
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelectively “size-excluding” water molecules to enable a highly reversible zinc metal anode†
Xiaowei
Shen‡
a,
Wanhao
Chen‡
bc,
Haocong
Wang
e,
Lifang
Zhang
b,
Baojiu
Hao
b,
Changhao
Zhu
b,
Xiuzhen
Yang
b,
Meizhu
Sun
b,
Jinqiu
Zhou
*b,
Xuejun
Liu
*c,
Chenglin
Yan
 de and
Tao
Qian
*b
de and
Tao
Qian
*b
aSchool of Electrical Engineering and Automation, Nantong University, Nantong 226019, China
bSchool of Chemistry and Chemical Engineering, Nantong University, Nantong 226019, China. E-mail: jqiuzhou@ntu.edu.cn; qiantao@ntu.edu.cn
cCollege of Chemistry and Chemical Engineering, State Key Laboratory of Bio-fibers and Eco-textiles, Qingdao University, Qingdao 266071, China. E-mail: xjliu@qdu.edu.cn
dSchool of Petrochemical Engineering, Changzhou University, Changzhou 213164, China
eKey Laboratory of Core Technology of High Specific Energy Battery and Key Materials for Petroleum and Chemical Industry, College of Energy, Soochow University, Suzhou 215006, China
First published on 17th May 2024
Abstract
Significant water-related side reactions hinder the development of highly safe, low-cost aqueous zinc metal batteries (AZMBs) for grid-scale energy storage. Herein, by regulating the length of alkyl chains, we successfully adjust interstitial voids between the polymer chains of a metal soap interface between 1.48 Å (size of a zinc ion) and 4.0 Å (size of a water molecule). Therefore, water molecules are selectively “size-excluded,” while smaller zinc ions are permitted to pass through. Consequently, water-related side reactions (including hydrogen evolution and corrosion) could be effectively inhibited. Furthermore, abundant zinc ion tunnels accompanied with zincophilic components facilitate the homogenization of the Zn2+ flux, thus preventing dendrite growth. Therefore, the Zn symmetric cell shows a lifespan of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 20 mA cm−2 and 1 mA h cm−2, and the Zn//Na5V12O32 (NVO) full cell delivers much better cycling stability with much higher capacity retention of around 93% after 2000 cycles at 2 A g−1 compared to its bare Zn counterpart (19%). This work provides valuable insights for the utilization of metal soap interfaces and regulation of their channel size between perpendicular alkyl chains to realize precise water shielding, which is not only applicable in ZMBs but also in other aqueous batteries.
000 cycles at 20 mA cm−2 and 1 mA h cm−2, and the Zn//Na5V12O32 (NVO) full cell delivers much better cycling stability with much higher capacity retention of around 93% after 2000 cycles at 2 A g−1 compared to its bare Zn counterpart (19%). This work provides valuable insights for the utilization of metal soap interfaces and regulation of their channel size between perpendicular alkyl chains to realize precise water shielding, which is not only applicable in ZMBs but also in other aqueous batteries.
Introduction
The use of water-based electrolytes can greatly improve safety, reduce costs, and enhance conductivity compared to most organic electrolytes.1–5 Rechargeable aqueous zinc metal batteries (AZMBs) with Zn2+ ions as charge carriers have garnered great attention for their potential to supplement lithium ion batteries (LIBs), owing to their large theoretical capacity of 820 mA h g−1, low equilibrium potential of −0.76 V vs. SHE and low cost of the Zn anode.6–11 However, the large-scale implementation of AZMBs still faces significant challenges due to the unique aqueous environment.12–14 Unlike the electrochemical process of LIB anodes in non-aqueous systems, water molecules with high polarity and a small radius (4 Å) can participate in solvation processes and thus cause corrosion and parasitic reactions on Zn anodes, which could exacerbate dendrite growth and electrolyte consumption, leading to reduced coulombic efficiency (CE) and shortened cycling lifespan.15–17Current works mainly focus on the “dendrite growth problem.” The optimization of anodes through artificial surface protective layers using various inorganic and organic materials is a promising research direction.18–20 To mitigate the issue of irregular Zn deposition caused by uneven ion concentration, artificial layers based on nanomaterials with selective pores, interlayer spacing, or ion channels (e.g., nano-CaCO3, metal–organic-framework-based materials, and kaolin) can be designed as Zn2+ flux regulators to induce uniform Zn deposition, enhance Zn2+ migration, and improve cycle stability.21–23 Moreover, the formation of a metal layer with Zn affinity on the electrode surface can enhance the zincophilic capacity, promote uniform zinc deposition, suppress the formation of zinc dendrites, and improve the cycling stability of zinc metal anodes.24–28 For example, Ho Seok Park's group has constructed in situ artificial interfacial layers (Sn, Sb, and Bi) with better Zn affinity via facile galvanic spontaneous replacement reactions to provide a stable interface for Zn deposition.29
Although various approaches aiming to protect zinc anodes by homogenizing Zn2+ flux have been proposed, they often fail to effectively prevent water-induced side reactions. The water-induced side reactions are equally significant, as they can result in notorious hydrogen evolution reactions and surface passivation at the unstable interface, thus exacerbating dendrite growth when the water-based electrolyte and zinc come into direct contact, because Zn2+/Zn has a lower redox potential than H+/H2 (0 V vs. SHE).30–32 Some hydrophobic material layers such as organic polymers (e.g., PVB33 and PAN34) are used to impede H2O molecules, but the physical barrier of these hydrophobic organic polymers may reduce ionic conductivity, leading to increased polarization and nucleation overpotentials. Electrolyte additives have been widely utilized to lock free water via strong multiple H-bonds or to tightly adsorb on the Zn surface to form a dense and robust interfacial complex layer, thus suppressing the activity of H2O.35,36In situ-formed robust solid electrolyte interphases derived from different well-designed electrolytes have also been reported to inhibit corrosion and the HER, as well as to enhance the reversible deposition of Zn.37,38
So far, however, artificial interfacial layers that can shield against the entry of water molecules but facilitate the deposition of Zn ions at the molecular level have not been reported. Therefore, it is urgent to regulate the molecular size to simultaneously realize precise shielding against the entry of water molecules and permit of Zn ion transport at the molecular level.39 Metal soaps, which are mainly composed of inorganic bonding between metal cations and the acid group of a hydrocarbon chain, are well-known due to their rich polymorphism and polymesomorphism. The corrosion of Cu, Fe, Zn, Pb and Mg in aqueous solutions could be inhibited by protection with a metallic soap film, and it has been concluded that the longer the carbon chain length, the more corrosion-resistant the coating.39 As early as the 1960s, Luzzati et al. correlated lattice parameters with the length of the hydrocarbon chain.40 The metal cations and functional groups from the organic part form an inorganic backbone, while long alkyl chains attached to the backbone form a lamellar structure with molecular-sized channels. K. V. Agrawal et al. confirmed that crystalline metal soap films are oriented with alkyl chains perpendicular to the porous substrate, and that different gap sizes between alkyl/alkyl groups or metal ion/carboxylate groups can favor the separation of gas molecules.41 This presents an opportunity to selectively “size-exclude” water molecules by adjusting channel gap sizes to allow precise shielding against the entry of water molecules at the molecular level. To the best of our knowledge, the application of metal soaps in metal anode batteries has not been reported.
Herein, we for the first time realize selective “size-exclusion” of water molecules while facilitating efficient transport of the zinc ions through precisely controlling the channel dimensions of a metal soap organic interface by adjusting the length of the alkyl chain.41 Specifically, by utilizing perpendicular decyl group chains, we have engineered an optimum channel size of approximately 3.40 Å, which results in a relatively high capability to selectively inhibit the ingress of desolvated water at the interface and facilitate the movement of the zinc ions, thus inhibiting hydrogen generation caused by water entry. Furthermore, the metal soap membrane, with abundant nanopores acting as ion tunnels, facilitates the uniform deposition of the zinc ions by homogenizing the Zn2+ ion flux, preventing dendrite formation and enabling long cycling stability in ZMBs. As anticipated, taking advantage of these synergistic effects, the anode can be stably cycled for more than 10000 cycles at 20 mA cm−2 and even 5000 cycles at a higher current density 50 mA cm−2. The assembled full cells exhibit a high capacity retention of 93% over 2000 cycles at 2 A g−1, demonstrating competitive advantages over general AZMBs. This innovative design holds significant potential in overcoming aforementioned challenges and contributes to the advancement of interface engineering in this field.
Results and discussion
Ingenious channel size regulation of metal soap interface to selectively shield water molecules
While the issue of dendrite formation has been prominently discussed, the impact of water-induced erosion is equally significant and is often overlooked in the performance evaluation of Zn anodes. During extended cycling, the unavoidable occurrence of water-induced H2 evolution leads to corrosion of the Zn surface and formation of hydroxide-based by-products. To achieve the precise exclusion of water molecules, we successfully synthesized a metal soap interface with an adjusted channel size between the perpendicular alkyl chains to realize precise water shielding. Three metal soaps, Cu(COOC7H15)2 (CuC7), Cu(COOC9H19)2 (CuC9) and Cu(COOC15H31)2 (CuC15) were prepared. Rietveld refinement of the X-ray powder diffraction (XRD) analysis was performed to investigate the crystal structures and lattice parameters of synthesized metal soaps CuCn, from which their channel sizes can be obtained (Fig. 1a–f and Table S1†). Clearly, the channel formed between the octyl group interstices of CuC7 is measured to be approximately 4.20 Å, which is larger than the size of the water molecule; thus, it is unable to inhibit the entry of water. The channels for CuC9 and CuC15 are measured to be about 3.40 Å and 2.95 Å, respectively, which are smaller than a water molecule and larger than a Zn2+ ion, making them capable of inhibiting the entry of water while allowing Zn2+ transfer. Obviously, the determining factor affecting channel size is the length of the alkyl chain. It can be seen from the lattice parameters that the shorter the carbon chains, the larger the channel size.42 In addition, from the perspective of polymer physics, longer alkyl chains with more flexibility have more conformations and may undergo bending, which would further lead to a shrunken channel size.43,44 To confirm the water shielding function, density functional theory (DFT) calculations were conducted (Fig. S1, S3† and 1g). The energy difference for introducing the water molecule (ΔEH2O) into the channel is positive, indicating that the channel rejects the entry of water molecules. In addition, the negative energy difference for the introduction of the zinc ions (ΔEZn2+) into the channels of CuC7 and CuC9 implies the unimpeded Zn2+ transfer. However, as the chain length increases, the channel size would decrease, and the values of energy differences for water and Zn2+ would both increase, which would indicate enhanced water shielding function and weakened Zn2+ transfer. After channel size modulation using three different alkyl chains, Zn//Zn symmetric cells with different metal soap membranes were further tested (Fig. 1h). Water-related side reactions occur on Zn@CuC7 because CuC7 cannot prevent water molecules from coming into contact with the zinc metal, and thus, the Zn@CuC7 symmetric cell exhibits a fluctuating polarization voltage profile. However, CuC9 and CuC15 with channel sizes smaller than 4.0 Å can effectively exclude water molecules and show excellent stability. Due to the smaller channel size of CuC15 compared to that of CuC9, the Zn@CuC15//Zn@CuC15 cell exhibits a larger overpotential than Zn@CuC9. Therefore, CuC9 was selected as the optimum metal soap membrane with relatively high capability to selectively inhibit the ingress of desolvated water and facilitate the movement of the zinc ions at the interface. Furthermore, the size characterization of the CuC9 metal soap was conducted via gas adsorption experiments including water vapor and N2 (Fig. S4†). However, it shows negligible uptake of water vapor and N2, which is consistent with the smaller pore size (3.40 Å) of CuC9 compared to the kinetic diameters of water vapor (4 Å) and N2 (3.64 Å).41,45 The above results demonstrate that the channel dimensions of metal soap can be regulated and “precise” shielding water can be modulated (Fig. 1i). Without the ingenious design of the channel size of the metal soap interface, the undesirable HER process and inhomogeneous Zn ion flux would result in an irreversible Zn anode (Fig. 1i, left), which ultimately leads to various battery failures, including short circuits, isolated zinc particles, electrode passivation, and battery swelling caused by gas formation. As shown in the schematic representation of the Zn anode with a metal soap membrane (Fig. 1i, right), the metal cations and functional groups generate a backbone, with the long alkyl chains forming a lamellar structure. The size-regulated channel formed between the decyl group interstices can selectively inhibit the ingress of desolvated water (size 4 Å) at the interface and facilitate the uniform deposition of the zinc ions (size 1.48 Å) under an applied electric field, thus contributing greatly to the suppressed HER and uniform Zn deposition.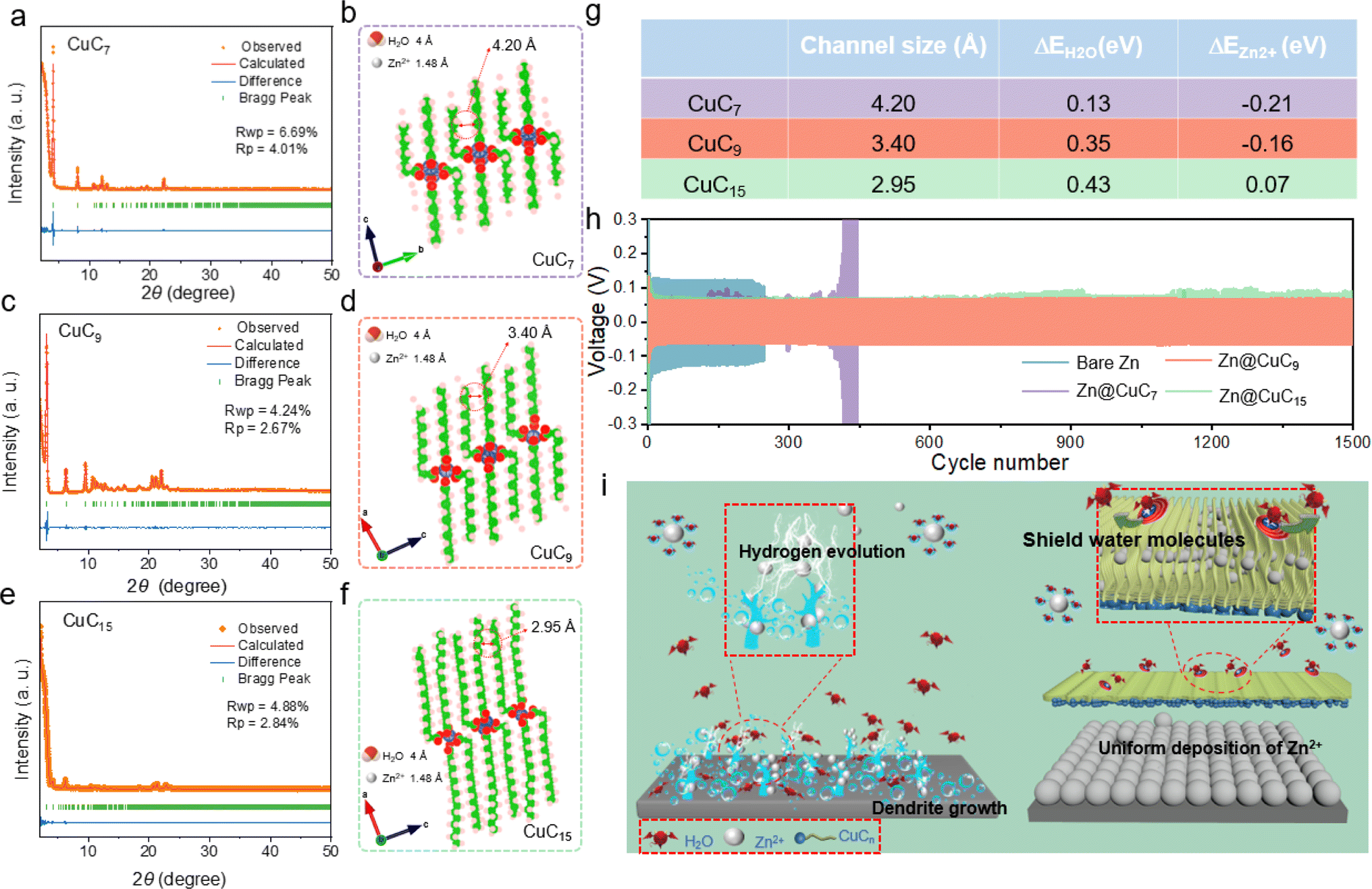 | ||
| Fig. 1 Rietveld refinement XRD analysis and crystal structure for (a and b) CuC7, (c and d) CuC9 and (e and f) CuC15. (g) Channel sizes and energy differences of insertion of a water molecule and zinc ion into the channel of CuC7, CuC9 and CuC15. (h) Symmetric half-cells of Zn@CuC7, Zn@CuC9 and Zn@CuC15 at 20 mA cm−2/1 mA h cm−2. (i) Schematic depiction of Zn deposition behavior on bare Zn and the Zn@CuC9 anodes. | ||
With the existence of the CuC9 film, the ionic conductivity is measured to be 10.7 mS cm−1, indicating that the added Cu soap film would not impede Zn2+ conduction. The relationship between the thickness of the CuC9 film and electrochemical cycling performance was estimated (Fig. S5†). With greater thickness, the water-shielding effect would be better. However, thicker film would increase the ion transfer pathway length and inevitably affect the rapid ion migration. As shown in Fig. S5,† a thickness of ∼10 μm is optimum and was used to conduct all the following tests. The uniformity of the distribution of the CuC9 metal soap membrane on the zinc surface was investigated, as it is vital to the modification effect (Fig. S6†). Detailed SEM scrutiny of the Zn@CuC9 sample reveals a comparatively flat metal soap membrane on the surface, displaying minimal irregularities and a thickness of 12.36 μm (Fig. S7a and b†). The EDS mapping in Fig. S7c† unequivocally portrays the uniform dispersion of the elements of Cu and C on the surface of the zinc plate.
Inhibition of hydrogen precipitation
Contact angle tests of 2 M ZnSO4 electrolyte on different anodes were conducted. On bare Zn foil, measurement reveals a contact angle of 87.7°, indicating a considerable degree of wettability of the electrolyte on the bare Zn foil (Fig. 2a). The Zn@CuC9 anode, in contrast, displays poor wettability and a high contact angle of 135.5°, which suggests apparent hydrophobic characteristics. For the HER reaction (2H2O + 2e− → H2↑ + 2OH−) to occur, it is a necessary condition that water molecules contact the conducting current collector to gain electrons. The resistances of Zn and Zn@CuC9 were measured to be 3.5 × 10−4 and 1.6 × 104 ohm, respectively, using a four-probe resistance experiment. It can be seen that the metal soap membrane is not only hydrophobic but also electronically insulating. Therefore, the CuC9 layer can not only efficiently separate the Zn metal from the bulk liquid electrolyte but can also block electron transfer from the Zn metal to water, thus restraining the electrochemical water decomposition (HER reactions) on the Zn@CuC9 electrode.46,47 We further conducted in situ optical microscopy to observe the Zn deposition process at various time intervals. As depicted in Fig. 2b, some bubbles are generated at 30 min on the surface of bare Zn. It is obvious that the interface is not hydrophobic; thus, the direct contact between zinc and water leads to the occurrence of the HER, which generates a large amount of hydrogen gas.48–50 Almost no bubble generation is observed on the Zn@CuC9 electrode from 0 to 30 minutes. The presence of the metal soap membrane, with its highly hydrophobic organic groups, prevents water molecules from easily coming into contact with zinc, ultimately inhibiting the HER. Subsequently, we applied linear scanning voltammetry (LSV) to examine the ability of the Zn@CuC9 electrode to restrict the undesired HER at a scan speed of 1 mV s−1 with 2 M Na2SO4 as the electrolyte. As obviously shown in Fig. 2c, the HER curves indicate that the current density of Zn@CuC9 (0.024 mA cm−2) is less than that of the bare Zn anode (0.049 mA cm−2). Based on LSV results, it can be inferred that CuC9 could significantly inhibit harmful interfacial parasitic reactions because it can efficiently block the entry of water molecules onto the zinc metal surface. | ||
| Fig. 2 (a) Electrolyte contact angles on bare Zn and the Zn@CuC9 electrode. (b) In situ optical observations of Zn deposition on bare Zn and Zn@CuC9. (c) Linear polarization curves in 2 M aqueous Na2SO4 electrolyte. (d) Schematic of the sealed bottle and (e) time–voltage curves of in situ EC-GC measurements. (f) Corresponding H2 production rates. | ||
In addition, in situ electrochemical gas chromatography (EC-GC) was used to detect the evolution of hydrogen during galvanization. A sealed electrolytic cell consisting of two electrodes (Zn or Zn@CuC9 foil as cathode and anode), an inlet tube (N2 inflow) and an outlet tube (hydrogen outflow to the GC) was designed to perform in situ EC-GC measurements (Fig. 2d). At a current of 2.5 mA cm−2, GC analysis of the hydrogen produced by the Zn plating process was performed at six-minute intervals (Fig. 2e). Using EC-GC profiles, the H2 production rate is calculated (Table S2†). As shown in Fig. 2f, the H2 generation average rate of the cell using bare Zn is 1.61 μg h−1, whereas the H2 generation rate of the system using Zn@CuC9 foil is only 0.11 μg h−1, which is 14.6 times higher than that of the latter. In situ EC-GC results further confirm the ability of the CuC9 metal soap interface to inhibit hydrogen generation.
Anti-corrosion effect
Subsequently, both anodes were immersed in a 2 M ZnSO4 aqueous electrolyte to evaluate their respective self-corrosion behaviors. Prior to immersion, the bare Zn surface exhibits a smooth texture with a brilliant metallic sheen, whereas the Zn@CuC9 electrode is coated with a blue layer. Following a 15 day immersion period, the bare Zn surface becomes notably rough, with the formation of surface accumulations identified as uneven flakes through SEM imaging; in contrast, no visible changes are observed on the surface of the Zn@CuC9 electrode (Fig. 3a). The chemical content of the byproduct on the electrode surface was identified using X-ray diffraction (XRD) patterns and was found to mostly be Zn4SO4(OH)6·3H2O. The intensity of the byproduct on the bare Zn electrode is significantly higher than that on the Zn@CuC9 electrode, indicating the particularly strong anti-corrosion effect of the CuC9 overlayer (Fig. 3b). Furthermore, the Tafel curves of bare Zn and Zn@CuC9 anodes were tested using three electrodes (Fig. 3c). As observed, the incorporation of CuC9 increases corrosion potentials in comparison to bare Zn, and the more positive corrosion potential indicates reduced tendency toward the corrosion reaction. Fig. 3d shows Nyquist plots with a semicircle in the high-frequency region corresponding to the charge transfer resistance (Rct). The EIS plots of the bare Zn symmetric cell clearly demonstrate a significant change in Rct before and after different cycles, while the Rct of the Zn@CuC9 symmetric cell in Fig. 3e remains small and more stable. Here, the possibility of short circuit is excluded. It can clearly be observed that enlarged voltage curves illustrated in Fig. S9a† for the symmetric cell always appear as slope curves, reaffirming that the cell is not in a short-circuited state. Furthermore, it should be determined whether a soft short occurs in the cycling.51 Therefore, Zn stripping/plating measurements of the cycled Zn//Zn cell were conducted at different low temperatures. The voltage profiles demonstrate enlarged voltage hysteresis at lower temperatures (Fig. S9a and b†), whereas a soft-short-circuited cell delivers stable or slightly dwindling profiles upon temperature decrease. The derived activation energy (Ea), which serves as a more direct indicator, was calculated to have a positive value (Fig. S9c†). These several pertinent test results eliminate the occurrence of soft shorts in our work. Collectively, these results offer compelling evidence for the exceptional capacity of the Zn@CuC9 anodes to suppress side reactions and resist corrosion in aqueous electrolytes.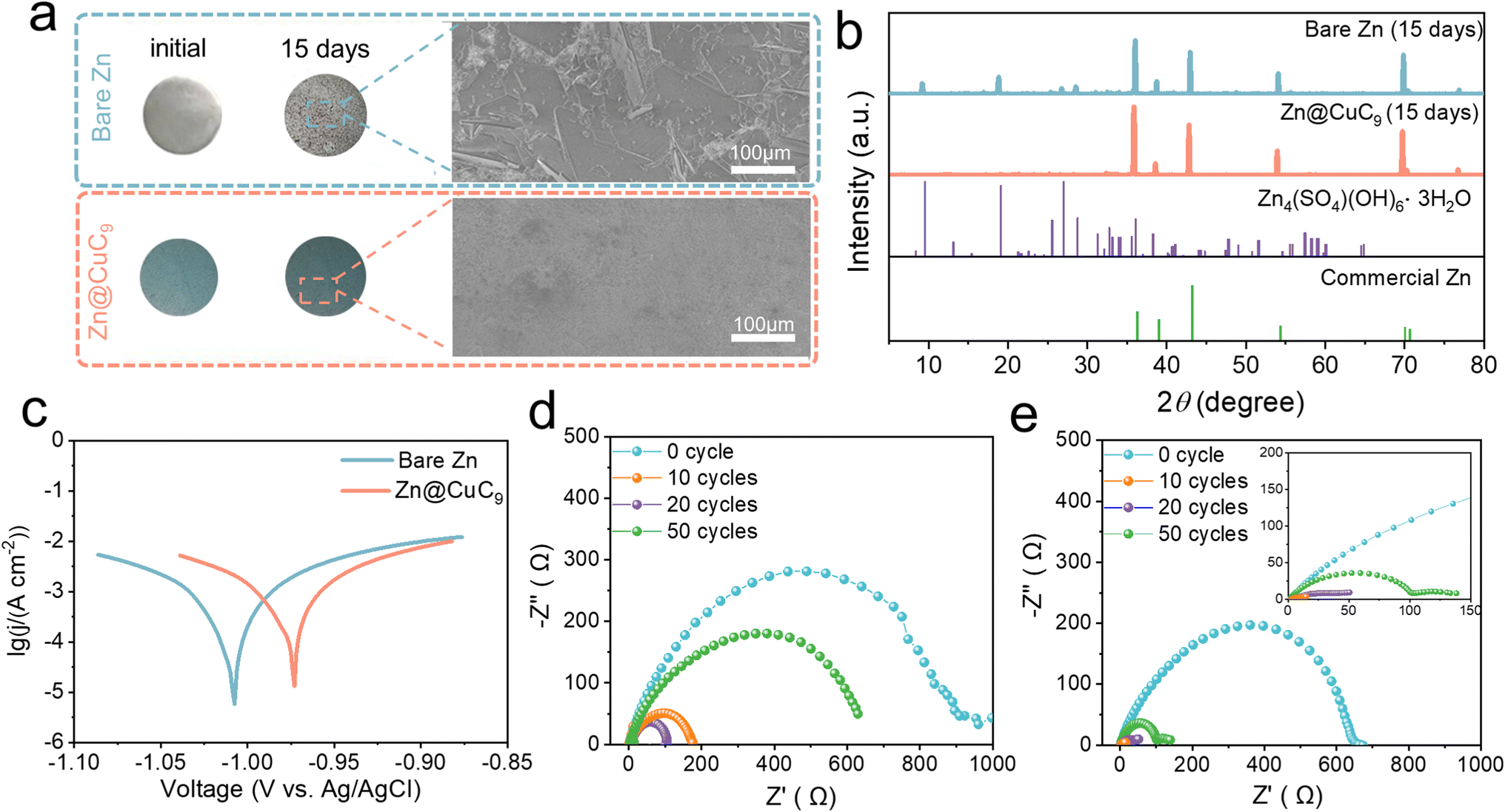 | ||
| Fig. 3 (a) Optical images of bare Zn and the Zn@CuC9 electrode before and after soaking in 2 M ZnSO4 electrolyte for 15 days, along with related SEM pictures. (b) XRD patterns of bare Zn and the Zn@CuC9 electrode after soaking for 15 days. (c) Tafel curves in 2 M ZnSO4 electrolyte with different anodes. Nyquist plots for (d) bare Zn and (e) Zn@CuC9 symmetric cells at various cycles. | ||
Stable Zn plating/stripping behavior
This metal soap membrane CuC9 was successfully synthesized and was not only highly hydrophobic but also possessed excellent ability to inhibit zinc dendrites. The coulombic efficiency (CE) is a critical metric for evaluating the electrochemical reversibility of AZMBs during repeated charge/discharge cycles. As shown in Fig. 4a, at 20 mA cm−2 for 1 mA h cm−2, the half-cell of bare Zn has a shortened cycle life, which suggests that the electrochemical reversibility of the bare Zn anode is unsatisfactory. In contrast, the half-cell of Zn@CuC9 shows remarkable durability and consistently retains a high CE (∼99.7%), highlighting its improved reversibility. The corresponding voltage profile analysis demonstrates a lower nucleation overpotential of 45.7 mV for the Zn@CuC9 electrode compared to the bare Zn electrode (82.3 mV) (Fig. 4b), indicating a reduced nucleation barrier. The channel size of the metal soap interface is precisely regulated between 1.48 Å (size of Zn ion) and 4.0 Å (size of water molecule), thus it can realize selective inhibition of water molecules while allowing the zinc ions to traverse freely in and out. Moreover, these channels provide a heightened number of uniform nucleation sites, and –COO– and Cu2+ have been proven to interact with Zn2+, assisting in accelerated, smooth and dense Zn2+ deposition.52,53 These all play a pivotal role in mitigating nucleation resistance during Zn2+ deposition, consequently lowering the nucleation overpotential of the zinc ions in Zn@CuC9 compared to bare Zn. Additionally, the Zn//Cu half-cell of bare Zn still exhibits superior cycling performance to improve the CE with a capacity of 50 mA cm−2 at 1 mA h cm−2 (Fig. S10†). To demonstrate the kinetic acceleration, we performed activation energy (Ea) testing of the Zn//Zn symmetric cells to examine the energy required for the interfacial redox reaction. The Rct of bare Zn and the Zn@CuC9 symmetric cells were tested at different temperatures from 0–60 °C, and Ea was calculated to be 15.07 kJ mol−1 for Zn@CuC9 and 30.93 kJ mol−1 for bare Zn using the Arrhenius equation (Fig. S11†). Chronoamperometry (CA) measurement is also conducted on the Zn//Zn symmetric cells with and without the CuC9 film (Fig. S12†). The linear current increase curve of bare Zn shows a 2D diffusion process and inhomogeneous growth of dendrites due to the “tip effect.” In contrast, the 2D diffusion process ends within 20 s for Zn@CuC9, and then, a stable 3D diffusion occurs in the subsequent process with a much lower steady-state current.54 The enhanced Zn2+ reaction kinetics and remarkable corrosion resistance of the Zn@CuC9 anode would contribute to the highly reversible Zn plating/stripping.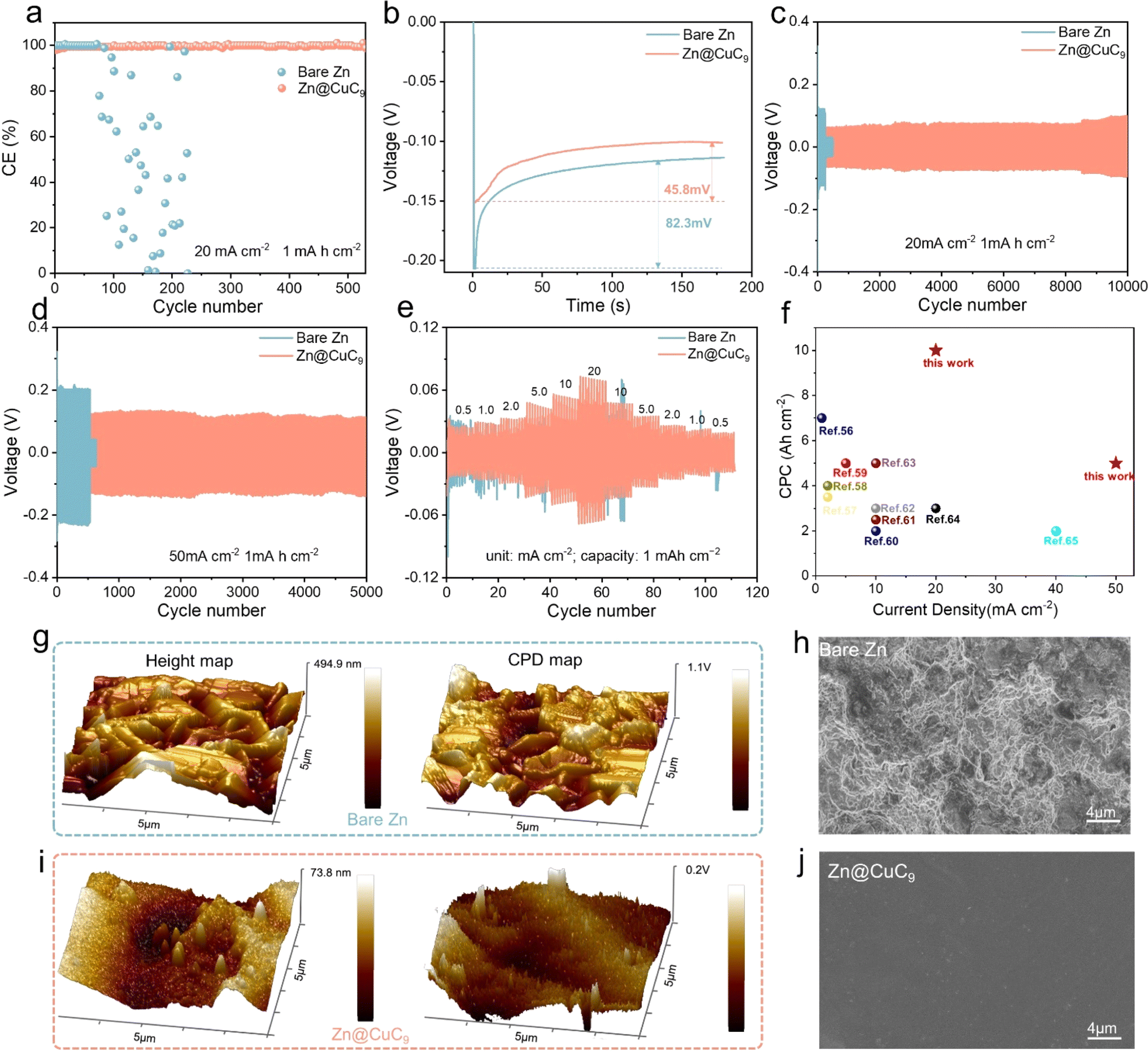 | ||
| Fig. 4 (a) Asymmetric Zn//Cu half-cells with and without the CuC9 interface. (b) Nucleation overpotentials of bare Zn and Zn@CuC9 electrodes at 20 mA cm−2. Symmetric Zn//Zn half-cells at (c) 20 mA cm−2/1 mA h cm−2 and (d) 50 mA cm−2/1 mA h cm−2. (e) Rate capability of symmetrical cells assembled using bare Zn and Zn@CuC9 anode. (f) CPC comparison of the Zn symmetric cell using a Zn@CuC9 electrode with other reported literature studies. AFM and KPFM images of (g) bare Zn and (i) Zn@CuC9 anode surfaces after the 50th cycle at 20 mA cm−2 with 1 mA h cm−2. SEM of cycled (h) bare Zn and (j) Zn@CuC9. | ||
We gradually increased the current density from 20 to 100 mA cm−2 to compare the cycling stability and polarization of the Zn//Zn and Zn@CuC9//Zn@CuC9 cells. Initially, under a current density of 20 mA cm−2 and cutoff capacity of 1 mA h cm−2 (Fig. 4c), the bare Zn symmetrical cell shows a larger polarization of approximately 155 mV. However, a smaller and stable polarization of about 60 mV is observed in the voltage profile of the Zn@CuC9//Zn@CuC9 symmetrical cell for continuous cycling of around 10000 cycles. Additionally, when the current density was increased to 50 and 100 mA cm−2 (Fig. 4d and S13†), the Zn@CuC9//Zn@CuC9 cells still show excellent cycling endurance compared to the bare Zn anode. The cycling performance of symmetric cells is also tested at a lower current density of 1 mA cm−2 (Fig. S14†). Apparently, Zn@CuC9 could remain stable for 1400 cycles, while the bare Zn cell deteriorates quickly. These results confirm the superior electrochemical reversibility of the well-designed Zn@CuC9 anode, highlighting its remarkable competitiveness. In addition, we evaluated the rate capability of symmetrical cells with a fixed plating/stripping capacity (1 mA h cm−2) under different current densities (0.5, 1, 2, 5, 10 and 20 mA cm−2). The symmetrical cell of Zn@CuC9 presents a smaller voltage hysteresis and better cycling stability, proving that the metal soap membrane is key to the stability of Zn metal anodes (Fig. 4e). For comparison, we summarize the cumulative plated capacity (CPC)55 and current density of recently reported aqueous Zn//Zn symmetric cells with a similar interfacial modification mechanism.56–65 As shown in Fig. 4f, the battery using Zn@CuC9 achieved an ultrahigh CPC of 10 A h cm−2 at 20 mA cm−2 and 5 A h cm−2 at 50 mA cm−2, which were comparable or even superior to most reported CPC and current density values. Furthermore, a Zn//Zn symmetric cell with a 50% Zn utilization rate was tested using ultrathin Zn foil (thickness of 0.01 mm). As remarkably illustrated in Fig. S15,† with the incorporation of CuC9, the symmetric cell exhibits stable cycling for a remarkable 300 cycles, showcasing the effectiveness of our design. Conversely, for the Zn anode lacking CuC9, the symmetric cell suffers breakdown after only 23 cycles, clearly highlighting the significant impact of CuC9.
To study the interfacial stability after long-term cycling, atomic force microscopy (AFM) and kelvin probe force microscopy (KPFM) were conducted to affirm the distinct advantage of incorporating CuC9 in achieving a stable Zn anode. As presented in Fig. 4g, the AFM analysis reveals an average height difference of 494.9 nm on the bare Zn anode surface and a large surface potential difference of 1.1 V as measured using KPFM, which is in agreement with the fractured and irregular surface in Fig. 4h. In contrast, the Zn@CuC9 anode exhibits a notably smoother surface, showcasing a smaller average height difference (73.8 nm) and a lower surface potential difference (0.2 V) (Fig. 4i). As shown in the top-view (Fig. 4j) and cross-sectional SEM (Fig. S16a†), the metallic soap membrane after cycling remains stable, similar to the morphology before cycling. The EDS mappings in Fig. S16b† unequivocally portray the uniform dispersion of Cu and C on the surface of the cycled zinc plate surface. The XRD spectra of the Zn@CuC9 anodes before and after cycling reveal no change, indicating that the crystal structure has not changed and further proving that the channel remains stable (Fig. S17†). These outcomes unequivocally demonstrate that the Zn@CuC9 anode significantly contributes to achieving uniform Zn deposition and inhibiting dendrite growth.64
Electrochemical performance of the full batteries
To demonstrate the practical applicability, full cells incorporating a Na5V12O32 (NVO) cathode were assembled (Fig. S18†).66 As shown in Fig. 5a, the Zn@CuC9-based full cell exhibits a smaller voltage gap in the cyclic voltammetry (CV) curve compared to bare Zn anode. Furthermore, for EIS spectra, compared to the bare Zn//NVO cell (Fig. 5b), the Zn@CuC9//NVO cell exhibits smaller semicircles in the high-frequency region with faster reaction kinetics (Fig. 5c). Fig. 5d illustrates the rate performance of the Zn//NVO cell at different current densities ranging from 0.1 to 5.0 A g−1. The Zn@CuC9//NVO cell exhibits significantly higher discharge capacity at various current densities. The improved rate performance in the Zn@CuC9 anode may be attributed to the improved charge transfer kinetics of the Zn anode (e.g., decreased activation energy of the zinc deposition process).67,68 In addition, the anti-self-discharge ability of Zn@CuC9 is examined (good corrosion resistance).69 As shown in Fig. 5e and f, after resting for 24 h in a fully charged state, the Zn@CuC9//NVO cell still maintains a high capacity retention of 97.8%, which is much better than that of the bare Zn anode (87.3%), confirming the enhanced anti-self-discharge ability of the Zn@CuC9 anode. The Zn@CuC9//NVO cell demonstrates significantly superior capacity retention and longer lifespan compared to the bare Zn//NVO full cell at a low current density of 0.5 A g−1 (Fig. S19†). Subsequently, the long-term cycling performance of both the cells was further examined at a higher current density of 2 A g−1 (Fig. 5g). The Zn@CuC9//NVO cell demonstrates significantly superior capacity retention and longer lifespan compared to the Zn//NVO full cell, maintaining up to 93% capacity and a stable CE close to 100% over 2000 cycles. In contrast, the bare Zn//NVO full cell exhibits rapid capacity degradation after approximately 500 cycles. CuC9 would not directly influence the zinc deposition on the cathode, but instead help to prolong the cycling performance of the cathode due to its positive function on the anode side. The metal soap membrane with a precisely regulated channel size can help to stabilize the anode interface and achieve uniform zinc deposition by impeding water molecules and facilitating zinc ion transport on the anode. As the battery is an integral structure composed of an anode and a cathode, a stable and homogeneous Zn2+ ion flux on the anode interface is also conducive to stabilization of the cathode by enabling uniform release and re-embedding of the zinc ions.70,71 In addition, deep cycling of the Zn//NVO full cell with a low capacity ratio between the anode and cathode (denoted as the N/P ratio) was conducted. As demonstrated in Fig. 5h, Zn//NVO with an N/P ratio of 5 (i.e., Zn utilization efficiency of 20%) exhibits excellent cycling stability over 1000 cycles, which could be ascribed to the highly reversible operation of Zn with the help of the CuC9 metal soap. Moreover, the Zn//NVO full cell could operate stably for 500 cycles with an N/P ratio of 1 (i.e., 100% Zn utilization) (Fig. 5i). Afterwards, we also analyzed the morphology variations of NVO cathodes cycled in Zn//NVO and Zn@CuC9//NVO. Compared to the initial morphology of the uncycled NVO (Fig. S20a†), the cycled NVO cathode with bare Zn exhibits cracks and breaks in the nanofiber morphology (Fig. S20b†). Conversely, the cathode morphology is well maintained in the Zn@CuC9//NVO cell (Fig. S20c†). These findings provide strong evidence of the feasibility of using the Zn@CuC9 anode as a substitute for bare zinc anodes, which offers improved electrochemical performance in terms of lifespan and capacity. | ||
| Fig. 5 (a) Cyclic voltammetry curves of Zn//NVO with a 1 mV s−1 scan rate. EIS plots of (b) bare Zn//NVO and (c) Zn@CuC9//NVO. (d) Rate performances. Self-discharge test of (e) bare Zn//NVO and (f) Zn@CuC9//NVO cells. (g) Cycle endurance of Zn//NVO full cells based on Zn@CuC9 and bare Zn anodes. Cycling stability of Zn//NVO full cells at 2 A g−1 with (h) N/P ratio = 5 and (i) N/P ratio = 1. | ||
Conclusions
In summary, metal soap membranes with regulated channel sizes have been designed on a zinc metal surface using a rapid and straightforward saponification reaction strategy; the membranes feature unique and selectively permeable channels. The CuC9 metal soap membrane, which has the optimum channel size, exhibits relatively high capability to selectively shield against water molecules while facilitating efficient passage of the zinc ions. These advantages enable the suppression of water-induced corrosion in mild electrolyte and homogeneity of the Zn ion flux, synergistically contributing to stable and improved Zn plating/stripping behaviors. Therefore, the Zn@CuC9-based symmetric cell demonstrates an exceptionally long lifespan of over 10000 cycles, accompanied by a significantly lower polarization voltage at 20 mA cm−2. The assembled Zn@CuC9//NVO cell also exhibits much better cycling stability over 2000 cycles at 2 A g−1 than the bare Zn//NVO cell.
It should be noted that the selection of metal ion for metal soap molecules was based on its zincophilic capacity. As shown in the Zn–Cu binary phase diagram (Fig. S21a†), there is a distinct solubility region of Cu in Zn metal, indicating the high zincophilicity of Cu.72,73 Cu2+ has been proven to interact with Zn2+, assisting in accelerated, smooth and dense Zn2+ deposition.53 As control experiments, we selected three other kinds of soap molecules: Ca (in the same period as Cu), Ag (in the same main group with Cu) and Zn soaps. As evidenced in binary alloy phase diagrams (Fig. S21b and c†), Ag also has solubility in Zn metal, while Ca shows no solubility in Zn. The sequence of zincophilicity is CuC9 ≅ AgC9 > ZnC9 > CaC9. As shown in the performance comparison of corresponding symmetric cells (Fig. S22†), Zn@AgC9 demonstrates better cycling performance and lower overpotential than Zn@ZnC9 and Ca@CaC9. It could be concluded that Ag soap could contribute to homogeneous deposition of the zinc ions such as Cu soap. This study presents valuable insights for the utilization of metal soap interfaces and regulation for their channel size between the perpendicular alkyl chains to realize selective water-shielding characteristics, which could be utilized not only in ZMBs but also in other aqueous batteries. For future research, the functions of different metal cations (such as Mg2+ and Y3+) and acid groups (–COOH, –SO3H, and –SO4H) of hydrocarbon chains (alkyl, enyl, and aryl, with different carbon numbers) could be carefully considered.
Data availability
Data will be made available on request.Author contributions
Xiaowei Shen, Wanhao Chen: investigation, writing-original draft preparation, writing-review & editing; Haocong Wang, Lifang Zhang: theoretical calculations; Baojiu Hao, Changhao Zhu, Xiuzhen Yang, Meizhu Sun: formal analysis; Jinqiu Zhou: conceptualization, writing-review & editing, funding acquisition; Xuejun Liu, Chenglin Yan: supervision, funding acquisition; Tao Qian: supervision, writing-review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the National Natural Science Foundation of China (grant numbers 52302270, 52002195, 52071226, and U21A20332), Natural Science Foundation of Jiangsu Province (grant number BK20220061), and Science and Technology Project of Nantong (grant number JC12022047), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX24_1994).References
- W. Yang, Y. Yang, H. Yang and H. Zhou, ACS Energy Lett., 2022, 7, 2515–2530 CrossRef CAS.
- C. Zhu, J. Zhou, Z. Wang, Y. Zhou, X. He, X. Zhou, J. Liu, C. Yan and T. Qian, Chem. Eng. J., 2023, 454, 140413 CrossRef CAS.
- M. Luo, C. Wang, H. Lu, Y. Lu, B. B. Xu, W. Sun, H. Pan, M. Yan and Y. Jiang, Energy Storage Mater., 2021, 41, 515–521 CrossRef.
- J. Zhou, L. Zhang, M. Peng, X. Zhou, Y. Cao, J. Liu, X. Shen, C. Yan and T. Qian, Adv. Mater., 2022, 34, 2200131 CrossRef CAS PubMed.
- J. Zheng, D. C. Bock, T. Tang, Q. Zhao, J. Yin, K. R. Tallman, G. Wheeler, X. Liu, Y. Deng, S. Jin, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi and L. A. Archer, Nat. Energy, 2021, 6, 398–406 CrossRef CAS.
- G.-L. Liu, T. Zhang, X.-J. Li, R.-P. Cao, J.-K. Shen, D.-L. Guo, N.-T. Wu, W.-W. Yuan, A. Cao and X.-M. Liu, Rare Met., 2023, 42, 3729–3740 CrossRef CAS.
- S. Tan, Y. Jiang, S. Ni, H. Wang, F. Xiong, L. Cui, X. Pan, C. Tang, Y. Rong and Q. An, Natl. Sci. Rev., 2022, 9, nwac183 CrossRef CAS PubMed.
- X. Zhang, J. P. Hu, N. Fu, W. B. Zhou, B. Liu, Q. Deng and X. W. Wu, InfoMat, 2022, 4, e12306 CrossRef CAS.
- M. Yan, C. Xu, Y. Sun, H. Pan and H. Li, Nano Energy, 2021, 82, 105739 CrossRef CAS.
- H. Kim, G. Jeong, Y. U. Kim, J. H. Kim, C. M. Park and H. J. Sohn, Chem. Soc. Rev., 2013, 42, 9011–9034 RSC.
- D. Yuan, J. Zhao, H. Ren, Y. Chen, R. Chua, E. T. J. Jie, Y. Cai, E. Edison, W. Manalastas Jr, M. W. Wong and M. Srinivasan, Angew. Chem., Int. Ed., 2021, 60, 7213–7219 CrossRef CAS PubMed.
- M. Chen, S.-C. Zhang, Z.-G. Zou, S.-L. Zhong, W.-Q. Ling, J. Geng, F.-A. Liang, X.-X. Peng, Y. Gao and F.-G. Yu, Rare Met., 2023, 42, 2868–2905 CrossRef CAS.
- C. Li, X. Xie, H. Liu, P. Wang, C. Deng, B. Lu, J. Zhou and S. Liang, Natl. Sci. Rev., 2022, 9, nwab177 CrossRef CAS PubMed.
- M. Wu, G. Zhang, H. Yang, X. Liu, M. Dubois, M. A. Gauthier and S. Sun, InfoMat, 2022, 4, e12265 CrossRef CAS.
- H. Yu, D. Chen, T. Zhang, M. Fu, J. Cai, W. Wei, X. Ji, Y. Chen and L. Chen, Small Struct., 2022, 3, 2200143 CrossRef CAS.
- K. Guan, L. Tao, R. Yang, H. Zhang, N. Wang, H. Wan, J. Cui, J. Zhang, H. Wang and H. Wang, Adv. Energy Mater., 2022, 12, 2103557 CrossRef CAS.
- M. Li, Q. He, Z. Li, Q. Li, Y. Zhang, J. Meng, X. Liu, S. Li, B. Wu, L. Chen, Z. Liu, W. Luo, C. Han and L. Mai, Adv. Energy Mater., 2019, 9, 1901469 CrossRef.
- H. Zhang, Q. Liu, D. Zheng, F. Yang, X. Liu and X. Lu, Nat. Commun., 2021, 12, 14 CrossRef CAS PubMed.
- C. Mao, Y. Chang, X. Zhao, X. Dong, Y. Geng, N. Zhang, L. Dai, X. Wu, L. Wang and Z. He, J. Energy Chem., 2022, 75, 135–153 CrossRef CAS.
- C. Cao, W. Du, C. C. Li, M. Ye, Y. Zhang, Y. Tang and X. Liu, J. Mater. Chem., 2023, 11, 14345–14355 RSC.
- Y. Xu, Y. Zhang, Z. Guo, J. Ren, Y. Wang and H. Peng, Angew. Chem., Int. Ed., 2015, 54, 15390–15394 CrossRef CAS PubMed.
- Y. Chu, S. Zhang, S. Wu, Z. Hu, G. Cui and J. Luo, Energy Environ. Sci., 2021, 14, 3609–3620 RSC.
- X. Liu, F. Yang, W. Xu, Y. Zeng, J. He and X. Lu, Adv. Sci., 2020, 7, 2002173 CrossRef CAS PubMed.
- S. Li, J. Fu, G. Miao, S. Wang, W. Zhao, Z. Wu, Y. Zhang and X. Yang, Adv. Mater., 2021, 33, e2008424 CrossRef PubMed.
- J. Yang, R. Zhao, Y. Wang, Z. Hu, Y. Wang, A. Zhang, C. Wu and Y. Bai, Adv. Funct. Mater., 2023, 33, 2213530 Search PubMed.
- S. Xie, Y. Li and L. Dong, J. Energy Chem., 2023, 76, 32–40 CrossRef CAS.
- Y. Tian, Y. An, C. Liu, S. Xiong, J. Feng and Y. Qian, Energy Storage Mater., 2021, 41, 343–353 CrossRef.
- Y. Chai, X. Xie, Z. He, G. Guo, P. Wang, Z. Xing, B. Lu, S. Liang, Y. Tang and J. Zhou, Chem. Sci., 2022, 13, 11656–11665 RSC.
- P. Xiong, Y. Kang, H. Yuan, Q. Liu, S. H. Baek, J. M. Park, Q. Dou, X. Han, W.-S. Jang, S. J. Kwon, Y.-M. Kim, W. Li and H. S. Park, Appl. Phys. Rev., 2022, 9, 011401 CAS.
- M. Wen, G. Li, H. Liu, J. Chen, T. An and H. Yamashita, Environ. Sci.: Nano, 2019, 6, 1006–1025 RSC.
- L. Cao, D. Li, T. Deng, Q. Li and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 19292–19296 CrossRef CAS PubMed.
- W. Lu, C. Zhang, H. Zhang and X. Li, ACS Energy Lett., 2021, 6, 2765–2785 CrossRef CAS.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS.
- S. Zhou, Y. Wang, H. Lu, Y. Zhang, C. Fu, I. Usman, Z. Liu, M. Feng, G. Fang and X. Cao, Adv. Funct. Mater., 2021, 31, 2104361 CrossRef CAS.
- K. Yang, H. Fu, Y. Duan, Z. Ma, D. Wang, B. Li, H. S. Park and D. Ho, ACS Energy Lett., 2024, 9, 209–217 CrossRef CAS.
- P. Xiong, C. Lin, Y. Wei, J.-H. Kim, G. Jang, K. Dai, L. Zeng, S. Huang, S. J. Kwon, S.-Y. Lee and H. S. Park, ACS Energy Lett., 2023, 8, 2718–2727 CrossRef CAS.
- P. Xiong, Y. Kang, N. Yao, X. Chen, H. Mao, W.-S. Jang, D. M. Halat, Z.-H. Fu, M.-H. Jung, H. Y. Jeong, Y.-M. Kim, J. A. Reimer, Q. Zhang and H. S. Park, ACS Energy Lett., 2023, 8, 1613–1625 CrossRef CAS.
- G. Wang, H. Fu, J. Lu, S. Huang, C. Pei, D. Min, Q. Zhang and H. S. Park, Adv. Energy Mater., 2024, 14, 2303549 CrossRef CAS.
- J. Peultier, E. Rocca and J. Steinmetz, Corros. Sci., 2003, 45, 1703–1716 CrossRef CAS.
- V. Luzzati, A. Tardieu, T. Gulik-Krzywicki, E. Rivas and F. Reiss-Husson, Nature, 1968, 220, 485–488 CrossRef CAS PubMed.
- Q. Liu, D. J. Babu, J. Hao, M. T. Vahdat, D. Campi and K. V. Agrawal, Adv. Funct. Mater., 2020, 31, 2303466 Search PubMed.
- M. Ramos Riesco, F. J. Martínez-Casado, J. A. R. Cheda, M. I. Redondo Yélamos, I. da Silva, T. S. Plivelic, S. López-Andrés and P. Ferloni, Cryst. Growth Des., 2015, 15, 2005–2016 CrossRef CAS.
- E. Forsyth, D. A. Paterson, E. Cruickshank, G. J. Strachan, E. Gorecka, R. Walker, J. M. Storey and C. T. Imrie, J. Mol. Liq., 2020, 320, 114391 CrossRef CAS.
- J.-R. Wang, M. Li, Q. Yu, Z. Zhang, B. Zhu, W. Qin and X. Mei, Chem. Commun., 2019, 55, 8532–8535 RSC.
- X. Suo, X. Cui, L. Yang, N. Xu, Y. Huang, Y. He, S. Dai and H. Xing, Adv. Mater., 2020, 32, 1907601 CrossRef CAS PubMed.
- Q. Li, H. Wang, H. Yu, M. Fu, W. Liu, Q. Zhao, S. Huang, L. Zhou, W. Wei and X. Ji, Adv. Funct. Mater., 2023, 3, 2303466 CrossRef.
- Z. Tao, Y. Zhu, Z. Zhou, A. Wang, Y. Tan, Z. Chen, M. Yu and Y. Yang, Small, 2022, 18, 2107971 CrossRef CAS PubMed.
- Q. Zhang, Y. Ma, Y. Lu, X. Zhou, L. Lin, L. Li, Z. Yan, Q. Zhao, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 133, 23545–23552 CrossRef.
- X. Bai, Y. Nan, K. Yang, B. Deng, J. Shao, W. Hu and X. Pu, Adv. Funct. Mater., 2023, 12, 2307595 CrossRef.
- M. He, J. Chen, A. Hu, Z. Yan, L. Cao and J. Long, Energy Storage Mater., 2023, 62, 102941 CrossRef.
- Q. Li, A. Chen, D. Wang, Z. Pei and C. Zhi, Joule, 2022, 6, 273–279 CrossRef.
- H. Dong, X. Hu, R. Liu, M. Ouyang, H. He, T. Wang, X. Gao, Y. Dai, W. Zhang and Y. Liu, Angew. Chem., Int. Ed., 2023, 22, e202311268 Search PubMed.
- J. Wu, C. Yuan, T. Li, Z. Yuan, H. Zhang and X. Li, J. Am. Chem. Soc., 2021, 143, 13135–13144 CrossRef CAS PubMed.
- B. Hao, H. Yang, Z. Wang, Z. Xie, C. Zhu, W. Chen, L. Zhang, J. Liu, C. Yan, J. Zhou and T. Qian, J. Phys. Chem. Lett., 2023, 14, 9986–9995 CrossRef CAS PubMed.
- C. Huang, X. Zhao, S. Liu, Y. Hao, Q. Tang, A. Hu, Z. Liu and X. Chen, Adv. Mater., 2021, 33, 2100445 CrossRef CAS PubMed.
- T. C. Li, C. Lin, M. Luo, P. Wang, D.-S. Li, S. Li, J. Zhou and H. Y. Yang, ACS Energy Lett., 2023, 8, 3258–3268 CrossRef CAS.
- J. Wang, B. Zhang, Z. Cai, R. Zhan, W. Wang, L. Fu, M. Wan, R. Xiao, Y. Ou and L. Wang, Sci. Bull., 2022, 67, 716–724 CrossRef CAS PubMed.
- A. Chen, C. Zhao, J. Gao, Z. Guo, X. Lu, J. Zhang, Z. Liu, M. Wang, N. Liu and L. Fan, Energy Environ. Sci., 2023, 16, 275–284 RSC.
- Y. Cui, Q. Zhao, X. Wu, X. Chen, J. Yang, Y. Wang, R. Qin, S. Ding, Y. Song and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 16594–16601 CrossRef CAS PubMed.
- Y. Qin, P. Liu, Q. Zhang, Q. Wang, D. Sun, Y. Tang, Y. Ren and H. Wang, Small, 2020, 16, 2003106 CrossRef CAS PubMed.
- Y. Xu, X. Zheng, J. Sun, W. Wang, M. Wang, Y. Yuan, M. Chuai, N. Chen, H. Hu and W. Chen, Nano Lett., 2022, 22, 3298–3306 CrossRef CAS PubMed.
- C. Li, Z. Sun, T. Yang, L. Yu, N. Wei, Z. Tian, J. Cai, J. Lv, Y. Shao and M. H. Rümmeli, Adv. Mater., 2020, 32, 2003425 CrossRef CAS PubMed.
- Y.-X. Song, J. Wang, X.-B. Zhong, K. Wang, Y.-H. Zhang, H.-T. Liu, L.-X. Zhang, J.-F. Liang and R. Wen, Energy Storage Mater., 2023, 58, 85–93 CrossRef.
- K. Zhu, L. Wu, C. Guo, J. Pu, Y. Liu, X. Chen, Y. Chen, P. Xue, J. Han and Y. Yao, Adv. Funct. Mater., 2023, 3, 2305098 CrossRef.
- Y. Lv, M. Zhao, Y. Du, Y. Kang, Y. Xiao and S. Chen, Energy Environ. Sci., 2022, 15, 4748–4760 RSC.
- X. Guo, G. Fang, W. Zhang, J. Zhou, L. Shan, L. Wang, C. Wang, T. Lin, Y. Tang and S. Liang, Adv. Energy Mater., 2018, 8, 1801819 CrossRef.
- K. Xie, K. Ren, C. Sun, S. Yang, M. Tong, S. Yang, Z. Liu and Q. Wang, ACS Appl. Energy Mater., 2022, 5, 4170–4178 CrossRef CAS.
- Y. Xin, K. Shen, T. Guo, L. Chen and Y. Li, Small, 2023, 21, 2300019 CrossRef PubMed.
- G. Chen, Y. Kang, H. Yang, M. Zhang, J. Yang, Z. Lv, Q. Wu, P. Lin, Y. Yang and J. Zhao, Adv. Funct. Mater., 2023, 12, 2300656 CrossRef.
- Q. Yang, Y. Guo, B. Yan, C. Wang, Z. Liu, Z. Huang, Y. Wang, Y. Li, H. Li and L. Song, Adv. Mater., 2020, 32, 2001755 CrossRef CAS PubMed.
- J. Zhou, M. Peng, X. Xia, S. Qian, Z. Wang, C. Zhu, X. Zeng, H. Ji, S. Wang, X. Zhou, J. Liu, X. Shen, Y. Cheng, T. Qian and C. Yan, Nano Lett., 2022, 22, 2898–2906 CrossRef CAS PubMed.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- Y. Zeng, P. X. Sun, Z. Pei, Q. Jin, X. Zhang, L. Yu and X. W. Lou, Adv. Mater., 2022, 34, 2200342 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06934f |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |