
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed regio- and stereo-selective phosphination of cyclic biarylsulfonium salts to access atropoisomeric phosphines†
Jinghui
Sun‡
,
Yifei
Yan‡
,
Xuanxuan
Chen
,
Zhiwei
Huang
and
Yinhua
Huang
 *
*
College of Materials, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, P. R. China. E-mail: yhhuang@hznu.edu.cn
First published on 9th April 2024
Abstract
A palladium-catalyzed regio- and stereo-selective phosphination of cyclic biarylsulfonium salts (racemic) with HPAr3Ar4 for straightforward synthesis of atropoisomeric phosphines (P,S-ligands) bearing a stereogenic axis or both a stereogenic axis and a P-stereogenic center is reported. The high reactivity and regio- and stereo-selectivity originate from the torsional strain release and palladium catalysis, and the construction of a P-stereogenic center is enabled by an efficient dynamic kinetic resolution. The high performance of the nascent P,S-ligands has been demonstrated in palladium-catalyzed asymmetric allylic substitutions, indicating the great potential of the present methodology.
Introduction
Chiral phosphines have demonstrated their high utility in synthetic chemistry and asymmetric catalysis, spurring the development in various areas including biomedicines, drugs and materials science.1 Among them, atropoisomeric phosphines represented by the iconic BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl)2 and MOP3 (monodentate phosphines) have attracted wide interest owing to their excellent axially chiral skeleton and unique properties. The development of efficient synthesis of chiral phosphines has been a long-standing goal. The conventional methods, relying on the use of either a stoichiometric quantity of chiral control elements or phosphine oxides/sulphides/boranes followed by reduction/deprotection, have been the dominant approaches.2–4 Despite the significant advances made in this area, catalytic, direct and enantioselective synthesis of chiral tertiary phosphines, particularly, chelating phosphines and P-stereogenic phosphines, remains an enduring challenge in organic chemistry. Herein, we report a straightforward synthesis of atropoisomeric phosphines, a type of C1-symmetric chelating P,S-ligand, potentially offering a high level of structural variations through modular assembly of P- and S-containing building blocks. The key process involves the palladium-catalyzed regio- and stereo-selective phosphination of cyclic biarylsulfonium salts (racemic) with HPAr3Ar4 to create a stereogenic axis (Ar3 = Ar4) or both a stereogenic axis and a P-stereogenic center (Ar3 ≠ Ar4) (Scheme 1c).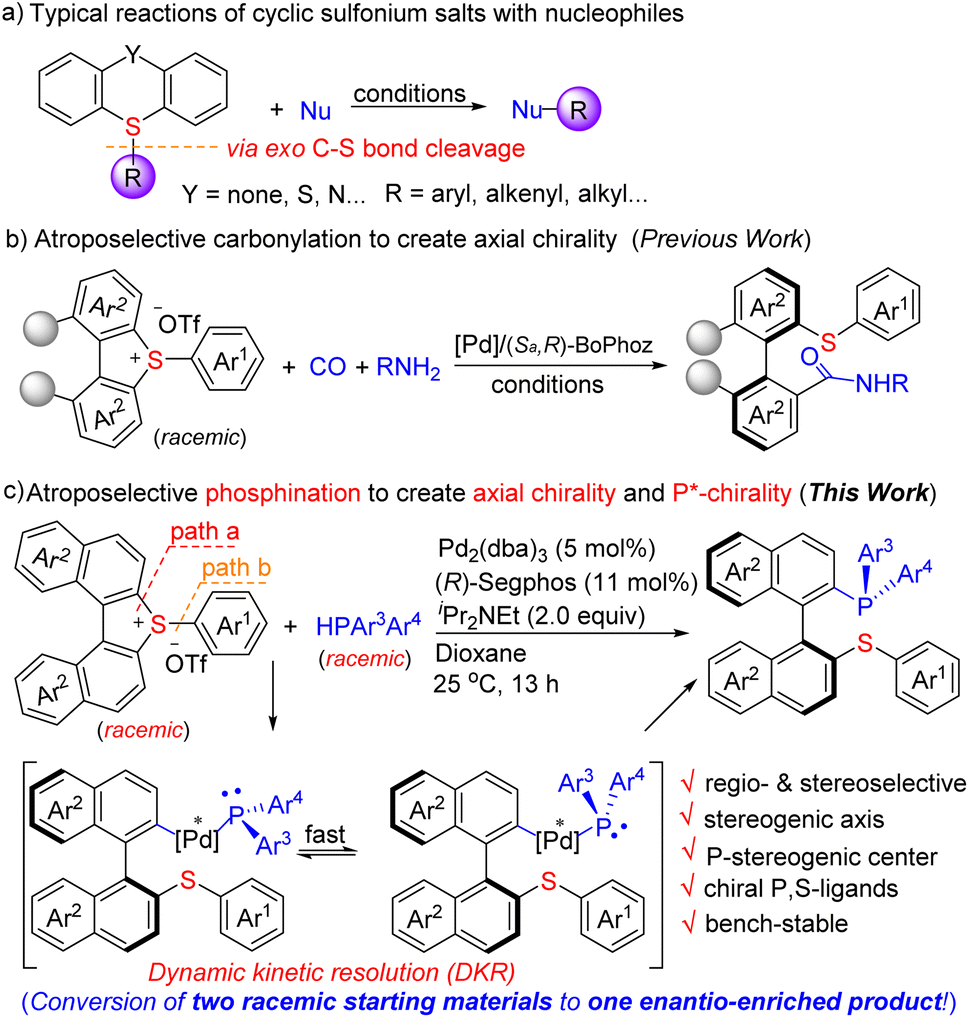 | ||
| Scheme 1 Regio- and stereo-selectivity of cyclic sulfonium salts. | ||
The key challenges of the present study are (1) the regio- and stereo-selectivity of the C–S bond cleavage5 of sulfonium salts (racemic), in which two types of C(Ar)–S bonds are to be discriminated (path a–b), (2) the nascent P,S-ligands have high affinity to transition metals, which could inhibit the reaction or exclude chiral induction, and (3) the chiral catalyst has to not only reach efficient atroposelectivity6 to create a stereogenic axis but also construct a P-stereogenic center via a dynamic kinetic resolution process (when racemic HPAr3Ar4 is used) in one step. Nevertheless, by taking the strategy of palladium-catalyzed phosphinative ring-opening of torsional cyclic biarylsulfonium salts, we successfully addressed these challenges. It is highlighted that the nascent chiral P,S-phosphine products are bench-stable and their enantiomeric purity can be further improved to an optically pure level (ee > 99%) just by a single recrystallization. The unique properties of P,S-ligands have been realized recently,7 indicating that the products have great potential as ligands for asymmetric catalysis.
Sulfonium salts, particularly, cyclic biarylsulfonium salts such as dibenzothiophium and thianthrenium salts, have been demonstrated as versatile electrophilic reagents, and it was generally observed that the cleavage of the exo C–S bond proceeded with predominant preference followed by the transfer of the exo group during the reaction (Scheme 1a).8 So far, most of them are limited to non-asymmetric reactions, asymmetric transformation of them being rarely explored. Only one asymmetric example (carbonylation), to the best of our knowledge, has been known (Scheme 1b).9 We have made continuous efforts in C–X (X = C, P, S, Si, B) bond formation10 by taking advantage of sulfonium salts and in asymmetric catalysis,11 which lead us to envision that the merge of the sulfonium salt chemistry and asymmetric catalysis may result in new solutions to construct desired chiral molecules.
Results and discussion
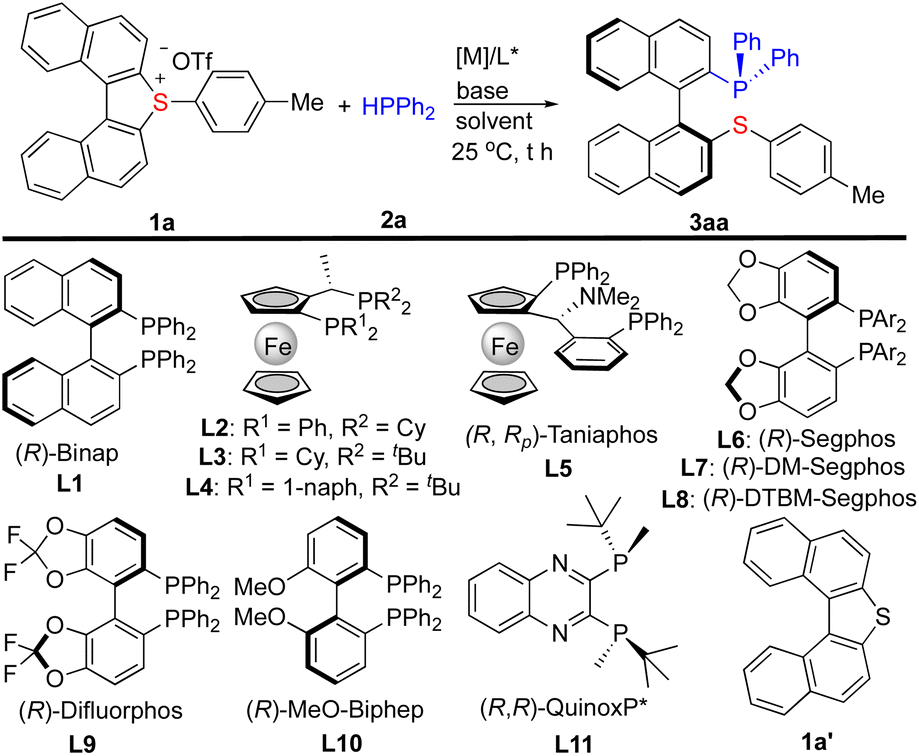
We began our investigation by the reaction of cyclic biarylsulfonium salt 1a with HPPh2 (2a). 1a is a newly synthesized compound for the present study and its X-ray diffraction analysis (CCDC-2303113)12 reveals that the two naphthyl rings are twisted by an angle of 27.07°, and that both (S)- and (R)-conformers exist in the crystal packing (cif). The preliminary investigation identified that Pd2(dba)3 can efficiently catalyse the reaction to furnish the ring-opening product 3aa. Subsequently, a variety of chiral ligands were evaluated for the asymmetric transformation. (R)-Binap (L1) smoothly facilitated the reaction to deliver 3aa in a 96% yield with 62% ee (entry 1, Table 1). A set of ferrocene bisphosphines (L2–L5) were also tested (entries 2–5), and it was found that taniaphos (L5) can give comparable results. Several chiral bisphosphines based on biaryl backbone (L6–L10) were also tested among which (R)-Segphos (L6) was found to give the best ee result (entry 6). The increased steric hindrance on the auxiliary (L7 and L8) did not improve the enantioselectivity (entries 8–9). The use of QuinoxP* (L11) resulted in a low yield with moderate enantioselectivity (entry 11). Next, bases and solvents were screened in order to improve the enantioselectivity (entries 13–19). It turned out that dioxane is a good solvent and iPr2NEt is a good base for this reaction. Finally, fine tuning of the conditions led to 3aa in a 95% yield with 93% ee (entry 21, standard conditions). Notably, the cleavage of the exo C–S bond was not observed under these conditions. The dinaphthothiophene 1a′, which was reported efficient for Ni-catalyzed asymmetric Grignard cross-coupling,5a is not reactive at all under the standard conditions (entry 22), indicating that the reactivity enabled by sulfonium is essential. The X-ray analysis of 3aa (CCDC-2303114)12 ambiguously revealed its (S)-configuration of the stereogenic axis. Chiral monophosphines which usually do not cause chelation with transition metals via catalytic asymmetric C–P bond formation have been documented;13 however, straightforward synthesis of atropoisomeric P,S-ligands which can readily cause chelation with transition metals, to the best of our knowledge, is not known before.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1 | L* | Solvent | Base | Yieldb (%) | eec (%) |
| a Reaction conditions: 1 (0.120 mmol), 2a (0.120 mmol), base (1.5 equiv.), and Pd2(dba)3 (2.5 mol%) in solvent (4.0 mL) at 25 °C for 13 h, unless otherwise noted. THF = tetrahydrofuran and MTBE = methyl tert-butyl ether. b The yields were obtained by 1H NMR analysis of the crude reaction mixture with the aid of Cl2CHCHCl2 as an internal standard. c The ee was determined by HPLC on a chiral stationary phase column. d The reaction was performed at 60 °C (no reaction at 25 °C). e Pd2(dba)3 (5 mol%). f 1a (0.132 mmol) and base (2.0 equiv.). | ||||||
| 1 | 1a | L1 | THF | Et3N | 96 | 62 |
| 2 | 1a | L2 | THF | Et3N | 42 | 53 |
| 3 | 1a | L3 | THF | Et3N | 58 | 5 |
| 4d | 1a | L4 | THF | Et3N | 53 | 0 |
| 5 | 1a | L5 | THF | Et3N | 93 | 65 |
| 6 | 1a | L6 | THF | Et3N | 58 | 79 |
| 7 | 1a | L7 | THF | Et3N | 72 | 52 |
| 8 | 1a | L8 | THF | Et3N | 87 | 0 |
| 9 | 1a | L9 | THF | Et3N | 39 | 58 |
| 10 | 1a | L10 | THF | Et3N | 80 | 63 |
| 11 | 1a | L11 | THF | Et3N | 10 | −64 |
| 12 | 1a | L6 | THF | Barton's base | 41 | <5 |
| 13 | 1a | L6 | THF | Cs2CO3 | 69 | 40 |
| 14 | 1a | L6 | THF | K3PO4 | 82 | 77 |
| 15 | 1a | L6 | Dioxane | K3PO4 | 62 | 87 |
| 16 | 1a | L6 | Toluene | K3PO4 | 85 | 64 |
| 17 | 1a | L6 | MTBE | K3PO4 | 91 | 60 |
| 18 | 1a | L6 | DCE | K3PO4 | 81 | 70 |
| 19e | 1a | L6 | Dioxane | K2HPO4 | 64 | 90 |
| 20e | 1a | L6 | Dioxane | iPr2NEt | 74 | 90 |
| 21e,f | 1a | L6 | Dioxane | iPr2NEt | 95 | 93 |
| 22e | 1a′ | L6 | Dioxane | iPr2NEt | 0 | n.a. |
Under the optimized conditions (Table 1, entry 21), a variety of cyclic biarylsulfonium salts reacted smoothly with 2a to give the desired products 3aa–3va in good to excellent yields with high enantioselectivities (Scheme 2). Various para, meta, and ortho mono- and disubstituted aromatic rings (Ar1) at the exo-position of 1 are well tolerated. Generally, the steric hindrance on Ar1 does not influence the reactivity and enantioselectivity. For example, the steric hindrance from the ortho methyl substituent of Ar1 (1b and 1o) and the tertiary butyl at the para-position (1h) smoothly delivered the corresponding products. A range of functional groups such as ether, halogens (F, Cl, and Br), CF3 were compatible with the conditions. The electronic properties of the substituents on Ar1 have an obvious effect on the reaction. The electron neutral (1d) and donating groups (1a–h and 1m) substituted on Ar1 furnished the desired products in good to excellent yields (51–97%) and enantioselectivities (84–94% ee). Comparatively, the electron-deficient ones with halogens (F, Cl, and Br) substituted on Ar1 (1i–1k) gave higher yields (91–97%) with good enantioselectivities (82–90% ee). 1l bearing a strong electron-deficient CF3 group resulted in a lower yield (74%) and enantioselectivity (48% ee). The introduction of disubstituents on Ar1 (1n and 1o) did not affect the reaction. The cyclic biarylsulfonium salts bearing two substituents (including Br, Ph, Tol, and 1-Ad) at the naphthyl rings (Ar2) (1p–u) were also amenable to this protocol and delivered the corresponding products 3p–u in 65–96% yields with 75–97% ee. It is worth noting that aryl halides including Br (1k, 1p, and 1u) and Cl (1j) can be tolerated under the reaction conditions, which employ palladium (0) as the catalyst. Interestingly, heteroaromatic rings such as 2-thienyl can also be tolerated to give the corresponding product 3va (53%, 89% ee).
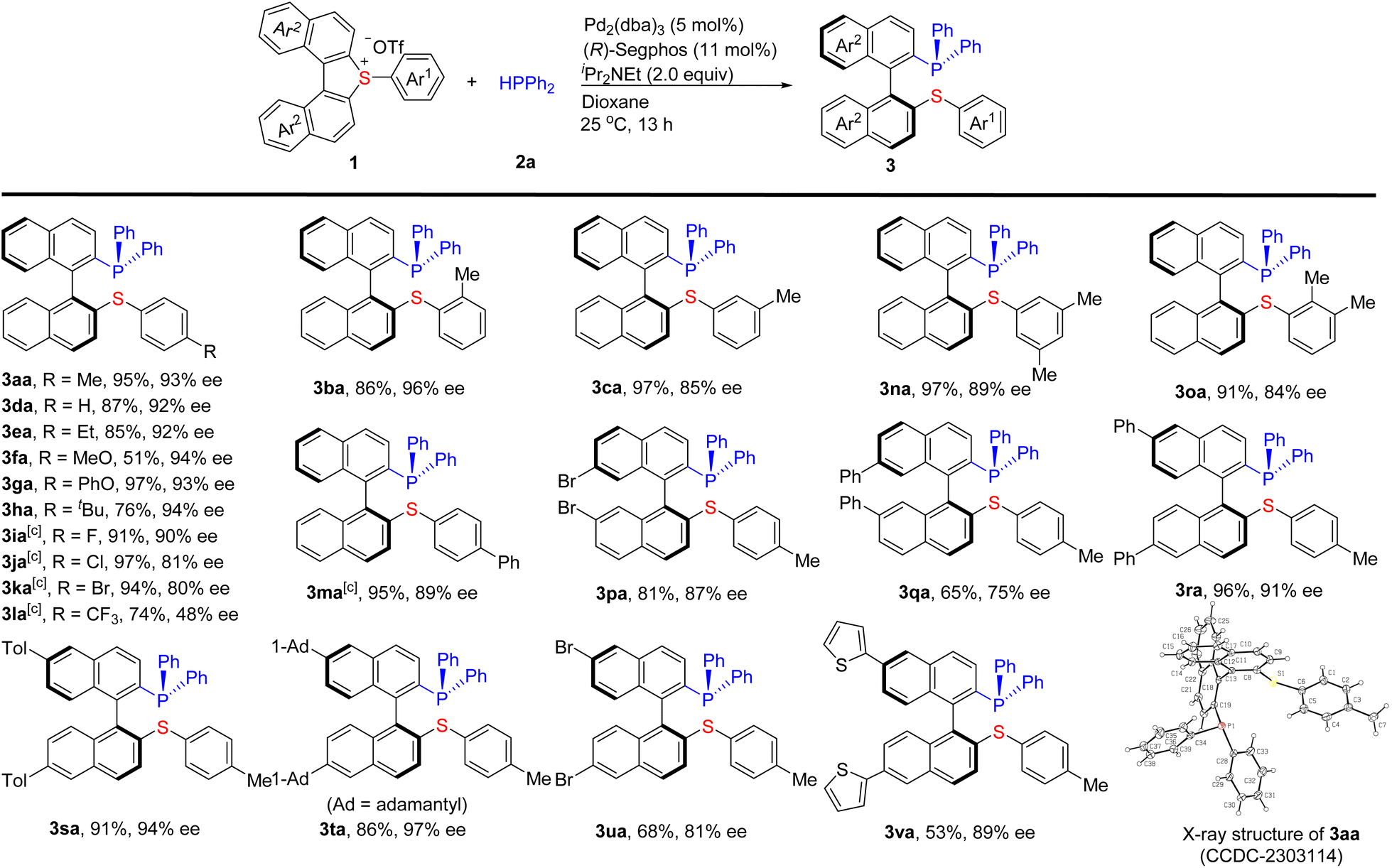 | ||
| Scheme 2 Substrate scope of cyclic biarylsulfonium salts. a Reaction conditions: 1 (0.132 mmol), 2a (0.120 mmol), iPr2NEt (2.0 equiv.), dioxane (4.0 mL), 25 °C, and 13 h. Isolated yield of 3 is given. b The ee% was determined by HPLC on a chiral stationary phase column. c 0–25 °C. | ||
Using the cyclic biarylsulfonium salt 1a, the scope of the secondary phosphine partners has also been investigated and the results are shown in Scheme 3. Firstly, several symmetrical diarylphosphines HPAr23 including 2b–2d were evaluated for this reaction. The results indicated that all of them are good partners for this transformation delivering the corresponding products 3ab–3ad in 83–93% yields with 81–96% ee. Motivated by the pivotal role of P-stereogenic phosphines,14 a series of unsymmetrical diarylphosphines HPAr3Ar4 (Ar3 ≠Ar4) were evaluated under the standard conditions. The substrates containing an ortho-substituent such as Me (2e and 2g–2i) on one aromatic ring (Ar4) reacted smoothly with 1a to give the corresponding products 3ae and 3ag–ai bearing both a stereogenic axis and a P-stereogenic center in 61–71% yields with 87–97% ee and ≥10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Comparatively, the 2-methoxyl substituted 2f was not so efficient, delivering the corresponding 3af in 64% yield with 50% ee and 6:1 dr. The absolute configuration of 3ae was unambiguously determined to be (Saxial,RP) by single-crystal X-ray diffraction analysis (CCDC-2306477).12 Other products yielded under the same chiral catalytic conditions are assumed to have identical configuration. Notably, all of the P,S-phosphine products produced by this protocol are bench-stable.
:1 dr. Comparatively, the 2-methoxyl substituted 2f was not so efficient, delivering the corresponding 3af in 64% yield with 50% ee and 6:1 dr. The absolute configuration of 3ae was unambiguously determined to be (Saxial,RP) by single-crystal X-ray diffraction analysis (CCDC-2306477).12 Other products yielded under the same chiral catalytic conditions are assumed to have identical configuration. Notably, all of the P,S-phosphine products produced by this protocol are bench-stable.
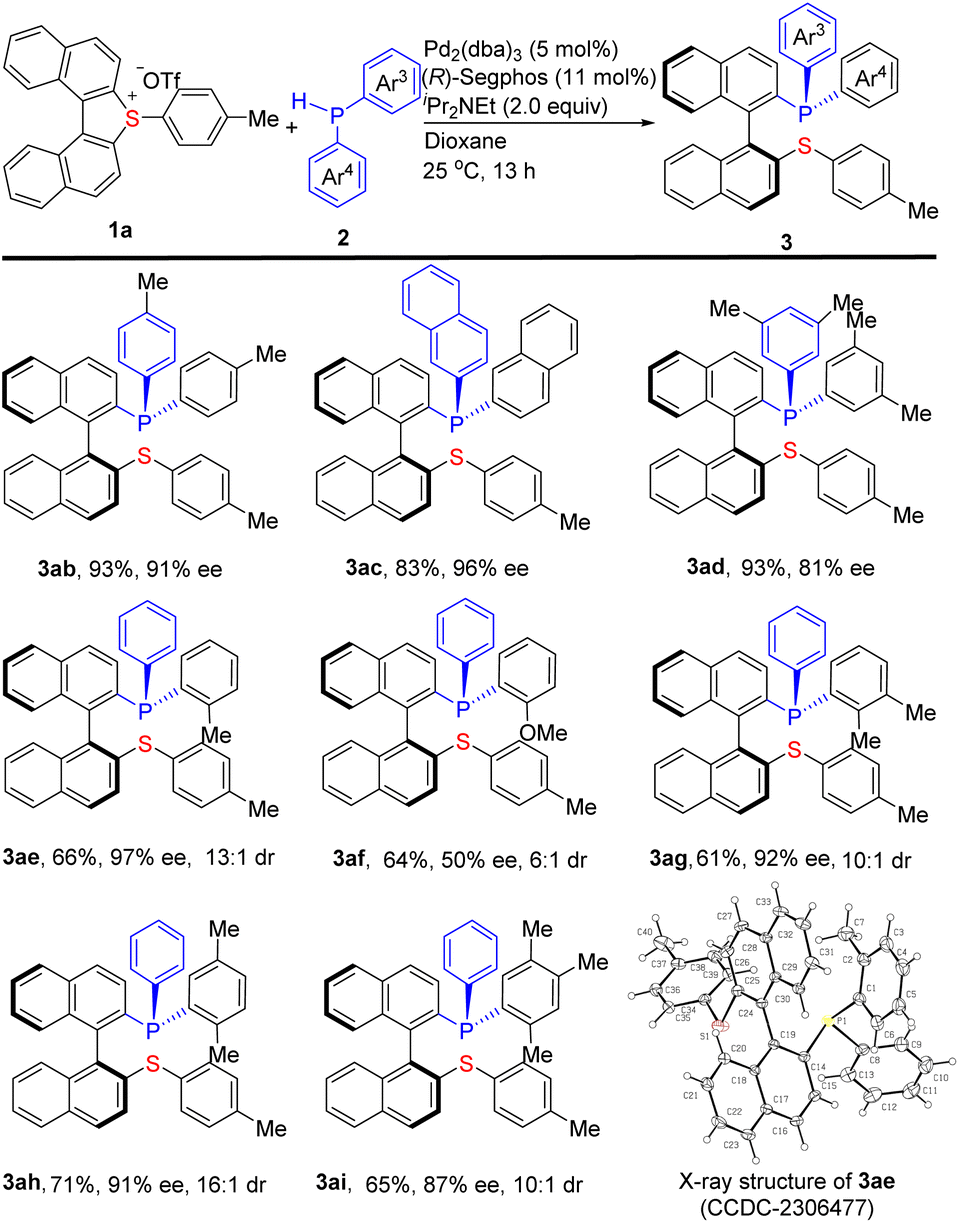 | ||
| Scheme 3 Substrate scope of secondary phosphines. a Reaction conditions: 1a (0.132 mmol), 2 (0.120 mmol), iPr2NEt (2.0 equiv.), dioxane (4.0 mL), 25 °C, and 13 h. Isolated yield of 3 is given. b The ee was determined by HPLC on a chiral stationary phase column. The dr ratio was determined by 31P NMR analysis of the crude reaction mixture. | ||
Scale-up (2 mmol) synthesis of 3aa (92%, 90% ee) and 3ae (62% ee, 97% ee) has been achieved (Scheme 4a). The yields and enantioselectivities are not obviously influenced, indicating the feasibility of scale-up synthesis. Importantly, a simple recrystallization (CHCl3/acetone) further improved the enantiomeric purity of 3aa (>99% ee) and 3ae (>99% ee) to an optically pure level, demonstrating the elegance of this protocol.
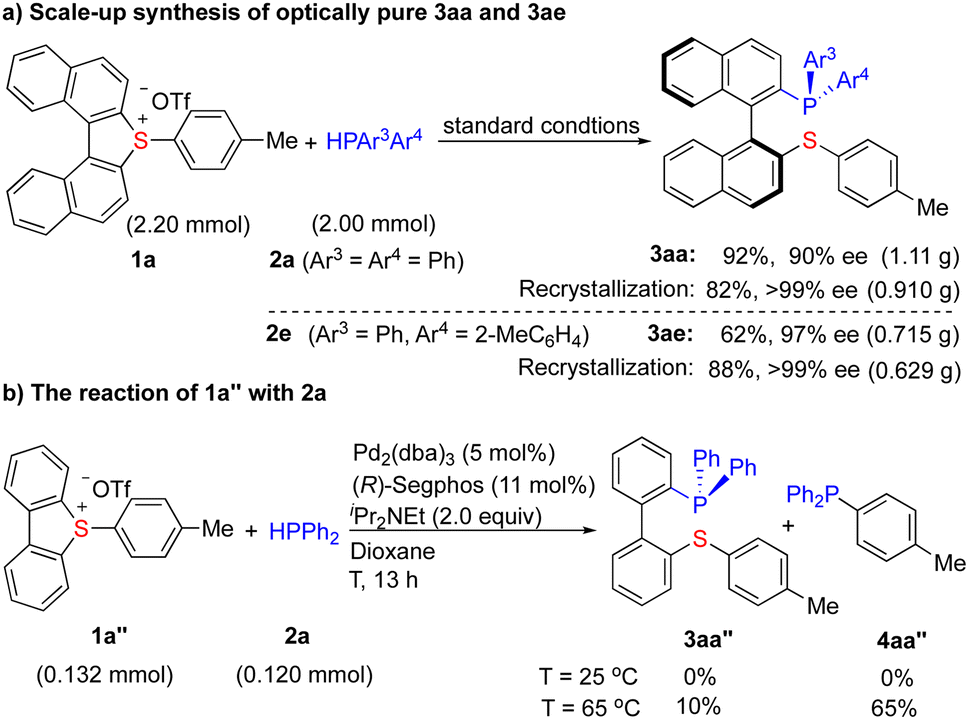 | ||
| Scheme 4 Scale-up synthesis of optically pure 3aa and 3ae. | ||
The use of a non-substituted dibenzothiophium salt 1a″, which lacks torsional strain, terminates the reaction completely under the standard conditions (Scheme 4b). The raise of temperature (65 °C) delivered 4aa″ (via exo C–S cleavage) as a major product (65%) rather than the expected 3aa″ (10%). These results demonstrate that the activation from both sulfonium and torsional strain is essential for this transformation.
To gain a deeper insight into the mechanism, radical trapping experiments have been conducted and the results excluded the radical pathway.12 A plausible catalytic cycle was proposed as depicted in Scheme 5. (Ra)-1 and (Sa)-1 are in rapid equilibrium5a,9 with each other via the rotation of the Caryl–Caryl single bond. The (R)-Segphos-ligated Pd(0) complex undergoes oxidative addition preferentially to (S)-1 cleaving an endo C–S bond to form biaryl [Pd]II species A. The subsequent coordination of 2 (B) leads to the formation of complex C followed by the abstraction of a proton to give the [Pd]II-phosphido species15D and E, which undergo a rapid equilibrium (when Ar3 ≠ Ar4). The subsequent reductive elimination of the more thermodynamic stable D furnishes the chiral phosphine product 3 and regenerates the [Pd]0 species, closing a catalytic cycle. The key point is that the equilibrium between D and E must be much faster than the reductive elimination in this dynamic kinetic resolution process in order to achieve high enantio-control of the P-stereogenic center. Notably, the existence of P,S-ligands which have high affinity to metals does not interrupt the catalytic cycle, presumably due to the stronger chelating ability of Segphos coordinated with palladium. It is the combination of the torsional strain release with the dynamic kinetic resolution process enabled by palladium catalysis that provides a straightforward synthesis of atropoisomeric chelating P-stereogenic phosphines that are otherwise difficult to achieve in a catalytic manner.
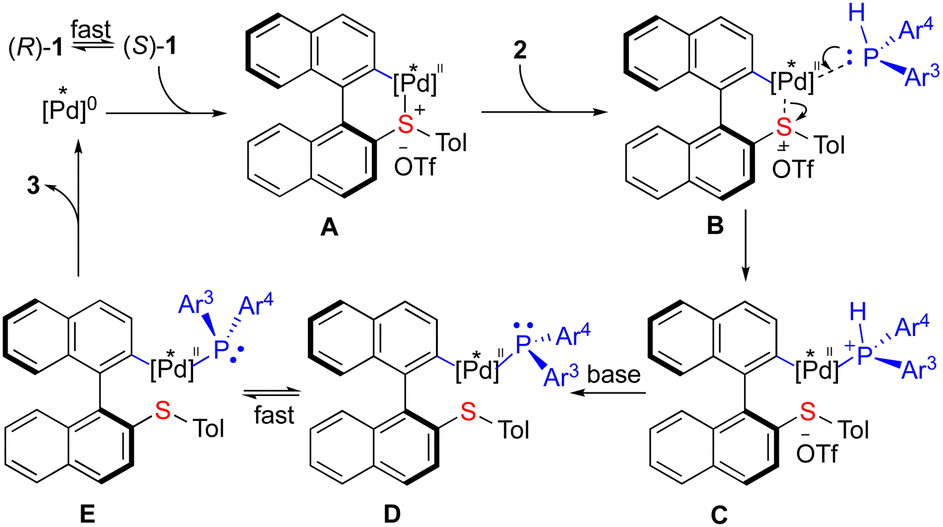 | ||
| Scheme 5 Proposed mechanism. | ||
Finally, to demonstrate the utility of the nascent P,S-phosphine products, preliminary investigations employing 3aa or 3ae as a chiral ligand for the palladium-catalyzed allylic substitution of 5 including alkylation (with 6a), etherification (with 6b), and amination (with 6c) under mild conditions have been carried out (Scheme 6).163aa or 3ae exhibited excellent reactivity (91–99% yields) and enantioselectivity (80–93% ee) in the alkylation and etherification and good reactivity (73%) and enantioselectivity (75% ee) in the amination.
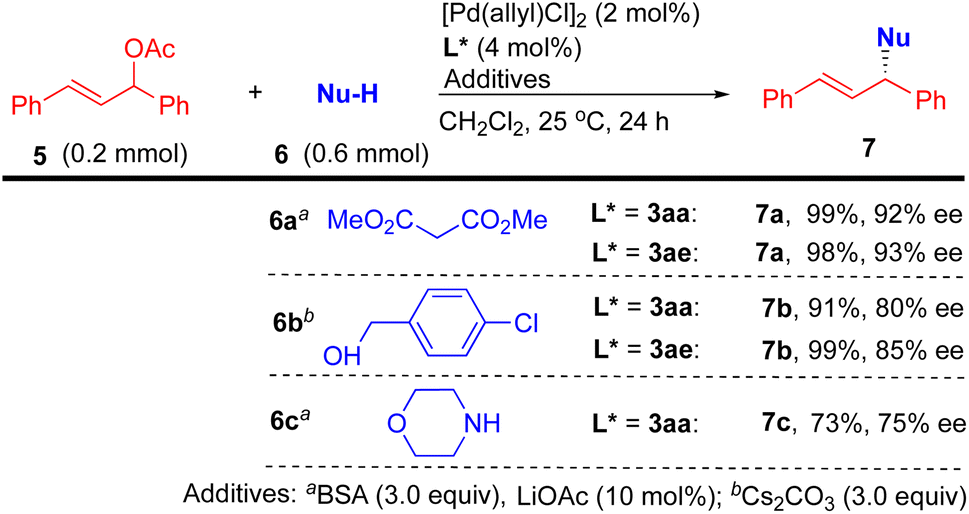 | ||
| Scheme 6 Application of P,S-ligands in Pd-catalyzed allylic substitutions. | ||
Conclusions
In conclusion, we have disclosed a palladium-catalyzed regio- and stereo-selective phosphination of cyclic biarylsulfonium salts with HPAr3Ar4. This protocol provides a general method for straightforward synthesis of P,S-ligands bearing a stereogenic axis, or both a stereogenic axis and a P-stereogenic center. The high performance of the nascent P,S-ligands has been demonstrated in palladium-catalyzed asymmetric allylic substitution reactions. Further mechanistic studies and applications of P,S-ligands in asymmetric catalysis are currently underway. We expect the versatile chiral P,S-ligands outlined herein to enable the related symmetric catalysis for the facile synthesis of known and new chiral compounds of interest.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
J. S. and Y. Y. carried out the synthetic and reaction studies, and they contributed equally. Y. H. managed the project and wrote the manuscript. All authors participated in the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of Zhejiang Province (ZJNSF) (LY24B020005) and Zhejiang Wanfeng Chemical Co., Ltd. for financial support. We are also grateful to Prof. T. Hayashi and P.-H. Leung for their insightful discussions which contributed to this work.Notes and references
- (a) Q.-L. Zhou Privileged Chiral Ligands and Catalysts, Wiley VCH, 2011, vol. 6 CrossRef; (b) V. Iaroshenko Organophosphorus Chemistry: from Molecules to Applications, Wiley VCH, Weinheim, 2019 CrossRef; (c) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed; (d) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 CrossRef CAS PubMed.
- T. Hayashi, Acc. Chem. Res., 2000, 33, 354–362 CrossRef CAS PubMed.
- (a) D. S. Glueck, J. Org. Chem., 2020, 85, 14276–14285 CrossRef CAS PubMed; (b) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC; (c) T. Imamoto, Proc. Jpn. Acad., Ser. B, 2021, 97, 520–542 CrossRef CAS PubMed.
- (a) T. Shimada, Y.-H. Cho and T. Hayashi, J. Am. Chem. Soc., 2002, 124, 13396–13397 CrossRef CAS PubMed; (b) D. Vasu, D. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2015, 54, 7162–7166 CrossRef CAS PubMed.
- (a) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS; (b) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (c) X. Zhang, K. Zhao and Z. Gu, Acc. Chem. Res., 2022, 55, 1620–1633 CrossRef CAS PubMed; (d) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (e) J. WencelDelord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (f) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC; (g) H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202311053 Search PubMed; (h) K. Zhao, L. Duan, S. Xu, J. Jiang, Y. Fu and Z. Gu, Chem, 2018, 4, 599–612 CrossRef CAS.
- (a) F. L. Lam, F. Y. Kwong and A. S. C. Chan, Chem. Commun., 2010, 46, 4649–4667 RSC; (b) S. Gladiali, S. Medici, G. Pirri, S. Pulacchini and D. Fabbri, J. Chem., 2001, 79, 670–678 CAS; (c) T. Hoshi, K. Sasaki, S. Sato, Y. Ishii, T. Suzuki and H. Hagiwara, Org. Lett., 2011, 13, 932–935 CrossRef CAS PubMed; (d) A. Berthelot-Bréhier, A. Panossian, F. Colobert and F. R. Leroux, Org. Chem. Front., 2015, 2, 634–644 RSC.
- (a) L. van Dalsen, R. E. Brown, J. A. Rossi-Ashton and D. J. Procter, Angew. Chem., Int. Ed., 2023, 62, e202303104 CrossRef CAS; (b) H. Meng, M.-S. Liu and W. Shu, Chem. Sci., 2022, 13, 13690–13707 RSC; (c) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS; (d) H. Yorimitsu, Chem. Rec., 2021, 21, 3356–3369 CrossRef CAS PubMed; (e) F. Berger and T. Ritter, Synlett, 2022, 33, 339–345 CrossRef CAS; (f) Á. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef; (g) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 2486–2500 CrossRef CAS.
- Q. Zhang, X. Xue, B. Hong and Z. Gu, Chem. Sci., 2022, 13, 3761–3765 RSC.
- (a) Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386–2391 CrossRef CAS PubMed; (b) R. Xie, J. Zhu and Y. Huang, Org. Chem. Front., 2021, 8, 5699–5704 RSC; (c) Y. Ye, J. Zhu, H. Xie and Y. Huang, Angew. Chem., Int. Ed., 2022, 61, e202212522 CrossRef CAS; (d) J. Zhu, Y. Ye and Y. Huang, Organometallics, 2022, 41, 2342–2348 CrossRef CAS; (e) J. Zhu, Y. Ye, Y. Yan, J. Sun and Y. Huang, Org. Lett., 2023, 25, 5324–5328 CrossRef CAS PubMed; (f) J. Zhu, J. Sun, Y. Yan, Z. Dong and Y. Huang, J. Org. Chem., 2023, 88, 15767–15771 CrossRef CAS PubMed.
- (a) Y. Huang and T. Hayashi, Chem. Rev., 2022, 122, 14346–14404 CrossRef CAS PubMed; (b) J. Li, J. Sun, W. Ren, J. Lei, R. Shen and Y. Huang, Org. Lett., 2022, 24, 2420–2424 CrossRef CAS PubMed; (c) X. Lu, J. Zhu and Y. Huang, Org. Lett., 2022, 24, 8816–8820 CrossRef CAS PubMed; (d) Y. Pan, X. Lu, H. Qiu, H. Tamio and Y. Huang, Org. Lett., 2020, 22, 8413–8418 CrossRef CAS PubMed; (e) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2015, 137, 7556–7559 CrossRef CAS PubMed; (f) Y. Huang and T. Hayashi, J. Am. Chem. Soc., 2016, 138, 12340–12343 CrossRef CAS PubMed.
- For more details, see the ESI†.
- (a) D. K. Wicht, I. V. Kourkine, B. M. Lew, J. M. Nthenge and D. S. Glueck, J. Am. Chem. Soc., 1997, 119, 5039–5040 CrossRef CAS; (b) A. D. Sadow, I. Haller, L. Fadini and A. Togni, J. Am. Chem. Soc., 2004, 126, 14704–14705 CrossRef CAS PubMed; (c) J.-J. Feng, X.-F. Chen, M. Shi and W.-L. Duan, J. Am. Chem. Soc., 2010, 132, 5562–5563 CrossRef CAS PubMed; (d) Y. Huang, S. A. Pullarkat, Y. Li and P.-H. Leung, Chem. Commun., 2010, 46, 6950–6952 RSC; (e) Y.-B. Li, H. Tian and L. Yin, J. Am. Chem. Soc., 2020, 142, 20098–20106 CrossRef CAS PubMed; (f) W.-J. Yue, J.-Z. Xiao, S. Zhan and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 7057–7062 CrossRef CAS PubMed; (g) J. M. Pérez, R. Postolache, M. C. Reis, E. G. Sinnema, D. Vargová, F. Vries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071–20076 CrossRef PubMed; (h) D. Ji, J. Jing, Y. Wang, Z. Qi, F. Wang, X. Zhang, Y. Wang and X. Li, Chem, 2022, 8, 3346–3362 CrossRef CAS; (i) Y. Zhang, Y. Jiang, M. Li, Z. Huang and J. Wang, Chem Catal., 2022, 2, 3163–3173 CrossRef CAS; (j) Z. Lu, H. Zhang, Z. Yang, N. Ding, L. Meng and J. Wang, ACS Catal., 2019, 9, 1457–1463 CrossRef CAS.
- (a) C. Luan, C.-J. Yang, L. Liu, Q.-S. Gu and X.-Y. Liu, Chem Catal., 2022, 2, 2876–2888 CrossRef CAS; (b) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477–7485 RSC; (c) D. S. Glueck, Chem.–Eur. J., 2008, 14, 7108–7117 CrossRef CAS PubMed; (d) D. S. Glueck, Synlett, 2007, 2627–2634 CrossRef CAS; (e) Y. Huang, Y. Li, P.-H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865–4868 CrossRef CAS PubMed; (f) C. Wang, K. Huang and W.-L. Duan, J. Am. Chem. Soc., 2021, 143, 5685–5690 CrossRef CAS PubMed; (g) S. Zhang, J.-Z. Xiao, Y.-B. Li, C.-Y. Shi and L. Yin, J. Am. Chem. Soc., 2021, 143, 9912–9921 CrossRef CAS PubMed; (h) Y.-B. Li, H. Tian, S. Zhang, J.-Z. Xiao and L. Yin, Angew. Chem., Int. Ed., 2022, 61, e202117760 CrossRef CAS PubMed; (i) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786–2787 CrossRef CAS PubMed; (j) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021–6032 CrossRef CAS PubMed.
- (a) N. F. Blank, J. R. Moncarz, T. J. Brunker, C. Scriban, B. J. Anderson, O. Amir, D. S. Glueck, L. N. Zakharov, J. A. Golen, C. D. Incarvito and A. L. Rheingold, J. Am. Chem. Soc., 2007, 129, 6847–6858 CrossRef CAS PubMed; (b) V. S. Chan, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 15122–15123 CrossRef CAS PubMed.
- (a) R. Shintani, W.-L. Duan, K. Okamoto and T. Hayashi, Tetrahedron: Asymmetry, 2005, 16, 3400–3405 CrossRef CAS; (b) J. Chen, F. Lang, D. Li, L. Cun, J. Zhu, J. Deng and J. Liao, Tetrahedron: Asymmetry, 2009, 20, 1953–1956 CrossRef CAS; (c) F. L. Lam, T. Au-Yeung, F. Y. Kwong, Z. Zhou, K. Y. Wong and A. S. C. Chan, Angew. Chem., Int. Ed., 2008, 47, 1280–1283 CrossRef CAS PubMed; (d) J.-H. Wu, S. Fang, X. Zheng, J. He, Y. Ma, Z. Su and T. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309515 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra, and crystallographic data. CCDC 2303113, 2303114 and 2306477. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00446a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |