily occurs. While some processes have capitalized on this phenomenon to produce new β-substituted α,β-unsaturated ketones, attempts to prepare β-amino ketones through conjugate addition are often plagued by formation of these compounds as side products.47–52 Thus, the successful implementation of this strategy requires carbanion equivalents of high nucleophilicity while simultaneously demanding relatively mild conditions to ensure that the desired adduct survives the reaction conditions (Scheme 6).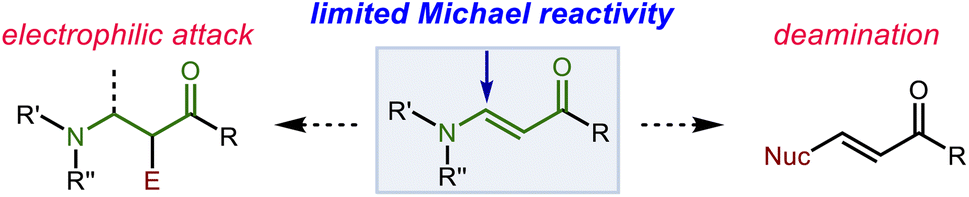 | ||
| Scheme 6 Side reactions in competition with 1,4-addition to enaminones. | ||
We began our investigation with the addition of allylbenzene to the phthalimido enaminone 6a. In contrast to the α,β-unsaturated imides explored earlier, BF3·Et2O was ineffective as the Lewis acid activator. Instead, we turned to silyl triflates as strong Lewis acids that irreversibly trap the incipient enolate ion intermediates as silyl enol ethers, which could be cleaved in a subsequent workup step to reveal the β-phthalimido ketone.36 We posited that this initial trapping as the silyl enol ether could also prevent the occurrence of deamination as a side reaction. In preliminary experiments, we found that an acidic workup (in contrast to fluoride-mediated or base-mediated desilylation) led to good yields of the ketone and minimal formation of the deaminated product.
Optimization of this reaction was carried out with careful attention to reagent stoichiometry. An increase of the reagents provided a corresponding increase in yield (Table 3, entry 2). The selection of TIPSOTf over TMSOTf was found to be highly beneficial to the reaction (entry 3). Other changes to the conditions, like decreasing the amount of LiNTf2 additive or the reaction temperature, led to decreased yields (entries 4 and 5).
|
|
||||
|---|---|---|---|---|
| Entry | Ratio (LA/base/LiNTf2) | Lewis acid | Temp. | Yielda (%) |
| a Yields determined by 1H NMR using 2,4-dinitrotoluene as the internal standard. b Isolated yield. Conditions: 1a (3.0 equiv.), 6a (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]− (20 mol%), TIPSOTf (2.5 equiv.), collidine (3.0 equiv.), LiNTf2 (0.6 equiv.), DCE [1.5 M], 80 °C, 16 h. Then 4 N HCl in dioxane. | ||||
| 1 | 1.5/2.0/0.6 | TMSOTf | 80 | 18 |
| 2 | 2.5/3.0/0.6 | TMSOTf | 80 | 37 |
| 3 | 2.5/3.0/0.6 | TIPSOTf | 80 | 90 (93)b |
| 4 | 2.5/3.0/0.3 | TIPSOTf | 80 | 73 |
| 5 | 2.5/3.0/0.6 | TIPSOTf | 60 | 85 |
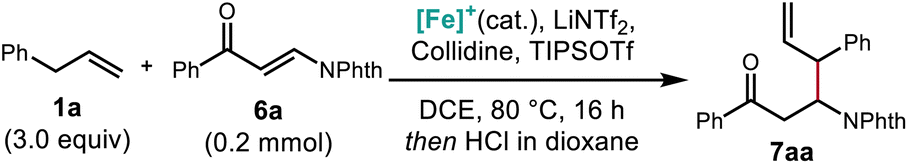
Using the optimized protocol, we investigated the scope of the alkene nucleophile. Allylbenzene coupling partners with a variety of substitution patterns provided excellent yields under the optimized conditions, including safrole (Scheme 7, 7ae) and allylpentafluorobenzene (7aj). Changing the conditions by using TMPH (4 equiv.) in place of 2,4,6-collidine (3 equiv.) allowed a variety of electronically unactivated alkenes to be utilized, including cyclopentene (7al) and a terminal alkene incorporating a pharmaceutical fragment (7aq). The improved yields using the stronger base TMPH over weaker pyridine bases is consistent with the decreased C–H acidity of the electronically unactivated substrates in relation to allylbenzene derivatives, further validating the importance of correctly identifying suitable combinations of base and Lewis acid for each pairing of alkene and electrophile. Alkenes bearing functional groups such as an alkyl p-toluenesulfonate ester (7am), a boronic acid pinacol ester (7ao), and a five membered heterocycle (7an), were also good coupling partners in this reaction. Finally, with slightly modified conditions, coupling with a phthalimide-protected allylic amine (7ap) provided the protected allylic 1,2-diamine in good yield.
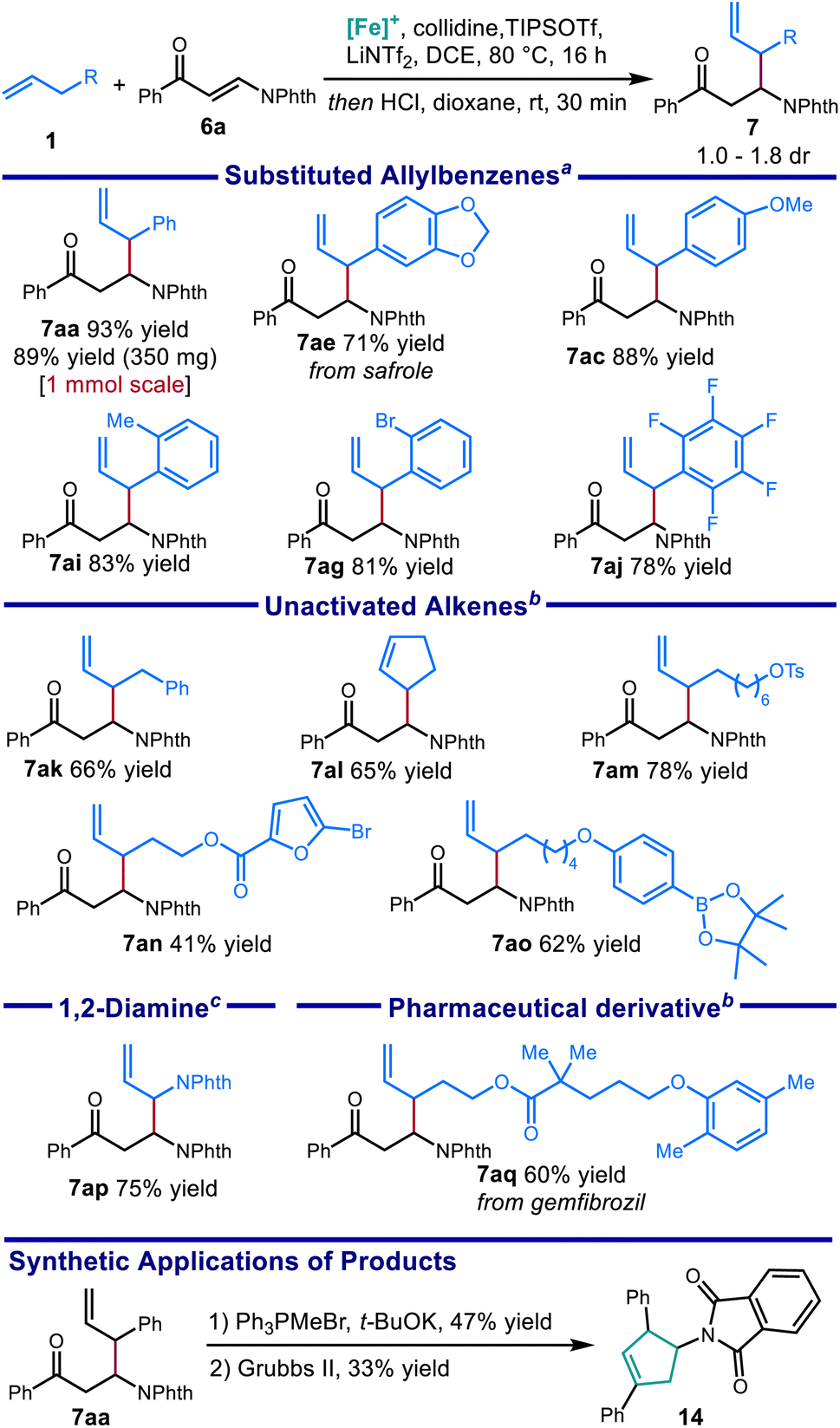 | ||
| Scheme 7 Scope of β-substituted β-phthalimido carbonyl compounds obtained from catalytic C–H allylic functionalization and synthetic applications of products. aConditions 1: 1 (3.0 equiv.), 6a (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]− (20 mol%), TIPSOTf (2.5 equiv.), collidine (3.0 equiv.), LiNTf2 (0.6 equiv.), DCE [1.5 M], 80 °C, 16 h. Then 4 N HCI in dioxane. bConditions 2: Base = 2,2,6,6-tetramethylpiperidine (4.0 equiv.). cConditions 3: LiNTf2 (0.8 equiv.). Isolated yields reported. | ||
The synthetic value of this method was further demonstrated through the elaboration of the product into a new scaffold: under Wittig conditions, the phthalimido protected β-amino ketone provided the β-amino-δ,ε-unsaturated diene, which was then cyclized through ring closing olefin metathesis to give 3-aminocyclopentene 14.53
Sulfonamide-protected vinylogous amide electrophiles
We then sought to expand this system to access β-substituted β-amino carbonyl compounds bearing a sulfonamide group on the nitrogen. Sulfonamide-bearing enaminones are a challenging substrate for this reaction due to the strong tendency toward deamination side reactions. This is reflected by the current lack of reported protocols to directly produce β-substituted β-amino ketones through 1,4-conjugate addition to sulfonyl-protected vinylogous amides. Optimizing the reaction conditions, we found that even milder conditions were necessary to avoid decomposition of the sensitive starting materials and products, including reduced reaction temperature and fewer equivalents of base (see the ESI for details†).These modifications allowed us to prepare a range of β-substituted β-sulfonamido ketones, including products derived from allylbenzene (9aa), an ester-containing eugenol derivative (9af), and an unactivated alkene (9ak). The protocol was also found to tolerate a wide variety of sulfonamides, including a p-nitrobenzenesulfonamide (9ba) and a sulfonamide derived from celecoxib (9da) (Scheme 8).
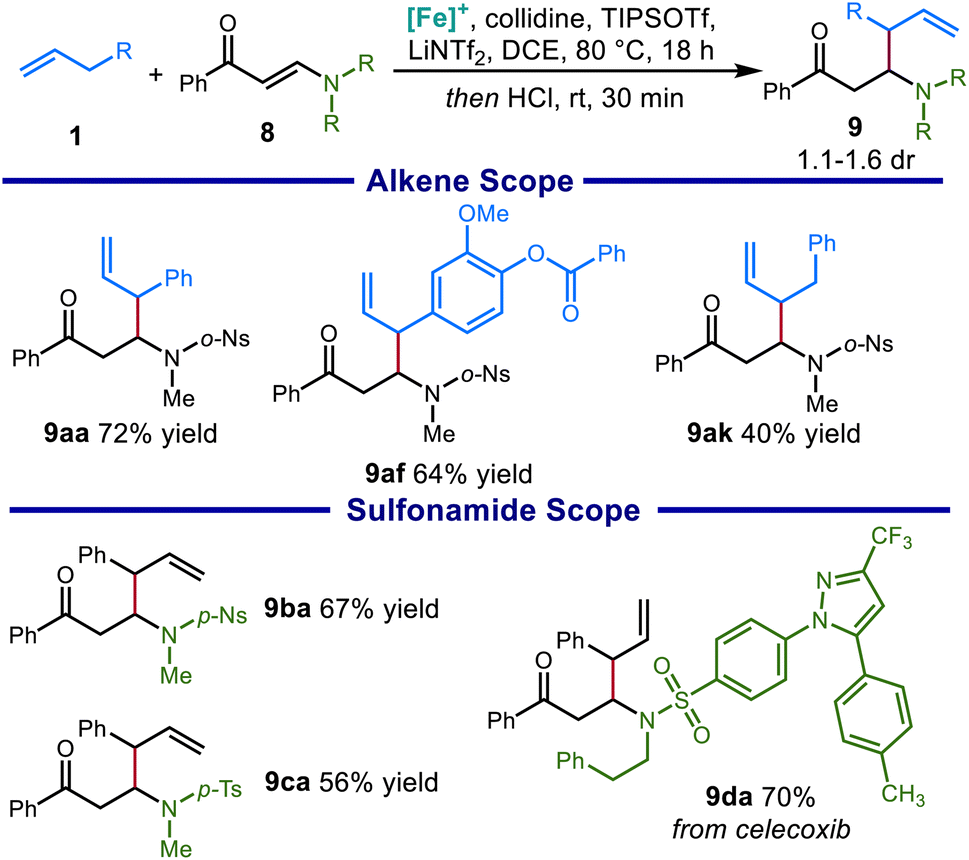 | ||
| Scheme 8 Scope of β-substituted β-sulfonamido carbonyl compounds obtained from catalytic C–H allylic functionalization. Conditions: 1 (3.0 equiv.), 8 (0.2 mmol), [Cp*Fe(CO)2(thf)]+[BF4]−(20 mol%), TIPSOTf (2.5 equiv.), collidine (2.5 equiv.), LiNTf2 (0.6 equiv.), DCE [1.5 M], 60 °C, 16 h. Then 4 N HCI in dioxane. Isolated yields reported. | ||
Conclusions
In summary, we have developed a series of iron-catalyzed allylic C–H functionalization reactions that provide allylated derivatives of nitrogen-containing carbonyl compounds in a general and modular approach. These diverse products were formed through the deprotonative generation of catalytic allyliron intermediates from allylbenzenes and other olefin derivatives which then undergo regiospecific addition to α,β-unsaturated carbonyl electrophiles. Further explorations of this strategy towards improved stereocontrol are ongoing in our laboratory and will be reported in due course.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
S. G. S. designed and performed the experiments, analyzed the data, and wrote the manuscript. Y.-M. W. conceptualized the project, analyzed the data, and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences, National Institutes of Health (R35GM142945). We thank Dr Jin Zhu (MIT) and Austin C. Durham for discussions and advice regarding the preparation of the manuscript. We thank Mason A. Semenick and Athanasios O. Tsoukaras for assistance in the preparation of some previously reported starting materials. We also thank Dr Damodaran K. Achary for helpful discussions on NMR spectroscopy of these compounds.References
- F. G.-C. Tatiana, J. H. Brandon, E. P. Ryan, B. M. Susan, A. E.-H. Raed, B. P. Aravinda, M. O. Crina, D. R. Damon, R. T. Boyd, X. Z. Michael, L. Chenglong, C. B. Stephen and B. M. Dennis, J. Pharmacol. Exp. Ther., 2009, 328, 504–515 CrossRef PubMed.
- N. Nakamura, A. Hirakawa, J.-J. Gao, H. Kakuda, M. Shiro, Y. Komatsu, C.-C. Sheu and M. Hattori, J. Nat. Prod., 2004, 67, 46–48 CrossRef CAS PubMed.
- S. C. Bergmeier, K. A. Ismail, K. M. Arason, S. McKay, D. L. Bryant and D. B. McKay, Bioorg. Med. Chem. Lett., 2004, 14, 3739–3742 CrossRef CAS.
- J. P. Roger, J. K. Penry, R. L. Joseph, E. N. Michael and J. K. Harvey, Neurology, 1979, 29, 1509–1513 CrossRef.
- J. C. Hubert, J. B. P. A. Wijnberg and W. N. Speckamp, Tetrahedron, 1975, 31, 1437–1441 CrossRef CAS.
- R. Aoun, J.-L. Renaud, P. H. Dixneuf and C. Bruneau, Angew. Chem., Int. Ed., 2005, 44, 2021–2023 CrossRef CAS.
- T. Shi, X. Wang, G. Yin and Z. Wang, Org. Chem. Front., 2022, 9, 1599–1603 RSC.
- B. Jana, M. Mondal, S. Halder, A. Mahata, S. Saurav and S. Paladhi, Asian J. Org. Chem., 2023, 12, e202300387 CrossRef CAS.
- S.-L. Liu, Y. Shi, C. Xue, L. Zhang, L. Zhou and M.-P. Song, Eur. J. Org Chem., 2021, 2021, 5862–5879 CrossRef CAS.
- R. Manoharan and M. Jeganmohan, Asian J. Org. Chem., 2019, 8, 1949–1969 CrossRef CAS.
- Y. Zhenhua, Z. Jianan, W. Caiyue, G. Yingxiang and Z. Shengyin, Chin. J. Org. Chem., 2019, 39, 2412–2427 CrossRef.
- A. Gómez-SanJuan, N. Sotomayor and E. Lete, Eur. J. Org Chem., 2013, 2013, 6722–6732 CrossRef.
- E. C. Izgu and T. R. Hoye, Chem. Sci., 2013, 4, 2262–2266 RSC.
- T. Sengoku, A. Shirai, A. Takano, T. Inuzuka, M. Sakamoto, M. Takahashi and H. Yoda, J. Org. Chem., 2019, 84, 12532–12541 CrossRef CAS.
- R. S. Coleman, M. C. Walczak and E. L. Campbell, J. Am. Chem. Soc., 2005, 127, 16038–16039 CrossRef CAS PubMed.
- T. Sengoku, Y. Murata, Y. Aso, A. Kawakami, T. Inuzuka, M. Sakamoto, M. Takahashi and H. Yoda, Org. Lett., 2015, 17, 5846–5849 CrossRef CAS PubMed.
- A. Chihab-Eddine, A. Daïch, A. Jilale and B. Decroix, Tetrahedron Lett., 2001, 42, 573–576 CrossRef CAS.
- G. Zhan, Q. He, X. Yuan and Y.-C. Chen, Org. Lett., 2014, 16, 6000–6003 CrossRef CAS PubMed.
- I. W. Harvey, E. D. Phillips and G. H. Whitham, Tetrahedron, 1997, 53, 6493–6508 CrossRef CAS.
- B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 1996, 118, 1209–1210 CrossRef CAS.
- A. P. Schaffner, K. Sarkunam and P. Renaud, Helv. Chim. Acta, 2006, 89, 2450–2461 CrossRef CAS.
- I. D. Cunningham, A. Brownhill, I. Hamerton and B. J. Howlin, Tetrahedron, 1997, 53, 13473–13494 CrossRef CAS.
- D. J. Bennett, P. L. Pickering and N. S. Simpkins, Chem. Commun., 2004, 12, 1392–1393 RSC.
- B. Daoust and J. Lessard, Tetrahedron, 1999, 55, 3495–3514 CrossRef CAS.
- A. R. Battersby and S. W. Westwood, J. Chem. Soc., Perkin Trans. 1, 1987, 1679–1687 RSC.
- T. Li and L. Zhang, J. Am. Chem. Soc., 2018, 140, 17439–17443 CrossRef CAS PubMed.
- J. Zhu, Y. Wang, A. D. Charlack and Y.-M. Wang, J. Am. Chem. Soc., 2022, 144, 15480–15487 CrossRef CAS.
- A. Cutler, D. Ehnholt, P. Lennon, K. Nicholas, D. F. Marten, M. Madhavarao, S. Raghu, A. Rosan and M. Rosenblum, J. Am. Chem. Soc., 1975, 97, 3149–3157 CrossRef CAS.
- A. C. Durham, C. R. Liu and Y.-M. Wang, Chem.–Eur. J., 2023, 29, e202301195 CrossRef CAS.
- R. Wang, Y. Wang, R. Ding, P. B. Staub, C. Z. Zhao, P. Liu and Y.-M. Wang, Angew. Chem., Int. Ed., 2023, 62, e202216309 CrossRef CAS.
- Y. Wang, S. G. Scrivener, X.-D. Zuo, R. Wang, P. N. Palermo, E. Murphy, A. C. Durham and Y.-M. Wang, J. Am. Chem. Soc., 2021, 143, 14998–15004 CrossRef CAS PubMed.
- Y. Wang, J. Zhu, A. C. Durham, H. Lindberg and Y.-M. Wang, J. Am. Chem. Soc., 2019, 141, 19594–19599 CrossRef CAS.
- (a) A. C. Durham, Y. Wang and Y.-M. Wang, Synlett, 2020, 31, 1747–1752 CrossRef CAS; (b) X. Yue and Y.-M. Wang, α-C–H Functionalization of π-Bonds Using Iron Complexes, In Handbook of CH-Functionalization, ed. D. Maiti, 2022, DOI:10.1002/9783527834242.chf0051.
- J. Zhu, A. C. Durham, Y. Wang, J. C. Corcoran, X.-D. Zuo, S. J. Geib and Y.-M. Wang, Organometallics, 2021, 40, 2295–2304 CrossRef CAS.
- Y. Wang, J. Zhu, R. Guo, H. Lindberg and Y.-M. Wang, Chem. Sci., 2020, 11, 12316–12322 RSC.
- S. G. Scrivener, Y. Wang and Y.-M. Wang, Org. Lett., 2023, 25, 1420–1424 CrossRef CAS PubMed.
- G. L. Beutner, B. M. Cohen, A. J. DelMonte, D. D. Dixon, K. J. Fraunhoffer, A. W. Glace, E. Lo, J. M. Stevens, D. Vanyo and C. Wilbert, Org. Process Res. Dev., 2019, 23, 1378–1385 CrossRef CAS.
- M. P. Sibi, H. Hasegawa and S. R. Ghorpade, Org. Lett., 2002, 4, 3343–3346 CrossRef CAS.
- J. M. Stevens, A. C. Parra-Rivera, D. D. Dixon, G. L. Beutner, A. J. DelMonte, D. E. Frantz, J. M. Janey, J. Paulson and M. R. Talley, J. Org. Chem., 2018, 83, 14245–14261 CrossRef CAS.
- Derivatization and comparison with spectral data for a known compound allowed for assignment of the relative stereochemistry of the major diastereomer of 5 Search PubMed(see the ESI†).
- M. Sato, S. Aoyagi, S. Yago and C. Kibayashi, Tetrahedron Lett., 1996, 37, 9063–9066 CrossRef CAS.
- D. B. Gotchev and D. L. Comins, J. Org. Chem., 2006, 71, 9393–9402 CrossRef CAS.
- S. V. Tsukanov and D. L. Comins, Angew. Chem., Int. Ed., 2011, 50, 8626–8628 CrossRef CAS.
- I. J. Amaye, R. D. Haywood, E. M. Mandzo, J. J. Wirick and P. L. Jackson-Ayotunde, Tetrahedron, 2021, 83, 131984 CrossRef CAS.
- B. Kaewmee, V. Rukachaisirikul and J. Kaeobamrung, Org. Biomol. Chem., 2017, 15, 7387–7395 RSC.
- C. Wang, C. Dong, L. Kong, Y. Li and Y. Li, Chem. Commun., 2014, 50, 2164–2166 RSC.
- F. Eiden and C. Herdeis, Arch. Pharm., 1978, 311, 287–293 CrossRef CAS PubMed.
- A. Alberola, L. A. Calvo, A. G. Ortega, M. C. Sañudo Ruíz, P. Yustos, S. G. Granda and E. García-Rodriguez, J. Org. Chem., 1999, 64, 9493–9498 CrossRef CAS.
- A. S. Devi, P. Helissey, R. L. Nongkhlaw and J. N. Vishwakarma, Synth. Commun., 2013, 43, 1653–1660 CrossRef CAS.
- J.-P. Wan, Y. Lin, K. Hu and Y. Liu, RSC Adv., 2014, 4, 20499–20505 RSC.
- S. Sundstrom, T. S. Nguyen and J. A. May, Org. Lett., 2020, 22, 1355–1359 CrossRef CAS PubMed.
- D. Chen, C. Wan, Y. Liu and J.-P. Wan, J. Org. Chem., 2023, 88, 4833–4838 CrossRef CAS PubMed.
- Coupling constant analysis and nOe experiments were performed on 14 to assign 1H NMR signals of 7 and 9 to the diastereomeric products Search PubMed(see the ESI†).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2314269. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00655k |
| This journal is © The Royal Society of Chemistry 2024 |