
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic isomerism in a heterometallic nickel–iron–sulfur cluster models substrate binding and cyanide inhibition of carbon monoxide dehydrogenase†
Luke C.
Lewis
 a,
José A.
Sanabria-Gracia
a,
Yuri
Lee
ab,
Adam J.
Jenkins
a and
Hannah S.
Shafaat
*ab
a,
José A.
Sanabria-Gracia
a,
Yuri
Lee
ab,
Adam J.
Jenkins
a and
Hannah S.
Shafaat
*ab
aDepartment of Chemistry and Biochemistry, The Ohio State University, Columbus, OH 43210, USA
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095, USA. E-mail: shafaat@ucla.edu
First published on 27th March 2024
Abstract
The nickel–iron carbon monoxide dehydrogenase (CODH) enzyme uses a heterometallic nickel–iron–sulfur ([NiFe4S4]) cluster to catalyze the reversible interconversion of carbon dioxide (CO2) and carbon monoxide (CO). These reactions are essential for maintaining the global carbon cycle and offer a route towards sustainable greenhouse gas conversion but have not been successfully replicated in synthetic models, in part due to a poor understanding of the natural system. Though the general protein architecture of CODH is known, the electronic structure of the active site is not well-understood, and the mechanism of catalysis remains unresolved. To better understand the CODH enzyme, we have developed a protein-based model containing a heterometallic [NiFe3S4] cluster in the Pyrococcus furiosus (Pf) ferredoxin (Fd). This model binds small molecules such as carbon monoxide and cyanide, analogous to CODH. Multiple redox- and ligand-bound states of [NiFe3S4] Fd (NiFd) have been investigated using a suite of spectroscopic techniques, including resonance Raman, Ni and Fe K-edge X-ray absorption spectroscopy, and electron paramagnetic resonance, to resolve charge and spin delocalization across the cluster, site-specific electron density, and ligand activation. The facile movement of charge through the cluster highlights the fluidity of electron density within iron–sulfur clusters and suggests an electronic basis by which CN− inhibits the native system while the CO-bound state continues to elude isolation in CODH. The detailed characterization of isolable states that are accessible in our CODH model system provides valuable insight into unresolved enzymatic intermediates and offers design principles towards developing functional mimics of CODH.
Introduction
Biological iron–sulfur (Fe–S) clusters perform a wide array of chemical functions across all kingdoms of life,1 from electron transfer to complex chemical reactions.2–6 A subset of iron–sulfur clusters employ a heterometallic (MFexSy) structure (M = Mo, V, Ni) to perform challenging biological transformations such as nitrogen fixation and carbon dioxide activation, driving great interest in their study.7–9 The carbon monoxide dehydrogenase (CODH) enzyme uses a cuboidal [NiFe4S4] cluster, known as the C-cluster,10 to catalyze the reversible reduction of carbon dioxide (CO2) to carbon monoxide (CO) at ambient temperatures and pressures.11 The enzyme performs this difficult interconversion with high turnover rates, low overpotential, and perfect selectivity,7,12–14 a set of characteristics that has yet to be replicated in any synthetic catalyst.15,16 Thus, understanding the enzymatic mechanism and elucidating key intermediates in the catalytic cycle has become an active area of research, with the overarching goals of applying these principles to the design of future synthetic catalysts for global carbon cycling and conversion.However, CODH is a large and complex homodimeric protein with two additional [Fe4S4] clusters per subunit required for electron transfer, complicating attempts to characterize intermediates within the catalytic cycle.17,18 At this point in time, few CODH states can be considered well-understood (Fig. 1). The Cred1, Cred2, and “Cred2-CO2” states have been isolated and characterized spectroscopically and crystallographically,10,19 while the Cred1 and Cred2 states have been further analyzed using electrochemical techniques.13,20 However, the electronic structure of the Cred2 state is still unknown, as two isoelectronic species have been proposed (Fig. 1).7,21 An EPR study indicating the presence of an EPR-silent state (Cint) between Cred1 and Cred2 has further convoluted the proposed mechanism, indicating two potential pathways to the “Cred2-CO2” state.22 Moreover, despite its importance, no CO-bound state of CODH has ever been isolated or observed. While an early structure of CO-exposed Methanosarcina barkeri CODH found coordination of a diatomic ligand at the nickel site, the identity of this ligand has since been attributed to a bound formyl species.23 Considering the high reactivity of CODH towards CO oxidation,12,20 understanding this intermediate will provide valuable insight into the electronic basis for catalysis.
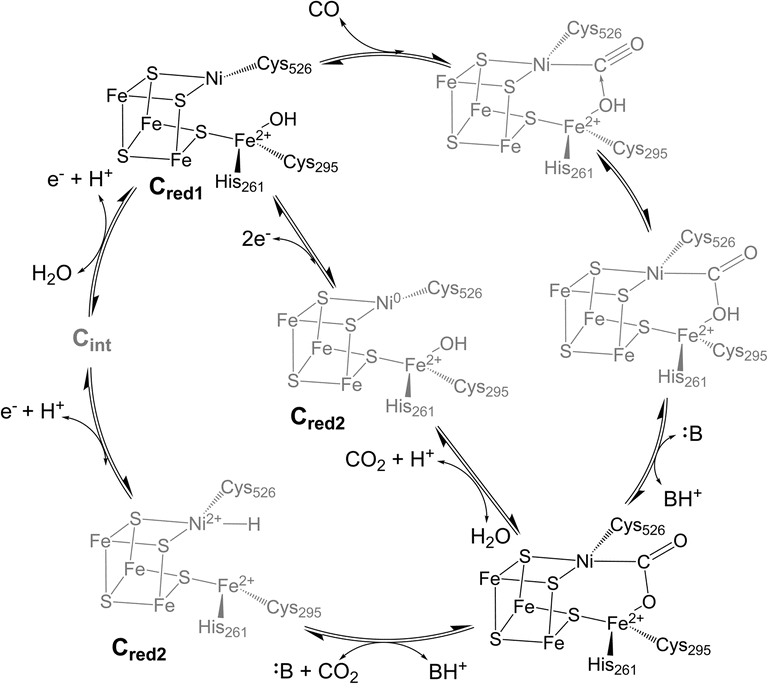 | ||
| Fig. 1 Proposed catalytic cycle for CO oxidation and CO2 reduction at the C-cluster of CODH showing two different hypothesized structures for Cred2. Amino acid numbering is from CODH IICh. Structures in gray indicate intermediate states that are not well-characterized. | ||
One strategy to elucidate structure–function relationships of complex metalloenzymes includes the development and study of model systems that replicate key structural motifs.24 The most popular method for modelling the active sites of enzymes has been to design complexes that mimic the primary coordination sphere within a synthetic ligand scaffold.7,25–28 While modelling the heterometallic iron–sulfur cubane of CODH has been achieved in select systems,29–33 and some of these model compounds also bind cyanide (CN−), a known inhibitor of CODH,29 none of the aforementioned structural models have demonstrated the propensity to bind CO2 or CO.7,34 Furthermore, these synthetic models are generated in aprotic organic solvents and typically utilize π-accepting ligands such as phosphines, which have significantly different bonding characteristics than the π-donating cysteine thiolate ligands of the native system. The development of protein-based models addresses many of these key differences, replicating exact geometric and electronic structural motifs while in some cases also mimicking the reactivity of the native system.35–39 Inspired by other model systems, we sought to develop a protein-based model of the C-cluster within a ferredoxin scaffold. By reproducing the iron–sulfur-cluster moeity, we hypothesized that we could access reactivity analogous to CODH and use spectroscopy to identify key electronic motifs promoting reactivity.
Our initial work has shown that incorporation of NiII into the site-differentiated iron–sulfur cluster of the Pyrococcus furiosus (Pf) ferredoxin (Fd) produced a stable, heterometallic [NiFe3S4]2+ cluster in Fd (NiFdox) capable of rapid, reversible electron transfer.40,41 The reduced [NiFe3S4]+ Fd (NiFdred) displayed the ability to bind both CN− and CO, as evidenced by EPR spectroscopy. The NiFd–CO species serves as the first model for the CO-bound state of CODH. Herein, we use an array of complementary spectroscopic techniques to resolve the electronic structures of the NiFdred, NiFdox, NiFd–CN, and NiFd–CO species, including variable-temperature electron paramagnetic resonance (EPR) spectroscopy, resonance Raman (rR) spectroscopy, X-ray absorption spectroscopy (XAS), and computational modelling. This study reveals distinct changes in the system upon reduction and binding of CO and CN−, suggesting a rearrangement of electron density within the cuboidal heterometallic iron–sulfur cluster depending on the identity of the ligand. The relationship between the NiFd–CO species and the “untrappable” Cred1-CO state in CODH is discussed, along with the role of electronic isomerization in driving inhibition of CODH activity by CN−. As the Cred1-CO state has yet to be observed in the native system, the in-depth characterization of the NiFd–CO species presented here provides valuable insight into the spin state, geometry, and complex electronic structure of this elusive Cred1-CO intermediate. Given the high catalytic rates and full reversibility of native CODH towards CO oxidation and CO2 reduction,12,20 elucidating the structure of this essential species will facilitate the development of functional models that reproduce this reactivity, with long-term implications in environmental remediation and energy storage.
Results
Isotopic labelling indicates spin is delocalized across Ni and Fe
Reduction of the EPR-silent nickel-incorporated NiFdox species to the NiFdred state gives a characteristic X-band EPR spectrum spanning ∼300 mT, with sharp peaks at gapp = 5.7 and gapp = 4.3 as well as broad peaks around gapp ∼ 2.7 and gapp ∼ 1.9 that are consistent with the formation of a single S = 3/2 species. This species had previously been simulated with spin Hamiltonian parameters of giso = 2.0 and |E/D| = 0.16 (Fig. 2A). Introduction of CN− results in quantitative conversion to a new S = 3/2 species, with peaks at gapp = 4.35, 3.9, 1.92 that had been simulated with spin Hamiltonian parameters of giso = 2.0 and |E/D| = 0.07 (Fig. 2B). A more complicated spectrum is obtained upon exposure to CO, as conversion to the new species is not quantitative. We attribute the residual NiFdred to the limited solubility of CO in aqueous solutions and weak binding affinity of NiFdred towards CO. However, the spectra are dominated by two new apparent sets of signals- one set starting around gapp ∼ 4 that would be consistent with an S = 3/2 species, and one set around gapp ∼ 2 that would be consistent with an S = 1/2 species (Fig. 2C). These signals were assumed to derive from a spin-coupling scheme within the cluster that involved all 4 metal centers with local high spin, analogous to conventional iron sulfur clusters. This intracluster coupling was implied to change upon ligand binding, giving rise to distinct spectra.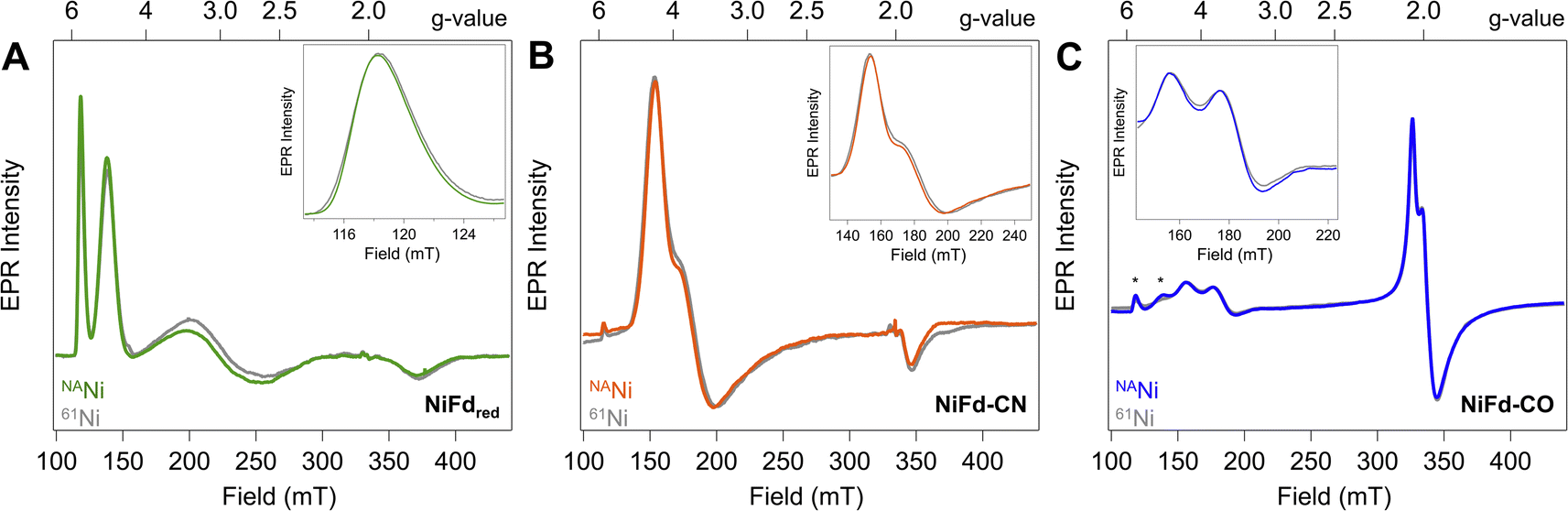 | ||
| Fig. 2 CW X-band EPR spectra (ν = 9.37 GHz, Pμw = 20 mW, T = 8.0 K) of natural abundance (colored traces) and 61Ni-isotopically labelled (gray traces) samples of (A) NiFdred, (B) NiFd–CN, and (C) NiFd–CO. (Insets) Zoomed in views of low-field turning points highlight 61Ni-induced broadening. * indicates residual NiFdred that remains after exposure to CO. | ||
To test this hypothesis and obtain element-specific information on the spin projection across each center in the EPR-active NiFdred, NiFd–CN, and NiFd–CO states, we incorporated 61Ni (I = 3/2) and 57Fe (I = 1/2) into the cluster. As previously observed for homometallic iron–sulfur clusters and other heterometallic clusters, 57Fe labeling resulted in global broadening of all species, consistent with a significant degree of spin delocalized across the Fe centers (Fig. S1†).42,43 The nickel contributions were more distinct across the NiFd species. In all cases, the broad linewidth precluded observation of well-defined hyperfine peaks from the I = 3/2 nucleus, which is typical of high-spin systems.40,44 A modest, 0.4 mT line broadening was observed for the gapp = 5.7 feature of NiFdred upon 61Ni incorporation (Fig. 2A), in good agreement with previously reported results.40 Similarly, upon isotopic labelling, the NiFd–CN and NiFd–CO exhibit 0.7 and 0.4 mT line broadening at the gapp = 4.35 and 3.65 features, respectively (Fig. 2B and C). Notably, there is no substantial broadening (<0.1 mT) of the feature at gapp = 2.05 for the NiFd–CO species (Fig. S2†). The measurable but small line broadening observed for all species indicates that spin density is distributed across the iron and nickel centers in the cluster, rather than being localized on a single ion, and highlights the integral role of nickel in the overall spin-coupling scheme of the cluster. Work is ongoing to obtain increased resolution of the electronic hyperfine coupling to 57Fe and 61Ni nuclei using variable-field Mössbauer and pulsed, high-frequency EPR techniques but is beyond the scope of this work.
Variable temperature CW-EPR suggests the presence of a single NiFd–CO species
To further evaluate the origin of the distinct EPR signals at gapp ∼ 4 and gapp ∼ 2 in NiFd–CO, variable temperature (VT) CW-EPR spectra were measured and compared to samples of NiFd–CN (Fig. 3). The VT-EPR lineshapes of NiFd–CN do not change significantly from 5.5 K to 40 K, suggestive of a single S = 3/2 species (Fig. 3A). On the other hand, the VT-EPR lineshapes of NiFd–CO display unusual behavior. The feature centered at gapp = 2.05 broadens substantially and decreases in intensity from 5.5 to 15.0 K, counter to what is typically observed in conventional mononuclear or spin-coupled S = 1/2 systems (Fig. S3–S5†). By 20 K, the signal centered at gapp = 2.05 is completely gone, while the low-field signal at gapp ≈ 4 can still be observed. Comparison of the individual signal intensities to the total integrated intensity across the temperature range suggests the gapp ∼ 2 species converts into the gapp ∼ 4 species at higher temperatures, rather than arising from independent signals (Fig. S6 and S7†).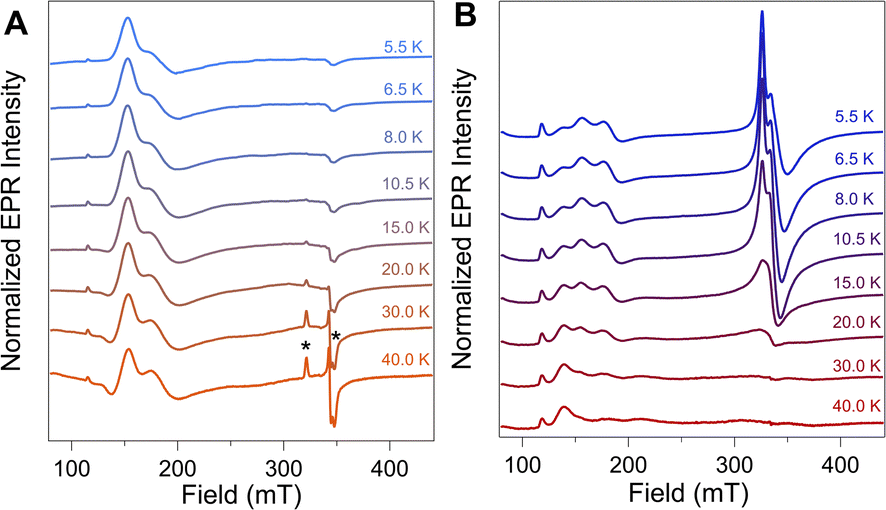 | ||
| Fig. 3 CW EPR spectra (ν = 9.37 GHz, Pμw = 20 mW) of (A) NiFd–CN and (B) NiFd–CO at the indicated temperatures. Spectra were normalized to temperature using a standard Curie dependence [I × T]. * denotes small (<5%) amount of contaminating S = 1/2 [Fe4S4]–CN Fd. | ||
The power saturation behavior of the NiFdred, NiFd–CN, and NiFd–CO was assessed from 5.5 to 15.0 K to estimate the energetic spacing of excited states. The P1/2 values of the NiFd–CO features were consistently larger than those found for the other NiFd species, suggestive of faster relaxation rates. Both the NiFdred and NiFd–CN thus appear to be well-isolated S = 3/2 spin systems (Fig. S8–S14†), while the NiFd–CO shows much faster relaxation. Collectively, the temperature-dependent lineshapes, Curie-corrected intensities, and relaxation properties suggest the NiFd–CO species possesses an S = 1/2 ground spin state with a low-lying S = 3/2 excited state.45,46
ν CO of NiFd–CO suggests a large degree of ligand activation
In prior work, Fourier transform infrared (FTIR) spectroscopy was used to probe the νCN mode frequency, which was observed at 2050 cm−1 and suggested a strong degree of σ donation from the CN− ligand and only weak π back-bonding from nickel.41 However, we were unable to observe an IR band for the NiFd–CO sample from 1800 to 2200 cm−1 that would be indicative of a bound CO ligand (Fig. S15†). The lack of signal was attributed to the low binding affinity of CO to NiFdred and limited CO solubility in aqueous solutions, which prevents measurements at high protein concentrations. Because we were unable to acquire vibrational information using FTIR spectroscopy, resonance Raman spectroscopy was employed.In addition to revealing the νCO mode frequency, this technique provides information on structure by resolving the low-frequency cluster vibrational modes. The resonance Raman spectrum of reduced [Fe3S4]0 Fd exhibits a strong vibrational band centered at 352 cm−1, which is attributed to the symmetric Fe–Sbridging stretching modes (Fig. 4). Weaker vibrational bands can be seen arising from the Fe–Sterminal modes.47 Upon incorporation of the Ni center into the cluster, the dominant vibrational band shifts to 333 cm−1. This bathochromic frequency shift is consistent with incorporation of a fourth metal center into the cluster.47 Additional weak bands can be observed for all other NiFd species (Fig. 4). The Raman spectrum does not change significantly upon CN− binding, with the major vibrational band from the M–Sbridging mode remaining at 333 cm−1. In contrast, CO binding to the NiFdred cluster significantly changes the low frequency region of the Raman spectrum. The dominant vibrational band representing the M–Sbridging mode shifts to 342 cm−1. The bands at 365 and 386 cm−1 shift slightly to lower energy in the presence of 13C-labeled CO, consistent with CO displacement coupling into these modes. As the shifts are substantially lower than those estimated for a local Ni–C or Fe–C oscillator, the CO motion in those bands must be coupled into other vibrational modes. Importantly, an additional isotopically sensitive band at 1964 cm−1 is present in the NiFd–CO species, which shifts to 1921 cm−1 for NiFd-13CO (Fig. 4 and S16†). This shift is nearly exactly as calculated for a local C–O oscillator, suggesting it corresponds to the νCO mode of the NiFd–CO species. This vibrational frequency is significantly lower than that of free CO (2170 cm−1), indicative of significant π back-donation and activation of the CO bond.
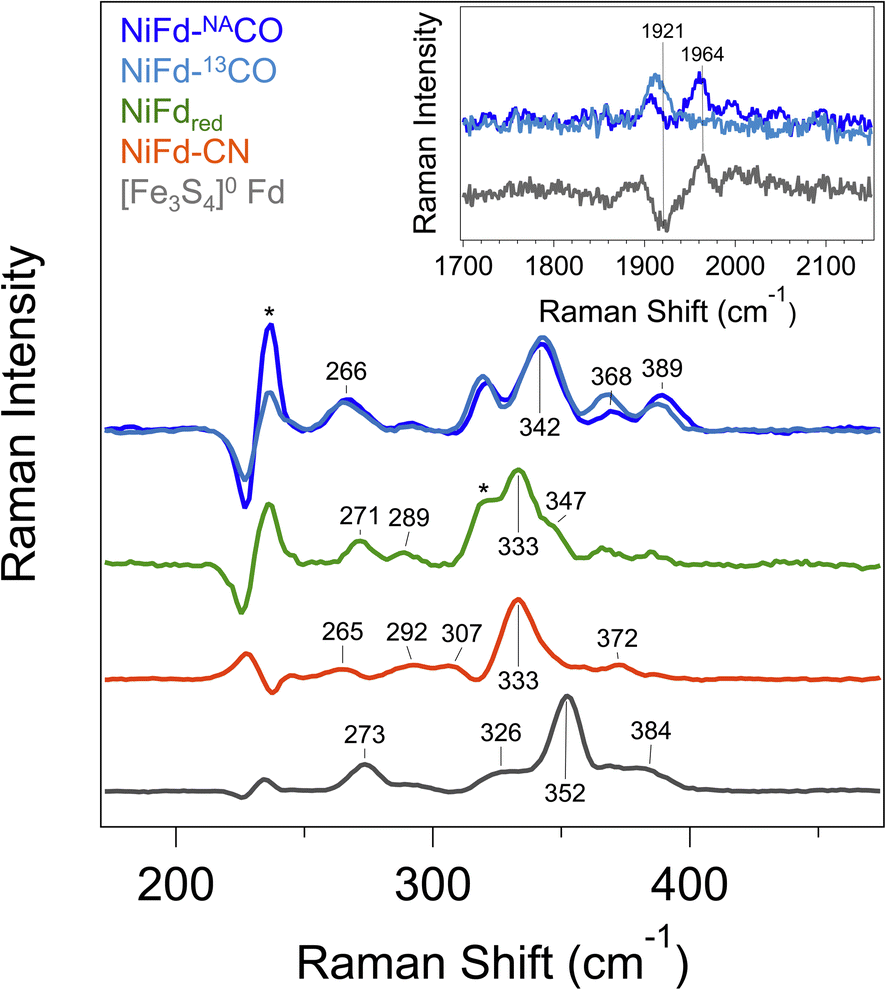 | ||
| Fig. 4 Resonance Raman spectra of [Fe3S4]0 Fd (gray), NiFdred (green), NiFd–CN (orange), and NiFd–CO (blue). Samples were collected at 77 K using an excitation wavelength of 407 nm, P = 8 mW. Residual features corresponding to buffer are indicated with an *. Bands arising from buffer, DT, and quartz were subtracted after collection. (Inset) High frequency region of the resonance Raman spectra of NiFd–CO prepared with natural abundance CO (dark blue) and 13CO (light blue), shown offset from the difference spectrum (gray). The band at 1906 cm−1 is present in both samples and independent of the CO isotope. | ||
Near edge X-ray absorption spectra of NiFd species highlight electronic structure changes localized at the Ni center
In order to obtain element-specific information on the electronic and geometric structure of the different NiFd species, both Ni and Fe K-edge X-ray absorption spectroscopies were used (Fig. 5). The edge transition energy of NiFdox occurs at 8343.8 eV, accompanied by a low-intensity, pre-edge feature located at 8333 eV. This feature arises from the formally dipole forbidden 1s–3d transition, which gains intensity from mixing of the 4p orbitals with the 3d t2 orbitals in non-centrosymmetric geometries (e.g., tetrahedral).48–51 Upon reduction of the cluster to the NiFdred state, the edge position shifts by −4.2 eV to 8339.6 eV, consistent with an increase of electron density at the nickel center. We note that making formal and physical oxidation state assignments for Ni ions using edge positions alone is challenging, as the edge position is not only dependent on the physical oxidation state but is also impacted strongly by metal–ligand covalency and geometry.48,51,52 The pre-edge transition of NiFdred occurs at a very similar energy and intensity as that of the oxidized cluster, suggesting the Ni center remains in a tetrahedral (or distorted tetrahedral) geometry. Interestingly, the XAS spectrum of NiFd–CN shows significant changes when compared to NiFdred. The edge position shifts higher in energy to 8342.2 eV, appearing at a similar energy as NiFdox. More importantly, the cyanide-bound cluster exhibits an intense pre-edge feature at 8336 eV and a very low intensity pre-edge feature at 8332.4 eV, suggesting a change in the geometric structure. Previous studies on model nickel compounds have also observed an intense pre-edge feature around 8336 eV, which is suggested to derive from a 1s–4p transition induced by substantial mixing of the d and p orbitals in a low-spin, square planar geometry.48,49 On the other hand, CO binding to NiFdred shifts the edge energy only slightly, to 8339.8 eV, with the appearance of a similarly intense pre-edge feature at 8334.8 eV. The second pre-edge feature observed in the spectrum at 8333 eV belongs to residual NiFdred that is not bound to CO. The relative edge shift between NiFd–CO and NiFdred suggests only a modest decrease in electron density at the Ni center upon CO binding.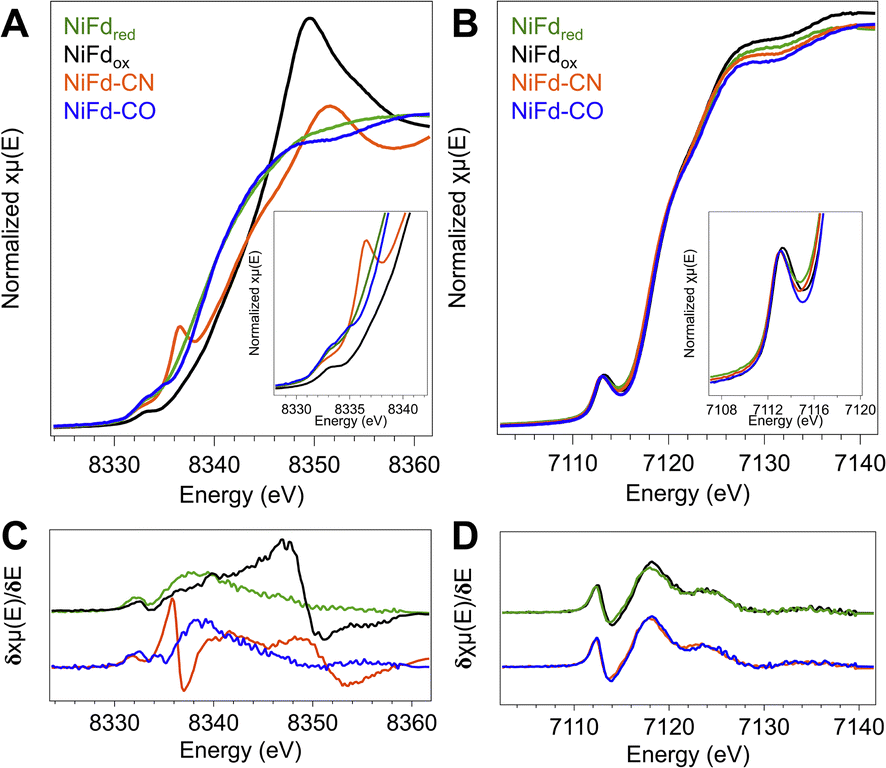 | ||
| Fig. 5 (A) Ni K-edge XANES of the four isolated forms of NiFd. (Inset) Zoom in on the pre-edge region. (B) Fe K-edge XANES of the four isolated forms of NiFd. (Inset) Zoom in on the pre-edge region. (C) Derivative of the Ni K-edge XANES from the traces in A. (D) Derivative of the Fe K-edge XANES from the traces in B. | ||
Ni K-edge EXAFS provides insight into structural changes at the Ni site of NiFd
In addition to information on electron density and local geometry, the Ni K-edge XAS spectra provide solution-phase structural information through analysis of the EXAFS region (Fig. 6). The best fit to the data for the NiFdox was obtained with a primary shell consisting of one N/O atom, three S atoms, and three Fe atoms at distances of 2.01, 2.22, and 2.65 Å, respectively (Table 1). The fit for the NiFdred is similar to that of the oxidized cluster, with contributions from one N/O, three S, and three Fe atoms at distances of 1.95, 2.27, and 2.65 Å, respectively. The distance along the S scattering pathways lengthens slightly, consistent with reduction of the Ni center and in line with previously observed trends for reduction of other [Fe4S4] clusters using Fe K-edge EXAFS.53 In both the reduced and oxidized states of NiFd, the EXAFS data suggest the nickel center retains coordination to three bridging sulfide ligands and a single water/aspartate residue (Fig. S20, S21 and Tables S1, S2†).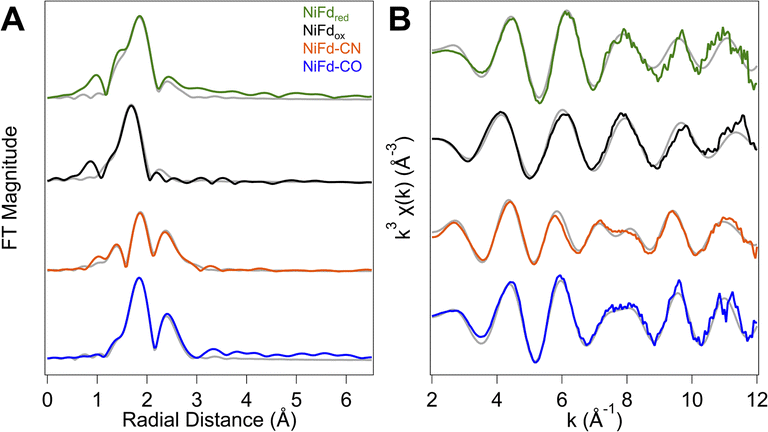 | ||
| Fig. 6 Ni K-edge EXAFS of the four isolated forms of NiFd. (A) Comparison of experimental Fourier transform (FT) EXAFS data (solid) for the four different forms of NiFd overlaid with the best fit (gray). (B) Comparison of experimental k3 EXAFS data (solid) for the four different forms of NiFd overlaid with the best fit (gray). | ||
| NiFdred | NiFdox | NiFd–CO | NiFd–CN | |
|---|---|---|---|---|
| Ni–O shell | ||||
| n | 1 | 1 | — | 1 |
| r (Å) | 1.95 | 2.01 | — | 1.87 |
| σ 2 × 103 (Å2) | 0.32 | 3.72 | — | 3.20 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Ni–C shell | ||||
| n | — | — | 1 | 1 |
| r (Å) | — | — | 1.80 | 1.88 |
| σ 2 × 103 (Å2) | — | — | 5.84 | 12.47 |
|
||||
| Ni–S shell | ||||
| n | 3 | 3 | 3 | 2 |
| r (Å) | 2.27 | 2.22 | 2.29 | 2.29 |
| σ 2 × 103 (Å2) | 3.48 | 6.94 | 6.35 | 5.76 |
|
||||
| Ni–Fe shell | ||||
| n | 3 | 3 | 3 | 3 |
| r (Å) | 2.65 | 2.65 | 2.73 | 2.75 |
| σ 2 × 103 (Å2) | 13.88 | 20.77 | 7.47 | 12.92 |
| ΔEo | 4.481 | −8.72 | 3.160 | 1.989 |
| R-factor | 0.074 | 0.054 | 0.042 | 0.039 |
| Reduced χ2 | 1552690 |
7349274 |
1001669 |
2848655 |
The NiFd–CN species showed significant changes in the XANES region, suggestive of substantial distortion towards a square planar structure. With this information, the preferred best-fit model of the experimental EXAFS data includes one O/N, one N/C, two S, and three Fe scattering pathways at distances of 1.87, 1.88, 2.29, and 2.73 Å, respectively, implicating loss of a bridging sulfide ligand. A significantly more intense peak at R + Δ = 2.3 Å in the R-space data is also observed. This feature is attributed to contributions from both a Ni–Fe single scattering pathway as well as the Ni–CN multiple scattering pathways (Fig. S22 and Table S3†). The EXAFS trace of NiFd–CO is similar to that of the NiFd–CN, including the intense peak in the R-space data at R + Δ = 2.3 Å. Including the parameters for residual NiFdred in a two-component fit to the EXAFS k- and R-space traces, the best fit to the data includes one C/N, three S, and three Fe pathways at distances of 1.80, 2.29, and 2.73 Å, respectively, suggesting that the three sulfide bridges remain intact with loss of the carboxylate ligand (Fig. S23 and Table S4†).
Fe K-edge XANES suggest minor changes at Fe across cluster oxidation states
The changes in the edge and pre-edge positions of the different NiFd species are significantly less pronounced in the Fe K-edge XAS spectra. The Fe K-edge XANES of the [Fe3S4] Fd and the [Fe4S4] Fd species were measured for comparison (Fig. S24†) and observed to follow the trends previously reported for other known biological and synthetic iron–sulfur clusters.50,53–55 The Fe edge positions of the four isolated forms of NiFd also lie within a narrow range, with values at ∼7119 eV for the NiFdox, NiFdred, NiFd–CN, and NiFd–CO, respectively (Fig. 5). While the differences are small (<1 eV), the edge position does follow a general trend across the series, where NiFd–CN < NiFd–CO ≈ NiFdred < NiFdox. Similarly, the pre-edge feature at 7113.2 eV does not change significantly when the cluster is reduced or bound to the small ligands. The insensitivity of the Fe K-edge XANES features across all species suggests that the electronic structure perturbations introduced by oxidation state changes and binding of small molecules are predominantly localized to the Ni center.DFT geometry optimization suggests substrate binding can occur with minimal perturbation to protein structure
At present, there are no published X-ray crystal structures of the WT Pf Fd in the [MFe3S4] state, and we were not able to obtain crystals of NiFd in any oxidation state.56,57 To gain some information on the interaction between the metallocofactor and protein environment, density functional theory calculations were used. A computational model was constructed that included the ligating residues and secondary coordination sphere (Fig. 7). Geometry optimizations were performed for the cluster assuming local and global high spin configurations, which overestimates bond lengths but allows us to qualitatively consider general trends across redox state changes and upon CO binding (Tables S7 and S8†).58 This method was not deemed appropriate for calculations on the NiFd–CN state because of the XAS evidence in favor of a low-spin NiII center (vide supra). Minimal structural perturbations were observed upon cluster reduction from the NiFdox to the NiFdred state, with all metal centers retaining a tetrahedral geometry (Fig. S25–27 and Table S7†). Two conformations of the Asp14 ligand could be optimized for the CO-bound state, one in which the aspartate remains ligated and a Ni–sulfide bond breaks, and another in which the aspartate rotates away from the cluster (Fig. S28–S30 and Table S8†). The EXAFS data and analyses led us to favor the latter geometry, in which the aspartate coordination to the cluster is broken (Fig. S29 and S30†), which preserves tetrahedral geometry at the nickel center and gives a near-linear Ni–C–O bond of 170°. No structures converged in which CO was bound to any of the Fe centers.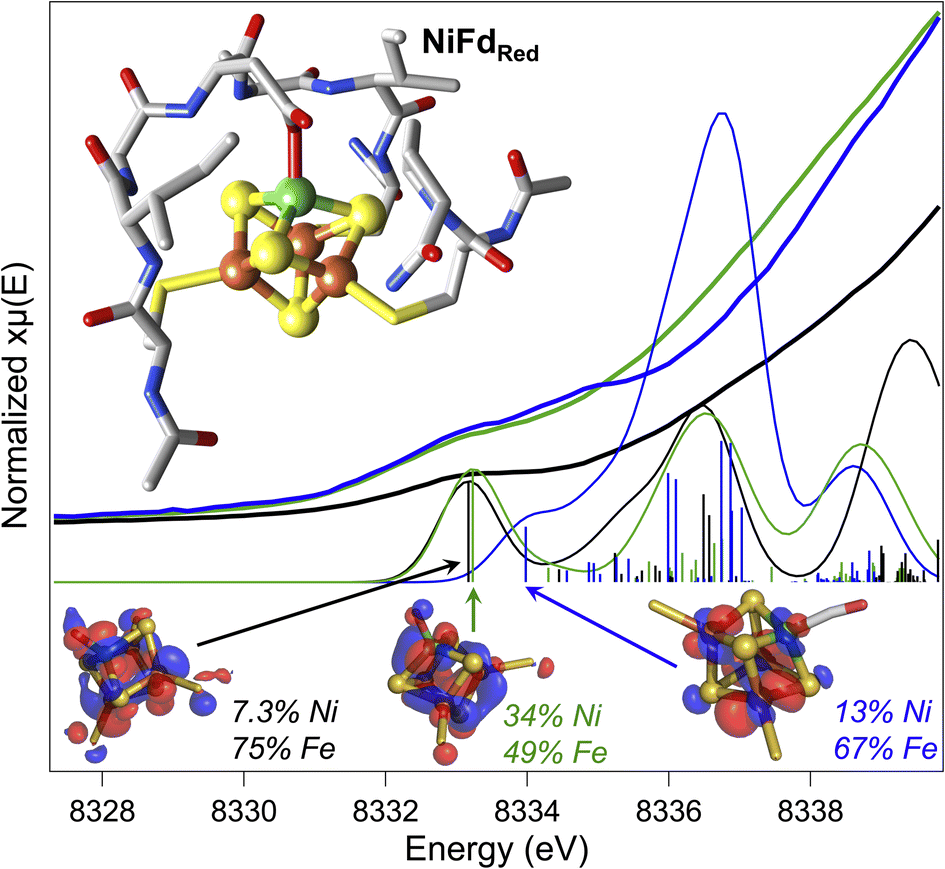 | ||
| Fig. 7 Experimental pre-edge Ni K-edge XANES spectra of NiFdred (green), NiFdox (black), and NiFd–CO (blue) with calculated TD-DFT contributions using the DFT geometry-optimized structures of the NiFd species (inset). Dominant contributing orbital to the indicated transition for each species shown at an isosurface value of 0.03 with distribution over Ni and the Fe atoms indicated. | ||
Preliminary validation of the optimized geometries was supported through comparison of the calculated and experimental Ni K-edge XAS pre-edge energies and intensities (Fig. 7). The calculated pre-edge transitions of NiFdox and NiFdred exhibit similar pre-edge energies and intensities while the NiFd–CO pre-edge transition is shifted to higher energy. In all cases, the dominant orbitals contributing to the transitions are delocalized across the Ni and the three Fe centers. The general agreement between the calculated and experimental data supports the premise that CO binding and redox state changes can occur without inducing large structural perturbations to the protein scaffold.
Discussion
A high density of states is present in the NiFd–CO species
Given the importance of the Cred1-CO state in CODH, we sought to obtain detailed information on the electronic and geometric structure of the CO-bound state(s) of NiFd. Through a holistic analysis of the results from several complementary spectroscopic studies, as reported above, we propose that the NiFd–CO species possesses a ground spin state of S = 1/2 with a low-lying, S = 3/2 excited state within a cuboidal [NiFe3S4]+–CO cluster. This hypothesis derives from several observations: (1) the temperature-dependent EPR spectra reveal that the gapp = 3.65 feature relaxes more slowly than the gapp = 2.05 feature (Fig. 3, S8 and S9†), in contrast to the expected behavior for a standard S = 1/2 species;59 even the spin-coupled S = 1/2 [Fe4S4]+Pf Fd species relaxes more slowly than the high-spin component (Fig. S3†).45 Moreover, the integrated intensities of the features belonging to the NiFd–CO display a linear relationship as a function of 1/T between the temperatures of 6.5–30 K (Fig. S7†), indicating that these two features belong to the same species, with the S = 1/2 feature interconverting to the S = 3/2 feature at higher temperatures. (2) The small but demonstrable EPR line broadening observed for the 61Ni and 57Fe labelling suggest both Ni and Fe contribute to the overall spin of the cluster, supporting an integral role of Ni in the cluster and spin scheme. (3) The high-frequency region of the resonance Raman spectrum of NiFd–CO displays a single isotopically sensitive band at 1964 cm−1, consistent with the presence of a single species. The low-frequency rR bands are consistent with a cubane-like system and show small shifts upon 13CO incorporation, providing further evidence for a CO-bound nickel–iron–sulfur cluster. Considering the XANES, EXAFS, resonance Raman, and EPR data together, we propose a cuboidal structure for NiFd–CO, with tetrahedral coordination at all metals and CO bound to the nickel center. The presence of a low-lying excited state in NiFd–CO may facilitate multistate reactivity in this model system and, by extension, native CODH (vide infra).Ligand binding triggers electron redistribution throughout the NiFd cluster: formal oxidation state assignments
The electronic structure of iron–sulfur clusters is of significant interest for the study of biological electron transfer and cluster reactivity. The most common configurations of [Fe4S4] clusters are generally considered well-understood: formally, the oxidized canonical [Fe4S4]2+ cluster is best described as two pairs of mixed-valent Fe2.5+ centers that are coupled through a double exchange pathway.60,61 Similarly, the reduced cluster can be described as a single mixed valent Fe2.5+ pair antiferromagnetically coupled to a pair of Fe2+ centers. However, a growing body of work suggests that the formal and physical oxidation states of the iron centers are dictated by the overall bonding environment of the cluster.32,62 One such example in biological systems is found in the radical SAM intermediate, Ω, where formation of the organometallic species upon reductive SAM cleavage results in a state that resembles the high-potential iron–sulfur cluster proteins (HiPIPs), with an excess ferric site.61,63,64 The physical electronic structure of Ω is still under debate; however, models of this intermediate suggest that the unique Fe bound to the SAM fragment may adopt a local FeIII electronic configuration.32 Similar electronic rearrangement has been observed in synthetic systems, where recent work from the Suess group has shown that binding of exogenous ligands to one unique site of a model [Fe4S4] cluster influences the oxidation states of the other irons within the cluster.32,62,65 This isomerization is likely linked to the close electronic communication between the metal centers through double exchange pathways within the cluster.Analogous to these prior examples in biological and synthetic [Fe4S4] clusters, binding of exogenous ligands to NiFd impacts the electronic distribution within the [NiFe3S4] cluster. The initial reduction event from NiFdox to NiFdred appears to be dominantly localized on the nickel center with minimal change at the iron centers, as evidenced by the XAS spectra. The similarity in the pre-edge features of NiFdox and NiFdred and comparison to well-defined synthetic compounds suggests a tetrahedral nickel center.48,49,51,52 While formal oxidation state assignments of metals provide only a rough connection to the physical oxidation states, particularly in systems with highly covalent bonding, the experimental evidence supports the assignment of a high-spin NiII in the NiFdox species and a NiI center in the NiFdred species (Fig. 8), with a formally assigned [Fe3S4]0 cluster fragment for both states respectively. The highly activated CO ligand (νCO = 1964 cm−1) in NiFd–CO is consistent with significant electron donation into the π* orbitals of the CO ligand and is in line with other known NiI–CO complexes.66,67 Typical four-coordinate NiII–CO and Ni0–CO compounds exhibit νCO values of ∼2060 and 1830 cm−1, respectively.68–71 Considering νCO along with the XAS edge energy, which is similar to that of NiFdred, a NiI–CO and [Fe3S4]0 configuration is suggested to be present in the NiFd–CO species. On the other hand, back-donation into the CN− π* orbitals is relatively small in NiFd–CN, as the νCN is observed at 2050 cm−1, a small shift from free cyanide (νCN = 2080 cm−1).41 This vibrational frequency is consistent with other NiII–CN complexes.72 The XANES spectra provide further evidence for differences in electronic structure at the nickel center. The edge position shifts to higher energies relative to NiFdred or NiFd–CO and instead overlaps with that of NiFdox, suggesting a significant loss of electron density from the nickel center. Additionally, the intense pre-edge feature that is observed at 8336 eV derives from the 1s–4p transition and is only prevalent in low-spin, square planar NiII species.48,49,52 Collectively, these observations suggest the presence of a low-spin, NiII oxidation state in NiFd–CN, with a [Fe3S4]− fragment (Fig. 8). A shift in physical oxidation state for NiFd–CN relative to NiFdred would suggest the electron density is redistributed back into the [Fe3S4] fragment. Indeed, careful analysis of the Fe K-edge XAS spectrum does show a shift of the NiFd–CN edge position to lower energy by approximately 0.2 eV, reflecting an average increase in electron density across the three iron centers (Fig. 5). Due to the highly delocalized nature of iron sulfur clusters, we would expect additional electron density to be further distributed across the bridging sulfide and terminal ligands. These analyses provide direct evidence for electronic communication and fluidity of electron density between the four metal centers, highlighting a potential mechanism for promoting multielectron chemistry within heterometallic clusters.
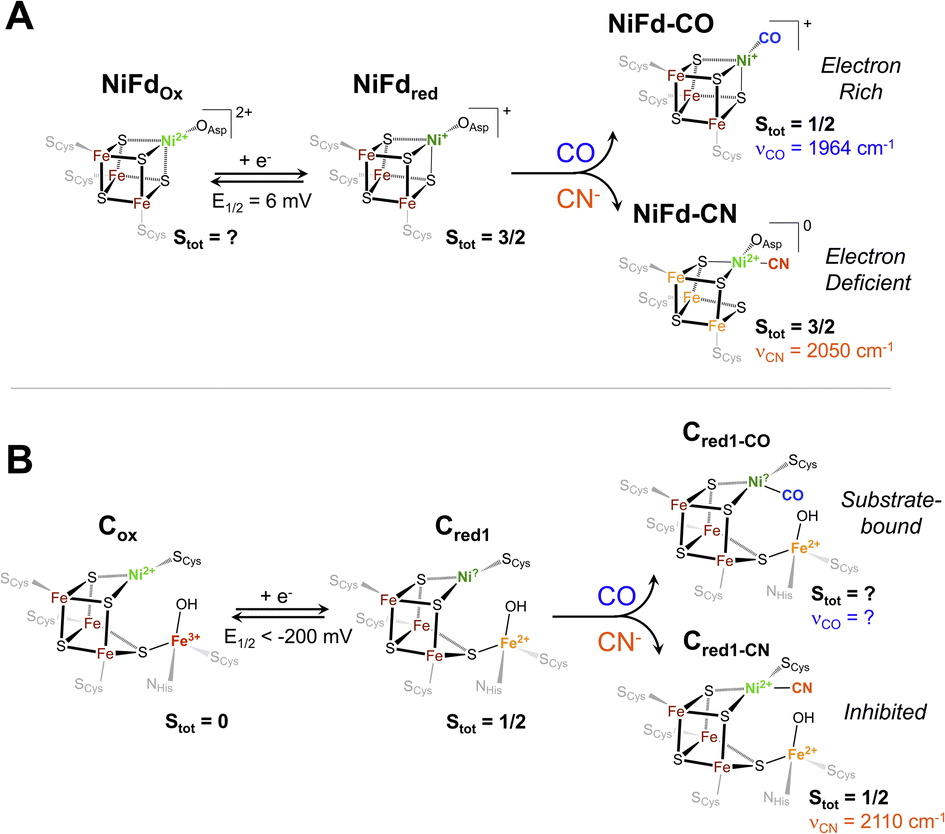 | ||
| Fig. 8 Proposed electronic and geometric structures of the (A) NiFdox, NiFdred, NiFd–CO, and NiFd–CN species with key experimental metrics indicated that have been obtained from this work, and (B) analogous Cox, Cred1, Cred1-CO, and Cred1-CN states of CODH with spectroscopic metrics obtained from ref. 17, 75 and 76. | ||
Implications for substrate and inhibitor binding in native CODH
The Ni K-edge XANES spectrum of NiFdred has remarkably similar features to previously observed spectra of reduced Rr CODH and other synthetic [NiFe3S4] clusters, including a weak pre-edge feature at 8333 eV.49 Oxidation of Rr CODH to the catalytically inactive Cox state does not cause significant changes to the Ni K-edge XAS spectra, suggesting the redox state changes are not localized to the Ni center; instead, Mössbauer and Fe K-edge XAS spectroscopy have suggested the initial reduction event to activate CODH occurs at the pendant iron center, generating the resting state Cred1 (Fig. 8).17,49,73 In the absence of an exogenous Fe ion, as in NiFd, the redox state changes are localized on the substrate-binding Ni center (Fig. 8), providing benchmarks for the limiting cases that might be observed throughout CODH catalysis (Fig. 5 and S24†). More parallels are observed in comparing the EXAFS data of reduced Rr CODH and NiFdred, as both species have an overall coordination number of four and similar distances to the bridging sulfide ligands. Like the spectra of Rr CODH, the NiFdred/NiFdox do not exhibit an intense Ni–Fe scattering feature that is present in the synthetic [NiFe3S4] cubane clusters. We have interpreted the latter observation to indicate that, in the absence of a small-molecule ligand, the NiFd cubane is not sufficiently rigid to resolve a clear Ni–Fe scattering vector.49 The Ni–Fe scattering pathway becomes more pronounced in the NiFd–CO and NiFd–CN samples, suggesting that binding of CO or CN− rigidifies the cluster (Fig. S22, S23 and Tables S3–S4†). This may be the case for the [NiFe3S4] subsite of CODH as well, allowing conformational flexibility prior to substrate binding.The EPR data show parallels to native CODH and other [NiFe3S4] synthetic models. The EPR spectra of Cred1 and the cyanide-inhibited state, Cred1-CN, both have total ground spin states of Stot = 1/2 with gavg = 1.82 and 1.72, respectively.17,74,75 From the Mössbauer and EPR data, this Stot = 1/2 ground state is suggested to arise from antiferromagnetic coupling between the S = 3/2 [NiFe3S4] subsite and the S = 2 exogenous Fe subsite. The NiFdred state has a ground spin state of S = 3/2, which is directly analogous to that of the [NiFe3S4] subsite of the Cred1 state.17,49 Additionally, the electronic parameters of the NiFdred species are similar to those of synthetic cubane [NiFe3S4]+ models by Holm and coworkers, which also have ground spin states of S = 3/2.34,49 In the case of NiFdred, we postulate this spin state likely arises from the coupling of an S = 1/2 NiI center to the [Fe3S4]0 fragment on the basis of the data presented here. Experiments using Mössbauer spectroscopy are currently underway to more precisely determine the local spin states and coupling scheme of Fe centers within the cluster. Additionally, like the native system, only small line-broadening is observed upon substitution of 61Ni,43 which may highlight the importance of electronic cooperativity and spin delocalization between the metal centers for reactivity in CODH. Binding of CN− to NiFdred preserves the S = 3/2 ground spin state but perturbs the EPR spectrum in a manner that is similar to the changes observed for synthetic cubane [NiFe3S4]+ models; the spin state of the cubane in native CODH is also suggested to remain S = 3/2 on the basis of coupling with the exogenous high-spin ferrous ion (Fig. 8).
The proposed geometry of the nickel center in NiFd–CN is also similar to that observed in the cyanide-bound structure of CODH IICh; however, the νCN (2110 cm−1) of the native enzyme is notably higher than that in the NiFd–CN state.76 We believe this discrepancy derives from the differences in hydrogen bonding environments for the two systems. In native CODH, proposed hydrogen bonding interactions with secondary sphere residues may promote greater σ-donating and less π-accepting character from the CN− ligand, giving rise to higher νCN vibrational frequencies.77
Though CO and CN− are isoelectronic and often considered interchangeable in the context of structure and bonding, it is evident that each of these ligands imparts unique behavior to the nickel-substituted iron–sulfur cluster in NiFd. It is thus worth considering the subtle distinctions in electronic structure between the two small ligands. Both CO and CN− bind to the NiI center in the stable NiFdred state and to the nickel center in Cred1,76,78 driving a natural comparison between the two systems across ligands. CO is a neutral ligand with strong π-accepting character, underpinning the tendency to bind to electron-rich metals,79 while the anionic CN− ligand has greater σ-donor ability and more capacity for hydrogen bonding.80 The carbon center in CN− is therefore more nucleophilic than in CO, which likely preferentially stabilizes the NiII oxidation state and drives the redistribution of electron density across the cluster in NiFd.62 This intracluster isomerization may hint at the mechanism through which CN− inhibits turnover of CODH. When CN− binds to the nickel center, which is suggested to be the binding site from multiple structural studies,76,78 the nucleophilicity of the carbon center may drive the nickel to adopt a formal divalent oxidation state, effectively trapping the system in a configuration that is more electron-deficient at the nickel center (Fig. 8). This electron-deficient state may stabilize the system in the presence of oxidants, as recent work suggests the CN-bound C-cluster is more resistant to O2 damage than the resting state, but requires reductive activation to re-enter the catalytic cycle.12,81 On the other hand, the electron-rich NiI center in NiFdred is stabilized upon CO binding. The large degree of activation of the substrate CO ligand suggests a similarly nucleophilic NiI state may be present in the catalytically active Cred1 and elusive Cred1-CO states (Fig. 8). The temperature-dependent EPR data on NiFd–CO may also provide insight into the absence of a known Cred1-CO signal. While the ground spin state of NiFd–CO is suggested to be Stot = 1/2, it is not well-isolated, as evidenced by the clear appearance of high-spin signals at cryogenic temperatures. We consider it possible that low-lying excited states of Cred1-CO may be present in native CODH that could complicate EPR observation. Moreover, the presence of the exogenous Fe center in the C-cluster is expected to affect the electronic structure of the spin system. The absence of a nearby nucleophilic site in NiFd–CO likely minimizes reactivity, permitting facile observation of the CO-bound state. Access to excited states in NiFd may also indicate potential for multistate reactivity in CODH, though this discussion would require advanced computational analysis that is beyond the scope of this work.45,82 The distinct differences observed between the CN− and CO underscore the importance of understanding the interactions between enzymes and their native substrates.
Conclusions
A nickel-substituted [NiFe3S4] ferredoxin (NiFd) protein was investigated as a model of the cubane subsite of carbon monoxide dehydrogenase (CODH). The NiFd system was prepared in several oxidation and substrate-bound states to mimic key states of native CODH. Through the use of EPR, rR, K-edge XANES, and EXAFS spectroscopies, we have assigned the redox processes observed to be formally centered on nickel and characterized the NiFd–CO species as a NiI–CO nickel center that has tetrahedral coordination. Cyanide binding induces redox isomerization, resulting in a low-spin, square planar nickel center and increased electron density spread across the iron atoms in the cluster. This is in line with phenomena previously observed within synthetic iron–sulfur clusters, providing evidence for the ability of heterometallic cubane clusters to shuffle electron density around within the cluster. Furthermore, extrapolating the results found using this protein-based model, we have postulated that similar phenomena may occur in native CODH, thus providing insight into the mechanism of inhibition by CN− in the native system. Ultimately, the results found from this study highlight the utility of developing protein-based models of enzymes.Data availability
All raw and processed data generated in this study, including data presented in the main manuscript and the ESI† are freely available upon request.Author contributions
Collection of EPR, FTIR, optical, resonance Raman, and Ni/Fe K-edge XAS spectroscopies along with all sample preparation was done by LCL. The computational DFT results were completed by JASG. Analysis of all data was performed by LCL and HSS. YL assisted in collecting XAS data. AJJ prepared the proposal for beamline time and assisted in collection of the XAS data. The manuscript was written by LCL and HSS. HSS was responsible for oversight and management of the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Energy, Basic Energy Sciences, Physical Biosciences program (DE SC-0018020 and SC-0023137). The Pf Fd plasmid (pTrc99aPfFd) was a generous gift from Professor Michael Adams (U. Georgia). LCL would like to acknowledge partial support from the Ohio State University and Lubrizol Foundation Fellowship. JASG would like to acknowledge partial support through the ACS Bridge Program. YL would like to acknowledge support from GM128852. The authors would like to acknowledge Dr Justin North for his help in preparation of high-pressure gaseous samples, Alina Yerbulekova, Yunqiao Gan, and Dr Adam Jenkins for laser assistance, and Dr Leland B. Gee and Leah Kelly for their help with collection of the X-ray spectroscopic data. X-ray spectroscopic data was collected at the Stanford Synchrotron Radiation Lightsource, Menlo Park, CA. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.Notes and references
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Structure, function, and formation of biological iron–sulfur clusters, Annu. Rev. Biochem., 2005, 74(1), 247–281, DOI:10.1146/annurev.biochem.74.082803.133518.
- H. Beinert, R. H. Holm and E. Münck, Iron–sulfur clusters: nature's modular, multipurpose structures, Science, 1997, 277(5326), 653–659, DOI:10.1126/science.277.5326.653.
- W. Yao, P. M. Gurubasavaraj and P. L. Holland, All-ferrous iron–sulfur clusters, in Molecular Design in Inorganic Biochemistry, ed. D. Rabinovich, Springer, Berlin, Heidelberg, Structure and Bonding, 2014; pp. 1–37. DOI:10.1007/430_2012_81.
- C. J. Walsby, D. Ortillo, J. Yang, M. R. Nnyepi, W. E. Broderick, B. M. Hoffman and J. B. Broderick, Spectroscopic approaches to elucidating novel iron−sulfur chemistry in the “radical-SAM” protein superfamily, Inorg. Chem., 2005, 44(4), 727–741, DOI:10.1021/ic0484811.
- E. Krahn, B. Weiss, M. Kröckel, J. Groppe, G. Henkel, S. Cramer, A. Trautwein, K. Schneider and A. Müller, The Fe-only nitrogenase from rhodobacter capsulatus: identification of the cofactor, an unusual, high-nuclearity iron–sulfur cluster, by Fe K-Edge EXAFS and 57Fe mössbauer spectroscopy, J. Biol. Inorg. Chem., 2002, 7(1–2), 37–45, DOI:10.1007/s007750100263.
- H. C. Angove, S. J. Yoo, E. Münck and B. K. Burgess, An all-ferrous state of the fe protein of nitrogenase: interaction with nucleotides and electron transfer to the MoFe protein, J. Biol. Chem., 1998, 273(41), 26330–26337, DOI:10.1074/jbc.273.41.26330.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Structure, function, and mechanism of the nickel metalloenzymes, CO dehydrogenase, and acetyl-CoA synthase, Chem. Rev., 2014, 114(8), 4149–4174, DOI:10.1021/cr400461p.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Nitrogenase MoFe-protein at 1.16 Å resolution: a central ligand in the FeMo-cofactor, Science, 2002, 297(5587), 1696–1700, DOI:10.1126/science.1073877.
- C. C. Lee, Y. Hu and M. W. Ribbe, Unique Features of the Nitrogenase VFe protein from Azotobacter vinelandii, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(23), 9209–9214, DOI:10.1073/pnas.0904408106.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster, Science, 2001, 293(5533), 1281–1285, DOI:10.1126/science.1061500.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation, Chem. Rev., 2013, 113(8), 6621–6658, DOI:10.1021/cr300463y.
- V. C.-C. Wang, S. W. Ragsdale and F. A. Armstrong, Investigations of two bidirectional carbon monoxide dehydrogenases from carboxydothermus hydrogenoformans by protein film electrochemistry, ChemBioChem, 2013, 14(14), 1845–1851, DOI:10.1002/cbic.201300270.
- A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, Rapid and efficient electrocatalytic CO2/CO interconversions by Carboxydothermus hydrogenoformans CO dehydrogenase I on an electrode, J. Am. Chem. Soc., 2007, 129(34), 10328–10329, DOI:10.1021/ja073643o.
- V. Svetlitchnyi, C. Peschel, G. Acker and O. Meyer, Two membrane-associated NiFeS–carbon monoxide dehydrogenases from the anaerobic carbon–monoxide-utilizing eubacterium Carboxydothermus hydrogenoformans, J. Bacteriol., 2001, 183(17), 5134–5144, DOI:10.1128/JB.183.17.5134-5144.2001.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Highly selective molecular catalysts for the CO2 -to-CO electrochemical conversion at very low overpotential. contrasting Fe vs. Co quaterpyridine complexes upon mechanistic studies, ACS Catal., 2018, 8(4), 3411–3417, DOI:10.1021/acscatal.7b04412.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Molecular catalysis of CO2 reduction: recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes, Chem. Soc. Rev., 2020, 49(16), 5772–5809, 10.1039/D0CS00218F.
- Z. Hu, N. J. Spangler, M. E. Anderson, J. Xia, P. W. Ludden, P. A. Lindahl and E. Münck, Nature of the C-cluster in Ni-containing carbon monoxide dehydrogenases, J. Am. Chem. Soc., 1996, 118(4), 830–845, DOI:10.1021/ja9528386.
- E. D. Carlson and E. T. Papoutsakis, Heterologous expression of the clostridium carboxidivorans CO dehydrogenase alone or together with the acetyl coenzyme a synthase enables both reduction of CO2 and oxidation of CO by clostridium acetobutylicum, Appl. Environ. Microbiol., 2017, 83(16), e00829, DOI:10.1128/AEM.00829-17.
- J.-H. Jeoung and H. Dobbek, Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase, Science, 2007, 318(5855), 1461–1464, DOI:10.1126/science.1148481.
- V. Wang, S. W. Ragsdale and F. A. Armstrong, Investigations of the efficient electrocatalytic interconversions of CO2 and CO by nickel-containing carbon monoxide dehydrogenases, Met. Ions Life Sci., 2014, 14, 71–97, DOI:10.1007/978-94-017-9269-1_4.
- P. A. Lindahl, The Ni-containing carbon monoxide dehydrogenase family: light at the end of the tunnel?, Biochemistry, 2002, 41(7), 2097–2105, DOI:10.1021/bi015932+.
- D. M. Fraser and P. A. Lindahl, Evidence for a proposed intermediate redox state in the CO/CO2 active site of acetyl-CoA synthase (carbon monoxide dehydrogenase) from clostridium thermoaceticum, Biochemistry, 1999, 38(48), 15706–15711, DOI:10.1021/bi990398f.
- W. Gong, B. Hao, Z. Wei, D. J. Ferguson, T. Tallant, J. A. Krzycki and M. K. Chan, Structure of the A;2ε2 Ni-dependent CO dehydrogenase component of the methanosarcina barkeri acetyl-CoA decarbonylase/synthase complex, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(28), 9558–9563, DOI:10.1073/pnas.0800415105.
- K. D. Karlin, Metalloenzymes, structural motifs, and inorganic models, Science, 1993, 261(5122), 701–708, DOI:10.1126/science.7688141.
- C. H. Arnett and T. Agapie, Activation of an open shell, carbyne-bridged diiron complex toward binding of dinitrogen, J. Am. Chem. Soc., 2020, 142, 10059–10068, DOI:10.1021/jacs.0c01896.
- C.-H. Wang and S. DeBeer, Structure, reactivity, and spectroscopy of nitrogenase-related synthetic and biological clusters, Chem. Soc. Rev., 2021, 50(15), 8743–8761, 10.1039/D1CS00381J.
- L. N. V. Le, G. A. Bailey, A. G. Scott and T. Agapie, Partial synthetic models of femoco with sulfide and carbyne ligands: effect of interstitial atom in nitrogenase active site, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(49), e2109241118, DOI:10.1073/pnas.2109241118.
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Hydrogenase enzymes and their synthetic models: the role of metal hydrides, Chem. Rev., 2016, 116(15), 8693–8749, DOI:10.1021/acs.chemrev.6b00180.
- S. Ciurli, S. B. Yu, R. H. Holm, K. K. P. Srivastava and E. Munck, Synthetic nickel–iron NiFe3Q4 cubane-type clusters (S = 3/2) by reductive rearrangement of linear [Fe3Q4(SEt)4]3- (Q = Sulfur, Selenium), J. Am. Chem. Soc., 1990, 112(22), 8169–8171, DOI:10.1021/ja00178a053.
- T. D. P. Stack and R. H. Holm, Subsite-differentiated analogs of biological [4Fe-4S]2+ clusters: synthesis, solution and solid-state structures, and subsite-specific reactions, J. Am. Chem. Soc., 1988, 110(8), 2484–2494, DOI:10.1021/ja00216a023.
- A. McSkimming and D. L. M. Suess, Selective synthesis of site-differentiated Fe4S4 and Fe6S6 clusters, Inorg. Chem., 2018, 57(23), 14904–14912, DOI:10.1021/acs.inorgchem.8b02684.
- A. McSkimming, A. Sridharan, N. B. Thompson, P. Müller and D. L. M. Suess, An [Fe4S4]3+–alkyl cluster stabilized by an expanded scorpionate ligand, J. Am. Chem. Soc., 2020, 142(33), 14314–14323, DOI:10.1021/jacs.0c06334.
- D. W. N. Wilson, M. S. Fataftah, Z. Mathe, B. Q. Mercado, S. DeBeer and P. L. Holland, Three-coordinate nickel and metal–metal interactions in a heterometallic iron–sulfur cluster, J. Am. Chem. Soc., 2024, 146(6), 4013–4025, DOI:10.1021/jacs.3c12157.
- S. Ciurli, P. K. Ross, M. J. Scott, S.-B. Yu and R. H. Holm, Synthetic nickel-containing heterometal cubane-type clusters with NiFe3Q4Cores (Q = sulfur, selenium), J. Am. Chem. Soc., 1992, 114(13), 5415–5423, DOI:10.1021/ja00039a063.
- J. W. Slater, S. C. Marguet, H. A. Monaco and H. S. Shafaat, Going beyond structure: nickel-substituted rubredoxin as a mechanistic model for the [NiFe] hydrogenases, J. Am. Chem. Soc., 2018, 140(32), 10250–10262, DOI:10.1021/jacs.8b05194.
- A. C. Manesis, B. W. Musselman, B. C. Keegan, J. Shearer, N. Lehnert and H. S. Shafaat, A Biochemical nickel(I) state supports nucleophilic alkyl addition: a roadmap for methyl reactivity in acetyl coenzyme a synthase, Inorg. Chem., 2019, 58(14), 8969–8982, DOI:10.1021/acs.inorgchem.8b03546.
- A. C. Manesis, A. Yerbulekova, J. Shearer and H. S. Shafaat, Thioester synthesis by a designed nickel enzyme models prebiotic energy conversion, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(30), e2123022119, DOI:10.1073/pnas.2123022119.
- D. J. DiPrimio and P. L. Holland, Repurposing metalloproteins as mimics of natural metalloenzymes for small-molecule activation, J. Inorg. Biochem., 2021, 219, 111430, DOI:10.1016/j.jinorgbio.2021.111430.
- K. Oohora, Y. Miyazaki and T. Hayashi, Myoglobin reconstituted with ni tetradehydrocorrin as a methane-generating model of methyl-coenzyme M reductase, Angew. Chem., Int. Ed., 2019, 58(39), 13813–13817, DOI:10.1002/anie.201907584.
- R. C. Conover, J. B. Park, M. W. W. Adams and M. K. Johnson, Formation and properties of an iron–nickel sulfide (NiFe3S4) cluster in Pyrococcus furiosus ferredoxin, J. Am. Chem. Soc., 1990, 112(11), 4562–4564, DOI:10.1021/ja00167a074.
- L. C. Lewis and H. S. Shafaat, Reversible electron transfer and substrate binding support [NiFe3S4] ferredoxin as a protein-based model for [NiFe] carbon monoxide dehydrogenase, Inorg. Chem., 2021, 60(18), 13869–13875, DOI:10.1021/acs.inorgchem.1c01323.
- J. Telser, M. J. Benecky, M. W. Adams, L. E. Mortenson and B. M. Hoffman, EPR and electron nuclear double resonance investigation of oxidized hydrogenase II (uptake) from clostridium pasteurianum W5. Effects of carbon monoxide binding, J. Biol. Chem., 1987, 262(14), 6589–6594, DOI:10.1016/S0021-9258(18)48282-8.
- P. J. Stephens, M. C. McKenna, S. A. Ensign, D. Bonam and P. W. Ludden, Identification of a Ni- and Fe-containing cluster in rhodospirillum rubrum carbon monoxide dehydrogenase, J. Biol. Chem., 1989, 264(28), 16347–16350, DOI:10.1016/S0021-9258(19)84710-5.
- E. D. Badding, S. Srisantitham, D. A. Lukoyanov, B. M. Hoffman and D. L. M. Suess, Connecting the geometric and electronic structures of the nitrogenase iron–molybdenum cofactor through site-selective 57Fe labelling, Nat. Chem., 2023, 15(5), 658–665, DOI:10.1038/s41557-023-01154-9.
- J. T. Henthorn, G. E. Cutsail, T. Weyhermüller and S. DeBeer, Stabilization of intermediate spin states in mixed-valent diiron dichalcogenide complexes, Nat. Chem., 2022, 14(3), 328–333, DOI:10.1038/s41557-021-00853-5.
- R. C. Conover, A. T. Kowal, W. G. Fu, J. B. Park, S. Aono, M. W. Adams and M. K. Johnson, Spectroscopic characterization of the novel iron–sulfur cluster in pyrococcus furiosus ferredoxin, J. Biol. Chem., 1990, 265(15), 8533–8541, DOI:10.1016/s0021-9258(19)38921-5.
- W. Fu, J. Telser, B. M. Hoffman, E. T. Smith, M. W. W. Adams, M. G. Finnegan, R. C. Conover and M. K. Johnson, Interaction of Tl+ and Cs+ with the [Fe3S4] cluster of pyrococcus furiosus ferredoxin: investigation by resonance raman, MCD, EPR, and ENDOR spectroscopy, J. Am. Chem. Soc., 1994, 116(13), 5722–5729, DOI:10.1021/ja00092a024.
- G. J. Colpas, M. J. Maroney, C. Bagyinka, M. Kumar, W. S. Willis, S. L. Suib, P. K. Mascharak and N. Baidya, X-ray spectroscopic studies of nickel complexes, with application to the structure of nickel sites in hydrogenases, Inorg. Chem., 1991, 30(5), 920–928, DOI:10.1021/ic00005a010.
- G. O. Tan, S. A. Ensign, S. Ciurli, M. J. Scott, B. Hedman, R. H. Holm, P. W. Ludden, Z. R. Korszun, P. J. Stephens and K. O. Hodgson, On the structure of the nickel/iron/sulfur center of the carbon monoxide dehydrogenase from rhodospirillum rubrum: an x-ray absorption spectroscopy study, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(10), 4427–4431, DOI:10.1073/pnas.89.10.4427.
- G. N. George, I. J. Pickering, R. C. Prince, Z. H. Zhou and M. W. W. Adams, X-ray absorption spectroscopy of pyrococcus furiosus rubredoxin, J. Biol. Inorg. Chem., 1996, 1(3), 226–230, DOI:10.1007/s007750050047.
- N. Baidya, M. M. Olmstead, J. P. Whitehead, C. Bagyinka, M. J. Maroney and P. K. Mascharak, X-ray absorption spectra of nickel complexes with N3S2 chromophores and spectroscopic studies on hydride and carbon monoxide binding at these nickel centers: relevance to the reactivity of the nickel site(s) in [FeNi] hydrogenases, Inorg. Chem., 1992, 31(17), 3612–3619, DOI:10.1021/ic00043a023.
- S. Hugenbruch, H. S. Shafaat, T. Krämer, M. U. Delgado-Jaime, K. Weber, F. Neese, W. Lubitz and S. DeBeer, In search of metal hydrides: an X-Ray absorption and emission study of [NiFe] hydrogenase model complexes, Phys. Chem. Chem. Phys., 2016, 18(16), 10688–10699, 10.1039/C5CP07293J.
- J. B. Solomon, K. Tanifuji, C. C. Lee, A. J. Jasniewski, B. Hedman, K. O. Hodgson, Y. Hu and M. W. Ribbe, Characterization of a nitrogenase iron protein substituted with a synthetic [Fe4Se4] cluster, Angew. Chem., Int. Ed., 2022, 61(19), e202202271, DOI:10.1002/anie.202202271.
- M. A. Blank, C. C. Lee, Y. Hu, K. O. Hodgson, B. Hedman and M. W. Ribbe, Structural models of the [Fe4S4] clusters of homologous nitrogenase Fe proteins, Inorg. Chem., 2011, 50(15), 7123–7128, DOI:10.1021/ic200636k.
- R. G. Shulman, P. Eisenberger, B. K. Teo, B. M. Kincaid and G. S. Brown, Fluorescence X-ray absorption studies of rubredoxin and its model compounds, J. Mol. Biol., 1978, 124(2), 305–321, DOI:10.1016/0022-2836(78)90301-7.
- M. S. Nielsen, P. Harris, B. L. Ooi and H. E. M. Christensen, The 1.5 Å resolution crystal structure of [Fe3S4]-ferredoxin from the hyperthermophilic archaeon pyrococcus furiosus, Biochemistry, 2004, 43(18), 5188–5194, DOI:10.1021/bi049942x.
- M. N. Løvgreen, M. Martic, M. S. Windahl, H. E. M. Christensen and P. Harris, Crystal structures of the all-cysteinyl-coordinated D14C variant of pyrococcus furiosus ferredoxin: [4Fe–4S] ↔ [3Fe–4S] cluster conversion, J. Biol. Inorg. Chem., 2011, 16(5), 763–775, DOI:10.1007/s00775-011-0778-7.
- F. Cappelluti, L. Bencivenni and L. Guidoni, Spin-symmetrised structures and vibrational frequencies of iron–sulfur clusters, Phys. Chem. Chem. Phys., 2020, 22(29), 16655–16664, 10.1039/D0CP01591A.
- J. Telser, H. Huang, H.-I. Lee, M. W. W. Adams and B. M. Hoffman, Site valencies and spin coupling in the 3Fe and 4Fe (S = 1/2) clusters of pyrococcus furiosus ferredoxin by 57Fe ENDOR, J. Am. Chem. Soc., 1998, 120(5), 861–870, DOI:10.1021/ja971546x.
- W. R. Hagen, EPR spectroscopy of iron—sulfur proteins, Adv. Inorg. Chem., 1992, 38, 165–222, DOI:10.1016/S0898-8838(08)60064-1.
- M.-E. Pandelia, N. D. Lanz, S. J. Booker and C. Krebs, Mössbauer spectroscopy of Fe/S proteins, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853(6), 1395–1405, DOI:10.1016/j.bbamcr.2014.12.005.
- A. C. Brown, N. B. Thompson and D. L. M. Suess, Evidence for low-valent electronic configurations in iron–sulfur clusters, J. Am. Chem. Soc., 2022, 144(20), 9066–9073, DOI:10.1021/jacs.2c01872.
- A. H. Priem, A. A. K. Klaassen, E. J. Reijerse, T. E. Meyer, C. Luchinat, F. Capozzi, W. R. Dunham and W. R. Hagen, EPR analysis of multiple forms of [4Fe–4S]3+ clusters in HiPIPs, J. Biol. Inorg. Chem., 2005, 10(4), 417–424, DOI:10.1007/s00775-005-0656-2.
- A. S. Byer, H. Yang, E. C. McDaniel, V. Kathiresan, S. Impano, A. Pagnier, H. Watts, C. Denler, A. L. Vagstad, J. Piel, K. S. Duschene, E. M. Shepard, T. P. Shields, L. G. Scott, E. A. Lilla, K. Yokoyama, W. E. Broderick, B. M. Hoffman and J. B. Broderick, Paradigm shift for radical S-Adenosyl-L-methionine reactions: the organometallic intermediate Ω is central to catalysis, J. Am. Chem. Soc., 2018, 140(28), 8634–8638, DOI:10.1021/jacs.8b04061.
- M. Ye, A. C. Brown and D. L. M. Suess, Reversible alkyl-group migration between iron and sulfur in [Fe4S4] clusters, J. Am. Chem. Soc., 2022, 144(29), 13184–13195, DOI:10.1021/jacs.2c03195.
- A. C. Manesis, M. J. O'Connor, C. R. Schneider and H. S. Shafaat, Multielectron chemistry within a model nickel metalloprotein: mechanistic implications for acetyl-CoA synthase, J. Am. Chem. Soc., 2017, 139(30), 10328–10338, DOI:10.1021/jacs.7b03892.
- J. D. Froehlich and C. P. Kubiak, The homogeneous reduction of CO2 by [Ni(Cyclam)]+: increased catalytic rates with the addition of a CO scavenger, J. Am. Chem. Soc., 2015, 137(10), 3565–3573, DOI:10.1021/ja512575v.
- M. Ito, T. Matsumoto and K. Tatsumi, Synthesis and reactions of mono- and dinuclear Ni(I) thiolate complexes, Inorg. Chem., 2009, 48(5), 2215–2223, DOI:10.1021/ic802276w.
- W.-F. Liaw, C.-H. Chen, C.-M. Lee, G.-H. Lee and S.-M. Peng, Distorted square planar nickel(II) carbonyl complexes containing terminal thiolate/selenolate ligands, J. Chem. Soc. Dalton Trans., 2001, 2, 138–143, 10.1039/b008902h.
- D. Sahoo, C. Yoo and Y. Lee, Direct CO2 addition to a Ni(0)–CO species allows the selective generation of a nickel(II) carboxylate with expulsion of CO, J. Am. Chem. Soc., 2018, 140(6), 2179–2185, DOI:10.1021/jacs.7b11074.
- C. Yoo, S. Oh, J. Kim and Y. Lee, Transmethylation of a four-coordinate nickel(I) monocarbonyl species with methyl iodide, Chem. Sci., 2014, 5(10), 3853–3858, 10.1039/C4SC01089B.
- T. G. Spiro, A. Terzis and K. N. Raymond, Structure of Ni(CN)53-. raman, infrared, and x-ray crystallographic evidence, Inorg. Chem., 1970, 9(11), 2415–2420, DOI:10.1021/ic50093a006.
- P. A. Lindahl, S. W. Ragsdale and E. Münck, Mössbauer study of CO dehydrogenase from clostridium thermoaceticum, J. Biol. Chem., 1990, 265(7), 3880–3888, DOI:10.1016/S0021-9258(19)39676-0.
- V. J. DeRose, J. Telser, M. E. Anderson, P. A. Lindahl and B. M. Hoffman, A multinuclear ENDOR study of the C-cluster in CO dehydrogenase from Clostridium thermoaceticum: evidence for HxO and histidine coordination to the [Fe4S4] center, J. Am. Chem. Soc., 1998, 120(34), 8767–8776, DOI:10.1021/ja9731480.
- M. E. Anderson, V. J. DeRose, B. M. Hoffman and P. A. Lindahl, Identification of a cyanide binding site in CO dehydrogenase from clostridium thermoaceticum using EPR and ENDOR spectroscopies, J. Am. Chem. Soc., 1993, 115(25), 12204–12205, DOI:10.1021/ja00078a085.
- A. Ciaccafava, D. Tombolelli, L. Domnik, J. Fesseler, J.-H. Jeoung, H. Dobbek, M. A. Mroginski, I. Zebger and P. Hildebrandt, When the inhibitor tells more than the substrate: the cyanide-bound state of a carbon monoxide dehydrogenase, Chem. Sci., 2016, 7(5), 3162–3171, 10.1039/C5SC04554A.
- H. Lee, J.-H. Choi and M. Cho, Vibrational solvatochromism and electrochromism of cyanide, thiocyanate, and azide anions in water, Phys. Chem. Chem. Phys., 2010, 12(39), 12658–12669, 10.1039/C0CP00214C.
- Y. Kung, T. I. Doukov, J. Seravalli, S. W. Ragsdale and C. L. Drennan, Crystallographic snapshots of cyanide- and water-bound C-clusters from bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase, Biochemistry, 2009, 48(31), 7432–7440, DOI:10.1021/bi900574h.
- P. T. Wolczanski, Flipping the oxidation state formalism: charge distribution in organometallic complexes as reported by carbon monoxide, Organometallics, 2017, 36(3), 622–631, DOI:10.1021/acs.organomet.6b00820.
- T. M. Gilbert, π Acceptor abilities of anionic ligands: comparisons involving anionic ligands incorporated into linear D10 [(NH3)Pd(A)]−, square planar D8 [(NN2)Ru(A)]−, and octahedral D6 [(AsN4)Tc(A)]− complexes, Inorg. Chem., 2023, 62(21), 8069–8079, DOI:10.1021/acs.inorgchem.2c03778.
- Y. Basak, J.-H. Jeoung, L. Domnik and H. Dobbek, Stepwise O2-induced rearrangement and disassembly of the [NiFe4(OH)(M3-S)4] active site cluster of CO dehydrogenase, Angew. Chem., Int. Ed., 2023, 62(32), e202305341, DOI:10.1002/anie.202305341.
- A. J. Redman, G. Moise, S. Richert, E. J. Peterson, W. K. Myers, M. J. Therien and C. R. Timmel, EPR of photoexcited triplet-state acceptor porphyrins, J. Phys. Chem. C, 2021, 125(21), 11782–11790, DOI:10.1021/acs.jpcc.1c03278.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental EPR, resonance Raman, and optical spectra; EXAFS fits and alternative fit parameters; description of selection of best EXAFS model; geometry optimized structures and geometric parameters; sample DFT input file. See DOI: https://doi.org/10.1039/d4sc00023d |
| This journal is © The Royal Society of Chemistry 2024 |