
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient red thermally activated delayed fluorescence emitters achieved through precise control of excited state energy levels†
BoHua
Zhang
a,
SiQi
Liu
a,
JiangXue
Pei
a,
MeiTing
Luo
a,
Yi
Chen
a,
QingYu
Jia
a,
ZhaoXin
Wu
 b and
DongDong
Wang
*a
b and
DongDong
Wang
*a
aSchool of Chemistry, Xi'an Jiaotong University, Xi'an 710049, P. R. China. E-mail: ddwang@mail.xjtu.edu.cn
bKey Laboratory for Physical Electronics and Devices of the Ministry of Education, Shaanxi Key Lab of Information Photonic Technique, School of Electronic and Information Engineering, Xi'an Jiaotong University, Xi'an 710049, P. R. China
First published on 12th March 2024
Abstract
The variety of highly efficient red/near-infrared (NIR) materials with thermally activated delayed fluorescence (TADF) feature is extremely limited so far, and it is necessary to expand the candidate pool of excellent red/deep-red emitters. However, how to control the energy level alignment of the 1CT (singlet charge transfer) state and the 3LE (triplet local excitation) state to improve the emission efficiency of materials remains a challenge. Herein, based on our previously reported green fluorescent material 67dTPA-FQ, three new donor–acceptor type TADF materials (TQ-oMeOTPA, TsQ-oMeOTPA and SQ-oMeOTPA) were designed by introducing 4,4′-dimethoxy triphenylamine (MeOTPA) as the donor, and introduced S atoms on the acceptors to enhance the spin–orbit coupling (SOC) and CT effects. The theoretical calculations showed that the newly introduced MeOTPA and S atom successfully enhanced the CT effect of the materials, not only shifting the luminescence peak to the deep red region but also effectively adjusting the energy level alignment of the excited state, accelerating the reverse intersystem crossing process. Finally, the organic light-emitting diodes based on SQ-oMeOTPA exhibit an external quantum efficiency of 19.1%, with an emission peak at 619 nm. This work not only expands the candidate inventory of red TADF materials, but also proves the feasibility of designing emitters by adjusting the excited state energy levels, greatly broadening the diversity of TADF emitters in design, and providing a powerful means for rapidly screening efficient emitters in the future.
1. Introduction
Organic light-emitting diodes (OLEDs) have become one of the most cutting-edge display technologies due to their advantages of low power consumption, fast response and flexibility.1–3 As the extremely important component of OLEDs, organic emitters are crucial for achieving excellent electroluminescence (EL) efficiency and long-term operational stability.4–6 Among multiple types of organic emitters, thermally activated delayed fluorescence (TADF) materials can make full use of singlet and triplet excitons and are expected to replace expensive phosphorescence materials containing precious metals and become the main materials for widely commercialized OLEDs in the future.7–9 For achieving efficient TADF emission, a sufficiently small energy splitting (ΔEST) is required between the lowest excited singlet (S1) and triplet (T1) states. At present, a mainstream TADF materials design principle is to construct a highly twisted molecular structure between the electron-donating and electron-accepting moieties, which can lead to significant charge transfer (CT) characteristics to achieve small ΔEST.10–12 Following this materials design principle, many OLEDs based on sky-blue to yellow TADF materials have shown extremely amazing performance, with high photoluminescence (PL) quantum yields (ΦPLs) of nearly 100% and external quantum efficiencies (EQE) exceeding 30%.13–15 However, for red to NIR region emitters, suitable strong donor–acceptor (D–A) pairs are additionally required to reduce the bandgap to the target range. According to the energy-gap laws, the especially narrow energy gaps between T1 and S0 states of red TADF materials will lead to severe non-radiative decay in the triplet states.16–18 Therefore, the variety of such excellent candidates is extremely limited so far, and it is necessary and significant to expand the candidate pool of excellent red emitters.Speeding up the reverse intersystem crossing (RISC) process of emitters through rational molecular design and ultimately gaining an advantage in the competition with non-radiative transitions are the key to solving the above problems.19–21 As we know, a small ΔEST is very important to achieve fast RISC processes.22,23 Generally, the ΔEST values are small enough for most D–A type TADF materials in order to realize an effective forbidden transition course. It is worth noting that Fermi's golden rules tell us that not only ΔEST, but enhanced spin–orbit coupling (SOC) interaction is also the key to achieving high-performance TADF materials.24,25 In this work, we have employed two approaches to enhance SOC interaction of materials. On the one hand, because TADF materials are completely organic, their SOC is typically small, commonly on the order of ≤1 cm−1.26–28 However, the SOC of materials can still be enhanced by replacing some atoms within the molecule with atoms having higher atomic numbers, such as replacing H atoms with F atoms and O atoms with S atoms.29 On the other hand, according to the conservation of angular momentum, for the 1CT and 3CT states where the orbital composition remains unchanged before and after the transition, the change in orbital angular momentum caused by the spin flip of the outermost electrons cannot be compensated, resulting in a relatively weak SOC interaction between these states.30–32 Due to the significant difference in orbital composition between the 1CT and 3LE states, the SOC interaction between them will naturally be stronger. If the energy level of 3LE can be precisely adjusted to effectively participate in the RISC process, its natural strong SOC advantage can be fully utilized, which can improve the efficiency of TADF.33–35 However, how to control the energy level alignment of the 1CT state and the 3LE state to accelerate the RISC process and improve the emission efficiency of TADF materials remains a major challenge.
In line with the previously mentioned design principles, as shown in Fig. 1, based on our previously reported green fluorescent material 67dTPA-FQ,36 we have designed a new material, named TQ-oMeOTPA. We chose 4,4′-dimethoxy triphenylamine (MeOTPA) with strong electron donor ability as the donor to adjust the singlet energy level of the material to achieve TADF.37 Simultaneously, upon substituting furan with thiophene on the acceptor skeleton, the newly introduced S atom is expected to enhance the SOC and CT effects of the material, resulting in more efficient and redder TADF emission. Furthermore, through in-depth theoretical calculations, we found that if the S atom on the acceptor skeleton is oxidized to sulfoxide (named TsQ-oMeOTPA) and sulfone (named SQ-oMeOTPA), the energy level alignment between the 1CT state and the 3LE state of TsQ-oMeOTPA and SQ-oMeOTPA can be precisely achieved, theoretically enabling a red-shift and more efficient TADF emission. Subsequently, we successfully synthesized these three materials, and the results of photophysical properties and device characterization proved that our design idea was correct. From TQ-oMeOTPA to TsQ-oMeOTPA to SQ-oMeOTPA, due to the enhancement of the CT effect of the acceptor, the PL peak in toluene gradually changes from yellow (580 nm) to red (621 nm) and eventually to deep-red (652 nm). More importantly, the introduction of the S atom effectively promotes the SOC interaction, and through reasonable 1CT, 3LE, and 3CT energy level regulation, the 3LE state of SQ-oMeOTPA can perfectly participate in the RISC process, ultimately enhancing the TADF performance. Compared to our previously reported material 67dTPA-FQ, these three new materials not only achieved TADF emission but also exhibited above-average EL performance. Specifically, the device based on SQ-oMeOTPA achieved pure red-light emission, with an EL peak at 619 nm and an EQE of 19.1%.
 | ||
| Fig. 1 Previously reported fluorescent emitter 67dTPA-FQ, as well as the design strategy and chemical structure of the three emitters in this work. | ||
2. Results and discussion
2.1 Computational simulation
For TADF materials, the objective of quantum chemical calculations is to achieve accurate singlet and triplet states energy level evaluations, and further determine their excitonic dynamics.38,39 In order to reliably describe the characteristics and energies of excited states, it is crucial to correctly select quantum chemical (QC) methods such as density functional theory (DFT)40 or wavefunction-based electron correlation methods.41,42 For time-dependent DFT (TD-DFT)43 calculations, non-hybrid functionals neglect the long-range Coulomb interaction between the separated hole and electron, which greatly underestimates the transition energy of CT states.44 For the pure Hartree–Fock (HF) functional, due to the high proportion of exchange terms, it encounters serious electronic correction problems and significantly overestimates the excitation energy of the system. In order to find the most suitable optimal HF% (OHF%) for this work, we employed a semi-empirical relationship formula, OHF% = 42q, based on the CT amount of the material to find the optimal functional.45,46 Detailed calculation procedures for this formula are provided in the ESI.†Initially, the ground state geometries of the three emitters were optimized at the B3LYP/6-31G* level.47,48 Based on the optimized S0 geometry, Multiwfn49 was employed to analyze the orbital compositions in order to obtain the CT amount of the emitters. Next, it was determined that the OHF% was close to 33% (shown in Table S1†). Finally, it was determined that the PBE0-1/3 functional (HF% = 33%) was the most suitable, and the S0 and S1 geometries of all emitters were re-optimized at the PBE0-1/3/6-31G* level.50 The optimized S0 and S1 geometries of all emitters are presented in Fig. 2, with concurrent listing of the root mean square displacements (RMSD).42 In addition, based on the optimized S0/S1 geometries, the absorption and emission data of the emitters in toluene solution obtained by the TD-DFT/PBE0-1/3 method are also provided in Table 1. As depicted in Fig. 2, all emitters manifest highly distorted structures, characterized by a twist angle between the donor and acceptor exceeding 50°. This distortion undoubtedly results in significant spatial separation of the HOMOs and LUMOs of the molecule, leading to a typical CT state. Moreover, to intuitively comprehend the geometric changes of the emitters during excitation, we also calculated the RMSD values between the S0 and S1 states. The calculated results were 0.477, 0.438, and 0.403, respectively. These RMSD values for the three emitters are essentially at the same level, indicating that the introduction of sulfone or sulfoxide groups on the acceptor skeleton did not significantly impact the geometric distortion of the emitters. However, as shown in Table 1, owing to the varying electron-withdrawing abilities of the acceptor skeletons, the three emitters exhibit noteworthy differences in absorption and emission. The progressively increasing CT effect induces red-shifts in the absorption and emission peaks of the three emitters. The luminescence color shifts from the yellow of TQ-oMeOTPA (548 nm) to the orange-red of TsQ-oMeOTPA (598 nm) and finally to the deep-red of SQ-oMeOTPA (653 nm). Concurrently, due to the gradual increase in the CT effect, the emission oscillator strengths (f) of the three emitters sequentially decrease. Nevertheless, their f values remain within the range where efficient luminescence is possible. Overall, by fine-tuning the electron-withdrawing ability of the acceptor skeleton while maintaining the rigidity of the skeleton, the designed SQ-oMeOTPA achieves deep-red emission while maintaining a certain oscillator strength, aligning with our expectations.
 | ||
| Fig. 2 Optimized ground state (S0) and excited state (S1) geometries from PBE0-1/3/6-31G* in toluene using the PCM model for the three emitters. The bottom geometries are intuitive pictures comparing the S0 (blue) and S1 (red) geometries with minimum root-mean-square deviation (RMSD). | ||
| Compound | Electronic transition | λ (nm) | f | Excitation energies (eV) | Configuration |
|---|---|---|---|---|---|
| TQ-oMeOTPA | S0 → S1 | 471 | 0.4592 | 2.63 | H → L (99%) |
| S1 → S0 | 548 | 0.4653 | 2.26 | H → L (99%) | |
| TsQ-oMeOTPA | S0 → S1 | 492 | 0.4010 | 2.52 | H → L (98%) |
| S1 → S0 | 598 | 0.3653 | 2.07 | H → L (99%) | |
| SQ-oMeOTPA | S0 → S1 | 516 | 0.3699 | 2.40 | H → L (99%) |
| S1 → S0 | 653 | 0.2941 | 1.90 | H → L (98%) |
As shown in Fig. 3a, the HOMO and LUMO of all emitters are mainly distributed on the donor and acceptor fragments, respectively. All emitters exhibit clear CT characteristics, which is consistent with our previous analysis in the geometric structure section. It's noteworthy that, owing to the identical donor segment MeO-TPA in the three emitters, their HOMO energy levels are almost the same. In comparison to TQ-oMeOTPA, both TsQ-oMeOTPA and SQ-oMeOTPA introduce sulfoxide and sulfone groups into the acceptor segment, respectively, enhancing their electron-withdrawing ability. Therefore, as shown in Fig. 3a, a portion of the LUMO electron cloud of TsQ-oMeOTPA and SQ-oMeOTPA is attracted by the sulfoxide and sulfone groups, resulting in a decrease in the LUMO energy level. Ultimately, TsQ-oMeOTPA and SQ-oMeOTPA obtain a smaller HOMO–LUMO energy gap compared to TQ-oMeOTPA, which well explains the reason for the red-shift of the emitter absorption and emission peaks.
 | ||
| Fig. 3 Calculated frontier orbital distributions and energy levels of the emitters (a). Frontier orbital distributions of TQ-oMeOTPA, TsQ-oMeOTPA and SQ-oMeOTPA (b). Calculated zero–zero energy levels based on excited geometries. | ||
Furthermore, in order to obtain the energy level arrangement of the emitters, the calculation of the zero–zero energy level (E0–0) is crucial. Following the Franck–Condon principle, the E0–0 (S1) energy level can be calculated from the average of EVA (S1) and EVE (S1) as eqn (1),51 while the E0–0 (3CT) and E0–0 (3LE) energy levels are calculated independently according to eqn (2) and (3).46 Based on the calculated E0–0 (3CT) and E0–0 (3LE) energy levels, the lower energy one is taken as the T1 state, and the value of ΔEST can be calculated based on the previously calculated S1 energy. This modified TD-DFT method has been proven to have good accuracy in the molecular design and prediction of TADF.52,53
 | (1) |
 | (2) |
| E0–0 (3LE) = EVA (T1)/EVA (S1,OHF)/EVA (S1,BLYP) | (3) |
The calculated energy level arrangement is illustrated in Fig. 3b, and the detailed calculation data are listed in Table S1.† The results show that all the E0–0 (3CT) energy levels of the emitters are below the E0–0 (3LE) energy level, indicating that their T1 states will be dominated by the 3CT state rather than the 3LE state. The calculated ΔEST of the emitters is 0.28 eV, 0.17 eV, and 0.18 eV, respectively. Such a small ΔEST makes the emitters possible to achieve TADF emission. Additionally, the E0–0 (3LE) energy is also a crucial consideration. The E0–0 (3LE) energies of the three emitters are 2.18 eV, 2.21 eV, and 2.22 eV, respectively. The 3LE state energies of the three materials are almost the same, which is due to their almost identical conjugation length and geometric conformation, and such energy is also comparable to the reported 3LE state energy of a similar furo[2,3-b]quinoxaline fragment.36,54 For TQ-oMeOTPA, the energy gap between the 3LE state and the 1CT state is 0.27 eV, whereas, for TsQ-oMeOTPA and SQ-oMeOTPA, the energy gap decreases to 0.09 eV and −0.07 eV, respectively. Clearly, in TsQ-oMeOTPA and SQ-oMeOTPA, the 3LE state, which has energy nearly equivalent to that of the 1CT state, will significantly participate in the RISC process. Especially for SQ-oMeOTPA, due to its slightly higher 3LE state energy than 1CT state, it can undergo a naturally faster energy down-conversion RISC (3LE → 1CT) process. More importantly, we also calculated the SOC between different excited states of the emitters (Fig. 3b). The results reveal that the SOC value between the 1CT and 3LE states is approximately twice that between the 1CT and 3CT states. This is because the orbital composition between the 1CT and 3LE states is different, (the hole–electron distribution of the excited state is shown in Fig. S1–S3†) and their SOC interaction will naturally be stronger. As a result, the RISC process involving the 3LE state will be significantly accelerated. In summary, the theoretical calculation results confirm that our molecular design concept is successful, and all three emitters have the potential to become efficient TADF materials, especially SQ-oMeOTPA, which according to theoretical predictions can exhibit deep-red emission and potentially have the fastest RISC rate among the three emitters, making it an excellent candidate for red emission TADF materials.
2.2 Synthesis, thermal and electrochemical properties
The synthetic routes of the three molecules are shown in Scheme S1.† Among them, TQ-oMeOTPA was synthesized by the Suzuki coupling reaction with the precursors, 2-phenyl-6,7-dibromothienoquinoxaline (6,7-dBr-TQ) prepared according to our previous published procedure,36 and the boronic acid intermediate, 4-methoxy-N-(4-methoxyphenyl)-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline, respectively. After obtaining TQ-oMeOTPA, it can be further oxidized to obtain TsQ-oMeOTPA and SQ-oMeOTPA. The three molecules were fully characterized and verified using 1H, 13C NMR spectra and high-resolution mass spectra. In addition, TsQ-oMeOTPA and SQ-oMeOTPA were also characterized by infrared spectroscopy.The cyclic voltammetry (CV) measurements were carried out to estimate the HOMO and the LUMO levels of these three compounds (Fig. S4†). As shown in CV curves, these three compounds show irreversible redox waves with forwarding scanning in CH2Cl2 solution, ascribing to the redox of the donors. From the half-wave onset potential of CV curves, the electrochemical HOMO energy levels are determined to be −5.24 eV (TQ-oMeOTPA), −5.25 eV (TsQ-oMeOTPA) and −5.25 eV (SQ-oMeOTPA), respectively. Correspondingly, their LUMO energy levels are calculated to be −2.85 eV, −2.99 eV, and −3.06 eV respectively, by combining the electrochemical HOMO and optical gap from violet-visible absorption spectra in toluene solution.
The thermal properties of the three compounds were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen atmosphere, and their curves are presented in Fig. S5.† The 5% weight loss temperatures (Td) for TQ-oMeOTPA, TsQ-oMeOTPA and SQ-oMeOTPA are 423 °C, 433 °C and 441 °C, respectively. The high Td indicate that all three compounds possess excellent thermal and morphological stability and are suitable for fabricating devices by the vacuum deposition method.
2.3 Photophysical properties
Although we have demonstrated the electronic cloud distribution of the three emitters through theoretical simulations and demonstrated the potential excellent TADF properties of the emitters through the regulation of excited state energy levels, more detailed photophysical experiments are still indispensable. Conducting comprehensive experiments to obtain additional information on the excited state of the material and comparing it with our previous quantum chemical calculations will provide a thorough understanding of the excitation state dynamics of the materials.Ultraviolet-visible (UV-vis) absorption and photoluminescence (PL) spectra of these three materials in dilute toluene solutions (concentration of 10−5 M) were measured to investigate their fundamental photophysical properties, as shown in Table 3 and Fig. 4. The higher energy absorption bands in the range of 290–350 nm are associated with the π–π* transition of the acceptor and donor fragments, while the absorption peaks at 375–410 nm belong to the n–π* transition absorption of the acceptor. And then, the absorption peaks at 459 nm (TQ-oMeOTPA), 481 nm (TsQ-oMeOTPA), and 488 nm (SQ-oMeOTPA) are associated with the intramolecular charge transfer (ICT) absorption from the donor to the acceptor for the three materials, respectively. The broad and structureless PL spectra of these three materials are gradually red-shifted with PL peaks moving from 580 nm for TQ-oMeOTPA to 621 nm for TsQ-oMeOTPA and 652 nm for SQ-oMeOTPA (Fig. 4b), indicating the enhancement of the CT effect. The S1 state of these materials was identified as a noticeable CT feature, which was further confirmed by the evident solvatochromic effect (Fig. S6a†). The absolute PLQY of these three materials in toluene solution was measured by using an integrating sphere. Among them, the PLQY in toluene solution for TQ-oMeOTPA is 66.7%, slightly higher than 51.1% for TsQ-oMeOTPA and 50.2% for SQ-oMeOTPA, which can be explained by the relative magnitude of the oscillator strengths discussed previously. Moreover, from the fluorescence and phosphorescence spectra at 77 K (Fig. 4c–e), the phosphorescence spectra of all materials exhibit a single wide emission band, implying the CT featured emission, namely, the low-lying 3CT state. Accordingly, the ΔEST of TQ-oMeOTPA, TsQ-oMeOTPA, and SQ-oMeOTPA was calculated to be 0.22 eV, 0.12 eV, and 0.17 eV, respectively. The small ΔEST is beneficial for up-conversion of excitons from the T1 state to the S1 state, suggesting that they are potential TADF candidates. The results of photophysical characterization are highly consistent with our theoretical calculations, which fully validates the reliability of our computational method.
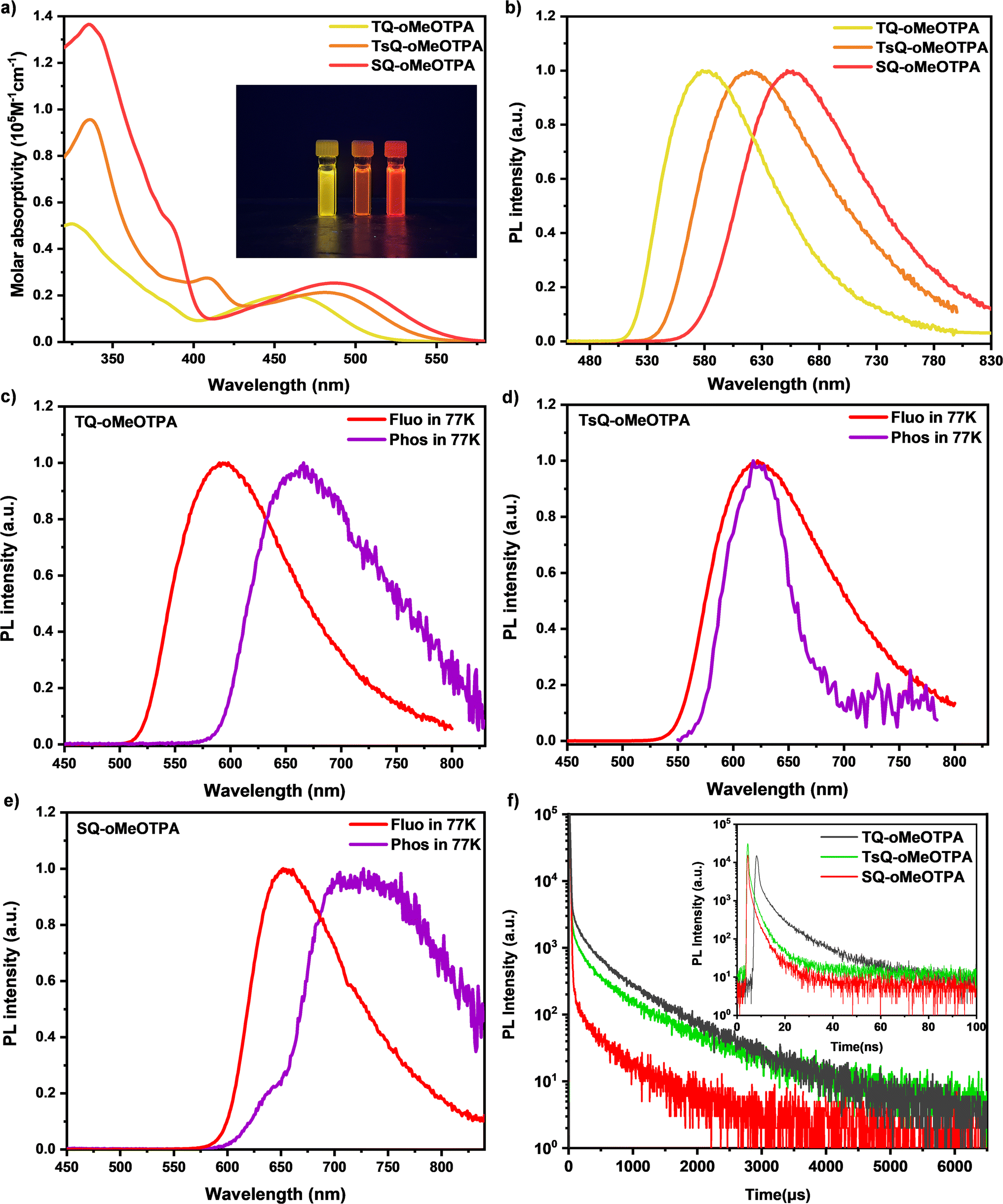 | ||
| Fig. 4 (a) UV-vis absorption of TQ-oMeOTPA, TsQ-oMeOTPA, and SQ-oMeOTPA in dilute toluene at 298 K, and the inset shows the luminescence of the three compounds in toluene. (b) Fluorescence spectra of TQ-oMeOTPA, TsQ-oMeOTPA, and SQ-oMeOTPA in dilute toluene at 298 K. (c) Photoluminescence spectra of TQ-oMeOTPA in dilute toluene. (d) Photoluminescence spectra of TsQ-oMeOTPA in dilute toluene. (e) Photoluminescence spectra of SQ-oMeOTPA in dilute toluene. (f) Transient characteristics of TQ-oMeOTPA, TsQ-oMeOTPA, and SQ-oMeOTPA in doped mCBP films (3 wt%). | ||
Next, in order to simulate the conditions associated with OLED applications, we selected low-polar 3,3′-di(9H-carbazol-9-yl)-1,1′-biphenyl (mCBP) as the host with a low doping concentration of 3 wt% and investigated the photophysical properties of the three materials in doped films. As shown in Fig. S7,† there is no emission peak from mCBP in the PL spectrum of the blended film of the three materials doped in the mCBP host, indicating that energy can be completely transferred from the host to the guest materials at a doping concentration of 3%. The emission peak of the 3% TQ-oMeOTPA: mCBP doped film (584 nm) is close to the emission peak of TQ-oMeOTPA in toluene solution (580 nm). However, the emission peak of the 3% TsQ-oMeOTPA: mCBP (593 nm) and SQ-oMeOTPA: mCBP doped film (615 nm) is blue-shifted nearly 28–37 nm compared with the emission peak of TsQ-oMeOTPA (621 nm) and SQ-oMeOTPA (652 nm) in toluene solution. This blue-shift effect of the TsQ-oMeOTPA and SQ-oMeOTPA doped film indicates that the relaxation process of the materials in the redder direction is inhibited in a rigid matrix. And the film environment may limit the deformation of the excited state towards the direction of elongation in the conjugated plane, resulting in a blueshift of the emission peaks of TsQ-oMeOTPA and SQ-oMeOTPA in mCBP. In addition, the absolute PLQY of these three materials in doped films is measured to be 68.1% (TQ-oMeOTPA), 66.4% (TsQ-oMeOTPA), and 66.8% (SQ-oMeOTPA), respectively, which were higher than that of the three materials in toluene solution. This also confirms the inhibitory effect of the rigidity of the doped film on non-radiative transitions.
The delayed fluorescence lifetimes (τds) are determined to be 484.2 μs for TQ-oMeOTPA, 389.7 μs for TsQ-oMeOTPA and 299.5 μs for SQ-oMeOTPA (Table 2 and Fig. 4f). As expected, the τds of SQ-oMeOTPA is significantly reduced relative to TQ-oMeOTPA and TsQ-oMeOTPA. We believe that the relatively short τds of SQ-oMeOTPA reveals the significant positive impact of the 3LE state on accelerating the RISC process, and provides strong evidence that an ideal energy level sequence can significantly improve TADF performance. To further determine the emission nature, temperature-dependent transient PL decay spectra are implemented with varied temperatures from 100 to 300 K. As shown in Fig. S8,† along with the increase of temperature, the delayed fluorescence intensity gradually increases and the delayed component gets higher, clearly indicating the participation of the triplet state in emission via the RISC process by thermal activation, and thus establishing their unquestionable TADF behaviors.
| Compounds | λ abs (nm) | λ em sola/filmb (nm) | S1c (eV) | T1c (eV) | ΔEstc (eV) | Φ PL sola/filmb (%) | τ p [ns] | τ d [μs] |
|---|---|---|---|---|---|---|---|---|
| a Measured in toluene solution (10−5 M) at R. T. b Measured in the 3 wt% doped mCBP film at R. T. c Estimated from the onsets of fluorescence and phosphorescence spectra of both compounds in 10−5 M toluene solution at 77 K, ΔEST = S1 − T1. | ||||||||
| TQ-oMeOTPA | 459 | 580/584 | 2.36 | 2.14 | 0.22 | 66.7/68.1 | 12.5 | 484.2 |
| TsQ-oMeOTPA | 481 | 621/593 | 2.25 | 2.13 | 0.12 | 51.1/66.4 | 8.2 | 389.7 |
| SQ-oMeOTPA | 488 | 652/615 | 2.08 | 1.91 | 0.17 | 50.2/66.8 | 6.5 | 299.5 |
Based on the above photophysical results and previous theoretical calculations, we can clearly describe the excitonic dynamics of these emitters under optical excitation. Upon photoexcitation, excitons form on these emitters, where a portion of the singlet excitons rapidly undergoes a radiative transition process back to the S0 states to produce fluorescence emission, while the other portion of the singlet excitons is converted into triplet excitons through intersystem crossing (ISC) channels. For SQ-oMeOTPA, the RISC process can occur not only between 3CT and 1CT but also through the VC&SOC (3CT → 3LE → 1CT) process to achieve rapid down-conversion of triplet excitons. Similar processes also occur in TsQ-oMeOTPA, but because its 3LE → 1CT process is an energy up-conversion, it results in a relatively longer τds. For TQ-oMeOTPA, due to the largest energy difference between its 1CT and 3LE levels, its 3LE → 1CT process will be relatively slow, resulting in the longest τds among the three emitters. Through the above experimental evidence, it is not difficult to find that in order to obtain an ideal emitter with short τds, it is necessary to ensure the smallest energy difference between 1CT and 3LE, and to achieve energy down-conversion of 3LE → 1CT as much as possible. In addition, the energy difference between 3LE and 3CT also needs to be maintained within a reasonable range to achieve effective VC.
2.4 Device characterization
After exhaustive investigation of the photophysical properties of the three materials, differences in device performance were further investigated. The EL performances of these materials were estimated using DMFL-CBP (9,9′-(9,9-dimethyl-9H-fluorene-2,7-diyl)bis(9H-carbazole)) as a host with the device structure, ITO/HAT-CN(5 nm)/TAPC(40 nm)/TCTA (5 nm)/DMFL-CBP: emitter (3%) (30 nm)/TmPyPB (40 nm)/LiF (1 nm)/Al (80 nm), in which 1,4,5,8,9,11-hexaazatriphenylenehexacarbonitrile (HAT-CN) and lithium fluoride (LiF) are used as hole and electron-injecting layers, respectively, 1,1-bis[(di-4-tolylamino)phenyl]cyclohexane (TAPC) and 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) acted as the hole/electron transporting layer, 4,4′,4′′-tri(N-carbazolyl)triphenylamine (TCTA) served as the electron blocking layer. Given that the aggregation-caused quenching (ACQ) phenomenon is generally severe in red emission materials because of the anabatic dipole–dipole interaction and π–π stacking induced non-adiabatic internal conversion, the doping concentrations of these three emitters were controlled at a low level (3%). The device architecture is shown in Fig. 5a. And the EL performances of these doped OLEDs are depicted in Fig. 5b, with the key EL parameters summarized in Table 4. | ||
| Fig. 5 (a) Device structure. (b) The EL spectra of the devices; (c) the current density–voltage–luminance (J–V–L) curves of the devices; (d) the EQE versus luminance relationships of the devices. | ||
| Emitting layer | V on (V) | L max (cd m−2) | CEmax (cd A−1) | PEmax (lm W−1) | EQEmax (%) | λ EL (nm) | CIE(x, y) |
|---|---|---|---|---|---|---|---|
| a Abbreviations: Von = turn-on voltage at 1 cd m−2; Lmax = maximum luminance; CEmax = maximum current efficiency; PEmax = maximum power efficiency; EQEmax = maximum values at 0.1 cd m−2 of external quantum efficiency; λEL = EL peak; CIE = Commission Internationale de I'Eclairage coordinates. | |||||||
| TQ-oMeOTPA | 4.8 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 820 820 |
23.2 | 26.4 | 14.5 | 588 | (0.53, 0.47) |
| TsQ-oMeOTPA | 4.8 | 10170 |
33.6 | 35.9 | 15.5 | 596 | (0.55, 0.44) |
| SQ-oMeOTPA | 4.8 | 26810 |
42.9 | 47.5 | 19.1 | 619 | (0.60, 0.40) |
The turn-on voltage of these devices is 4.8 V, and this may be related to the high LUMO energy level of host DMFL-CBP. The peak luminance values of these devices were 10820, 10170, and 26810 cd m−2, respectively. Furthermore, no emission peak from the DMFL-CBP host was observed in the EL spectra of all devices. The devices based on TQ-oMeOTPA, TsQ-oMeOTPA and SQ-oMeOTPA at 3 wt% doping concentration radiate yellow to red lights with EL peaks at 588, 596 and 619 nm, corresponding to Commission Internationale de I'Eclairage (CIE) coordinates of (0.53, 0.47), (0.55, 0.44) and (0.60, 0.40), respectively. The EL spectra of these OLEDs show a small red-shift (∼6 nm) with respect to the PL spectra of the EMLs, which is related to the electronic polarization effect, and optical microcavity effect in the devices. Just as we expected, the SQ-oMeOTPA based devices exhibited positive EL performances with maximum EQE (EQEmax), current efficiency (CEmax) and power efficiency (PEmax) of 19.1%, 42.9 cd A−1 and 47.5 lm W−1, significantly higher than those of TQ-oMeOTPA -based (14.5%, 23.2 cd A−1 and 26.4 lm W−1) and TsQ-oMeOTPA -based (15.5%, 33.6 cd A−1 and 35.9 lm W−1) devices. Unfortunately, like most red to deep-red TADF OLEDs, the efficiency roll-offs of these devices are still large. The sensitizing technique may be an effective alternative to solve this problem for red and deep-red TADF materials. But in comparison with the TQ-oMeOTPA, TsQ-oMeOTPA and SQ-oMeOTPA have much smaller efficiency roll-offs. This can be attributed to their much faster RISC processes, which can reduce the non-radiative loss of triplet excitons and thus alleviate the decrease of the EQE.
3. Conclusion
In summary, herein we reported a series of novel yellow, orange-red, and red emitters. Before synthesis, we used a functional with optimized Hartree–Fock percentage (OHF%) to accurately predict the 1CT, 3CT, and 3LE energy level relationships of the emitters, and proposed a strategy that fully utilizes the 3LE state to improve the utilization of triplet excitons in TADF materials. After that, we successfully synthesized this series of emitters and conducted in-depth evaluations of their photophysical properties and device performance. The experimental results showed that the corresponding devices based on TQ-oMeOTPA, TsQ-oMeOTPA, and SQ-oMeOTPA achieved emission peaks of 588 nm, 596 nm, and 619 nm, with EQEmax of 14.5%, 15.5%, and 19.1%, respectively. The experimental results are highly consistent with theoretical predictions, fully confirming that the SOC effect and the energy level alignment of 1CT, 3LE, and 3CT have a significant impact on the RISC process of long-wavelength organic emitters. In short, this study not only strongly demonstrates the importance and necessity of the energy level alignment of 3LE in achieving the utilization of triplet excitons in TADF materials during photoluminescence and electroluminescence processes, but also proves the feasibility of designing emitters by adjusting the excitation energy level through preliminary theoretical calculations, greatly broadening the diversity of TADF emitters in design, and providing a very powerful means for rapid screening of high-efficiency molecules in the future.Data availability
All data supporting the findings of this study are presented in the article and ESI.† Additional data are available from the corresponding author upon reasonable request.Author contributions
B. H. Zhang carried out most parts of the experiments and DFT calculations and wrote the original manuscript. S. Q. Liu contributed to performing the fabrication and characterization studies of the OLEDs. J. X. Pei, M. T. Luo, Y. Chen, Q. Y. Jia and Z. X. Wu analyzed the data and provided comments. D.D. Wang supervised the project, analyzed the data and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 62075178). We are grateful to the High-Performance Computing Center and Instrumental Analysis Center of Xi'an Jiaotong University for its support.References
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao and J. Zhang, Adv. Sci., 2021, 8, e2102970 CrossRef PubMed.
- M. C. Tang, M. Y. Chan and V. W. Yam, Chem. Rev., 2021, 121, 7249–7279 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J. L. Bredas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- R. Noriega, E. S. Barnard, B. Ursprung, B. L. Cotts, S. B. Penwell, P. J. Schuck and N. S. Ginsberg, J. Am. Chem. Soc., 2016, 138, 13551–13560 CrossRef CAS PubMed.
- S. Y. Yang, Y. K. Wang, C. C. Peng, Z. G. Wu, S. Yuan, Y. J. Yu, H. Li, T. T. Wang, H. C. Li, Y. X. Zheng, Z. Q. Jiang and L. S. Liao, J. Am. Chem. Soc., 2020, 142, 17756–17765 CrossRef CAS PubMed.
- T. Zhang, Y. Xiao, H. Wang, S. Kong, R. Huang, V. Ka-Man Au, T. Yu and W. Huang, Angew. Chem., Int. Ed., 2023, 62, e202301896 CrossRef CAS PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 CrossRef CAS PubMed.
- X. Chen, X. Zhang, X. Xiao, Z. Wang and J. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202216010 CrossRef CAS PubMed.
- X. K. Chen, Y. Tsuchiya, Y. Ishikawa, C. Zhong, C. Adachi and J. L. Bredas, Adv. Mater., 2017, 29, 1702767 CrossRef PubMed.
- R. K. Konidena and J. Y. Lee, Chem. Rec., 2019, 19, 1499–1517 CrossRef CAS.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- G. Meng, H. Dai, Q. Wang, J. Zhou, T. Fan, X. Zeng, X. Wang, Y. Zhang, D. Yang, D. Ma, D. Zhang and L. Duan, Nat. Commun., 2023, 14, 2394 CrossRef CAS.
- X. F. Luo, S. Q. Song, H. X. Ni, H. Ma, D. Yang, D. Ma, Y. X. Zheng and J. L. Zuo, Angew. Chem., Int. Ed., 2022, 61, e202209984 CrossRef CAS PubMed.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- J. H. Kim, J. H. Yun and J. Y. Lee, Adv. Opt. Mater., 2018, 6, 1800255 CrossRef.
- A. Zampetti, A. Minotto and F. Cacialli, Adv. Funct. Mater., 2019, 29, 1807623 CrossRef.
- R. Jiang, N. Qiu, J. Li, X. Wu, N. Zheng, B. Z. Tang and Z. Zhao, Innovations Mater., 2023, 1, 100012 CrossRef.
- W. Xie, M. Li, X. Peng, W. Qiu, Y. Gan, Z. Chen, Y. He, W. Li, K. Liu, L. Wang, Q. Gu and S.-J. Su, Chem. Eng. J., 2021, 425, 131510 CrossRef CAS.
- H. Wang, J. X. Chen, Y. Z. Shi, X. Zhang, L. Zhou, X. Y. Hao, J. Yu, K. Wang and X. H. Zhang, Adv. Mater., 2023, 2307725 Search PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X.-K. Chen, CCS Chem., 2020, 2, 1256–1267 CrossRef CAS.
- X.-C. Fan, K. Wang, Y.-Z. Shi, Y.-C. Cheng, Y.-T. Lee, J. Yu, X.-K. Chen, C. Adachi and X.-H. Zhang, Nat. Photonics, 2023, 17, 280–285 CrossRef CAS.
- A. Pershin, D. Hall, V. Lemaur, J. C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- J. Li, T. Tian, D. Guo, T. Li, M. Zhang and H. Zhang, J. Mater. Chem. C, 2023, 11, 6119–6129 RSC.
- C. Li, A. K. Harrison, Y. Liu, Z. Zhao, C. Zeng, F. B. Dias, Z. Ren, S. Yan and M. R. Bryce, Angew. Chem., Int. Ed., 2022, 61, e202115140 CrossRef CAS.
- J. Xu, X. Wu, J. Li, Z. Zhao and B. Z. Tang, Adv. Opt. Mater., 2022, 10, 2102568 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- Y. H. Lee, Y.-S. Shin, T. Lee, J. Jung, J.-H. Lee and M. H. Lee, Chem. Eng. J., 2021, 423, 130224 CrossRef CAS.
- U. Balijapalli, Y. T. Lee, B. S. B. Karunathilaka, G. Tumen-Ulzii, M. Auffray, Y. Tsuchiya, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2021, 60, 19364–19373 CrossRef CAS.
- J. X. Chen, Y. F. Xiao, K. Wang, D. Sun, X. C. Fan, X. Zhang, M. Zhang, Y. Z. Shi, J. Yu, F. X. Geng, C. S. Lee and X. H. Zhang, Angew. Chem., Int. Ed., 2021, 60, 2478–2484 CrossRef CAS.
- X. Wang, Y. Zhang, Z. Yu, Y. Wu, D. Wang, C. Wu, H. Ma, S. Ning, H. Dong and Z. Wu, J. Mater. Chem. C, 2022, 10, 1681–1689 RSC.
- J. S. Ward, R. S. Nobuyasu, M. A. Fox, J. A. Aguilar, D. Hall, A. S. Batsanov, Z. Ren, F. B. Dias and M. R. Bryce, J. Org. Chem., 2019, 84, 3801–3816 CrossRef CAS PubMed.
- R. Gomez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120–1127 CrossRef CAS PubMed.
- T. Huang, Q. Wang, G. Meng, L. Duan and D. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202200059 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. T. Yang, Science, 2008, 321, 792–794 CrossRef CAS.
- B. H. Zhang, J. A. Li, M. Wang, A. M. Ren, T. F. He, P. P. Lin, Y. L. Zhang, X. Y. Xi and L. Y. Zou, Phys. Chem. Chem. Phys., 2021, 23, 5652–5664 RSC.
- J. Autschbach, T. Ziegler, S. J. A. van Gisbergen and E. J. Baerends, J. Chem. Phys., 2002, 116, 6930–6940 CrossRef CAS.
- T. Korzdorfer and J. L. Bredas, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- Q. Zhang, H. Kuwabara, W. J. Potscavage Jr, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS PubMed.
- S. Huang, Q. Zhang, Y. Shiota, T. Nakagawa, K. Kuwabara, K. Yoshizawa and C. Adachi, J. Chem. Theory Comput., 2013, 9, 3872–3877 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 2006, 27, 209–214 CrossRef.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- C. A. Guido, E. Bremond, C. Adamo and P. Cortona, J. Chem. Phys., 2013, 138, 021104 CrossRef PubMed.
- D. Jacquemin, A. Planchat, C. Adamo and B. Mennucci, J. Chem. Theory Comput., 2012, 8, 2359–2372 CrossRef CAS PubMed.
- X. Cai, D. Chen, K. Gao, L. Gan, Q. Yin, Z. Qiao, Z. Chen, X. Jiang and S. J. Su, Adv. Funct. Mater., 2017, 28, 1704927 CrossRef.
- L. Gan, Z. Xu, Z. Wang, B. Li, W. Li, X. Cai, K. Liu, Q. Liang and S. J. Su, Adv. Funct. Mater., 2019, 29, 1808088 CrossRef.
- X. Wang, Y. Li, Y. Wu, K. Qin, D. Xu, D. Wang, H. Ma, S. Ning and Z. Wu, New J. Chem., 2022, 46, 18854–18864 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00535j |
| This journal is © The Royal Society of Chemistry 2024 |