
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining geometric constraint and redox non-innocence within an ambiphilic PBiP pincer ligand†
Peter
Coburger
a,
Ana Guilherme
Buzanich
 b,
Franziska
Emmerling
bc and
Josh
Abbenseth
*c
b,
Franziska
Emmerling
bc and
Josh
Abbenseth
*c
aDepartment of Inorganic Chemistry, Technische Universität München, Lichtenbergstr. 4, 85747 Garching, Germany
bDepartment of Materials Chemistry, Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany
cInstitut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: josh.abbenseth@hu-berlin.de
First published on 18th March 2024
Abstract
The synthesis of the first pincer ligand featuring a strictly T-shaped group 15 element and its coordination behaviour towards transition metals is described. The platform is itself derived from a trianionic redox non-innocent NNN scaffold. In addition to providing a rigid coordination environment to constrain a Bi centre in a T-shaped geometry to manipulate its frontier molecular orbital constitution, the NNN chelate displays highly covalent bonding towards the geometrically constrained Bi centre. The formation of intriguing ambiphilic Bi–M bonding interactions is demonstrated upon formation of a pincer complex as well as a multimetallic cluster. All compounds are comprehensively characterised by spectroscopic methods including X-ray Absorption Near Edge Structure (XANES) spectroscopy and complemented by DFT calculations.
Introduction
Tridentate pincer ligands have emerged as highly versatile tools to tune the reactivity of transition metal centres in the context of small molecule activation reactions and catalysis.1–5 While central donor functionalities based on 2nd and 3rd row elements are well investigated and utilised in various applications, the chemistry of ligand systems involving heavier elements is comparably underdeveloped. In particular, studies on pincer ligands featuring elements of the 6th period are scarce and their chemistry remains largely unexplored.6–13 In this context, Bi stands out as a particularly fascinating element because of its high natural abundance, low toxicity, multiple accessible oxidation states, and metallic character. These attributes have been effectively leveraged in catalytic applications in recent years.14–20 PBiP pincer ligands featuring a neutral Bi(III)R3 moiety have mainly been reported to act as L-type or X-type ligands to transition metal centres utilising Bi(6s) or Bi(6p) orbitals, respectively (Fig. 1).21,22 Late transition metal complexes of an ambiphilic PBi(Cl)P ligand, however, were reported to exhibit dominant Z-type bonding caused by metal donation into a Bi–Cl(σ*)-antibonding orbital, highlighting the unique bonding properties of Bi derived ligands.7,8 Besides varying the substituents to manipulate the donor and acceptor properties of Bi, the donor capabilities of group 15 elements can further be tuned upon induction of geometric constraint.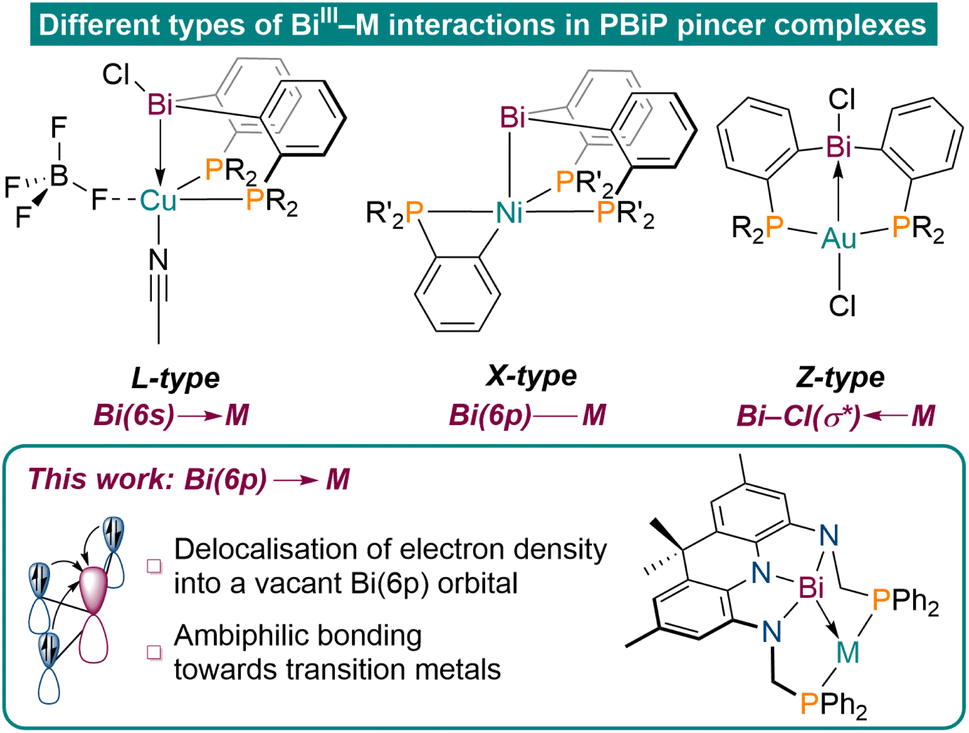 | ||
| Fig. 1 Representative examples of established bonding modes of Bi(III)R3 derived transition metal pincer complexes (top, R = Ph, R′ = iPr);7–9,12 our results on realizing ambiphilic Bi(6p)–M bonding (bottom). | ||
Non-VSEPR geometries of PnR3 (Pn = Pnictogen) species have proven to unlock unique transformations such as (reversible) E–H bond scission, two-electron redox cycles and various small molecule activation reactions, even leading to efficient metal-free catalytic applications.23–28 Upon geometric perturbation of PnR3 species from tetrahedral C3v symmetric structures towards bent or fully T-shaped (C2v) geometries, frontier molecular orbital rearrangement occurs, furnishing Lewis-acidic group 15 centres that feature vacant p-orbitals. The latter geometry necessitates rigid ligand platforms to enforce planarity rendering such species challenging to access, especially in the case of the lighter pnictogens. While various examples of geometrically constrained pnictogens have been reported over the last decade, studies on their properties as ligands remain scarce. Geometrically distorted phosphorus donor moieties were shown to display electrophilic behaviour, engage in P–M cooperative substrate activation reactions, P-centred catalysis as well as insertion into M–E bonds.24,28–36 To date, polydentate ligand platforms that feature strictly T-shaped group 15 centres remain elusive but hold large promise for innovative ligand design due to the unique orbital constitution present within these exotic species. In this context, Bi derived ligand platforms would be particularly suited to realise intriguing heavy metal–metal bonding interactions upon geometric distortion. Chitnis and co-workers showed that planarised trisamidobismuthanes exhibit Lewis-acidic reactivity, however, coupling with redox active supports was proposed to enable Bi(6p)→M L-type bonding due to electron delocalisation into the vacant Bi(6p) orbital originating from planarisation.37–40 These unique electronic features might allow to access Bi donor species that exhibit ambiphilic bonding properties towards transition metals centres which are both mediated via the partially occupied Bi(6p) orbital. While spectroscopic evidence and theoretical studies of such Bi complexes have been reported, structural authentication of this bonding mode, reminiscent of Bi(I) coordination chemistry, as well as its implementation into a pincer ligand, remains elusive.41–47 We aimed to construct an ambiphilic PBiP pincer ligand that features a T-shaped Bi donor functionality which itself is embedded within a redox non-innocent scaffold to realise such a bonding interaction. Recently, we reported the synthesis of rigid acridane-derived NNN pincer ligands that display pronounced redox non-innocence.48,49 Our synthetic approach extends this ligand platform by additional phosphine donor moieties to access a bespoke PBiP pincer ligand featuring a central T-shaped Bi trisamide. Instead of typical L-type bonding utilising the relativistically contracted Bi(6s) orbital, as commonly observed for neutral ligands featuring Bi–C bonds, Bi(6p)→M bonding should consequently be enforced via coupling planarised Bi with the redox non-innocent acridane scaffold.21,50–54
Results and discussion
Synthesis and characterisation of the ambiphilic PBiP pincer ligand
The protioligand 1 can be accessed upon addition of 2,7,9,9-tetramethyl-9,10-dihydroacridine-4,5-diamine48 to diphenyl-phosphinomethanol in dichloromethane (DCM) in moderate yields (Scheme 1). We developed a reliable procedure for the large-scale synthesis of PPh2(CH2OH) that affords a brittle white solid in excellent yield (91%, see ESI†). 1 displays overall C2v symmetry on the NMR timescale indicated by the equivalency of the phosphorus atoms, the flanking amine groups as well as the methyl groups of the acridane unit (31P{1H} NMR: δ = −18.6 ppm). The molecular structure of 1 in the solid state obtained via single crystal X-ray diffraction (SCXRD)55 confirms the expected symmetric structure which features a planar acridane unit and displays almost identical bond metrics when compared to our recently reported acridane derived NNN ligands (Scheme 1).48,49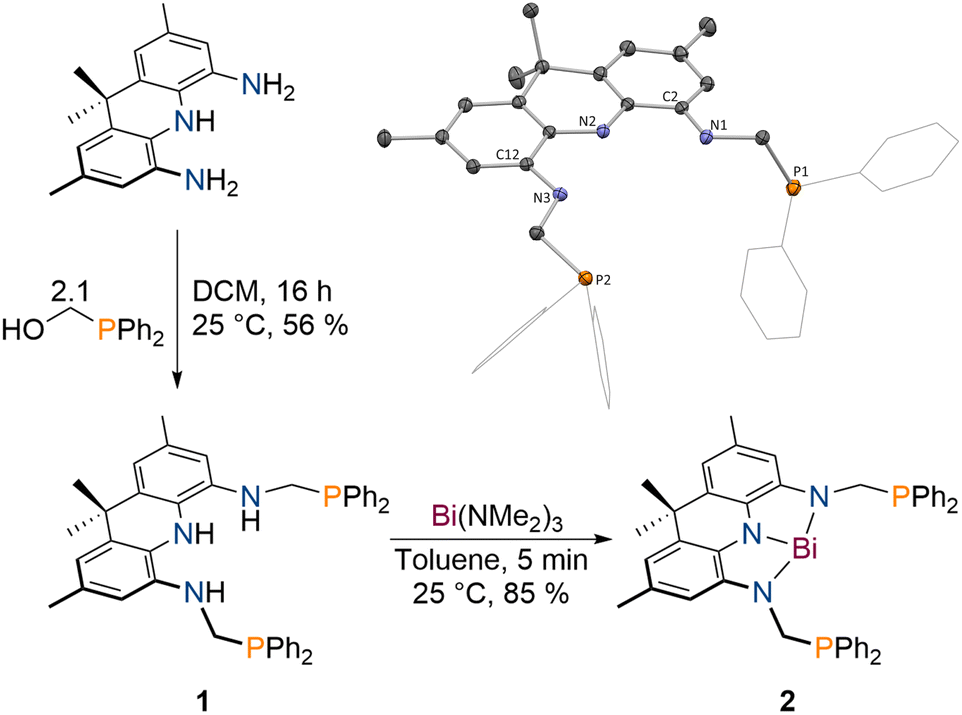 | ||
| Scheme 1 Synthesis of 1 upon reaction of 2,7,9,9-tetramethyl-9,10-dihydroacridine-4,5-diamine with diphenylphosphinomethanol and subsequent formation of 2 upon addition of Bi(NMe2)3 to produce 2; inset: molecular structure of 1 in the solid state obtained via SCXRD, ellipsoids at 50% probability level; H atoms omitted for clarity. | ||
1 was further analysed by IR spectroscopy and combustion analysis, both fully in line with the expected structure and composition of the protioligand. When 1 is reacted with Bi(NMe2)3 in toluene at room temperature, an instant colour change from colourless to deep purple is observed. The Bi trisamide 2 can be obtained in high isolated yields in form of a dark blue powder (Scheme 1). As observed for 1, 2 displays overall C2v symmetry on the NMR timescale. Upon incorporation of Bi, the 31P{1H} NMR signal undergoes a minor low-field shift suggesting no significant interaction between the Bi centre and the phosphine donors (31P{1H} NMR: δ = −22.2 ppm). The resonances of the pincer arms' methylene protons further exhibit a significant downfield shift caused by Bi complexation (Δδ = 2.27 ppm). SCXRD reveals formation of a strictly planar Bi trisamide in which the Bi centre adopts a slightly contracted T-shaped geometry (Fig. 2, N1–Bi1–N2: 71.29(13)°; N2–Bi1–N3: 72.03(13)°). Contraction of the C2/12–N1/3 bonds from 1.4133(14) and 1.4182(14) Å in 1 towards 1.353(5), 1.359(5), 1.365(5) and 1.355(5) Å upon Bi incorporation suggests an increased quinone character of the NNN ligand and by that depletion of electron density from the acridane ligand scaffold upon formation of 2 due to electron delocalisation into the vacant Bi(6p) orbital. This compares well with previous studies on tantalum(V) complexes of an acridane derived NNN pincer ligand that showed similar bond metrics upon ligand oxidation.48 The accurate description of the Bi redox state in geometrically constrained trisamides featuring redox active substituents has been debated and ligand mediated reduction towards Bi(I) has been proposed, while a recent report by Andrada, Salvador and co-workers points towards a more accurate description as Bi(III) featuring delocalisation of electron density from the redox active substituents into the vacant Bi(6p) orbital to allow for Lewis basic reactivity to occur.38,41 The computed molecular orbitals of 2 indicate highly covalent bonding between the Bi centre and the NNN scaffold.
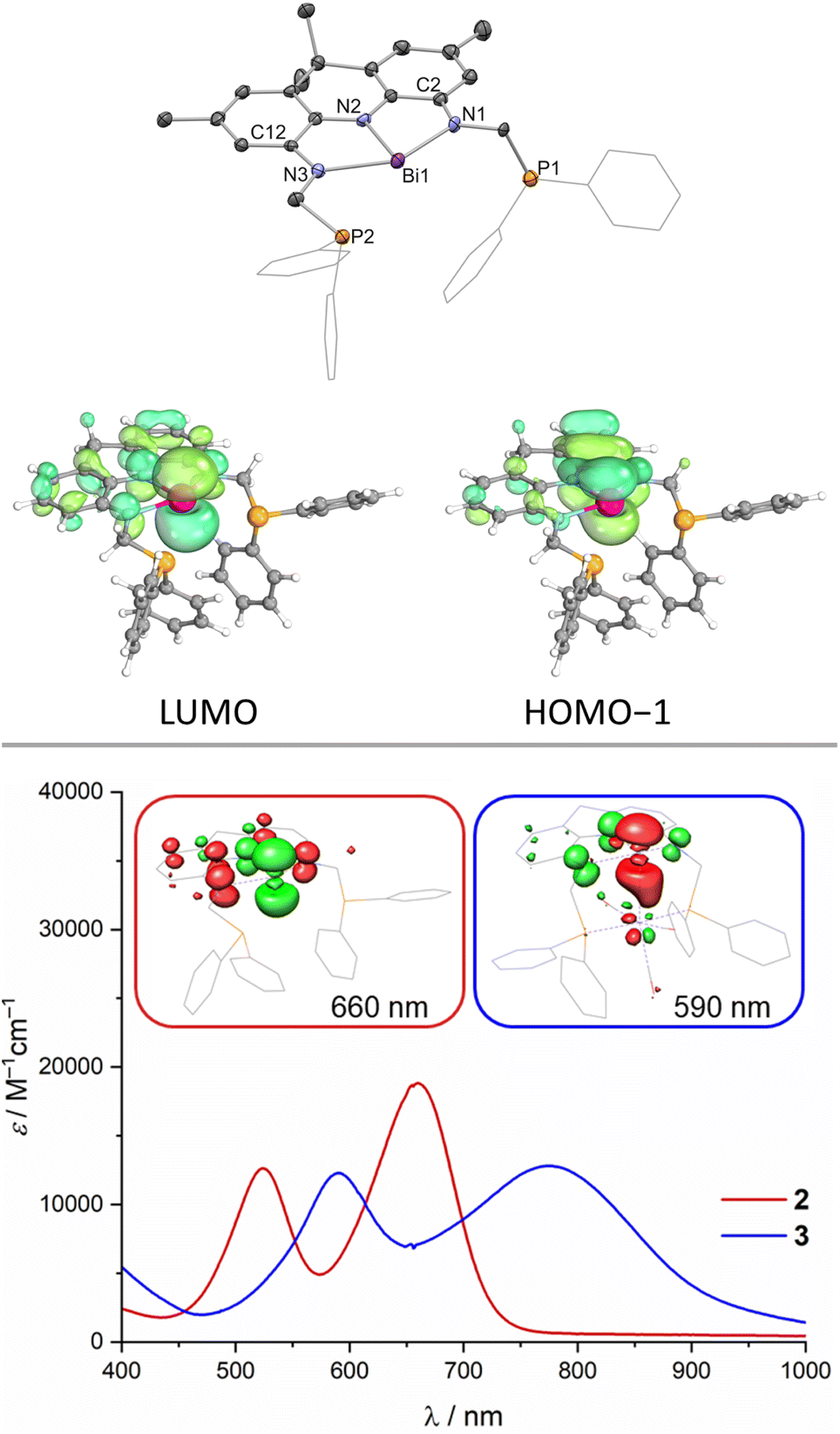 | ||
| Fig. 2 Molecular structure of 2 in the solid state obtained via SCXRD, ellipsoids at 50% probability level; H atoms are omitted for clarity, one molecule of the asymmetric unit shown and selected computed frontier molecular orbitals (top, Me groups of the acridane ligand truncated to H); UV/vis spectra of 2 and 3 (DCM, 25 °C, inset: calculated differences densities of selected transitions, depletion of electron density shown in red and accumulation in green). | ||
While the HOMO is mainly based on the NNN backbone, the HOMO−1 features significant Bi(6p) orbital contribution with in-phase bonding towards the occupied NNN scaffold's central N(2p) orbital (Fig. 2). The LUMO is represented by the antibonding combination. A high degree of covalency in the bonding between the Bi centre and the NNN chelate is further evidenced by Complete Active Space Self Consistent Field (CASSCF) calculations that reveal the presence of a 2c–2e π bond between Bi and the central N of the NNN pincer with very minor polarisation towards nitrogen and an occupation of the Bi(6p) orbital with 0.91 e− thus preventing the unequivocal assignment of a Bi(III) or Bi(I) redox state for 2. Time-dependent DFT calculations reveal that the intense purple colour of 2 can be described as HOMO−1 → LUMO and HOMO → LUMO excitations associated with bands at λ = 524 nm and 660 nm, respectively, observed in the UV/vis spectrum in DCM at room temperature, in line with prior reports on T-shaped pnictogen species (Fig. 2, see ESI†).38,56 Tan, Ruan, Wang an co-workers recently reported the successful isolation of T-shaped P, As and Sb radical anions upon one-electron reduction of the respective T-shaped trisamides.56 The cyclic voltammogram of 2 features a quasi-reversible reductive event at E1/2 = −1.98 V vs. Fc0/+ that becomes increasingly irreversible at lower scan rates (see ESI†). This points towards the possible isolation of T-shaped Bi radical anions when sterically more demanding ligand scaffolds are employed. Furthermore, ill-defined oxidative events at E ⪆ 0 V vs. Fc0/+ accompanied by immediate fouling of the glassy carbon working electrode and deposition of insoluble black material on the electrode surface indicate disintegration of the molecular structure upon oxidation (see ESI†). The Lewis-acidity of 2 was assessed via the Gutmann–Beckett method upon comparing 31P NMR shift differences of free Et3PO with an equimolar mixture of 2 and Et3PO.57,58 An acceptor number (AN) of ≤20 was obtained in dichloromethane which represents an upper limit due to the intrinsic Lewis-acidity of the employed solvent (ANDCM = 20.4). Lichtenberg and co-workers introduced an alternative scale for soft Bi Lewis-acids upon comparing the 31P NMR shift of free Me3PS against 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts with the respective Lewis-acid in dichloromethane.592 features a low acceptor number of 8, comparable with BiPh3 and in line with results obtained for similar systems reported by Chitnis and co-workers.37 The calculated Fluoride Ion Affinity (FIA) of 61 kcal mol−1 supports the notion of 2 being a very weak Lewis-acid.60
:1 adducts with the respective Lewis-acid in dichloromethane.592 features a low acceptor number of 8, comparable with BiPh3 and in line with results obtained for similar systems reported by Chitnis and co-workers.37 The calculated Fluoride Ion Affinity (FIA) of 61 kcal mol−1 supports the notion of 2 being a very weak Lewis-acid.60
To further elucidate the bonding situation, we probed the Bi oxidation state via X-ray Absorption Near Edge Structure (XANES) spectroscopy. Bi L1-edge X-ray absorption spectra of 2, BiPh3 and Bi foil are depicted in Fig. 3. 2 features a low energy edge position (16388.6 eV), lower than the classical Bi(III) compound BiPh3 (16387.9 eV), and closer to metallic Bi (16388.0 eV). This becomes even more pronounced when comparing the whiteline shape and energy position. For 2 this is at 16394.0 eV, considerably lower than the one observed for BiPh3 (16397.9 eV) and closer to metallic Bi foil (16393.5 eV). The same trend can be observed for the Bi L3-edge (see ESI†). These spectroscopic results, in combination with our theoretical studies, demonstrate the highly covalent bonding between the Bi centre and the NNN scaffold that results in partial occupation of the Bi(6p) orbital, quenching its expected Lewis-acidity stemming from planarisation and complicating unequivocal assignment of a Bi redox state. Consequently, subsequent reactivity studies are required to obtain a more comprehensive understanding of 2's donor/acceptor properties stemming from this unusual electronic structure, in particular its capability to form transition metal complexes displaying ambiphilic bonding interactions.
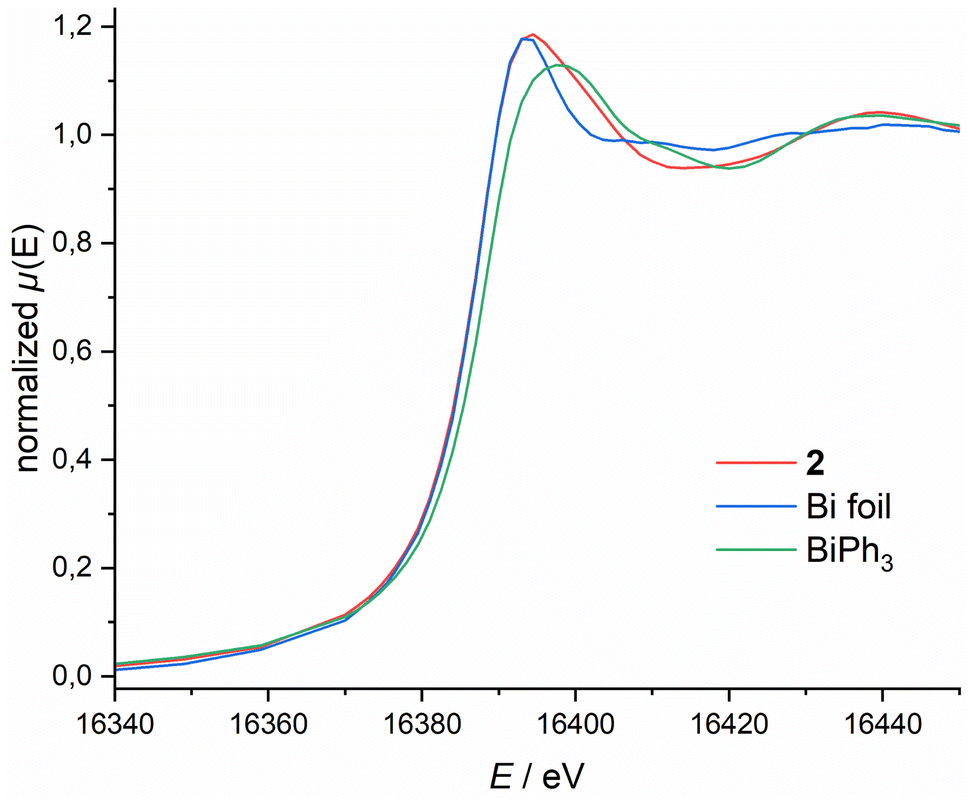 | ||
| Fig. 3 Bi L1-edge XANES spectra of 2, Bi foil and BiPh3. | ||
Complexation of transition metal centres by 2
When 2 is reacted with [W(CO)3(MeCN)3] in DCM, an instant colour change from dark purple towards dark blue is observed. A single new signal is detected in the 31P{1H} NMR spectrum at δ = 42.3 ppm which features 183W satellites (1JP–W = 288 Hz) suggesting incorporation of tungsten into the PBiP pincer. The 1H NMR spectrum features significantly broadened resonances except for the equatorial benzylic and the aromatic protons of the acridane moiety. Cooling the sample to lower temperatures (−60 °C), however, results in sharpening of all signals and an overall Cs symmetry on the NMR timescale is revealed which indicates that the tungsten centre binds to the ligand via the Bi(6p) orbital (see ESI†). The structure of the formed complex could be clarified via SCXRD (Fig. 4), IR spectroscopy and combustion analysis confirming the successful incorporation of a W(CO)3 fragment into the PBiP pincer ligand in near quantitative yield (3, Fig. 4). Comparing the carbonyl stretching frequencies of 3 with values reported for structurally related tungsten tricarbonyl PNP pincer complexes that feature a meridional coordination mode and flanking PPh2 donor moieties reveals that the Bi centre's donor capabilities are comparable to pyridine.61,623 slowly degrades in solution and in the solid state at room temperature but can be stored for weeks at −30 °C in the dark. The Bi centre slightly bends out of the NNN ligand's plane accompanied by a further shortening of the C2/12–N1/3 bonds (ca. 0.02 Å) within the ligand when compared to 2, thus demonstrating further depletion of electron density from the acridane support to facilitate metal binding. The Bi–W bond length (2.8059(9) and 2.8049(5) Å) is slightly shorter compared to a recently reported Bi(I)–W(CO)5 complex (2.933(7) and 2.944(6) Å) or Ph3Bi–W(CO)5 (2.829(3) Å), likely owing due to the chelating nature of the employed ligand scaffold.46,63 The observed dynamic behaviour in solution is attributed to a rocking motion of the phosphine ligated W centre around the Bi(NNN) plane. This is supported by theoretical calculations which predict a low barrier for this process (ΔG‡DFT = 12 kcal mol−1) in line with observed broadened NMR signals at room temperature that sharpen upon cooling (see ESI†). The acceptor numbers of 3versus Et3PO and Me3PS are identical to 2 suggesting only a minor influence of metal incorporation on the Bi Lewis-acidity as further supported by a calculated FIA of 56 kcal mol−1. In contrast to 2, 3 features well-defined redox events in the cyclic voltammogram (see ESI†). An irreversible oxidative event (v = 100–800 mV s−1) is observed in dichloromethane at E ≈ −0.2 V vs. Fc0/+ likely due to metal decarbonylation upon one-electron oxidation. A quasi-reversible reduction is detected at E1/2 = −1.39 V vs. Fc0/+. The reaction of 3 with decamethylcobaltocene in dichloromethane, however, resulted in unselective decomposition. To elucidate the bonding between W and Bi a number of theoretical techniques were utilised. Distinct L-type binding of the W centre by the central Bi donor is suggested by the calculated frontier molecular orbitals. In line with the contraction observed in the C2/12–N1/3 bonds with regards to 2, the HOMO−4 of 3 clearly displays Bi(6p)→W donation while the LUMO is mainly located on the NNN backbone in contrast to 2 (Fig. 4). 3 features two intense transitions in the UV/vis spectrum at λ = 590 nm and 774 nm (Fig. 3).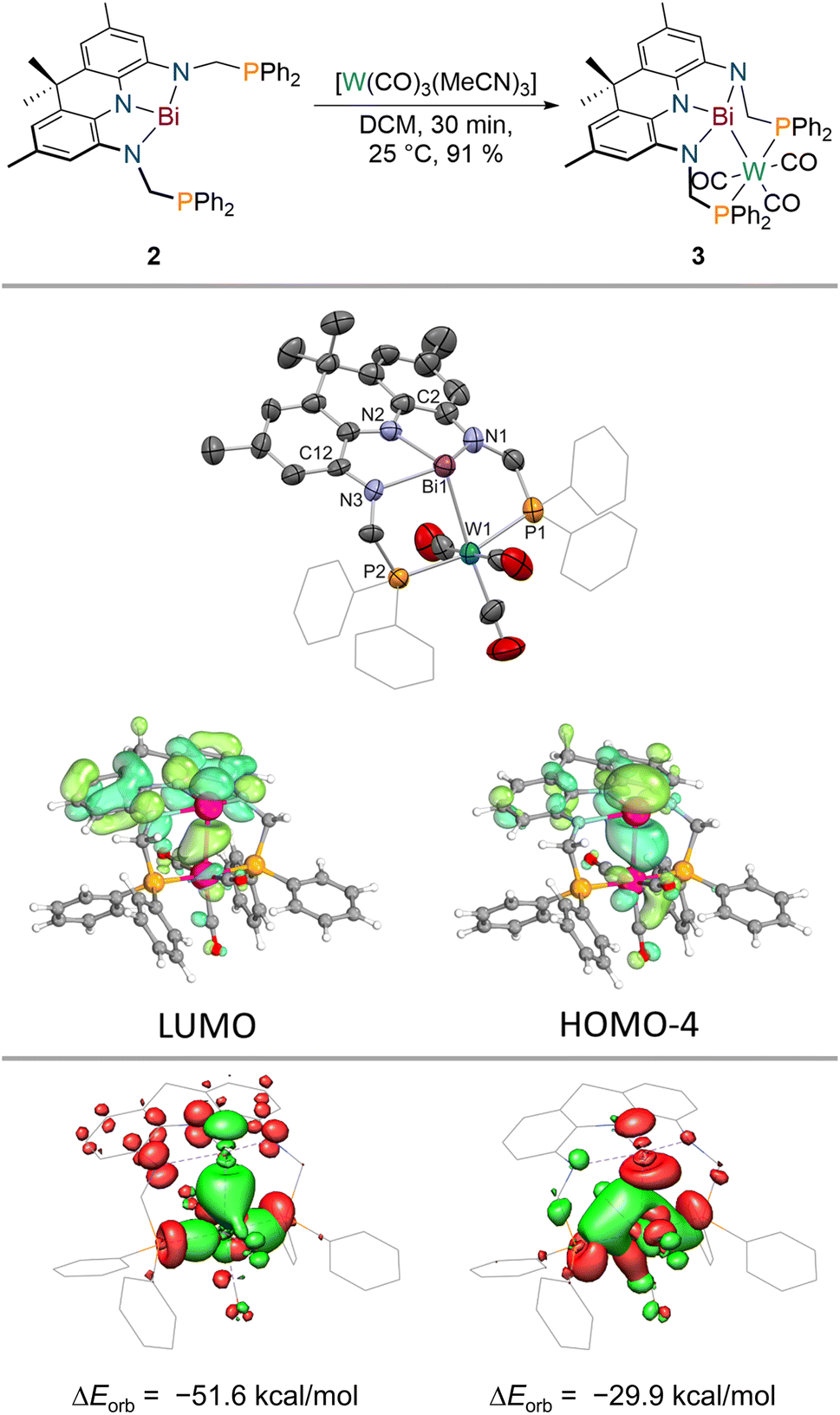 | ||
| Fig. 4 Synthesis of 3 upon addition of [W(CO)3(MeCN)3] to 2 (top); molecular structure of 3 in the solid state obtained via SCXRD, ellipsoids at 50% probability level; H atoms are omitted for clarity, one molecule of the asymmetric unit shown; and selected computed frontier molecular orbitals (middle, Me groups of the acridane ligand truncated to H); selected donor (red)/acceptor (green) orbital pairs derived via energy decomposition analysis showcasing the ligand's redox activity as well as the ambiphilic bonding between tungsten and bismuth (bottom). | ||
Calculation of the difference densities shows that these transitions are drastically different in nature when compared to 2. While the low-energy transition stems from an acridane π–π* transition, the other band can be interpreted as a Bi(6p) → acridane(π*) electron transfer, in full agreement with the proposed population of the formally vacant Bi(6p) orbital. The calculated Electron Localization Function (ELF; see ESI†)64,65 showcases significant accumulation of electron density along the Bi–W bond complemented by a calculated Mayer bond order of 0.59 which compares well with values obtained for Bi(I)–W(CO)5 complexes showcasing the successful mimicry of typical Bi(I) coordination behaviour enabled by coupling of Bi planarisation and the redox non-innocence of the NNN scaffold.46 The characteristic energy and electron densities at the bond critical point derived via Quantum Theory of Atoms in Molecules (QTAIM)66 calculations are in full agreement with a dative metal–metal interaction (ρ(r) = 0.044; ∇2ρ(r) = 0.052; H(r) = −0.011; see ESI†).67 Energy Decomposition Analysis (EDA)68 between the PBiP and W(CO)3 fragments further reveals the unique ambiphilic nature of the PBiP pincer ligand system (Fig. 4). Bi(6p)→W as well as W→Bi(6p) donor/acceptor interactions can be unequivocally deduced while Bi(6s)→W L-type bonding could not be identified (see ESI†). Consequently, the implementation of Bi within a rigid, redox non-innocent supports allows for the realisation of ambiphilic Bi–W donor/acceptor interactions in which both interactions are mediated by the Bi(6p) orbital due the highly covalent bonding within the Bi(NNN) unit. This contrasts with prior reports on related systems which facilitate ambiphilic Bi–M bonding via two different orbitals, e.g., Bi(6s) and Bi–Cl(σ*) orbitals in PBiP pincer ligands that are not redox non-innocent.7,8
Next, we aimed to investigate the bonding of 2 towards an electron-rich d10 metal centre to clarify if the donor properties of 2 are dependent on the Lewis-acidity and electron configuration of the utilised metal. Gold complexes of Bi(III) derived ligands are still extremely scarce since transmetallation instead of coordination can easily occur, necessitating tailored substituents at Bi to prevent undesired side reactions (Fig. 1).6–8,69,70 When 2 is reacted with [AuCl(SMe2)] in DCM at −30 °C, a colour change towards turquoise (intense absorption at λ = 744 nm) is observed accompanied by the quantitative formation of a new signal in the 31P{1H} NMR spectrum at δ = 59.3 ppm. The isolated product features significantly reduced signal broadening in contrast to 3 in the 1H NMR spectrum at room temperature. While the methyl groups of the acridane pincer scaffold are magnetically equivalent on the NMR timescale, the methylene protons of the phosphine donor arms display geminal coupling (2JH–H = 12 Hz, Δδ = 1.53 ppm) pointing towards an overall C2v symmetry of the reaction product in solution in which complexation of two gold centres by 2 occurs. This is further corroborated by diffusion ordered NMR spectroscopy (DOSY-NMR) which reveals that the formed metal complex is significantly larger in solution than 1–3 (1.7 times the size of 3, see Table S1†). SCXRD revealed that a multi-metallic cluster has been formed (Fig. 5).
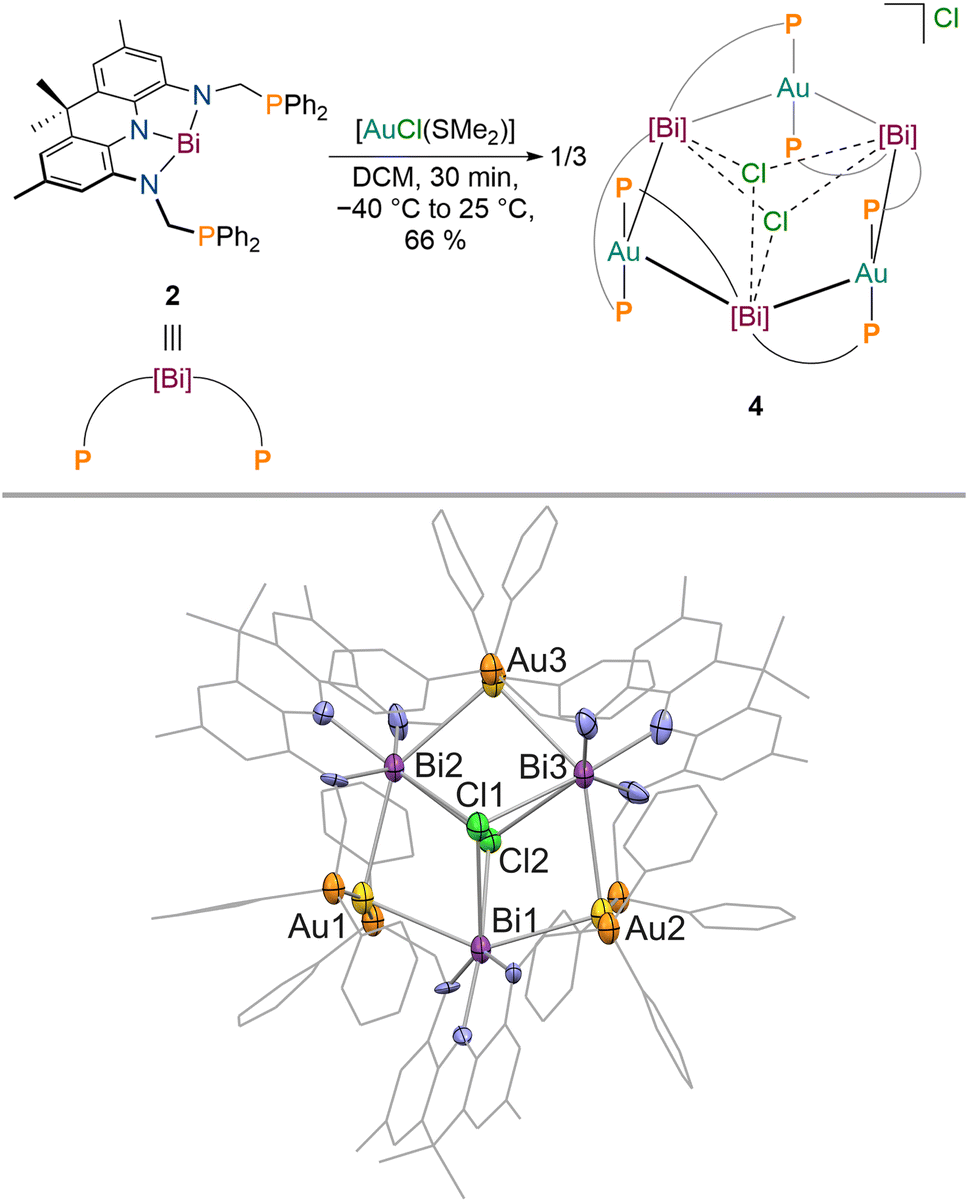 | ||
| Fig. 5 Synthesis of 4 upon addition of [AuCl(SMe2)] to 2 (top); molecular structure of 4 in the solid state obtained via SCXRD, ellipsoids at 50% probability level; H atoms, the chloride anion and solvent molecules are omitted for clarity (bottom). | ||
The gold centres are linearly coordinated by two phosphine donors stemming from different PBiP ligands and additionally feature Bi–Au interactions resulting in the formation of a six-membered Au3Bi3 ring that is capped by two chlorides to give the monocationic complex 4 (Fig. 5). 4 is stable for hours at room temperature in DCM but immediately decomposes when exposed to strongly donating or aromatic solvents such as tetrahydrofuran, toluene, benzene or chlorobenzene. The molecular structure of 4 shows that all Bi–Cl and Bi–Au bond distances are below the sum of their respective van der Waals radii suggesting the presence of Bi–Au orbital interactions. This is corroborated by the calculated Mayer bond orders that indicate Bi–Au (0.24) and Bi–Cl (0.14) bonding to be present, while Bi–Bi, Au–Au and Au–Cl bonding is negligible (all below 0.1). The bonding within the cluster was further analysed via EDA calculations, since inspection of the frontier molecular orbitals revealed little information due to the size and delocalisation of the studied system. As for 3, the phosphine–gold interactions are dominant. In addition, electron flow between the redox non-innocent ligand towards the Bi–Au bonds as well as gold stabilisation via the ambiphilic Bi(6p) orbital is evident, albeit less pronounced when compared to 3 (Fig. 6). Bi–Cl bonding interactions indicate an essential role of the capping chlorides to stabilise the cluster (see ESI†).
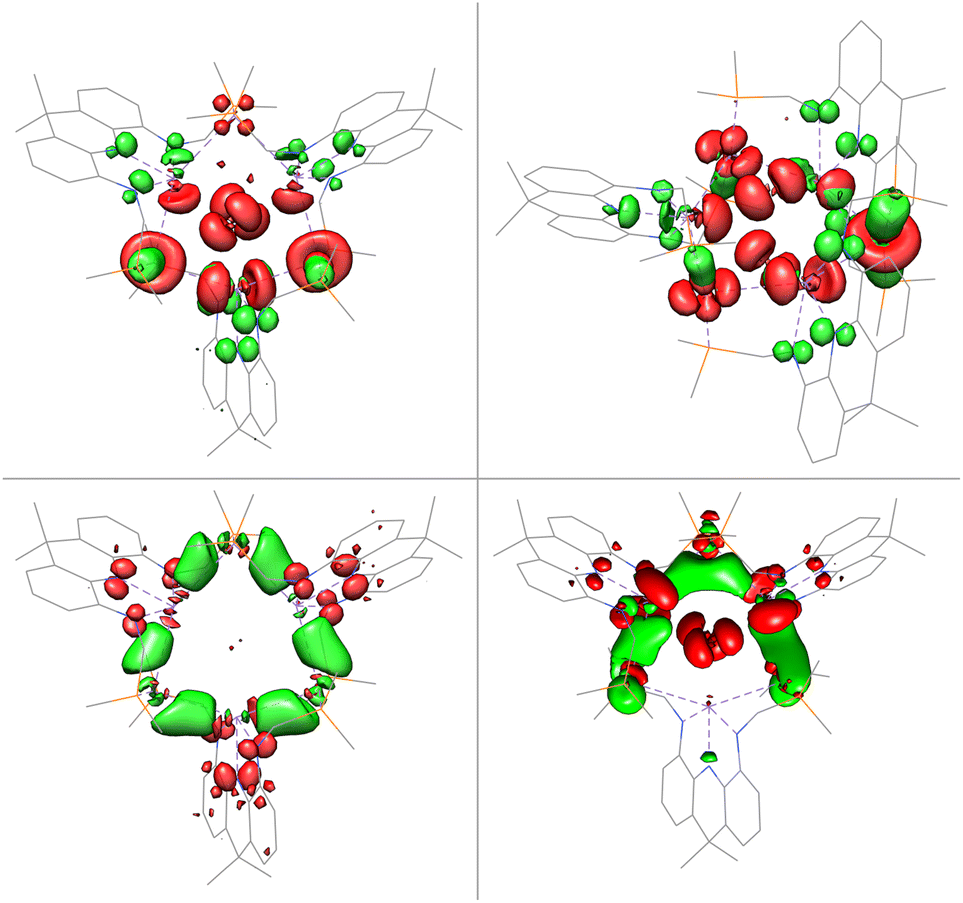 | ||
| Fig. 6 Selected donor and acceptor orbital pairs derived by EDA of 4 (PPh2 truncated to PMe2 and equatorial acridane methyl groups to H; donor orbitals in red and acceptor orbitals in green). | ||
This showcases that 2 is a competent ligand for early as well as late transition metals and allows not only to form discrete pincer complexes with unique bonding characteristics but also large multi-metallic clusters due to its flexible phosphine side-arms. In both of these structure types ambiphilic Bi–M binding is realised originating from a coupling of Bi planarisation and employment of a redox-active support.
Conclusions
In conclusion, we report the first synthesis of a PBiP pincer ligand in which transition metals engage in rare ambiphilic Bi(6p)–M bonding enabled by a combination of geometric constraint on the Bi centre and a redox active NNN scaffold. The highly covalent bonding between these two fragments allows Bi to act as an electron relay for the NNN chelate when bound to transition metals resulting in the formation of pincer complexes as well as multi-metallic clusters. Future study will target to exploit these unique bonding characteristics in the context of small molecule activation reactions via heavy atom – transition metal cooperativity and the controlled construction of larger cluster compounds.Data availability
Synthetic procedures, spectroscopic results and details as well as computational details can be found in the ESI.† Crystallographic datasets have been deposited at the CCDC under 2320292, 2320297 and 2320304.Author contributions
P. C. performed the theoretical analysis of all presented compounds. A. G. B. performed the XANES measurements and interpreted the data under the supervision of F. E. J. A. conceptualised the project, performed the synthetic work, interpreted the spectroscopic results, performed the SCXRD studies and wrote the original draft of the manuscript. The final version was edited and approved by all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. C. and J. A. thank the Fond der Chemischen Industrie e.V. for a Liebig-Scholarship and J. A. further thanks Prof. Christian Limberg for his continuous support. Dr André Dallmann and the whole analytical department of the Humboldt-Universität zu Berlin are acknowledged for their technical support. We extend our gratitude to M.Sc. Kristin Klaue and the Hecht group at Humboldt-Universität zu Berlin for their support in conducting fluorescence measurements. XANES experiments were performed at the BAMline at the BESSY-II storage ring (Helmholtz Center Berlin). We thank the Helmholtz-Zentrum Berlin für Materialien und Energie for the allocation of synchrotron radiation beamtime.Notes and references
- G. van Koten and D. Milstein, Organometallic Pincer Chemistry, Springer, 2012 Search PubMed.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018 Search PubMed.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- Y. Li, J. D. Collett and H. Guan, in Comprehensive Coordination Chemistry III, ed. E. C. Constable, G. Parkin and L. Que Jr, Elsevier, Oxford, 2021, pp. 505–606 Search PubMed.
- C. Tschersich, B. Braun, C. Herwig and C. Limberg, Organometallics, 2015, 34, 3782–3787 CrossRef CAS.
- C. Tschersich, C. Limberg, S. Roggan, C. Herwig, N. Ernsting, S. Kovalenko and S. Mebs, Angew. Chem., Int. Ed., 2012, 51, 4989–4992 CrossRef CAS PubMed.
- T.-P. Lin, I.-S. Ke and F. P. Gabbaï, Angew. Chem., Int. Ed., 2012, 51, 4985–4988 CrossRef CAS PubMed.
- A. C. Brannan, D. Yoo, J. Choi and Y. Lee, Inorg. Chem., 2023, 62, 8589–8597 CrossRef CAS PubMed.
- V. K. Greenacre, W. Levason and G. Reid, Coord. Chem. Rev., 2021, 432, 213698 CrossRef CAS.
- L. Dostál and R. Jambor, in Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018, pp. 47–65 Search PubMed.
- C. Tschersich, B. Braun, C. Herwig and C. Limberg, J. Organomet. Chem., 2015, 784, 62–68 CrossRef CAS.
- K. Materne, S. Hoof, N. Frank, C. Herwig and C. Limberg, Organometallics, 2017, 36, 4891–4895 CrossRef CAS.
- A. I. Poddel'sky and V. V. Sharutin, J. Organomet. Chem., 2022, 957, 122152 CrossRef.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- T. Ollevier, Org. Biomol. Chem., 2013, 11, 2740–2755 RSC.
- E. Lopez, S. C. Thorp and R. S. Mohan, Polyhedron, 2022, 222, 115765 CrossRef CAS.
- Y. Pang, N. Nöthling, M. Leutzsch, L. Kang, E. Bill, M. van Gastel, E. Reijerse, R. Goddard, L. Wagner, D. SantaLucia, S. DeBeer, F. Neese and J. Cornella, Science, 2023, 380, 1043–1048 CrossRef CAS PubMed.
- X. Yang, E. J. Reijerse, N. Nöthling, D. J. SantaLucia, M. Leutzsch, A. Schnegg and J. Cornella, J. Am. Chem. Soc., 2023, 145, 5618–5623 CrossRef CAS PubMed.
- M. Mato and J. Cornella, Angew. Chem., Int. Ed., 2024, 63, e202315046 CrossRef CAS PubMed.
- J. Ramler and C. Lichtenberg, Dalton Trans., 2021, 50, 7120–7138 RSC.
- J. Ramler, K. Radacki, J. Abbenseth and C. Lichtenberg, Dalton Trans., 2020, 49, 9024–9034 RSC.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- S. Kundu, Chem.–Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- L. Greb, F. Ebner, Y. Ginzburg and L. M. Sigmund, Eur. J. Inorg. Chem., 2020, 2020, 3030–3047 CrossRef CAS.
- T. J. Hannah and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 RSC.
- A. J. I. Arduengo and C. A. Stewart, Chem. Rev., 1994, 94, 1215–1237 CrossRef CAS.
- M. J. Drance, A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2022, 144, 20243–20248 CrossRef CAS PubMed.
- Q. J. Bruch, A. Tanushi, P. Müller and A. T. Radosevich, J. Am. Chem. Soc., 2022, 144, 21443–21447 CrossRef CAS PubMed.
- A. Brand and W. Uhl, Chem.–Eur. J., 2019, 25, 1391–1404 CrossRef CAS PubMed.
- S. J. Hwang, A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 21285–21291 CrossRef CAS PubMed.
- K. Lee, A. V. Blake, A. Tanushi, S. M. McCarthy, D. Kim, S. M. Loria, C. M. Donahue, K. D. Spielvogel, J. M. Keith, S. R. Daly and A. T. Radosevich, Angew. Chem., Int. Ed., 2019, 58, 6993–6998 CrossRef CAS PubMed.
- G. T. Cleveland and A. T. Radosevich, Angew. Chem., Int. Ed., 2019, 58, 15005–15009 CrossRef CAS PubMed.
- A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 8114–8118 CrossRef CAS PubMed.
- N. Beims, T. Greven, M. Schmidtmann and J. I. van der Vlugt, Chem.–Eur. J., 2023, 29, e202302463 CrossRef CAS PubMed.
- T. J. Hannah, W. M. McCarvell, T. Kirsch, J. Bedard, T. Hynes, J. Mayho, K. L. Bamford, C. W. Vos, C. M. Kozak, T. George, J. D. Masuda and S. S. Chitnis, Chem. Sci., 2023, 14, 4549–4563 RSC.
- M. B. Kindervater, K. M. Marczenko, U. Werner-Zwanziger and S. S. Chitnis, Angew. Chem., Int. Ed., 2019, 58, 7850–7855 CrossRef CAS PubMed.
- K. M. Marczenko, S. Jee and S. S. Chitnis, Organometallics, 2020, 39, 4287–4296 CrossRef CAS.
- K. M. Marczenko, J. A. Zurakowski, M. B. Kindervater, S. Jee, T. Hynes, N. Roberts, S. Park, U. Werner-Zwanziger, M. Lumsden, D. N. Langelaan and S. S. Chitnis, Chem.–Eur. J., 2019, 25, 16414–16424 CrossRef CAS PubMed.
- M. Gimferrer, S. Danés, D. M. Andrada and P. Salvador, Inorg. Chem., 2021, 60, 17657–17668 CrossRef CAS PubMed.
- L. Dostál, R. Jambor, M. Aman and M. Hejda, ChemPlusChem, 2020, 85, 2320–2340 CrossRef PubMed.
- M. Kořenková, V. Kremláček, M. Erben, R. Jirásko, F. D. Proft, J. Turek, R. Jambor, A. Růžička, I. Císařová and L. Dostál, Dalton Trans., 2018, 47, 14503–14514 RSC.
- M. Kořenková, V. Kremláček, M. Erben, R. Jambor, Z. Růžičková and L. Dostál, J. Organomet. Chem., 2017, 845, 49–54 CrossRef.
- I. Vránová, M. Alonso, R. Jambor, A. Růžička, M. Erben and L. Dostál, Chem.–Eur. J., 2016, 22, 7376–7380 CrossRef PubMed.
- I. Vránová, V. Kremláček, M. Erben, J. Turek, R. Jambor, A. Růžička, M. Alonso and L. Dostál, Dalton Trans., 2017, 46, 3556–3568 RSC.
- I. Vránová, T. Dušková, M. Erben, R. Jambor, A. Růžička and L. Dostál, J. Organomet. Chem., 2018, 863, 15–20 CrossRef.
- J. Underhill, E. S. Yang, T. Schmidt-Räntsch, W. K. Myers, J. M. Goicoechea and J. Abbenseth, Chem.–Eur. J., 2023, 29, e202203266 CrossRef CAS PubMed.
- A. J. King, J. Abbenseth and J. M. Goicoechea, Chem.–Eur. J., 2023, 29, e202300818 CrossRef CAS PubMed.
- J. P. Desclaux and Y.-K. Kim, J. Phys. B: At. Mol. Phys., 1975, 8, 1177 CrossRef CAS.
- K. S. Pitzer, Acc. Chem. Res., 1979, 12, 271–276 CrossRef CAS.
- P. S. Bagus, Y. S. Lee and K. S. Pitzer, Chem. Phys. Lett., 1975, 33, 408–411 CrossRef CAS.
- P. Pyykko, Chem. Rev., 1988, 88, 563–594 CrossRef CAS.
- P. Pyykko and J. P. Desclaux, Acc. Chem. Res., 1979, 12, 276–281 CrossRef CAS.
- Deposition numbers 2320292 (1), 2320297 (2), and 2320304 (3) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.†.
- M. K. Mondal, L. Zhang, Z. Feng, S. Tang, R. Feng, Y. Zhao, G. Tan, H. Ruan and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 15829–15833 CrossRef CAS PubMed.
- U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- J. Ramler and C. Lichtenberg, Chem.–Eur. J., 2020, 26, 10250–10258 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- W. Schirmer, U. Flörke and H.-J. Haupt, Z. Anorg. Allg. Chem., 1987, 545, 83–97 CrossRef CAS.
- Ö. Öztopcu, C. Holzacker, M. Puchberger, M. Weil, K. Mereiter, L. F. Veiros and K. Kirchner, Organometallics, 2013, 32, 3042–3052 CrossRef PubMed.
- N. J. Holmes, W. Levason and M. Webster, J. Organomet. Chem., 1997, 545–546, 111–115 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808–1832 CrossRef CAS.
- P. Fuentealba, E. Chamorro and J. C. Santos, in Theoretical and Computational Chemistry, ed. A. Toro-Labbé, Elsevier, 2007, vol. 19, pp. 57–85 Search PubMed.
- C. F. Matta and R. J. Boyd, in The Quantum Theory of Atoms in Molecules, John Wiley & Sons, Ltd, 2007, pp. 1–34 Search PubMed.
- R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360–2366 CrossRef CAS PubMed.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed.
- O. Schuster, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 4079–4083 CrossRef CAS.
- C. Silvestru, in Modern Supramolecular Gold Chemistry, ed. A. Laguna, John Wiley & Sons, Ltd, 2008, pp. 181–293 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, theoretical analysis and crystallographic information. CCDC 2320292 (1), 2320297 (2) and 2320304 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc00197d |
| This journal is © The Royal Society of Chemistry 2024 |