
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCascade reactions of HDDA-benzynes with tethered cyclohexadienones: strain-driven events originating from ortho-annulated benzocyclobutenes†
Bhavani Shankar
Chinta
,
Dorian S.
Sneddon
 and
Thomas R.
Hoye
*
and
Thomas R.
Hoye
*
Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455, USA. E-mail: hoye@umn.edu
First published on 30th April 2024
Abstract
Intramolecular net [2 + 2] cycloadditions between benzyne intermediates and an electron-deficient alkene to give benzocyclobutene intermediates are relatively rare. Benzynes are electrophilic and generally engage nucleophiles or electron-rich π-systems. We describe here reactions in which an alkene of a tethered enone traps thermally generated benzynes in a variety of interesting ways. The number of atoms that link the benzyne to C4 of a cyclohexa-2,5-dienone induces varying amounts of strain in the intermediates and products. This leads to a variety of different reaction outcomes by way of various strain-releasing events that are mechanistically intriguing. This work demonstrates an underappreciated class of strain that originates from the adjacent fusion of two rings to both C1–C2 and C2–C3 of a benzenoid ring – i.e. ‘ortho-annulation strain’. DFT computations shed considerable light on the mechanistic diversions among various reaction pathways as well as allow more fundamental evaluation of the strain in a homologous series of ortho-annulated carbocycles.
Introduction
Reactive intermediates possess, inherently, a relatively high amount of potential energy. Arynes, one of the most versatile of all such intermediates, are well known to engage alkenes in net [2 + 2] cycloaddition reactions to produce benzocyclobutenes.1,2 Simple alkenes and their more electron rich analogs such as enol ethers and enamines comprise the vast majority of such transformations. In contrast, there are very few reports of the reaction of an aryne with an electron-deficient alkene in an analogous fashion, a reflection no doubt of the electrophilic nature of an aryne.3A significant feature of the work reported here is the way in which an atypical and underappreciated type of strain can lead to unusual modes of reactivity. In particular, we describe species in which the strain and novel reactivity originate from the simultaneous fusion of two adjacent rings to the C1–C2 and C2–C3 bonds of an aromatic ring. This contrasts with the more classical and well-studied classes of strained ring systems (e.g., propellanes, bicyclobutanes, cyclophanes, cyclic alkynes, etc.).4,5
Results and discussion
We set out to explore reactions of benzynes with a conjugated enone that was tethered to the benzyne in such a fashion as to allow for benzocyclobutene formation. We envisioned using a triyne-containing cyclohexadienone substrate such as 1 to thermally produce the benzyne 2, which would allow us to assess its ability to generate the benzocyclobutene derivative 3 (Fig. 1a). We carried out an initial set of DFT computations using the simplified tethered benzyne 4 to learn about the energetics of such a process (Fig. 1b). The intermediate diradical 5 can exist as either a cis- or trans-fused diastereomer. The calculations show a large difference in the barriers leading to each (ΔΔG‡ = 17.9 kcal mol−1). The kinetically less favorable formation of the trans-isomer is reflective of the higher torsional strain necessary for proper orbital overlap in the TS. Closure of the cis-isomer of diradical 5 to the all-cis isomer of the benzocyclobutene derivative 6 occurs via a very low-barrier collapse of the diradical. This simple study suggested that there should be a substantial preference for formation of only one of the diastereomeric, cis-fused products (i.e., structure 6 with both Hs up rather than with both Hs down).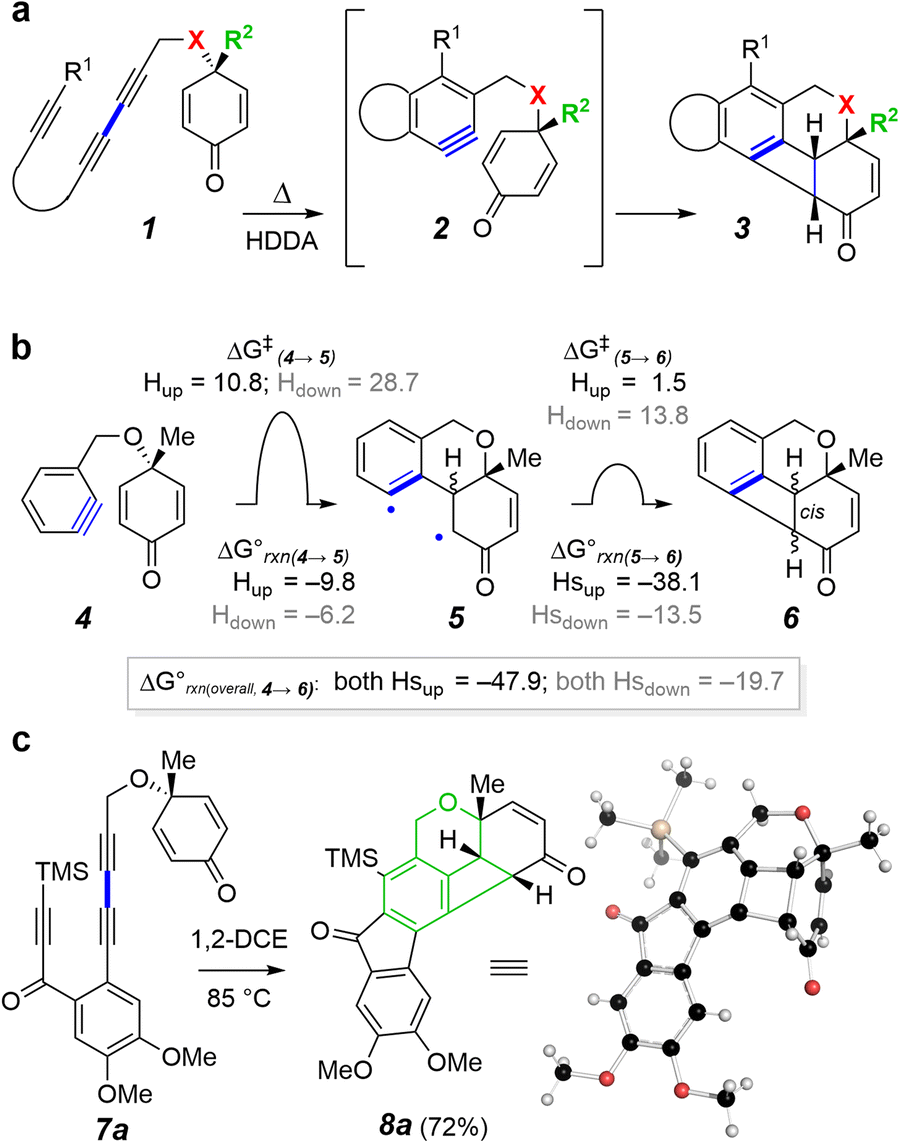 | ||
| Fig. 1 (a) Concept of trapping a HDDA benzyne by the electron-poor alkene in a tethered cyclohexadienone. (b) DFTa relative energies of the stationary points converting model benzyne 4 to the benzocyclobutene 6 (via the diradical 5). (c) First example of a benzocyclobutene-forming reaction from a cyclohexadienone. a[(U)B3LYP-GD3BJ/6-311++G(d,p), SMD: dichloroethane]; we have previously used unrestricted B3LYP with the D3BJ dispersion correction in the optimization of other singlet diradical structures in the context of aryne chemistry.6,7 | ||
Encouraged, we synthesized the triyne 7a8 and observed its efficient conversion to the isomeric, hexacyclic benzocyclobutene product 8a when heated at 85 °C in 1,2-dichloroethane (1,2-DCE) (Fig. 1c). The structure of this product was initially deduced from NMR analyses and later corroborated by an X-ray diffraction study. The presence of the tricyclo[5.3.1.0 (ref. 3 and 8)]oxadodecatriene subunit in 8a (green bonds), comprises three contiguously fused (ortho-annulated) rings and includes three adjacent sp2-hybridized benzenoid carbons.
We wondered how common this type of substructural motif might be. Structure searching the literature identified only three types of such skeletons (9a–b, Fig. 2a), all fully carbocyclic (our search allowed for O- and N-heterocyclic variants as well, but none were found).9–16 Moreover, we could locate no examples of a skeleton in which the saturated 6-membered ring in 9 is, instead, a contracted 5-membered cycle. This implies that these sorts of fused polycycles house a relatively rare type of strain. One indication of the severity of the ring strain in 8a was observed by the rapid onset of dark color of the crystalline sample almost immediately after it was melted (218–220 °C). Thermal lability of 8a was also suggested by its partial injector temperature-dependent conversion to a new product (see below, Fig. 5a) during GC analysis.
 | ||
| Fig. 2 (a) The three known classes of compounds containing a [6/6/4]tricyclic substructure. (b) Differences in DFTa free energies of all three sets of minima for the conversion of 11 + ethane (10) to 13via12 + ethane (all energies within each series of homologs have been referenced to the energy of the tethered alkene 11 + ethane). a[MN15/6-311+G(d,p)]. | ||
To probe how much strain might reside within tricyclic skeletons related to the one present in 8a and 9, we performed a set of DFT computations of the energetic minima for ethane (10) and the five sets of model compounds 11–13 (Fig. 2b).17 Here, we will call the skeletons present in 12 as [6/(n + 3)/4] tricycles; we varied n (the number of intervening methylene units) from 0 to 4. The energy of the starting benzyne 11 for each of the five sets of homologs was set to be 0 (all energies in kcal mol−1). A minimum could not be located for the most highly strained [6/3/4] tricycle 12 (n = 0). The calculation instead optimized to a ring-cleaved species. The ΔG° between the starting benzyne and the tricycle 12 for each of the successively larger rings (4- to 7-membered) progressively became more exergonic – an indication of the relative strain in the homologous series of [6/(n + 3)/4] tricycles. To evaluate the strain in each of 12 more directly, we calculated the homodesmotic reaction in which the C(sp3)–C(sp3) bond in ethane was exchanged for the C(sp3)–C(sp3) bond in the four-membered ring in each of 12.18,19 The energetic changes (ΔG° values in blue) for these ethanolyses reflect the amount of strain released by cleavage of the smallest ring in each ortho-annulated tricycle.20
In view of the substantial strain in these [6/(n + 3)/4] tricycles, it occurred to us that it might be possible to “store” the inherent strain energy of a benzyne within such a tricyclic subunit and subsequently utilize it to drive additional interesting transformations. If so, these benzocyclobutenes could be viewed as primed for further dissipation of the potential energy brought to the reaction via the three alkynes in the benzyne precursors. It is relevant that the triyne to benzyne conversion via a hexadehydro-Diels–Alder (HDDA) cyclization is computed to be, typically, 40–50 kcal mol−1 exergonic.21 This overall idea seemed most promising to pursue for the case where n = 3, especially in light of the fact that we already had in hand the knowledge of the existence of the readily handleable [6/6/4] tricyclic derivative 8a.
We present here an array of benzocyclobutene derivatives prepared by intramolecular reaction between an HDDA benzyne and a tethered cyclohexadienone (Fig. 3 and 4). We then show a variety of strain-driven transformations, some quite unexpected, that these strained species were observed to undergo (Fig. 5–8). At the end of the manuscript (Fig. 9), we will return to the case of a potential (and more highly strained) [6/5/4] tricyclic intermediate.
 | ||
| Fig. 3 [6/6/4]-Benzocyclobutenes 8b–h produced from triynone precursors 7b–h. | ||
 | ||
| Fig. 4 Triyne substrates 15a–c containing different triyne tethers also cyclize to [6/6/4]-benzocyclobutenes. | ||
A number of additional types of substrates will participate in this enone cycloaddition chemistry to produce isolable benzocyclobutene derivatives. Those based on the dimethoxyphenyl ketone linker are shown in Fig. 3. In substrates 7b and 7c, the oxygen atom in the oxymethylene tether present in 7a has been replaced by an NTs or a CH2 group, respectively. Substrates 7d–h, in which the quaternary methyl group in 7a is replaced with a variety of different substituents, demonstrate the compatibility of an array of common functional groups with the transformation. The structures of 8b–h were assigned based on their analogous proton NMR characteristics vis-à-vis8a for key portions of the spectra. Additionally, the structure of the estrone-derived product 8h was subjected to DP4+22 probability analysis,17 which showed definitive agreement with the assigned structure. In no instance did we detect the presence of an alternative diastereomeric benzocyclobutene (cf.6-Hsupvs.6-Hsdown) when analyzing the crude product mixtures.
We also examined three additional benzyne substrates that had different linkers between the diyne and dienophile (Fig. 4). Each proceeded smoothly to provide the lactone- and carbazole-containing products 16a, and 16b, respectively.
We next investigated whether some of these strained intermediates would undergo further transformation by thermal processes. The first hint of that possibility came in the form of the decomposition upon melting 8a, as mentioned earlier. This observation prompted us to explore the fate of 8a upon being heated to a temperature higher than that used to create it (i.e., >85 °C). When a solution of 8a was heated at 130 °C in 1,2-dichloroethane (1,2-DCE), it gave rise to an isomeric product, the structure of which was not immediately apparent. More than one candidate structure was under consideration even after fairly detailed analysis of the 1D and 2D NMR data, although it was clear that an isolated alkene was present in the molecule. Therefore, we sought to make a crystalline derivative and turned to the very useful Rychnovsky OsO4·TMEDA method, which involves in situ derivatization of alkenes and crystallization of their osmate esters.23 The newly formed isomer of 8a was treated with 1.0 equivalent of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.1 mixture of OsO4 and TMEDA in dichloromethane resulting in efficient formation of a 1:1 adduct. Vapor diffusion (DCM vs. pentane) gave black crystals characteristic of an osmate ester (see photo in Fig. 5a). An X-ray diffraction analysis was performed, and we were amazed to see that the osmate had been formed by addition to a pair of adjacent carbons on the aromatic ring to give the product 18, leaving the alkene intact! Accordingly, we could then definitively assign the structure of the rearranged [6/6/4]-benzocyclobutenes 8a as 17.
:1.1 mixture of OsO4 and TMEDA in dichloromethane resulting in efficient formation of a 1:1 adduct. Vapor diffusion (DCM vs. pentane) gave black crystals characteristic of an osmate ester (see photo in Fig. 5a). An X-ray diffraction analysis was performed, and we were amazed to see that the osmate had been formed by addition to a pair of adjacent carbons on the aromatic ring to give the product 18, leaving the alkene intact! Accordingly, we could then definitively assign the structure of the rearranged [6/6/4]-benzocyclobutenes 8a as 17.
 | ||
| Fig. 5 (a) First indication of a strain-driven rearrangement of a [6/4/6] benzocyclobutene (8a) and confirmation of its structure by an unusual osmylation of the arene ring in 17 (see page S305 in the ESI† for a 3D rendering of the X-ray diffraction structure). (b) Proposed reaction pathway using the truncated model compound 19 and passing through o-xylylene and epoxide intermediates 20 and 21, respectively. (c) A DFTa potential energy surface (PES) suggesting that the observed pathway is viable. a[MN15/6-311++G(d,p), SMD: dichloroethane]24. | ||
How did 17 arise? The essential aspects of the proposed mechanism are shown using the truncated structures 19–23 (Fig. 5b). We envisioned a pathway in which 19 proceeds to the o-xylylene 20 and epoxide 21 enroute to 23. We again turned to DFT computations to further evaluate this thinking (Fig. 5c). Our goal here was to demonstrate the viability of various stationary states required for this overall PES. Because of the absence of structural features (especially the conjugated polyyne tether) that would be expected to alter the energies of these states, perhaps dramatically, the various ΔG and ΔG‡ specific values should be taken as qualitative. Nonetheless, this did demonstrate the mechanistic viability of (i) a conrotatory electrocyclic ring-opening of 19 to 20, (ii) an unorthodox, aromatization-driven, concerted cyclization in which the carbonyl oxygen atom captures benzylic cation character in the forming epoxide ring in 21 and (iii) an unusual 1,2-vinyl migration with concomitant epoxide ring-opening leading to 23.
With this mechanistic thinking in mind, we explored the thermal chemistry of the benzocyclobutenes derived from 7a (and some of its analogs) in the presence of several types of trapping agents that might engage one of the reactive species on this manifold (Fig. 6a). Initially, a mixture of 7a and N-phenylmaleimide (24a) was heated at 130 °C in 1,2-DCE. Again, to our surprise, the principal product, 25a, was one in which the maleimide had engaged the arene ring as a dienophile in a [4 + 2] cycloaddition reaction. This mode of reaction was then observed in reactions of additional maleimide derivatives with the triyne substrates 7a, 7b and 7c, leading to 25b, 25c and 25d, respectively.
 | ||
| Fig. 6 Unexpected Diels–Alder adduct formation between the benzocyclobutenes derived from 7a–c and 15b and (a) dienophilic maleimides and (b) dimethyl acetylenedicarboxylate (DMAD). | ||
We then used dimethyl acetylenedicarboxylate (DMAD) as a trapping agent with the triyne substrates 7a and 15b (Fig. 6b). Again, each underwent preferential [4 + 2] cycloaddition across the aromatic ring in the intermediate benzocyclobutenes (cf.26). However, this was now followed by a rapid retro-Diels–Alder event under the reaction conditions to rearomatize the benzenoid ring in each of the final products 27a and 27b. All of the reactions in Fig. 6 reflect a degree of distortion of the arene ring that renders it susceptible to [4 + 2] cycloaddition events. Indeed, the crystal structure of 8a shows that the ortho-annulated benzenoid ring has three internal dihedral angles (among the six) within the arene ring distorted to the extent of 13–19° and that the internuclear distance of the para carbons that engage in the DA reactions25 (and give rise to C3a and C6b in 25 and 26) is 2.65 Å while the other two pairs of para carbon internuclear distances are both 2.85 Å.
We have found several additional reactions in which cleavage of the cyclobutene was the dominant strain-relieving driving force (Fig. 7), as first seen in the conversion of 8a to 17 (Fig. 5a). First, when 7b was heated in 1,2-DCE containing 10 equivalents of acetic acid, the rearranged acetate ester 28 was the only isolated product (Fig. 7a). We infer that this arises by an acid-catalyzed opening of the epoxide intermediate 21′. Second, when triyne 7b was heated in 1,2-DCE containing DMAD, the 1,3-dipolar cycloaddition product 29a was the only isolated product. This provides clear evidence for the carbonyl ylide 22′ as a species on the reaction pathway. Similar 1,3-dipolar cycloaddition products were obtained starting from the triyne 15a using DMAD or N-methylmaleimide (24c), leading to the formation of 29b or 29c, respectively (Fig. 7b). These experimental outcomes support the key aspects of the model computations described in Fig. 5c: namely, the intermediacy of epoxide analogs of both 21 and 1,3-dipole analogs of 22.
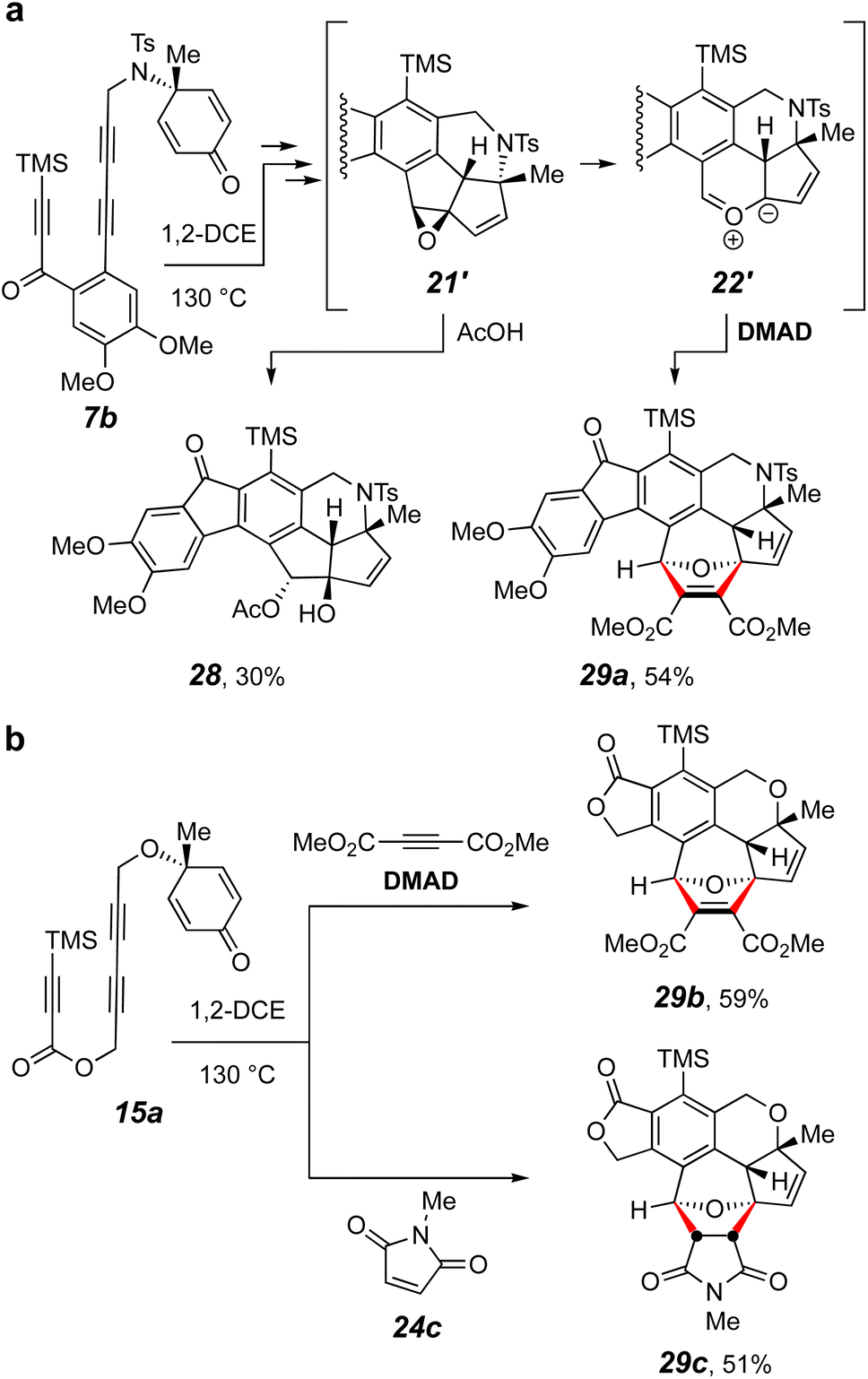 | ||
| Fig. 7 (a) Trapping reactions in which either the intermediate epoxide 21′ or carbonyl ylide 22′ was intercepted, providing experimental support of the computed PES shown in Fig. 5b and c. (b) Analogous trapping reactions of a related 1,3-dipolar intermediate. | ||
We also explored the behavior of substrates 30a and 30b, each containing a spirocyclic ketal moiety within the linker (Fig. 8). If these were to have followed the 2 + 2 reaction pathway, the ortho-annulated intermediate benzocyclobutene would have contained an 8-membered ring (i.e., a [6/8/4] substructure). Instead, and perhaps not surprisingly, the reactions took a different, although still interesting, course. Namely, 30a gave rise to 34a and 35, two unusual polycycloisomerization products. The structure of each was assigned by extensive 1D and 2D NMR spectroscopic studies, and that of the cyclopropane-containing compound 35 was further verified by an X-ray diffraction study. Likewise, the ester-linked triyne 30b gave the phenol derivative 34b, an analog of 34a, as the sole isolated product (Fig. 8b).
 | ||
| Fig. 8 Novel polycycloisomerization products arising from substrates containing a ketal within the cyclohexadienone-to-triyne linker. | ||
The formation of these polycyclic products can be rationalized by attack of the distal ketal oxygen to the electrophilic benzyne carbon atom in, e.g., 31. Cyclization of the 1,3-zwitterion 32 by conjugate addition to an enone would produce the new zwitterion 33, which simultaneously houses both an enolate anion and oxonium cation. This could bifurcate via eliminative opening shown with red arrows to give, ultimately, the phenol derivative 34a or through the cyclopropane-forming event indicated with the blue arrows. Since there are a number of diastereomeric possibilities for structure 33, it is certainly possible that certain one(s) are preferentially leading to 34avs.35. A control experiment demonstrated that isolated 35 is very stable when heated independently at 85 °C for 20 h in CDCl3, ruling it out as a precursor to 34a. Finally, it is also notable that in these reactions the alkynes in 30a fueled not only the formation of the benzyne but, in turn, the production of a compound containing the most classic of all strained cyclic hydrocarbons – a cyclopropane.
We were, of course, interested in learning what would happen using a substrate with a shorter, single-atom tether between the triyne and cyclohexadienone. Would that be capable of giving rise to a yet more highly strained [6/5/4] ortho-annulated benzocyclobutene skeleton? Thus, we prepared the substrate 36 and heated it to induce benzyne-formation (Fig. 9a). A very clean conversion to the unexpected pentacyclic product 39, which contains a newly formed benzochromene subunit was observed. It was difficult to envision a mechanistic pathway that involved initial addition of the proximal benzyne carbon to Cβ of the enone to account for this outcome. We then considered the alternative addition of the proximal benzyne carbon to Cα of the enone to form 37 (in truncated form) this mechanism would proceed from 40, via41 and 42, to 43 (Fig. 9b). The final step involves a pseudo-6π-electrocyclic ring closure of 42 to 43 (cf., 38 to 39) in which there is a concomitant hydrogen atom migration. Notable features of this overall mechanism are (i) the β-cleavage within diradical 37 (or 41), which gives rise to the α,3-dehydrotoluene species 38 (or 42) and (ii) the unusual, concerted migration of a hydrogen atom that accompanies the electrocyclization of 38 to 39 (or 42 to 43), which is further seen in TS7. These unorthodox events are indicated to be energetically feasible by the DFT PES shown in Fig. 9c (cf. EactsviaTS5–TS7).
 | ||
| Fig. 9 (a) An unexpected reaction outcome starting from triyne 36 having a one-atom tether. (b) A mechanistic rationale involving initial C–C bond formation to Cα of the enone (40 to 41), β-cleavage to an α,3-dehydrotoluene derivative (41 to 42), and an unusual electrocyclization with concomitant hydrogen atom migration (42 to 43). (c) DFTa results showing the energetic feasibility of this pathway. a[(U)B3LYP-GD3BJ/6-311++G(d,p), SMD: dichloroethane]. | ||
Conclusions
Because alkynes are of relatively high potential energy, their transformation into products often occurs with a high degree of exothermicity. In this work we have shown reaction sequences that not only use three alkynes to fuel the formation and trapping of benzyne intermediates21 to produce ortho-annulated benzocyclobutenes, but that these polycycles temporarily house strain that can then be released to give rise to a variety of additional unorthodox transformations. These include osmylation of an arene π-bond, Diels–Alder additions to a distorted arene, rearrangements to epoxide and 1,3-dipolar intermediates, and uncatalyzed 1,2-alkene migration of an epoxide.The work highlights the importance of an underappreciated type of strain: namely, one arising from the presence of two rings fused, adjacently, to C1–C2 and C2–C3 of a parent benzenoid ring. We refer to that here as ortho-annulation strain. The amount of strain energy embedded within this type of substructure is demonstrated by DFT computations of homodesmotic reactions between ethane and a series of tricyclic hydrocarbons (12, Fig. 2b).
Finally, the studies also demonstrate that the electron-deficient alkene of an enone can undergo initial bond formation at either its β- or α-carbons, depending on the length of the tether between the benzyne and enone. The former proceeds to net 2 + 2 cycloadducts, a rare process for electron-poor alkenes with arynes, whereas the latter undergoes an unusual process involving β-scission of a diradical leading to yet another strained reactive intermediate, an α,3-dehydrotoluene derivative. Energy release from thermal reactions of alkynes can be captured and then progressively liberated to drive atypical sequences of reactions.
Experimental
General procedure for HDDA cyclization reactions
The triyne substrate (1 equiv.) was dissolved in 1,2-dichloroethane and placed in a screw-capped culture tube. The capped tube was heated at 85 °C for 14 hours. The solvent was removed under reduced pressure, and the residue was purified by MPLC (hexanes:EtOAc, 4:1) to provide the benzocyclobutene product.
Data availability
The data upon which the conclusions in this manuscript are based are provided in the ESI† document and in a .zip file of a master Mestrenova file of all NMR spectra.Author contributions
B. S. C. performed the majority of the experimental work. D. S. S. performed some of the experimental work and all of the computational studies; all authors interpreted the data and D. S. S. and T. R. H. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by a research grant from the National Institutes of General Medical Sciences (R35 GM127097), part of the U.S. Department of Health and Human Services. D. S. S. was a Wayland E. Noland Excellence Fellowship holder. Some of the NMR spectral data were collected using an instrument partially funded by the Shared Instrumentation Grant program (S10OD011952) of the National Institutes of Health. ESI HRMS data were collected at the Masonic Cancer Center (Analytical Biochemistry Shared Resource laboratory) at the University of Minnesota. The instrumentation there was partially funded by the National Cancer Institute (Cancer Center Support Grant CA-77598). The X-ray diffraction data were obtained using an instrument purchased with the support of the National Science Foundation (NSF/MRI 1229400). The DFT computational studies were carried out using the resources of the University of Minnesota Supercomputing Institute (MSI). Victor G. Young Jr and Alex Lovstedt (Department of Chemistry, University of Minnesota) performed the X-ray diffraction analyses.Notes and references
- See various subsections in a number of chapters in Modern Aryne Chemistry, ed. A. Biju, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2021 Search PubMed.
- T. Hamura, in Comprehensive Aryne Synthetic Chemistry, ed. H. Yoshida, Elsevier, Amsterdam, 2022, ch. 3, pp. 267–330 Search PubMed.
- N. G. Rondan, L. N. Domelsmith and K. N. Houk, The relative rates of electron-rich and electron-deficient alkene cycloadditions to benzyne. Enhanced electrophilicity as a consequence of alkyne bending distortions, Tetrahedron Lett., 1979, 35, 3237–3240 CrossRef.
- K. B. Wiberg, in Reactive Intermediate Chemistry, ed. R. A. Moss, M. S. Platz and M. Jones Jr, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2004, ch. 15, pp. 717–740 Search PubMed.
- Strained Hydrocarbons: Beyond the Van't Hoff and Le Bel Hypothesis, ed. H. Dodziuk, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed.
- D. J. Marell, L. R. Furan, B. P. Woods, X. Lei, A. J. Bendelsmith, C. J. Cramer, T. R. Hoye and K. T. Kuwata, Mechanism of the intramolecular hexadehydro-Diels−Alder reaction, J. Org. Chem., 2015, 80, 11744–11754 CrossRef CAS PubMed.
- Q. Xu and T. R. Hoye, Electronic character of α,3-dehydrotoluene intermediates generated from isolable allenyne-containing substrates, Angew. Chem., Int. Ed., 2022, 61, e202207510 CrossRef CAS PubMed.
- Triyne 7a was prepared by a sequence in which (i) p-cresol was trapped by propargyl alcohol in the presence of phenyliodine diacetate (PIDA), (ii) alkyne bromination, (iii) Cadiot–Chodkiewicz cross-coupling with a diyne, and (iv) final oxidation (MnO2) to the ynone. Similar strategies were used to construct all of the polyyne HDDA substrates used in this study. See the ESI for details†.
- F. E. Friedli and H. Shechter, Aromatic substitution and addition reactions of 1H-cyclobuta[de]naphthalenes, Tetrahedron Lett., 1985, 26, 1157–1158 CrossRef CAS.
- J. K. Kendall, T. A. Engler and H. Shechter, Preparative methodology and pyrolytic behavior of anthrylmonocarbenes: synthesis and chemistry of 1H-cyclobuta[de]anthracene, J. Org. Chem., 1999, 64, 4255–4266 CrossRef CAS.
- B. Salem, P. Klotz and J. Suffert, Cyclocarbopalladation: formation of bicyclic 1,2-cyclobutanediols through a rare 4-exo-dig cyclization, Org. Lett., 2003, 5, 845–848 CrossRef CAS PubMed.
- C. Bour, G. Blond, B. Salem and J. Suffert, 4-exo-dig and 5-exo-dig Cyclocarbopalladations: an expeditious solution toward molecular complexity?, Tetrahedron, 2006, 62, 10567–10581 CrossRef CAS.
- G. Blond, C. Bour, B. Salem and J. Suffert, A new Pd-catalyzed cascade reaction for the synthesis of strained aromatic polycycles, Org. Lett., 2008, 10, 1075–1078 CrossRef CAS PubMed.
- J. Petrignet, A. Boudhar, G. Blond and J. Suffert, Step-economical synthesis of taxol-like tricycles through a palladium-catalyzed domino reaction, Angew. Chem., Int. Ed., 2011, 50, 3285–3289 CrossRef CAS PubMed.
- E. Fillion, R. J. Carson, V. E. Trépanier, J. M. Goll and A. A. Remorova, palladium-catalyzed carbon-carbon bond-forming 1,2-ligand migration of organoalanes, J. Am. Chem. Soc., 2004, 126, 15354–15355 CrossRef CAS PubMed.
- G. Blond and J. Suffert, in Strategies and Tactics in Organic Synthesis 4-exo-dig-Cyclocarbopalladation, ed. M. Harmata, Elsevier, Amsterdam, 2021, ch. 10, vol. 15, pp. 366–416 Search PubMed.
- See the ESI for details†.
- S. E. Wheeler, K. N. Houk, P. v. R. Schleyer and W. D. Allen, A hierarchy of homodesmotic reactions for thermochemistry, J. Am. Chem. Soc., 2009, 131, 2547–2560 CrossRef CAS PubMed.
- D. B. Magers, A. K. Magers and D. H. Magers, The s-homodesmotic method for the computation of conventional strain energies of bicyclic systems and individual rings within these systems, Int. J. Quantum Chem., 2019, 119, 1–14 CrossRef.
- We also computed the strain energies of a series of homologous tricyclic hydrocarbons, these having a three-membered cyclopropene ring replacing the cyclobutene in 12. The strain release measured by the ethanolyses of these benzocyclopropene-containing polycycles showed parallel trends to those described in Fig. 2b for the cyclobutene analogs [see Fig. S4† and discussion in Section IVc of the ESI (homodesmotic reaction energies for the [6/n/3] series of homologs)].
- L. L. Fluegel and T. R. Hoye, Hexadehydro-Diels–Alder reaction: Benzyne generation via cycloisomerization of tethered triynes, Chem. Rev., 2021, 121, 2413–2444 CrossRef CAS PubMed.
- M. O. Marcarino, S. Cicetti, M. M. Zandari and A. M. Sarotti, A critical review on the use of DP4+ in the structural elucidation of natural products: the good, the bad and the ugly. A practical guide, Nat. Prod. Rep., 2022, 58–76 RSC.
- A. S. Burns, C. Dooley III, P. R. Carlson, J. W. Ziller and S. D. Rychnovsky, Relative and absolute structure assignments of alkenes using crystalline osmate derivatives for x-ray analysis, Org. Lett., 2019, 21, 10125–10129 CrossRef CAS PubMed.
- We used the MN15 functional because of inferential information that suggests it is effective in locating converged transition structures. N. Mardirossian and M. Head-Gordon, Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- For examples in which curvature of arenes accelerates cycloadditions to the distorted polycyclic arenes see (a) S. Osuna and K. N. Houk, Cycloaddition reactions of butadiene and 1,3-dipoles to curved arenes, fullerenes, and nanotubes: theoretical evaluation of the role of distortion energies on activation barriers, Chem.–Eur. J., 2009, 15, 13219–13231 CrossRef CAS PubMed; (b) Y. García-Rodeja, M. Solà, F. M. Bickelhaupt and I. Fernández, Reactivity and selectivity of bowl-shaped polycyclic aromatic hydrocarbons: relationship to C60, Chem.–Eur. J., 2016, 22, 1368–1378 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2302618–2302621. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00571f |
| This journal is © The Royal Society of Chemistry 2024 |