
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCustomizing catalyst surface/interface structures for electrochemical CO2 reduction
Xin
Tan†
a,
Haojie
Zhu†
a,
Chang
He
a,
Zewen
Zhuang
 c,
Kaian
Sun
c,
Chao
Zhang
*b and
Chen
Chen
*a
c,
Kaian
Sun
c,
Chao
Zhang
*b and
Chen
Chen
*a
aEngineering Research Center of Advanced Rare Earth Materials, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cchen@mail.tsinghua.edu.cn
bMOE International Joint Laboratory of Materials Microstructure, Institute for New Energy Materials and Low-Carbon Technologies, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: czhang@email.tjut.edu.cn
cCollege of Materials Science and Engineering, Fuzhou University, Fuzhou 350108, China
First published on 27th February 2024
Abstract
Electrochemical CO2 reduction reaction (CO2RR) provides a promising route to converting CO2 into value-added chemicals and to neutralizing the greenhouse gas emission. For the industrial application of CO2RR, high-performance electrocatalysts featuring high activities and selectivities are essential. It has been demonstrated that customizing the catalyst surface/interface structures allows for high-precision control over the microenvironment for catalysis as well as the adsorption/desorption behaviors of key reaction intermediates in CO2RR, thereby elevating the activity, selectivity and stability of the electrocatalysts. In this paper, we review the progress in customizing the surface/interface structures for CO2RR electrocatalysts (including atomic-site catalysts, metal catalysts, and metal/oxide catalysts). From the perspectives of coordination engineering, atomic interface design, surface modification, and hetero-interface construction, we delineate the resulting specific alterations in surface/interface structures, and their effect on the CO2RR process. At the end of this review, we present a brief discussion and outlook on the current challenges and future directions for achieving high-efficiency CO2RR via surface/interface engineering.
 Xin Tan | Xin Tan received his BS degree from the School of Materials Science and Engineering at Wuhan University of Technology in 2019. He is now a PhD candidate in Prof. Chen Chen's group in the Department of Chemistry, Tsinghua University. His research interests are focused on developing new catalysts for electrochemical CO2 reduction. |
 Chen Chen | Chen Chen is a professor at Department of Chemistry in Tsinghua University. He received his B.S. in 2006 from Beijing Institute of Technology, and PhD in 2011 from Tsinghua University. He worked as a postdoctoral researcher at Lawrence Berkeley National Laboratory and UC Berkeley from 2011 to 2014, and joined the faculty of Tsinghua University in 2015. His research interest is focused on inorganic materials, catalysis, renewable and sustainable energy. Prof. Chen has published over 100 scientific papers with more than 20 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations and an H-index of over 60. He was designated a Highly Cited Researcher (2021–2023) by Clarivate.
000 citations and an H-index of over 60. He was designated a Highly Cited Researcher (2021–2023) by Clarivate.1. Introduction
In recent years, the global annual CO2 emission due to fossil fuel consumption has been on an alerting high level. Approximately half of the emission could be neutralized by land and ocean via the natural carbon cycle, and the other half accumulates in the Earth's atmosphere, resulting in pressing issues relating to environment and sustainability.1–3 According to statistics from the Energy & Climate Intelligence Unit, more than 130 countries and regions have proclaimed the goal of “zero carbon” or “carbon neutrality”, and thus effective measures have to be taken to lower the atmospheric CO2 concentration.4,5 To this end, CO2 conversion and utilization has been proposed as a potential solution to carbon neutrality. In particular, CO2 could be electrochemically reduced into fuels and commodity chemicals; when integrated with renewable electricity (such as solar and wind power), this electrochemical route is expected to close the carbon cycle.6 However, CO2 is notorious for its chemical inertness, the activation of CO2 molecules generally has to overcome a high energy barrier, and the practical implementation of electrochemical CO2 reduction reaction (CO2RR) faces a number of challenging issues, such as high overpotentials, competing side reactions (most notably, hydrogen evolution reaction, HER), and diverse reduction products.7–9 As a result, one of the major topics in CO2RR is to design and construct catalytic materials with optimized activities and selectivities.The customization of catalyst surface/interface refers to the approach that resorts to design and modulate the structures and properties of catalyst surface/interface, so as to optimize the catalytic performance.10–12 In the past few decades, with the emergence of advanced methodologies in both experimental characterization and theoretical simulation, the research focus of catalysis has shifted from observing and documenting the macroscopic catalytic phenomena to monitoring and deciphering catalytic processes on the atomic/molecular level.2,13–15 The idea of structure/mechanism-oriented catalyst design has thus emerged, which offers immense opportunities for developing high-performance CO2RR catalysts. Some catalytic systems with specific structures and functions can now be constructed via rational design; with the surface/interface reaction processes ideally customized, catalysts with superior activities, selectivities and stabilities can be obtained eventually. Such customization strategies include manipulating the crystallographic structures of the catalysts so as to expose specific atomic sites,16,17 altering the coordination configurations so as to optimize the electronic structures of active centers (in particular, the coordination number and configuration of metal active centers),18,19 modifying the surfaces/interfaces so as to regulate the microenvironment,13,20 and constructing hetero-interfaces so as to promote the adsorption and activation of substrate molecules and intermediate species.21,22 In addition, only by looking deep into the surface/interface structures of catalysts can we establish the structure–performance relationship for CO2RR catalysis, which is of fundamental importance for developing more advanced electrocatalysts.2,23
In this review, we start with an overview of the advanced methodologies for probing the structures of catalyst surfaces/interfaces (including synchrotron-radiation-based X-ray absorption spectroscopy, atomic-resolution electron microscopy, and infrared spectroscopy), which could offer more in-depth insights into catalytic structures at the atomic level. Subsequently, we present a systematic overview on the recent progress in customizing the surfaces/interfaces of CO2RR electrocatalysts, including atomic-site catalysts (e.g., single-atomic-site catalysts, diatomic-site catalysts), metal catalysts, and metal/oxide catalysts. From the perspectives of coordination engineering, atomic interface design, surface modification, and hetero-interface construction, we delineate the resulting specific alterations in surface/interface structures, and their effect on the CO2RR process, including (but not limited to) microenvironment modulation, enrichment of substrate molecules, adsorption/desorption of intermediate species. At the end of this review, we give a discussion on the challenges currently faced in the field of surface/interface engineering of CO2RR electrocatalysts, and present an outlook on the possible future directions in this field (Fig. 1).
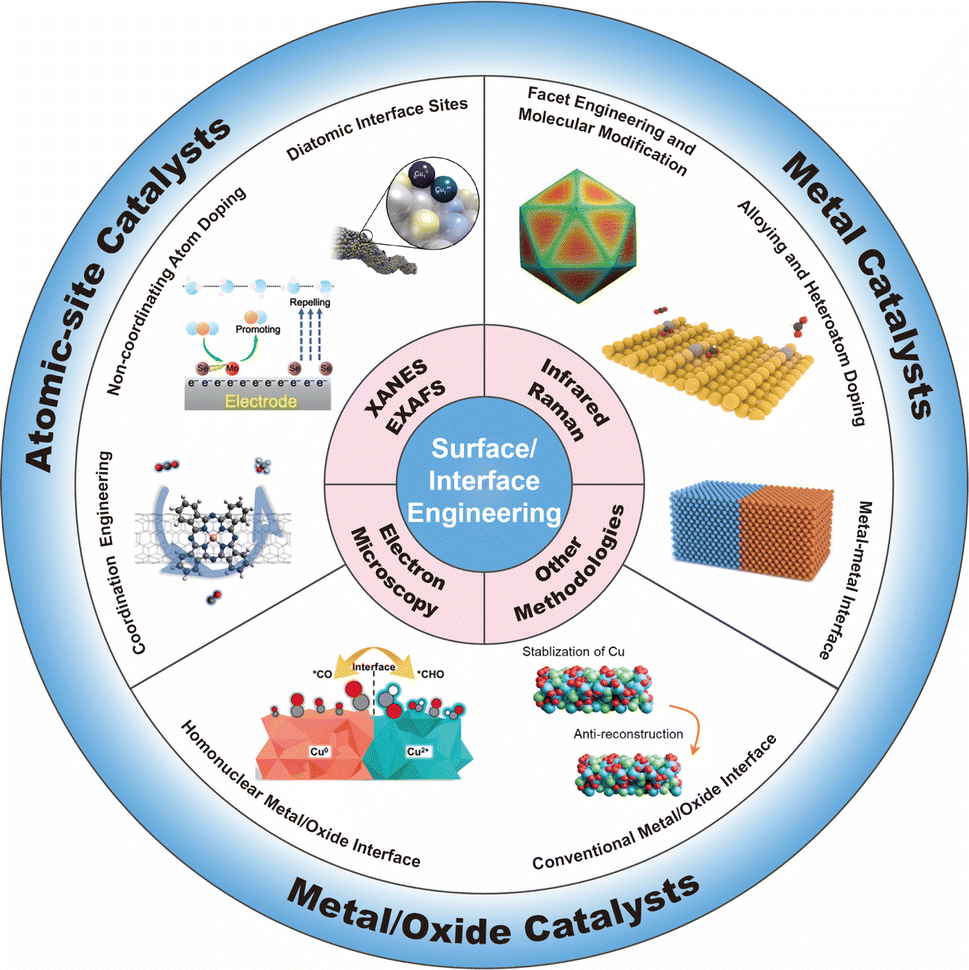 | ||
| Fig. 1 Overview of the surface/interface engineering for CO2RR catalysts. | ||
2. Advanced methodologies for probing the structures of catalyst surfaces/interfaces
In the realm of surface/interface engineering, a profound exploration for advanced structural characterization methodologies is imperative. By employing advanced techniques, one can gain a more comprehensive and in-depth understanding of the microscopic structural features of catalyst surfaces and interfaces. In this section, we will discuss advanced characterization techniques and corresponding in situ and operando techniques under CO2RR test conditions, aiming to unveil the unique structural characteristics of catalyst surfaces/interfaces and to lay the groundwork for achieving efficient CO2RR.2.1 X-ray absorption spectroscopy
X-ray absorption spectroscopy (XAS) has become one of the most popular methodologies for probing information regarding the structures of catalyst surfaces/interfaces.24 This characterization technique has achieved notable success in probing the surface/interface site structure of single-atomic catalysts. For instance, one can determine the local coordination configuration of synthesized catalysts, such as the FeN4 structure or FeN4O structure, through fitting the spectra.25,26 It is important to note that distinguishing ligand atoms (such as C, N and O) using XAS is quite challenging, and often requires complementary techniques such as X-ray emission spectroscopy (XES) for a more comprehensive assessment.25 At the solid–liquid interface in electrocatalysis, the dynamically evolving catalyst surface, where reactants, intermediates, and product species coexist, is often referred to as having disordered features rather than ordered crystalline properties.27 In this regard, operando XAS is one of the characterization techniques capable of revealing the real dynamic configuration of the structures of catalyst surface/interface. Timoshenko et al. utilized time-resolved operando XAS to investigate the impact of the duration of pulse oxidation potential and reduction potential (Δta and Δtc) on the surface oxidation state of copper catalysts (Fig. 2(a)).28 The results indicate that both surface defects and oxidation states can enhance ethanol production. On the basis of these results, a coordinated optimization of Δta and Δtc was employed to balance the oxidation state and morphology, leading to the identification of a region that significantly promotes ethanol generation (Fig. 2(b)).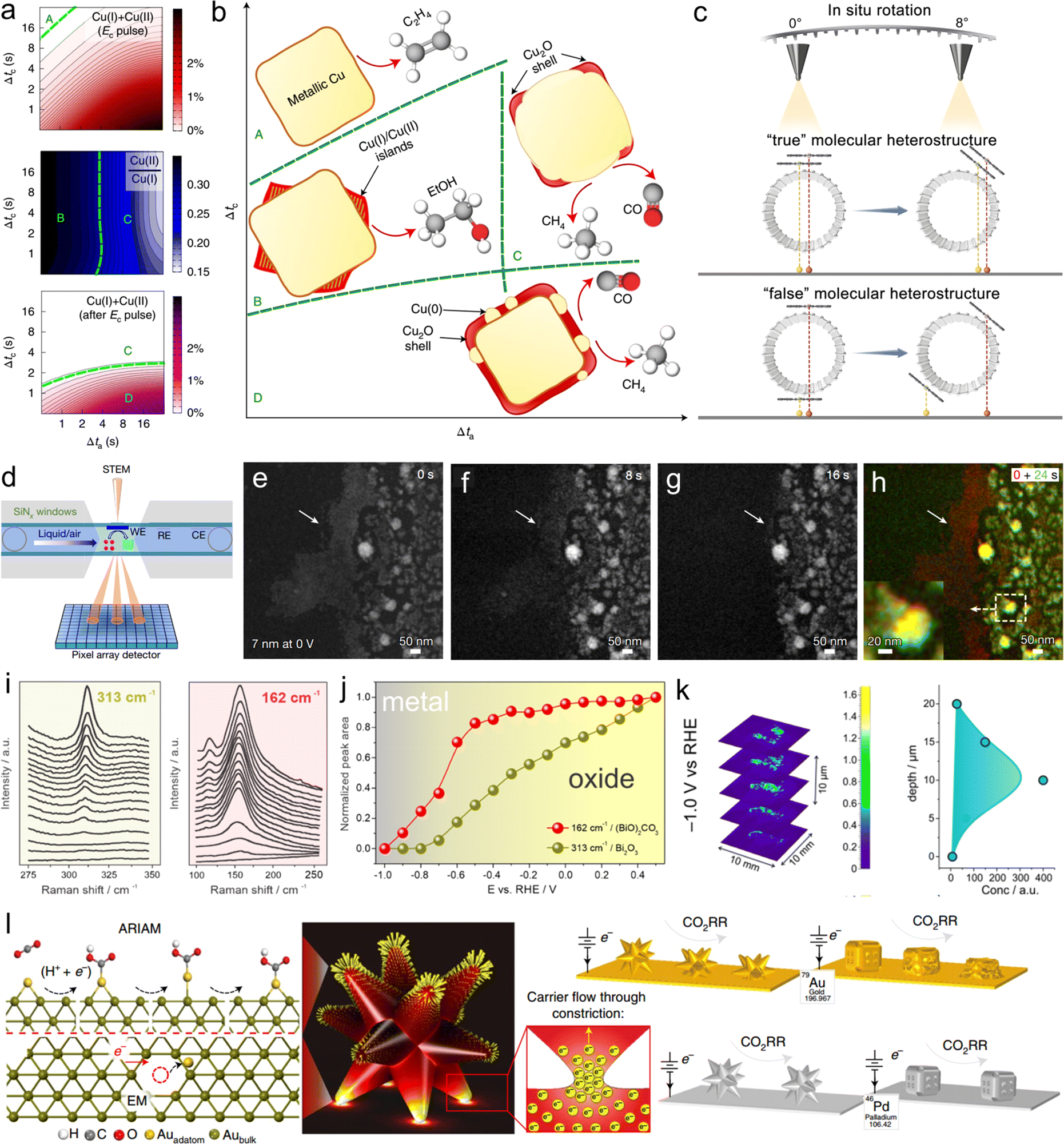 | ||
| Fig. 2 (a) The results obtained from the analysis of XAS data for copper catalyst during pulsed CO2RR with Eanode = 0.6 V, showing the average concentration of the oxide species present during the cathodic pulse. (b) Evolution of the copper catalyst structure and composition during a cathodic pulse extracted from XAS and XRD data. Reproduced with permission from ref. 28. Copyright 2022, Springer Nature. (c) In situ rotation in AC-HAADF-STEM characterization to differentiate the “true” and “false” dual-atomic sites. Reproduced with permission from ref. 30. Copyright 2023, American Chemical Society. (d) Schematic of operando EC-STEM and 4D-STEM. Morphological evolution of 7 nm NPs at 0 V vs. RHE at 0 (e), 8 (f), 16 (g) and 24 s (h). Reproduced with permission from ref. 23. Copyright 2023, Springer Nature. (i) Potential-dependent changes of the Raman peak at 313 cm−1 and 162 cm−1 used as the fingerprint for the presence of Bi2O3 and Bi2O2CO3, respectively. (j) Potential-dependent integrated intensities of the Raman peaks shown in (i). Reproduced with permission from ref. 34. Copyright 2021, American Chemical Society. (k) XRD-CT scans of the catalyst layer after CO2RR (1 h) at −1 V vs. RHE. Reproduced with permission from ref. 35. Copyright 2022, American Chemical Society. (l) Illustration of the factors driving atomic migration in electrocatalysts. Reproduced with permission from ref. 36. Copyright 2021, Springer Nature. | ||
2.2 Atomic-resolution electron microscopy
Advancements in materials science and catalysis research demand an in-depth understanding of catalyst surfaces and interfaces at the atomic level. Electron microscopies have evolved significantly, enabling scientists to probe catalyst surfaces with unparalleled precision, uncovering details crucial for optimizing catalytic performance. The work of Chung et al. serves as a groundbreaking example; they pioneered the use of aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) combined with electron energy loss spectroscopy (EELS) to directly observe the configuration of Fe–N4 for the first time.29 This study provides crucial guidance for subsequent structural analyses of M–N–C catalysts at the atomic level and their exploration in various catalytic applications. Additionally, our research group identified “true” dual-atomic sites through in situ rotation manipulation using AC-HAADF-STEM (Fig. 2(c)).30 Operando electrochemical liquid-cell scanning transmission electron microscopy (EC-STEM) enables imaging with high spatial resolution at the nanometer scale within a liquid environment under electrochemical reaction conditions. This technique has been employed to track the changes in the surface/interface structure of nanocatalysts under CO2RR conditions. Employing this approach, Yang et al. used 7–18 nm copper nanoparticles as model catalysts to comprehensively reveal the evolution of copper nanocatalysts during the reaction process (Fig. 2(d–h)).23 On the basis of EC-STEM results, the team further developed a four-dimensional scanning transmission electron microscopy (4D-STEM) structural characterization technique to unveil the evolution processes of copper catalysts.2.3 Other methodologies
Raman and infrared spectroscopies could serve as powerful tools for monitoring surface-adsorbed molecules and intermediate species involved in electron/proton transfer and bond formation under CO2RR conditions.31,32 Moreover, they also demonstrate great potential in probing the structures of catalyst surfaces/interfaces. For instance, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) of CO adsorption has been conducted to determine the distribution state of Rh in CuRh alloy, where Rh single atoms and Rh clusters exhibited distinctly different CO adsorption peak shapes.33 To investigate the structural evolution of the Bi2O3 catalyst during the CO2RR, Broekmann et al. utilized Raman spectroscopy to identify Bimetal, Bi2O3, and Bi2O2CO3 phases and proposed two pathways for formate production based on experimental results: the “sub-carbonate” pathway at low overpotentials and the Bimetal–O pathway at high overpotentials (Fig. 2(i and j)).34 Additionally, the team employed X-ray diffraction computed tomography (XRD-CT) to analyze the spatial distribution of the chemical composition after the reaction with a relatively high spatial resolution (40 μm), confirming that the distribution of Bi2O2CO3 and Bimetal phases is rather homogeneous as a function of the catalyst layer depth (Fig. 2(k)).35The combination of experimental and theoretical approaches offers a novel avenue to investigating the structures of catalyst surfaces/interfaces. Klinkova et al. utilized ex situ high-resolution scanning electron microscopy (SEM) and electrochemical surface analysis through underpotential deposition (UPD) to explore the structural behavior of well-defined gold and palladium core cages and branched nanoparticles during CO2RR processes.36 On the basis of experimental trends and the integration of density functional theory (DFT) and finite element method (FEM) calculations, the team proposed a mechanistic description of their structural dynamics (Fig. 2(l)). Specifically, the team unveiled the interaction of electrochemical effects (atomic migration facilitated by adsorption reaction intermediates) and electronic effects (electron migration). Both of these effects were identified as driving forces for structural transformations in the materials. The theoretical foundation established in this work serves as a predictive method for assessing the electrochemical surface/interface structural behaviors of novel materials. In summary, these pioneering methodologies offer robust support for future catalyst design and optimization, enabling us to precisely control the structures of catalyst surfaces/interfaces and promote the development of CO2RR.37,38
3. Surface/interface engineering for atomic-site catalysts
The advancements in both preparation methods and characterization techniques have sparked significant interest in atomic-site catalysts. For one thing, atomic-site catalysts theoretically offer the highest atomic utilization efficiency, aligning with the principles of green chemistry. For another thing, they not only possess distinctive structural features and high activity similar to homogeneous catalysts but also exhibit ease of separation from reaction system. By precisely manipulating the coordination environment of atomic sites, researchers can alter the adsorption/desorption processes of key intermediates in the CO2RR process, thus modulating the activity and selectivity of the catalyst.6 Moreover, the electrocatalytic process of CO2RR involves the participation of multiple molecules such as water and carbon dioxide, making the design of synergistic catalytic systems or reaction microenvironments crucial in addition to directly modulating the intrinsic activity of ASCs.39,40 This section summarizes the rational design strategies for ASCs interfaces focused on coordination engineering, non-coordinating atom doping, and construction of diatomic interface sites.3.1 Coordination engineering
The two most direct influencing factors of the coordination environment are the identity of the coordinating atoms and the coordination number. Therefore, this section primarily emphasizes two coordination engineering strategies: coordinating atom engineering and coordination number engineering.Another notable example for coordinating atom engineering is the Ga1 site in CO2RR. Ga nanomaterials are capable of thermally reducing CO2 to generate solid carbon, whereas Ga metal and Ga2O3 exhibit poor CO2RR performance.42 Zhang et al. reported the development of a Ga-N3S-PC catalyst by downsizing the active center of the main group element gallium to the atomic level. By incorporating S atoms in the first coordination shell and P atoms in the external coordination shell, the electronic environment of Ga1 sites is significantly altered, resulting in an increase in the Gibbs free energy barriers for HER and a decrease in those for CO2RR. Consequently, the Ga-N3S-PC catalyst demonstrates a FECO of approximately 92% at −0.3 V vs. RHE. Theoretical simulations revealed the presence of an adaptive dynamic transition of Ga, during which the continual reconfiguration of Ga–S and Ga–P bonds can optimize the adsorption energy of the *COOH intermediate and regenerate the active sites in a timely manner, leading to significantly enhanced CO2RR selectivity and stability compared with Ga–N4 (Fig. 3(a)).43 Therefore, manipulating the coordinating atoms (even in the external coordination shell) can lead to better mimicking of molecular catalysts. Similarly, in addition to the two-electron reduction product CO, by altering the adsorption mode and strength of intermediates, Zn and Co atomic sites have the potential to produce a more diverse array of products in CO2RR, including methanol and methane.40,44
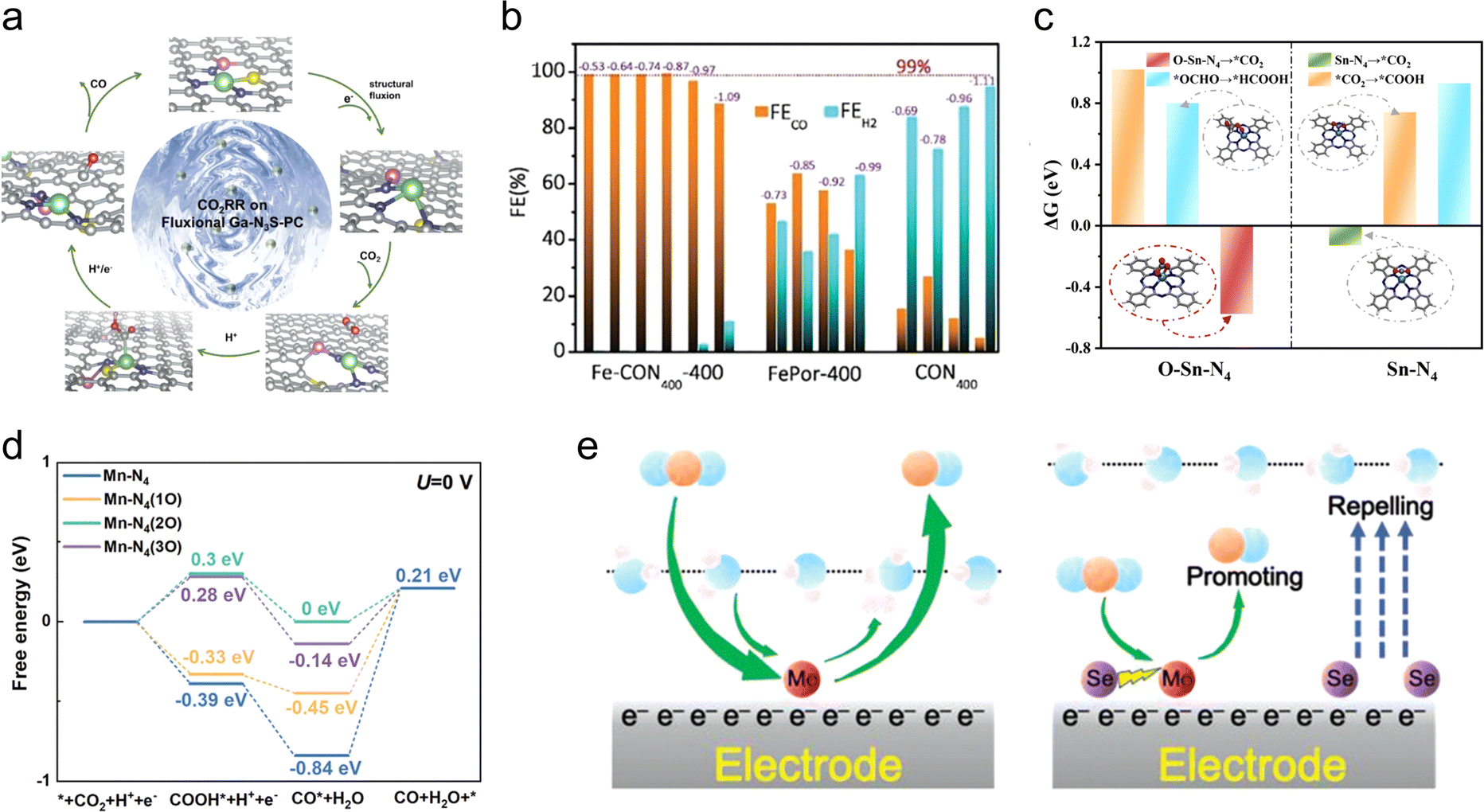 | ||
| Fig. 3 (a) Schematic diagram of the catalytic CO2RR process on fluxional GaN3S-PC catalyst. Reproduced with permission from ref. 43. Copyright 2023, Wiley-VCH. (b) FECO and FEH2 of Fe-CON400-400, FePor-400 and CON400 at different potentials. Reproduced with permission from ref. 26. Copyright 2021, The Royal Society of Chemistry. (c) Calculated ΔG for CO2RR over O–Sn–N4 and Sn–N4 sites. Reproduced with permission from ref. 18. Copyright 2023, American Chemical Society. (d) Calculated ΔG for CO2RR to CO on the Mn–N4, Mn–N4(O), Mn–N4(2O), and Mn–N4(3O) surfaces. Reproduced with permission from ref. 53. Copyright 2023, Wiley-VCH. (e) Schematic diagram of the reaction mechanism of CO2RR on the MoSA–SeSA catalyst. Reproduced with permission from ref. 55. Copyright 2022, Wiley-VCH. | ||
In addition to CO selectivity, coordination number engineering could also enhance current density and catalyst durability. Huang et al. developed a single-atom nanozyme (Ni–N5–C) with a square-pyramidal Ni–N5 site.46 Because of the axial N coordination, the energy levels of the dz2 and dxz/yz orbitals of the Ni–N5 site were increased and decreased, respectively, compared with that for the Ni–N4 site. This results in a decrease in the *CO2-to-*COOH activation barrier and enhanced CO desorption, thereby boosting the kinetic activation process and catalytic performance. In catalytic tests for CO2 reduction, the Ni–N5–C catalyst demonstrated an impressive FECO of 99.6%, with a turnover frequency of 69.7 s−1, under an ultra-high current density of 1.23 A cm−2. Furthermore, the catalyst displayed a remarkable stability over a 100 hour period, surpassing numerous other reported single-atom catalysts. Moreover, for main group elements, the axial coordination has proved to be a crucial factor in imparting catalytic activity. Deng et al. developed a series of well-defined Sn single-atom catalysts as model systems to investigate their electrochemical reactivity for CO2RR.18 Through a combination of DFT calculations and in situ characterization experiments, the team demonstrated that the key site for CO2 reduction into formic acid involves an axial O coordination in the O–Sn–N4 structure. This site facilitates the CO2-to-HCOOH conversion by promoting the formation of surface-bound bidentate tin carbonate species (Fig. 3(c)). Alto sum up, axial coordination engineering provides a versatile approach to regulating the coordination environment of single-atom sites, offering insights that are poised to deepen our understanding of the catalytic pathways and mechanisms involved in CO2RR.
In addition to coordinating atom engineering and coordination number engineering, the dynamic structural evolution of atomic-site catalysts under reaction conditions significantly influences their catalytic performance.47 In particular, the phenomena of reversible transformation between isolated atoms, metal clusters, and metal nanoparticles are of great importance for understanding the catalyst surface/interface structures and the true active centers for CO2RR. For instance, Hsu et al. utilized in situ electrochemical transmission electron microscopy (EC-TEM) to image the dynamic changes of atomic-site catalysts during CO2RR for the first time.48 This structural reconfiguration can generate low-coordinated Cu SACs, which exhibit significantly high selectivity and activity to CO production. Similarly, NiN2 sites originated from nitrogen vacancy induced coordinative reconstruction also demonstrate enhanced CO2RR catalytic activity.49 Furthermore, Cu SACs with CuN4 sites were found to efficiently convert CO2 to ethanol, with a faradaic yield as high as 55%.50 Operando XAS experiments indicate that under reaction conditions, the reconstruction of CuN4 sites forms metallic Cu nanoparticles, which are the truly catalytically active species. Interestingly, such reconstruction behavior is reversible, rendering the material a stable catalyst. With the advancement of characterization techniques, the reconstruction phenomenon of Cu SACs has garnered widespread attention in recent years. Other factors such as the applied potentials and the affinity between isolated Cu sites and coordinating atoms are considered to be closely related to this evolution process,51 offering the possibility of further understanding and optimizing the surface/interface structure of atomic-site catalysts.
3.2 Non-coordinating atom doping
In addition to direct coordination interactions with active metal atoms, the presence of non-coordinating atoms doped within the catalyst support could also play a significant role in regulating the performance of atomic-site catalysts. On one hand, non-coordinating dopant atoms can alter the electronic structures of active sites through long-range interactions, thereby regulating the adsorption/desorption processes of intermediates and in turn modulating the catalytic performance.52,53 On the other hand, doping of foreign atoms within the support is crucial in altering the microenvironment of the catalyst surface.25 In the case of CO2RR, where HER acts as a notable competing reaction, the presence of interfacial water greatly would impact the activity and selectivity of the catalyst. Thus, non-coordinating atom doping provides an additional means of tuning the atomic sites, particularly at the catalyst surface microenvironment, including the interfacial water.Following a similar approach, Wang et al. investigated the relationship between the catalytic activity and the local structures of Mn–N–C catalysts through DFT calculations.53 The results revealed that the introduction of different amounts of oxygen atoms in the second coordination shell can effectively improve the CO desorption from the Mn–N4 surface to varying degrees (Fig. 3(d)). On the basis of this finding, they successfully introduced epoxy groups near the Mn–N4 sites through a versatile nanoemulsion assembly approach. The resulting catalyst exhibited an enhanced electrocatalytic performance, achieving a high FECO of 94.5% at a low overpotential of 0.44 V in aqueous environments. Further calculations indicated that the adjacent epoxy groups to the Mn–N4 sites could lower the d-band center of the Mn site, thus weakening the binding strength with CO. This decrease in the energy barrier for CO desorption significantly promoted the CO2RR kinetics. It is worth mentioning that through the doping of non-coordinating oxygen atoms, this catalyst also demonstrated high performance in aprotic Li–CO2 batteries, exhibiting an improved reversibility and a long cycling life.
In addition to directly manipulating the non-coordinating atoms in the support, doping the support with a second element is another approach to regulating the interfacial water of single-atomic-site catalysts. Inspired by the Mo-dependent formate dehydrogenase found in nature, researchers infer that Mo single-atomic sites hold great potential in the field of CO2RR.54 Theoretical simulations have also confirmed the strong interaction between Mo atoms and *COOH. However, owing to the strong adsorption of CO on Mo single-atomic sites, these sites are often easily poisoned during CO2RR. Additionally, the thermodynamically neutral nature of the hydrogen evolution reaction intermediate (H*) on Mo single-atomic sites suggests a strong competition from the HER as a side reaction. To address these two issues, Sun et al. co-deposited atomically dispersed Se atoms in the MoNC catalyst.55 On one hand, Se atoms (electron configuration, 4s2 4p4) have more valence electrons than C (2s2 2p2) and N atoms (2s2 2p3), allowing for more effective regulation of the d-electronic structure of Mo atoms. On the other hand, the additional valence electrons help repel water molecules from approaching the catalyst surface, hindering the HER progress (Fig. 3(e)). As a result, the obtained catalyst exhibited a FECO over 90% in the potential range from −0.4 to −1.0 V.
3.3 Construction of diatomic interface sites
As mentioned earlier, both coordination engineering and non-coordinating atom doping provide the atomic-site catalysts with rich adjustability. However, owing to the limitations of spatial constraints and electronic quantum states of single-atomic sites, the structures of these single-metal species stabilized on solid supports may not be suitable for catalyzing complex molecular transformations. Diatomic catalysts (DACs) almost perfectly inherit the advantages of single-atomic catalysts, also featuring near 100% utilization of their active atoms and the adjustable coordination environment of the metal centers. The design of diatomic interface sites is crucial for enhancing the rates, conversions, and selectivities of chemical reactions.56 Additionally, the interatomic electronic interactions, co-adsorption, and synergistic catalytic effects of diatomic sites can surpass the limitations of intrinsic activity for single-atomic sites, thus showcasing the remarkable potential in the field of CO2RR.On the basis of these considerations, Jiao et al. constructed a Cu atom-pair catalyst (Cu-APC) from Pd10Te3 alloy nanowires.14 The Pd10Te3 nanowire itself exhibits only minimal HER activity. In contrast, the synthesized Cu-APC demonstrates over 92% faradaic efficiency for CO production, with the competing HER almost completely suppressed. AC-HAADF STEM, XAS, and theoretical calculations collectively confirmed that the key catalytic sites are the Cu10–Cu1x+ units (Fig. 4(a)), which play a crucial role in CO2 reduction through a “diatomic-activating-bimolecular” mechanism. Cu1x+ acts as a Lewis acid, adsorbing water molecules, while Cu10 acts as a Lewis base, adsorbing CO2 molecules. The synergistic proton-coupled electron transfer between them greatly promotes the activation of CO2. This work provides a novel and effective approach to constructing functional interfaces at the atomic level, and serves as an exemplary model for building diatomic catalytic interfaces for CO2RR. Although positively charged Cu sites have been shown to significantly enhance the electrochemical CO2RR to C2 products, it is rather challenging to stabilize positively charged copper atoms under high cathodic potentials. To stabilize Cuδ+ sites, Zhang et al. designed and synthesized a Pdδ−Cu3N catalyst, which contains charge-separated Pdδ−–Cuδ+ atomic pairs.57 Compared with Cu3N, the Pdδ−Cu3N catalyst demonstrated a 14-fold increase in faradaic efficiency for C2 products, reaching up to 78.2%. In situ characterizations and DFT calculations demonstrate that this diatomic site has an exceptional CO binding capability, and the synergistic effect between Pd and Cu plays a significant role in promoting the CO dimerization step for the production of C2 products (Fig. 4(b)). This work presents a new strategy for the design and synthesis of diatomic interfaces and atomic modulation of relatively unstable Cuδ+ sites in CO2RR.
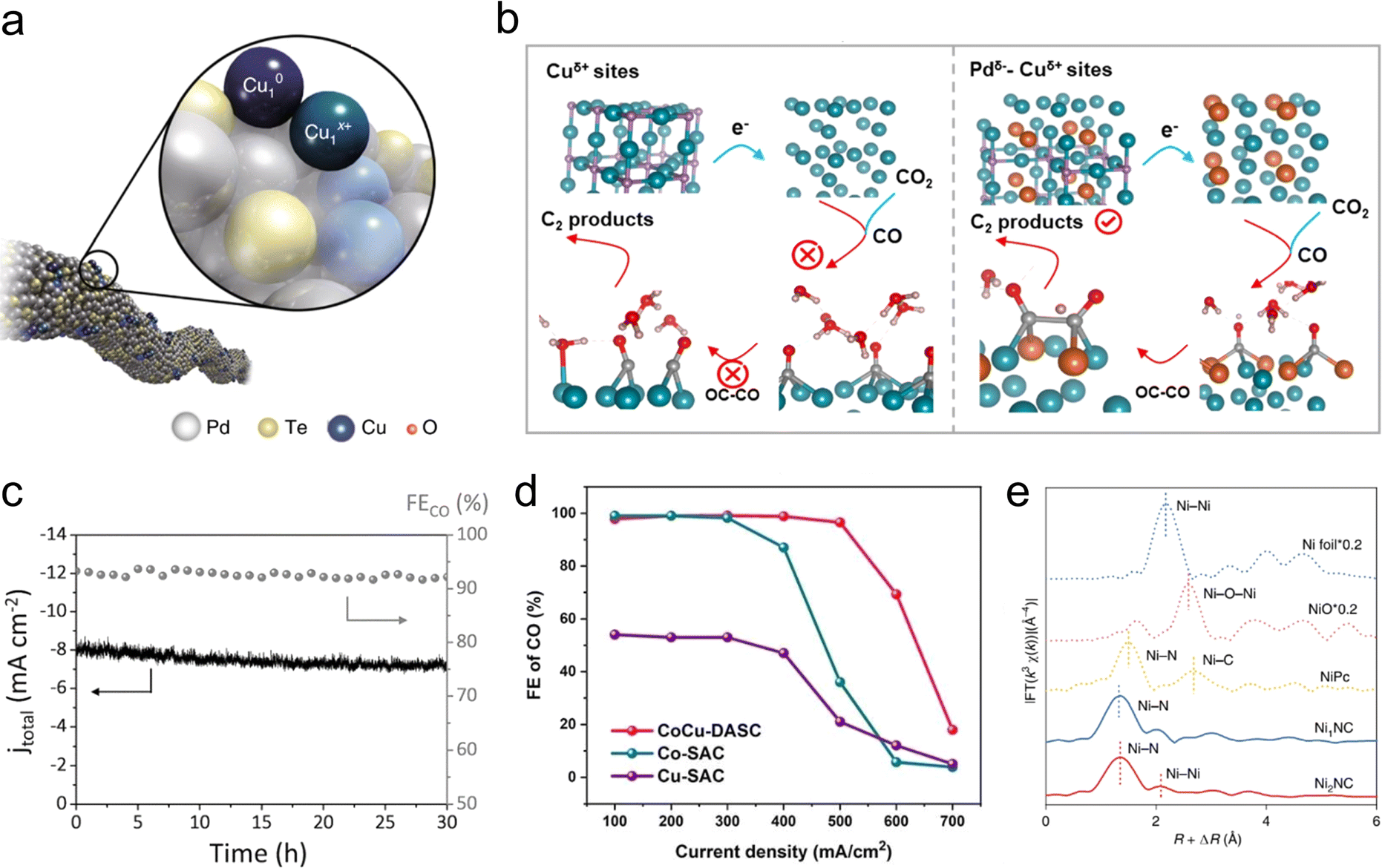 | ||
| Fig. 4 (a) Schematic diagram of the Cu atom pair anchored on Pd10Te3 nanowires. Reproduced with permission from ref. 14. Copyright 2019, Springer Nature. (b) Schematic diagram of the catalytic CO2RR process to C2 products on Pdδ−–Cuδ+ atom pairs. Reproduced with permission from ref. 57. Copyright 2023, American Chemical Society. (c) Stability test for the Ni/Fe–N–C catalyst. Reproduced with permission from ref. 60. Copyright 2019, Wiley-VCH. (d) FECO of CoCu-DASC, Co-SAC and Cu-SAC. Reproduced with permission from ref. 61. Copyright 2022, Wiley-VCH. (e) Fourier transforms (FT) of k3-weighted Ni K edge EXAFS experimental data for Ni2NC, Ni1NC, NiPc, NiO and Ni foil. Reproduced with permission from ref. 20. Copyright 2022, Springer Nature. | ||
The strategy of constructing diatomic interfaces has also been employed in catalyst design on N-doped C materials. The electronic effects between adjacent heteroatomic sites promote the formation and/or adsorption of specific intermediates, exhibiting CO2RR activities superior to isolated monatomic counterparts.58,59 For instance, Ren et al. reported the NiFe/NC catalyst with diatomic NiFe sites, which achieved a FECO above 90% within the range from −0.5 to −0.9 V vs. RHE, and the selectivity was barely changed after 30 h electrolysis (Fig. 4(c)), demonstrating an excellent durability.60 Yi et al. synthesized CoCu/NC heterogeneous diatomic catalysts, where the CO selectivity remained above 95% across a broad range of current densities from 100 mA cm−2 to 500 mA cm−2 (Fig. 4(d)).61 Previous effort was focused mainly on constructing heteronuclear diatomic sites on N-doped C supports. However, the synthesis of homonuclear diatomic sites using simple pyrolysis methods remains challenging. Hao et al. synthesized Ni2NC catalysts through the co-pyrolysis of Ni–citric acid complexes adsorbed on carbon supports and dicyandiamide.20 AC-HAADF-STEM and XAS spectroscopy confirmed the atomic dispersion of Ni atom pairs (Fig. 4(e)). In addition, in situ environmental scanning transmission electron microscopy (ESTEM) revealed that the formation of Ni2 diatomic sites were mediated by Ostwald ripening. The hydroxyl-induced Ni diatomic site (Ni2N6OH) under reaction conditions served as an electron-rich active centres, significantly lowering the energy barrier for *COOH intermediate formation and *CO desorption. The authors further successfully synthesized a few other homonuclear diatomic sites such as Mn2NC, Pd2NC, and Zn2NC using this synthesis method, demonstrating the universality of this strategy for the preparation of homonuclear diatomic sites.
What's more, different from single-atom catalysts, diatomic catalysts have demonstrated superior performance and advantages in C–C coupling due to the synergistic effect between adjacent metal sites.62 For instance, Yang et al. reported a MOF with low-coordinated Cu diatomic sites, exhibiting exceptional current density of 0.9 A cm−2 and a high faradaic efficiency of 71% for C2 products.63In situ characterization and theoretical calculations revealed that the key to C–C coupling at the diatomic sites is the stable adsorption of two *CO intermediates on the adjacent Cu sites. Furthermore, to enhance the selectivity towards specific C2 products, Zhao et al. rationally designed a novel heterometallic Sn⋯Cu dual-site catalyst.64 The active centers of this catalyst are defined as SnN2O2 and CuN4 sites bridged by N atoms. Mechanistic studies indicated that due to the higher oxygen affinity of the SnN2O2 sites, the heterometallic Sn⋯Cu dual sites are more favorable for asymmetric C–C coupling between *CO and *OCH2, leading to the predominant production of ethanol. Ultimately, this catalyst achieved a current density of 68 mA cm−2 at a low potential of −0.57 V (vs. RHE), with an FEethanol of 56%, and maintained considerable stability. In summary, the development of diatomic catalysts provides a potential pathway for efficient C–C coupling in CO2RR. These studies provide reliable support for understanding the interfacial information of diatomic catalysts.
4. Surface/interface engineering for metal catalysts
Due to the complex surface structure and relatively concentrated active sites, metal catalysts exhibit significant selectivity towards various products of CO2RR. And the controllable synthesis of nanocrystals provides great convenience for the modelization of metal catalysts, facilitating the exploration and optimization of their surface structures. After years of research, many metal catalysts demonstrate high catalytic activity, enabling efficient conversion of CO2 to value-added products at relatively low overpotentials, which is crucial for energy efficiency. For example, Pd-, Ag-, and Au-based catalysts are effective in catalyzing the conversion from CO2 to CO; Bi- and Sn-based catalysts can help selectively reduce CO2 to HCOOH; Cu is widely used for the high-selectivity conversion of CO2 into various multi-carbon products.65–70 However, the electrocatalytic CO2RR system is rather complex, with many reaction pathways involving different intermediates. Because of the formation of similar surface adsorption bonds, the binding energies of different intermediates are correlated to each other, a phenomenon known as the scaling relationship.71 Simple metal catalysts usually have a uniform surface structure, making it difficult to achieve specific adsorption of particular intermediates, resulting in low intrinsic catalytic activities. Therefore, it is necessary to engineer the surfaces/interfaces of metal catalysts to decouple the binding of different intermediates, thereby improving the selectivity and activity for CO2RR.4.1 Facet engineering and molecular modification
The different facets of nanocrystal catalysts have distinct electronic structures, leading to varying adsorption strengths for different CO2RR intermediates, which significantly impacts catalytic performance. Therefore, the most straightforward method to regulate metal catalysts is to maximize the proportion of dominant crystal facets in the nanostructure, thus achieving more effective adsorption of specific intermediates. Numerous studies have confirmed the effectiveness of this strategy. Compared with octahedral facets exposing (111) crystal planes and truncated ditetragonal prisms exposing (310) crystal planes, rhombohedral facets exposing (110) crystal planes in gold nanocrystals exhibit higher CO2RR activity and selectivity, especially at low overpotentials.66 Liu et al. synthesized triangular Ag nanoplates, and demonstrated that the Ag(100) facet significantly lowers the required ΔG for the formation of COOH*.67 Triangular Ag nanoplates, with more exposed Ag(100) crystal planes compared with similarly sized Ag nanoparticles and bulk Ag, show an enhanced current density, significantly improved faradaic efficiency (96.8%) for CO and energy efficiency (61.7%), as well as a considerable stability. For the same crystal facet, the performance of electrocatalysts can also be influenced by the differences in electronic structure resulting from surface strain. Huang et al. demonstrated that tensile strain on the surface of Pd icosahedra (Fig. 5(a)) shifts the d-band center upward, enhancing the adsorption of crucial intermediate COOH* and thereby achieving higher CO2RR activity to CO than Pd octahedra do.17 Facet control is also important in CO2RR to multi-carbon products. With the dominant exposed Cu(100) and Cu(111) crystal facets, metal copper nanocrystals derived from cuprous chloride precursors can effectively adsorb key *C1 and *C2 intermediates, demonstrating a considerable yield for propylene (Fig. 5(b)).16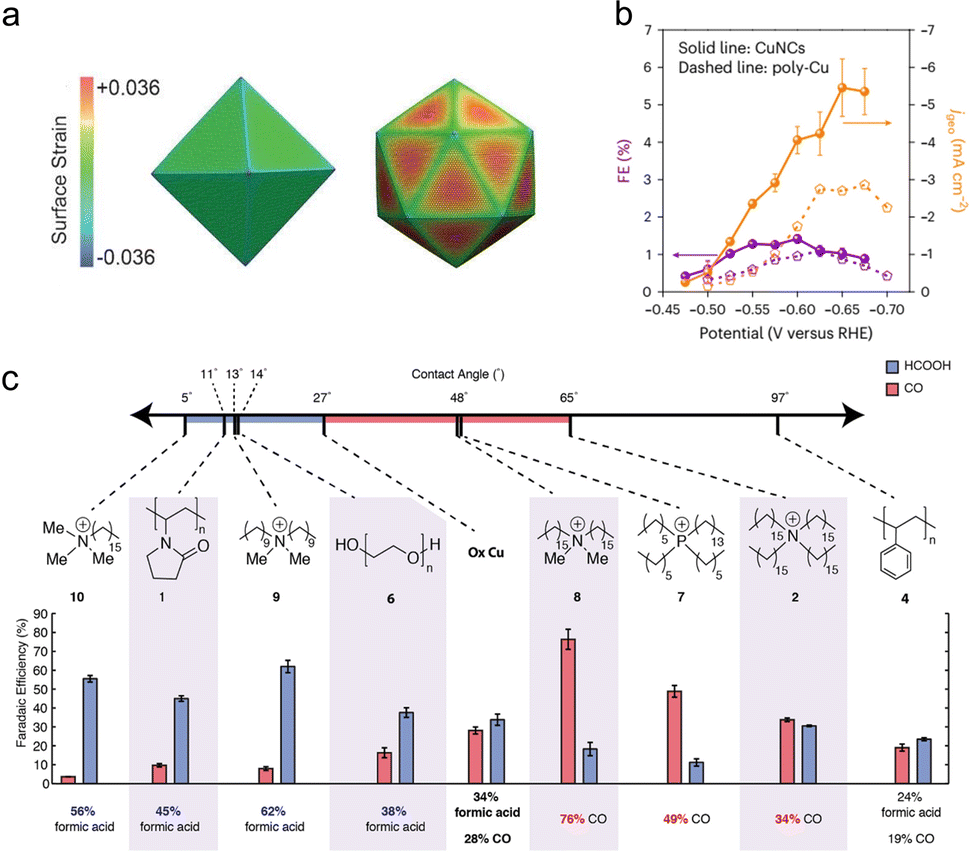 | ||
| Fig. 5 (a) Schematic diagram of the surface strain fields of the Pd icosahedron and octahedron. Reproduced with permission from ref. 17. Copyright 2017, Wiley-VCH. (b) Partial current density and FE for CO2RR to propylene on polycrystalline copper and CuNCs. Reproduced with permission from ref. 16. Copyright 2023, Springer Nature. (c) The contact angles, FECO and FEHCOOH of organic molecular modified Cu surfaces. Reproduced with permission from ref. 73. Copyright 2019, American Chemical Society. | ||
Organic small molecules/polymers have great potential in further regulating the surface structure of metal catalysts. On one hand, molecular modification can improve the adsorption of intermediates on the catalyst surface. Li et al. deposited a series of differently substituted N-arylpyridinium salts on a porous polytetrafluoroethylene gas diffusion layer, forming an organic–metal interface with a sputtered Cu layer.72 This strategy effectively enhanced the stability of ‘atop-bound’ CO, which is the key intermediate in CO2RR to ethylene. In liquid-electrolyte flow cell tests, this system exhibited a 72% ethylene faradaic efficiency and a partial current density of 230 mA cm−2. On the other hand, organic molecule/polymer modification can regulate the microenvironment (including local pH, interfacial water et al.) near the catalyst interface, thus optimizing the mass transport and local concentrations of carbon dioxide and water. Buckley et al. investigated the effects of a range of polymers and molecular modifiers on the selectivity of Cu catalysts for CO2RR.73 They found that protic polymers could provide abundant protons for Cu, enhancing the HER activity. Among aprotic polymers, cationic hydrophobic polymers were found to improve the selectivity for CO, while hydrophilic polymers promoted the formation of formic acid (Fig. 5(c)). Similarly, imidazolium-based ionomers were shown to regulate the competing HER on Ag surfaces, thereby controlling the efficiency of CO2RR.74 Surface-bound dihexadecyldimethylammonium bromide greatly increased the faradaic efficiency for CO (from 25% to 97%) by promoting the mass transport of carbon dioxide and water near the Ag foil surface.75
Metal catalysts, especially copper-based catalysts, often exhibit low selectivity and poor stability owing to severe surface restructuring. In situ reduction of Cu(II)Pc precursors on conductive carbon nanoparticles (CNP) can constrain the size of Cu clusters, generating low-coordination copper sites. However, within one hour, the faradaic efficiency of CO2RR to methane rapidly decreases (from 46% to 25%) because of the continuous leaching and aggregation of copper ions. To address this issue, Fan et al. prepared ligand-modified CuPc/CNP composite catalysts.76 Ethylenediaminetetraacetic acid (EDTA), with its strong coordinating ability, captures and stabilizes free copper ions, thereby preventing the leaching and growth of copper clusters, and greatly enhancing the catalyst's stability. This strategy demonstrates that surface modification with organic molecules can also regulate the catalytic performance by controlling the structure evolution of metal catalysts.
4.2 Metal–metal interface
In contrast to single-component metal catalysts, the catalytic performance of bimetallic catalysts is highly dependent on the atomic distribution. The interactions occurring at the interface of the two metals are significantly different from those of bulk metals. The effect could alter the adsorption energy of intermediates or suppress competing reactions, and thus constructing bimetallic catalysts provide an effective and promising approach to modulating the selectivity and activity of CO2RR.71Ab initio calculations have identified that the Sn/Bi bimetallic interface would favor HCOOH formation. On the basis of this result, Ren et al. constructed a rich Sn–Bi interface on three-dimensional highly porous carbon fabrics (3D CF) through in situ electrodeposition (ED) and evolution under CO2RR conditions.68 Compared with pure Sn and Sn–Bi bulk alloy, this bimetallic interface structure has proven to enhance the p-band center of Sn because of charge transfer from Sn to Bi (Fig. 6(a)). This weakens the Sn–C hybridization of competing COOH* adsorption, while strengthening the Sn–O hybridization of HCOO* adsorption. The resulting Sn–Bi bimetallic catalyst exhibits a faradaic efficiency >90% for formic acid over a wide potential window (from −0.74 to −1.14 V vs. RHE), with a partial current density up to 140 mA cm−2, and maintains its stability for 160 h. Similarly, Wu et al. successfully synthesized a Sn–Bi-based bimetallic aerogel with abundant interfaces and channels, which demonstrated excellent conversion rates for CO2RR to formic acid.69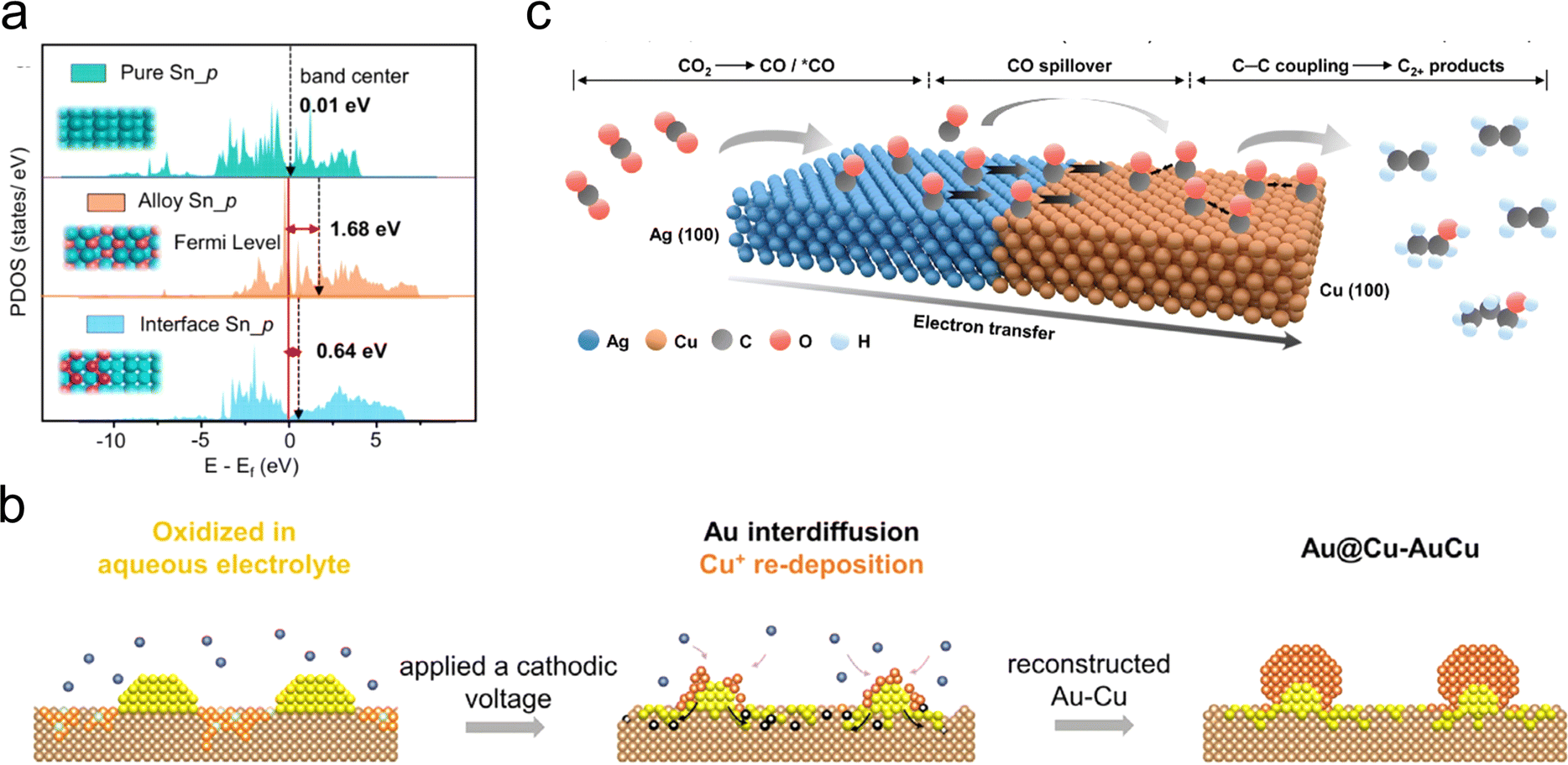 | ||
| Fig. 6 (a) Partial density of states (PDOS) of Sn 5p orbitals of interface Sn, alloy Sn and pure Sn. Reproduced with permission from ref. 68. Copyright 2022, Springer Nature. (b) Schematic diagram of the proposed structural reconstruction and phase transformation of Au–Cu catalyst during CO2RR. Reproduced with permission from ref. 79. Copyright 2022, Elsevier. (c) Schematic diagram of a plausible CO2RR mechanism on Ag65–Cu35 JNS-100 catalyst. Reproduced with permission from ref. 81. Copyright 2022, Wiley-VCH. | ||
Copper is the only known metal capable of catalyzing the generation of multi-carbon products from CO2RR with notable efficiencies. However, copper catalysts are often limited by poor selectivity.77 Compared with pure copper, copper-based bimetallic materials can optimize the reaction kinetics through charge transfer, spillover effects, and other mechanisms, thereby improving the selectivity of CO2RR.11 Unlike Cu, the overpotential for CO2 reduction over Pd systems is lower, and CO or formic acid is typically the main product. However, by controlling the metal ratio of PdCu aerogels, Lu et al. prepared a Pd83Cu17 aerogel catalyst with a faradaic efficiency up to 80.0% for methanol production, with a current density of 31.8 mA cm−2. The excellent catalytic performance of this aerogel is attributed mainly to the synergistic effects between Pd0 and CuI, which facilitates the efficient adsorption and stabilization of the ˙CO2− radical anions at the interface.70
In addition to C1 products, some metal–metal interfaces are believed to promote the generation of multi-carbon products through a tandem catalysis mechanism. Morales-Guio et al. employed physical vapor deposition to deposit gold nanoparticles on the surface of polycrystalline copper foil, and obtained an Au/Cu bimetallic catalyst.78 Catalytic testing and characterization results indicate that the gold nanoparticles could increase the local concentration of CO at the Au/Cu interface, leading to CO spillover from the Au surface to the Cu surface. This effectively increases the probability of C–C coupling on the Cu surface. The catalyst exhibits significantly enhanced efficiency in converting carbon dioxide to >2e− reduction products, surpassing that of individual gold or copper by more than two orders of magnitude. Employing high-resolution transmission electron microscopy characterization, Zhu et al. revealed the reconstruction/phase transition of bimetallic electrocatalysts during the tandem CO2RR.79 The oxidation/reduction of Cu during electrocatalysis drives the transition from phase-separated bimetallic AuCu to alloy-supported core–shell nano-clusters (Fig. 6(b)). In situ infrared spectroscopy experiments in combination with theoretical simulations further confirm that the accumulation of *CO at the metal–metal interface is a key factor in the continuous production of C2+ alcohols.
In contrast to Au–Cu bimetallic catalysts, the primary product of CO2RR at the Ag–Cu interface is ethylene. The key structure responsible for the enhancement in the faradaic efficiency for C2H4 is also the Ag–Cu interface. The Cu/Ag mass ratio has a significant impact on the catalytic performance. Compared with Cu nanoparticles of a similar size and shape, the synthesized Ag1–Cu1.1 nanodimers demonstrated a 3.4-fold increase in the FE for ethylene and an overall 2-fold enhancement in CO2RR activity.80 For Ag–Cu catalysts, the improvement in performance is attributed to electron transfer at the metal–metal interface and CO spillover (Fig. 6(c)).81 In order to further investigate the dynamic catalytic sites of Ag–Cu catalysts during CO2RR and achieve a rational design of AgCu catalysts, Chen et al. synthesized mixed and phase-separated AgCu nanoparticles on a carbon paper electrode.82 Through time-sequential electron microscopy and elemental mapping, the migration behavior of Cu in AgCu under CO2RR conditions was observed for the first time, ultimately leading to the identification of thermodynamically stable Ag-rich and Cu-rich grains, namely Ag0.88Cu0.12 and Ag0.05Cu0.95. Additionally, operando high-energy-resolution X-ray absorption spectroscopy confirmed that the metallic state of Cu in AgCu serves as the catalytically active site during the CO2RR process. The above-mentioned research is crucial for a more in-depth understanding of the structure–activity relationship of bimetallic catalysts, and it is beneficial for guiding the design of more efficient Ag/Cu catalysts.
4.3 Alloying and heteroatom doping
Alloying provides a novel approach to disrupting the scaling relationship. Interactions with heteroatoms can induce geometric and/or electronic effects to alter surface adsorption properties and reactivity.11 For example, the high-index (211) face of face-centered cubic (fcc) Au exhibits superior current density for the electrochemical CO2RR to CO, approximately 20 times higher than the low-index (100) face of fcc Au. However, owing to the thermodynamically unfavorable properties, further optimization of interface architecture, composition, and defects is still required to achieve stable and superior catalytic performance. Recently, Zhou et al. synthesized hexagonal close-packed (2H-type)/fcc-heterophase AuCu alloy nanostructures using a simple wet-chemical method.83In situ differential electrochemical mass spectroscopy (DEMS) and attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) demonstrate that the unique structural features of the synthesized 2H/fcc Au99Cu1, with appropriate copper alloying on high-index surfaces, play a crucial role in enhancing CO2RR performance. For metallic species that are difficult for alloying, a controllable electrodeposition approach is typically used to construct alloy catalysts. For example, alloying metals with high oxygen affinity into Cu can enhance the binding of O. This strategy can increase the adsorption energy of *CHxO and improve methane production. The La5Cu95 synthesized using this method exhibited high performance in a flow cell, with a faradaic efficiency for methane of 64.5% and a partial current density of 193.5 mA cm−2.84 Similarly, using a co-electrodeposition method with suppressed galvanic replacement, Qi et al. prepared an AgCu alloy catalyst with exceptional selectivity for 2-propanol.85 Dispersed Ag atoms in Cu could weaken the surface binding of intermediates in the middle position of the alkyl chain and strengthen the C–O bonds. Furthermore, it is worth noting that GaCu alloy not only exhibits superior catalytic activity for CO2RR,86 but also greatly enhances the stability of the catalyst because Ga could suppress Cu oxidation at open-circuit potential and impart significant electronic interactions with Cu.87In order to fundamentally understand the atomic structure of active sites and thereby achieve more precise control over alloy catalysts, Wang et al. synthesized a series of PdAu catalysts with varying Pd content (Fig. 7(a)).88 The study reveals a non-linear relationship between the catalytic activity for CO2 reduction to CO and the Pd content, which is due to the different adsorption energies of CO and carboxyl on Pd sites in different size ensembles. Pd dimers, in particular, exhibited the optimal catalytic performance as they could activate CO2 with lower energy barriers and avoid strong binding with CO intermediates. Consequently, single-atom alloy (SAA) catalysts have been developed to optimize the catalytic performance for CO2RR by controlling the catalyst's electronic structure to regulate the adsorption of intermediates.89 For instance, Cao et al. reported that the BiCu-SAA could effectively catalyze CO2RR to multi-carbon (C2+) products due to enhanced CO2 adsorption, *CO2 protonation and C–C coupling (Fig. 7(b)).90 Selective production of formate via CO2RR using Cu catalysts is often challenging because it is rather difficult to suppress the C–C coupling process. CO2 molecules become adsorbed and activated on the catalyst surface, forming HCOO* or COOH* intermediates. The former is a key intermediate for formate production, whereas the latter (and its subsequent CO/CO* derivatives) leads to C1 and C2+ products other than formate. Therefore, for the selective conversion of CO2 to formate over Cu-based catalysts, it is essential to maximize the generation of HCOO* intermediates rather than COOH*. In this regard, Zheng et al. reported a Pb1Cu single-atom alloy catalyst that can convert CO2 into formate with a faradaic efficiency of ∼96% and a partial current density of over 1 A cm−2 (Fig. 7(c)).91 The Pb1Cu electrocatalyst converts CO2 to formate at the modulated Cu sites rather than on isolated Pb. Both in situ spectroscopic evidence and theoretical calculations indicate that the atomically dispersed Pb lowers the energy barrier for the first protonation step of the CO2RR to HCOO*, thus achieving efficient formate production.
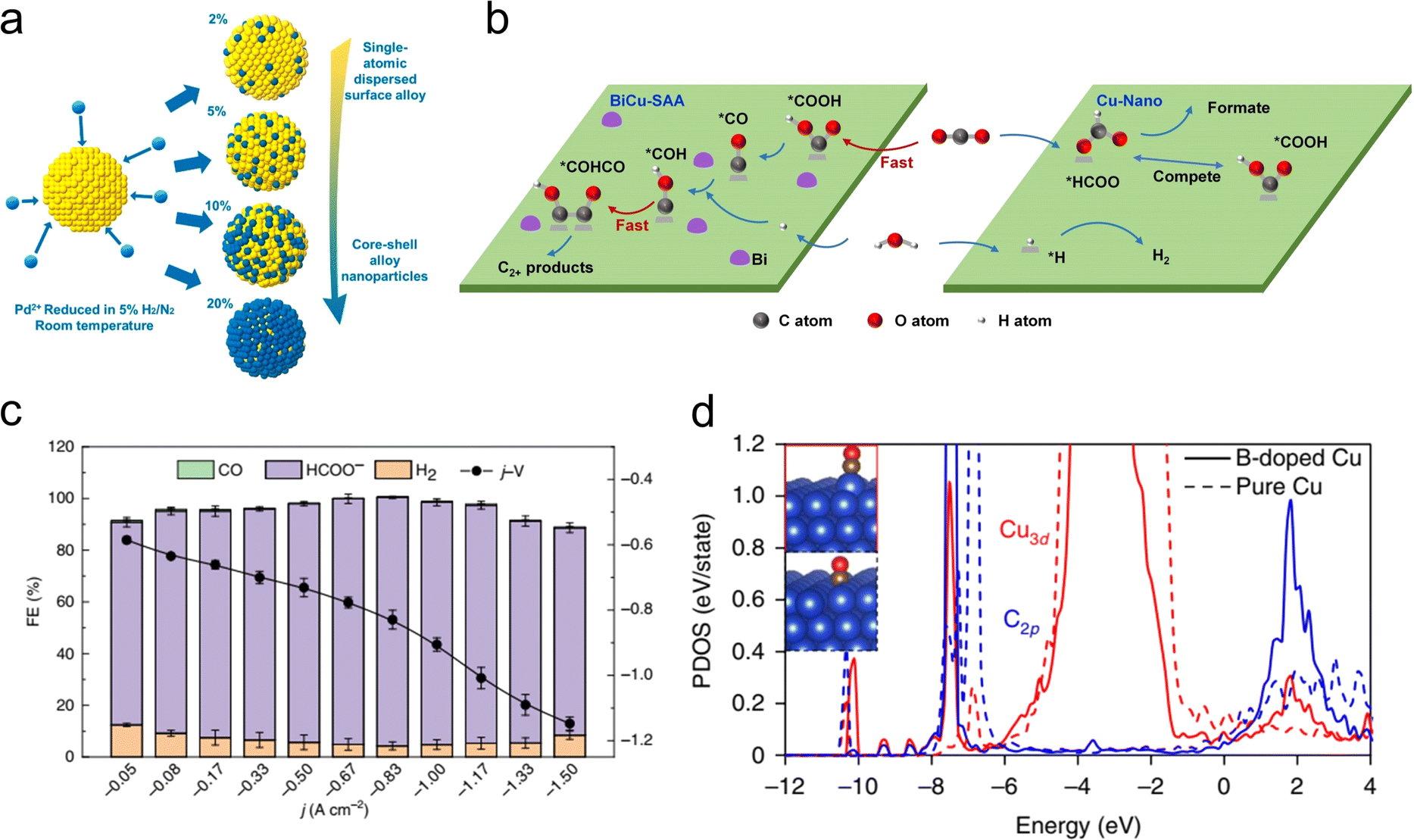 | ||
| Fig. 7 (a) Schematic diagram of the preparation process for the Pd@Au nanoparticles with different dose of Pd. Reproduced with permission from ref. 88. Copyright 2019, American Chemical Society. (b) Schematic diagram of the proposed reaction mechanism of CO2RR on the BiCu(111)-SAA and Cu(111)-Nano. Reproduced with permission from ref. 90. Copyright 2023, Wiley-VCH. (c) FEs of all CO2RR products at different current densities and the corresponding j–V curve of Pb1Cu catalyst. Reproduced with permission from ref. 91. Copyright 2021, Springer Nature. (d) Partial density of states (PDOS) of Cu3d and C2p orbitals in boron-doped copper and pure copper catalysts. Reproduced with permission from ref. 92. Copyright 2018, Springer Nature. | ||
Compared with metallic dopants, non-metal dopants, such as B, S, and halogens, are expected to exhibit greater modulation capabilities on the electronic structures of metal catalysts on account of their higher electronegativity.92–95 In copper catalysts, surface Cuδ+ sites are considered active for CO2RR. Previously, Cuδ+ has been introduced by employing oxygen-containing species, derived from copper oxide catalysts. However, under CO2RR conditions, especially at high cathodic potentials required for the formation of C2 products, Cuδ+ species can be easily reduced to Cu0, resulting in poor stability of the copper catalysts. Theoretical calculations indicate that boron is more prone to diffuse into the subsurface of Cu(111) crystal planes, whereas oxygen tends to remain on the surface. This suggests that boron doping has the potential to enhance the stability of Cuδ+ species at high potentials and thus improve the stability of copper catalysts. Additionally, compared with pure copper, the d-band center of copper atoms near boron is shifted away from the Fermi level, indicating a greater positive charge on the neighboring copper atoms. This results in stronger binding energy of CO on boron-doped copper surfaces (Fig. 7(d)). On the basis of these considerations, Zhou et al. synthesized boron-doped copper catalysts, which exhibited approximately 80% C2 faradaic efficiency and retained the stability for over 40 hours.92 Furthermore, halogen atoms have even higher electronegativities and can enhance CO adsorption by increasing surface Cuδ+ sites. Ma et al. synthesized a series of halogen-doped Cu catalysts, among which fluorine-modified Cu demonstrated the highest catalytic activity, with a current density of up to 1.6 A cm−2 and 80% C2+ (mainly ethylene and ethanol) faradaic efficiency.93 This is attributed to the ability of halogen-modified Cu surfaces to activate water molecules and facilitate *CHO intermediate formation; on the basis of these findings, the team proposed a novel hydrogen-assisted C–C coupling mechanism.
5. Surface/interface engineering for metal/oxide catalysts
The unique oxidation state structure, abundant surface acidic sites, and variable valence capability of oxides provide a broad platform for tailoring the surface/interface structures of catalysts, thereby offering immense new opportunities to enhance catalytic performance. For instance, by tailoring the meta/oxide (M/O) interface, one can obtain stable adsorption sites for key reaction intermediates, thereby facilitating the activation of CO2 and the selective generation of specific products.21,96 Recent studies have also indicated that some oxidation states of active sites, which are difficult to stabilize during CO2RR, can be effectively stabilized through the design of M/O interface.97,98 In this section, considering the diversity in design approaches, we categorize metal/oxide catalyst interfaces into two major groups: namely, homonuclear M/O interfaces and conventional M/O interfaces, and discuss them separately.5.1 Homonuclear M/O interfaces
In the design of homonuclear M/O interfaces, particular attention needs to be focused on the unique active interface sites formed between the metal active centers and their corresponding oxidized-state sites. This synergistic interaction could potentially promote the activation of CO2 and enhance the selectivity. Constructing homonuclear M/O interfaces can be achieved by two means: one involves the preparation of a metal (such as Cu,99 Co,100etc.) and the subsequent implementation of an in situ oxidation method to convert the surface metal atoms into their corresponding metal oxides. The other means is to first synthesize compounds with a certain oxidation state (such as metal oxides,101,102 metal phosphates,103etc.), and then obtain homonuclear M/O interfacial sites through an in situ electrochemical reconstruction process.Xie et al. conducted a series of investigations on atomic layer catalysts related to Co for the conversion of CO2 into formate.100 They prepared two types of catalysts, each consisting of four atomic layers (pure cobalt metal and a domain where cobalt metal coexists with cobalt oxide), aiming to elucidate the impact of their inherent oxides on electrocatalytic activity. The team found that the surface Co atoms in the atomic-thin layers exhibited a higher intrinsic activity and selectivity for formate compared with the surface Co atoms in bulk samples. Additionally, the intrinsic activity of partially oxidized atomic layers was further enhanced. This research underscores the critical role of atomic-scale structures and the presence of oxides on metal catalysts in intrinsic activity and selectivity. Copper, as one of the most intriguing catalysts on account of its unique capability in producing hydrocarbons, has been widely studied for its M/O interface. Chou et al. conducted in situ SEIRAS to investigate the interaction of different oxidation states of Cu surface with surface intermediates.99 They observed that electro-deposited copper electrodes exhibited a higher Cu+ content, with surface CO intermediates predominantly existing in the configuration of top adsorption (COatop). During further reduction processes, COatop led to the generation of C1 hydrocarbon products, such as methane. At the oxidized state surface of Cu0 bridge-bonded CO (CObridge) are readily formed, suppressing the formation of hydrocarbon products. In contrast, Cu electrodes treated with cyclic voltammetry (CV) exhibited a coexisting M/O interface of Cu(I) and Cu(0). During CO2RR, this interface resulted in the formation of both COatop and CObridge, thus enhancing the selectivity for C2 products (Fig. 8(a and b)).
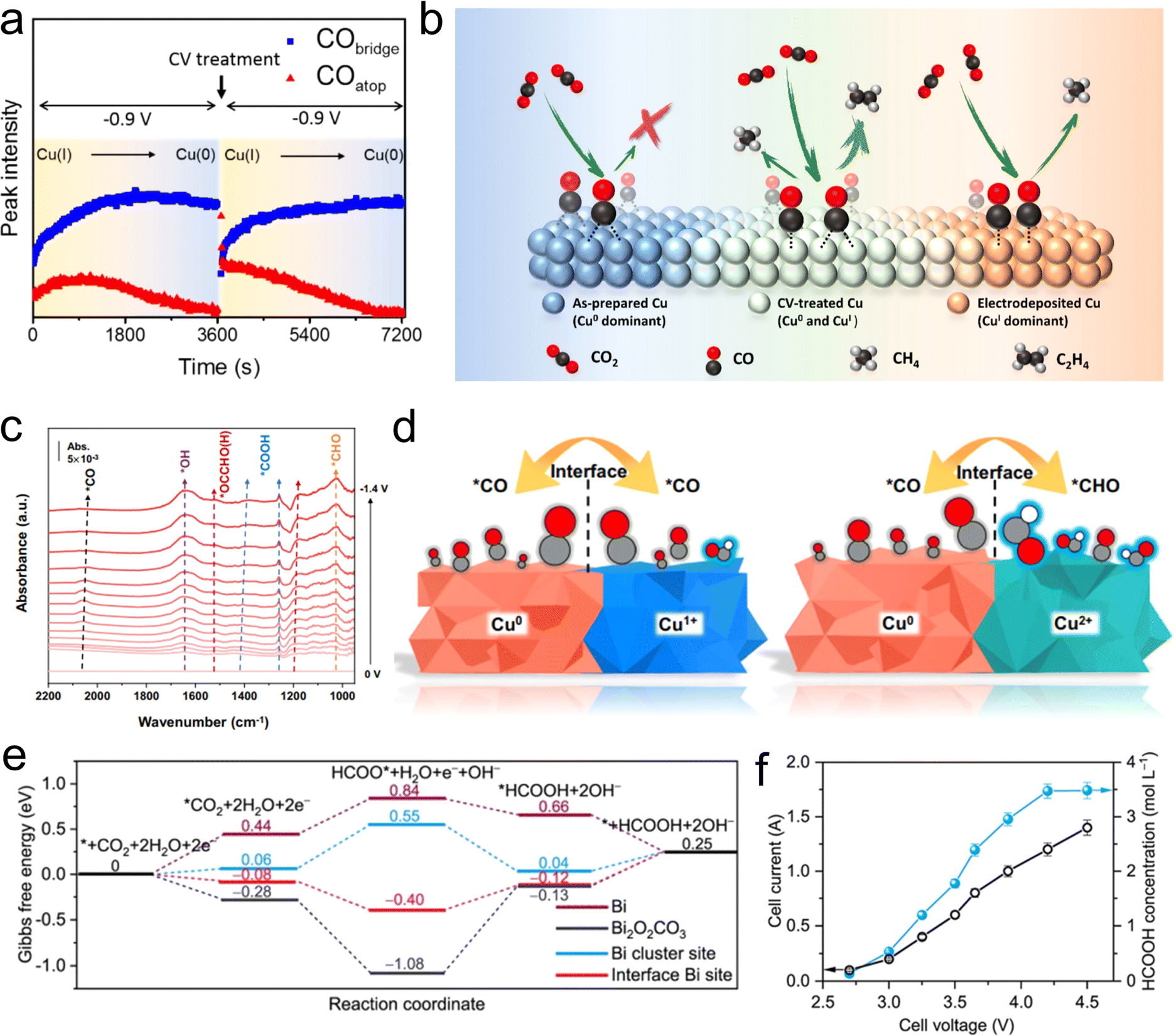 | ||
| Fig. 8 (a) Time dependence of the CObridge- and COatop-associated peak intensities at −0.9 V. (b) Schematic Illustration of the CO2RR on the Cu surface. Reproduced with permission from ref. 99. Copyright 2020, American Chemical Society. (c) In situ SEIRAS spectra of the Cu3(PO4)2 in 0.1 M KHCO3 electrolyte in real-time condition. (d) Schematic diagram of different intermediates on the Cu0/Cu+ and Cu0/Cu2+ interfaces. Reproduced with permission from ref. 103. Copyright 2023, Springer Nature. (e) Gibbs free energy diagram for CO2RR to HCOOH on Bi, Bi2O2CO3, Bi cluster site of Bi3/Bi2O2CO3, and interfacial Bi site of Bi3/Bi2O2CO3. (f) The total current and the concentration of formic acid at different cell voltages in the solid-electrolyte cell. Reproduced with permission from ref. 114. Copyright 2023, Wiley-VCH. | ||
Oxide-derived catalysts (OD-catalysts) represent an important category of catalysts for CO2RR and serve as a crucial means to obtain homonuclear M/O interfaces. Kanan et al. systematically investigated a range of OD-catalysts, such as OD-Cu, OD-Au, and OD-Sn, for CO2RR, revealing enhanced electrochemical activity and selectivity.101,102,104,105 As the research progressed, scientists developed other electrochemical reconstruction-derived catalysts to obtain homonuclear M/O interfaces, including metal phosphate derivatives, metal silicate derivatives, and more, aiming for more stable electrochemical active M/O interfaces and superior performances.103,106,107 As mentioned earlier, the synergistic effect between Cu0 and Cu+ promotes the generation of C2 products. However, obtaining stable Cu+ species during the CO2RR process has been a daunting challenge. To address this issue, Yuan et al. synthesized CuO/CuSiO3 composite catalysts, where Cu2+ species were in situ reduced to Cu0 and Cu+ during the electrochemical process.106 By adjusting the loading amount of Cu, the authors achieved continuous tunability of the Cu0/Cu+ ratio. During the CO2RR process, Cu0 facilitated CO2 activation by lowering the thermodynamic activation energy barrier, whereas Cu+ enhanced the adsorption of the intermediate *CO. The synergistic interaction at this M/O interface enhanced the dynamics of C–C coupling. Notably, the catalyst with 20% Cu/CuSiO3 exhibited a remarkable 51.8% selectivity for C2H4. Recent studies have indicated that asymmetric coupling between *CO and *CHO or *COH has lower energy barriers than direct *CO–*CO coupling.108–110 The key challenge lies in designing interface sites to achieve asymmetric coupling. Apart from the extensively studied Cu+ species, Cu2+ species with a higher oxidation state exhibit structural features that facilitate the binding of CO or H2O.111,112 This enhanced CO adsorption is expected to promote further protonation of *CO to *CHO (or *COH). Thus, the rational design of Cu0/Cu2+ interface sites holds promise for facilitating the CO–CHO coupling process. However, obtaining stable Cu2+ during the CO2RR is rather difficult. To address this issue, Zhang et al. utilized the Materials Project Database and DFT calculations, and screened 83 compounds containing Cu2+ species, eventually identifying Cu2+ phosphorus oxysalts (CuPO) as the most promising candidate for stable Cu0/Cu2+ interface under electroreduction conditions.103 Subsequently, the team experimentally synthesized the CuPO catalyst, which exhibited a faradaic efficiency of 90.9% for C2+ products with a C2+ partial current density of over 300 mA cm−2 in a flow cell. In situ SEIRAS further demonstrated that Cu2+ sites favored the formation of *CHO, which could be readily coupled with *CO on the Cu0 surface to form *OCCHO intermediates, thereby achieving efficient CO2-to-C2+ conversion (Fig. 8(c and d)).
The design of homonuclear M/O interface has been widely applied to other metal active sites as well. For instance, bismuth compounds (such as Bi2O3, Bi-MOF) often undergo a dynamic restructuring process from Bi3+ to Bi2O2CO3 during the CO2RR.34,113 This process becomes more pronounced in alkaline-electrolyte flow cells, and the inherent capability of oxidative state retention provides a foundation for designing the Bi/Bi3+ interface.35 In their theoretical investigations, Lin et al. found that the activation of CO2 and the formation of HCOO* proceed slowly on the surface of metallic Bi, despite the favorable energy landscape for the subsequent reduction to *HCOOH and its subsequent desorption. In the case of Bi2O2CO3, strong Coulomb interactions between CO2 and positively charged surface Bi atoms facilitate the activation of CO2 and the subsequent formation of HCOO*, but the subsequent protonation of HCOO* and the desorption of *HCOOH become challenging. In light of this, the team designed and prepared a stable Bi0 cluster and Bi2O2CO3 nanosheets as the active phase by using Bi2S3 as a precursor in an alkaline flow cell.114 Experimental and theoretical results indicate that this Bi0/Bi3+ interface exhibits optimized CO2 activation and subsequent intermediate protonation capabilities, demonstrating excellent selectivity for formate (Fig. 8(e)). It achieves a 93% selectivity for formic acid at a high current density of 2 A cm−2, and the electrochemical stability can be maintained for over 100 hours. Furthermore, employing solid-state electrolyte devices, the team successfully achieved continuous production of 3.5 mol L−1 pure formic acid (Fig. 8(f)). These results indicate that establishing homonuclear M/O interfaces to achieve synergistic interactions between metals and their oxidized species is a highly promising strategy for the design of advanced CO2RR catalysts.
5.2 Conventional M/O interfaces
In contrast to homonuclear M/O interfaces, the design of conventional metal/oxide catalysts typically involves the composite of active metals with electrochemically relatively inert metal oxides to regulate the properties of the M/O interfaces. This approach has achieved significant success in thermocatalytic CO2 hydrogenation.115,116 In recent years, with the continuous development of electrocatalytic CO2RR technology, researchers have come to realize that constructing suitable M/O interface can effectively stabilize reactive species or regulate processes such as the adsorption and desorption of reaction intermediates.21,117 This, in turn, directly influences the activity and selectivity of CO2RR.Ceria has been widely employed in constructing active M/O interface for CO2RR by virtue of its unique physicochemical properties, including outstanding stability, and tunable oxidation states. Liu et al. synthesized a CeO2–SnO2 heterostructure catalyst using a facile electrospinning method, and achieved a maximum formate partial current density of 500 mA cm−2 and a faradaic efficiency of 87.1% during CO2RR.118 Through in situ SEIRAS and XPS analysis, the team proposed a dynamic CeO2-mediated Sn0/Snδ+ redox cycling mechanism (Fig. 9(a)). The oxygen vacancies generated on the CeO2 surface facilitate water decomposition to produce *OH and *H species. The former oxidizes Sn0 to active Snδ+, promoting the conversion of CO2 to a crucial intermediate, *OCHO, with the assistance of the *H. The strategy of utilizing CeO2 to stabilize the oxidation state of active centers has also been applied to stabilize Cu2+ species by Qiao's research group.98 The majority of Cu-based catalysts would undergo electrochemical reduction from Cu2+ to Cu0 or Cu+, often accompanied by phase restructuring and the formation of new active centers. The team employed CeO2 as a sacrificial component to protect the Cu2+ active species (Fig. 9(b)). Through in situ Raman spectroscopy experiments, they observed a significant increase in Ce3+ concentration in Cu–CeOx after CO2RR, demonstrating that electrons would preferentially reduce Ce4+ to Ce3+ in Cu–Ce-Ox. Simultaneously, the newly generated Ce3+/Ce4+ pairs act as a conductive network, enhancing the overall conductivity of the solid solution, accelerating electron transfer, and thereby inhibiting the electron accumulation at Cu2+ sites and preventing their self-reduction. As a result, the stabilized Cu2+ enhances CO adsorption and further hydrogenation to produce CH4 rather than dimerization to form C2 products. Additionally, M/O between metals such as Au, Bi, Cu, and ceria has shown significant potential in promoting the adsorption and activation of CO2, as well as stabilizing crucial reaction intermediates.96,117,119 For instance, Yin et al. demonstrated substantial improvements in CO2 adsorption and activation capabilities by constructing a Cu/CeO2 interface compared with commercial Cu without the M/O interfaces. Through precise adjustment of the Cu content, the team also effectively controlled the ratio of low-frequency binding of *COLFB to high-frequency binding of *COHFB at the M/O interface. This manipulation resulted in distinct reaction pathways for C1 and C2+ products (Fig. 9(c)).119
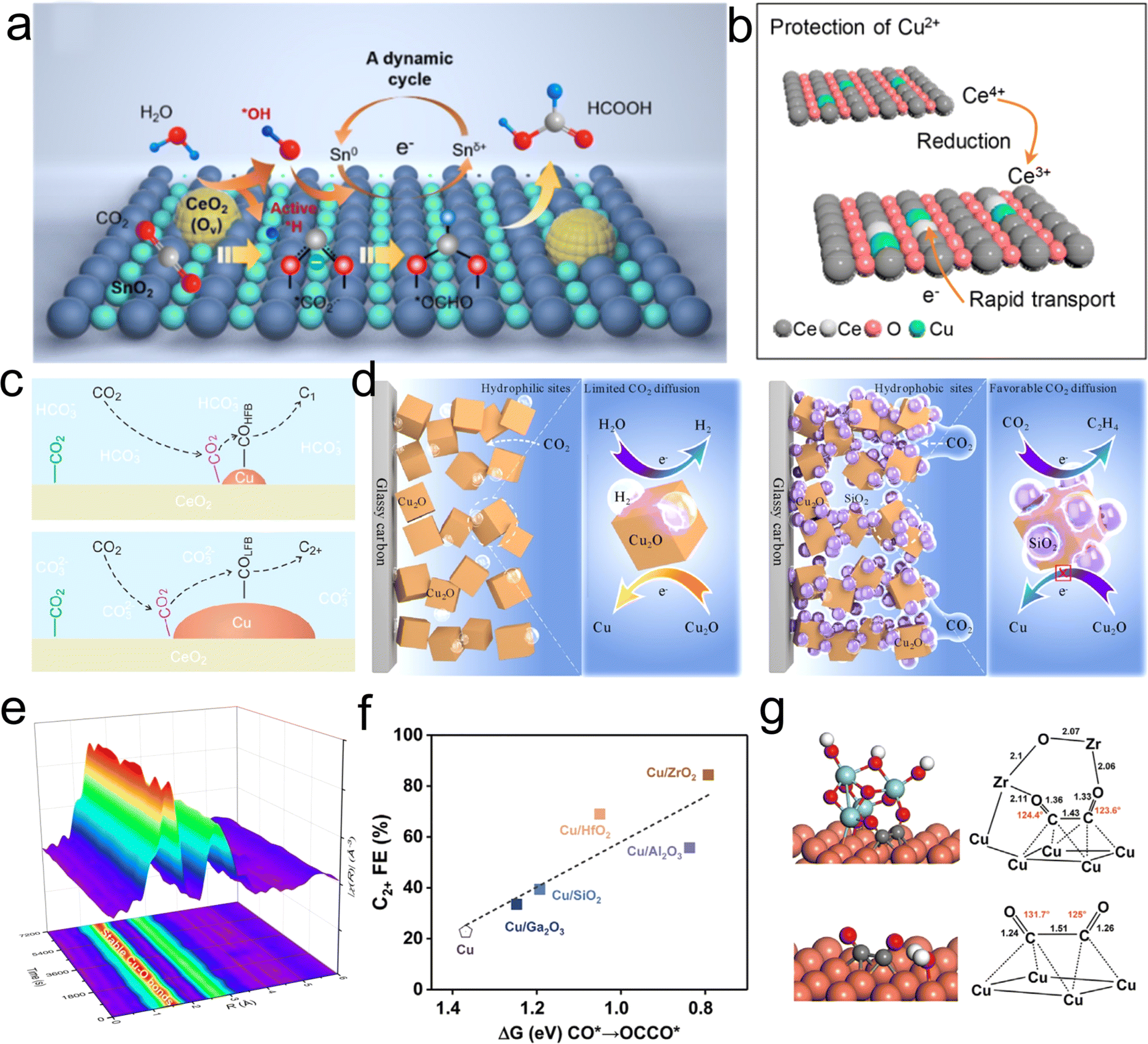 | ||
| Fig. 9 (a) Proposed reaction mechanism of CO2 to HCOOH on the Sn–CeO2 surface. Reproduced with permission from ref. 118. Copyright 2023, American Chemical Society. (b) Scheme of the self-sacrifice mechanism to protect Cu2+. Reproduced with permission from ref. 98. Copyright 2022, American Chemical Society. (c) Proposed CO2RR mechanism at the Cu and CeO2 interface. Reproduced with permission from ref. 119. Copyright 2022, American Chemical Society. (d) Schematic depiction of different catalytic microenvironments of Cu2O and Cu2O/SiO2. Reproduced with permission from ref. 123. Copyright 2023, American Chemical Society. (e) In situ EXAFS data of CuSiOx during CO2RR reaction at −1.4 V vs. RHE. Reproduced with permission from ref. 97. Copyright 2023, American Chemical Society. (f) The relationship between ΔG of *CO to *OCCO and the Faraday efficiency of C2+ products for Cu and Cu/oxide catalysts. (g) The optimized configurations for *OCCO on Cu and Cu-ZrO2 heterostructure. Reproduced with permission from ref. 22. Copyright 2022, Elsevier. | ||
Silica is an exceptionally stable oxide capable of effectively adsorbing and capturing CO2.120 In addition, it exhibits a strong affinity with copper. Its unique catalytic interface with copper has achieved significant success in thermocatalytic hydrogenation reactions.121,122 Therefore, the modification of copper with silica to construct an active M/O interface offers a novel approach to the design of catalysts for CO2RR. Zhao et al. utilized inorganic SiO2 aerosols as hydrophobic additives to modulate the catalytic environment of Cu2O cubes for CO2RR.123 The results indicate that compared with Cu2O, Cu2O/SiO2 enhances the selectivity of multi-carbon products (faradaic efficiency for C2+ increases from 52.4% to 75.6%) and suppresses hydrogen generation (faradaic efficiency for H2 decreases from 30.0% to 9.6%). Through a combination of in situ spectroscopic analysis, electrocatalytic experiments, and molecular dynamics (MD) simulations, the team comparatively studied the interactions between Cu2O/SiO2 catalysts and reaction intermediates, revealing that the weakened buffering effect, enhanced mass transport of CO2, increased CO2 capture by SiO2 aerosols are the primary factors contributing to the improved catalytic performance (Fig. 9(d)). Using DFT calculations, Sinton et al. introduced an oxide modulation strategy, wherein silica is incorporated onto the copper surface to create active Cu–SiOx interface sites.124 This M/O interface significantly lowers the formation energy of OCOH* and OCCOH*, that is, the crucial intermediates in the ethylene generation pathway. Subsequently, the team synthesized the Cu–SiOx catalyst using a one-pot co-precipitation method. The results indicated that the MEA electrolyzer based on the Cu–SiOx catalyst exhibited an impressive 65% ethylene faradaic efficiency at a high partial current density of 215 mA cm−2. Furthermore, the catalyst demonstrated outstanding stability with continuous operation for over 50 hours. The strong metal–support affinity was also harnessed to stabilize electrocatalysts for the CO2RR. One of our recent works indicated that the deactivation and reconstruction issues of copper-based catalysts under cathodic potentials could be effectively addressed through the construction of an atomic Cu–O–Si interface.97In situ XAS characterization revealed that the synthesized amorphous CuSiOx catalyst exhibited exceptionally high electrochemical stability in CO2RR (Fig. 9(e)). Active Cu species showed no signs of reduction or aggregation, demonstrating an outstanding resistance against reconstruction that far surpassed that of commercial CuO catalysts. DFT calculations and AIMD simulations further suggested that the interaction with silica enhanced the strength of the Cu–O bonds. Furthermore, Cu–O–Si interface sites were more conducive to the protonation pathway from *CO to *COH, resulting in methane production rather than hydrogen evolution or C–C coupling.
The design of active M/O interface for CO2RR catalysts has also been extended to other oxides, such as BaO, Al2O3, ZrO2, etc.21,22,125 Sargent et al. expanded the modification of Cu catalysts with different alkaline earth metal oxides (Ba, Sr, Ca).21 They found that BaO significantly improved the faradaic efficiency for C2+ alcohols. Mechanistic studies indicated that the increased alcohol selectivity originated from the M/O interface between Cu and alkaline earth metal oxides. The introduction of M/O would favor the formation of C2 intermediates containing hydroxyl groups (*HCCHOH) rather than hydrocarbon intermediates (*HCC). The study by Wu et al. has demonstrated that coupling oxides with Cu to construct Cu/oxide heterogeneous interface could break the constraints of the original scaling relationship between intermediate adsorption on Cu catalysts.22 This effectively regulated the product distribution in the CO2RR process (Fig. 9(f)). Specifically, introducing oxides exhibiting strong CO2*/CO* adsorption on the Cu surface facilitated the activation of CO2 molecules through Lewis acid–base interactions. Furthermore, the constructed M/O interface could lower the reaction barrier for the C–C coupling step, thereby promoting the generation of multi-carbon products. Notably, the ZrO2 modified Cu electrode exhibited an 85% selectivity for multi-carbon products (Fig. 9(g)). Generally, the fine construction of a metal/oxide interface holds great promise for enhancing the stability of catalytic active centers, stabilizing crucial reaction intermediates, and optimizing the catalytic microenvironment, which is important for improving the CO2RR performance.
6. Summary and outlook
Surface/interface engineering has demonstrated its feasibility in developing high-performance electrocatalysts for CO2RR. In this review, we have summarized the advanced techniques for high-precision characterizations of the catalyst surfaces/interfaces, and systematically discussed the progress in developing various CO2RR electrocatalysts. As the understanding on atomic-level catalytic structures further deepens, new strategies for customizing catalyst surfaces/interfaces have emerged aiming to optimize the CO2RR process, such as constructing diatomic-site catalysts for synergistic catalysis, and employing small molecules to precisely manipulate the configurations of surface-adsorbed intermediates. However, surface/interface engineering is facing challenges such as limited manipulation at the atomic scale, insufficient precision in characterization techniques (especially in monitoring the dynamic evolution of surface/interface structures during electrochemical processes to identify real catalytic active sites), and limitations in theoretical models, which calls for more effort to be devoted in this field.6.1 Atomic-level fine manipulation on the catalyst surfaces/interfaces
As the methodologies in material synthesis and characterization grow more powerful, a major research direction in electrocatalytic CO2RR is to pursue atomic-level fine manipulation on the catalyst surfaces/interfaces. The most notable example is, arguably, the single-atomic-site catalysts, which feature relatively simple and well-defined structures, and therefore are of great significance for deciphering the complex catalytic processes. However, the CO2RR process involves the activation of multiple molecular species, and a single active site may not be ideal for activating multiple molecules and mediating the coupling between different intermediates.14 Therefore, it is desirable to develop more sophisticated synthesis methodologies and to construct multi-atomic-site catalysts, over which CO2 and H2O molecules could be synergistically activated and potentially afford complex multi-carbon reduction products. For instance, Chen et al. achieved CO2 activation by constructing Cu10–Cu1x+ interfaces, where Cu1x+ served as a Lewis acid, adsorbing water molecules, while Cu10 acted as a Lewis base, adsorbing CO2 molecules. This synergistic proton-coupled electron transfer, also known as the “diatomic-activating-bimolecular” mechanism, greatly enhanced CO2 activation.14 Liao et al. rationally designed a novel MOF with unprecedented heterometallic Sn–Cu dual-site sites. Due to the high oxygen affinity of the SnN2O2 site, the Sn–Cu dual sites are more conducive to the asymmetric C–C coupling between *CO and *OCH2, leading to the predominant production of ethanol.64 In addition, the atomic-level fine manipulation on catalyst surfaces/interfaces could be extended to other catalytic systems, such as metal catalysts and metal/oxide catalysts. This would surely enrich the library of catalysts available for mediating the CO2RR process with higher efficiency and controllability.6.2 High-precision characterization techniques for clarifying the structure–performance relationship
In the CO2RR process, the catalyst surface/interface, the reaction intermediates, and the microenvironment are all dynamic, and these dynamic components are strongly interacting, which adds to the complexity of the reaction systems and to the difficulty for identifying the structure–performance relationship. In this regard, in situ or operando characterization techniques could be of great help. For instance, operando XAS has enabled the probing of the surface/interface status and coordination structures under real operational conditions, which helps to unveil the active role of metal centers during catalysis.27 Our recent study, employing in situ XAS, provides evidence that Cu2+ species within CuO exhibit limited electrochemical stability, prone to rapid reduction to Cu0. Introducing silica to form Cu–O–Si interfaces effectively reinforces the strength of Cu–O bonds, thereby markedly enhancing the stability of Cu2+ species.97 EC-STEM and 4D-STEM have enabled the visualization of surface/interface structures during real reaction processes, and thus directly provide the images of catalyst evolution during CO2RR.23 Chen et al. provided the first imaging evidence of Cu cluster formation in Cu single-atom catalysts during the CO2RR process using EC-TEM. This also confirms the finding that small clusters can freely migrate on graphene-like support, attributed to the breaking of C–N and Cu–N bonds.48In situ/operando infrared spectroscopy and surface-enhanced Raman spectroscopy could monitor the dynamics of reaction intermediates and the microenvironment near the catalytic interface.31,32 Shao et al. employed in situ infrared spectroscopy to unveil the mechanism behind the enhanced CO2 reduction activity on OD-Cu. In situ SEIRAS results indicate that the pre-oxidation process can significantly enhance overall CO2RR activity by (a) enhancing CO2 activation, (b) increasing CO uptake, and (c) promoting C–C coupling.126 In the future, these techniques need to go for higher temporal/spatial resolutions so as to pursue superior accuracy and precision. For example, X-ray free-electron laser (XFEL) could be employed as the beam source to conduct XAS and IR measurements, which could afford useful information with higher quality within shorter test times than do synchrotron-radiation beam sources.2 In addition, the combination of multiple in situ/operando techniques could also help to improve the accuracy and efficiency of characterization by simultaneously gathering information from the catalyst surface/interface, the reaction intermediates and the microenvironment. For example, the combination of operando infrared spectroscopy with time-of-flight mass spectrometry may offer more precise intermediate information for CO2RR.127 Needless to say, such integration of multiple techniques depends on the well-coordinated collaboration between the software and hardware sectors, such as information recording, signal processing, and specifically designed reactors.6.3 Advanced theoretical modelling and simulation on CO2RR process at the surface/interface
Thus far, with the aid of theoretical computation, scientists have gained some in-depth understanding on the basic steps of electrochemical CO2RR process and the reaction pathways, from which some consensuses have be derived. For example, it has been recognized that Cu0/Cu+ interface sites could be conducive to C–C coupling, and Cu2+ sites would favor *CO binding and further protonation to give methane.98,106 Despite the remarkable success achieved thus far in theoretical computation, when the local concentrations of relevant species at the catalyst surface and the adsorption/desorption of different intermediates are taken into consideration, the accuracy/precision and the efficiency of theoretical simulation are still far from ideal. Moreover, the catalyst surface/interface generally features a dynamic and non-equilibrated state, rather than the (over)simplified stationary state that is often presumed in computation. Therefore, advanced theoretical models covering multiple time/space scales are needed for simulating the CO2RR process with higher precision. In this regard, advanced descriptions based on atomistic simulations (such as autonomous workflows and machine learning) could help to tackle these challenges and to establish a clearer image of the electrified interfaces. Autonomous workflows facilitate detailed mechanistic studies by automatically assessing surface states and reaction energies. Machine learning offers opportunities to transform workflows and atomic-level understanding of electrified interfaces, such as generating pertinent geometries for high-throughput computing and autonomous workflows, automating decision-making in workflows (e.g., selecting relevant intermediates and transition states), and replacing costly DFT computations with faster energy evaluations.128Author contributions
C. Z. and C. C. conceived the topic of the review. X. T. and H. Z. wrote the original draft of the manuscript. All authors reviewed and revised the original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2023YFB4005100), National Natural Science Foundation of China (21925202, U22B2071, 22302042), Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AO370017), International Joint Mission On Climate Change and Carbon Neutrality, Pilot Group Program of the Research Fund for International Senior Scientists (22250710676).Notes and references
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- L. Li, S. Wang, C. Wan, C. Xu, M. Zuo, Y. Sun and Y. Xie, Next Energy, 2023, 1, 100027 CrossRef.
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, X. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y. M. Yiu, T. K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Nature, 2023, 617, 724–729 CrossRef CAS.
- L. Li, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- X. Tan, Z. Zhuang, Y. Zhang, K. Sun and C. Chen, Chem. Commun., 2023, 59, 2682–2696 RSC.
- C. J. Chang, Y. A. Lai, Y. C. Chu, C. K. Peng, H. Y. Tan, C. W. Pao, Y. G. Lin, S. F. Hung, H. C. Chen and H. M. Chen, J. Am. Chem. Soc., 2023, 145, 6953–6965 CrossRef CAS PubMed.
- Y. Zhang, X.-Y. Zhang and W.-Y. Sun, ACS Catal., 2023, 13, 1545–1553 CrossRef CAS.
- Y. Jia, H. S. Hsu, W. C. Huang, D. W. Lee, S. W. Lee, T. Y. Chen, L. Zhou, J. H. Wang, K. W. Wang and S. Dai, Nano Lett., 2023, 23, 2262–2268 CrossRef CAS.
- Z. Zhang, L. Bian, H. Tian, Y. Liu, Y. Bando, Y. Yamauchi and Z.-L. Wang, Small, 2022, 18, 2107450 CrossRef CAS.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- X. Hu, J. Hu, S. Zheng, Y. Fan, H. Li, S. Zhang, W. Liu, B. Zha, F. Huo and F. Saleem, Chem.–Asian J., 2022, 17, e202200990 CrossRef CAS.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- J. Jiao, R. Lin, S. Liu, W. C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S. F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- H. Xu, D. Rebollar, H. He, L. Chong, Y. Liu, C. Liu, C.-J. Sun, T. Li, J. V. Muntean, R. E. Winans, D.-J. Liu and T. Xu, Nat. Energy, 2020, 5, 623–632 CrossRef CAS.
- J. Gao, A. Bahmanpour, O. Krocher, S. M. Zakeeruddin, D. Ren and M. Gratzel, Nat. Chem., 2023, 15, 705–713 CrossRef CAS PubMed.
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 3594–3598 CrossRef CAS PubMed.
- Y. Deng, J. Zhao, S. Wang, R. Chen, J. Ding, H. J. Tsai, W. J. Zeng, S. F. Hung, W. Xu, J. Wang, F. Jaouen, X. Li, Y. Huang and B. Liu, J. Am. Chem. Soc., 2023, 145, 7242–7251 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Q. Hao, H.-X. Zhong, J.-Z. Wang, K.-H. Liu, J.-M. Yan, Z.-H. Ren, N. Zhou, X. Zhao, H. Zhang, D.-X. Liu, X. Liu, L.-W. Chen, J. Luo and X.-B. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef.
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- X. Li, Q. Liu, J. Wang, D. Meng, Y. Shu, X. Lv, B. Zhao, H. Yang, T. Cheng, Q. Gao, L. Li and H. B. Wu, Chem, 2022, 8, 2148–2162 CAS.
- Y. Yang, S. Louisia, S. Yu, J. Jin, I. Roh, C. Chen, M. V. Fonseca Guzman, J. Feijóo, P.-C. Chen, H. Wang, C. J. Pollock, X. Huang, Y.-T. Shao, C. Wang, D. A. Muller, H. D. Abruña and P. Yang, Nature, 2023, 614, 262–269 CrossRef CAS PubMed.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, e2100102 CrossRef.
- C. Liu, Y. Wu, K. Sun, J. Fang, A. Huang, Y. Pan, W.-C. Cheong, Z. Zhuang, Z. Zhuang, Q. Yuan, H. L. Xin, C. Zhang, J. Zhang, H. Xiao, C. Chen and Y. Li, Chem, 2021, 7, 1297–1307 CAS.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun, X. Tan, J. Zhang, D. Liu, Y. Zhang, P. Jiang, Y. Pan, C. Chen, Q. Peng and Y. Li, Energy Environ. Sci., 2021, 14, 3430–3437 RSC.
- J. Wang, C.-S. Hsu, T.-S. Wu, T.-S. Chan, N.-T. Suen, J.-F. Lee and H. M. Chen, Nat. Commun., 2023, 14, 6576 CrossRef CAS.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- C. Chen, Y. Li, A. Huang, X. Liu, J. Li, Y. Zhang, Z. Chen, Z. Zhuang, Y. Wu, W.-C. Cheong, X. Tan, K. Sun, Z. Xu, D. Liu, Z. Wang, K. Zhou and C. Chen, J. Am. Chem. Soc., 2023, 145, 21273–21283 CrossRef CAS.
- S. Zhu, T. Li, W.-B. Cai and M. Shao, ACS Energy Lett., 2019, 4, 682–689 CrossRef CAS.
- C. Long, X. Liu, K. Wan, Y. Jiang, P. An, C. Yang, G. Wu, W. Wang, J. Guo, L. Li, K. Peng, Q. Li, C. Cui, S. Liu, T. Tan and Z. Tang, Sci. Adv., 2023, 9, eadi6119 CrossRef CAS.
- J. Zhang, P. Yu, C. Peng, X. Lv, Z. Liu, T. Cheng and G. Zheng, ACS Catal., 2023, 13, 7170–7177 CrossRef CAS.
- A. Dutta, I. Zelocualtecatl Montiel, K. Kiran, A. Rieder, V. Grozovski, L. Gut and P. Broekmann, ACS Catal., 2021, 11, 4988–5003 CrossRef CAS.
- I. Zelocualtecatl Montiel, A. Dutta, K. Kiran, A. Rieder, A. Iarchuk, S. Vesztergom, M. Mirolo, I. Martens, J. Drnec and P. Broekmann, ACS Catal., 2022, 12, 10872–10886 CrossRef CAS.
- F. Li, X. V. Medvedeva, J. J. Medvedev, E. Khairullina, H. Engelhardt, S. Chandrasekar, Y. Guo, J. Jin, A. Lee, H. Thérien-Aubin, A. Ahmed, Y. Pang and A. Klinkova, Nat. Catal., 2021, 4, 479–487 CrossRef CAS.
- R. Amirbeigiarab, J. Tian, A. Herzog, C. Qiu, A. Bergmann, B. Roldan Cuenya and O. M. Magnussen, Nat. Catal., 2023, 6, 837–846 CrossRef CAS.
- M. Munz, J. Poon, W. Frandsen, B. R. Cuenya and C. S. Kley, J. Am. Chem. Soc., 2023, 145, 5242–5251 CrossRef CAS PubMed.
- D. Zhao, K. Yu, P. Song, W. Feng, B. Hu, W.-C. Cheong, Z. Zhuang, S. Liu, K. Sun, J. Zhang and C. Chen, Energy Environ. Sci., 2022, 15, 3795–3804 RSC.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS.
- J. Gu, C. S. Hsu, L. Bai, H. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- K. Zuraiqi, A. Zavabeti, J. Clarke-Hannaford, B. J. Murdoch, K. Shah, M. J. S. Spencer, C. F. McConville, T. Daeneke and K. Chiang, Energy Environ. Sci., 2022, 15, 595–600 RSC.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- C. Chen, J. Li, X. Tan, Y. Zhang, Y. Li, C. He, Z. Xu, C. Zhang and C. Chen, EES Catal., 2024, 2, 71–93 RSC.
- J. R. Huang, X. F. Qiu, Z. H. Zhao, H. L. Zhu, Y. C. Liu, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, e202210985 CrossRef CAS.
- L. Liu and A. Corma, Trends Chem., 2020, 2, 383–400 CrossRef CAS.
- C.-S. Hsu, J. Wang, Y.-C. Chu, J.-H. Chen, C.-Y. Chien, K.-H. Lin, L. D. Tsai, H.-C. Chen, Y.-F. Liao, N. Hiraoka, Y.-C. Cheng and H. M. Chen, Nat. Commun., 2023, 14, 5245 CrossRef CAS PubMed.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang, S. Zheng, F. Pan and C. Zhao, Adv. Funct. Mater., 2021, 31, 2107072 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvao Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- L. Zhang, X. Yang, Q. Yuan, Z. Wei, J. Ding, T. Chu, C. Rong, Q. Zhang, Z. Ye, F.-Z. Xuan, Y. Zhai, B. Zhang and X. Yang, Nat. Commun., 2023, 14, 8311 CrossRef.
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS.
- M. Wang, Y. Yao, Y. Tian, Y. Yuan, L. Wang, F. Yang, J. Ren, X. Hu, F. Wu, S. Zhang, J. Wu and J. Lu, Adv. Mater., 2023, 35, e2210658 CrossRef PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- K. Sun, K. Yu, J. Fang, Z. Zhuang, X. Tan, Y. Wu, L. Zeng, Z. Zhuang, Y. Pan and C. Chen, Adv. Mater., 2022, 34, e2206478 CrossRef.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4201–4215 RSC.
- Z. Zhang, S. Chen, J. Zhu, C. Ye, Y. Mao, B. Wang, G. Zhou, L. Mai, Z. Wang, X. Liu and D. Wang, Nano Lett., 2023, 23, 2312–2320 CrossRef CAS.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS.
- W. Zhu, L. Zhang, S. Liu, A. Li, X. Yuan, C. Hu, G. Zhang, W. Deng, K. Zang, J. Luo, Y. Zhu, M. Gu, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2020, 59, 12664–12668 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- J. D. Yi, X. Gao, H. Zhou, W. Chen and Y. Wu, Angew. Chem., Int. Ed., 2022, 61, e202212329 CrossRef CAS PubMed.
- S. Li, A. Guan, C. Yang, C. Peng, X. Lv, Y. Ji, Y. Quan, Q. Wang, L. Zhang and G. Zheng, ACS Mater. Lett., 2021, 3, 1729–1737 CrossRef CAS.
- K. Yang, Y. Sun, S. Chen, M. Li, M. Zheng, L. Ma, W. Fan, Y. Zheng, Q. Li and J. Duan, Small, 2023, 19, 2301536 CrossRef CAS PubMed.
- Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2023, 145, 26783–26790 CrossRef CAS.
- R. Wang, J. Liu, Q. Huang, L. Z. Dong, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 19829–19835 CrossRef CAS.
- S. Jeong, M.-H. Choi, G. S. Jagdale, Y. Zhong, N. P. Siepser, Y. Wang, X. Zhan, L. A. Baker and X. Ye, J. Am. Chem. Soc., 2022, 144, 12673–12680 CrossRef CAS PubMed.
- S. Liu, H. Tao, L. Zeng, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, J. Am. Chem. Soc., 2017, 139, 2160–2163 CrossRef CAS PubMed.
- B. Ren, G. Wen, R. Gao, D. Luo, Z. Zhang, W. Qiu, Q. Ma, X. Wang, Y. Cui, L. Ricardez-Sandoval, A. Yu and Z. Chen, Nat. Commun., 2022, 13, 2486 CrossRef CAS PubMed.
- Z. Wu, H. Wu, W. Cai, Z. Wen, B. Jia, L. Wang, W. Jin and T. Ma, Angew. Chem., Int. Ed., 2021, 60, 12554–12559 CrossRef CAS PubMed.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 57, 14149–14153 CrossRef CAS PubMed.
- X. Li, X. Wu, X. Lv, J. Wang and H. B. Wu, Chem Catal., 2022, 2, 262–291 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141, 7355–7364 CrossRef CAS.
- D. M. Koshy, S. A. Akhade, A. Shugar, K. Abiose, J. Shi, S. Liang, J. S. Oakdale, S. E. Weitzner, J. B. Varley, E. B. Duoss, S. E. Baker, C. Hahn, Z. Bao and T. F. Jaramillo, J. Am. Chem. Soc., 2021, 143, 14712–14725 CrossRef CAS PubMed.
- A. K. Buckley, T. Cheng, M. H. Oh, G. M. Su, J. Garrison, S. W. Utan, C. Zhu, F. D. Toste, W. A. Goddard and F. M. Toma, ACS Catal., 2021, 11, 9034–9042 CrossRef CAS.
- M. Fan, R. K. Miao, P. Ou, Y. Xu, Z. Y. Lin, T. J. Lee, S. F. Hung, K. Xie, J. E. Huang, W. Ni, J. Li, Y. Zhao, A. Ozden, C. P. O'Brien, Y. Chen, Y. C. Xiao, S. Liu, J. Wicks, X. Wang, J. Abed, E. Shirzadi, E. H. Sargent and D. Sinton, Nat. Commun., 2023, 14, 3314 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- C. Zhu, L. Zhou, Z. Zhang, C. Yang, G. Shi, S. Zhao, H. Gu, J. Wu, X. Gao, Y. Li, K. Liu, S. Dai and L. Zhang, Chem, 2022, 8, 3288–3301 CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- Y. Ma, J. Yu, M. Sun, B. Chen, X. Zhou, C. Ye, Z. Guan, W. Guo, G. Wang, S. Lu, D. Xia, Y. Wang, Z. He, L. Zheng, Q. Yun, L. Wang, J. Zhou, P. Lu, J. Yin, Y. Zhao, Z. Luo, L. Zhai, L. Liao, Z. Zhu, R. Ye, Y. Chen, Y. Lu, S. Xi, B. Huang, C. S. Lee and Z. Fan, Adv. Mater., 2022, 34, 2110607 CrossRef CAS.
- P. C. Chen, C. Chen, Y. Yang, A. L. Maulana, J. Jin, J. Feijoo and P. Yang, J. Am. Chem. Soc., 2023, 145, 10116–10125 CrossRef CAS PubMed.
- X. Zhou, A. Zhang, B. Chen, S. Zhu, Y. Cui, L. Bai, J. Yu, Y. Ge, Q. Yun, L. Li, B. Huang, L. Liao, J. Fu, Q. Wa, G. Wang, Z. Huang, L. Zheng, Y. Ren, S. Li, G. Liu, L. Zhai, Z. Li, J. Liu, Y. Chen, L. Ma, C. Ling, J. Wang, Z. Fan, Y. Du, M. Shao and H. Zhang, Adv. Mater., 2023, 35, 2304414 CrossRef CAS PubMed.
- J. Zhao, P. Zhang, T. Yuan, D. Cheng, S. Zhen, H. Gao, T. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2023, 145, 6622–6627 CrossRef CAS PubMed.
- K. Qi, Y. Zhang, N. Onofrio, E. Petit, X. Cui, J. Ma, J. Fan, H. Wu, W. Wang, J. Li, J. Liu, Y. Zhang, Y. Wang, G. Jia, J. Wu, L. Lajaunie, C. Salameh and D. Voiry, Nat. Catal., 2023, 6, 319–331 CrossRef CAS.
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
- V. Okatenko, A. Loiudice, M. A. Newton, D. C. Stoian, A. Blokhina, A. N. Chen, K. Rossi and R. Buonsanti, J. Am. Chem. Soc., 2023, 145, 5370–5383 Search PubMed.
- Y. Wang, L. Cao, N. J. Libretto, X. Li, C. Li, Y. Wan, C. He, J. Lee, J. Gregg, H. Zong, D. Su, J. T. Miller, T. Mueller and C. Wang, J. Am. Chem. Soc., 2019, 141, 16635–16642 Search PubMed.
- M. Chhetri, M. Wan, Z. Jin, J. Yeager, C. Sandor, C. Rapp, H. Wang, S. Lee, C. J. Bodenschatz, M. J. Zachman, F. Che and M. Yang, Nat. Commun., 2023, 14, 3075 Search PubMed.
- Y. Cao, S. Chen, S. Bo, W. Fan, J. Li, C. Jia, Z. Zhou, Q. Liu, L. Zheng and F. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202303048 CrossRef CAS PubMed.
- T. Zheng, C. Liu, C. Guo, M. Zhang, X. Li, Q. Jiang, W. Xue, H. Li, A. Li, C. W. Pao, J. Xiao, C. Xia and J. Zeng, Nat. Nanotechnol., 2021, 16, 1386–1393 CrossRef CAS PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS.
- T. Wang, J. Chen, X. Ren, J. Zhang, J. Ding, Y. Liu, K. H. Lim, J. Wang, X. Li, H. Yang, Y. Huang, S. Kawi and B. Liu, Angew. Chem., Int. Ed., 2023, 62, e202211174 CrossRef CAS PubMed.
- L. Lv, R. Lu, J. Zhu, R. Yu, W. Zhang, E. Cui, X. Chen, Y. Dai, L. Cui, J. Li, L. Zhou, W. Chen, Z. Wang and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202303117 CrossRef CAS.
- Y. X. Duan, Y. T. Zhou, Z. Yu, D. X. Liu, Z. Wen, J. M. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8798–8802 CrossRef CAS.
- X. Tan, K. Sun, Z. Zhuang, B. Hu, Y. Zhang, Q. Liu, C. He, Z. Xu, C. Chen, H. Xiao and C. Chen, J. Am. Chem. Soc., 2023, 145, 8656–8664 CAS.
- X. Zhou, J. Shan, L. Chen, B. Y. Xia, T. Ling, J. Duan, Y. Jiao, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed.
- T.-C. Chou, C.-C. Chang, H.-L. Yu, W.-Y. Yu, C.-L. Dong, J.-J. Velasco-Vélez, C.-H. Chuang, L.-C. Chen, J.-F. Lee, J.-M. Chen and H.-L. Wu, J. Am. Chem. Soc., 2020, 142, 2857–2867 CrossRef CAS PubMed.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68–71 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS PubMed.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS.
- X. Y. Zhang, Z. X. Lou, J. Chen, Y. Liu, X. Wu, J. Y. Zhao, H. Y. Yuan, M. Zhu, S. Dai, H. F. Wang, C. Sun, P. F. Liu and H. G. Yang, Nat. Commun., 2023, 14, 7681 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- Y. Chen and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 1986–1989 CrossRef CAS.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS.
- X. Wang, Y. Jiang, K. Mao, W. Gong, D. Duan, J. Ma, Y. Zhong, J. Li, H. Liu, R. Long and Y. Xiong, J. Am. Chem. Soc., 2022, 144, 22759–22766 CrossRef CAS.
- C. Chen, S. Yu, Y. Yang, S. Louisia, I. Roh, J. Jin, S. Chen, P.-C. Chen, Y. Shan and P. Yang, Nat. Catal., 2022, 5, 878–887 CrossRef CAS.
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed.
- K. Yao, J. Li, H. Wang, R. Lu, X. Yang, M. Luo, N. Wang, Z. Wang, C. Liu, T. Jing, S. Chen, E. Cortés, S. A. Maier, S. Zhang, T. Li, Y. Yu, Y. Liu, X. Kang and H. Liang, J. Am. Chem. Soc., 2022, 144, 14005–14011 CrossRef CAS.
- J. S. Woertink, P. J. Smeets, M. H. Groothaert, M. A. Vance, B. F. Sels, R. A. Schoonheydt and E. I. Solomon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18908–18913 CrossRef CAS.
- S. Xu, S. Chansai, C. Stere, B. Inceesungvorn, A. Goguet, K. Wangkawong, S. F. R. Taylor, N. Al-Janabi, C. Hardacre, P. A. Martin and X. Fan, Nat. Catal., 2019, 2, 142–148 CrossRef CAS.
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS.
- L. Lin, X. He, X. G. Zhang, W. Ma, B. Zhang, D. Wei, S. Xie, Q. Zhang, X. Yi and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214959 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nat. Catal., 2022, 5, 818–831 CrossRef CAS.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed.
- H. Liu, B. Li, Z. Liu, Z. Liang, H. Chuai, H. Wang, S. N. Lou, Y. Su, S. Zhang and X. Ma, ACS Catal., 2023, 13, 5033–5042 CrossRef CAS.
- J. Yin, Z. Gao, F. Wei, C. Liu, J. Gong, J. Li, W. Li, L. Xiao, G. Wang, J. Lu and L. Zhuang, ACS Catal., 2022, 12, 1004–1011 CrossRef CAS.
- P. Goyal, M. J. Purdue and S. Farooq, Ind. Eng. Chem. Res., 2019, 58, 19611–19622 CrossRef CAS.
- C. Xu, G. Chen, Y. Zhao, P. Liu, X. Duan, L. Gu, G. Fu, Y. Yuan and N. Zheng, Nat. Commun., 2018, 9, 3367 CrossRef.
- J. Sun, J. Yu, Q. Ma, F. Meng, X. Wei, Y. Sun and N. Tsubaki, Sci. Adv., 2018, 4, 3275 CrossRef.
- T. Zhao, J. Li, J. Liu, F. Liu, K. Xu, M. Yu, W. Xu and F. Cheng, ACS Catal., 2023, 13, 4444–4453 CrossRef CAS.
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang, Y. Xu, D.-H. Nam, J. Wicks, B. Chen, X. Wang, M. Luo, M. Graetzel, F. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS PubMed.
- S. Chen, B. Wang, J. Zhu, L. Wang, H. Ou, Z. Zhang, X. Liang, L. Zheng, L. Zhou, Y. Q. Su, D. Wang and Y. Li, Nano Lett., 2021, 21, 7325–7331 CrossRef CAS PubMed.
- E. P. Delmo, Y. Wang, Y. Song, S. Zhu, H. Zhang, H. Xu, T. Li, J. Jang, Y. Kwon, Y. Wang and M. Shao, J. Am. Chem. Soc., 2024, 146, 1935–1945 CrossRef CAS.
- H. Ren, M. Kovalev, Z. Weng, M. Z. Muhamad, H. Ma, Y. Sheng, L. Sun, J. Wang, S. Rihm, W. Yang, A. A. Lapkin and J. W. Ager, Nat. Catal., 2022, 5, 1169–1179 CrossRef CAS.
- S. N. Steinmann and Z. W. Seh, Nat. Rev. Mater., 2021, 6, 289–291 CrossRef CAS.
Footnote |
| † Xin Tan and Haojie Zhu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |