
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA marine bacteria-inspired electrochemical regulation for continuous uranium extraction from seawater and salt lake brine†
Linsen
Yang
a,
Yongchao
Qian
a,
Zhehua
Zhang
ab,
Tingyang
Li
ab,
Xiangbin
Lin
ab,
Lin
Fu
a,
Shengyang
Zhou
a,
Xiang-Yu
Kong
 ab,
Lei
Jiang
ab and
Liping
Wen
*ab
ab,
Lei
Jiang
ab and
Liping
Wen
*ab
aCAS Key Laboratory of Bio-inspired Materials and Interfacial Science, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: wen@mail.ipc.ac.cn
bSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 23rd February 2024
Abstract
Oceans and salt lakes contain vast amounts of uranium. Uranium recovery from natural water not only copes with radioactive pollution in water but also can sustain the fuel supply for nuclear power. The adsorption-assisted electrochemical processes offer a promising route for efficient uranium extraction. However, competitive hydrogen evolution greatly reduces the extraction capacity and the stability of electrode materials with electrocatalytic activity. In this study, we got inspiration from the biomineralisation of marine bacteria under high salinity and biomimetically regulated the electrochemical process to avoid the undesired deposition of metal hydroxides. The uranium uptake capacity can be increased by more than 20% without extra energy input. In natural seawater, the designed membrane electrode exhibits an impressive extraction capacity of 48.04 mg-U per g-COF within 21 days (2.29 mg-U per g-COF per day). Furthermore, in salt lake brine with much higher salinity, the membrane can extract as much uranium as 75.72 mg-U per g-COF after 32 days (2.37 mg-U per g-COF per day). This study provides a general basis for the performance optimisation of uranium capture electrodes, which is beneficial for sustainable access to nuclear energy sources from natural water systems.
Introduction
The development of nuclear power is indispensable for global clean energy transitions to cope with current carbon emissions and future fossil-fuel exhaustion.1–3 Annual nuclear capacity additions to 2050 are expected to grow four times in the Net Zero Emissions by 2050 Scenario (NZE) announced by the International Energy Agency (IEA).4 Uranium is a critical element in the nuclear industry. Owing to the limited reserves and uneven distribution of uranium resources on land, it is important to look for alternative uranium sources to ensure a sufficient supply.5 Oceans are estimated to contain more than 4.5 billion tons of uranium, which can ensure continued nuclear energy delivery.6 However, extracting uranium at a concentration of 3.3 μg L−1 from the hypersaline and complex marine environment is an enormous challenge.7–11From a cost perspective, chemical adsorption has long been recognised as the preferred method for uranium extraction from seawater (UES).12–14 This method typically usually relies on the number of accessible binding sites and their affinity for uranium, which is limited by adsorption thermodynamics.15–17 However, in environments with higher salinity, such as seawater desalination rejects or salt lake brine, adsorbents perform poorly because of increased competition from other ions, even if these brine samples have significantly higher uranyl concentrations.18–22 Electrochemical methods are a substitute for traditional chemical adsorption for UES.23–26 Highly efficient electrodes for electrocatalytic uranium deposition, which directly determines external energy consumption, are critical in this process. Hydrogen evolution under high salinity is a major obstacle to continuous uranium extraction. A half-wave pulse method has been reported to prevent water splitting at the electrode.23 However, when using electrocatalytic materials as the cathode to further improve the efficiency of uranium extraction, the possible enhancement of hydrogen evolution would cause stronger competition between uranium reduction and water splitting, especially when the metal concentration is extremely limited. Accordingly, the precipitation of hydroxides resulting from hydrogen evolution will seriously reduce the uranium uptake capacity and durability of the electrode.27
Marine bacteria are single-celled organisms that live in the ocean and account for more than twenty percent of ocean biomass.28 Some of these bacteria could reduce heavy metals to lower valence states, being not affected by the high salinity of marine environments.29–31 Drawing inspiration from this special biological phenomenon, exquisitely regulating the process of electrochemical uranium capture is expected to avoid hydroxide deposition under a high salinity background when using electrocatalytic materials, further improving the extraction efficiency and operation life of electrode materials (Fig. 1).
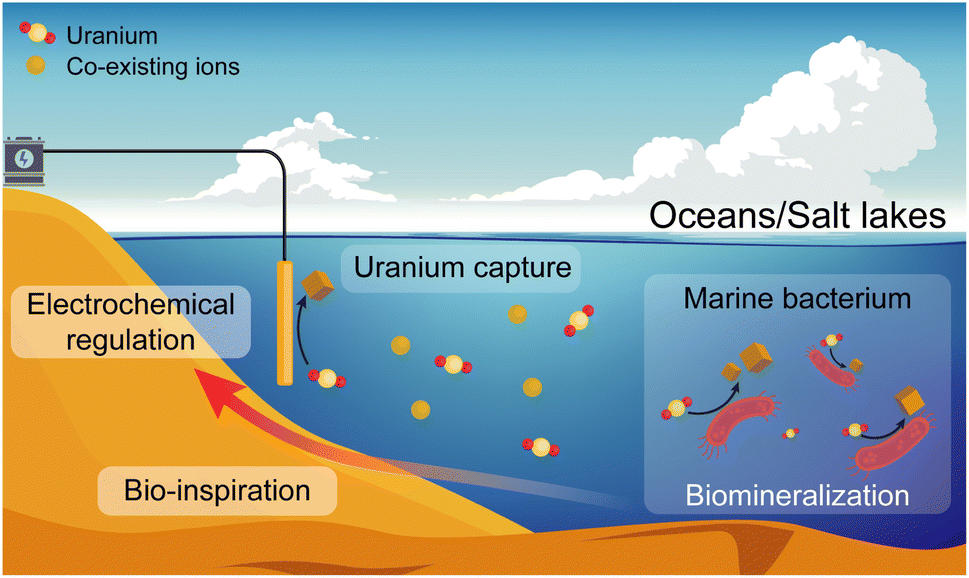 | ||
| Fig. 1 Schematic of the biological reduction/mineralization of marine bacteria and the bio-inspired electrochemical regulation for continuous uranium capture from oceans and salt lakes. | ||
Here, we report a membrane electrode (denoted as S-COF membrane) composed of single-walled carbon nanotubes (SWCNTs) and sp2 carbon-conjugated covalent organic framework fibers (AO-g-C34N6-COF) for efficient uranium extraction from seawater and salt lake brine. The superior chemical stability of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds enables amidoxime functionalisation in alkaline solutions for introducing specific uranium-binding sites. The high specific surface area, abundant selective uranium-binding sites, and spontaneous electron transfer from SWCNTs to the fully π-conjugated frameworks synergistically endow the membrane with an efficient electrocatalytic reduction of uranium. In addition, for the serious hydroxide deposition caused by the competitive hydrogen evolution, we mimic the biological reduction/mineralization of marine bacteria under high salinity and assess the effect of different electrical signal parameters to realize biomimetic mineralization. By avoiding the deposition of metal hydroxides, we successfully increase the uranium uptake capacity and durability of the electrodes. The membrane electrode exhibited a high capacity of 48.04 mg-U per g-COF (2.29 mg-U per g-COF per day) during a long-term test for up to 21 days in natural seawater. Furthermore, it demonstrated outstanding performance in salt lake brine and exhibited a capacity of 75.72 mg-U per g-COF within 32 days (2.37 mg-U per g-COF per day), and the uranium capture performance remains stable.
C bonds enables amidoxime functionalisation in alkaline solutions for introducing specific uranium-binding sites. The high specific surface area, abundant selective uranium-binding sites, and spontaneous electron transfer from SWCNTs to the fully π-conjugated frameworks synergistically endow the membrane with an efficient electrocatalytic reduction of uranium. In addition, for the serious hydroxide deposition caused by the competitive hydrogen evolution, we mimic the biological reduction/mineralization of marine bacteria under high salinity and assess the effect of different electrical signal parameters to realize biomimetic mineralization. By avoiding the deposition of metal hydroxides, we successfully increase the uranium uptake capacity and durability of the electrodes. The membrane electrode exhibited a high capacity of 48.04 mg-U per g-COF (2.29 mg-U per g-COF per day) during a long-term test for up to 21 days in natural seawater. Furthermore, it demonstrated outstanding performance in salt lake brine and exhibited a capacity of 75.72 mg-U per g-COF within 32 days (2.37 mg-U per g-COF per day), and the uranium capture performance remains stable.
Results and discussion
Synthesis and characterization
The synthesis of g-C34N6-COF and its amidoxime functionalization are described in the Methods section. The Fourier transform infrared spectra of the monomers and g-C34N6-COF in Fig. S1 (ESI)† confirm the generation of the carbon–carbon double bond, whose characteristic peak is at 1627 cm−1. The stretching vibration of trans –HCCH– in the fingerprint region at 975 cm−1 demonstrates the trans-configurations of olefin linkages. The COF demonstrates a feature of full π-conjugation along with CC bond connection, leading to improved electrochemical activity. Compared to common imine-linked COFs, g-C34N6-COF linked by olefinic bonds has better chemical stability, making it possible to convert cyano groups into amidoxime groups in alkaline solutions (Fig. 2a and S2, ESI†).32–34 As shown in Fig. 2b, the disappearance of the nitrile peak at 2221 cm−1 and the emergence of both the CN peak at 1643 cm−1 and the N–O peak at 871 cm−1 illustrate the successful functionalization of g-C34N6-COF. We characterised this process using energy-dispersive spectroscopy (EDS) elemental line scans. There was an apparent oxygen signal on the AO-g-C34N6-COF fibres, which was related to the introduction of amidoxime groups (Fig. 2c).
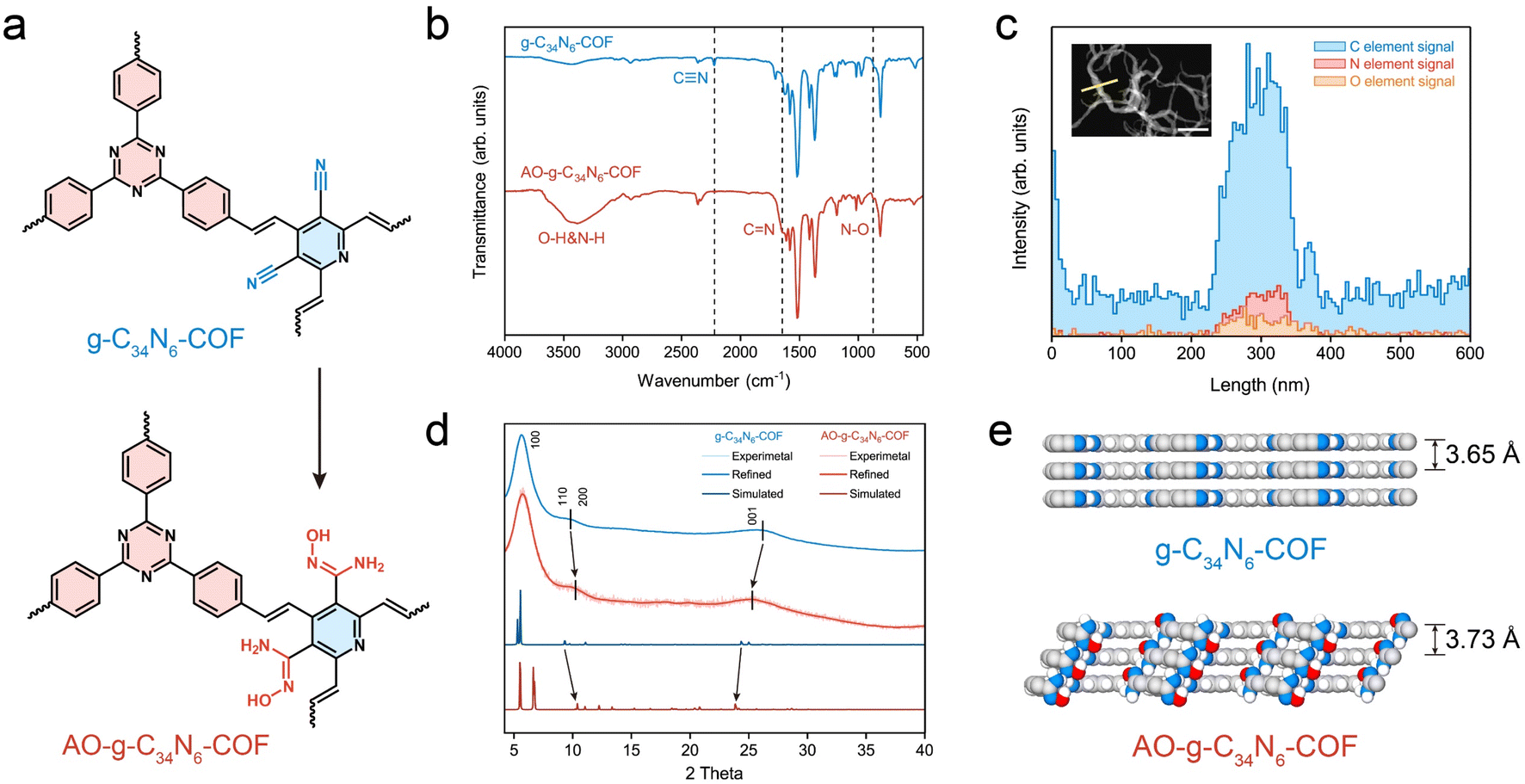 | ||
| Fig. 2 Amidoxime functionalization of g-C34N6-COF. (a) Structural unit of g-C34N6-COF and AO-g-C34N6-COF. (b) Fourier transform infrared spectra of g-C34N6-COF and AO-g-C34N6-COF. The disappearance of the nitrile peak and the appearance of new characteristic peaks related to amidoxime confirm the successful functionalization of g-C34N6-COF. (c) Energy dispersive spectroscopy elemental line scan of AO-g-C34N6-COF. The signal of oxygen proved the introduction of amidoxime groups. The inset shows the transmission electron microscopy of AO-g-C34N6-COF and the length of elemental analysis. The scale bar is 500 nm. (d) PXRD patterns for g-C34N6-COF and AO-g-C34N6-COF. The peak shifts demonstrate that amidoxime functionalization leads to lattice changes. (e) Side views of final optimized models of g-C34N6-COF and AO-g-C34N6-COF. The interlayer shifting and interlayer spacing variation could be observed. | ||
Scanning electron microscopy was used to investigate the topography of the synthesised COFs. The fibrous g-C34N6-COF showed a uniform diameter of ∼100 nm and a length of several microns, which is consistent with previous literature (Fig. S3, ESI†). After amidoxime functionalization, the fibrous morphology was preserved (Fig. S4, ESI†), which makes it easy to fabricate membranes via vacuum filtration. Gas adsorption measurements were performed to investigate the porosities of the powdered g-C34N6-COF and AO-g-C34N6-COF samples. N2 adsorption measurements (at 77 K) revealed permanent porosities. The Brunauer–Emmett–Teller surface areas of g-C34N6-COF and AO-g-C34N6-COF were 719 and 584 m2 g−1, respectively (Fig. S5, ESI†). The pore size distribution curves were obtained from the adsorption branches using the QSDFT method (Fig. S6, ESI†). Gas adsorption measurements demonstrated that after functionalization, the specific surface area remained high, and the pore structure was preserved. Given the extremely low concentration of uranium in oceans, a high surface area is necessary to provide abundant adsorption sites during the early stages of electrodeposition, which is an advantage of COFs. Thermogravimetric analysis was performed to study the thermal stabilities of the two COFs (Fig. S7, ESI†). Compared with g-C34N6-COF, AO-g-C34N6-COF had an additional step starting at approximately 200 °C before the degeneration of the backbone, corresponding to the degradation of the amidoxime group. Overall, thermal stabilisation was preserved, enabling it to cope with temperature fluctuations during uranium recovery.
Powder X-ray diffraction (PXRD) measurements were used to evaluate the crystallinity of the two COFs and the possible influence of amidoxime functionalization on the crystal structure (Fig. 2d). In the experimental PXRD profile of g-C34N6-COF, a strong peak at 5.69° along with relatively weaker peaks at 9.05°, 10.83° and 26.10° were assigned to (100), (110), (200), and (001) planes, respectively. The experimental PXRD pattern is in good agreement with the Pawley refinement of the AA-eclipsed layer stacking model, with reliability parameters Rwp = 0.99% and Rp = 0.78%. After converting the cyano groups into amidoxime, the PXRD pattern of AO-g-C34N6-COF was comparable to that of g-C34N6-COF. The (110) and (200) diffraction peaks shifted slightly to higher angles, whereas the (001) diffraction peak shifted to a lower angle. This indicates that the steric hindrance arising from the introduction of the functional group resulted in slight interlayer shifting and interlayer spacing variation. Geometric optimisation of the model of AO-g-C34N6-COF also displayed a degree of lattice change (Fig. S8, S9, Tables S1 and S2, ESI†). The calculated layer spacing increased from 3.65 Å to 3.73 Å (Fig. 2e). Interlayer shifting correlated with the introduction of groups has recently been reported. This lattice deformation can lead to a quasi-AA stacking structure that is similar, but not identical, to AA stacking.35
Electronic properties and membrane fabrication
The electronic properties of the two COFs were investigated using photophysical and electrochemical methods. Their ultraviolet-visible diffuse reflectance spectra displayed broad absorption bands in the UV and visible regions. The spectrum of AO-g-C34N6-COF showed a slight blueshift of the adsorption edge compared with that of g-C34N6-COF, indicating that amidoxime functionalization affected the π-conjugated structure to some extent. However, overall, the extended π-conjugated skeleton was well preserved (Fig. S10†). Accordingly, the optical band gaps were calculated using the Kubelka–Munk function, and g-C34N6-COF demonstrated an optical band gap of 1.38 eV, while AO-g-C34N6-COF had an optical band gap of 1.64 eV (Fig. 3a). Mott–Schottky measurements were conducted to further estimate the semiconductor behaviour of AO-g-C34N6-COF.36 The positive slopes indicated typical n-type semiconductor characteristics, suggesting that the majority carriers are electrons (Fig. 3b). Density functional theory (DFT) calculations using the Perdew–Burke–Ernzerhof functional predicted that AO-g-C34N6-COF had an indirect bandgap of 1.58 eV, which was close to the measured optical band gap (Fig. 3c). The DFT-calculated band structures had large band dispersions along the out-of-plane direction (Γ–X, Γ–L, Γ–N, and Γ–R) in both the valence and conduction bands.37,38 In contrast, the in-plane direction (Γ–Y, Γ–Z, and Γ–M) exhibited different band dispersions. These results suggest that the charge transport in AO-g-C34N6-COF might be anisotropic. The corresponding partial density-of-states curves illustrate that the bands mainly originate from the 2p-orbitals. Collectively, these results indicate that AO-g-C34N6-COF is a narrow band gap organic semiconductor. According to a recent study, n-type semiconductors have a universal self-gating characterization that favours cathodic reactions,39,40 implying that AO-g-C34N6-COF has the potential for electrocatalytic uranium extraction from seawater or salt-lake brine.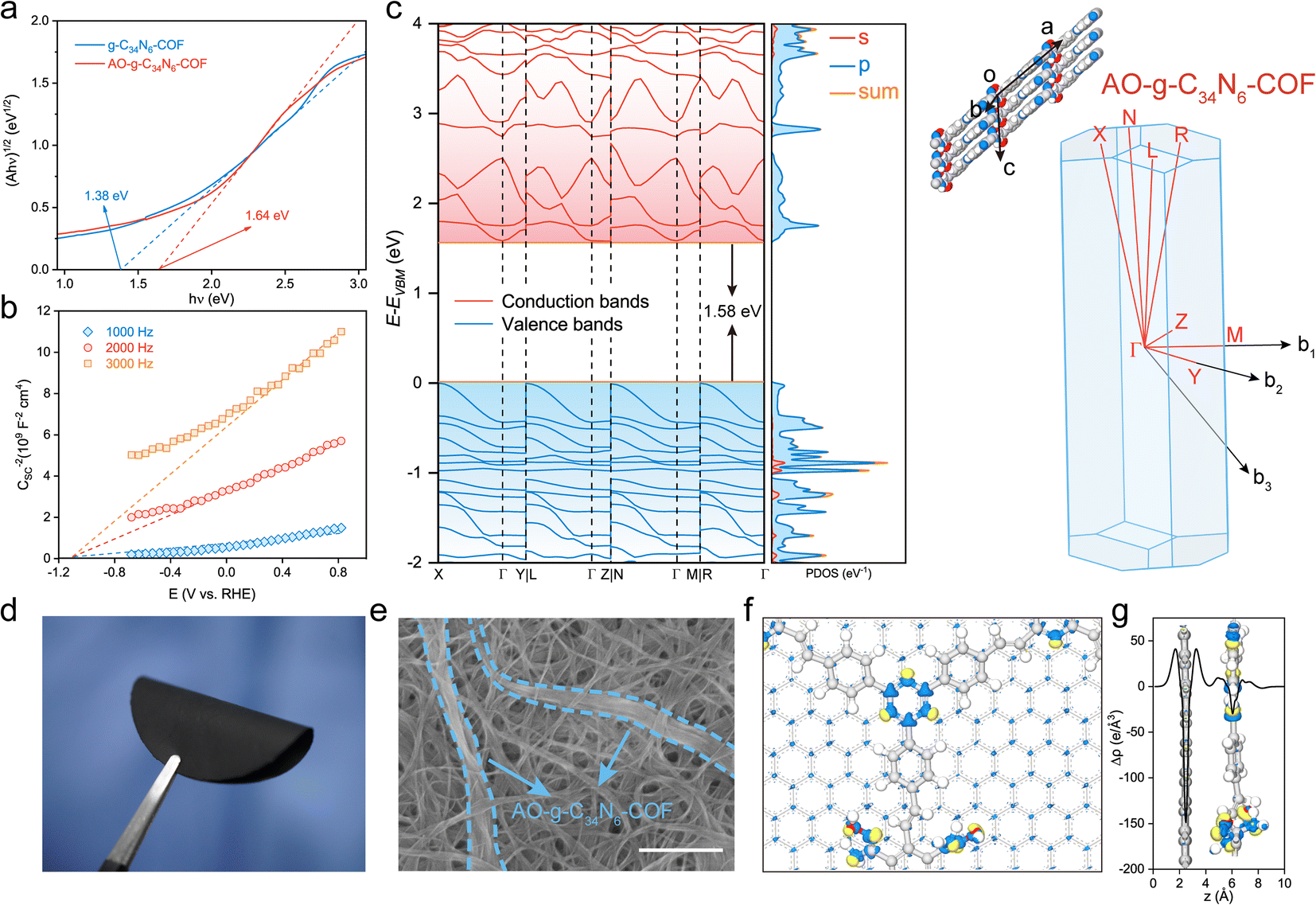 | ||
| Fig. 3 Electronic properties and characterization. (a) Band gaps of g-C34N6-COF and AO-g-C34N6-COF determined from the Kubelka–Munk-transformed reflectance spectra. The amidoxime functionalization process led to the widening of the band gap. (b) Mott–Schottky plots for g-C34N6-COF and AO-g-C34N6-COF measured in 0.2 M Na2SO4 (pH 6.8) with an Ag/AgCl reference electrode. The positive slopes suggested that the COF is a typical n-type semiconductor. (c) Electronic band structures and corresponding partial density-of-states of AO-g-C34N6-COF determined using DFT calculations. The Brillouin zone is shown on the right. (d) Digital photograph of a free-standing S-COF membrane. The membrane presented impressive flexibility and can be highly curved. (e) Scanning electron microscopy image showing that the AO-g-C34N6-COF fibres are tightly tangled in the nanotubes. The scale bar is 500 nm. (f) Differential charge density distribution of the AO-g-C34N6-COF/graphene heterointerface calculated using DFTB+. Yellow and blue areas represent charge density increase and decrease, respectively. (g) Plane-average electron difference perpendicular to the AO-g-C34N6-COF/graphene heterointerface. | ||
Given its fibrous morphology, AO-g-C34N6-COF can be easily mixed with commercially available carboxylated single-walled carbon nanotubes (SWCNTs) and dispersed in organic solvents. A self-supporting S-COF membrane was prepared on a nylon filter using vacuum-assisted filtration. The content (20%) of COF is a balance between its uranium extraction capacity and membrane mechanical properties. More COF would result in the membrane becoming brittle (Fig. S11, ESI†). As shown in Fig. 3d, the S-COF membrane exhibited excellent flexibility. Hydrogen bonds between the carboxyl and amidoxime groups can improve the mechanical strength of the membrane. The top-view SEM image shows that AO-g-C34N6-COF was tightly tangled in the carbon nanotubes (Fig. 3e). To estimate the electron transport performance of the composite membrane, we used the density-functional tight-binding (DFTB+) method, which is an approximate density functional theory method based on the tight-binding approach, to analyse the electronic interactions. For computational expediency, the SWCNTs were simplified to a periodic graphene substrate, on which the geometry-optimised AO-g-C34N6-COF was modeled.41,42 As shown by the differential charge density distribution in Fig. 3f and plane-average electron difference in Fig. 3g, the graphene substrate (electron-deficient, blue colour) injects delocalised electrons into AO-g-C34N6-COF (electron-rich, yellow colour), resulting in an n-doping effect on the COF. Delocalised electrons serving as excessive carriers can change the electrochemical activity of the COF.43 This process can be mainly attributed to the support effects in carbon-supported catalysts, which is the reason why we choose carbon nanotubes as the component.44
Extraction performance in uranium-spiked seawater
Cyclic voltammetry (CV) was performed to measure the uranium reduction activities. The CV scan curves at a scan rate of 10 mV s−1 of natural seawater spiked with 50 ppm and 100 ppm uranyl nitrate at pH 8.2 are shown in Fig. 4a. In comparison with seawater without extra uranium(VI), additional reduction peaks at −0.65 VSCE and anodic peaks at −0.46 VSCE were observed, corresponding to uranium reduction/oxidation reactions. It is worth mentioning that this membrane electrode based on AO-g-C34N6-COF has a more positive reduction potential than those previously reported for semiconductor/carbon-based electrodes for uranium extraction from seawater measured under similar experimental conditions.23–25,45 This means that energy consumption can be further reduced when an electric field is applied.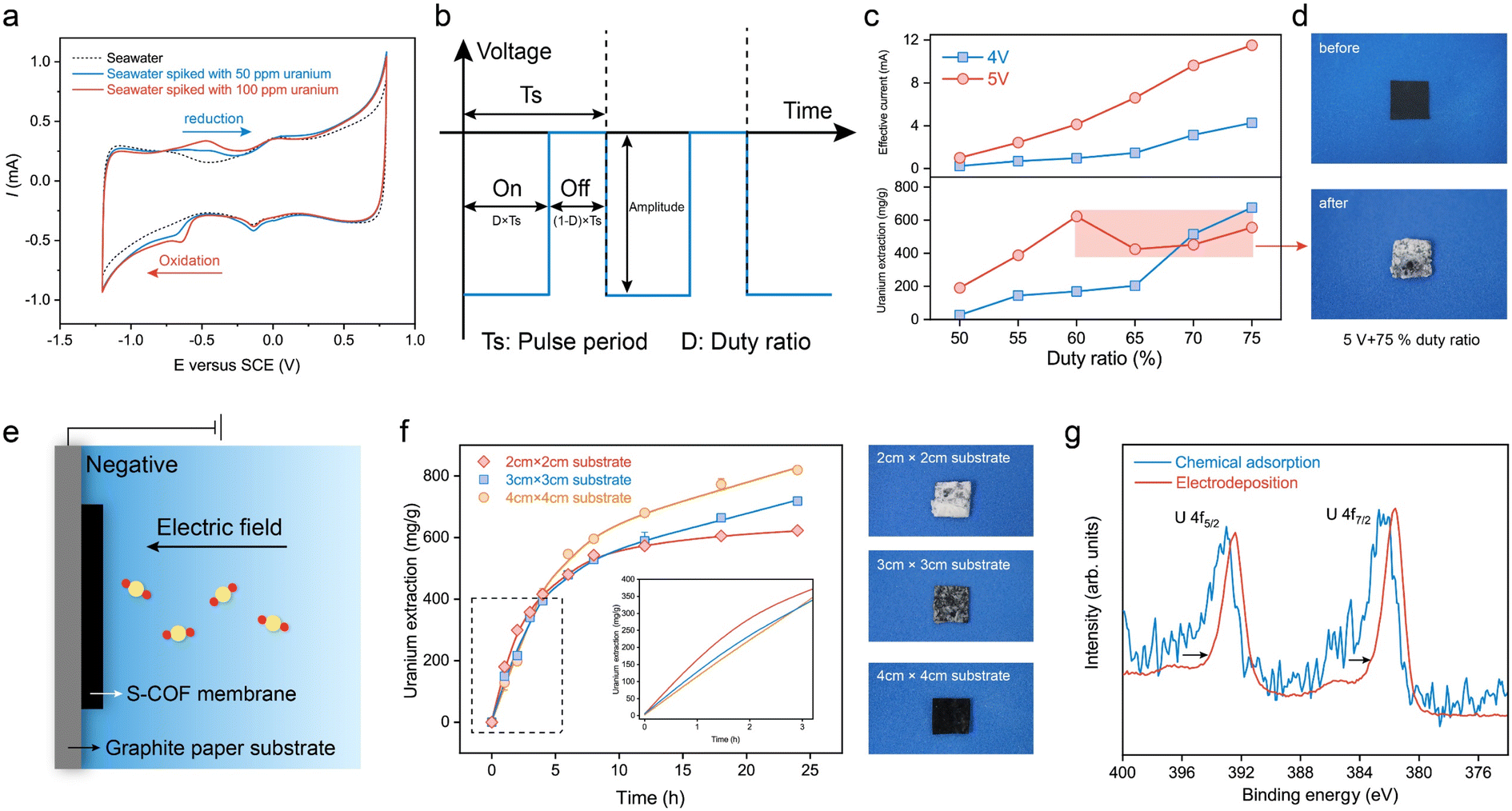 | ||
| Fig. 4 Uranium extraction performance of the S-COF membrane in spiked seawater. (a) Cyclic voltammograms of uranium-spiked natural seawater with concentrations of 50 ppm and 100 ppm compared to unspiked seawater. (b) Schematic of the pulse applied to the working electrode. The amplitude and the duty ratio were adjustable parameters. (c) Effective values of current and the corresponding uranium extraction amount of the electrode when pulses were applied with different amplitudes and duty ratios. (d) Digital photographs of the S-COF membrane before (top) and after (bottom) extracting uranium at 5 V voltage with a 75% duty ratio. The membrane was wrapped tightly by white deposits. (e) Schematic of the current density regulation strategy. The S-COF membrane was bonded to graphite papers of different sizes to control the current density. (f) Uranium extraction from spiked seawater using the S-COF membrane with varying substrate sizes. The error bars represent the standard deviation (n = 3). The digital photographs correspond to the S-COF membrane with substrate sizes of 2 cm × 2 cm (top panel), 3 cm × 3 cm (middle panel), and 4 cm × 4 cm (bottom panel) after extracting uranium for 24 h. (g) High-resolution X-ray photoelectron spectroscopy profile of the S-COF membrane used for uranium uptake by chemical adsorption and electrodeposition. The 4f5/2 and U 4f7/2 peaks shifted to lower binding energy, indicating the reduction reaction of U(VI). | ||
In order to investigate the role of selective adsorption and spontaneous electron transfer in adsorption-assisted electrochemical enrichment, a series of control experiments were carried out. Two pieces of control membranes with g-C34N6-COF (SWNT-COOH membrane with g-C34N6-COF) and without COF (SWNT-COOH membrane) were prepared. Three membranes were tested in seawater with various uranium concentrations by applying a square-wave pulse with a frequency of 400 Hz and a duty ratio of 75% for 24 h (Fig. S12, ESI†). When the uranium concentration is high, three membranes all showed the ability to extract uranium and the membrane having AO-g-C34N6-COF had the highest capacity. The facilitation effect of COFs gradually increased with the uranium concentration decreased. When the uranium concentration was low at 1 ppm, the SWNT-COOH membrane could hardly extract uranium from seawater. The SWNT-COOH membrane with g-C34N6-COF retained some extraction capacity and the S-COF membrane still performed robustly. The results indicate that spontaneous electron transfer cooperates with selective adsorption in complex environments to promote effective uranium extraction from seawater through electrochemical methods.
To further evaluate the uranium uptake performance, the S-COF membrane was cropped into 1 cm × 1 cm pieces and clamped using a glassy carbon clip as the working electrode. A graphite rod was used as the counter electrode. A series of square-wave pulses at a frequency of 400 Hz were applied to the membrane electrode. In these experiments, the voltage amplitude and duty ratio were set as adjustable parameters (Fig. 4b). We recorded the instantaneous current using a multimeter when the electrode worked for 5 min to evaluate the variations in current density caused by parameter adjustment. The uranium extraction system was operated continuously in uranium-spiked natural seawater (∼8 ppm) for 24 h, and the extraction amount was determined based on changes in the concentration of uranium in the solution. The uranium concentrations were measured from ultraviolet-visible absorption spectra (Fig. S13, ESI†). As shown in Fig. 4c, the measured current was approximately proportional to the duty ratio. When the voltage amplitude was 4 V, the uranium extraction amount increased with increasing duty ratio and reached a maximum value of 675.9 mg-U per g-membrane at a 75% duty cycle. This phenomenon is reasonable because higher duty cycles imply higher input energy. When the voltage amplitude was 5 V, the extracted uranium continued to increase as the duty ratio increased from 50% to 60%. However, the extraction amount decreased anomalously at higher duty ratios. To determine the reason for this decline in electrode performance, we recorded the macroscopic changes at the electrode surface over time. When a voltage of 5 V (75% duty ratio) was applied, the electrode was quickly wrapped by large amounts of white deposits in the first hour (Fig. 4d). In contrast, deposition on the electrode at 4 V (75% duty ratio) was significantly slower. Its wrapping state was not as compact as that at 5 V until electrolytic deposition continued for 24 h (Fig. S14, ESI†). This suggests that although high-frequency pulses could reduce water splitting to some extent and there were no visible bubbles on the electrode, hydrogen evolution was still not completely avoided. Fast electron transfer depletes the uranyl cations near the electrode and causes concentration polarisation. Further polarisation of the electrode promotes a competitive hydrogen evolution reaction and changes the pH near the electrode, leading to the deposition of alkaline earth metal hydroxides. These compact deposits prevent uranyl cations from migrating to the electrode, thereby reducing uranium extraction.
To address the negative effects of undesired deposition on the electrochemical process, we tried to further decrease the operating voltage. However, we found it difficult to achieve a balance between keeping high uptake capacity and avoiding hydroxide deposition. Although the surface deposition gradually slowed down, the uranium extraction capacity of the electrode also decreased substantially, diminishing its practicality. Hence, we attempted to regulate the current density of the membrane without changing the working voltage. We used graphite paper as the substrate to support the S-COF membrane (Fig. 4e and S15, ESI†). The introduction of the inert area would lower the local current density, decreasing the reaction rate of the electrode. In this way, the concentration polarization of uranium could be decreased, reducing the competition between uranium reduction and hydrogen evolution. The cumulation and expulsion of hydroxide ions could gradually reach an equilibrium, averting the generation of deposition. The current density was controlled by varying the substrate size to 2 cm × 2 cm, 3 cm × 3 cm, and 4 cm × 4 cm, and the size of the S-COF membrane was fixed at 1 cm × 1 cm. The test device is shown in Fig. S16 (ESI).† The possible influence of the substrate on the uranium extraction amount was excluded using graphite paper alone as the working electrode. Fig. S17 (ESI)† shows that the uranium concentration in the spiked seawater remained virtually unchanged under the same conditions. The current changes also demonstrated this phenomenon. The measured instantaneous current was only slightly enhanced with an increase in the substrate (Fig. S18, ESI†). These slight variations were mainly due to an increase in the non-faradaic current. We studied the dynamics of composite electrodes with different substrate sizes during uranium extraction (Fig. 4f). The inset shows the partially enlarged kinetic curves. It can be seen that the kinetics of uranium extraction on the electrode with a size of 2 cm × 2 cm was the most rapid during the first 5 h because it had the maximum current density. However, the electrode was gradually wrapped by undesired deposits, and the amount of extracted uranium slowed. There was almost no increase in this amount for the remaining time. The digital photograph in Fig. 4f (top panel) shows the S-COF membrane to which dense deposits were attached after the electrode was operated for 24 h. When the substrate size was 3 cm × 3 cm (middle panel), the amount of deposition was less than that of the electrode with a 2 cm × 2 cm substrate, indicating that some space remained in the membrane for uranium transport and further extraction. Correspondingly, the electrode had a higher uranium extraction amount of 718.72 mg-U per g-membrane after running for 24 h. The kinetic curve also maintained a gradual increase, indicating that electrodeposition continued. When a 4 cm × 4 cm substrate was used, no deposits were observed on the surface of the membrane (bottom panel). Although this composite electrode had the slowest uranium extraction rate during the first 5 h, it exhibited a maximal extraction amount of 819.35 mg-U per g-membrane at 24 h, and the enrichment process did not stop. The excellent uranium uptake capacity was approximately 17 times that of chemical adsorption (Fig. S19, ESI†). The scanning electron microscope (SEM) images of the membrane surface reveal that, with regulated current density, the micro-voids on the surface are maintained, while without current density regulation, these voids become clogged with deposits (Fig. S20, ESI†).The above experiments suggest that an appropriate current density reduction for decreasing the electrode reaction rate could increase the uranium extraction amount by avoiding undesired deposits, which could also increase the life of the electrode.
High-resolution X-ray photoelectron spectroscopy (XPS) and X-ray diffraction were used to characterise the uranium species produced during the electrocatalytic process. The S-COF membrane was used to extract uranium from uranium aqueous solutions (50 ppm) via traditional chemical adsorption and electrodeposition for 12 h. The electrical uranium extraction was performed under a nitrogen atmosphere after deoxygenation of the solution to eliminate the impacts of oxygen reduction. As shown in Fig. 4g, the U 4f peaks of uranium on the electrocatalytic membrane were shifted more significantly to lower binding energies than those of the adsorption membrane, indicating an decrease in the valence state of U.46 Raman spectroscopy was also used to identify the uranium species (Fig. S21, ESI†). It can be seen that the sample extracting uranium under the N2 atmosphere showed characteristic peaks from UO2 at ∼230 cm−1 and 445 cm−1.47 The generation of U(IV) is generally considered to come from the disproportionation of unstable U(V) intermediates:48
| 2UVO2+ + 4H+ → U4+ + UVIO22+ + H2O |
To further investigate the uranium species generated in natural seawater, we performed the extraction process in real seawater spiked with 1000 ppm uranium. A large amount of yellow powder was produced, which was attached to the membrane (Fig. S22, ESI†). The collected powder was identified as a Na2O(UO3·H2O)x species according to the PXRD result (Fig. S23, ESI†). Similar results have been reported in related work.24,49
Uranium extraction performance in unspiked natural water
The uranium extraction performance of the S-COF membrane was investigated using natural seawater and salt lake brine (Fig. 5a). Given the extremely low concentration of uranium in seawater and the possible concentration fluctuation caused by the pre-filtration step, we monitored the concentration variation in seawater and salt lake water during each operational period. For experiments using seawater, 2 mg of the membrane was composited with a 5 × 5 cm substrate as the working electrode. Seawater (2 L) was treated with the electrode for 24 h during each period and then replaced with fresh seawater. This process lasted 21 days. As shown in Fig. 5b, the initial concentration of uranium fluctuated slightly near 3.3 ppb, and apparent decreases in concentration were observed during each extraction period. In the long term, the magnitude of the decline became progressively smaller, perhaps because the performance attenuation of the electrode was more evident in an environment containing a low concentration of uranium.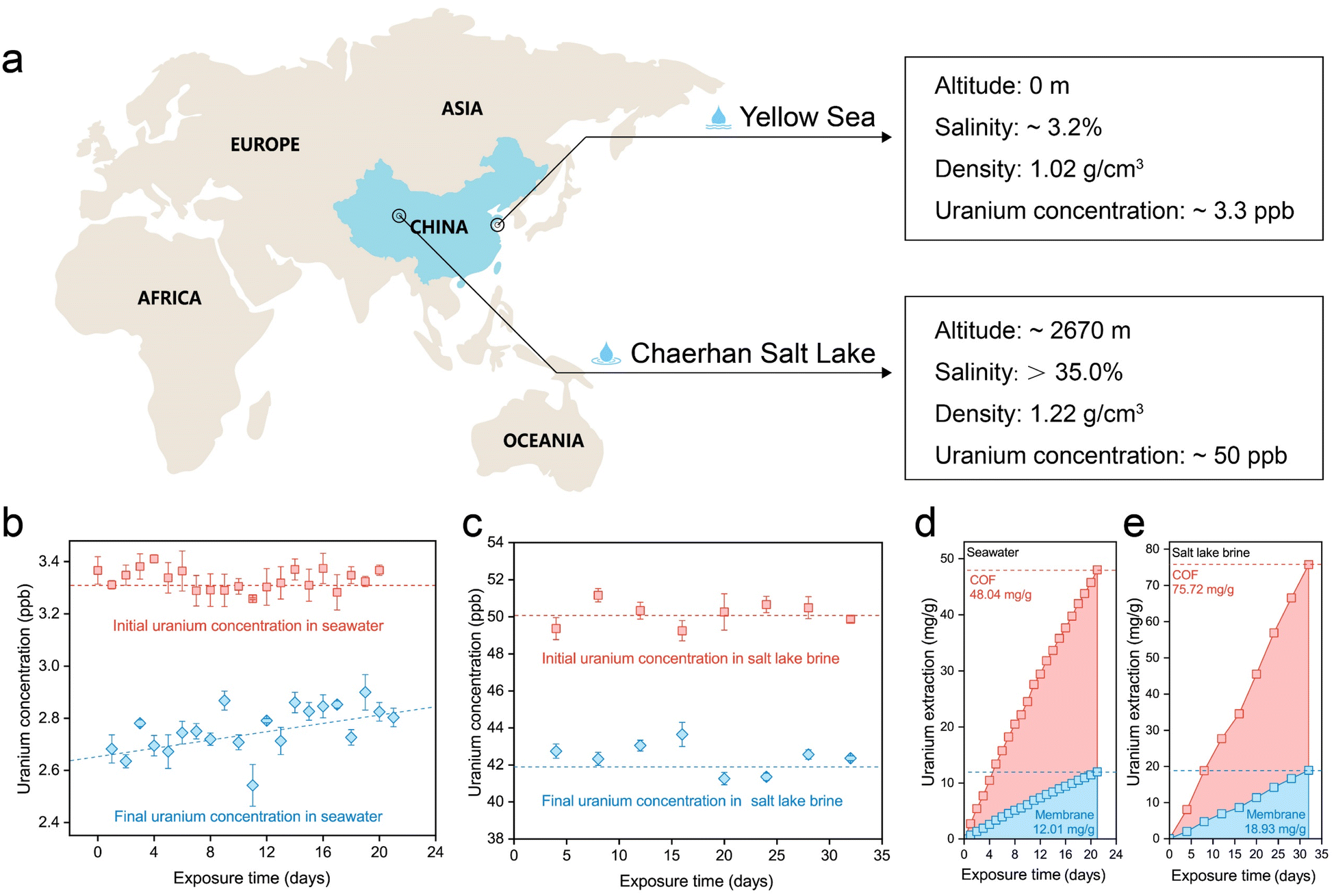 | ||
| Fig. 5 Uranium extraction performance of the S-COF membrane in natural seawater and salt lake brine. (a) Schematic illustration showing the sampling locations and physicochemical properties of natural seawater and salt lake brine. (b) Initial and final uranium concentrations in seawater during each extraction period. 2 L of seawater was treated by the electrode for 24 h per treatment. The whole process lasted 21 days. The error bars represent the standard deviation (n = 3). (c) Initial and final uranium concentrations in the salt lake brine during each extraction period. The electrode treated 500 mL of brine for 4 days per treatment. The error bars represent the standard deviation (n = 3). (d) Cumulative uranium extraction amount of the S-COF electrode from seawater. (e) Cumulative uranium extraction amount of the S-COF electrode from salt lake brine. | ||
Encouraged by the outstanding uranium-extraction properties of the S-COF membrane, we further tested its performance in real salt-lake brine from Chaerhan Salt Lake, which has a salinity much higher than seawater. The membrane weighed 2 mg, and the substrate was maintained at 5 × 5 cm. During each period, the electrode was treated with 500 mL salt lake brine for four days, and the entire process lasted 32 days. Although the uranium concentration was approximately 50.0 ppb, the extremely high alkali metal content in the brine occupied the adsorption sites, leading to unsatisfactory uranium extraction performance using the traditional chemical adsorption method (Fig. S24, ESI†). However, when the S-COF membrane was employed as the working electrode with voltage pulses, the uranium concentration in the brine showed a clear decreased during each extraction period (Fig. 5c). The cumulative extraction amount was calculated according to the uranium concentration variations (Fig. 5d and e), which was 12.01 mg-U per g-membrane (48.04 mg-U per g-COF, 2.29 mg-U per g-COF per day) for seawater and 18.93 mg-U per g-membrane (75.72 mg-U per g-COF, 2.37 mg-U per g-COF per day). The extraction capability of this S-COF membrane far exceeded that of previously reported chemical adsorbents or photocatalysts based on olefin-linked COFs. At the same time, this is currently the longest running time based on the electrochemical method for continuous uranium extraction from seawater compared to what has been reported so far (Table S3, ESI†). There were no deposits on the S-COF membrane surface after the uranium extraction process in natural water and salt lake brine (Fig. S25, ESI†), indicating long-term service performance. The continuous uranium capture performance and the long-term stability of the electrodes prove that this biomimetic electrochemical regulation is an effective strategy to promote the application potential of electroactive materials in harsh environments with high salinity. We also evaluate the economic viability of the electrode along with the electrical extraction process. The cost of preparing the S-COF membrane was estimated to be ≈52 USD per g. The electricity cost was calculated to be ≈3.40 × 10−3 USD per g-U using seawater and 2.15 × 10−3 USD per g-U using salt lake brine. Given the continuous electrical power consumption of the electrochemical method, future research should focus on the higher working efficiency of electrodes and cheaper electrical energy.
Conclusions
In summary, to improve the extraction capacity and the durability of uranium-capture electrodes with electro-catalytic activity, we got inspiration from the biomineralization of marine bacteria in high-salinity environments, and biomimetically regulated the electrochemical process. This regulation could reduce the competitive reaction between uranium reduction and hydrogen evolution, preventing the deposition of earth metal hydroxides. In this way, the uranium uptake capacity of the designed S-COF membrane could be increased by more than 20% without extra energy input. This process also endowed the membrane electrode with stability. In a long-term test, the S-COF membrane showed an excellent extraction capacity of 48.04 mg-U per g-COF in seawater within 21 days and 75.72 mg-U per g-COF in salt lake brine within 32 days, which is impossible to achieve using physicochemical adsorption. This way of optimization provides a new strategy for mining uranium from unconventional resources.Data availability
The data supporting the findings can be found in the article and ESI,† and are available from the authors upon reasonable request.Author contributions
Linsen Yang: conceptualization, methodology, writing – original draft, writing – review & editing, visualization, validation, formal analysis, investigation, resources, and data curation. Yongchao Qian: investigation and resources. Zhehua Zhang: investigation and resources. Tingyang Li: investigation and resources. Xiangbin Lin: investigation and resources. Lin Fu: investigation and resources. Shengyang Zhou: investigation and resources. Xiang-Yu Kong: investigation and resources. Lei Jiang: conceptualization and supervision. Liping Wen: conceptualization, supervision and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R & D Program of China (grant no. 2022YFB3805904 and 2022YFB3805900). We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant no. 22122207) and CAS Project for Young Scientists in Basic Research (grant no. YSBR-039). We gratefully acknowledge the financial support from the China Postdoctoral Science Foundation funded project (grant no. 2022TQ0345 and 2022M723229) and the National Natural Science Foundation of China (grant no. 22305256). The authors are grateful to Prof. Min Wang (Qinghai Institute of Salt Lakes, Chinese Academy of Sciences) for providing natural salt lake brine.References
- A. Goldthau and S. Tagliapietra, Nature, 2022, 612, 627–630 CrossRef CAS PubMed
.
- L. Duan, R. Petroski, L. Wood and K. Caldeira, Nat. Energy, 2022, 7, 260–269 CrossRef
- J. D. Jenkins, M. Luke and S. Thernstrom, Joule, 2018, 2, 2498–2510 CrossRef
-
IEA, World Energy Outlook 2022, OECD, 2022 Search PubMed
- NEA/IAEA, Uranium 2020: Resources, Production and Demand, OECD Publishing, Paris, 2021, DOI:10.1787/d82388ab-en.
- T.-L. Ku, G. G. Mathieu and K. G. Knauss, Deep-Sea Res., 1977, 24, 1005–1017 CrossRef CAS
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed
- S. Dai, Chem, 2021, 7, 537–539 CAS
- A. I. Wiechert, A. P. Ladshaw, L.-J. Kuo, H.-B. Pan, J. Strivens, N. Schlafer, J. R. Wood, C. Wai, G. Gill, S. Yiacoumi and C. Tsouris, Ind. Eng. Chem. Res., 2020, 59, 13988–13996 CrossRef CAS
- A. P. Ladshaw, A. I. Wiechert, S. Das, S. Yiacoumi and C. Tsouris, Materials, 2017, 10, 1268 CrossRef PubMed
- P.-l. Liang, L.-y. Yuan, H. Deng, X.-c. Wang, L. Wang, Z.-j. Li, S.-z. Luo and W.-q. Shi, Appl. Catal., B, 2020, 267, 118688 CrossRef CAS
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed
- L. Yang and L. Wen, Cell Rep. Phys. Sci., 2022, 3, 101167 CrossRef
- L. Yang, H. Xiao, Y. Qian, X. Zhao, X.-Y. Kong, P. Liu, W. Xin, L. Fu, L. Jiang and L. Wen, Nat. Sustain., 2022, 5, 71–80 CrossRef
- H. Li and S. Wang, Chem, 2021, 7, 279–280 CAS
- Y. Yuan, Q. Yu, M. Cao, L. Feng, S. Feng, T. Liu, T. Feng, B. Yan, Z. Guo and N. Wang, Nat. Sustain., 2021, 4, 708–714 CrossRef
- Y. Yuan, Y. Yang, X. Ma, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1706507 CrossRef PubMed
- A. I. Wiechert, A. P. Ladshaw, G. A. Gill, J. R. Wood, S. Yiacoumi and C. Tsouris, Ind. Eng. Chem. Res., 2018, 57, 17237–17244 CrossRef CAS
- J. Bai, X. Yin, Y. Zhu, F. Fan, X. Wu, W. Tian, C. Tan, X. Zhang, Y. Wang, S. Cao, F. Fan, Z. Qin and J. Guo, Chem. Eng. J., 2016, 283, 889–895 CrossRef CAS
- Y. Wang, J. Wang, J. Wang, J. Liang, D. Pan, P. Li and Q. Fan, J. Mol. Liq., 2020, 308, 113007 CrossRef CAS
- J. Bai, J. Chu, X. Yin, J. Wang, W. Tian, Q. Huang, Z. Jia, X. Wu, H. Guo and Z. Qin, Chem. Eng. J., 2020, 391, 123553 CrossRef CAS
- Z. Jia, Y. Cui, J. Chu, Q. Huang, J. Wang, W. Tian, X. Wu, Z. Qin and J. Bai, Environ. Technol. Innovation, 2022, 25, 102214 CrossRef CAS
- C. Liu, P.-C. Hsu, J. Xie, J. Zhao, T. Wu, H. Wang, W. Liu, J. Zhang, S. Chu and Y. Cui, Nat. Energy, 2017, 2, 17007 CrossRef CAS
- H. Yang, X. Liu, M. Hao, Y. Xie, X. Wang, H. Tian, G. I. N. Waterhouse, P. E. Kruger, S. G. Telfer and S. Ma, Adv. Mater., 2021, 33, 2106621 CrossRef CAS PubMed
- F. Chi, S. Zhang, J. Wen, J. Xiong and S. Hu, Ind. Eng. Chem. Res., 2018, 57, 8078–8084 CrossRef CAS
- D. Chen, Y. Li, X. Zhao, M. Shi, X. Shi, R. Zhao and G. Zhu, ACS Cent. Sci., 2023, 9, 2326–2332 CrossRef CAS PubMed
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed
- Y. M. Bar-On and R. Milo, Cell, 2019, 179, 1451–1454 CrossRef CAS PubMed
- X. Zhang, Y. Guo, G. Liu, Y. Liu, M. Song, J. Shi, L. Hu, Y. Li, Y. Yin, Y. Cai and G. Jiang, Environ. Sci. Technol., 2021, 55, 14258–14268 CrossRef CAS PubMed
- D. L. Cologgi, S. Lampa-Pastirk, A. M. Speers, S. D. Kelly and G. Reguera, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15248–15252 CrossRef CAS PubMed
- Z. Wu, R. Zheng, G. Liu, R. Liu, S. Wu and C. Sun, Environ. Microbiol., 2020, 23, 3541–3553 CrossRef PubMed
- X.-F. Luan, C.-Z. Wang, Q.-Y. Wu, J.-H. Lan, Z.-F. Chai, L.-S. Xia and W.-Q. Shi, Dalton Trans., 2022, 51, 11381–11389 RSC
- C.-Z. Wang, J.-H. Lan, Q.-Y. Wu, Q. Luo, Y.-L. Zhao, X.-K. Wang, Z.-F. Chai and W.-Q. Shi, Inorg. Chem., 2014, 53, 9466–9476 CrossRef CAS PubMed
- L. Wang, H. O. Badr, Y. Yang, J. H. Cope, E. Ma, J. Ouyang, L. Yuan, Z. Li, Z. Liu, M. W. Barsoum and W. Shi, Chem. Eng. J., 2023, 474, 145635 CrossRef CAS
- C. Kang, Z. Zhang, A. K. Usadi, D. C. Calabro, L. S. Baugh, K. Chai, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2022, 144, 20363–20371 CrossRef CAS PubMed
- S. Bi, C. Yang, W. Zhang, J. Xu, L. Liu, D. Wu, X. Wang, Y. Han, Q. Liang and F. Zhang, Nat. Commun., 2019, 10, 2467 CrossRef PubMed
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- W. Setyawan and S. Curtarolo, Comput. Mater. Sci., 2010, 49, 299–312 CrossRef
- Y. He, Q. He, L. Wang, C. Zhu, P. Golani, A. D. Handoko, X. Yu, C. Gao, M. Ding, X. Wang, F. Liu, Q. Zeng, P. Yu, S. Guo, B. I. Yakobson, L. Wang, Z. W. Seh, Z. Zhang, M. Wu, Q. J. Wang, H. Zhang and Z. Liu, Nat. Mater., 2019, 18, 1098–1104 CrossRef CAS PubMed
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS
- C. Choi, X. Wang, S. Kwon, J. L. Hart, C. L. Rooney, N. J. Harmon, Q. P. Sam, J. J. Cha, W. A. Goddard, M. Elimelech and H. Wang, Nat. Nanotechnol., 2023, 160–167 CrossRef CAS PubMed
- C. Liu, F. Liu, H. Li, J. Chen, J. Fei, Z. Yu, Z. Yuan, C. Wang, H. Zheng, Z. Liu, M. Xu, G. Henkelman, L. Wei and Y. Chen, ACS Nano, 2021, 15, 3309–3319 CrossRef CAS PubMed
- Z. Wang, B. Yang, Y. Wang, Y. Zhao, X. M. Cao and P. Hu, Phys. Chem. Chem. Phys., 2013, 15, 9498–9502 RSC
- I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS PubMed
- Y. K. Kim, S. Lee, J. Ryu and H. Park, Appl. Catal., B, 2015, 163, 584–590 CrossRef CAS
- E. S. Ilton, Y. Du, J. E. Stubbs, P. J. Eng, A. M. Chaka, J. R. Bargar, C. J. Nelin and P. S. Bagus, Phys. Chem. Chem. Phys., 2017, 19, 30473–30480 RSC
- E. A. Stefaniak, A. Alsecz, I. E. Sajó, A. Worobiec, Z. Máthé, S. Török and R. V. Grieken, J. Nucl. Mater., 2008, 381, 278–283 CrossRef CAS
- C. Hennig, A. Ikeda-Ohno, F. Emmerling, W. Kraus and G. Bernhard, Dalton Trans., 2010, 39, 3744–3750 RSC
- X. Liu, Y. Xie, M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Adv. Sci., 2022, 9, 2201735 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc00011k |
| This journal is © The Royal Society of Chemistry 2024 |