
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifaceted nucleic acid probing with a rationally upgraded molecular rotor†
Tuan-Khoa
Kha
 ,
Qi
Shi
,
Nirali
Pandya
and
Ru-Yi
Zhu
*
,
Qi
Shi
,
Nirali
Pandya
and
Ru-Yi
Zhu
*
Department of Chemistry, National University of Singapore, 4 Science Drive 2, 117544, Singapore. E-mail: rzhu@nus.edu.sg
First published on 7th March 2024
Abstract
Probing the sequence alterations, structures, interactions, and other important aspects of nucleic acids serves as the cornerstone of understanding nucleic acid-mediated biology and etiology, as well as the development of nucleic acid-based therapeutics. New strategies capable of accommodating these imperatives without necessitating specialized instrument or skills and potentially complementing existing methods are highly desired. Herein, we describe a rationally designed molecular rotor CCVJ-H ((9-(2-carboxy-2-cyanovinyl)julolidine-hydrazide)) and its superior performances compared to the universal base excision reporter probe CCVJ-1 in applications such as nucleic acid detection and DNA glycosylase assays. Furthermore, we showcase that the CCVJ-H probe accurately profiles the interactions between nucleic acids and small molecules, providing binding affinity and binding site information in a single reaction. We subsequently demonstrate the feasibility of applying the CCVJ-H system in high-throughput screening to identify nucleic acid-binding small molecules such as DNA CTG repeat expansion binders, potentially providing therapeutic interventions for myotonic dystrophy type 1. Finally, we profile the recognition difference between DNA/DNA and DNA/RNA against a library of small molecules, uncovering two drug-like molecules that preferentially bind DNA/RNA. We anticipate the versatile CCVJ-H probe will be a useful tool for both fundamental and translational nucleic acid research and application.
Introduction
Techniques capable of probing sequence alterations, structures, interactions, among other important aspects, of nucleic acids play a pivotal role in advancing our understanding of nucleic acid-mediated biological processes, and the development of improved diagnostic strategies or therapeutic interventions based on nucleic acids.1–4 Nucleic acid sequencing or polymerase chain reaction stands as the gold standard for precise identification of mutations or the presence of pathogens in disease diagnosis, which are complemented by various fluorescence-based detection methods.5–10 Crystallography,11 nuclear magnetic resonance spectroscopy,12 isothermal titration calorimetry (ITC),13 chemical probing,1–3 among others, serve as useful tools for structural determinations or profiling interactions of nucleic acids. Several of these techniques have been harnessed to probe interactions between nucleic acids and small molecules, with the aspiration of identifying more efficacious small-molecule therapeutics targeting nucleic acids.14–16 In spite of the availability of these established methodologies, novel approaches capable of accommodating these imperatives without necessitating specialized instrumentation or expertise are highly desirable. In particular, fluorescent molecular rotors have been widely utilized for DNA sensing and probing.17–19 Drawing inspiration from the molecular rotor CCVJ (9-(2-carboxy-2-cyanovinyl)julolidine),20,21 especially the recent work done by Kool and co-workers for measuring DNA glycosylase activity and DNA labelling, we surmise that this fluorescent probe might offer a convenient avenue to meet the aforementioned requisites (Fig. 1A).22–24 Indeed, the same lab recently demonstrated that CCVJ-1 can be employed to detect complementary DNA sequences with single-nucleotide specificity, albeit with relatively low sensitivity and long incubation time.24 The suboptimal efficacy observed in utilizing CCVJ-1 for such purposes could potentially be attributed to the inherent structural characteristics of CCVJ-1. Additionally, although targeting nucleic acids with small molecules for therapeutic interventions has been a hot and rapid growing area,14–16 fluorescence-based methods capable of profiling nucleic acid-small molecule interaction are lacking. Herein, we present the rational design of an upgraded molecular rotor and its applications in nucleic acid detection, structure probing, and exploration of nucleic acid interactions with DNA glycosylase and small molecules.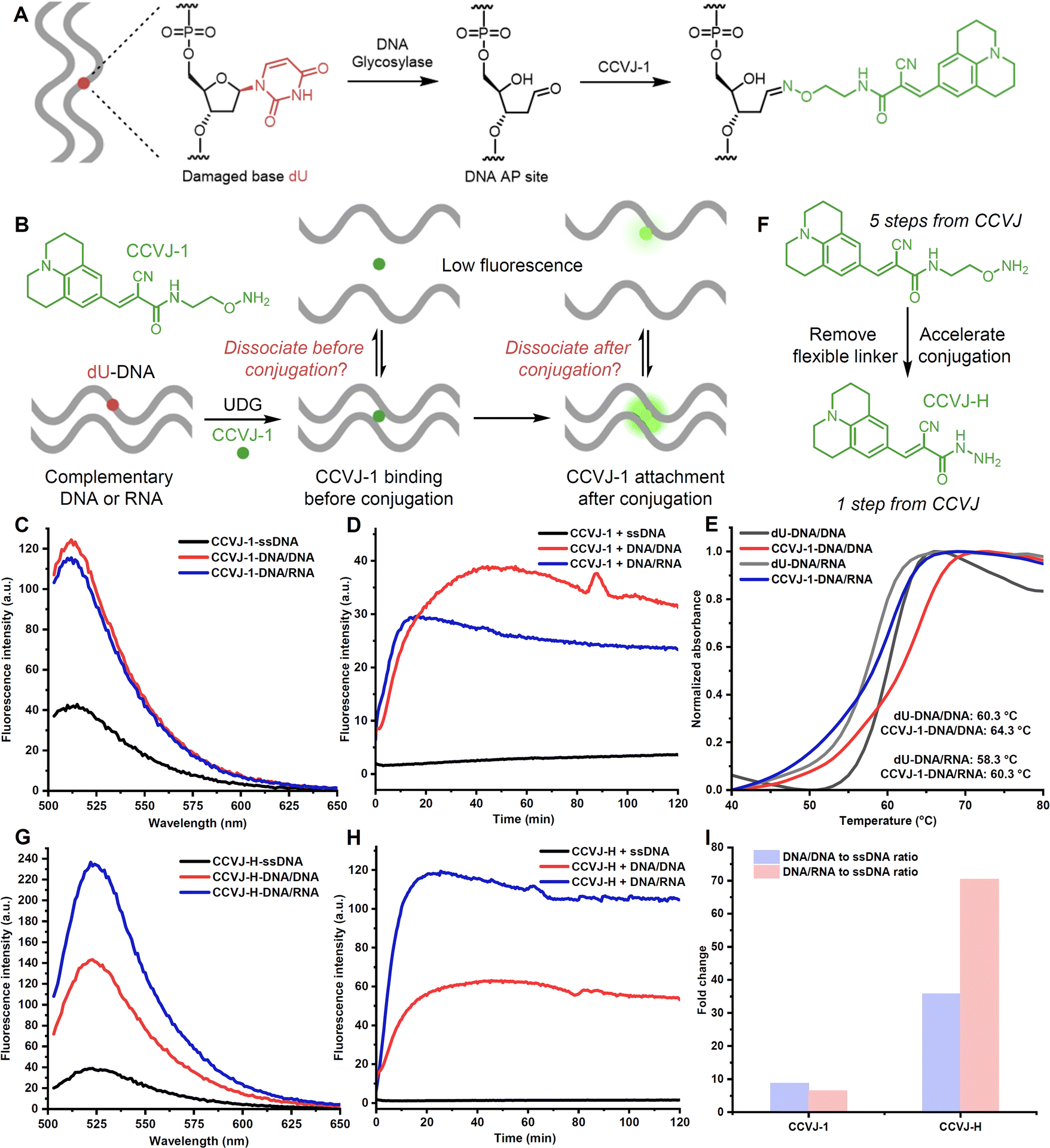 | ||
| Fig. 1 Rational design of an improved molecular rotor dye for nucleic acid sensing. (A) Reaction of CCVJ-1 with DNA abasic site for measuring DNA glycosylase activity and DNA labelling. (B) Possible CCVJ-1-induced destabilization pathways. (C) Emission spectra of CCVJ-1-conjugated DNA/DNA, DNA/RNA, and ssDNA. (D) Fluorescence real-time response of DNA/RNA, DNA/DNA and ssDNA with CCVJ-1 and UDG. (E) Tm measurement of CCVJ-1-conjugated DNA/DNA, DNA/RNA, and their unmodified counterparts (5.0 μM) in PBS buffer. (F) Design of an upgraded molecular rotor CCVJ-H. (G) Emission spectra of CCVJ-H-conjugated DNA/DNA, DNA/RNA, and ssDNA. (H) Fluorescence real-time response of DNA/RNA, DNA/DNA, and ssDNA with CCVJ-H and UDG. (I) Fluorescence fold change of each type of duplexes when reacted with probes (CCVJ-1 and CCVJ-H) and compared with ssDNA after 2 hours. (C–E and G–I) The excitation wavelength and emission wavelength were 485 nm and 530 nm, respectively. Fluorescence spectra were recorded with 2.0 μM nucleic acid in 50 mM Tris pH 7.0, 100 mM NaCl at room temperature. The reactions were conducted with 2.0 μM nucleic acid, 20 μM CCVJ-H, 50 U mL−1 UDG in 50 mM Tris pH 7.0, 100 mM NaCl at 37 °C. 23-mer 1 dU-DNA and its complementary strand, 23-mer DNA and 23-mer RNA were used. | ||
Results and discussion
The efficient oxime formation facilitated by the close association of planar and hydrophobic CCVJ-1 to a DNA apurinic/apyrimidinic site (AP site), which is generated by a DNA glycosylase, is critical for achieving high fluorescent signals and fast responses.22,25 The julolidine core of CCVJ-1 is larger than canonical DNA bases, potentially causing destabilization of the DNA duplex upon CCVJ-1 binding before or after the oxime formation. Both scenarios may contribute to the dissociation of the DNA duplex, thereby dampening fluorescence signal (Fig. 1B). This phenomenon could potentially elucidate the prevalent utilization of CCVJ-1 in highly stable duplexes, such as hairpin DNA (hpDNA) or long duplexes. Indeed, the Kool lab demonstrated that a single-stranded 33-mer DNA, labelled with four CCVJ-1 moieties, could not hybridize with its complementary DNA.24 To investigate the destabilization effect of CCVJ-1 in application relevant models, we sought to examine the destabilization effect and the resulting fluorescence change on shorter DNA/DNA and DNA/RNA with one CCVJ-1 label. We began the study by converting a single deoxyuridine (dU) containing 23-mer single stranded DNA (ssDNA) with E. coli uracil DNA glycosylase (UDG) to a DNA AP site, which was subsequently reacted with excess CCVJ-1 overnight to ensure full conversion. The synthesized conjugate (2 μM) was annealed with its complementary DNA or RNA to afford the CCVJ-1 labeled DNA/DNA or DNA/RNA, respectively. The intensities of the emission fluorescence for CCVJ-1 labeled ssDNA, DNA/DNA, and DNA/RNA were measured (Fig. 1C and S1†). Concurrently, the same ssDNA, DNA/DNA, and DNA/RNA constructs containing a single dU were treated with UDG and CCVJ-1 (20 μM) respectively, and the mixtures were incubated and the fluorescence intensities were monitored over the course of 2 hours. The fluorescence signals of DNA/DNA and DNA/RNA plateaued within 40 and 15 minutes, respectively. In contrast, the fluorescence signal from ssDNA exhibited little increase over the 2 hours period (Fig. 1D). In all cases, the fluorescence intensities of the kinetic reactions were significantly lower than those of annealed stoichiometric conjugates, suggesting CCVJ-1-mediated destabilization subsequent to oxime formation is not the predominant factor for reduced signals. In fact, the melting temperatures (Tm) of the CCVJ-1-embeded DNA/DNA and DNA/RNA are slightly higher than the unmodified counterparts, clearly showing a stabilization effect of CCVJ-1 after oxime formation (Fig. 1E). This is likely due to the enhanced stacking interactions between CCVJ-1 and neighboring bases within the duplex. These results collectively suggest that the dissociation triggered by CCVJ-1 binding likely precedes the oxime formation.To address the low conversion issues for DNA/DNA and DNA/RNA with CCVJ-1 and to enhance fluorescence signals, we proposed two strategies: the reduction of the julolidine core size or acceleration of the conjugation step between the DNA AP site and a molecular rotor. However, any structural changes to the julolidine core could compromise its binding affinity to the DNA AP site and perturb fluorescence characteristics. Thus, we contend that accelerating the conjugation step through structural modifications of CCVJ-1, without affecting its binding affinity and fluorescence properties, offers a more viable avenue. Through careful examination of the computational model depicting the binding of CCVJ-1 bound to the DNA AP site, we reason that removing the flexible linker connecting the CCVJ moiety to the oxyamine group might expedite the conjugation process.22 We therefore synthesized CCVJ-hydrazide (CCVJ-H) one step compared to a 5-step synthesis for CCVJ-1 from CCVJ (Fig. 1F).21,25,26 The stochiometric conjugates of CCVJ-H in the same ssDNA, DNA/DNA, and DNA/RNA were constructed as described and their respective emission fluorescence intensities were measured (Fig. 1G and S1†). Similar kinetic experiments were conducted for CCVJ-H and the fluorescence signals were recorded over the course of 2 hours (Fig. 1H and I). Intriguingly, the utilization of the CCVJ-H label on the DNA/RNA conjugate yielded a twofold enhancement in fluorescence intensity compared to its CCVJ-1 labeled counterpart presumably due to the A form structure of DNA/RNA restricts the bond rotation of CCVJ-H more effectively than that of CCVJ-1.27 Concurrently, the fluorescence intensities exhibited by CCVJ-1 and CCVJ-H when applied to ssDNA or DNA/DNA conjugates were found to be comparable, which might be attributed to similar conformational restrictions for CCVJ-H and CCVJ-1 in these contexts. Furthermore, it is imperative to highlight that CCVJ-H exhibited markedly superior fluorescence responses and conversions in kinetic experiments, particularly when probing DNA/RNA hybrid. This pronounced enhancement in sensitivity, as afforded by CCVJ-H, holds substantial relevance for a diverse range of applications involving nucleic acid probing. It is worth noting that CCVJ-H consistently outperformed CCVJ-1 in terms of fluorescence responses, even in the hairpin DNA (Fig. S1 and 2†), and long DNA specifically optimized for CCVJ-1 (Fig. S3†). The successful incorporation of CCVJ-H to DNA was confirmed by MALDI-TOF and HPLC analyses (Fig. S4†).
To further amplify fluorescence intensities, two CCVJ-H labels were incorporated, and the impact of opposite bases and the inter-dye distance was investigated (Fig. 2A). In comparison to the single CCVJ-H-labeled DNA/RNA, the introduction of an additional CCVJ-H label, positioned opposite adenine and separated by three bases, resulted in a substantial augmentation of fluorescence signal (Fig. 2B). Employing two CCVJ-H labels in a manner that they were both positioned opposite adenine and separated by nine bases, yielding markedly reduced fluorescence output, likely attribute to steric hindrance induced destabilization and dissociation. Notably, the use of smaller uracil opposite CCVJ-H labels led to even higher fluorescence intensity, underscoring the significance of opposite base selection in achieving optimal signal enhancement. To assess the nucleic acid detection capability, a mixture of two spectator RNAs along with a specific target RNA were utilized (Fig. 2C). With the optimal DNA probe and CCVJ-H, robust and reproducible fluorescent signal was obtained (Fig. 2D). In the absence of the target RNA, no fluorescence enhancement was observed over a period of 2 hours. It is noteworthy that the presence of spectator RNAs had no discernible impact on the fluorescence response, thereby underscoring the marked specificity of the CCVJ-H detection system. To demonstrate the utility of this detection methodology in a context relevant to disease diagnosis, we selected the wild-type KRAS (WT-KRAS) DNA and the G12V-KRAS DNA segments, differed by a single nucleotide (Fig. 2E).28–30 A short 11 mer dU-containing DNA (dU-KRAS-cDNA) was designed to have T:C and U:T mismatches for WT-KRAS and only one U:T mismatch for G12V-KRAS (Fig. 2F). The melting temperature analysis revealed that no duplex formation occurred at 37 °C for the WT-KRAS DNA with the dU-KRAS-cDNA (Fig. S5A†). Consequently, minimal fluorescence enhancement was observed comparable to dU-KRAS-cDNA only. In contrast, the G12V-KRAS DNA segment hybridized with dU-KRAS-cDNA more effectively, leading to a rapid increase in fluorescence upon CCVJ-H incorporation, followed by a subsequent decrease in fluorescence, which is likely due to CCVJ-H incorporation-induced destabilization and strand separation (Fig. S5B†). Notably, the DNA point mutation can be determined in the first 5 min with a 10-fold fluorescence enhancement, featuring a rapid diagnosis process. Even after 2 hours, there is still a 4-fold fluorescence increase, large enough for point mutation differentiation. In conclusion, the CCVJ-H system exhibited a remarkable capability to sensitively and rapidly detect DNA point mutations in the KRAS gene fragment. A linear response was observed for DNA or RNA sensing with single-nucleotide specificity, high sensitivity, and short detection time, underscoring the great potential of CCVJ-H in various nucleic acid sensing applications (Fig. S6†).
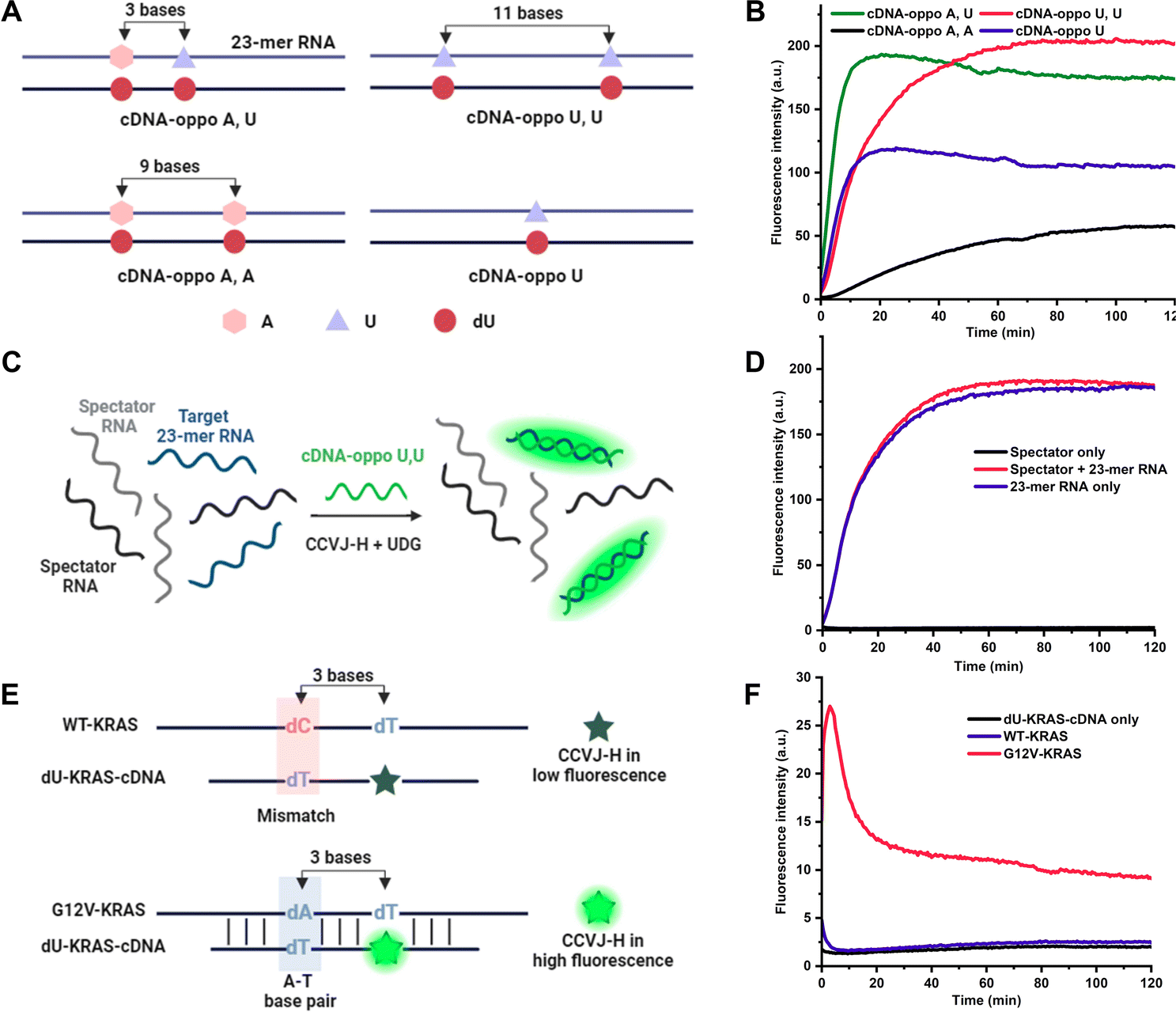 | ||
| Fig. 2 Applications of CCVJ-H in nucleic acid detection. (A) Relative positions of opposite bases in the 2 dU 23-mer RNA (A and/or U) and corresponding complementary dU-DNA. (B) Fluorescence kinetics of 2 dU-containing DNA/RNA duplexes with CCVJ-H and UDG. (C) Selective detection of RNA by CCVJ-H. (D) Fluorescence kinetics of cDNA-oppo U,U with CCVJ-H and UDG in the presence and absence of target 23-mer RNA. (E) Detection of single nucleotide polymorphism (SNP) in KRAS DNA (WT-KRAS and G12V-KRAS). (F) Fluorescence kinetics of KRAS/dU-cDNA duplex with CCVJ-H and UDG. (B, D and F) [DNA/RNA] or [DNA/DNA] = 2.0 μM, [CCVJ-H] = 20 μM, [UDG] = 50 U mL−1. The excitation wavelength and emission wavelength were 485 nm and 530 nm, respectively. The reactions were conducted in 50 mM Tris pH 7.0, 100 mM NaCl at 37 °C. | ||
Elucidating and uncovering the substrate preference and scope of DNA glycosylase is important for the understanding of the DNA base excision repair pathways.31,32 Conventional approaches rely on radiation/fluorescence-based electrophoresis assays, which are time consuming, laborious and low-throughput. Interestingly, UDG has displayed comparable efficacy in recognizing and repairing dU in both DNA/DNA and DNA/RNA contexts, opening the door to the intriguing possibility of DNA repair during transcription involving DNA glycosylases.24,33,34 In pursuit of assessing the prospect with human DNA glycosylases, DNA-3-methyladenine glycosylase (MPG) was examined using a 23-mer deoxyinosine (dI)-containing DNA hybridized with its complementary DNA or RNA (i.e., cDNA or cRNA) (Fig. 3A).31,35 Time-dependent experiments revealed that MPG exhibited superior dI repair efficiency in DNA/DNA compared to DNA/RNA (Fig. 3B). Nevertheless, dI excision by MPG in DNA/RNA, albeit less efficient, was demonstrated. This result indicates that DNA/DNA is a better substrate for MPG than DNA/RNA, which agrees with a recent finding.36 Notably, CCVJ-H produced notably higher fluorescence signals than CCVJ-1 in the context of DNA/RNA, further confirming the superiority of CCVJ-H with a different DNA glycosylase (Fig. 3C and D). Notably, these data directly compared the enzymatic activity of MPG on DNA/DNA and DNA/RNA substrates for the first time. The conventional MPG glycosylase assay necessitates alkaline cleavage of the AP site followed by electrophoresis, a method characterized by its indirect, labor-intensive, and low-throughput nature. To overcome these limitations, we leveraged the CCVJ-H system, which enabled the real-time reporting of substrate preferences, aligning seamlessly with the results obtained from conventional electrophoresis-based fluorescent assays (Fig. 3E).37 In addition, this assay allows direct comparison of enzyme activity among different DNA glycosylases, such as UDG and MPG, matching with the previous report.38 Owing to the working mechanism of CCVJ-H, the probe can be applied to any other DNA glycosylase, such as the human uracil DNA glycosylase SMUG1, providing reliable and rapid results on enzyme activity in real time (Fig. S7†).39 Taken together, we believe the CCVJ-H system is a valuable tool for the comprehensive assessment of substrate scope and preferences for DNA glycosylases conveniently, rapidly, and cost-effectively.
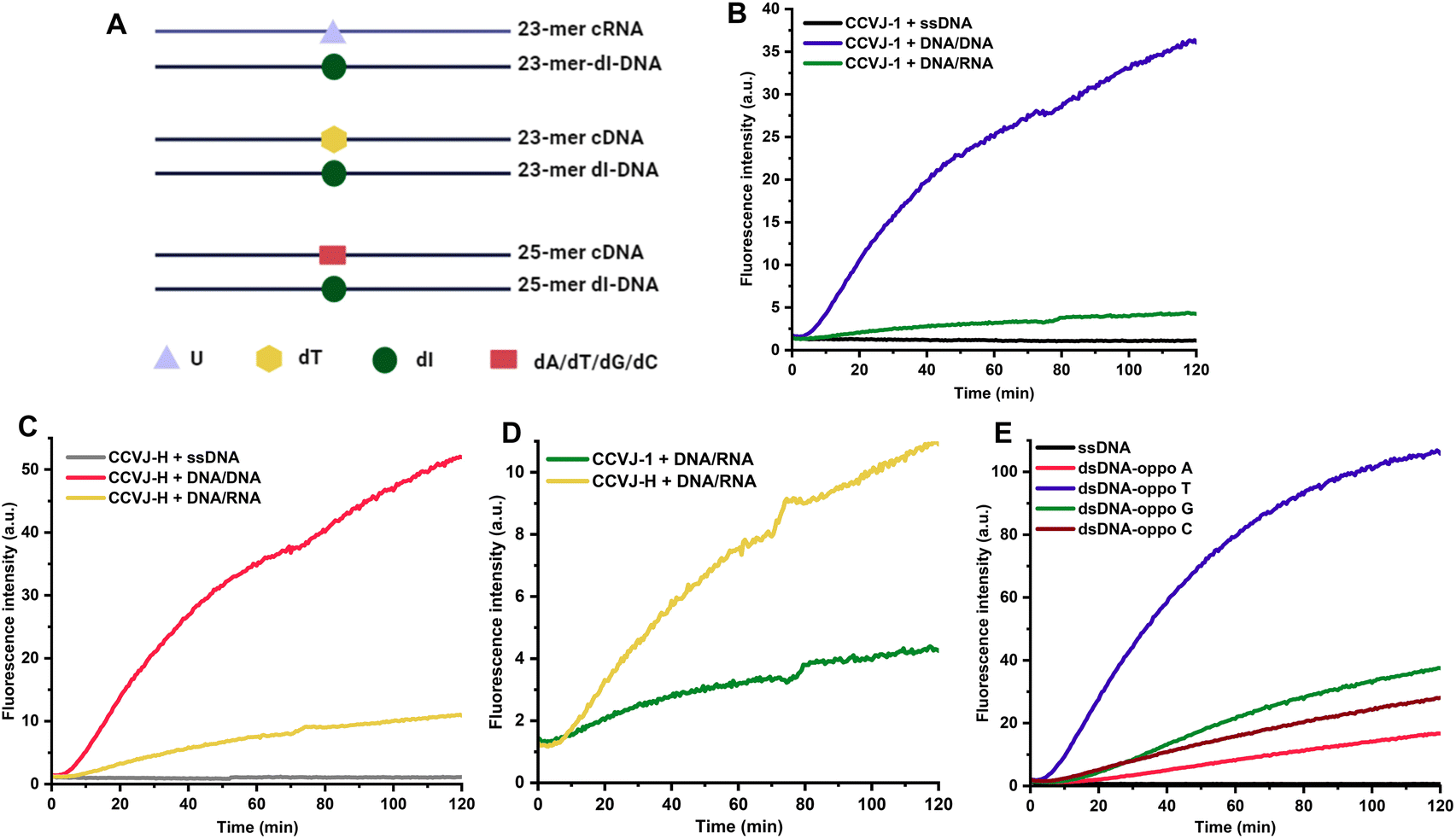 | ||
| Fig. 3 Evaluation of DNA-3-methyladenine glycosylase (MPG) substrate preference with CCVJ-H. (A) Sequence of dI-containing DNA and its complementary DNA or RNA. (B) Real-time fluorescent response of CCVJ-1 for different dI-containing DNA substrates. (C) Real-time fluorescent response of CCVJ-H for different dI-containing DNA substrates. (D) Comparison of the fluorescence response between CCVJ-H and CCVJ-1 for DNA/RNA. (E) Effect of opposite bases for MPG substrates. (B–E) Reaction conditions were [DNA/DNA] or [DNA/RNA] = 2 μM, [probe] = 20 μM, [MPG] = 50 U mL−1 in 50 mM Tris pH 7.0, 100 mM NaCl. Excitation wavelength and emission wavelength was 485 and 530 nm, respectively. The reactions were conducted at 37 °C. (B–D) dI 23-mer DNA and its complementary DNA or RNA (23-mer DNA and 23-mer RNA) were used. (E) 25-mer dI-DNA and its complementary DNA with different opposite bases (25-mer cDNA oppo A, T, G, C) were used. | ||
In addition to the aforementioned applications of CCVJ-H, we envision that the CCVJ-H system holds the potential for profiling interactions between nucleic acids and small molecules, as well as for identifying small molecules that bind to nucleic acids via high-throughput screening (HTS).40 A notable fluorescent intercalator displacement assay suitable for HTS was developed by Boger using strong DNA intercalators ethidium bromide (EtBr) and Thiazole Orange (TO).41 Weaker DNA binders may be overlooked especially at low concentrations due to high DNA-binding affinity of EtBr or TO. Small molecules bind DNA via non-intercalation and/or bind at different locations may not displace EtBr or TO and yield fluorescence reduction. In this regard, we hypothesize that small molecules binding to or near the DNA damage site (i.e., dU) will compete with UDG for binding and processing, resulting in a diminished fluorescence signal (Fig. 4A). This decrease in fluorescence should correlate with the binding affinity and inversely correlate to the binding distance from the damage site. Notably, dU differs from thymidine only by a single methyl group at the 5-position and should induce minimal perturbations in the original DNA sequence and structure. To demonstrate the feasibility of employing CCVJ-H for the facile and sensitive probing of DNA-small molecule interactions, along with the quantitative assessment of binding affinity, we assembled a panel of well-known DNA-binding compounds exhibiting various interaction modes (e.g., intercalation, grove binding, electrostatic attraction, or combinations thereof).42 Five compounds, each at concentrations of 2 and 20 μM, were incubated with a 17-mer dU-hpDNA (2 μM), prior to the addition of CCVJ-H (20 μM) and UDG at 37 °C (Fig. 4B and S8†). Since binding affinity inversely correlates to fluorescence intensity, this assay facilitated a convenient ranking of binding affinities for the five compounds assessed at 20 μM, with the order being as follows: 9-aminoacridine > ethidium bromide (EtBr) ≈ daunorubicin oxime ≈ ellipticine > Hoechst. Notably, this ranking aligns well with the ITC data, underscoring the reliability of CCVJ-H in comparing the binding affinities of small molecules with diverse structures and binding modes for a given DNA sequence (Fig. S9†). Furthermore, we demonstrated the capability of CCVJ-H to precisely identify binding sites with base resolution (Fig. 4C). We designed two 33-mer dU-hpDNAs containing six consecutive AT base pairs (AT-rich region). In the “dU-near” hpDNA, we substituted the central T with dU. In the “dU-far” hpDNA, we replaced the T located two bases away from the AT-rich region with dU. As Hoechst is known for its preference in binding to AT-rich regions in the minor grove,43 we anticipated that Hoechst binding to the “dU-near” hpDNA would inhibit UDG processing to a greater extent than its binding to the “dU-far” hpDNA. Indeed, minimal fluorescence increase was observed, even at a Hoechst concentration of 2 μM for the “dU-near” hpDNA, while the fluorescence response remained relatively unaffected in the “dU-far” hpDNA (Fig. S10†). EtBr is a well-established potent and non-selective DNA intercalator as evidenced by its equivalent fluorescence responses when interacting with both the “dU-near” and the “dU-far” hpDNAs, thereby indicating minimal sequence preference for binding. Moreover, the CCVJ-H system was employed in a more complex setting to probe the interactions between a DNA aptamer and its small-molecule ligand (Fig. 4D). Four Ts located in different regions were substituted separately by dU for subsequent UDG processing and CCVJ-H conjugation. Small molecule binding to or near the damage site should result in a decrease in fluorescence. We found that dopamine likely interacts more closely to regions 1 and 2 as their fluorescence intensities were reduced in the presence of dopamine.44 Regions 3 and 4 are likely far away from the dopamine binding site. These findings correlate well with the ITC-based mutational assays, underscoring the reliability of the CCVJ-H assay for probing the interactions between complex DNA structures and small molecules.45 To the best of our knowledge, the CCVJ-H system stands as the sole method capable of providing both quantification of binding affinity and detailed binding site information for nucleic acid-small molecule interactions in a single experiment.
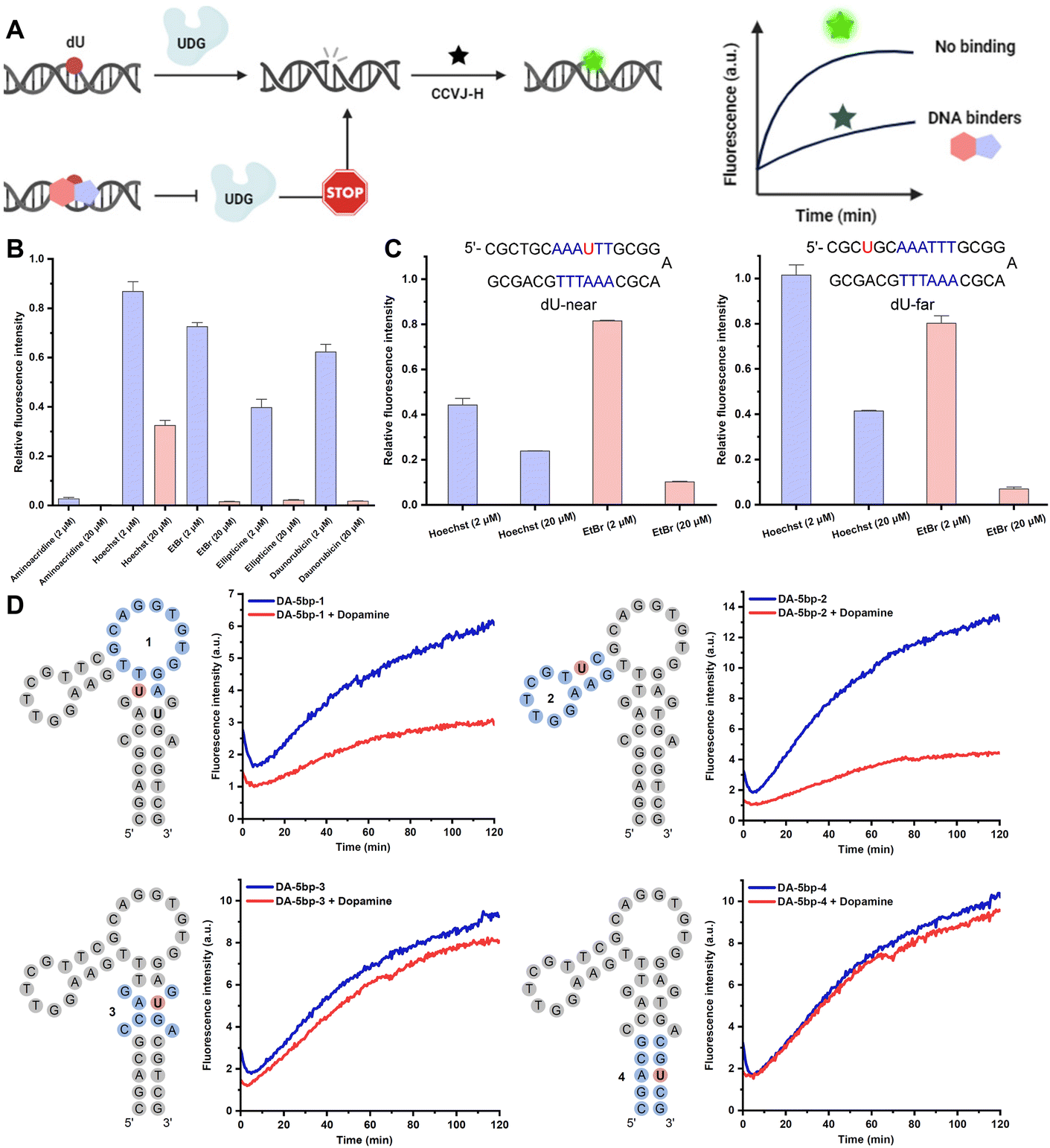 | ||
| Fig. 4 Profiling interactions between nucleic acids and small molecules with CCVJ-H. (A) Schematic illustration of DNA-binder identification assay. (B) Assay validation with known DNA-binding small molecules. dU 17-mer hpDNA (oppo C) were used. (C) Effect of Hoechst and EtBr on fluorescence response with dU placed near (dU-near) or far (dU-far) from AT-rich region. (D) Binding site probing for dopamine-binding DNA aptamer with CCVJ-H. (B–D) The excitation wavelength and emission wavelength were 485 nm and 530 nm, respectively. All reactions occurred between DNA (2.0 μM) and CCVJ-H (20 μM) in the presence of UDG (50 U mL−1) at 37 °C. (B and C) The reactions were conducted in 20 mM Tris pH 8.0, 1.0 mM DTT, 1.0 mM EDTA with the indicated small molecule concentration (2.0 μM or 20 μM). (D) The reactions were conducted in 50 mM Tris pH 7.0, 100 mM NaCl, 5.0 mM MgCl2 with 50 μM dopamine. | ||
To demonstrate the amenability of the CCVJ-H system for HTS in a disease relevant-model, we set out to identify small-molecule ligands targeting DNA CTG repeat expansion (Fig. 5A). Repeat expansion disorders, primarily affect the nervous system, are a class of genetic diseases that are caused by expansions in DNA repeats.46,47 Expanded trinucleotide repeat, such as CAG, GCG, CTG, and CGG, both in coding and non-coding sequences in distinct genes results in a diverse group of diseases with mechanisms linked to protein levels or toxicity, RNA, and/or both.48 Although targeting RNA repeat expansion with small molecules is well documented,49,50 the endeavour to target DNA repeat expansion has garnered much less attention. For a proof-of-concept purpose, we selected a seven-unit CTG repeat expansion with the central T replaced by dU for HTS, aimed at identifying small-molecule binders for this specific DNA motif. The CTG repeat expansion motif occurs in myotonic dystrophy type 1 (DM1), which is transcribed to the CUG RNA expansion that sequesters the alternative-splicing regulator muscleblind-like protein, leading to splicing defects and disease symptoms.51,52 In a 384-well microplate, DNA (2.0 μM) was incubated with small molecules (20 μM) at room temperature for 15 minutes. Subsequently, CCVJ-H (20 μM) and UDG (5.0 U mL−1) were introduced into the mixture. The real-time fluorescence intensities were collected immediately following UDG addition and extended over a 2 hours period. The relative fluorescence ratio, comparing the presence of small molecules to a DMSO-only control, was calculated to evaluate the binding affinity. Among the 80 small molecules examined at 20 μM, six potent compounds exhibited a fluorescence ratio of less than 0.2 were selected for further validation (Fig. S11†). In a subsequent round of screening, the entire process was repeated at 2 μM of small molecules, and all of the selected compounds exhibited binding affinity to DNA, with the fluorescence ratio ranging from 0.2 to 0.6 (Fig. 5B and S12†). Notably, Actinomycin D (G2) and Mitoxantrone (A9) have been previously shown to reduce the level of CTG repeats in DM1 model (Fig. 5C).53,54
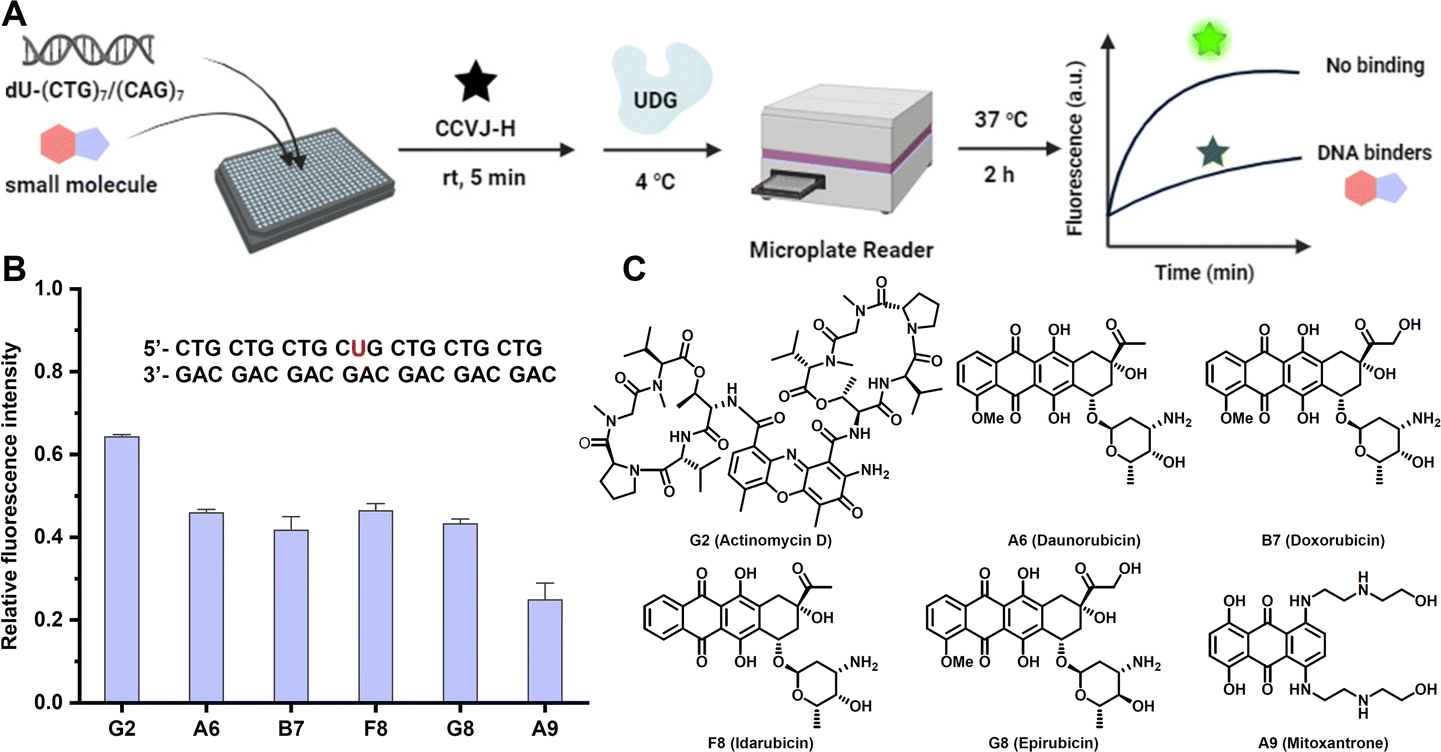 | ||
| Fig. 5 Identification of DNA repeat expansion binding small molecules via HTS. (A) Schematic representation of the HTS assay. (B) Effect of potential DNA-binding small molecules (2.0 μM) to the fluorescence response. (C) Chemical structures of potential (CTG)7/(CAG)7 binders. (B) The reactions were conducted at 37 °C with DNA duplex (2.0 μM) and CCVJ-H (20 μM) in the presence of UDG (5.0 U mL−1) at 20 mM Tris pH 8.0, 1.0 mM DTT, 1.0 mM EDTA. The excitation and emission wavelength were 485 nm and 530 nm, respectively. | ||
DNA/RNA is structurally and topologically distinct from DNA/DNA. Understanding the differences in terms of how they interact with small molecules is important for basic and translational research.55 Persistent DNA/RNA structures in cells, known as R-loops, are formed during transcription, wherein the nascent RNA strand bases pair with its template DNA, displacing the non-template strand.56 These R-loops play diverse roles in a number of physiological processes and may contribute to the development of diseases. Particularly prevalent at promoters, R-loops modulate gene expression through different mechanisms.57 Consequently, targeting R-loops emerges as a potentially promising strategy for disease treatment. However, there is no method capable of profiling the interaction between DNA/RNA and a large number of small molecules.55 Thus, we embarked on an HTS involving a 23-mer DNA/RNA sequence and 80 small molecules, comparing the binding profile with its DNA/DNA counterpart (Fig. 6A). The majority of tested compounds displayed negligible binding affinity for both DNA/DNA and DNA/RNA. Notably, five molecules, all of which are apparent nucleic acid binders, demonstrated potent binding affinities towards both DNA/DNA and DNA/RNA, without exhibiting a binding preference. Anti-cancer drugs C10 and B11 exhibited a preference for binding with DNA/RNA over DNA/DNA, marking the first example of discovering drug-like small molecules with a binding preference for DNA/RNA (Fig. 6B, D, S13B and C†). Compound G2, a strong DNA/DNA binder, intriguingly displayed weak binding with DNA/RNA, showcasing a 5-fold binding difference (Fig. 6C and S13A†). Taken together, the CCVJ-H probe presents a valuable tool for in vitro screening, enabling the discovery of small molecules that bind DNA repeat expansions and DNA/RNA hybrid thus holding promise for therapeutic interventions in the context of repeat expansion disorders and diseases associated with R-loops.
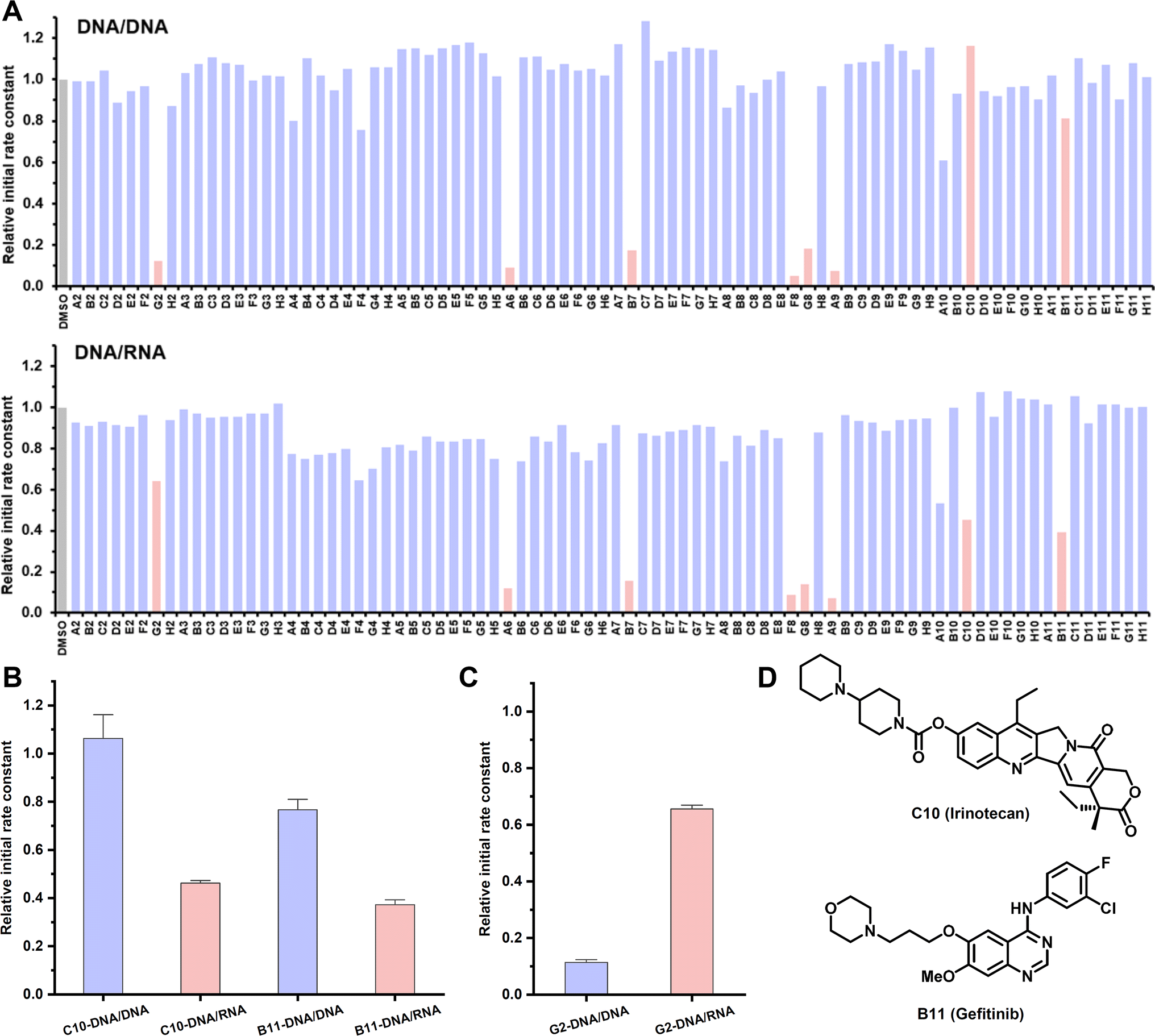 | ||
| Fig. 6 Profiling interactions between DNA/RNA and small molecules with CCVJ-H. (A) Binding profile of 80 small molecules with DNA/RNA comparing with DNA/DNA counterpart. (B) Small molecules with binding preference with DNA/RNA over DNA/DNA. (c) Small molecules with binding preference with DNA/DNA over DNA/RNA. (D) Chemical structures of small molecules with binding preference with DNA/RNA over DNA/DNA. (A–C) The reactions were conducted at 37 °C with DNA/DNA or DNA/RNA duplex (2.0 μM), CCVJ-H (20 μM), UDG (5.0 U mL−1) and small molecules (20 μM) at 20 mM Tris pH 8.0, 1.0 mM DTT, 1.0 mM EDTA. The excitation and emission wavelength were 485 nm and 530 nm, respectively. 23-mer 1 dU-DNA and its complementary strand, 23-mer DNA or 23-mer RNA were used. | ||
Conclusions
In summary, we have employed a rational design approach to develop the molecular rotor CCVJ-H, which reacted rapidly with DNA AP sites, resulting in a remarkable enhancement of fluorescence signals. Notably, CCVJ-H consistently outperformed the UBER probe CCVJ-1 across all the applications we investigated. Furthermore, CCVJ-H was synthesized through a one-step chemical synthesis from CCVJ without chromatography, in contrast to the five-step synthesis required for CCVJ-1 from the same starting material. The utility of the CCVJ-H probe was extended to the specific and sensitive detection of nucleic acids at a single-nucleotide resolution within a matter of minutes, illustrated by the precise differentiation between WT KRAS and G12V KRAS in five minutes. Additionally, we harnessed the CCVJ-H probe for real-time profiling of MPG substrates, revealing significantly reduced enzymatic activity in DNA/RNA. Importantly, this approach seamlessly aligned with conventional gel-based assays for substrate preferences for opposite bases. Finally, the versatility of the CCVJ-H probe was demonstrated by profiling the interactions between nucleic acids and small molecules. This probe stands as a unique method, offering both quantification of binding affinity and comprehensive information about binding sites in a single experiment. Its compatibility with HTS facilitated the identification of small-molecule binders for DNA CTG repeat expansion and selective binders for DNA/RNA.Data availability
All the data supporting this study are included in the main text and the ESI.†Author contributions
Tuan-Khoa Kha: chemical probe synthesis, investigation, data curation, methodology, validation, writing—original draft. Qi Shi: formal analysis, data curation. Nirali Pandya: formal analysis, methodology. Ru-Yi Zhu: conceptualization, funding acquisition, project administration, writing—original draft, writing—review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the National University of Singapore for the Presidential Young Professorship start-up grant (A-0008363-00-00 to R. Y. Z.), white space funding (A-0008363-01-00 to R. Y. Z.), and Singapore Ministry of Education for the Academic Research Fund Tier 1 grant (A-8000476-00-00 to R. Y. Z.). We also would like to thank the National Cancer Institute for providing compound libraries.References
- B. Y. Michel, D. Dziuba, R. Benhida, A. P. Demchenko and A. Burger, Front. Chem., 2020, 8, 112 CrossRef CAS PubMed.
- R. C. Spitale and D. Incarnato, Nat. Rev. Genet., 2023, 24, 178–196 CrossRef CAS PubMed.
- M. Kubota, C. Tran and R. C. Spitale, Nat. Chem. Biol., 2015, 11, 933–941 CrossRef CAS PubMed.
- K. B. Turner, R. G. Brinson, H. Y. Yi-Brunozzi, J. W. Rausch, J. T. Miller, S. F. Le Grice, J. P. Marino and D. Fabris, Nucleic Acids Res., 2008, 36, 2799–2810 CrossRef CAS PubMed.
- A. C. Yu, G. Vatcher, X. Yue, Y. Dong, M. H. Li, P. H. Tam, P. Y. Tsang, A. K. Wong, M. H. Hui, B. Yang, H. Tang and L. T. Lau, Front. Med., 2012, 6, 173–186 CrossRef PubMed.
- D. Khodakov, C. Wang and D. Y. Zhang, Adv. Drug Delivery Rev., 2016, 105, 3–19 CrossRef CAS PubMed.
- M. Smith, Front. Mol. Biosci., 2017, 4, 24 CrossRef PubMed.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- R. M. Franzini and E. T. Kool, J. Am. Chem. Soc., 2009, 131, 16021–16023 CrossRef CAS PubMed.
- O. Kohler, D. V. Jarikote and O. Seitz, ChemBioChem, 2005, 6, 69–77 CrossRef PubMed.
- X. Zuo, G. Cui, K. M. Merz Jr, L. Zhang, F. D. Lewis and D. M. Tiede, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3534–3539 CrossRef CAS PubMed.
- M. Lukin and C. de los Santos, Chem. Rev., 2006, 106, 607–686 CrossRef CAS PubMed.
- E. Freire, O. L. Mayorga and M. Straume, Anal. Chem., 1990, 62, 950A–959A CrossRef CAS.
- J. L. Childs-Disney, X. Yang, Q. M. R. Gibaut, Y. Tong, R. T. Batey and M. D. Disney, Nat. Rev. Drug Discovery, 2022, 21, 736–762 CrossRef CAS PubMed.
- T. Yamada, K. Furuita, S. Sakurabayashi, M. Nomura, C. Kojima and K. Nakatani, Nucleic Acids Res., 2022, 50, 9621–9631 CrossRef CAS PubMed.
- M. D. Shortridge and G. Varani, ACS Med. Chem. Lett., 2021, 12, 1253–1260 CrossRef CAS PubMed.
- H. Zipper, H. Brunner, J. Bernhagen and F. Vitzthum, Nucleic Acids Res., 2004, 32, e103 CrossRef PubMed.
- J. Mohanty, N. Barooah, V. Dhamodharan, S. Harikrishna, P. I. Pradeepkumar and A. C. Bhasikuttan, J. Am. Chem. Soc., 2013, 135, 367–376 CrossRef CAS PubMed.
- R. E. Johnson, M. T. Murray, L. J. Bycraft, S. D. Wetmore and R. A. Manderville, Chem. Sci., 2023, 14, 4832–4844 RSC.
- S. Sawada, T. Iio, Y. Hayashi and S. Takahashi, Anal. Biochem., 1992, 204, 110–117 CrossRef CAS PubMed.
- C. Rumble, K. Rich, G. He and M. Maroncelli, J. Phys. Chem. A, 2012, 116, 10786–10792 CrossRef CAS PubMed.
- D. L. Wilson and E. T. Kool, J. Am. Chem. Soc., 2019, 141, 19379–19388 CrossRef CAS PubMed.
- Y. W. Jun, E. Albarran, D. L. Wilson, J. Ding and E. T. Kool, Angew. Chem., Int. Ed., 2022, 61, e202111829 CrossRef CAS PubMed.
- Y. W. Jun, E. M. Harcourt, L. Xiao, D. L. Wilson and E. T. Kool, Nat. Commun., 2022, 13, 5043 CrossRef CAS PubMed.
- D. K. Kolmel and E. T. Kool, Chem. Rev., 2017, 117, 10358–10376 CrossRef CAS PubMed.
- X. Zhang, M. Breslav, J. Grimm, K. Guan, A. Huang, F. Liu, C. A. Maryanoff, D. Palmer, M. Patel, Y. Qian, C. Shaw, K. Sorgi, S. Stefanick and D. Xu, J. Org. Chem., 2002, 67, 9471–9474 CrossRef CAS PubMed.
- Y. Xiong and M. Sundaralingam, Structure, 1998, 6, 1493–1501 CrossRef CAS PubMed.
- S. Olmedillas Lopez, D. C. Garcia-Olmo, M. Garcia-Arranz, H. Guadalajara, C. Pastor and D. Garcia-Olmo, Int. J. Mol. Sci., 2016, 17, 484 CrossRef PubMed.
- K. Nguyen, Y. Wang, W. E. England, J. C. Chaput and R. C. Spitale, J. Am. Chem. Soc., 2021, 143, 4519–4523 CrossRef CAS PubMed.
- S. Kim and A. Misra, Annu. Rev. Biomed. Eng., 2007, 9, 289–320 CrossRef CAS PubMed.
- J. T. Stivers and Y. L. Jiang, Chem. Rev., 2003, 103, 2729–2760 CrossRef CAS PubMed.
- A. L. Jacobs and P. Schar, Chromosoma, 2012, 121, 1–20 CrossRef CAS PubMed.
- M. J. Lucey, D. Chen, J. Lopez-Garcia, S. M. Hart, F. Phoenix, R. Al-Jehani, J. P. Alao, R. White, K. B. Kindle, R. Losson, P. Chambon, M. G. Parker, P. Schar, D. M. Heery, L. Buluwela and S. Ali, Nucleic Acids Res., 2005, 33, 6393–6404 CrossRef CAS PubMed.
- N. P. Montaldo, D. L. Bordin, A. Brambilla, M. Rosinger, S. L. Fordyce Martin, K. O. Bjoras, S. Bradamante, P. A. Aas, A. Furrer, L. C. Olsen, N. Kunath, M. Otterlei, P. Saetrom, M. Bjoras, L. D. Samson and B. van Loon, Nat. Commun., 2019, 10, 5460 CrossRef PubMed.
- M. Hedglin and P. J. O'Brien, Biochemistry, 2008, 47, 11434–11445 CrossRef CAS PubMed.
- Y. Liu, Y. Rodriguez, R. L. Ross, R. Zhao, J. A. Watts, C. Grunseich, A. Bruzel, D. Li, J. T. Burdick, R. Prasad, R. J. Crouch, P. A. Limbach, S. H. Wilson and V. G. Cheung, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 20689–20695 CrossRef CAS PubMed.
- D. M. Lyons and P. J. O'Brien, J. Am. Chem. Soc., 2009, 131, 17742–17743 CrossRef CAS PubMed.
- E. D. Olmon and S. Delaney, ACS Chem. Biol., 2017, 12, 692–701 CrossRef CAS PubMed.
- H. E. Krokan, F. Drablos and G. Slupphaug, Oncogene, 2002, 21, 8935–8948 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- D. L. Boger, B. E. Fink, S. R. Brunette, W. C. Tse and M. P. Hedrick, J. Am. Chem. Soc., 2001, 123, 5878–5891 CrossRef CAS PubMed.
- A. Paul and S. Bhattacharya, Curr. Sci., 2012, 102, 212–231 CAS.
- J. Portugal and M. J. Waring, Biochim. Biophys. Acta, Gene Struct. Expression, 1988, 949, 158–168 CrossRef CAS PubMed.
- N. Nakatsuka, K.-A. Yang, J. M. Abendroth, K. M. Cheung, X. Xu, H. Yang, C. Zhao, B. Zhu, Y. S. Rim, Y. Yang, P. S. Weiss, M. N. Stojanovi and A. M. Andrews, Science, 2018, 362, 319–324 CrossRef CAS PubMed.
- X. Liu, Y. Hou, S. Chen and J. Liu, Biosens. Bioelectron., 2020, 173, 112798 CrossRef PubMed.
- I. Malik, C. P. Kelley, E. T. Wang and P. K. Todd, Nat. Rev. Mol. Cell Biol., 2021, 22, 589–607 CrossRef CAS PubMed.
- J. R. Gatchel and H. Y. Zoghbi, Nat. Rev. Genet., 2005, 6, 743–755 CrossRef CAS PubMed.
- C. T. McMurray, Nat. Rev. Genet., 2010, 11, 786–799 CrossRef CAS PubMed.
- S. Wagner-Griffin, M. Abe, R. I. Benhamou, A. J. Angelbello, K. Vishnu, J. L. Chen, J. L. Childs-Disney and M. D. Disney, J. Med. Chem., 2021, 64, 8474–8485 CrossRef CAS PubMed.
- A. J. Angelbello, S. G. Rzuczek, K. K. McKee, J. L. Chen, H. Olafson, M. D. Cameron, W. N. Moss, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7799–7804 CrossRef CAS PubMed.
- K. L. Taneja, M. McCurrach, M. Schalling, D. Housman and R. H. Singer, J. Cell Biol., 1995, 128, 995–1002 CrossRef CAS PubMed.
- J. D. Brook, M. E. McCurrach, H. G. Harley, A. J. Buckler, D. Church, H. Aburatani, K. Hunter, V. P. Stanton, J.-P. Thirion, T. Hudson, R. Sohn, B. Zemelman, R. G. Snell, S. A. Rundle, S. Crow, J. Davies, P. Shelbourne, J. Buxton, C. Jones, V. Juvonen, K. Johnson, P. S. Harper, D. J. Shaw and D. E. Housman, Cell, 1992, 68, 799–808 CrossRef CAS PubMed.
- R. B. Siboni, M. Nakamori, S. D. Wagner, A. J. Struck, L. A. Coonrod, S. A. Harriott, D. M. Cass, M. K. Tanner and J. A. Berglund, Cell Rep., 2015, 13, 2386–2394 CrossRef CAS PubMed.
- V. I. Hashem, M. J. Pytlos, E. A. Klysik, K. Tsuji, M. Khajavi, T. Ashizawa and R. R. Sinden, Nucleic Acids Res., 2004, 32, 6334–6346 CrossRef CAS PubMed.
- R. T. Wheelhouse and J. B. Chaires, in Methods in Molecular Biology, ed. K. R. Fox, Humana Press, Totowa, NJ, 2010, vol. 613, pp. 55–70 Search PubMed.
- J. M. Santos-Pereira and A. Aguilera, Nat. Rev. Genet., 2015, 16, 583–597 CrossRef CAS PubMed.
- N. Elsakrmy and H. Cui, Int. J. Mol. Sci., 2023, 24, 7064 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional results. See DOI: https://doi.org/10.1039/d4sc00141a |
| This journal is © The Royal Society of Chemistry 2024 |