
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced synthesis of multiblock copolymers via acid-triggered RAFT polymerization†
Maria-Nefeli
Antonopoulou
,
Nghia P.
Truong
 and
Athina
Anastasaki
*
and
Athina
Anastasaki
*
Laboratory of Polymeric Materials, Department of Materials, ETH Zürich, Zürich, Switzerland. E-mail: athina.anastasaki@mat.ethz.ch
First published on 7th March 2024
Abstract
The synthesis of multiblock copolymers has emerged as an efficient tool to not only reveal the optimal way to access complex structures and investigate polymer properties but also to ascertain the end-group fidelity of a given polymerization methodology. Although reversible addition–fragmentation chain-transfer (RAFT) polymerization is arguably the most dominant strategy employed, its success is often hampered by the unavoidable and excessive use of radical initiators which results in increased termination and loss of end-group fidelity. In this work, we employ acid in RAFT polymerization to enhance the synthesis of multiblock copolymers. By the addition of a small amount of acid, a 4-fold decrease in the overall required radical initiator concentration was achieved, enabling the synthesis of a range of well-defined multiblock copolymers with various degrees of polymerization (DP) per block. The acid enhances the propagation rate, minimizing the initiator concentration. In all cases, near-quantitative monomer conversion was obtained (>97%) for every iterative block formation step. Notably, and in contrast to conventional RAFT approaches, the tailing to low molecular weight was significantly suppressed and the dispersity was maintained nearly constant (i.e. in most cases Đ = 1.1–1.2), thus indicating minor termination events and side reactions during acid-enhanced synthesis. The possibility to synthesize multiblocks consisting of methacrylates, acrylates, and acrylamides was also demonstrated. This work presents an advancement in the synthesis of well-defined multiblock copolymers and more broadly, RAFT polymers with high end-group fidelity.
Introduction
With the advent of reversible deactivation radical polymerization, commonly referred to as controlled radical polymerization, it is now possible to prepare well-defined polymers with advanced characteristics including controlled architecture, dispersity, functionality, and sequence under relatively mild and user-friendly conditions.1–9 A particularly exciting aspect of controlled radical polymerization explored over the last decade is the possibility of synthesizing sequence-controlled multiblock copolymers.10–13 On one hand, such synthesis offers a unique approach to explore the effect of sequence on polymer properties including self-assembly, antimicrobial activity, folding and microphase separation.14–22 On the other hand, the preparation of multiblock copolymers consists of a unique strategy to indirectly evaluate the livingness of a given polymerization system. For instance, multiblock copolymers with an overall lower dispersity and suppressed low and high molecular weight shoulders are more “living” and as such would be the preferred choice to make these complex structures with low number of terminated chains that influence the material properties. It is also noted that such optimization in the synthesis also enables access to the efficient preparation of simpler and more commonly utilized materials such as diblocks and triblocks. As a result, competition has sparked between various groups to target the highest number of blocks possible without compromising the final dispersity.13In atom transfer radical polymerization (ATRP), Whittaker first demonstrated the synthesis of multiblock copolymers consisting of very short blocks (i.e. DP = 2 per block) in the presence of zerovalent copper.23 Although this original work inspired the field of sequence-controlled multiblock copolymers, targeting longer blocks appeared to be a formidable challenge.24–27 For instance, when a decablock copolymer with ∼DP = 20 per block was attempted, a final dispersity of 1.72 was reported accompanied by significant low molecular weight tailing. Alternative approaches by the groups of Haddleton, Junkers and Hawker employing photo-induced ATRP subsequently expanded the scope of materials while resulting in fast polymerizations and improved dispersities.26–34 However, pronounced termination was still clearly visible by size exclusion chromatography (SEC) leading to the gradual accumulation of Cu(II) and the inevitable cessation of the polymerization, thus severely limiting the number of blocks.
From a mechanistic perspective, reversible addition–fragmentation chain-transfer (RAFT) polymerization would perhaps be a more promising alternative as the incorporation of an additional radical initiator would, in principle, allow for the continuation of polymerization, thus potentially overcoming the obstacle of the limited block numbers.35–37 However, conventional RAFT suffers from the inherent limitation that external termination (due to added the radical initiator) is inevitable in the RAFT process and as such RAFT polymerization, in theory, exhibits a higher extent of termination than ATRP.38 For a successful RAFT polymerization, one has to maintain a high fraction of living chains and this challenge becomes particularly difficult when targeting multiblock copolymers as an additional radical initiator must be added upon each monomer addition. Perrier and co-workers showed in a series of publications that the successful synthesis of even icosablock copolymers was feasible by ensuring a sufficiently high propagation rate.39–45 To minimize the amount of required radical initiator, a high kp monomer class (i.e. acrylamides) and a polar solvent (i.e. water) were selected, yielding high livingness for such demanding architectures. Yet, dispersity continually increased during the synthesis and significant low molecular weight tailing was evident by SEC. For example, in a dodecablock copolymer, a final dispersity of ∼1.6 was reported with the dispersity of the homopolymer being as low as 1.2.41 Upon targeting higher molecular weights per block (i.e. DP = 100), higher dispersity could already be observed in the pentablock copolymer (Đ = 1.5). More recently, significant progress has been made by the groups of Zetterlund, Boyer, and Hartlieb who have employed emulsion, photoinduced electron/energy transfer RAFT and photoiniferter polymerizations respectively to prepare a range of multiblock copolymers.11,34,46–51 Despite these recent achievements, the vast majority of these strategies still exhibit broad molar mass distributions and visible termination, exemplified by non-uniform molar mass distributions (Fig. 1). At the same time, thermal RAFT remains an attractive and versatile polymerization method and as such, alternative approaches that further suppress the required radical concentration would be highly advantageous.37
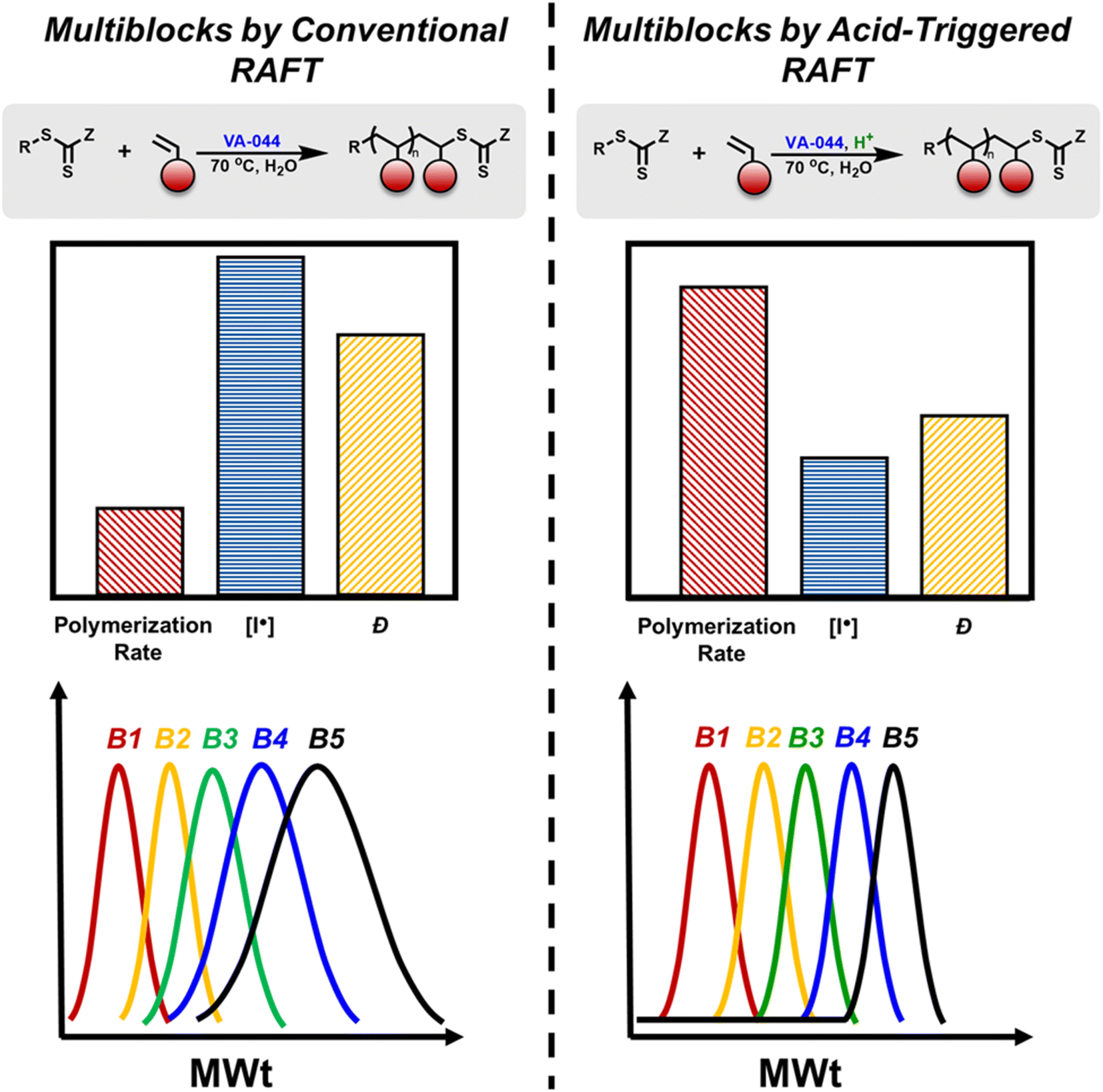 | ||
| Fig. 1 Comparison of the synthesis of multiblock copolymers via conventional RAFT polymerization versus acid-triggered RAFT polymerization. Top left: schematic representation of a conventional RAFT polymerization. Middle left: graphical representation of polymerization rate, initiator concentration and dispersity in a conventional RAFT process. Bottom left: illustration of SEC traces of a pentablock copolymer synthesized via a conventional RAFT process. Top right: schematic representation of acid-triggered RAFT polymerization. Middle right: graphical representation of polymerization rate, initiator concentration and dispersity in acid-triggered RAFT process. Bottom right: illustration of SEC traces of a pentablock copolymer synthesized via acid-triggered RAFT. | ||
Our group recently reported the acid-triggered polymerization of vinyl monomers in which a highly abundant acid (e.g. sulfuric acid) was shown to trigger a successful RAFT polymerization in the absence of conventional radical initiators.52 Interestingly, the acid was capable of not only initiating the polymerization but also increasing the reaction rate thus allowing the system to operate with an overall lower radical concentration. Although the synthesis of diblock and triblock copolymers was successful with only acid (i.e. no radical initiator was required), rather slow polymerization rates were observed (i.e. 20 h per block), thus limiting the possible total number of blocks and the ability to synthesize well-defined multiblock copolymers. In this work, we hypothesize that the synergy of an acid and a free radical initiator will enable the rapid synthesis of multiblock copolymers while significantly suppressing termination events.
Results and discussion
To perform a conventional RAFT polymerization, dimethylacrylamide (DMA) was used as a monomer, 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) as a radical initiator, 2-(((butylsulfanyl)carbothioyl)sulfanyl)propanoic acid (PABTC) as the chain-transfer agent (CTA 1) and water as the solvent. We targeted an initial DP = 500. In the presence of 0.02 equiv. of radical initiator, a conventional thermal RAFT polymerization took place at 70 °C (Fig. 2a, e( ) and 2f–h, Table S1†). Within 20 minutes, 30% of conversion by H1 nuclear magnetic resonance (NMR) was achieved and no further monomer consumption could be observed at prolonged reaction times suggesting that all the radical initiator had been consumed. In addition, a broad molar mass distribution was observed (Đ = 1.53) which was attributed to the low CTA consumption at this relatively early stage of polymerization. Indeed, a significant amount of remaining CTA was confirmed by the SEC UV-detector (Fig. S2†). Typically, a higher equivalent of initiator (>0.02) can be used to increase conversion and CTA consumption. However, this approach would also lead to increased termination.
) and 2f–h, Table S1†). Within 20 minutes, 30% of conversion by H1 nuclear magnetic resonance (NMR) was achieved and no further monomer consumption could be observed at prolonged reaction times suggesting that all the radical initiator had been consumed. In addition, a broad molar mass distribution was observed (Đ = 1.53) which was attributed to the low CTA consumption at this relatively early stage of polymerization. Indeed, a significant amount of remaining CTA was confirmed by the SEC UV-detector (Fig. S2†). Typically, a higher equivalent of initiator (>0.02) can be used to increase conversion and CTA consumption. However, this approach would also lead to increased termination.
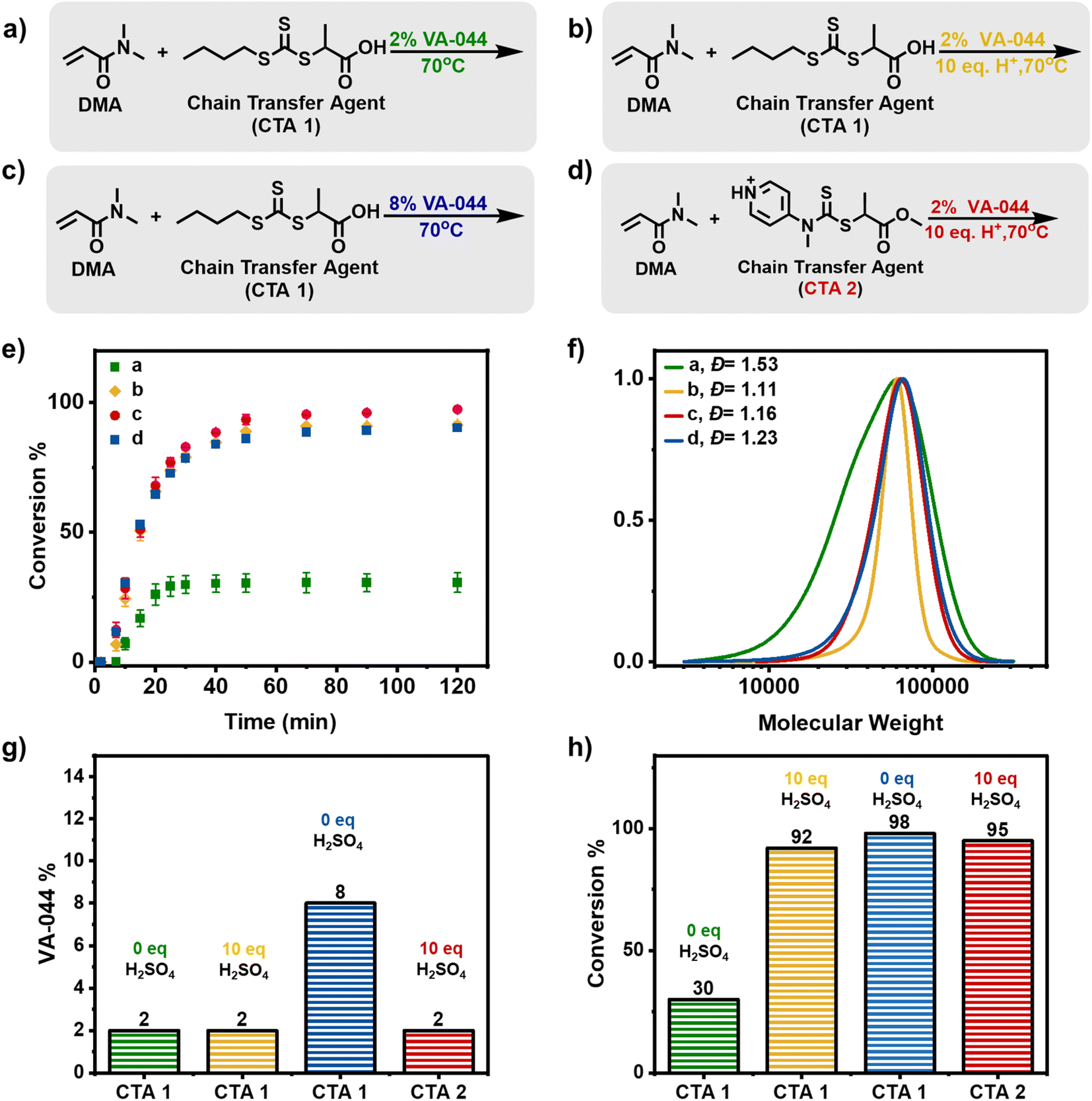 | ||
| Fig. 2 (a) Scheme of DMA polymerization with CTA 1 (TTC) in the absence of acid and 2% initiator. (b) Scheme of DMA polymerization with CTA 1 (TTC) with 10 equiv. of acid and 2% initiator. (c) Scheme of DMA polymerization with CTA 1 (TTC) in the absence of acid and 8% initiator. (d) Scheme of DMA polymerization with CTA 2 (DTC) with 10 equiv. of acid and 2% initiator. (e) Kinetic analysis of the polymerization of DMA with DP = 500 under the conditions described in (a–d). (f) SEC traces for the final time points of the reactions conducted in (e). (g and h) Initiator percentage and final conversion of reactions (a–d) accordingly. | ||
Inspired by our recent work in acid-triggered RAFT polymerization,52 we hypothesized that the addition of acid would result in both faster reaction rates and overall higher monomer conversions. To examine this scenario, sulfuric acid (10 equiv.) was added and the reaction was repeated under otherwise identical conditions (Fig. 2b, e ( ) and 2f–h, Table S1†). Detailed kinetic analysis revealed a two-fold increase in the monomer conversion within the first 20 minutes (i.e. 60% versus 30% that was observed in the absence of acid) and the polymerization further progressed to 92% conversion within 1 hour. These results suggest that the presence of acid can indeed have a profound effect on the reaction rate and subsequently on the final conversion reached. Notably, narrow molar mass distributions (Đ = 1.11) were obtained at near-quantitative conversions indicating that a much faster incorporation of the RAFT agent was achieved, leading to better control over the molecular weight.
) and 2f–h, Table S1†). Detailed kinetic analysis revealed a two-fold increase in the monomer conversion within the first 20 minutes (i.e. 60% versus 30% that was observed in the absence of acid) and the polymerization further progressed to 92% conversion within 1 hour. These results suggest that the presence of acid can indeed have a profound effect on the reaction rate and subsequently on the final conversion reached. Notably, narrow molar mass distributions (Đ = 1.11) were obtained at near-quantitative conversions indicating that a much faster incorporation of the RAFT agent was achieved, leading to better control over the molecular weight.
As a control experiment, the optimal amount of added radical initiator that could achieve high monomer conversions in the absence of acid was also explored (Fig. 2c, e ( ) and 2f–h). It was found that to achieve a similarly high monomer conversion (i.e. ≥90%), 0.08 equiv. of the radical initiator was essential leading to 90% conversion within 1 h and a final dispersity of 1.24. It is noted that the concentration of the utilized radical initiator has been increased by four-fold to achieve similar conversions when compared to the experiment in the presence of acid. Another interesting observation here is that we observe a slower CTA consumption (Fig. S3†) as opposed to the acid-triggered RAFT experiment (Fig. S4†) which suggests that the presence of acid not only accelerates the rate and yields higher monomer conversions but also results in improved dispersities due to the faster CTA consumption.
) and 2f–h). It was found that to achieve a similarly high monomer conversion (i.e. ≥90%), 0.08 equiv. of the radical initiator was essential leading to 90% conversion within 1 h and a final dispersity of 1.24. It is noted that the concentration of the utilized radical initiator has been increased by four-fold to achieve similar conversions when compared to the experiment in the presence of acid. Another interesting observation here is that we observe a slower CTA consumption (Fig. S3†) as opposed to the acid-triggered RAFT experiment (Fig. S4†) which suggests that the presence of acid not only accelerates the rate and yields higher monomer conversions but also results in improved dispersities due to the faster CTA consumption.
Although CTA 1 is an excellent candidate to trigger an acid-triggered RAFT polymerization, we were also interested in examining a more water-soluble CTA, namely methyl 2-(methyl(4-pyridinyl)carbamothioylthio)propionate (CTA 2). CTA 2 is a switchable RAFT agent that in acidic media is a high-activity RAFT agent.53 When CTA 1 was replaced by CTA 2 in the presence of 10 equiv. of sulfuric acid, a well-controlled polymerization took place reaching near-quantitative conversion (i.e. 98%) and a dispersity of 1.16 (Fig. 2d, e ( ) and 2f–h). Considering the improved solubility and high monomer conversion attained, CTA 2 was subsequently selected as the model CTA for the synthesis of the multiblock copolymers. As CTA 2 is a switchable RAFT agent and therefore would yield high dispersity polymers in the absence of acid, CTA 1 was also selected as an alternative RAFT agent for all the control experiments.
) and 2f–h). Considering the improved solubility and high monomer conversion attained, CTA 2 was subsequently selected as the model CTA for the synthesis of the multiblock copolymers. As CTA 2 is a switchable RAFT agent and therefore would yield high dispersity polymers in the absence of acid, CTA 1 was also selected as an alternative RAFT agent for all the control experiments.
Encouraged by the homopolymerization results, a pentablock copolymer with a DP = 100 per block was set out as the initial multiblock target considering that when either ATRP or RAFT polymerization methodologies were previously employed for this synthesis, rather broad final molar mass distributions (Đ > 1.5) and/or severe low and high molecular weight termination were reported.25,41
In particular, the synthesis of two pentablock copolymers were performed; the first in the absence of acid using CTA 1 and the second in the presence of acid utilizing CTA 2. Aside from the acid content and CTA choice, all other conditions were identical and are summarized in ESI (Fig. S5, Tables S2 and S3†). In the absence of acid, the first block was easily attained with a very narrow molar mass distribution (Đ = 1.09) (Fig. 3a–c). Upon reaching near-quantitative monomer conversion (>95% by 1H NMR), an aliquot of the second acrylamide monomer was injected in situ together with another batch of free radical initiator to ensure the continuation of the polymerization. At approximately 98% conversion SEC revealed a slight increase in the dispersity (Đ = 1.13) which became even more pronounced for the third block (Đ = 1.20) and was accompanied by obvious low molecular weight tailing. Perhaps not surprisingly, blocks 4 and 5 exhibited further termination and broadening of the molar mass distributions yielding a tetrablock and pentablock copolymer with dispersities of 1.28 and 1.38 respectively (Fig. 3a–c). It is also noted that severe low molecular weight tailing is visible by SEC. This result is rather expected and is attributed to the necessity of adding further radical initiator at each iterative monomer addition. Specifically, to ensure high monomer conversions at each step approximately 0.02 equiv. of radical initiator was added per block equating to an overall 0.11 equiv. utilized for the entire multiblock synthesis. Following the livingness equation previously developed by Perrier and co-workers37,41 the total livingness was calculated at 89.6%.
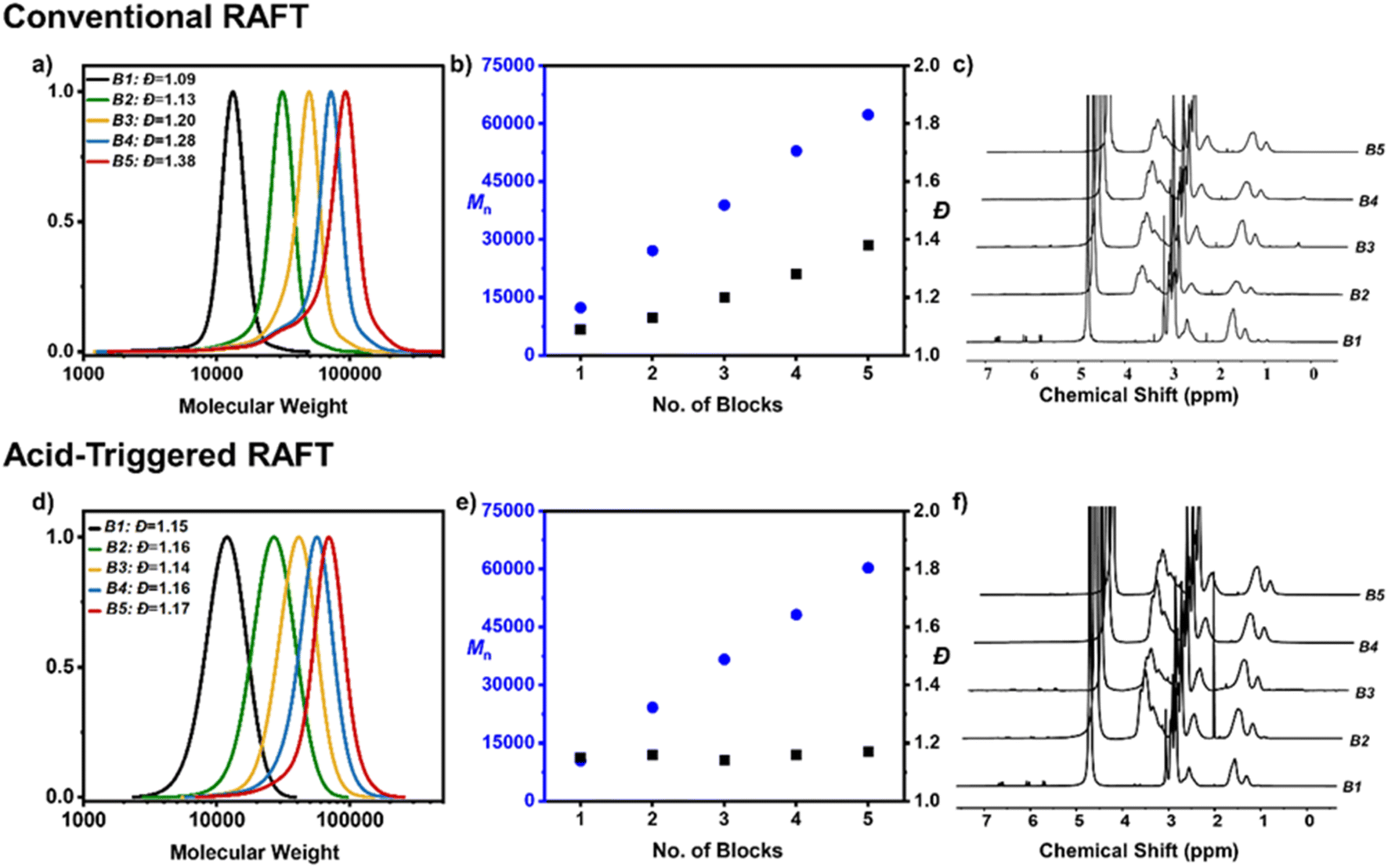 | ||
| Fig. 3 (a) SEC traces illustrating the molar mass distributions for consecutive blocks during the synthesis of a pentablock copolymer synthesized via conventional RAFT polymerization (RI detector). (b) Evolution of Mn (blue) and Đ (black) of the pentablock copolymer via conventional RAFT polymerization. (c) 1H NMR spectra illustrating the monomer conversion of each block. (d) SEC traces illustrating the molar mass distributions for consecutive blocks during the synthesis of a pentablock copolymer synthesized via acid-triggered RAFT polymerization (RI detector). (e) Evolution of Mn (blue) and Đ (black) of the pentablock copolymer via acid-triggered RAFT polymerization. (f) 1H NMR spectra illustrating the monomer conversion of each block. | ||
Instead, in the presence of sulfuric acid, a completely different evolution of the molar mass distributions was observed. While the first block exhibited slightly broader molar mass distributions than when conventional RAFT polymerization was employed (Đ = 1.15 versus Đ = 1.09), the presence of acid had a remarkable effect for the subsequent blocks and the overall dispersity remained relatively constant. In particular, the diblock yielded a dispersity of 1.16, followed by 1.14 for the triblock, 1.16 for the tetrablock and 1.17 for the final pentablock copolymer. It is highlighted that near-quantitative monomer conversions were also reached throughout the synthesis (>97% for each block) and the extent of termination at either low or high molecular weight was significantly suppressed, exemplified by a monomodal final molar mass distribution (Fig. 3d–f).
Thanks to the rate acceleration offered by the addition of acid, a much lower overall amount of radical initiator was employed (i.e. a total of 0.05 equiv.) As such, the acid-triggered RAFT polymerization led to the efficient synthesis of a pentablock copolymer with an overall livingness of 95.1%. It is noted that the small amount of radicals generated by autoinitiation has not been considered to ease the calculations. In other words, acid-triggered RAFT polymerization yielded a two-fold decrease in the overall termination when compared to conventional RAFT polymerization (4.9% of terminated chains versus 10.4% of terminated chains). A pentablock copolymer (DP = 100 per block, Mn = 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300, Đ = 1.23) was successfully synthesized in the presence of added acid, suggesting that acid can trigger polymerization without the need for a radical initiator (Table S4†).
300, Đ = 1.23) was successfully synthesized in the presence of added acid, suggesting that acid can trigger polymerization without the need for a radical initiator (Table S4†).
Considering the initial success of multiblock copolymer synthesis via acid-triggered RAFT polymerization, we subsequently decided to further challenge our system by attempting the synthesis of pentablock copolymers consisting of an even higher DP per block (DP = 200). In the presence of acid, the first block yielded a well-defined homopolymer with a symmetrical and monomodal molar mass distribution (Đ = 1.15) (Fig. 4a and b, Table S5†). Upon chain extension at high monomer conversion, a diblock copolymer with low dispersity was also attained (Đ = 1.18). This process was repeated two more times with SEC analysis confirming a clear shift to higher molecular weights after each addition resulting in the near-quantitative synthesis (i.e. >96% of conversion per block) of a triblock and a tetrablock copolymer with dispersities of 1.17 and 1.19 respectively. Notably, even the pentablock copolymer possessed rather narrow molar mass distributions (Đ = 1.22) and an overall livingness of 95% (Table S5, Fig. S6†).
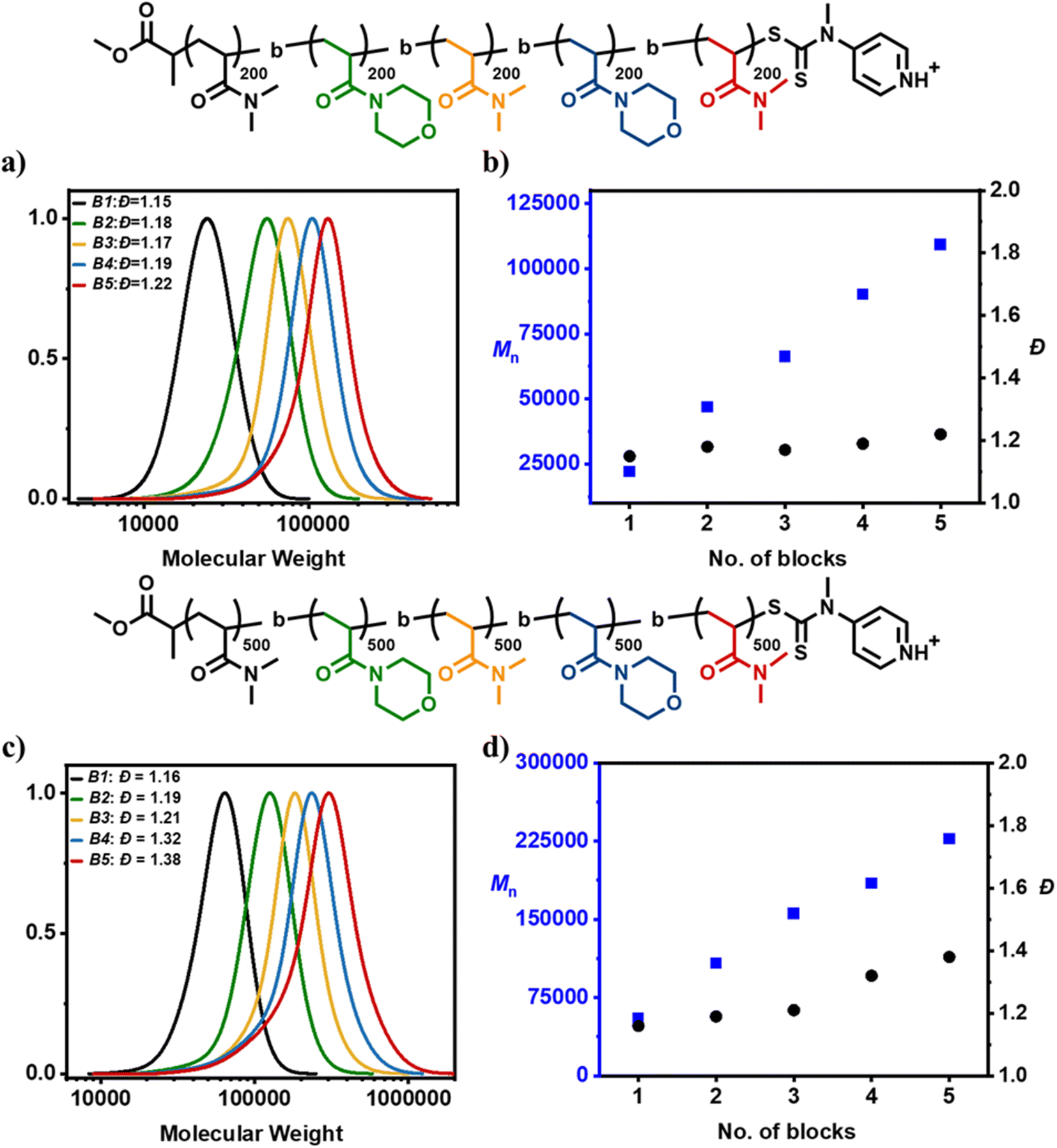 | ||
| Fig. 4 Sequence controlled multiblocks. (a) SEC traces illustrating the molecular weight distributions for consecutive cycles during the synthesis of a pentablock with DP = 200 and CTA 2 (RI detector). (b) Evolution of dispersity (black) and the measured molecular weight (blue) during this multiblock synthesis. (c) SEC traces illustrating the molecular weight distributions for consecutive cycles during the synthesis of a pentablock with DP = 500 and CTA 2 (RI detector). (d) Evolution of dispersity (black) and the measured molecular weight (blue) during this multiblock synthesis. | ||
Encouraged by these findings, a demanding pentablock copolymer with DP = 500 per block was subsequently attempted. For the first three blocks low dispersity values were maintained (Đ = 1.16 for block 1, Đ = 1.19 for block 2 and Đ = 1.21 for block 3) despite the very high monomer conversions reached prior to the next monomer addition (Fig. 4c and d, Table S6†). However, upon further monomer additions, the dispersity rose to 1.32 for the tetrablock and 1.38 for the pentablock copolymer with an overall livingness of 90.6% (Table S6, Fig. S7†). Although SEC reveals clear termination events, these results represent the most successful one-pot synthesis of high molecular weight multiblock copolymers. Acid-triggered RAFT polymerization yielded a pentablock copolymer (DP = 500 per block) with slightly higher overall theoretical livingness than the corresponding lower molecular weight pentablock copolymer (DP = 100 per block) previously synthesized with conventional RAFT polymerization (90.6% versus 89.6%). This is also reflected in the comparable final dispersities achieved despite the large difference in the molecular weight of each pentablock (i.e. Đ = 1.38 for both the acid-synthesized DP = 500 per block pentablock and the conventional RAFT-synthesized DP = 100 per block pentablock). When conventional RAFT polymerization (i.e. through the exclusive use of radical initiator) was employed to synthesize an equally high molecular weight pentablock (i.e. DP = 500 per block), the overall dispersity attained rose to 1.75 (Fig. S8 and S9, Table S7†). Furthermore, the total livingness recorded was reduced to just 64.5%, meaning that 35.5% of chains are terminated (Fig. S9, Table S7†). Instead, the corresponding multiblock synthesized by acid-triggered RAFT polymerization possessed only 9.4% terminated chains (Table S6†). As such, it can be concluded that acid-triggered RAFT polymerization can reduce termination up to 4-fold by reducing the overall radical initiator concentration required. Such a large difference in the overall livingness may be significant not only for the synthesis of multiblock copolymers but also for applications where post-polymerization modification is essential to install desired functionality at the chain-end.
Although the preparation of high molecular weight polymers represents one of the most challenging targets in the synthesis of multiblock copolymers, previous literature also focused on the synthesis of lower molecular weight multiblock copolymers to achieve the highest number of blocks possible. For instance, Perrier and co-workers reported an impressive icosablock copolymer comprised of low molecular weight blocks (DP = 3 per block).41 Although the initial dispersity recorded was as low as 1.10, this value gradually increased during the multiblock synthesis with a final dispersity of 1.36 while SEC showed clearly visible low molecular weight tailing. Harrisson and co-workers then convincingly demonstrated the limits of precision monomer placement suggesting a minimum DP of 6 in order to minimize the percentage of defective chains.43 In line with this recommendation, we set out to target an icosablock copolymer with DP = 6 per block despite the fact that it would, in principle, require higher radical initiator concentration as opposed to a multiblock with only DP = 3 per block. Acid-triggered RAFT polymerization started with a relatively high dispersity of 1.25 and this value gradually decreased down to 1.14 for the octablock copolymer while reaching high monomer conversions throughout the synthesis (Fig. 5a and b, Table S8†). Unlike the previous synthesis of multiblock copolymers via either ATRP or RAFT polymerization methodologies, the dispersity was maintained for the next monomer additions yielding an icosablock copolymer with an overall dispersity of 1.15.25,41 To date, this is the lowest reported dispersity for such a complex architecture and further supports the high livingness obtained via acid-triggered RAFT polymerization. A minimum conversion of at least 97% was ensured prior to the next monomer addition for each block.
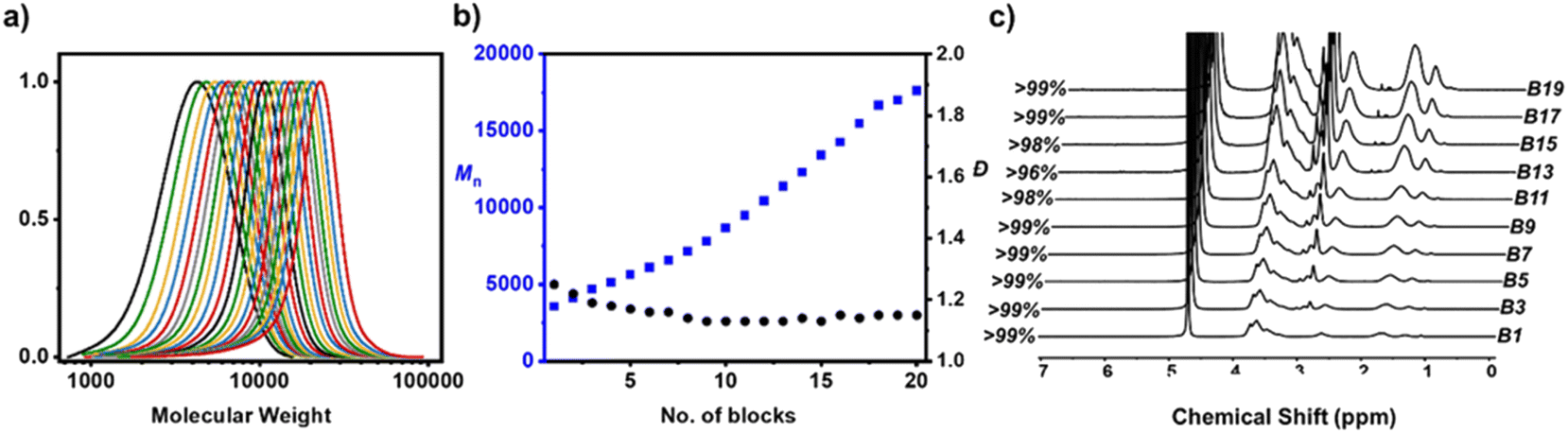 | ||
| Fig. 5 Sequence-controlled multiblocks. (a) SEC traces illustrating the molar mass distributions of consecutive blocks during the synthesis of an icosablock copolymer with DP = 6 (RI detector). (b) Evolution of dispersity (black) and the measured molecular weight (blue) during this multiblock synthesis. (c) Representative 1H NMR spectra illustrating the monomer conversion. | ||
Last but not least, we were also interested in synthesizing a more complex multiblock copolymer simultaneously consisting of methacrylate, acrylate, and acrylamide-based monomers. In particular, glycerol monomethacrylate (GMA), poly(ethylene glycol) methyl ether acrylate (PEGA480), DMA and NAM were employed to construct the pentablock copolymers under the following sequence: PGMA-b-PPEGA-b-PDMA-b-PNAM-b-PDMA. In line with our previous synthesis, acid-triggered RAFT polymerization enabled access to a well-defined material with narrow molar mass distributions (Đ = 1.23) and an Mn of 51000 (Fig. S10, Table S9†). Instead, our control experiment of the identical multiblock using a radical initiator resulted in significantly lower molecular weight tailing and an overall higher dispersity (Đ = 1.46). These results highlight the compatibility of our approach to other monomer classes such as methacrylates and acrylates (Fig. S10, Table S10†).
Conclusions
To summarize, in this work we employed acid in RAFT polymerization to prepare a range of multiblock copolymers with variable DPs per block. The presence of the acid substantially increased the polymerization rate thereby minimizing the quantity of required initiator employed to achieve quantitative monomer conversion. The lower amount of radical initiator utilized consequently suppressed the extent of termination events and preserved the end-group fidelity, yielding multiblock copolymers with high livingness. When compared to conventional RAFT polymerization, a four-fold decrease in the proportion of terminated chains was calculated. Notably, the synthesis of very high molecular weight multiblock copolymers was achieved with each block set at either DP = 100, DP = 200 or DP = 500. In all cases, the livingness exceeded 90% thus leading to the preparation of well-defined materials. Finally, an icosablock copolymer was synthesized where the dispersity was maintained at approximately 1.16 throughout the iterative monomer additions thus indicating a highly living multiblock copolymer. Taken together, acid-triggered RAFT polymerization enables the advanced synthesis of multiblock copolymers with high livingness by significantly suppressing termination events.Data availability
All the data supporting this article have been included in the main text and the ESI.†Author contributions
A. A. managed and directed the overall project and supervised the experimental work. N. P. T. also co-supervised part of the experimental work. M.-N. A. performed the experiments. M.-N. A. analysed the data with input from A. A. and N. P. T. M.-N. A. and A. A. co-wrote the manuscript with input from N. P. T. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A. acknowledges ETH Zürich (Switzerland) for financial support. N. P. T. acknowledges the award of a DECRA Fellowship from the ARC (DE180100076). M. L. C. acknowledges support from Flinders University, an ARC Laureate Fellowship from the ARC (FL170100041) and supercomputer time on the National Facility of Australian National Computational Infrastructure.Notes and references
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595 CrossRef CAS.
- E. Harth, A. Bosman and C. Hawker, Chem. Rev., 2001, 101, 3661–3688 CrossRef PubMed.
- K. Parkatzidis, H. S. Wang, N. P. Truong and A. Anastasaki, Chem, 2020, 6, 1575–1588 CAS.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Chem. Sci., 2019, 10, 8724–8734 RSC.
- J. De Neve, J. J. Haven, L. Maes and T. Junkers, Polym. Chem., 2018, 9, 4692–4705 RSC.
- G. K. Clothier, T. R. Guimarães, S. W. Thompson, J. Y. Rho, S. Perrier, G. Moad and P. B. Zetterlund, Chem. Soc. Rev., 2023, 52, 3438–3469 RSC.
- H. S. Wang, K. Parkatzidis, S. Harrisson, N. P. Truong and A. Anastasaki, Chem. Sci., 2021, 12, 14376–14382 RSC.
- V. P. Beyer, J. Kim and C. R. Becer, Polym. Chem., 2020, 11, 1271–1291 RSC.
- S. M. Barbon, N. P. Truong, A. G. Elliott, M. A. Cooper, T. P. Davis, M. R. Whittaker, C. J. Hawker and A. Anastasaki, Polym. Chem., 2020, 11, 84–90 RSC.
- P. R. Judzewitsch, T. K. Nguyen, S. Shanmugam, E. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559–4564 CrossRef CAS PubMed.
- A. Kuroki, P. Sangwan, Y. Qu, R. Peltier, C. Sanchez-Cano, J. Moat, C. G. Dowson, E. G. Williams, K. E. Locock and M. Hartlieb, ACS Appl. Mater. Interfaces, 2017, 9, 40117–40126 CrossRef CAS PubMed.
- R. Namivandi-Zangeneh, E. H. Wong and C. Boyer, ACS Infect. Dis., 2021, 7, 215–253 CrossRef CAS PubMed.
- J.-F. Lutz, J.-M. Lehn, E. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- B. V. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed.
- M. Zamfir, P. Theato and J.-F. Lutz, Polym. Chem., 2012, 3, 1796–1802 RSC.
- M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917–924 CrossRef CAS PubMed.
- F. S. Bates, M. A. Hillmyer, T. P. Lodge, C. M. Bates, K. T. Delaney and G. H. Fredrickson, Science, 2012, 336, 434–440 CrossRef CAS PubMed.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- C. Boyer, A. Derveaux, P. B. Zetterlund and M. R. Whittaker, Polym. Chem., 2012, 3, 117–123 RSC.
- C. Boyer, A. H. Soeriyadi, P. B. Zetterlund and M. R. Whittaker, Macromolecules, 2011, 44, 8028–8033 CrossRef CAS.
- Y.-M. Chuang, A. Ethirajan and T. Junkers, ACS Macro Lett., 2014, 3, 732–737 CrossRef CAS PubMed.
- B. Wenn, A. Martens, Y.-M. Chuang, J. Gruber and T. Junkers, Polym. Chem., 2016, 7, 2720–2727 RSC.
- A. Anastasaki, V. Nikolaou, N. W. McCaul, A. Simula, J. Godfrey, C. Waldron, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 1404–1411 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, Q. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3542 RSC.
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS PubMed.
- A. Anastasaki, B. Oschmann, J. Willenbacher, A. Melker, M. H. Van Son, N. P. Truong, M. W. Schulze, E. H. Discekici, A. J. McGrath, T. P. Davis, C. M. Bates and C. J. Hawker, Angew. Chem., Int. Ed., 2017, 56, 14483–14487 CrossRef CAS PubMed.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 CrossRef CAS PubMed.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Chem.–Asian J., 2013, 8, 1634–1644 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- N. P. Truong, G. R. Jones, K. G. Bradford, D. Konkolewicz and A. Anastasaki, Nat. Rev. Chem, 2021, 5, 859–869 CrossRef CAS PubMed.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. b. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed.
- L. Martin, G. Gody and S. Perrier, Polym. Chem., 2015, 6, 4875–4886 RSC.
- P. B. Zetterlund, G. Gody and S. Perrier, Macromol. Theory Simul., 2014, 23, 331–339 CrossRef CAS.
- G. K. Clothier, T. R. Guimarães, M. Khan, G. Moad, S. Perrier and P. B. Zetterlund, ACS Macro Lett., 2019, 8, 989–995 CrossRef CAS PubMed.
- G. K. Clothier, T. R. Guimarães, G. Moad and P. B. Zetterlund, Macromolecules, 2021, 54, 3647–3658 CrossRef CAS.
- G. K. Clothier, T. R. Guimarães, L. T. Strover, P. B. Zetterlund and G. Moad, ACS Macro Lett., 2023, 12, 331–337 CrossRef CAS PubMed.
- M. Khan, T. R. Guimarães, K. Choong, G. Moad, S. Perrier and P. B. Zetterlund, Macromolecules, 2021, 54, 736–746 CrossRef CAS.
- A.-C. Lehnen, J. Gurke, A. M. Bapolisi, M. Reifarth, M. Bekir and M. Hartlieb, Chem. Sci., 2023, 14, 593–603 RSC.
- A.-C. Lehnen, J. A. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537–1546 RSC.
- M.-N. Antonopoulou, G. R. Jones, A. A. Kroeger, Z. Pei, M. L. Coote, N. P. Truong and A. Anastasaki, Nat. Synth., 2024 DOI:10.1038/s44160-023-00462-9.
- D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2011, 44, 6738–6745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00399c |
| This journal is © The Royal Society of Chemistry 2024 |