
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable Pt–NiO interaction-induced efficient electrocatalytic water oxidation and methanol oxidation†
Fenglin
Wang
ab,
Zhicheng
Zheng
b,
Dan
Wu
b,
Hao
Wan
 *a,
Gen
Chen
b,
Ning
Zhang
b,
Xiaohe
Liu
*ab and
Renzhi
Ma
*c
*a,
Gen
Chen
b,
Ning
Zhang
b,
Xiaohe
Liu
*ab and
Renzhi
Ma
*c
aZhongyuan Critical Metals Laboratory, Zhengzhou University, Zhengzhou 450001, P. R. China. E-mail: wanhao@zzu.edu.cn; liuxh@csu.edu.cn
bSchool of Materials Science and Engineering, Key Laboratory of Electronic Packaging and Advanced Functional Materials of Hunan Province, Central South University, Changsha, Hunan 410083, P. R. China
cResearch Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan. E-mail: MA.Renzhi@nims.go.jp
First published on 27th May 2024
Abstract
Metal–support interaction engineering is considered an efficient strategy for optimizing the catalytic activity. Nevertheless, the fine regulation of metal–support interactions as well as understanding the corresponding catalytic mechanisms (particularly those of non-carbon support-based counterparts) remains challenging. Herein, a controllable adsorption–impregnation strategy was proposed for the preparation of a porous nonlayered 2D NiO nanoflake support anchored with different forms of Pt nanoarchitectures, i.e. single atoms, clusters and nanoparticles. Benefiting from the unique porous architecture of NiO nanosheets, abundant active defect sites facilitated the immobilization of Pt single atoms onto the NiO crystal, resulting in NiO lattice distortion and thus changing the valence state of Pt, chemical bonding, and the coordination environment of the metal center. The synergy of the porous NiO support and the unexpected Pt single atom–NiO interactions effectively accelerated mass transfer and reduced the reaction kinetic barriers, contributing to a significantly enhanced mass activity of 5.59 A mgPt−1 at an overpotential of 0.274 V toward the electrocatalytic oxygen evolution reaction (OER) while 0.42 A mgPt−1 at a potential of 0.7 V vs. RHE for the methanol oxidation reaction (MOR) in an alkaline system, respectively. This work may offer fundamental guidance for developing metal–loaded/dispersed support nanomaterials toward electrocatalysis through the fine regulation of metal–support interactions.
Introduction
Tremendous energy conversion technologies have been directed towards renewable energy sources and carbon neutralization, including water electrolysis and direct methanol fuel cells (DMFCs).1 Hydrogen energy, the most eco-friendly renewable energy source, can be sustainably generated by conducting water electrolysis. However, the anodic multi-electron water oxidation (i.e. oxygen evolution reaction, OER) possesses sluggish kinetics manifested in a high energy barrier and overpotential,2 which significantly affects the efficiency of the overall water splitting. Compared to hydrogen energy, renewable methanol serves as another commonly utilized fuel that can be easily stored and transported with existing infrastructure of petrol.3 DMFCs represent a kind of remarkable energy conversion device for the high-efficiency utilization of reproducible methanol through the oxidization of fuel, realizing the direct conversion of chemical energy into electric energy.4,5 Nevertheless, the development of effective anodic electrocatalysts for the sluggish methanol oxidation reaction (MOR, CH3OH + H2O → CO2 + 6H+ + 6e−) remains the bottleneck of DMFCs.6,7 In addition, the MOR process involves a complex 6-electron transfer mechanism and multiple pathways, such as the CO-containing pathway or others, which severely depend on catalysts.6,7 In this regard, it is challenging and of scientific significance to design electrocatalysts with high activities toward the OER and MOR.8,9Precious platinum (Pt) has attracted great interest in the oxidation reaction field due to its intriguing physicochemical properties and unprecedented catalytic efficiency.10,11 Strategies such as constructing ingenious geometrical nanostructures and modulating the Pt valence state are aimed at enhancing Pt utilization and electrocatalytic activity, but still remain challenging. Single-atom catalysts (SACs) have emerged as novel nanostructures with unique advantages in precisely controlling active sites, which is attributed to the exposure of numerous surface atoms, the tunable electronic structure and the coordinative environment.12 The typical synthetic methods such as impregnation, co-precipitation, sputtering and alloying have been employed for the doping of single atoms, showcasing the versatility and potential of SACs in catalysis.13–16
Notably, the catalytic activity of an entire SAC is closely dependent on the anchored substrate. Besides paying attention to single-atom center species, it is necessary to regulate the surrounding environment to achieve the optimal electronic structure and coordination by tuning the chemical bonds between single atoms and the substrate.17–21 For example, Pt single atoms supported by carbon black or carbon nanotubes exhibit inert catalytic activity, whereas Pt single atoms immobilized on RuO2 show superior catalytic mass activity and stability.12,22 Thereby, the selection of substrates for SACs implies a complex and multi-perspective challenge in boosting the SAC activity toward the OER and MOR, such as alloys,23 MoS2,15 MXene nanosheets,24,25 metal nanoparticles16,26 and transition metal oxides.13 Notably, NiO is a promising substrate due to its facile phase transformation into highly active NiOOH with a layered double hydroxide structure during the catalytic oxidation process,27 which is conducive to providing a migration channel for OER intermediates and thus facilitating the oxidative removal of COads on the activity sites in the MOR.
Recently, metal–support interactions, mainly those between metals and carbon substrates, have been massively studied for enhancing the electrocatalytic performance. For instance, Pt single atoms loaded on thiolated multiwalled carbon nanotubes are barely significant to the MOR catalytic activity, while Pt clusters possess a small MOR overpotential,28 attributed to the requirement of at least three Pt atoms to form a combined active site for the MOR.29 The dispersion and size of Pt are vital factors in determining the metal–support interactions and thus optimizing the electrocatalytic activity. Considering that single atoms predominantly anchor on the surface of materials, the charge state and distribution of SACs on the support directly influence Gibbs free energies of the crucial intermediates involved in the catalytic process, thereby tuning the rate-determining steps involved in the OER and MOR. Hence, regulating the noble metal–NiO support interactions by controlling the metal size and nanoarchitecture is an effective approach to modulate the electronic structure and bonding state around single atom-based catalysts.
Herein, a facile adsorption–impregnation strategy was employed to adjust the light condition and reaction time, facilitating the immobilization of different Pt nanostructures (single atoms, clusters, and nanoparticles) on the porous NiO nanoflake substrate, and thus achieving NiO–Pt single atoms (NiO–PtSA), NiO–Pt clusters (NiO–Ptcluster) and NiO–Pt particles (NiO–Ptparticle) respectively. The porous architecture of 2D nonlayered NiO offered abundant defects and accelerated mass transfer. The metal–NiO interactions and their effects on the catalytic performance were systematically studied. Remarkably, the immobilized Pt single atoms effectively reduced the reaction kinetic barrier and enhanced the catalytic activity in an alkaline system. As a result, the as-constructed NiO–PtSA catalyst displayed high OER performance with an enhanced mass activity of 5.59 A mgPt−1 at a current density of 10 mA cm−2, and an enhanced MOR peak mass activity of 0.42 A mgPt−1 at 0.7 V vs. RHE.
Results and discussion
Diverse Pt nanostructures (i.e. single atoms, clusters or nanoparticles) immobilized on a porous NiO nanosheet substrate were synthesized by a facile adsorption–impregnation method, as illustrated in Fig. 1A. First, α-type layered nickel hydroxide intercalated with anionic surfactant dodecyl sulfate (denoted as α-Ni(OH)2-DS) was prepared via an oil bath process. The basal reflection series of the product well matched with a typical α-Ni(OH)2-DS layered structure with an interlayer spacing of 2.6 nm (Fig. 1B(I)). A substantial quantity of uniform nanoflakes was observed in the representative scanning electron microscopy (SEM) image (Fig. 1C). Second, the influence of calcination temperature on the morphology and structure of the calcined product was investigated in detail. Interlayer anionic DS− could not be completely removed at 600 °C (Fig. S1 and S2†), while pores would form and lead to morphological defects at a calcined temperature of 800 °C (Fig. S3 and S4†). Therefore, the optimal NiO with standard crystal (JCPDS No. 65-2901, Fig. 1B(II)) and porous nanosheet morphology (Fig. S5†) was obtained through calcination at an ambient temperature of 700 °C. The porous feature of the NiO nanoflakes was further confirmed by transmission electron microscopy (TEM) (Fig. 1D). A high-resolution TEM image (HRTEM) indicated clear lattice fringes with a lattice space of 0.24 nm, corresponding to the (111) planes of NiO (Fig. 1F). The selected area electron diffraction (SAED) pattern, as shown in Fig. 1E, revealed the polycrystalline nature of NiO nanoflakes.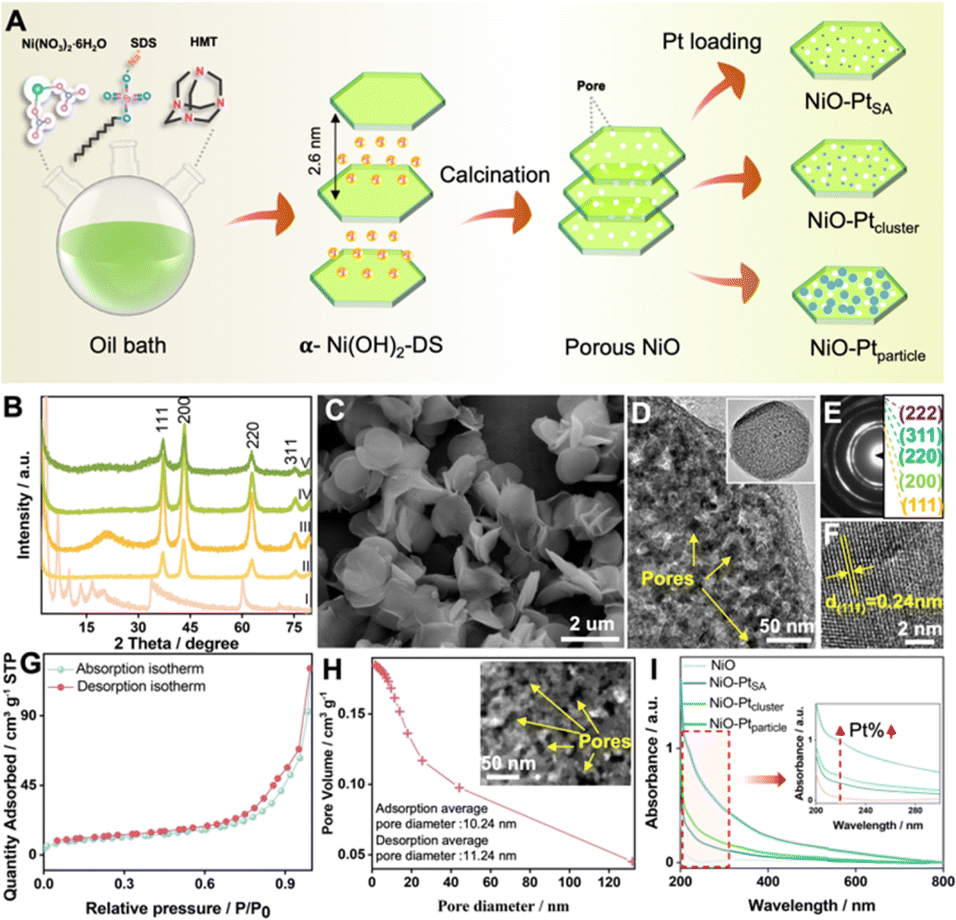 | ||
| Fig. 1 Compositional and structural characterization of the porous NiO substrate and NiO–PtSA, NiO–Ptcluster, NiO–Ptparticle. (A) Schematic illustration of the fabrication procedure for porous NiO–PtSA, NiO–Ptcluster and NiO–Ptparticle catalysts. (B) XRD patterns of (I) α-Ni(OH)2-DS, (II) porous NiO (calcined at 700 °C), (III) NiO–PtSA, (IV) NiO–Ptcluster, and (V) NiO–Ptparticle. (C) SEM image of a-Ni(OH)2. (D) TEM image, (E) SAED pattern and (F) HRTEM image of NiO. (G) BET isotherm of porous NiO. (H) BJH-plot of pore volume vs. pore diameter, the inset is a HAADF image of porous NiO. (I) UV-visible absorption spectra of NiO and the three NiO/Pt samples. | ||
The surface properties of NiO were investigated by Brunauer–Emmett–Teller (BET) characterization. The isotherms of NiO showed a hysteresis loop with high relative pressure (P/P0) in the range of 0 to 1 according to the N2 adsorption–desorption isotherms (Fig. 1G), and the BET surface area for NiO was thus estimated as 36.99 m2 g−1. Based on the related pore size distribution plot by the Barrett–Joyner–Halenda (BJH) method, the pore size of NiO nanoplates was predominantly distributed between 1 and 20 nm. The average pore diameter for adsorption and desorption was 10.24 nm and 11.24 nm, respectively, as shown in Fig. 1H. The porous feature morphology of NiO may be convenient to supply pores, defects and abundant active sites, making it an outstanding substrate. In the final step, the NiO–PtSA, NiO–Ptcluster, and NiO–Ptparticle were prepared by immersing porous NiO into the deionized water containing chloroplatinic acid (H2PtCl6) solution for spontaneous adsorption. The evolution in the morphology and structure of the different Pt nanostructure decorated catalysts was created by tuning the synthetic reaction time and light condition. No obvious differences and crystal diffraction peaks of Pt for the three catalysts were observed in the XRD patterns, which may be due to the low Pt content (Fig. 1B(III)–(V)). However, the Pt peaks show a gradually increased tendency according to the UV-visible absorption spectra (Fig. 1I), indicating that the Pt content increased in the sequence of NiO–PtSA, NiO–Ptcluster, and NiO–Ptparticle.
No significant differences were observed in the lamellar morphologies of NiO–PtSA and NiO–Ptcluster catalysts based on the visualized SEM images (Fig. S6A and S6C†), while the surface of NiO–Ptparticle nanoflakes was modified by some small nanoparticles (Fig. S6E†). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. S6B and S6D†) further confirmed the porous nature of NiO–PtSA and NiO–Ptcluster nanoflakes. The white highlights recorded in the HAADF-STEM observation of NiO–Ptparticle might be ascribed to Pt nanoparticles (Fig. S6F†). The corresponding STEM-EDX maps demonstrated uniform dispersion of Ni and Pt elements throughout NiO nanoflakes. To further confirm the component of light spots, SAED and HRTEM of the three catalysts were conducted, as displayed in Fig. S7–S9,† respectively. The lattice fringes and SAED patterns of NiO–PtSA and NiO–Ptcluster catalysts matched well with NiO crystals, verifying the substrate as NiO. In contrast, the lattice fringes of 0.22 nm were assigned to the Pt (111) plane for the nanoparticles on the NiO surface, affirming the bright spots as Pt nanoparticles. Additionally, for comparison, pure Pt nanoparticles were fabricated under the same conditions as those for NiO–Ptparticle without the addition of NiO (Fig. S10–S12†).
To intuitively investigate the dispersion state of Pt, aberration-corrected high-angle annular darkled-scanning transmission electron microscopy (AC HAADF-STEM) was used to unambiguously contrast the sub-nanometer metal particles. Fig. 2A and B display the AC-HAADF-STEM images of NiO–PtSA, showing brighter spots (i.e. Pt atoms, marked by the green circles) that were atomically dispersed. Meanwhile, no bright Pt atoms were observed in between any two adjacent lattice fringes, revealing the absence of Pt atoms in the interstitial sites of the lattices. It was thus speculated that Pt atoms substituted Ni sites, i.e., achieved the doping behavior in NiO lattices. The Pt single atom loading was further revealed by ICP-OES as 0.73 wt%, which may change the electronic structure and coordination of the center atoms due to the substitution into the crystal lattices, as illustrated in Fig. 2C. When the Pt loading increased up to 1.22 wt%, not only highly dispersed Pt single atoms but also some bright clusters were found in the AC-HAADF-STEM images of NiO–Ptcluster (Fig. 2D and E), indicating that Pt atoms could be gradually nucleated on the surface of NiO nanoflakes and grew into Pt clusters at such an increased Pt content, as shown in Fig. 2F. In contrast, aggregated spots of ∼3 nm were observed in the AC-HAADF-STEM images of NiO–Ptparticle with a high Pt loading of 3.68 wt%, corresponding to Pt nanoparticles (Fig. 2G and H). The above results show that Pt may be nucleated from single atoms to nanoparticles through clusters on the surface of NiO nanoflakes, as displayed in Fig. 2I.
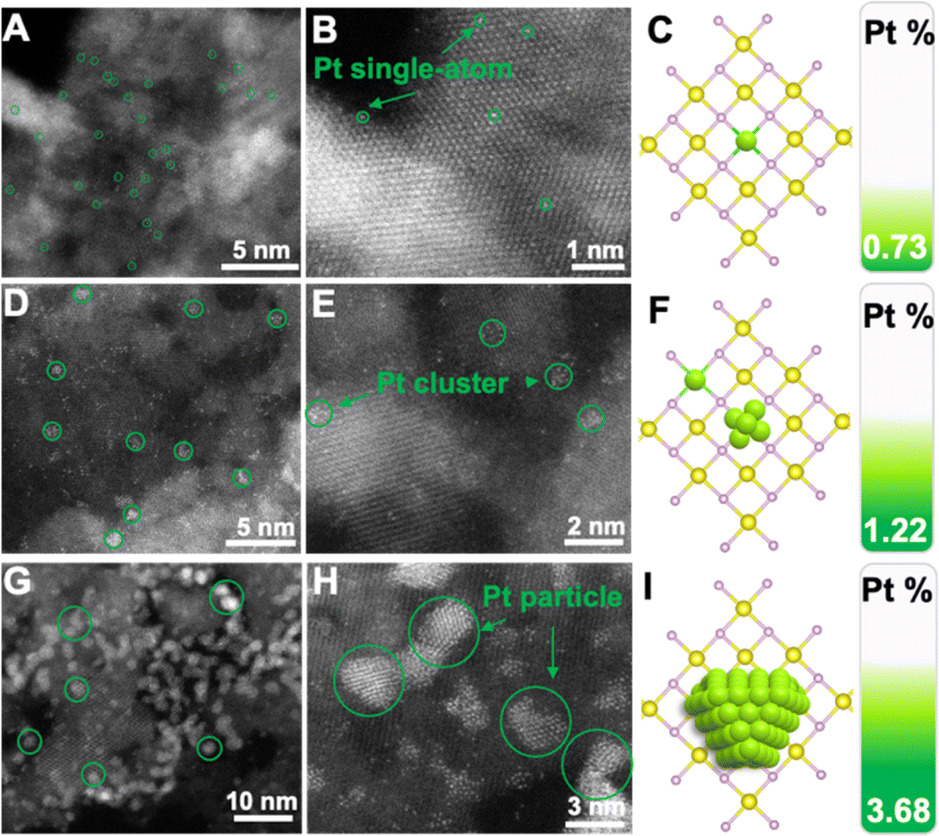 | ||
| Fig. 2 Atomic structure characterization of the as-prepared NiO–PtSA, NiO–Ptcluster, and NiO–Ptparticle. (A and B) AC-HAADF-STEM images of NiO–PtSA. (C) Schematic illustration and ICP-OES of NiO–PtSA. (D and E) AC-HAADF-STEM images of NiO–Ptcluster. (F) Schematic illustration and ICP-OES of NiO–Ptcluster. (G and H) AC-HAADF-STEM images of NiO–Ptparticle. (I) Schematic illustration and ICP-OES of NiO–Ptparticle. | ||
The electronic state and chemical composition of the Pt atoms in different NiO/Pt catalysts were investigated by X-ray photoelectron spectroscopy (XPS), as shown in Fig. 3A–C. In Pt 4f spectra, three characteristic states of Pt were deconvoluted, which were ascribed to metallic Pt0 (70.9 eV, 74.2 eV), Pt2+ (72.9 eV, 76.4 eV) and Pt4+ (78.35 eV), respectively. In particular, Pt atoms in NiO–PtSA mainly existed in the valence state of Pt2+, indicating that Pt was coordinated with O in the NiO crystal. Besides the Pt2+ coordinations, the signal of metallic Pt0 was also detected for NiO–Ptcluster, indicating that Pt was nucleated and grew into clusters. In contrast, the Pt nanoparticles in the NiO–Ptparticle catalyst were formed as Pt0, Pt2+ and Pt4+ species; herein Pt4+ might be derived from the adsorbed PtCl62− ions on the surface of the samples.30,31 The XPS results of Pt were consistent with the AC-HAADF-STEM observations. Fig. 3B depicts the high-resolution Ni 2p spectrum, in which the peaks at 855.8 eV and 873.3 eV along with two weak satellite peaks at 861.0 eV and 879.4 eV corresponded to the Ni 2p3/2 and Ni 2p1/2 signals of NiO, respectively. This was assigned to the valence state of Ni2+ by forming the Ni–O bond in the NiO crystal. The O 1s spectra of the three catalysts were divided into two characteristic peaks of lattice oxygen (Olat, 529.3 eV) and oxygen vacancies (Ovan, 532.3 eV), in which the lattice oxygen was formed by M–O (M = Ni and/or Pt) and the oxygen vacancies originate from the porous interfacial defects in the NiO nanoplates.32
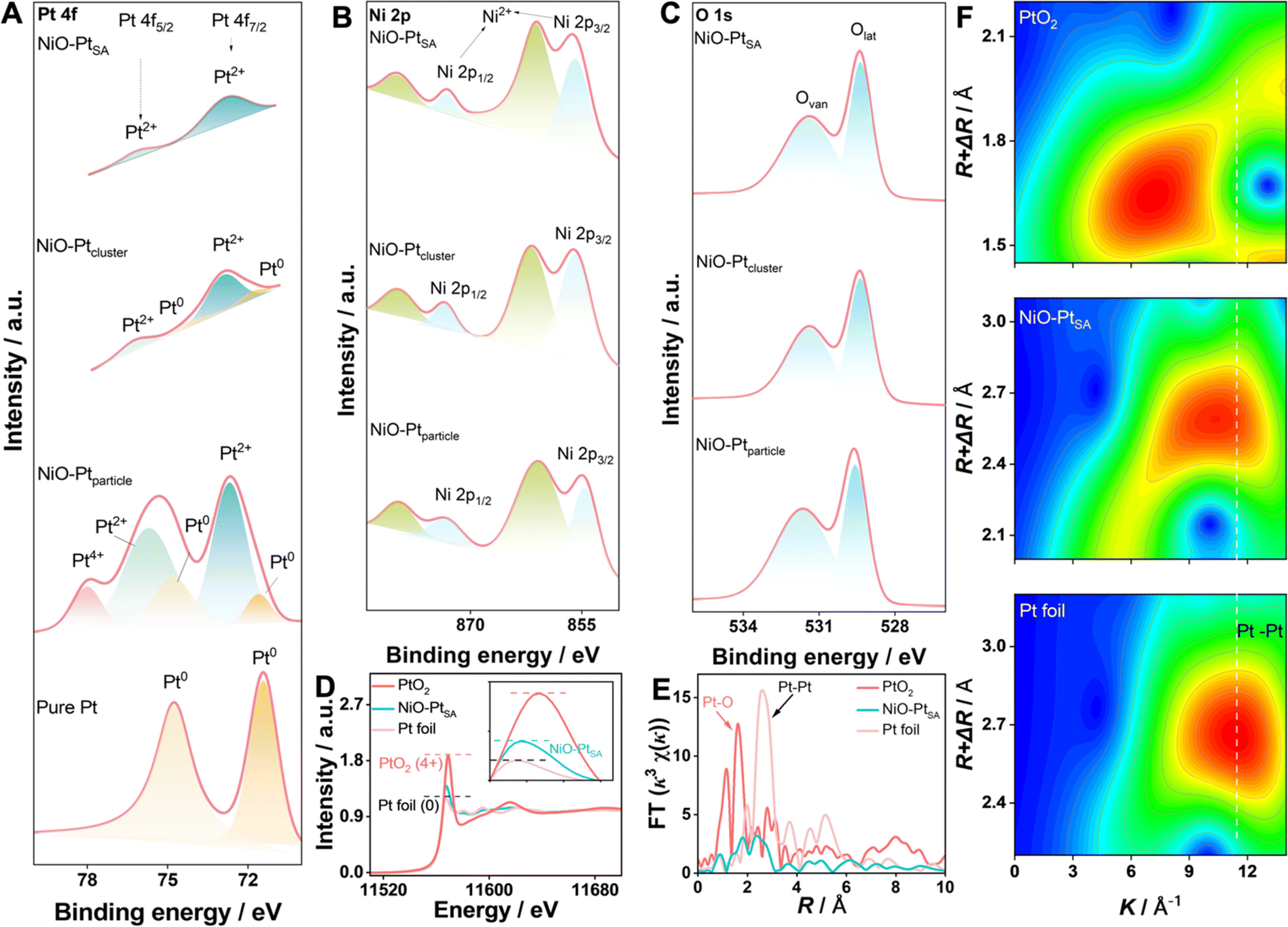 | ||
| Fig. 3 Electronic state and coordination characterization. XPS spectra of (A) Pt 4f, (B) Ni 2p and (C) O 1s for NiO–PtSA, NiO–Ptcluster, and NiO–Ptparticle. (D) XANES spectra, (E) the corresponding FT-EXAFS curves and (F) EXAFS wavelet transform plots of NiO–PtSA, Pt foil and PtO2. | ||
For the in-depth study of the coordination environment and electronic configuration of Pt single atoms in the NiO–PtSA, X-ray absorption fine structure (XAFS) spectroscopy was carried out. The Pt L3-edge normalized X-ray absorption near-edge structure (XANES) spectrum of NiO–PtSA is significantly distinguished from those of Pt foil and PtO2 (Fig. 3D), in which the intensity of white-line peaks at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 568 eV corresponded to the transfer of the Pt 2p3/2 core electron to 5d states, and thus was used as an indicator of Pt 5d band occupancy.33,34 The decreased white-line intensity corresponded to the increased Pt 5d occupancy. Thus, lower 5d occupancy indicated the less charge loss of the Pt single atoms after coordinating with the substrate in comparison with Pt foil. This implied that Pt in NiO–PtSA was positively charged due to the electron transfer from Pt to the surrounding O atoms in the NiO substrate.24,35 The charge state of Pt in NiO–PtSA was closely affected by the coordination with the substrate. The atomic coordination configuration of Pt was further revealed by the extended XAFS (EXAFS) at the Pt L3-edge, as shown in Fig. 3E. The absence of the typical Pt–Pt peak at 2.65 Å for NiO–PtSA strongly indicated that Pt is in the atomically dispersed single atom state, matching well with the AC-HAADF-STEM observations. Specifically, the dominant peak of Pt–O coordination for NiO–PtSA slightly shifted to 1.83 Å in comparison with 1.62 Å of PtO2 in the R-space spectrum, indicating the formed Pt–O as Pt2+ instead of Pt4+, which is consistent with the XPS results. As shown in Fig. 3F, the wavelet-transform plots of NiO–PtSA showed a maximum of about 10.5 Å which was lower than 11.4 Å of Pt foil but higher than 7.4 Å of PtO2, further confirming the coordination conditions of Pt–O for Pt single atoms in the NiO–PtSA sample.
568 eV corresponded to the transfer of the Pt 2p3/2 core electron to 5d states, and thus was used as an indicator of Pt 5d band occupancy.33,34 The decreased white-line intensity corresponded to the increased Pt 5d occupancy. Thus, lower 5d occupancy indicated the less charge loss of the Pt single atoms after coordinating with the substrate in comparison with Pt foil. This implied that Pt in NiO–PtSA was positively charged due to the electron transfer from Pt to the surrounding O atoms in the NiO substrate.24,35 The charge state of Pt in NiO–PtSA was closely affected by the coordination with the substrate. The atomic coordination configuration of Pt was further revealed by the extended XAFS (EXAFS) at the Pt L3-edge, as shown in Fig. 3E. The absence of the typical Pt–Pt peak at 2.65 Å for NiO–PtSA strongly indicated that Pt is in the atomically dispersed single atom state, matching well with the AC-HAADF-STEM observations. Specifically, the dominant peak of Pt–O coordination for NiO–PtSA slightly shifted to 1.83 Å in comparison with 1.62 Å of PtO2 in the R-space spectrum, indicating the formed Pt–O as Pt2+ instead of Pt4+, which is consistent with the XPS results. As shown in Fig. 3F, the wavelet-transform plots of NiO–PtSA showed a maximum of about 10.5 Å which was lower than 11.4 Å of Pt foil but higher than 7.4 Å of PtO2, further confirming the coordination conditions of Pt–O for Pt single atoms in the NiO–PtSA sample.
The OER electrocatalytic activities of as-fabricated NiO/Pt catalysts were measured in 1.0 M KOH electrolyte, as displayed in Fig. 4. The NiO–PtSA catalyst possessed the highest OER activity with a rather low overpotential of 0.274 V delivering 10 mA cm−2 in comparison with the other catalysts (Fig. 4A). Furthermore, the NiO–PtSA catalyst exhibited a smaller Tafel slope of 83 mV dec−1 and an enhanced double-layer capacitance (Cdl) of 58 mF cm−2 compared to the hydroxide precursor and the other two NiO/Pt catalysts featuring different Pt nanostructures (Fig. 4B and C), suggesting that atomically dispersed Pt single atoms on the NiO substrate could supply more accessible active sites for the OER. Moreover, when normalized with the Pt loading mass, NiO–PtSA delivered a higher current of 5.59 A mgPt−1 at an overpotential of 0.274 V, which was 2.64 and 9.63 times that of NiO–Ptcluster (2.11 A mgPt−1) and NiO–Ptparticle (0.58 A mgPt−1), respectively (Fig. 4D), indicating a much higher mass activity for NiO–PtSA. The above results highlight that the incorporation of NiO with a minimal amount of Pt single atoms can extremely maximize the mass activity toward alkaline OER, significantly reducing the Pt cost while increasing its utilization. Moreover, the NiO–PtSA possessed a higher turnover frequency (TOF) and O2 production rate compared with the other as-prepared catalysts according to the calculations and analyses (Fig. 4E and F). Furthermore, the NiO–PtSA catalyst demonstrated high durability in continuous electrocatalysis, with negligible potential fluctuation detected over 20 h during the OER at current densities of both 10 mA cm−2 and 50 mA cm−2 (Fig. 4G). The structural and morphological characterization of the NiO–PtSA catalyst after the stability test at 10 mA cm−2 suggested negligible changes after a long-term alkaline OER process (Fig. S13 and S14†). Meanwhile, the charge transfer resistance (Rct) of NiO–PtSA was lower than those of precursors and Pt-based catalysts (Fig. 4H and Table S1†), which was mainly due to the optimized electronic structure and coordination environment originating from the atomically Pt dopant in the NiO crystal. Fig. 4I shows the cyclic voltammetry (CV) curve of NiO–PtSA, in which a pair of redox peaks were observed before the OER region, revealing that the catalyst was electro-oxidized before the OER process. To probe the interface electron transfer reactions, operando EIS was conducted to obtain the Nyquist (Fig. 4J) and Bode plots (Fig. 4K) during the OER for NiO–PtSA. The charge transfer resistance significantly decreased with the increase of potential, accompanied by distinct electro–oxidation reaction and OER. The equivalent circuit diagram and model, as presented in Fig. 4L, represent the situations before and/or after electro–oxidation reaction.36,37 Based on the intrinsic consistency of the equivalent resistance with the reaction and current density of the NiO–PtSA catalyst, it was speculated that the electro-oxidation reaction (in the potential windows higher than 1.295 V) took place at the high-frequency interface while the OER (in the potential region higher than 1.470 V) occurred at the low-frequency interface.
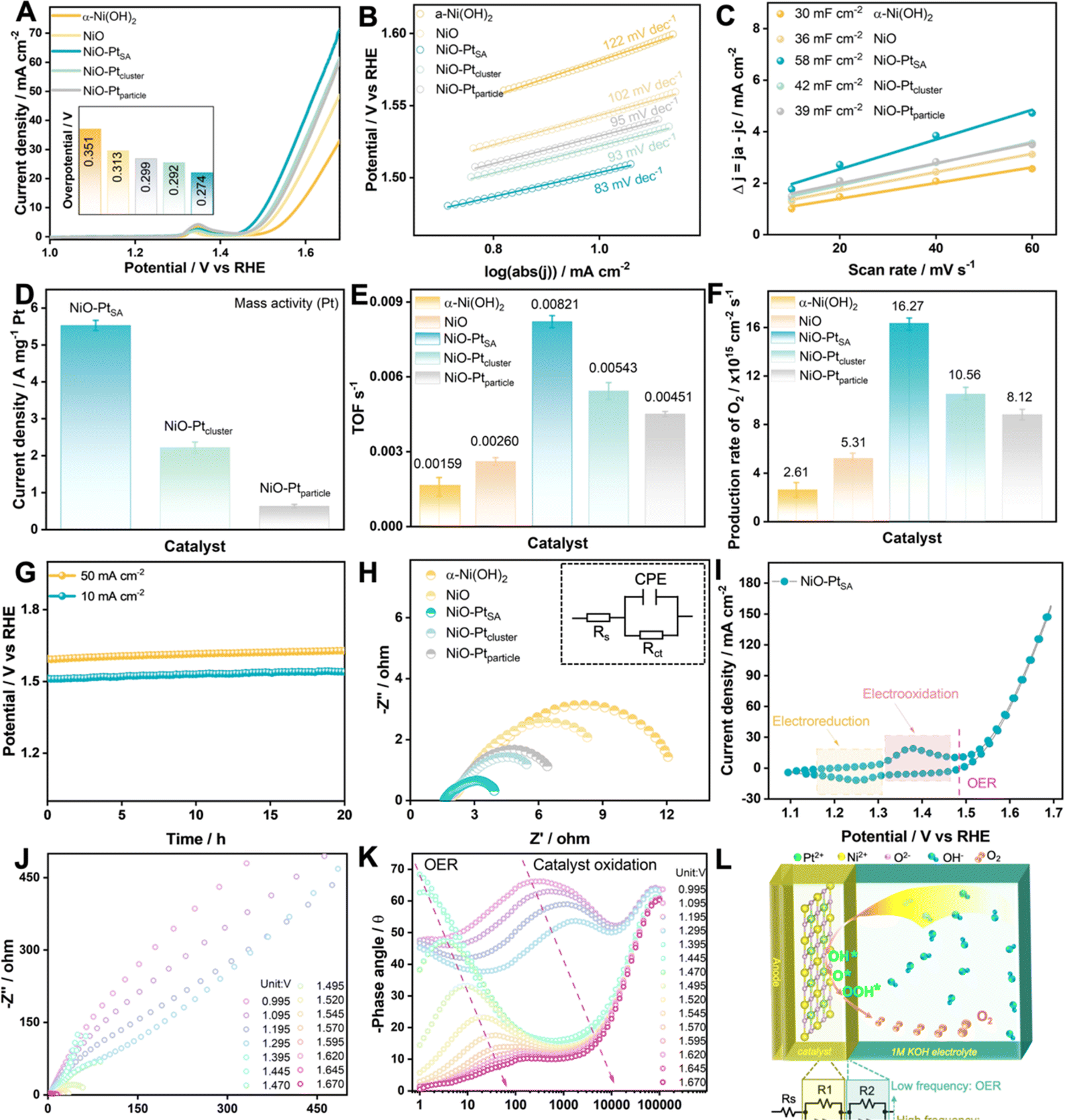 | ||
| Fig. 4 Electrocatalytic alkaline OER performance of the catalysts in 1.0 M KOH electrolyte. (A) OER polarization curves of α-Ni(OH)2, NiO and kinds of NiO/Pt catalysts, the inset shows the overpotentials of different catalysts delivering a current density of 10 mA cm−2. (B) Tafel slopes. (C) Estimation of Cdl by fitting a linear regression at different scan rates. (D) The mass activity of NiO/Pt catalysts. (E) TOFs and (F) production rate of O2. (G) Chronoamperometric curves of NiO–PtSA. (H) Nyquist plots of the catalysts, the inset shows the fitting equivalent circuit. (I) CV curve of NiO–PtSA at 5 mV s−1. (J) Nyquist plots of NiO–PtSA at different potentials. (K) Bode plots for NiO–PtSA during the OER. (L) Schematic illustration showing the relationship between surface species, interfaces and reactions for NiO–PtSA during the OER. | ||
To deeply understand the structural transition during the OER, in situ Raman electrochemical investigation was conducted on NiO–PtSA during the overpotential sweep from 0 to 400 mV (Fig. 5A). Five characteristic peaks appeared at 539, 735, 909, 1091 and 1507 cm−1 under no bias condition, which match well with the first-order longitudinal optical (LO), transverse optical (2TO), LO+TO, 2LO and 2 M modes of NiO, respectively.38,39 At an overpotential of 0 V (1.23 V vs. RHE), additional peaks slightly appeared at 477 and 558 cm−1, corresponding to the γ-NiOOH phase.40,41 With increased overpotentials, the signal intensity of γ-NiOOH intensified, while that for NiO decreased, verifying that introducing Pt single atoms facilitates a structural transition at a lower potential and plays the key role in boosting subsequent OER kinetics.
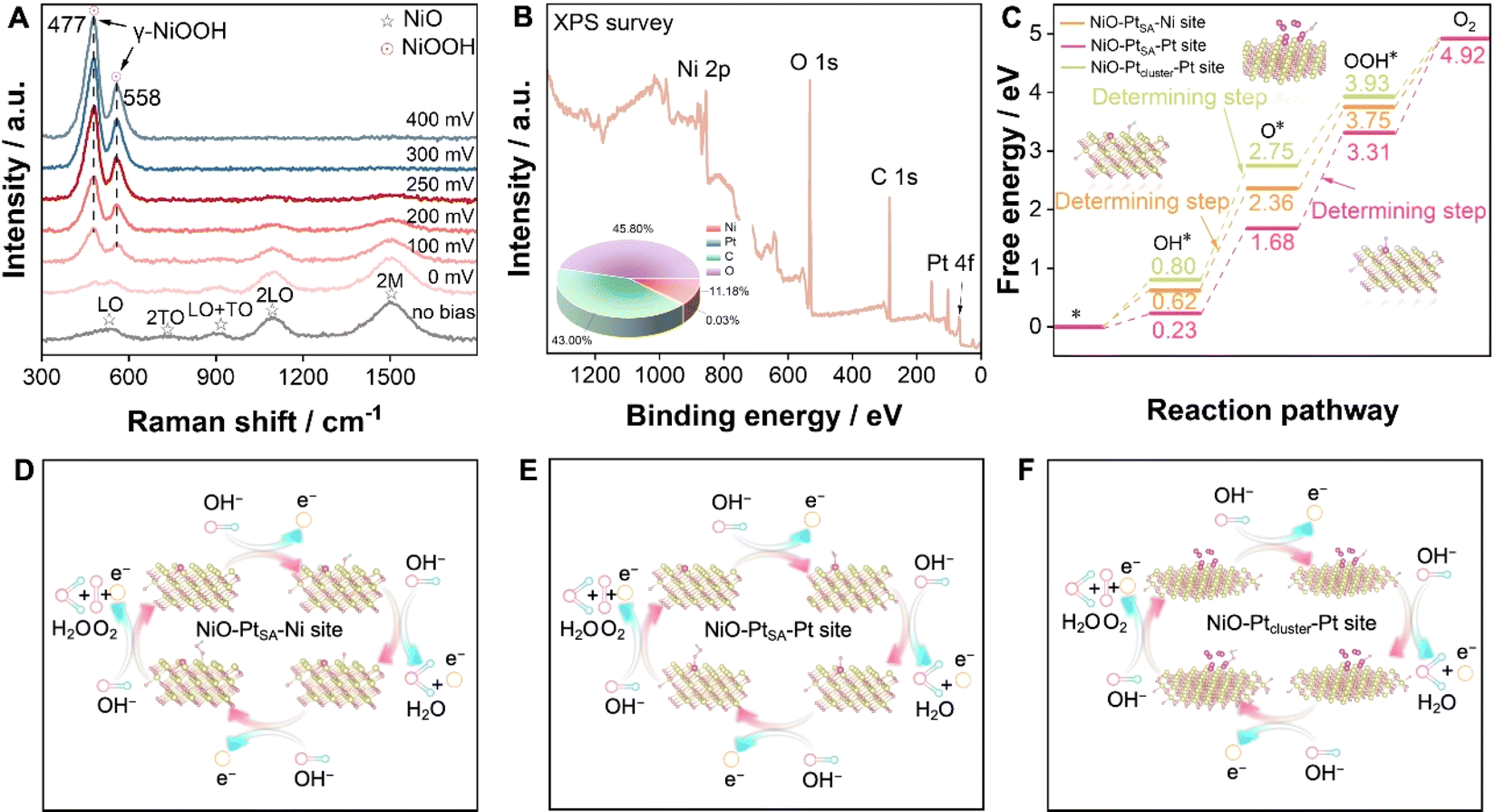 | ||
| Fig. 5 Mechanism and theoretical calculations on the OER. (A) The in situ Raman spectra of NiO–PtSA under various applied overpotentials. (B) The survey XPS of NiO–PtSA after the OER stability test. The inset is the atomic content of Ni, Pt, C and O in the NiO–PtSA catalyst after the OER stability test, obtained from the XPS. (C) Free energy diagrams. Reaction pathways for the OER on the (D) Ni site in NiO–PtSA, (E) Pt site in NiO–PtSA and (F) Pt site in NiO–Ptcluster. | ||
XPS was engaged to analyze the surface chemical states and element components of NiO–PtSA after the OER stability test (Fig. 5B and S15†). The survey XPS spectrum identified Ni, Pt, C and O with 11.18, 0.03, 43.00 and 45.8 at%, respectively (the inset of Fig. 5B). Accordingly, the proportions of Pt and Ni were 0.35 at% and 99.65 at% (Table S2†), respectively, indicating that the Pt content did not show significant loss after OER measurement compared to that in the initial catalyst (0.73 wt%, 0.34 at%). The O 1s spectrum still exhibited two peaks for the oxygen lattice and vacancies, respectively. The Ni 2p spectrum also deconvoluted to two sets of Ni 2p3/2 and Ni 2p1/2 assigned to Ni2+, indicating that the structural transformation of the NiOOH phase into initial NiO is highly reversible. The Pt 4f spectrum was assigned to Pt2+ without any other peaks, indicating that Pt single atoms are relatively robust and no agglomeration occurs due to the absence of metallic Pt peaks after the OER.
The energy barrier is a key descriptor to evaluate the rate-determining step for the OER. Density functional theory (DFT) calculations were introduced to calculate the Gibbs free energy on different active sites for pure NiO, NiO–PtSA, and NiO–Ptcluster. Fig. S16† exhibits the intermediate transformation pathway (OH*, O* and OOH*) in the OER process for Ni sites of the pure NiO catalyst, through which the rate-determining step of pure NiO was determined as the deprotonation of OH*. Moreover, the reaction dynamics on the Ni and Pt active sites of NiO–PtSA were further investigated (Fig. 5C–E). The rate-determining step was ascribed to the adsorption of OH* to form OOH* with an energy barrier of 1.62 eV on the Pt sites of NiO–PtSA for the OER, which was lower than 1.75 eV for the deprotonation of OH* on Ni sites, indicating that Pt single atoms in NiO–PtSA tended to be the active sites for the OER and were more favorable for O2 generation. When the Pt atoms aggregated into a cluster state, the rate-determining step of the NiO–Ptcluster on Pt sites changed back to the deprotonation of OH* with an energy barrier of 1.95 eV (Fig. 5C and F), which was higher than the Pt single atom state, suggesting that the Pt clusters might be less conducive to the OER process. Therefore, it could be concluded that tunable metal–support interactions originating from the size and dispersion of Pt directly affected the OER process. In particular, Pt single atoms in NiO nanoflakes reduced the energy barrier and thus promoted the OER process.
The MOR performance of the as-prepared catalysts was tested in a mixed 1.0 M KOH + 1.0 M CH3OH aqueous electrolyte. The catalytic activities were evaluated from CV curves at a scan rate of 50 mV s−1, as recorded in Fig. 6A and S17.† Pure NiO exhibited no obvious current response toward the MOR, while all three NiO/Pt catalysts showed a significant current increase when using NiO as the substrate. The NiO–PtSA catalyst displayed the lowest onset oxidation potential (0.519 V vs. RHE) compared with NiO–Ptcluster (0.541 V vs. RHE) and NiO–Ptparticle (0.615 V vs. RHE), indicating that Pt single atoms enabled NiO–PtSA to be an efficient MOR electrocatalyst compared to the other catalysts. The lowest onset oxidation potential of NiO–PtSA revealed the formation of weakly adsorbed COads on Ni and/or Pt active sites, which affected the key step of the co-adsorption of *CO + *OH in the MOR to avoid self-poisoning and deactivation of the catalyst.42,43 In addition, the ratios of the forward peak current density (If) to backward peak current density (Ib), i.e. If/Ib, were estimated to be 5.13, 4.30, 3.52, and 1.93 for NiO–PtSA, NiO–Ptcluster, NiO–Ptparticle and pure Pt catalysts, respectively. The prominent If/Ib of NiO–PtSA indicated a more efficient MOR process with less residual intermediate species which were generated as a result of the incomplete electrooxidation of CH3OH. Moreover, as presented in Fig. 6B, the mass activity of NiO–PtSA (0.422 A mgPt−1) was 2.62 and 18.35 times that of NiO–Ptcluster (0.161 A mgPt−1) and NiO–Ptparticle (0.023 A mgPt−1), respectively. Higher peak mass activity and smaller peak potential of NiO–PtSA indicated a lower activation barrier. Such a prominent mass activity was also superior to those of previously reported Pt-based catalysts.44–49 The effect of Ib on methanol oxidation was assessed by comparing the typical forward direction with the reverse direction (Fig. 6C). Two curves were highly overlapped, which revealed that backward oxidation was almost not influenced by the forward or reverse reactions. The EIS data, as shown in Fig. 6D and Table S3,† indicated that NiO–PtSA presented a smaller semicircle radius of impedance than those of NiO–Ptcluster and NiO–Ptparticle, which implied that electron transfer was significantly facilitated because of the Pt single atoms–NiO support interaction. Chronoamperometry was carried out at 0.70 V vs. RHE to further evaluate the durability toward the MOR (Fig. 6E). It should be noted that the final current density of NiO–PtSA after 10000 s was remarkedly higher than those of NiO–Ptcluster and NiO–Ptparticle. In addition, NiO–PtSA maintained a superior capacity retention of 95% after 500 cycles for the MOR (Fig. S18†). The outstanding electrocatalytic stability and durability of the NiO–PtSA further verified its excellent anti-poisoning properties.
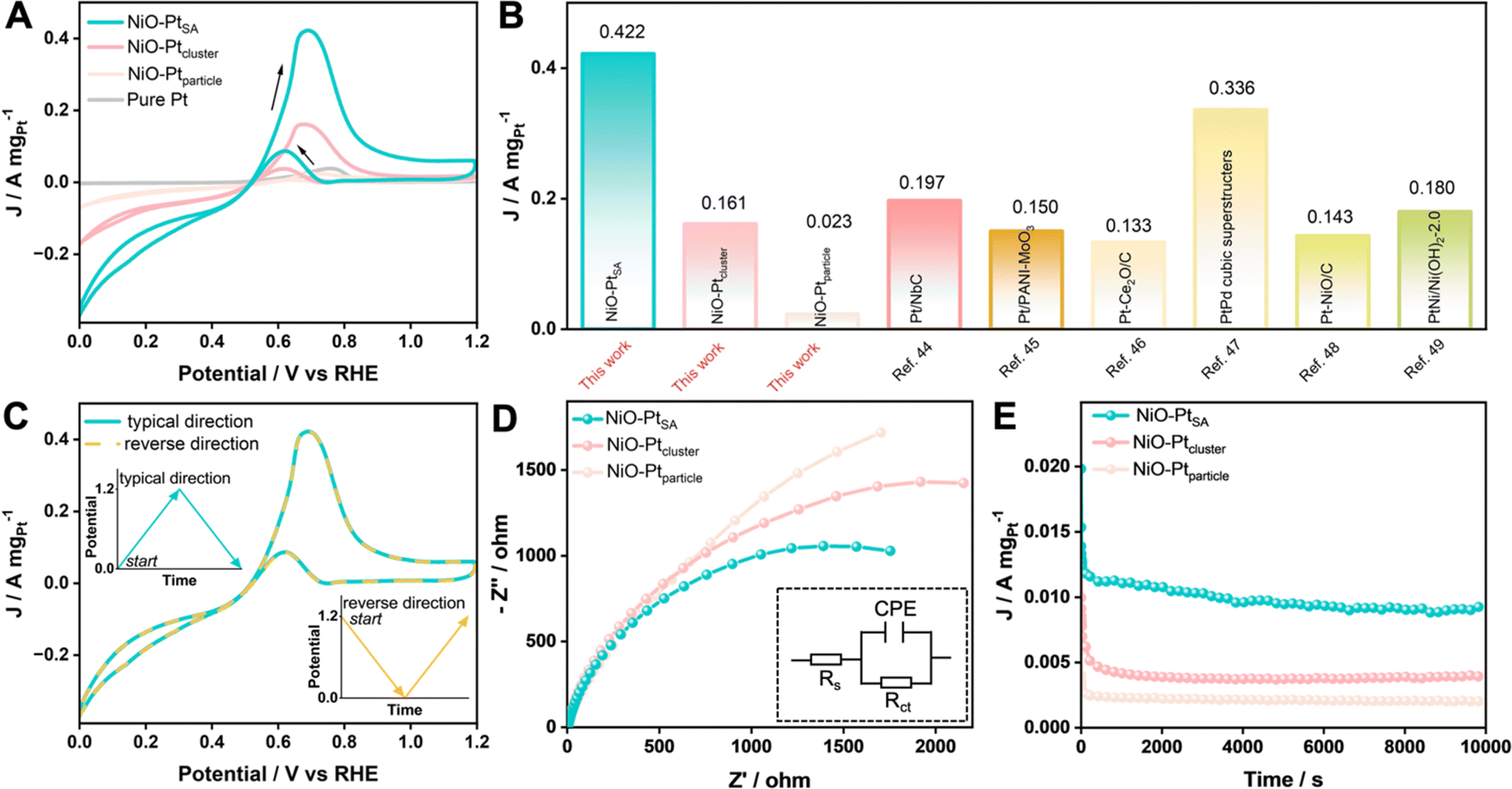 | ||
| Fig. 6 MOR performance in a mixed electrolyte of 1.0 M KOH and 1.0 M CH3OH. (A) The mass-normalized CV curves and (B) a comparison showing mass activities at peak potentials for as-prepared catalysts at 50 mV s−1 and the previously reported Pt-based catalysts. (C) CV results of NiO–PtSA with the typical direction and reverse direction (blue line: typical direction, yellow dashed line: reverse direction). (D) Nyquist plots, the inset shows the simulated circuit. (E) Chronoamperometry curves for the NiO/Pt catalysts at 0.70 V for 10000 s. | ||
Conclusions
A series of NiO/Pt catalysts with different Pt nanostructures (single atoms, clusters, and nanoparticles) were prepared using a simple adsorption–impregnation strategy by adjusting the light condition and reaction prolongation, and further applied to the electrocatalytic OER and MOR. According to experimental analyses and theoretical calculations, Pt single atoms in the Pt2+ valence state are mainly coordinated with the surrounding O, optimizing the electronic structure and coordination of the metal centers, i.e. the metal–support interactions. This strategy combined the advantages of porous nanoflake structures of the NiO substrate with the Pt dopant to optimize the noble metal–NiO support interactions. As a result, the NiO–PtSA catalyst showed the merits of low Pt loading for high Pt utilization, and improved transmission of active species with a remarkably enhanced catalytic activity of 5.59 A mgPt−1 for the OER and 0.42 A mgPt−1 for the MOR, which significantly surpassed the NiO–Ptcluster and NiO–Ptparticle. This work will offer an approach of carefully modulating metal–non-carbon support interactions to develop highly active hybrid catalysts for energy-related technologies.Data availability
The theoretical calculations and experimental procedure details have been provided within the manuscript and ESI.† The data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
F. W. and H. W. conceived and coordinated the project. H. W., G. C., N. Z., X. L., and R. M. supervised the project. F. W. performed the experiments, analyzed the data, and wrote the original draft. Z. Z. assisted with the DFT calculations and analysis. D. W. helped with the OER measurements and data analyses. The manuscript was finished through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (U20A20123 and 51874357). H. W. acknowledges the support from the Project of Zhongyuan Critical Metals Laboratory (GJJSGFYQ202336). This work was supported in part by the High-Performance Computing Center of Central South University.Notes and references
- Z. X. Xia, X. M. Zhang, H. Sun, S. L. Wang and G. Q. Sun, Recent advances in multi-scale design and construction of materials for direct methanol fuel cells, Nano Energy, 2019, 65, 104048 CrossRef CAS
.
- H. Chen, J. Chen, P. Ning, X. Chen, J. Liang, X. Yao, D. Chen, L. Qin, Y. Huang and Z. Wen, 2D Heterostructure of Amorphous CoFeB Coating Black Phosphorus Nanosheets with Optimal Oxygen Intermediate Absorption for Improved Electrocatalytic Water Oxidation, ACS Nano, 2021, 15, 12418–12428 CrossRef CAS PubMed
- E. Sarwar, T. Noor, N. Iqbal, Y. Mehmood, S. Ahmed and R. Mehek, Effect of Co-Ni Ratio in Graphene Based Bimetallic Electro-catalyst for Methanol Oxidation, Fuel Cells, 2018, 18, 189–194 CrossRef CAS
- A. Carne-Sanchez, F. J. Carmona, C. Kim and S. Furukawa, Porous materials as carriers of gasotransmitters towards gas biology and therapeutic applications, Chem. Commun., 2020, 56, 9750–9766 RSC
- C. Liu, Y. Shen, J. F. Zhang, G. Li, X. R. Zheng, X. P. Han, L. Y. Xu, S. Z. Zhu, Y. A. Chen, Y. D. Deng and W. B. Hu, Multiple Twin Boundary-Regulated Metastable Pd for Ethanol Oxidation Reaction, Adv. Energy Mater., 2022, 12, 2103505 CrossRef CAS
- Y. W. Zhou, Y. F. Chen, K. Jiang, Z. Liu, Z. J. Mao, W. Y. Zhang, W. F. Lin and W. B. Cai, Probing the enhanced methanol electrooxidation mechanism on platinum-metal oxide catalyst, Appl. Catal., B, 2021, 280, 119393 CrossRef CAS
- W. C. Geng, Y. J. Zhang, L. Yu, J. J. Li, J. L. Sang and Y. J. Li, Integrating Pt(16) Te Nanotroughs and Nanopillars into a 3D "Self-Supported" Hierarchical Nanostructure for Boosting Methanol Electrooxidation, Small, 2021, 17, e2101499 CrossRef PubMed
- L. L. Zhang, P. P. Lu, Y. R. Luo, J. Y. Zheng, W. Ma, L. X. Ding and H. H. Wang, Graphene-quantum-dot-composited platinum nanotube arrays as a dual efficient electrocatalyst for the oxygen reduction reaction and methanol electro-oxidation, J. Mater. Chem. A, 2021, 9, 9609–9615 RSC
- X. M. Zhao, X. Liu, B. Y. Huang, P. Wang and Y. Pei, Hydroxyl group modification improves the electrocatalytic ORR and OER activity of graphene supported single and bi-metal atomic catalysts (Ni, Co, and Fe), J. Mater. Chem. A, 2019, 7, 24583–24593 RSC
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J. Y. Ma, D. Su and X. Huang, Biaxially strained PtPb/Pt core/shell nanoplate boosts oxygen reduction catalysis, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed
- X. Li, C. Deng, Y. Kong, Q.-H. Huo, L.-R. Mi, J.-J. Sun, J.-Y. Cao, J.-X. Shao, X.-B. Chen, W.-L. Zhou, M.-Y. Lv, X.-Y. Chai, H.-P. Yang, Q. Hu and C.-X. He, Unlocking the transition of electrochemical water oxidation mechanism induced by heteroatom doping, Angew. Chem., Int. Ed., 2023, 62, e202309732 CrossRef CAS PubMed
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim, T. Zhao and F. Ciucci, Single-atom catalyst for high-performance methanol oxidation, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed
- X.-J. Li, H.-K. Zhang, Q. Hu, W.-L. Zhou, J.-X. Shao, X.-X.-X. Jiang, C. Feng, H.-P. Yang and C.-X. He, Amorphous NiFe oxide-based nanoreactors for efficient electrocatalytic water oxidation, Angew. Chem., Int. Ed., 2023, 62, e202300478 CrossRef CAS PubMed
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y. R. Lu, T. S. Chan, F. M. F. de Groot and Y. Tan, Single platinum atoms embedded in nanoporous cobalt selenide as electrocatalyst for accelerating hydrogen evolution reaction, Nat. Commun., 2019, 10, 1743 CrossRef PubMed
- Q. Hu, K.-R. Gao, X.-D. Wang, H.-G. Zheng, J.-Y. Cao, L.-R. Mi, Q.-H. Huo, H.-P. Yang, J.-H. Liu and C.-X. He, Subnanometric Ru clusters with upshifted D band center improve performance for alkaline hydrogen evolution reaction, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed
- F. Kong, X. Liu, Y. Song, Z. Qian, J. Li, L. Zhang, G. Yin, J. Wang, D. Su and X. Sun, Selectively Coupling Ru Single Atoms to PtNi Concavities for High-Performance Methanol Oxidation via d-Band Center Regulation, Angew. Chem., Int. Ed., 2022, 61, e202207524 CrossRef CAS PubMed
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Regulating the Coordination Environment of MOF-Templated Single-Atom Nickel Electrocatalysts for Boosting CO2 Reduction, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed
- R. Qin, K. Liu, Q. Wu and N. Zheng, Surface Coordination Chemistry of Atomically Dispersed Metal Catalysts, Chem. Rev., 2020, 120, 11810–11899 CrossRef CAS PubMed
- C. Tang, Y. Jiao, B. Shi, J. N. Liu, Z. Xie, X. Chen, Q. Zhang and S. Z. Qiao, Coordination Tunes Selectivity: Two-Electron Oxygen Reduction on High-Loading Molybdenum Single-Atom Catalysts, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed
- Q. Hu, S. Qi, Q.-H. Huo, Y.-X. Zhao, J.-J. Sun, X.-B. Chen, M.-Y. Lv, W.-L. Zhou, C. Feng, X.-Y. Chai, H.-P. Yang and C.-X. He, Designing efficient nitrate reduction electrocatalysts by identifying and optimizing active sites of Co-based spinels, J. Am. Chem. Soc., 2024, 146, 2967–2976 CrossRef CAS PubMed
- Z.-Y. He, J. Zhang, Z.-L. Gong, H. Lei, D. Zhou, N. Zhang, W.-J. Mai, S.-J. Zhao and Y. Chen, Activating lattice oxygen in NiFe-based (oxy) hydroxide for water electrolysis, Nat. Commun., 2022, 13, 2191 CrossRef CAS PubMed
- Y. T. Kim, K. Ohshima, K. Higashimine, T. Uruga, M. Takata, H. Suematsu and T. Mitani, Fine size control of platinum on carbon nanotubes: from single atoms to clusters, Angew. Chem., Int. Ed., 2006, 45, 407–411 CrossRef CAS PubMed
- Y. Ding, B. Q. Miao, Y. C. Liu, N. Hou, Y. Yang, P. J. Jin, S. B. Yin and Y. Chen, Rhodium-Cobalt Alloy Nanotubes Toward Methanol Oxidation Reaction, Small Struct., 2022, 3, 2200046 CrossRef CAS
- J. Q. Zhang, Y. F. Zhao, X. Guo, C. Chen, C. L. Dong, R. S. Liu, C. P. Han, Y. D. Li, Y. Gogotsi and G. X. Wang, Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction, Nat. Catal., 2018, 1, 985–992 CrossRef CAS
- D. B. Liu, X. Y. Li, S. M. Chen, H. Yang, C. D. Wang, C. Q. Wu, Y. A. Haleem, S. Duan, J. L. Lu, B. H. Ge, P. M. Ajayan, Y. Luo, J. Jiang and L. Song, Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution, Nat. Energy, 2019, 4, 512–518 CrossRef CAS
- L. Hui, Y. R. Xue, C. Y. Xing, Y. X. Liu, Y. C. Du, Y. Fang, H. D. Yu, C. Zhang, F. He and Y. L. Li, Atomic alloys of nickel-platinum on carbon network for methanol oxidation, Nano Energy, 2022, 95, 106984 CrossRef CAS
- C. Lin, Y. Zhao, H. Zhang, S. Xie, Y. F. Li, X. Li, Z. Jiang and Z. P. Liu, Accelerated active phase transformation of NiO powered by Pt single atoms for enhanced oxygen evolution reaction, Chem. Sci., 2018, 9, 6803–6812 RSC
- Y. T. Kim, K. Ohshima, K. Higashimine, T. Uruga, M. Takata, H. Suematsu and T. Mitani, Fine size control of platinum on carbon nanotubes: from single atoms to clusters, Angew. Chem., Int. Ed., 2006, 45, 407–411 CrossRef CAS PubMed
- B. D. McNicol, Electrocatalytic problems associated with the development of direct methanol-air fuel cells, J. Electroanal. Chem. Interfacial Electrochem., 1981, 118, 71–87 CrossRef CAS
- K. L. Zhou, C. Wang, Z. Wang, C. B. Han, Q. Zhang, X. Ke, J. Liu and H. Wang, Seamlessly conductive Co(OH)2 tailored atomically dispersed Pt electrocatalyst with a hierarchical nanostructure for an efficient hydrogen evolution reaction, Energy Environ. Sci., 2020, 13, 3082–3092 RSC
- A. Romanchenko, M. Likhatski and Y. Mikhlin, X-ray Photoelectron Spectroscopy (XPS) Study of the Products Formed on Sulfide Minerals Upon the Interaction with Aqueous Platinum (IV) Chloride Complexes, Minerals, 2018, 8, 578 CrossRef CAS
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Dynamic oxygen adsorption on single-atomic Ruthenium catalyst with high performance for acidic oxygen evolution reaction, Nat. Commun., 2019, 10, 4849 CrossRef PubMed
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu, G. A. Botton and X. Sun, Platinum single-atom and cluster catalysis of the hydrogen evolution reaction, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed
- S. Dai, J. P. Chou, K. W. Wang, Y. Y. Hsu, A. Hu, X. Pan and T. Y. Chen, Platinum-trimer decorated cobalt-palladium core-shell nanocatalyst with promising performance for oxygen reduction reaction, Nat. Commun., 2019, 10, 440 CrossRef CAS PubMed
- J. Zhang, T. Wang, P. Liu, Z. Liao, S. Liu, X. Zhuang, M. Chen, E. Zschech and X. Feng, Efficient hydrogen production on MoNi4 electrocatalysts with fast water dissociation kinetics, Nat. Commun., 2017, 8, 15437 CrossRef CAS PubMed
- R. L. Doyle and M. E. G. Lyons, Kinetics and Mechanistic Aspects of the Oxygen Evolution Reaction at Hydrous Iron Oxide Films in Base, J. Electrochem. Soc., 2013, 160, H142–H154 CrossRef CAS
- M. E. Lyons, R. L. Doyle and M. P. Brandon, Redox switching and oxygen evolution at oxidized metal and metal oxide electrodes: iron in base, Phys. Chem. Chem. Phys., 2011, 13, 21530–21551 RSC
- L. Zhao, Y. Zhang, Z.-L. Zhao, Q.-H. Zhang, L.-B. Huang, L. Gu, G. Lu, J.-S. Hu and L.-J. Wan, Steering
elementary steps towards efficient alkaline hydrogen evolution via size-dependent Ni/NiO nanoscale heterosurfaces, Natl. Sci. Rev., 2020, 7, 27–36 CrossRef CAS PubMed
- J. Al Boukhari, M. Noun and R. Awad, Raman spectroscopic investigations of pure, (Mg, Cu), and (Mg, Ru) codoped NiO nanoparticles, Chem. Phys. Lett., 2024, 836, 141038 CrossRef CAS
- M. Liu, K.-A. Min, B. Han and L. Y. S. Lee, Interfacing or doping? Role of Ce in highly promoted water oxidation of NiFe-layered double hydroxide, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS
- Z. Qiu, C.-W. Tai, G.-A. Niklasson and T. Edvinsson, Direct observation of active catalyst surface phases and the effect of dynamic self-optimization in NiFe-layered double hydroxides for alkaline water splitting, Energy Environ. Sci., 2019, 12, 572–581 RSC
- D.-J. Chen and Y.-J. Tong, Irrelevance of carbon monoxide poisoning in the methanol oxidation reaction on a PtRu electrocatalyst, Angew. Chem., Int. Ed., 2015, 54, 9394–9398 CrossRef CAS PubMed
- Q. Lu, X. Zhao, R. Luque and K. Eid, Structure-activity relationship of tri-metallic Pt-based nanocatalysts for methanol oxidation reaction, Coord. Chem. Rev., 2023, 493, 215280 CrossRef CAS
- O.-T. Ajenifujah, A. Nouralishahi, S. Carl, S.-C. Eady, Z. Jiang and L.-T. Thompson, Platinum supported on early transition metal carbides: efficient electrocatalysts for methanol electro-oxidation reaction in alkaline electrolyte, Chem. Eng. J., 2021, 406, 126670 CrossRef CAS
- Q. Zhang, J.-N. Lv, X.-Y. Hu, Y.-L. He, H.-F. Yang, D.-S. Kong and Y.-Y. Feng, Polyaniline decorated MoO3 nanorods: synthesis, characterization and promoting effect to Pt electrocatalyst, Int. J. Hydrogen Energy, 2018, 43, 5603–5609 CrossRef CAS
- C. Xu, R. Zeng, P.-K. Shen and Z. Wei, Synergistic effect of CeO2 modified Pt/C catalysts on the alcohols oxidation, Electrochim. Acta, 2005, 51, 1031–1035 CrossRef CAS
- L. N. Zhou, X. T. Zhang, Z. H. Wang, S. Guo and Y. J. Li, Cubic superstructures composed of PtPd alloy nanocubes and their enhanced electrocatalysis for methanol oxidation, Chem. Commun., 2016, 52, 12737–12740 RSC
- P.-K. Shen, C. Xu, R. Zeng and Y. Liu, Electro-oxidation of methanol on NiO-promoted Pt/C and Pd/C catalysts, Electrochem. Solid-State Lett., 2005, 9, A39 CrossRef
- K.-M. Yu, G.-Q. Ning, J.-T. Yang, Y. Wang, X. Zhang, Y.-C. Qin, C.-L. Luan, L. Yu, Y. Jiang, X.-B. Luan, Z. Dong, H. Wang and X.-P. Dai, Restructured PtNi on ultrathin nickel hydroxide for enhanced performance in hydrogen evolution and methanol oxidation, J. Catal., 2019, 375, 267–278 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information, DFT calculations, XRD and SEM data, HAADF-STEM image, SAED pattern, HRTEM image, mass-normalized CV curves for the MOR. See DOI: https://doi.org/10.1039/d4sc00454j |
| This journal is © The Royal Society of Chemistry 2024 |