
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTetryliumylidene ions in synthesis and catalysis
Sebastian
Stigler
 ,
Shiori
Fujimori
,
Arseni
Kostenko
and
Shigeyoshi
Inoue
*
,
Shiori
Fujimori
,
Arseni
Kostenko
and
Shigeyoshi
Inoue
*
TUM School of Natural Sciences, Department of Chemistry, WACKER-Institute of Silicon Chemistry and Catalysis Research Center, Technical University of Munich, Lichtenbergstraße 4, 85748 Garching bei München, Germany. E-mail: s.inoue@tum.de
First published on 20th January 2024
Abstract
Tetryliumylidene ions ([R–E:]+), recognised for their intriguing electronic properties, have attracted considerable interest. These positively charged species, with two vacant p-orbitals and a lone pair at the E(II) centre (E = Si, Ge, Sn, Pb), can be viewed as the combination of tetrylenes (R2E:) and tetrylium ions ([R3E]+), which makes them potent Lewis ambiphiles. Such electronic features highlight the potential of tetryliumylidenes for single-site small molecule activation and transition metal-free catalysis. The effective utilisation of the electrophilicity and nucleophilicity of tetryliumylidenes is expected to stem from appropriate ligand choice. For most of the isolated tetryliumylidenes, electron donor- and/or kinetic stabilisation is necessary. This minireview highlights the developments in tetryliumylidene syntheses and the progress of research towards their reactivity and applications in catalytic reactions.
 Sebastian Stigler | Sebastian Stigler received his MSc degree in chemistry in 2021 from the Technical University of Munich under the supervision of Prof. Shigeyoshi Inoue on the application of NHC-stabilised silyliumylidene ions in hydroboration and hydrosilylation catalysis. Since then he has continued his stay in the group of Prof. Shigeyoshi Inoue at the Technical University of Munich as a PhD student. His current research interests focus on the reactivity of silyliumylidene ions and their utilisation in catalytic reactions. |
 Shiori Fujimori | Shiori Fujimori obtained her MSc and PhD degrees in chemistry in 2019 at Kyoto University under the supervision of Prof. Norihiro Tokitoh on the synthesis of novel aromatic compounds containing heavier group 14 elements. In October 2019, she was awarded a Eurotech-Marie Curie Fellowship and joined the group of Prof. Inoue at the Technical University of Munich. She continued her stay at the TUM as an Alexander von Humboldt Fellow where she pursued her interest in low-valent silicon chemistry. |
 Arseni Kostenko | Arseni Kostenko obtained his bachelor's degree in Molecular Biochemistry at the Technion Israel Institute of Technology, subsequently pursuing a Master's degree and a PhD in the field of silicon chemistry under the supervision of Prof. Yitzhak Apeloig. In 2016, he received the Alexander von Humboldt fellowship and joined the group of Prof. Matthias Driess at the Technical University of Berlin. Since 2021, Dr Kostenko has been working in the group of Prof. Shigeyoshi Inoue at the Technical University of Munich, computationally studying the structure and reactivity of low-valent main group element-based compounds. |
 Shigeyoshi Inoue | Shigeyoshi Inoue obtained his PhD from the University of Tsukuba in 2008 supervised by Prof. Akira Sekiguchi. Following postdoctoral study at the TU Berlin with Prof. Matthias Driess, he established an independent research group within the framework of the Sofja Kovalevskaja program in 2010. Since 2015 he has been on the faculty at the Technical University of Munich (TUM). His research interests include the synthesis and reactivity of main-group compounds with unusual structures and unique electronic properties, with the goal of finding novel applications in synthesis, catalysis and materials science. |
Introduction
Recent developments in the chemistry of low-valent/oxidation state main-group compounds have revealed their inherent electronic properties. These allow them to mimic the behaviour of transition-metal complexes, with regard to their ability to form coordination complexes, engage in multiple bonding, undergo redox reactions, and exhibit catalytic activity.1–5 An important class of low-valent main group species capable of such behaviour are tetrylenes [R2E:] (E = Si, Ge, Sn, Pb) – the heavier analogues of carbenes (R2C:). Numerous representatives of these species are able to activate small molecules under mild conditions and can be used as ligands in catalytic organic reactions due to their ambiphilic nature.6–12 The heavier congeners of carbonium ions, namely tetrylium cations [R3E]+ (E = Si, Ge, Sn, Pb), have also garnered significant interest in recent decades attributed to their pronounced electrophilic nature, leading to various applications in catalysis and bond activation chemistry.13–16 A related and less explored class of compounds are the tetryliumylidene ions (E = Si, Ge, Sn, Pb), which exhibit distinctive electronic properties, merging the strong electrophilicity observed in tetrylium cations with the Lewis ambiphilic character found in tetrylenes. In their idealised form, these mono-substituted cations have two vacant degenerate p orbitals and a lone pair at the E(II) centre (Fig. 1).17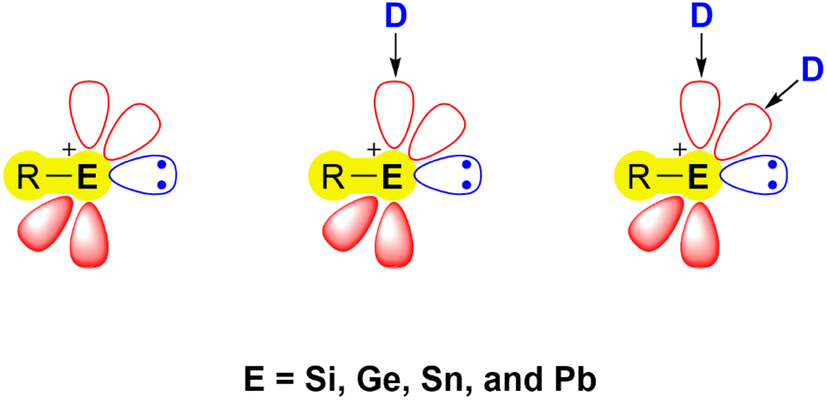 | ||
| Fig. 1 Frontier orbitals of free (left) and donor-stabilised tetryliumylidene ions (centre, right) (D = Lewis base). | ||
The bonding situation in parent tetryliumylidenes can be examined from the point of view of the valent ns and np orbitals and their proclivity to hybridise. The inert s-pair effect, which reflects the situation where the diminishing hybridisation between ns and np orbitals makes only np orbitals available for bonding, has a great influence on the electronic structure of p-block element species. Consideration of the relative roles of s and p orbitals can be done using Weinhold's natural bond orbital (NBO) scheme,18 through which natural atomic orbital (NAO) hybridisations of natural localised molecular orbitals (NLMOs) can be obtained.19 It is known that tetrylenes exhibit higher tendency to the inert pair effect as we go down the period – the heavier elements tend to retain a low oxidation state, and forming [R2E:] species becomes easier – thus, lead, for example, prefers PbIIR2 species over PbIVR4. Calculations (at the B3LYP20–23/def2-TZVP24 level of theory using Gaussian 16 (ref. 25) and NBO7 (ref. 26) software) show that the inert s-pair effect is significantly more pronounced in tetryliumylidenes in comparison with tetrylenes, even for lighter elements. In [H2E:] series, there is a high p character of the lone pair in the parent carbene (0.68), and a significant drop in the p/s ratio in the silylene (0.30) (Fig. 2, top). The ratio gradually decreases when going down the period, and is 0.13 in [H2Pb:]. In the case of [H–E:]+, even the methylidyne ion lone pair has a s/p ratio (0.13) similar to that of [H2Pb:]. The decrease is less significant in [H–Si:]+, and only a slight change is observed when going down the period.
 | ||
| Fig. 2 Lone pair (top) and E–H bond (bottom) NAO/NLMO p/s hybridisation ratios in tetrylenes and tetryliumylidenes at the B3LYP/def2-TZVP level of theory. | ||
An analogous situation is observed when comparing the E–H bonds in tetrylenes and tetryliumylidenes (Fig. 2, bottom). The C–H bond in [H2C:] has a low p/s ratio of 3.67 which increases to 13.61 in plumbylene. In contrast, in the tetryliumylidene series, the methylidyne ion already has a high p/s ratio of 7.68, which is comparable with that of [H2Ge:]. Overall, when going down the period the p composition of the hybrid orbital increases from 87.8% in [H–C:]+ to 95.4% in [H–Pb:]+. These results indicate that while hybridisation is increasingly more difficult in the case of tetrylenes when going down the periodic table, in tetryliumylidenes the effect is less dramatic, since sp hybridisation is difficult to begin with.
[R–E:]+ have the potential for versatile synthetic applicability due to their unique electronic structure with multiple reactive sites, originating from the two electrophilic vacant p-orbitals and one nucleophilic lone pair. Such arrangement should in principle allow the formation of up to three new bonds in a single reaction.27 Thus, tetryliumylidene ions are expected to be highly reactive, and in general, electron donor stabilisation by inter- or intramolecular coordination of Lewis bases is required to isolate these species as stable compounds.28
In the pioneering studies, Jutzi and co-workers reported tetryliumylidene ions [Cp*E:]+ (E = Ge, Sn, Pb; Cp* = η5-C5Me5) stabilised by the hyper-coordinating cyclopentadienyl group.29,30 Following these discoveries, researchers have successfully isolated a range of tetryliumylidene ions.31 This was made possible by employing sterically demanding ligands for kinetic stabilisation, including bulky amide groups,32 β-diketiminato,33 aminotroponiminate,34 and cyclophane groups.35 Alternatively, Lewis basic ligands, such as carbenes,36,37 imines,38 or weakly coordinating arenes,39 have been utilised for the enhancement of the thermodynamic stability. Several decades after the discovery of Jutzi's heavier tetryliumylidene ions [Cp*E:]+ (E = Ge, Sn, Pb), in 2004, the same group succeeded in the synthesis of the first isolable silyliumylidene ion, the pentamethylcyclopentadienyl-silicon(II) cation [Cp*Si:][B(C6F5)4].40 Since these seminal discoveries, the reactivity of tetryliumylidene ions has been studied to some extent. It has been demonstrated that these species can be used as synthons for the isolation of novel low-valent compounds containing a heavier group 14 element. For example, a new class of fascinating main-group compounds, tetrylones, which are two-coordinate E0 species (L:→E←:L; E = Si,41 Ge42), were synthesised from the reduction of the corresponding chloro-tetryliumylidenes. In addition, the ambiphilic character of tetryliumylidene ions, which mimics the frontier d-orbitals found in transition metals, enables them to activate small molecules (e.g., CO2, N2O, H2S, H2O, and heavier chalcogens).43–47 Furthermore, they have been successfully applied in several catalytic transformations without transition metals.48–51 Various sterically bulky electron donor ligands have been developed in the last few decades, allowing the preparation of donor-stabilised hydro-tetryliumylidenes [H–E:]+ with superior potency for widespread applications. For instance, hydrosilylation and hydroboration reactions were catalysed by [H–E:]+.52–54 In this minireview, we have summarised the recent developments in the chemistry of tetryliumylidene ions, including their reactivity towards small molecules, as precursors to a variety of additional low-valent species and catalytic applications. While some tetryliumylidene ions were isolated and employed in catalysis by taking advantage of cooperative effects between transition metals and group 14 elements,55–66 this review highlights only the main-group examples.
Silyliumylidenes
The first synthesis of a silyliumylidene ions in 2004 by Jutzi et al. was achieved by the reaction of decamethylsilicocene, (Cp*)2Si, with the proton transfer reagent [Cp*H2][B(C6F5)4] to form [Cp*Si:][B(C6F5)4] 1a – the first isolable silyliumyidene ion.40 In a similar fashion, in 2018, the group of Filippou showed the successful synthesis of 1a by protonation of (Cp*)2Si with one equivalent of [H(Et2O)2][B(C6F5)4].57 Another procedure to form this compound was reported more recently, in 2019, by Fritz-Langhals with the hydride abstraction of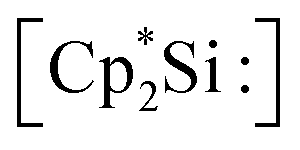 by a tritylium salt, resulting in tetramethylfulvene and 1a on a technical scale.48 Jutzi et al. also reported the formation of penta-iso-propylcyclopentadienylsilicon(II) cation iPr5C5Si+1b by reacting the mixed silicocene (iPr5C5)(Me5C5)Si with H(OEt2)2+Al(OtBuF)4− (Fig. 3).67
by a tritylium salt, resulting in tetramethylfulvene and 1a on a technical scale.48 Jutzi et al. also reported the formation of penta-iso-propylcyclopentadienylsilicon(II) cation iPr5C5Si+1b by reacting the mixed silicocene (iPr5C5)(Me5C5)Si with H(OEt2)2+Al(OtBuF)4− (Fig. 3).67
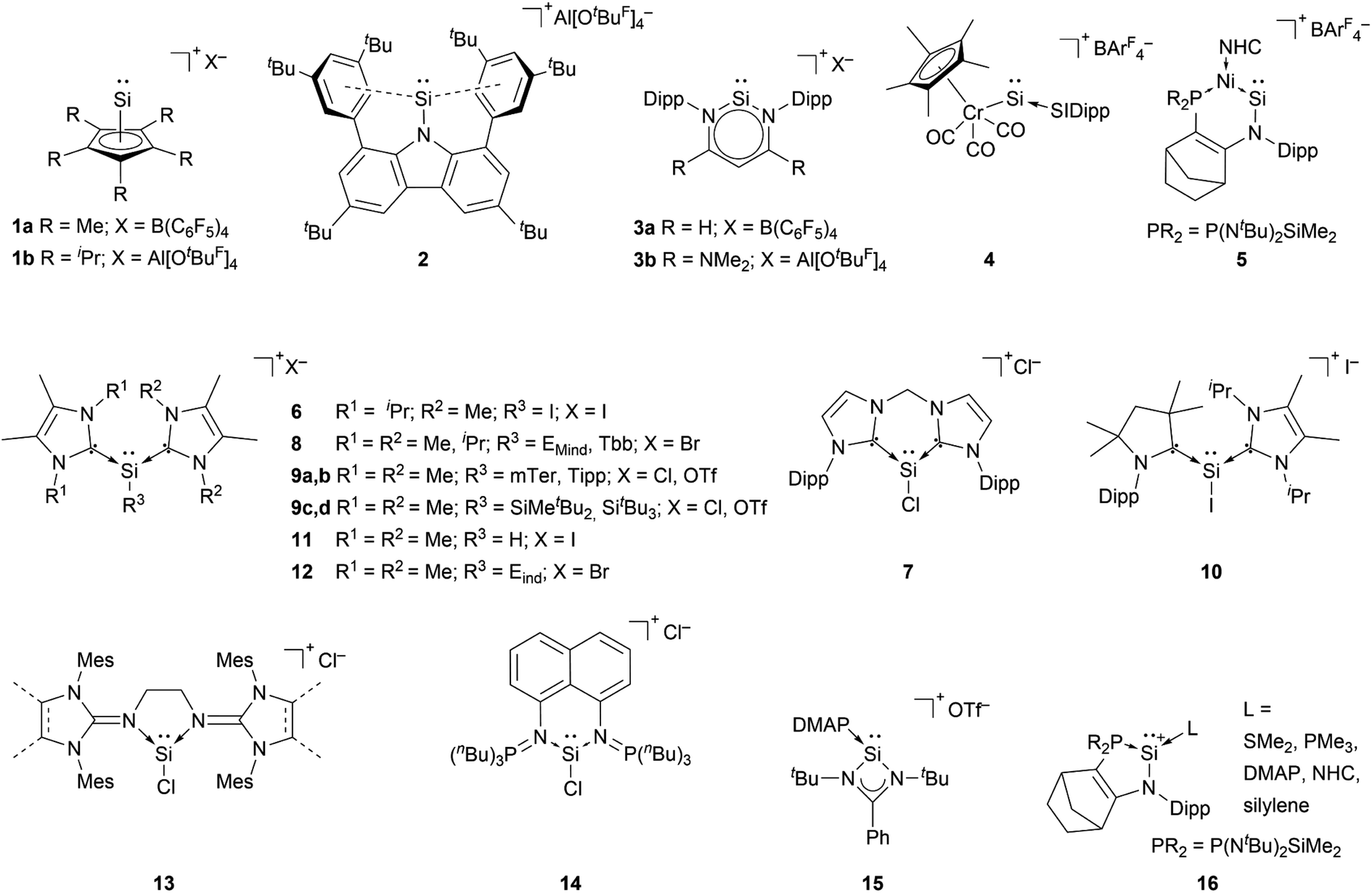 | ||
| Fig. 3 Reported silyliumylidene ions. (OtBuF = OC(CF)3; Dipp = 2,6-iPr2C6H3; SIDipp = 1,3-bis(Dipp)imidazolidin-2-ylidene; BAr4F = B[C6H3-3,5-(CF3)2]4; NHC = 1,3-diisopropyl-4,5-dimethyl-imidazol-2-ylidene; EMind = 1,1,7,7-tetraethyl-3,3,5,5-tetramethyl-s-hydrindacen-4-yl; Tbb = ((5-(tert-butyl)-1,3-phenylene)bis(methanetriyl))tetrakis(trimethylsilane); mTer = 2,6-Mes2C6H3; Mes = 2,4,6-Me3C6H2; Tipp = 2,4,6-iPr3C6H2; Eind = 1,1,3,3,5,5,7,7-octaethyl-s-hydrindacen-4-yl; DMAP = 4-(N,N-dimethylamino)pyridine). | ||
Cyclopentadienyls and their derivatives are 2σ, 4π electron donor ligands and are routinely used for stabilisation of electron-deficient species. Their proclivity to afford hyper-coordinated centres allowed, in this case, for stabilisation of highly reactive silyliumylidene ions. Due to this, 1a and 1b cannot be considered true mono-substituted silyliumylidene ion complexes. It took almost two decades after the isolation of 1a, when Hinz reported the first achievements towards the mono-substituted silicon(II) cation [RSi:]+ (R = bulky carbazolyl substituent) 2 by halide abstraction from a base-free halosilylene with Ag[Al(OtBuF)4]. Even though there are arene interactions between the Si atom and the carbazolyl scaffold, this silyliumylidene bears no other σ-donors except for the carbazolyl substituent.68
The first two-coordinated silyliumylidene ion 3a was reported by Driess et al. in 2006. Here, the authors protonated the sterically demanding β-diketiminate ligand backbone of the corresponding zwitterionic N-heterocyclic silylene with Jutzi's oxonium acid [H(Et2O)2][B(C6F5)4].69 More recently, the group of Aldridge reported the silyliumylidene ion 3b stabilised by a β-diketiminate ligand featuring a backbone with NMe2 groups.70 In 2014, Filippou et al. reacted a chromium silylidyne complex salt, containing the first Cr–Si triple bond, with CO to achieve the four-legged piano–stool complex cation 4.43 Very recently, in 2022, Kato et al. showed the formation of the Ni(0)-stabilised Si(II) species 5. The dative Ni → Si σ-interaction and π-donations from the amino- and Ni-moieties stabilise the highly electrophilic Si centre.71
Most of the known silyliumylidene ions are three-coordinated, stabilised kinetically, via sterically demanding ligands, and electronically, via donation to the silicon centre by Lewis bases. A majority of reported silyliumylidene ions use stabilisation by N-heterocyclic carbenes (NHCs). The first example of this class of compounds was reported by the group of Filippou in 2013.72 Here, the NHC stabilised silicon(II) diiodide was reacted with a sterically more demanding N-heterocyclic carbene to get to the silyliumylidene ion 6. At essentially the same time, Driess et al. isolated the chloro-silyliumylidene ion 7 stabilised by a cyclic bis-NHC.41 In 2014, Tokitoh et al. and our group independently used NHCs to synthesise aryl-substituted silyliumylidene ions. The group of Tokitoh treated dibromodisilenes with an excess of NHCs forming the bis-NHC adduct [ArSi(NHC)2][Br] 8.73 Meanwhile, our group utilised dichloroarylsilanes and three equivalents of NHCs in a facile one-pot reaction to obtain the corresponding silyliumylidene ions 9a,b.74 In 2016, the group of So showed the formation of the silyliumylidene iodide 10, stabilised by both an NHC and a cyclic alkyl(amino) carbene (cAAC), by reacting the corresponding cAAC stabilised silicon(II) diiodide with NHC.75 One year later, the same group isolated the first NHC-stabilised parent-silyliumylidene ion 11 by reacting an NHC–iodosilicon(I) dimer with four equivalents of NHC.76 Matsuo et al. reported a similar approach to Tokitoh in 2018. Here, they used Eind-substituted 1,2-dibromodisilene to form with four equivalents of NHCs the corresponding bis-NHC adduct of the formal aryl-silyliumylidene ion 12.77 In 2019, utilising the already reported approach, our group could show the formation of the silyl-substituted NHC-stabilised silyliumylidene ions 9c,d.78 Additionally, very recently we reported the synthesis of a neutral bidentate NHI ligand (NHI = N-heterocyclic imine) with a saturated imidazoline backbone and the formation of the corresponding chloro-silyliumylidene ion 13.79,80
Apart from NHCs, the group of Driess utilised a bis(iminophosphorane) chelate ligand to form the chloro-silyliumylidene 14. The two N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PnBu3 ylide moieties can act as very strong Brønsted and Lewis bases.81 In 2013, So et al. isolated the silyliumylidene ion 15 stabilised by an amidinate ligand and 4-dimethylaminopyridine (DMAP).82 More recently, Kato et al. reported in 2022 the use of different Lewis bases, such as SMe2, PMe3, and DMAP, and even observed silylene-stabilised silyliumylidene complexes 16.83
PnBu3 ylide moieties can act as very strong Brønsted and Lewis bases.81 In 2013, So et al. isolated the silyliumylidene ion 15 stabilised by an amidinate ligand and 4-dimethylaminopyridine (DMAP).82 More recently, Kato et al. reported in 2022 the use of different Lewis bases, such as SMe2, PMe3, and DMAP, and even observed silylene-stabilised silyliumylidene complexes 16.83
Reactivity of silyliumylidenes and their use in catalytic reactions
Among tetryliumylidenes, silyliumylidene ions show the widest diversity in reactivity reported so far. The first stable silyliumylidene 1 reacts with the metalate Na[TpMeMo(CO)2(PMe3)] to afford the silylidyne complex [TpMe(CO)2MoSi(η3-Cp*)] (TpMe = κ3-N,N′,N′′-hydridotris(3,5-dimethyl-1-pyrazolyl)borate) featuring a delocalised Mo–Si bond with a partial triple bond character.57 Despite the high steric demand of the carbazolyl moiety, pseudo-mono-coordinated silyliumylidene ion 2 retains high reactivity and reacts with an amine to form three bonds at the silicon atom in one reaction 17 – justifying being called a “supersilylene” (Scheme 1).27,68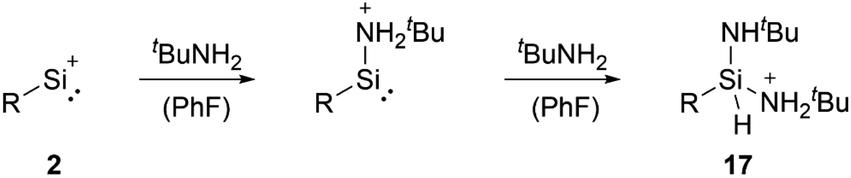 | ||
| Scheme 1 Reaction of 2 with two equivalents of amines to form 17 (R = bulky carbazolyl ligand). | ||
β-Diketiminate stabilised silyliumylidene 3b can react with NH2tBu via formal N–H oxidative addition to form a Si(H)(NHtBu) moiety.70 When two-coordinated silyliumylidene ion 4 is exposed to an N2O atmosphere, rapid formation of metallosilanone occurs.435 shows multiple examples of activation of small molecules, such as MeOTf 18, DMAP 19, H220, diphenylacetylene 21, and 2,3-dimethyl-1,3-butadiene 22 (Scheme 2).71
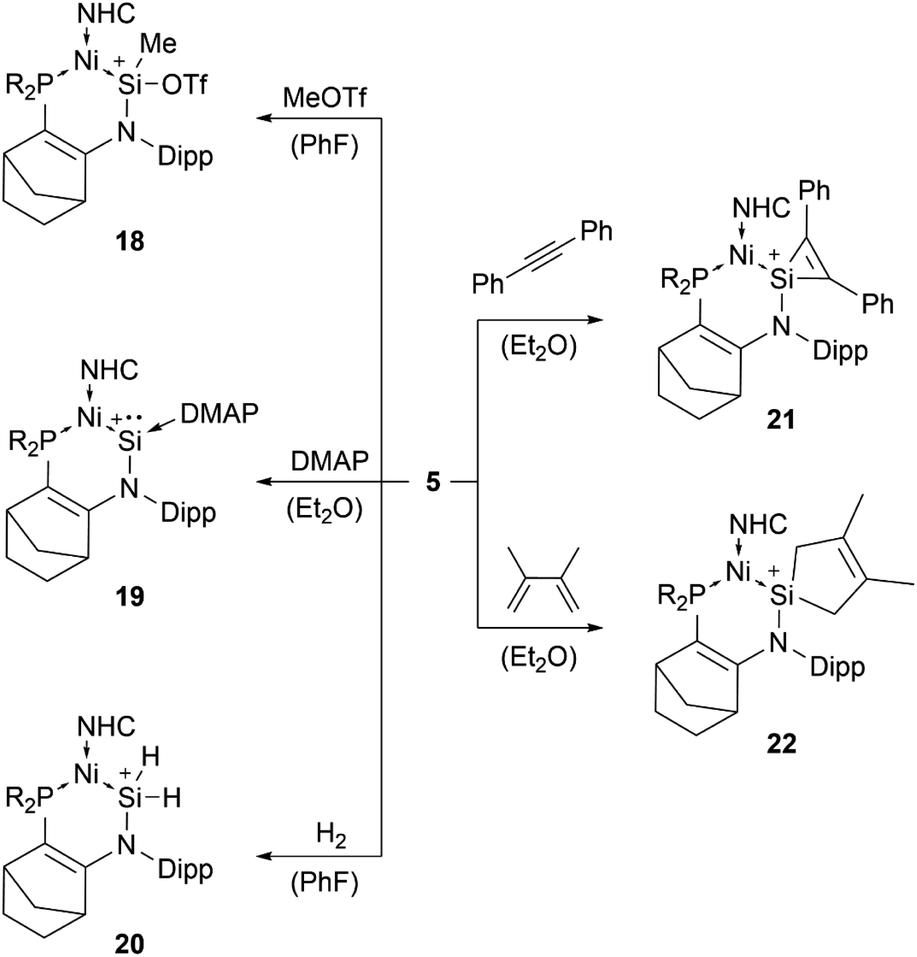 | ||
| Scheme 2 Multiple examples for small molecule activations of 5 (R2P = P(NtBu)2SiMe2, NHC = 1,3-diisopropyl-4,5-dimethyl-imidazol-2-ylidene). | ||
Silyliumylidenes could be used as precursors for the formation of additional exotic low-valent species. Thus, dehalogenation of chloro-silyliumylidene 7 yielded the cyclic bis-NHC stabilised silylone 23 (Scheme 3),41 and mixed NHC–cAAC stabilised silyliumylidene 10 could be reduced with KC8 to form the bent silaallene 33 (Scheme 4).75
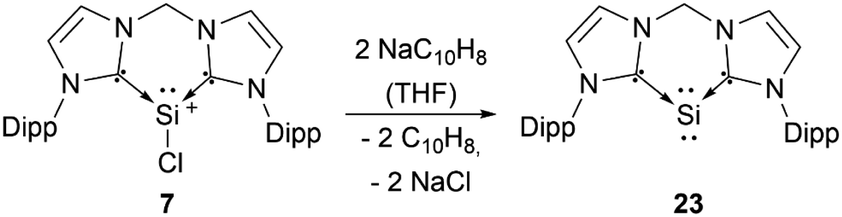 | ||
| Scheme 3 Reductive dechlorination of 7. | ||
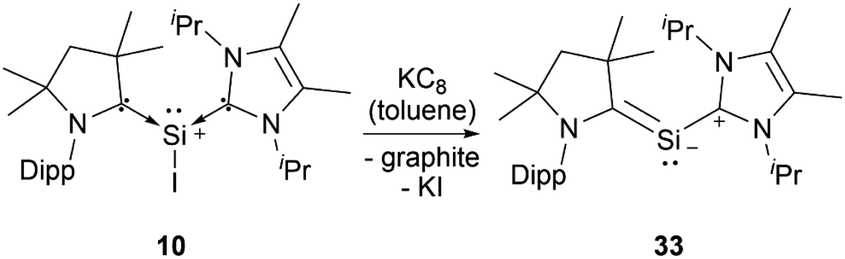 | ||
| Scheme 4 Reductive dehalogenation of 10. | ||
Aryl- and silyl-substituted silyliumylidene ions 9 showed a plethora of reactivities with small molecules, such as the C–H insertion reaction with three equivalents of phenylacetylene to form 1-alkenyl-1,1-dialkynylsilane 24.749 can activate CO2 to form the silaacylium ion 25,44 as well as the NHC-stabilised heavier silaacylium ions 26 with sulphur, selenium, and tellurium via chalcogen-atom transfer.45 Moreover, 9 can activate the S–H bond of hydrogen sulfide to form the thiosilaaldehyde 27,46 react with GaCl3 and water to afford the silaaldehyde 28,47 and convert to the Si(IV) complex 29 by reaction with triflic acid (Scheme 5).84 In some of these reactions, one NHC acts as a H–Cl scavenger, which is a substantial thermodynamic driving force for the reaction progress. Furthermore, compound 9 derivatives are capable of forming transition metal complexes. They can either react with coinage metals, such as copper, silver, and gold, to form the metal chloride adducts 30,85 or transition metal carbonyls (M = Cr, Wo, W, Fe) to form the M(CO)n (n = 4 or 5) silyliumylidene adducts 31.86 Additionally, 9 can undergo a facile insertion into M–Cl bonds (M = Ru, Rh), forming the chlorosilylene transition-metal complexes 32 (Scheme 5).87
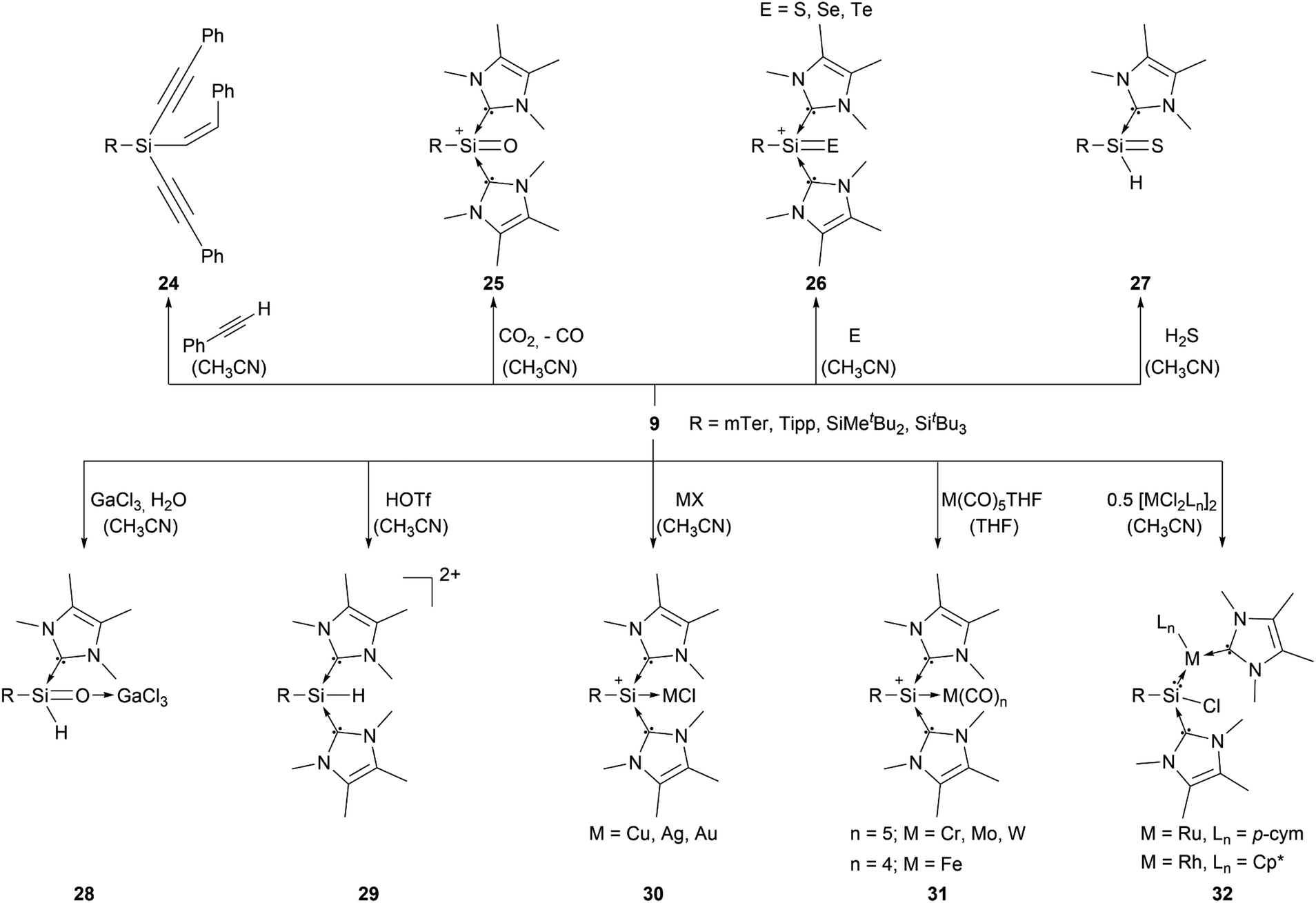 | ||
| Scheme 5 Reported reactivity of 9 (X = Cl, OTf; p-cym = 1-Me-4-iPr-benzene). | ||
When reacted with elemental sulphur, the bis(iminophosphorane) coordinated chloro-silyliumylidene 14 is oxidised to the chlorosilathionium salt 33 (Scheme 6).81 This reactivity is similar to the reactivity of 9 with chalcogens.
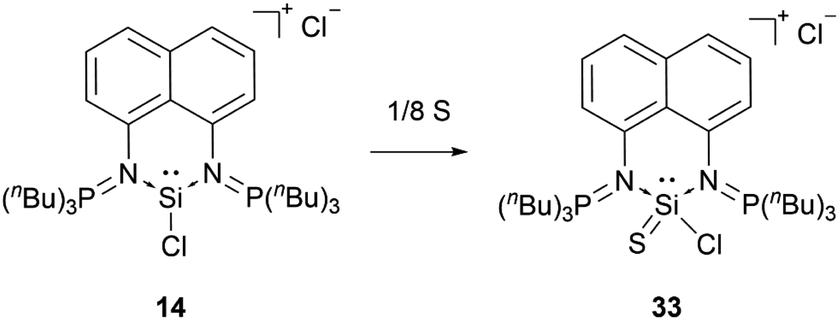 | ||
| Scheme 6 Oxidation of 14 with elemental sulphur. | ||
Bis-NHI stabilised silyliumylidene 13 shows the formation of the heavier silaacylium ions (E = S, Se, Te) 34 in reaction with chalcogens, coinage metal complexes 35 with CuCl, AgCl, and (Me2S)AuCl, and the heterobimetallic gold–iron complex 36 in reaction with a gold–chloride complex in the presence of tetracarbonylferrate (Scheme 7).79
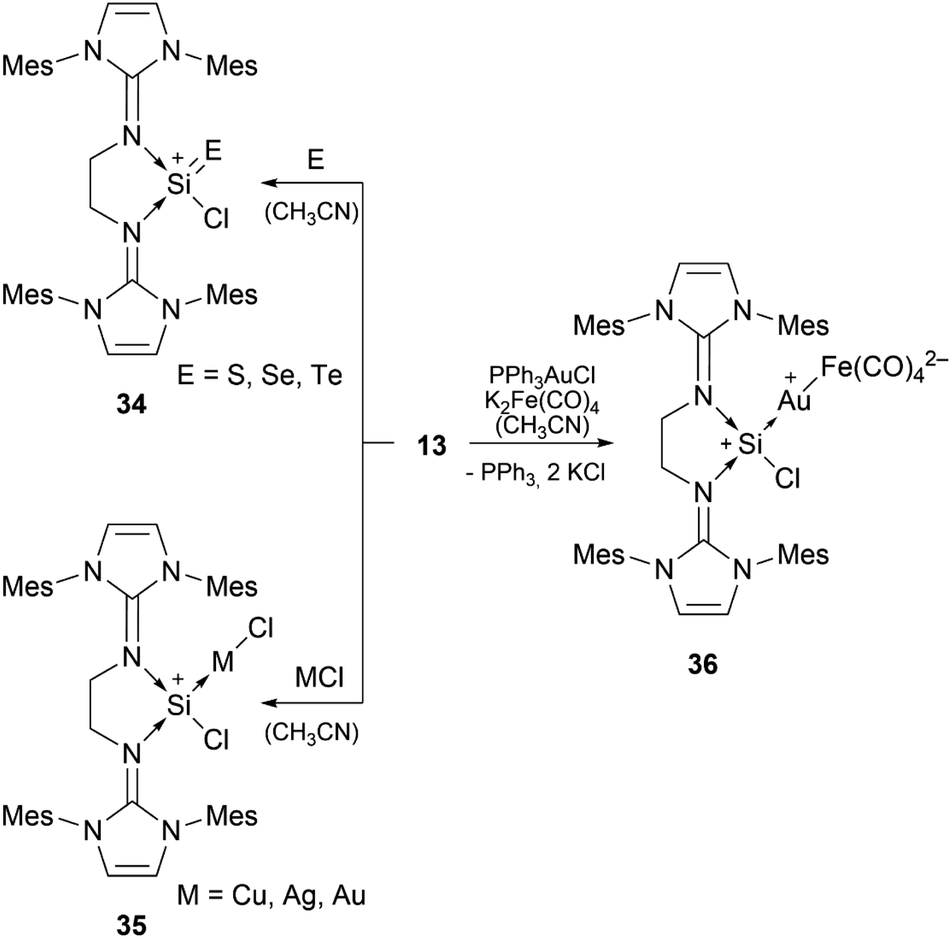 | ||
| Scheme 7 Reported reactivity of 13. | ||
Amidinate-substituted silyliumylidene 15 can undergo oxidative addition at the silicon centre with the amidinate silicon(I) dimer to form the disilylenylsilylium triflate 37, react with nucleophilic reagents, such as K[HB(iBu)3], to afford the corresponding silylsilylene 38, and form the silanethionium triflate 39 with elemental sulphur (Scheme 8).82
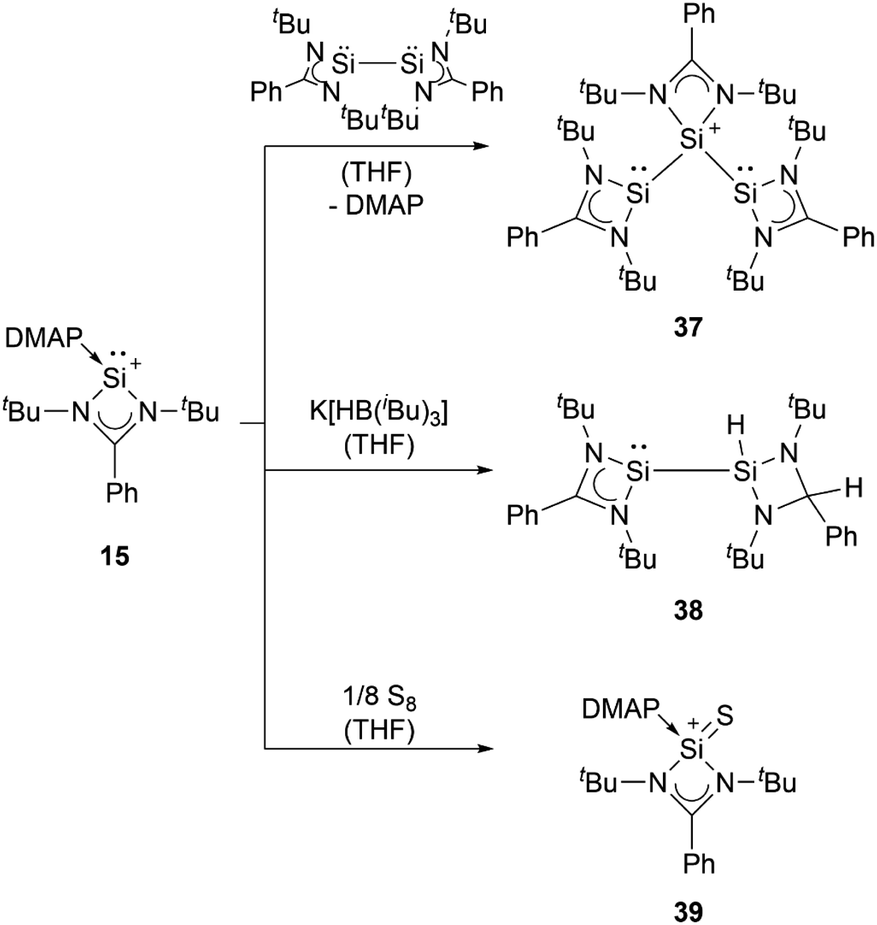 | ||
| Scheme 8 Reported reactivity of 15. | ||
Lewis base stabilised silyliumylidene 16 is able to undergo the [1+2]-cycloaddition with ethylene at the silicon centre to form the silacyclopropyl cation 40, [4+1]-cycloaddition with 2,3-dimethyl-1,3-butadiene (41), [2+1]-cycloaddition with diphenylacetylene (42), oxidation of the Si(II) centre with CO2 to generate a cationic silanone 43, and oxidative addition of triethylsilane, diphenylphosphine, or pinacolborane to give the corresponding silylium ion 44 (Scheme 9).83,88
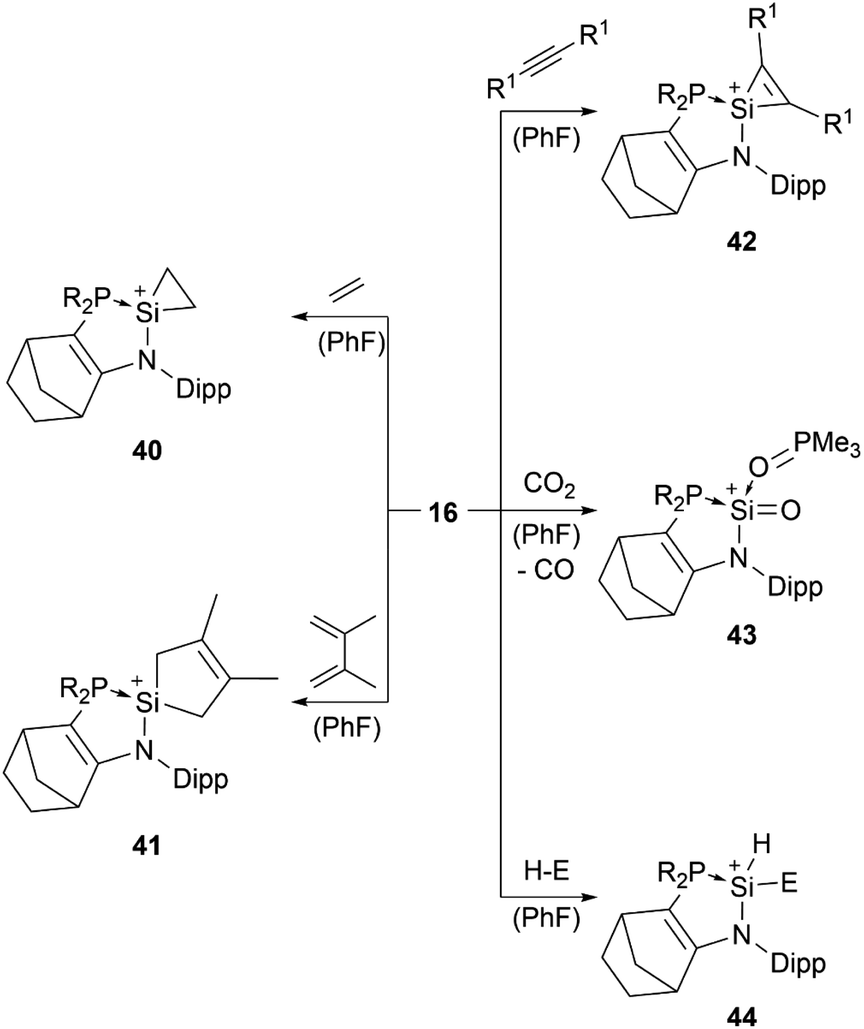 | ||
| Scheme 9 Reported reactivity of 16 (R1 = Ph, SiMe3, Mes; H–E = HSiEt3, HPPh2, HBpin; R2P = P(NtBu)2SiMe2). | ||
While there are a multitude of examples of silyliumylidene ion reactivities, to date there are only several examples of their use as catalysts. The first isolable silyliumylidene 1 proved to be an efficient non-metallic catalyst for the hydrosilylation of unsaturated carbon–carbon bonds at low catalyst loadings of <0.01 mol% and the Piers–Rubinsztajn reaction.48 In the proposed mechanism for the hydrosilylation, the silyliumylidene first activates the silicon hydrogen bond to form the hydrogen-bridged complex 45. The substrate can insert into the silicon–hydrogen bond of the silane to form 46. Subsequent elimination of the hydrosilylated product regenerates the catalyst (Scheme 10).
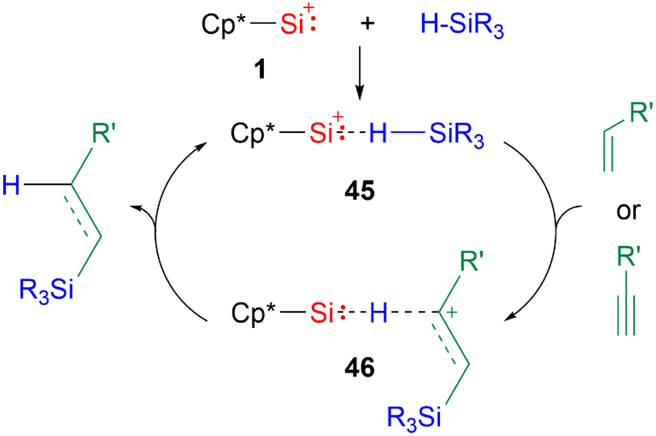 | ||
| Scheme 10 Proposed mechanism of the hydrosilylation of carbon–carbon multiple bonds (R3 = Me2OTMS, Me(OTMS)2, Et3, Me2Ph, Me2Cl, MeCl2; R′ = nC4H9, SiMe3, Ph, cyclo-hexene, norbornene). | ||
The NHC-stabilised parent-silyliumylidene ion [NHC2SiH]+11 shows unprecedented abilities in the catalytic hydroboration of CO2, aldehydes, ketones, and pyridines with a catalyst loading of 10 mol%, as well as isocyanates with a catalyst loading of 1 mol% (Scheme 11).52,54
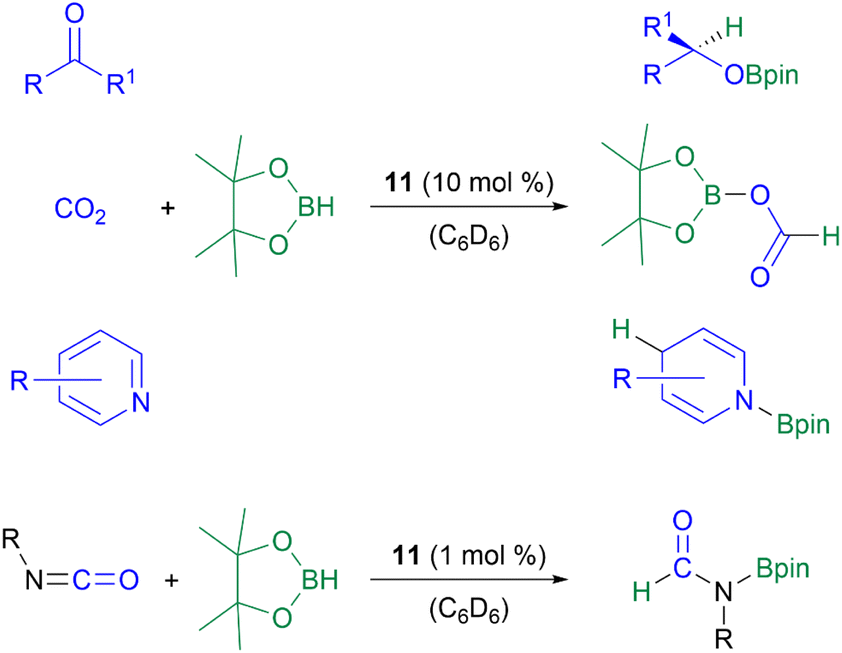 | ||
| Scheme 11 Hydroboration of aldehydes, ketones, CO2, pyridines, and isocyanates with pinacolborane. | ||
Furthermore, 11 catalyses the chemoselective N-formylation of amines using CO2 and phenylsilane with a catalyst loading of 5 mol%. The reaction mechanism was studied using density-functional theory (DFT) calculations. The silyliumylidene sequentially activates first CO2, then simultaneously phenylsilane and amines via transition state 47. Following a dihydrogen elimination mechanism, formamides, siloxanes, dihydrogen gas, and the regenerated catalyst are formed (Scheme 12).53
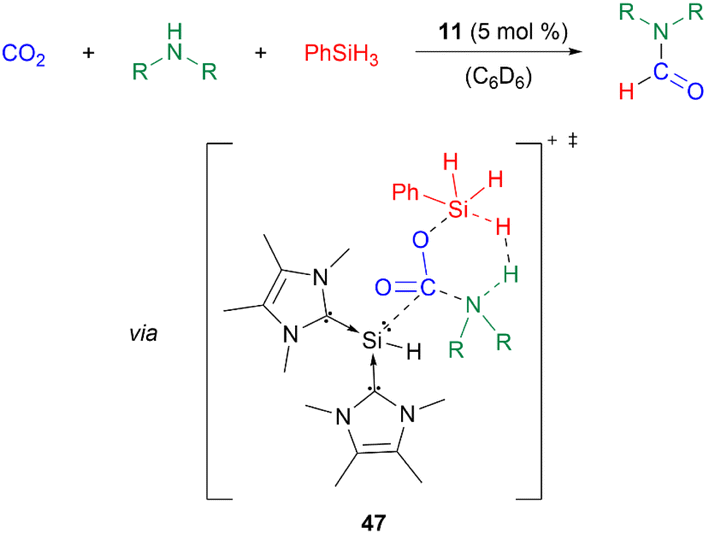 | ||
| Scheme 12 N-Formylation of amines using CO2 and phenylsilane (HNR2 = primary and secondary amines). | ||
Germyliumylidenes
The richest chemistry among tetryliumylidenes, in terms of the number of different reported examples, is that of germyliumylidenes. Over two decades before the silicon analogue, the cyclopentadienyl moiety was utilised by Jutzi's group to achieve the synthesis of the first isolable germyliumylidene ion 48 by reaction of decamethylgermanocene (Cp*)2Ge with HBF4.29 Following this outstanding work, numerous germyliumylidenes have been reported. There are several synthetic strategies for the isolation of Ge(II)-derived monocations, most of them utilising neutral ancillary ligands (Fig. 4).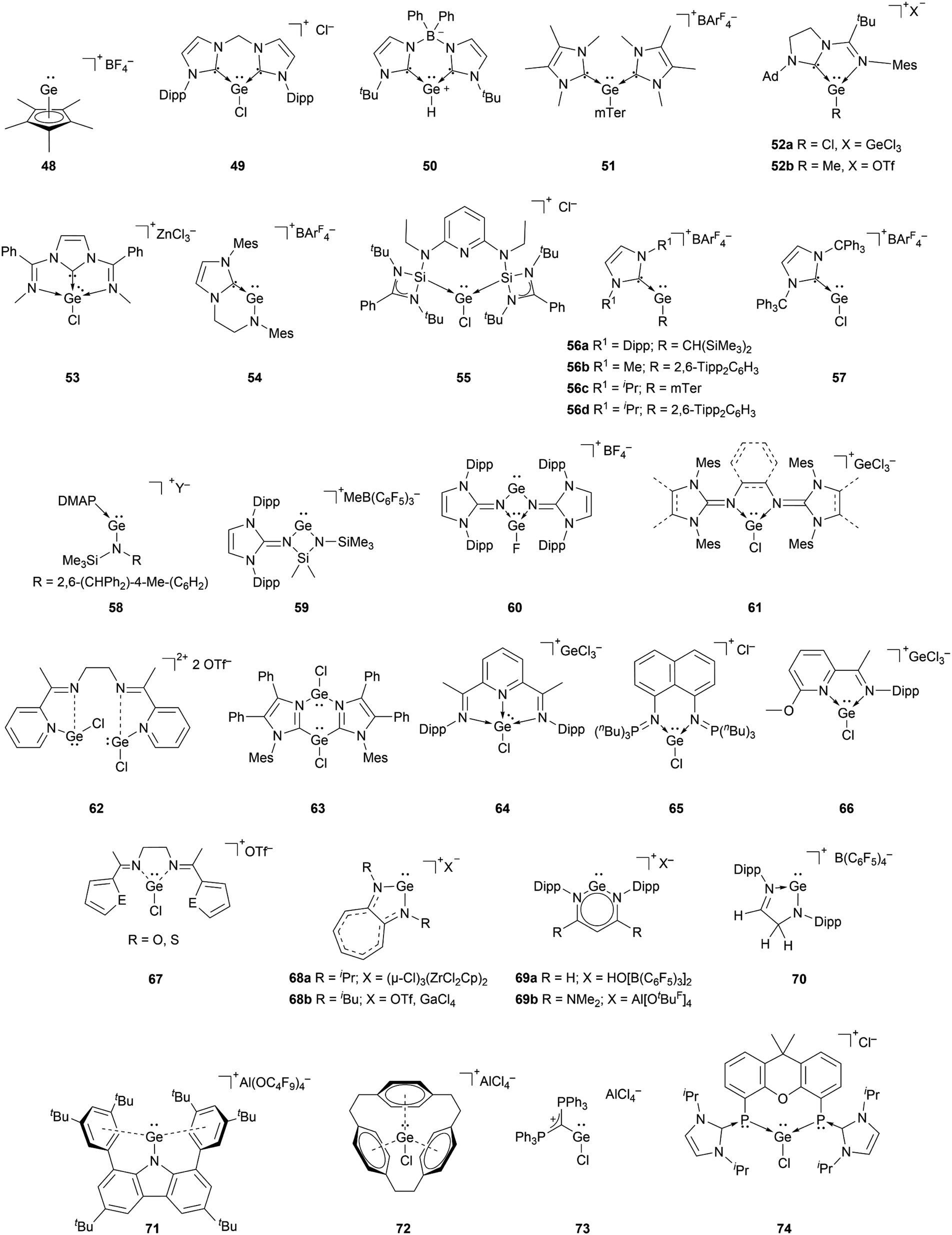 | ||
| Fig. 4 Reported germyliumylidene ions. | ||
For example, N-heterocyclic carbenes (NHCs) have been used extensively to stabilise germyliumylidenes. Driess et al. reported the formation of bis-NHC stabilised chloro-germyliumylidene 49 by reacting GeCl2·dioxane with the corresponding ligand.42 They also isolated parent-germyliumylidene hydride 50 stabilised by a bis(NHC)borate ligand.37 By the addition of two equivalents of NHC to an aryl chloro-germylene, our group could isolate the corresponding three-coordinate germanium cation 51.89
Utilising an imino-N-heterocyclic carbene ligand, Kinjo et al. obtained chloro- and methyl-substituted germyliumylidene 52a,b, respectively.90,91 More recently, a diimino-carbene was utilised by Nikonov et al. to stabilise the germyliumylidene 53.92 The groups of Glorius and Hahn reported the first intramolecularly NHC-stabilised germyliumylidene 54.93 Utilisation of a bis(N-heterocyclic silylenyl)pyridine pincer ligand led to the formation of chloro-germyliumylidene 55 reported by Driess et al.94
In 2016, it was demonstrated that a single NHC with adequate steric bulk together with a sterically more demanding substituent is also sufficient for germyliumylidene stabilisation. Thus, Rit, Aldridge, et al. isolated the two-coordinate germyliumylidene 56a bearing a CH(SiMe3)2 substituent, stabilised by a Dipp-substituted NHC (Dipp = 2,6-diisopropylphenyl).95 More recently, the same group reported NHC-stabilised germyliumylidenes 56b–d with smaller NHCs, but larger substituents at the Ge centre.96 Rivard et al. utilised the steric protection provided by the extremely bulky trityl (CPh3)–NHC to prepare the chloro-germyliumylidene 57.97 Using a single Lewis base, Krossing, Jones, and co-workers reported the bulky amido germanium(II) monocation 58 stabilised by either weak intramolecular arene interactions or DMAP.32
NHIs can also be used for germyliumylidene stabilisation. By utilising Dipp–NHIs, we could isolate the four-membered amino(imino)germyliumylidene 59 and the germylene-germyliumylidene 60 borate salts, respectively.98–100 Computational analysis indicated the latter to have a considerable bis(germyliumylidene) character. In 2020, we used bidentate bis(N-heterocyclic imine) ligands to stabilise the three-coordinate chloro-germyliumylidenes 61.80,101
A similar approach to [R–Ge:]+ stabilisation is the use of imino ligands. Recently, Majumdar et al. reported bis(chlorogermyliumylidene) stabilised within a flexible tetra-dentate 2,7-bis(2-pyridyl)-3,6-diazaocta-2,6-diene ligand 62 (ref. 102) and within bifunctional PNNP ligand frameworks.103 In 2018, Kinjo et al. reported the bis(imidazolyl) supported chloro-germyliumylidene 63 in which the Ge2N2C2 six-membered ring possesses two Ge–Cl units.104 In 2012, the groups of Roesky and Stalke used the substituted Schiff base 2,6-diacetylpyridinebis(2,6-diisopropylanil) as Lewis base, which mediated the autoionisation of GeCl2 to the germyliumylidene 64.38 The group of Driess isolated the bis(iminophosphorane) chelate stabilised chloro-germyliumylidene 65 similar to the structurally equivalent silyliumylidene 3.105 In 2013, Jambor et al. showed the synthesis of the chloro-germyliumylidene 66 by treating a neutral 2-[C(CH3)N(C6H3-2,6-iPr2)]-6-(CH3O)C6H3N ligand with GeCl2, which spontaneously dissociates.106 Majumdar and co-workers reported acyclic flexible diiminodi(furan) and diiminodi(thiophene) ligands in which the two imino nitrogen coordinating sites can stabilise the chloro-germyliumylidene ion 67.107
Another type of germyliumylidene stabilisation is the use of monoanionic ligands that form cyclic compounds, such as the aminotroponiminate derivatives 68a,b utilised by Dias et al. in 1997,34 and, more recently, by the group of Nagendran in 2019.49 Power et al. used a Dipp-substituted β-diketiminate ligand to form the two-coordinated germyliumylidene 69a.33 In 2020, Aldridge et al. utilised a β-diketiminate ligand bearing two NMe2 groups to form the germyliumylidene 69b.70 The structurally similar Ge(II) cation 70 was isolated by Müller and co-workers by protonating the corresponding germylene.108 Similar to silyliumylidene 2, stabilised by arene coordination, Hinz showed the formation of the carbazolyl-substituted pseudo-one-coordinate germyliumylidene 71 by halide abstraction.109
Schmidbaur reported a related approach, for which he reacted cyclophane with one equivalent of GeCl2 to form the [2.2.2]-paracyclophane chloro-germyliumylidene complex 72.35
Two additional examples of methods to stabilise germyliumylidenes were provided by the groups of Alcarazo and Driess.110,111 Alcarazo et al. isolated the two-coordinated chloro-germyliumylidene 73 stabilised by σ- and π-donation from a monodentate carbodiphosphorane;110 while Hadlington, Driess et al. reported the bisphosphinidine stabilised chloro-germyliumylidene 74.111
Reactivity of germyliumylidenes and their use in catalytic reactions
Several examples of the reactivity of germyliumylidene ions have been reported, while only a few catalytic applications are known. Similarly to the silicon analogue, the chloro-germyliumylidene 49 can be dehalogenated (by sodium naphthalenide) to form the cyclic bis(NHC) Ge(0) complex 75 (Scheme 13).42 The reduction of 52a with KC8 leads to the formation of the corresponding germylone 76 (Scheme 13), similar to germyliumylidene 64.90,92 Germyliumylidene 51 reacts with N2O to form the germa-acylium ion 77 (Scheme 14).112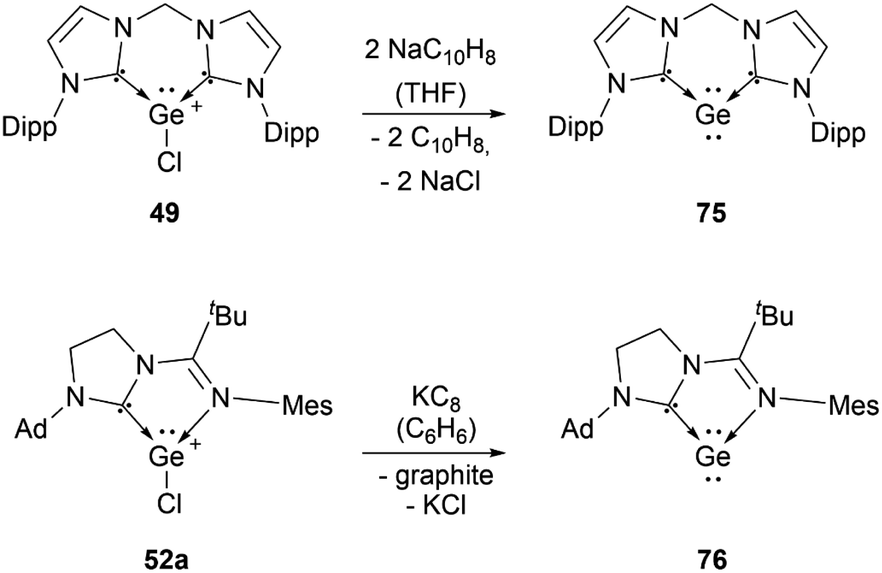 | ||
| Scheme 13 Dechlorination of 49 and 52a to form the corresponding germylone. | ||
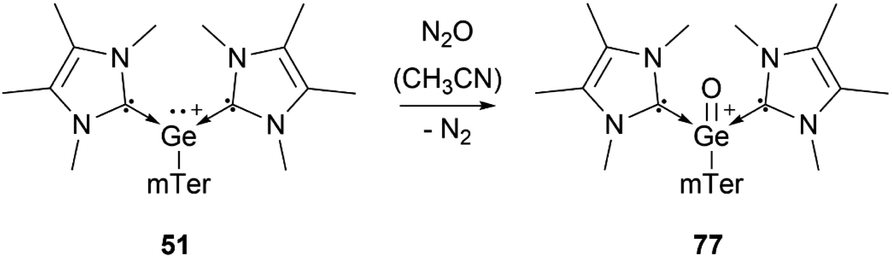 | ||
| Scheme 14 Formation of germa-acylium ion 77. | ||
By reacting it with K2[Fe(CO)4], germyliumylidene 55 forms the germylone–iron carbonyl complex 78 stabilised by a bis(NHSi)pyridine pincer ligand (Scheme 15).94
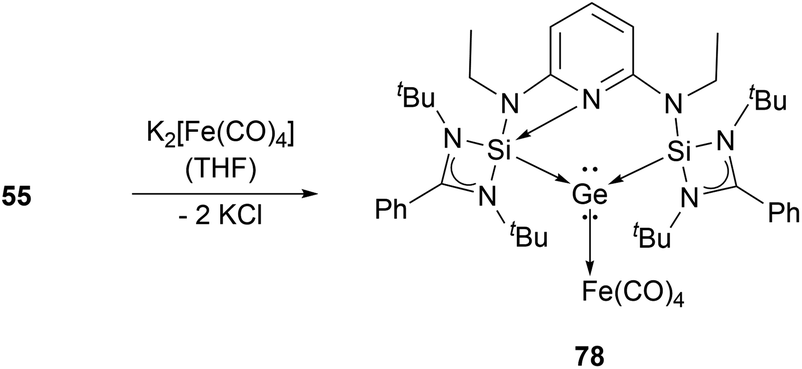 | ||
| Scheme 15 Reaction of 55 with K2[Fe(CO)4]. | ||
56a can undergo oxidative addition of dichloromethane (79), [2+1] cycloaddition with phenylacetylene (80), and formation of an imido complex (81) by reacting with Me3SiN3 (Scheme 16).95
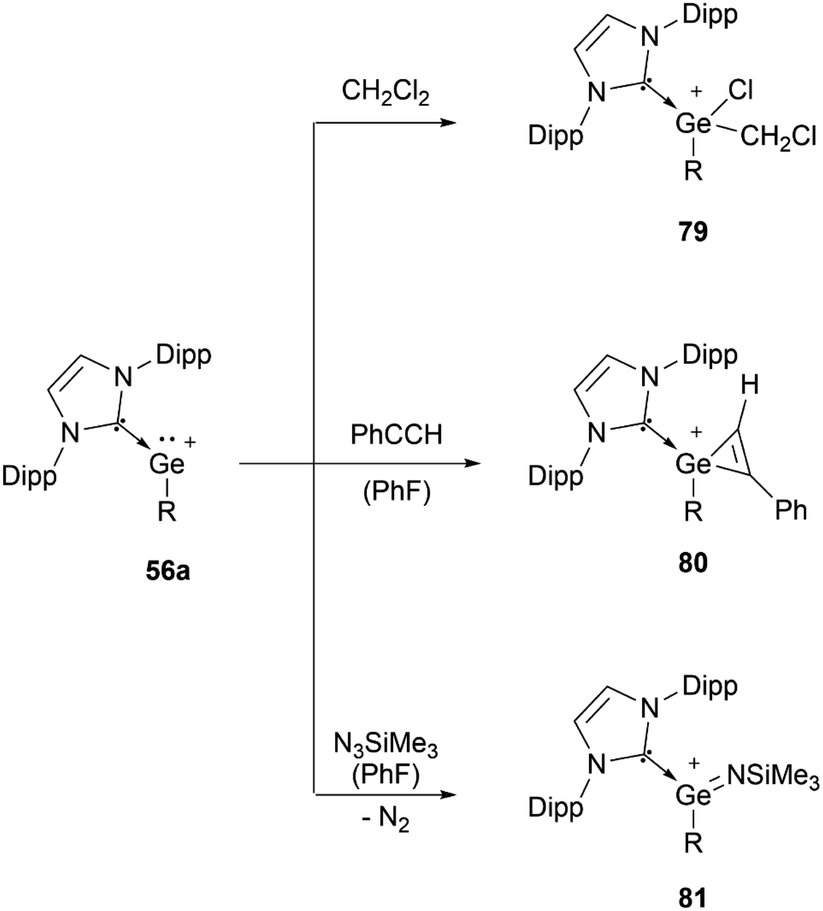 | ||
| Scheme 16 Reported reactivity of 56a. | ||
56b can insert into the Si–H bond of phenylsilane.96 Germylene-germyliumylidene 60 reacts with Me3SiOTf to form a bis(triflate).100 Bis(NHI)-stabilised chloro-germyliumylidene 61 can react with NaBH4 to the hydridogermyliumylidene–BH3 adduct 82 and with LiAlH4 to the corresponding aluminium dihydride 83 (Scheme 17).101
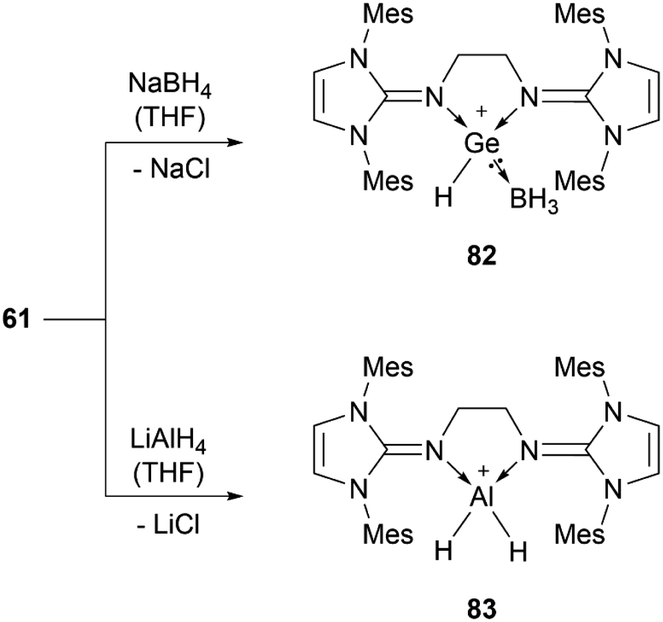 | ||
| Scheme 17 Reactions of 61 with NaBH4 and LiAlH4. | ||
By using organosilicon reductants bis(chlorogermyliumylidene) 62 can undergo reductive cyclisation to form the 2,3-di(pyridin-2-yl)-substituted piperazine 84 with high diastereoselectivity (Scheme 18).102
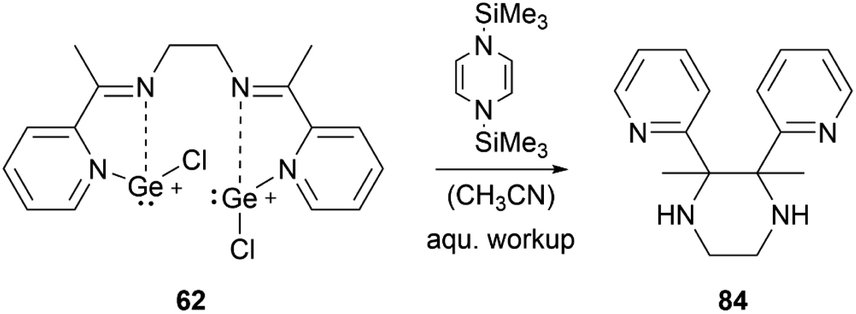 | ||
| Scheme 18 Reductive cyclisation of 62. | ||
65 can undergo oxidation with elemental sulphur to form a chloro-germathionium salt.105
In contrast to its Si analogue, germyliumylidene 69b does not undergo oxidative addition but reacts with NH2tBu to form the simple adduct.70 Two-coordinated chloro-germyliumylidene 73 can form a DMAP adduct and reacts with elemental sulphur to form a dimeric SGe double-bond species.110
In terms of catalysis, germyliumylidenes have only rarely been applied in catalytic reactions. As already mentioned, 51 can form the germa-acylium ion 77, which is used as a precatalyst for the germanium-catalysed N-functionalisation of amines with CO2.11251 can catalyse the reduction of CO2 with amines and phenylsilane, the cyanosilylation of carbonyls with TMSCN, and the hydroboration of aldehydes with pinacolborane (HBpin) (Scheme 19).50
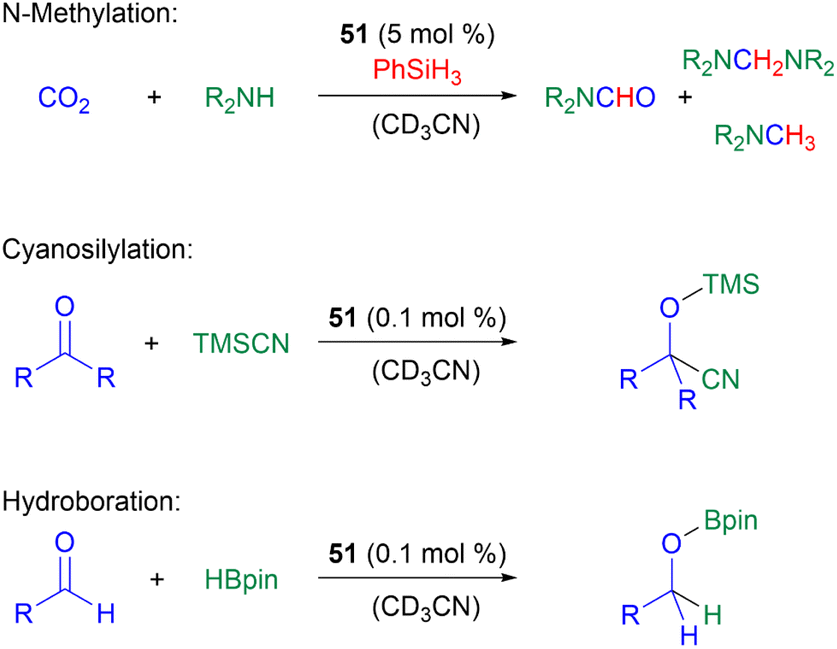 | ||
| Scheme 19 Multiple reactions catalysed by germyliumylidene 51. | ||
The proposed mechanism of the hydroboration catalysed by 51 shows the initial substitution of IMe4 (IMe4 = 1,3,4,5-tetramethyl-1H-imidazole-3-ium-2-ide) by an aldehyde to form 86via transition state 85. The subsequent B–H bond activation in HBpin is mediated by the previously released IMe4. The formed adduct transfers the hydride to the carbonyl carbon in 86, while the boron coordinates to the oxygen centre to form intermediate 88. Subsequently, the IMe4 re-associates with the Ge centre to regenerate the catalyst and obtain the hydroboration product (Scheme 20).
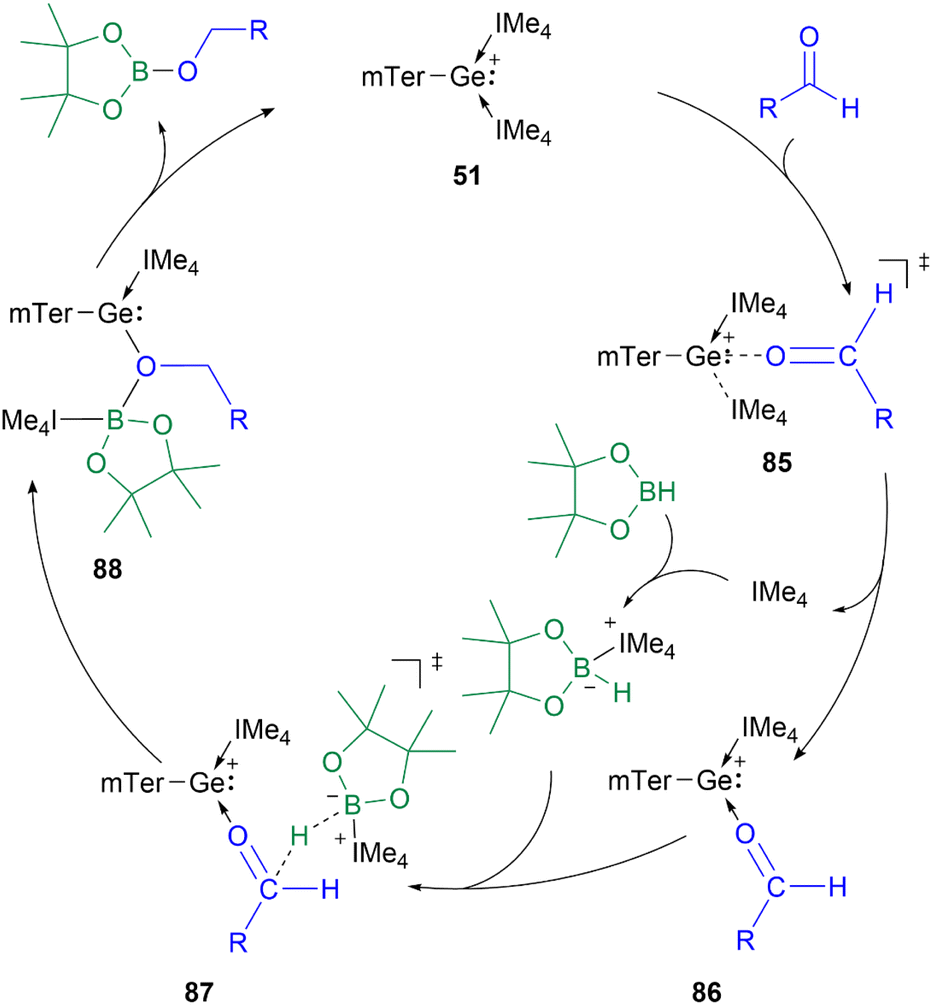 | ||
| Scheme 20 Proposed mechanism for the hydroboration of aldehydes catalysed by 51. | ||
Germyliumylidene 68b can act as a precatalyst in the catalytic hydroboration of aldehydes and ketones with pinacolborane.49 In the proposed mechanism, 68b reacts with HBpin to form the hydridogermylene 89 as the active catalyst. Subsequent reaction with the substrate via a four-membered heterocyclic transition state leads to formation of the germylene alkoxide intermediate 90. Through σ-bond metathesis with HBpin the catalyst is regenerated and the hydroboration product is formed (Scheme 21).
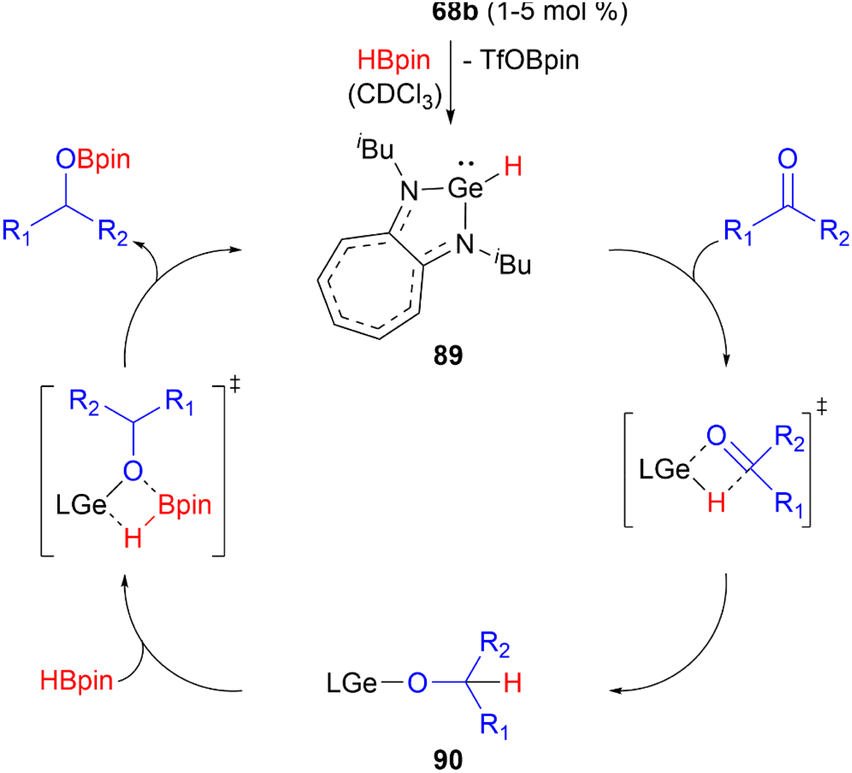 | ||
| Scheme 21 Proposed mechanism of the hydroboration of carbonyls catalysed by 68b (L = aminotroponiminate ligand). | ||
Stannyliumylidenes
The chemistry of stannyliumylidenes [R–Sn:]+ was commenced with the first isolation of [Cp*Sn:]+[BF4]−91, reported by Jutzi and co-workers in 1980.2991, like the other first examples of tetryliumylidenes was stabilised by the hyper-coordinating pentamethylcyclopentadienyl. Since then, several stannyliumylidenes have been isolated by sterically demanding ligands (kinetic stabilisation) and/or electronically stabilising ligands based on heteroatom substituents. Halogen-substituted stannyliumylidenes [X–Sn:]+ (X = halogen) are crucial intermediates since they have been employed as synthons for the isolation of novel low-valent tin compounds such as stannyliumylidene derivatives [R′–Sn:]+ and Sn(0) through substitution reactions and halide abstraction, respectively.Unlike germyliumylidene, where several examples of stabilisation using carbenes were reported, in the case of tin, there is only one reported mixed NHC–cAAC-substituted species 92.113
Our group could isolate the N-heterocyclic imine (NHI) stabilised stannyliumylidenes 93,11494,101 and 95.115 Similar to germanium, neutral chelating ligands with nitrogen atoms have attracted attention for stannyliumylidene stabilisation due to the tunability of the steric and/or electronic nature by changing the substituents on the nitrogen atoms. Thus, iminopyridine 96,106 diiminopyridine (DIMPY) 97,38 and bis(α-iminopyridine) 98 (ref. 116) have been utilised.
Coordination by bisphosphinidene 99,111 mixed phosphine-imino 100117 and ferrocene-bridged N-heterocyclic carbene-phosphinidene (NHCP) 101 (ref. 118) ligands have been reported.
Stannyliumylidene ions stabilised by monoanionic ligands such as aminotroponiminate 102,119 β-diketiminate 103a,b,70,120 extremely hindered amide 104,32 bis(oxazoline) 105,121 organo-phophane oxide 106,122 and bulky carbazolyl 107 (ref. 109) were isolated. Schmidbaur et al. utilised a [2.2.2]–paracyclophane ligand to stabilise stannyliumylidene 108 (Fig. 5).35
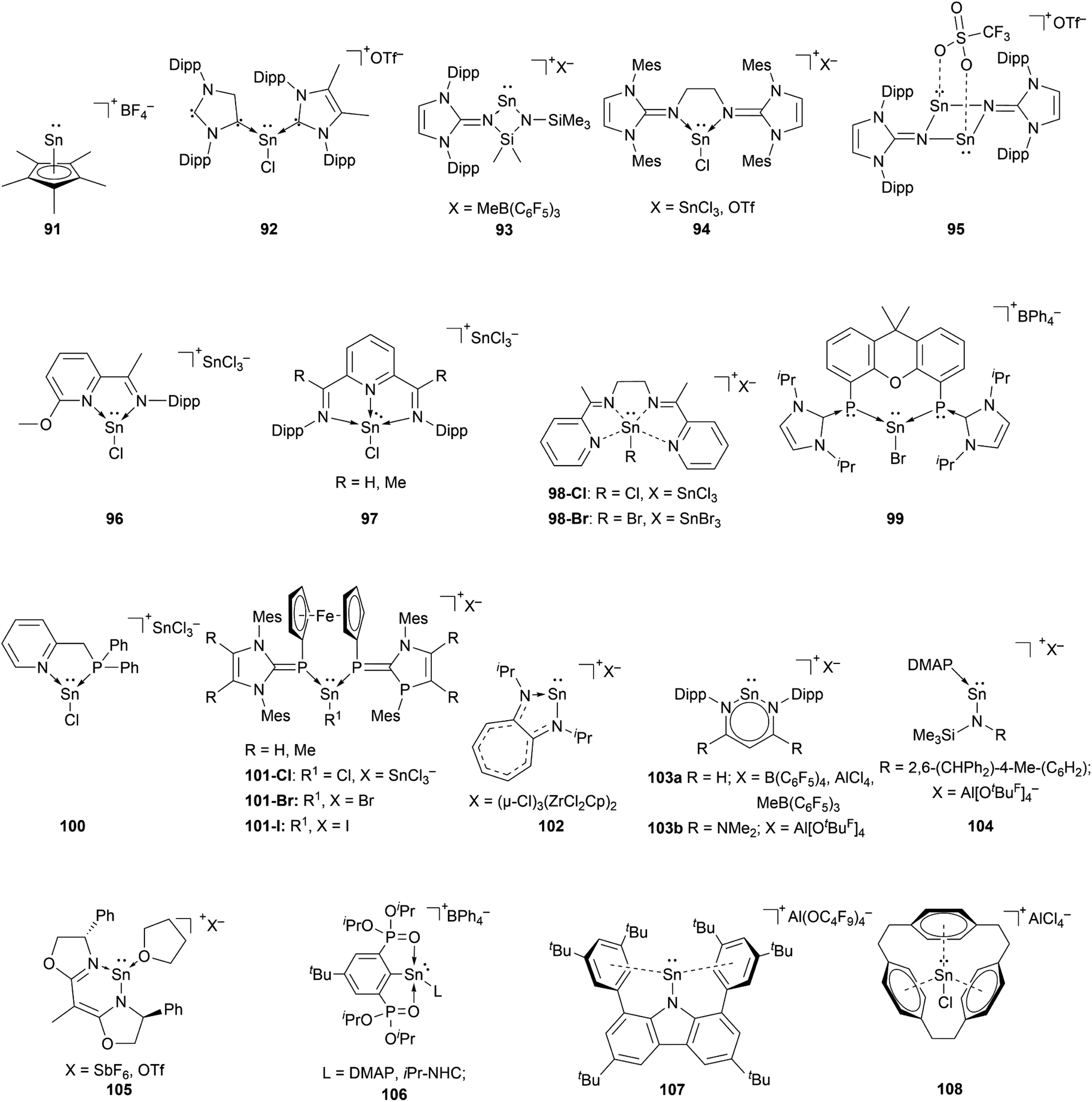 | ||
| Fig. 5 Reported stannyliumylidenes. | ||
Reactivity of stannyliumylidenes
The reactivity of stannyliumylidenes is scarcely studied, and only a few examples of their clearly identified transformations have been reported so far. The β-diketiminate stabilised stannyliumylidene 103b forms, like its germanium congener, with NH2tBu the simple adduct.70 The organo-phosphane oxide substituted stannyliumylidene 106 reacts with elemental sulphur to form a heavier acylium ion intermediate, which upon head-to-tail dimerisation yields the corresponding dication 109 containing a four-membered Sn–S–Sn–S ring (Scheme 22).122 This type of reactivity would be expected from heavy ketone derivatives. The NHI-substituted stannyliumylidene has been demonstrated to undergo transmetalation reactions. Thus, in the reaction of 94 with NaBH4 in a mixture of THF and 1,2-difluorobenzene (DFB), selective transformation to form the bisNHI-stabilised dihydroboronium 110 was observed. The precipitation of elemental tin accompanied this process. Analogously, treatment of 94 with LiAlH4 resulted in the formation of the aluminium dihydride 111. In a similar fashion, transmetalation of 94 with an equivalent of GeCl2·(dioxane) in acetonitrile at room temperature furnished the germyliumylidene cation 61 (Scheme 23).101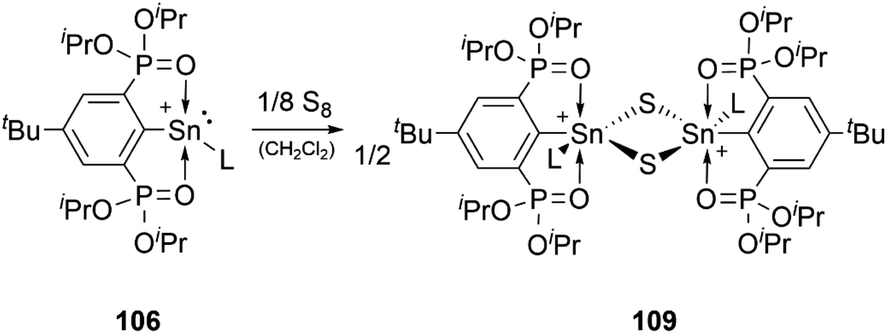 | ||
| Scheme 22 Reaction of stannyliumylidene 106 with S8 (L = DMAP). | ||
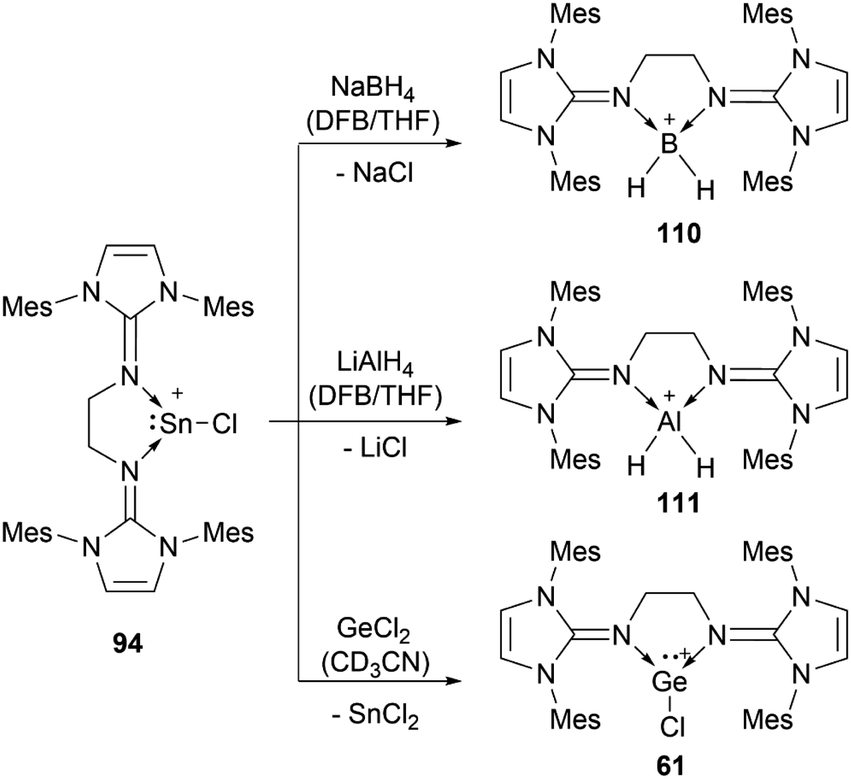 | ||
| Scheme 23 Transmetalation reactions of stannyliumylidene 94 (DFB = 1,2-difluorobenzene). | ||
Like the NHI-substituted counterpart, NHCP-stabilised stannyliumylidene 101 could also be transmetalated. 101-I reacts with CuCl to provide the transmetalation product 112 (Scheme 24).118 In addition, chloro-stannyliumylidene 101-Cl was utilised to transfer the Sn(II) synthon from the bisphosphinidene to a bisimine. Treatment of the bisNHCP-supported 101-Cl with bisNHI in THF-d8 gave a bisNHI-stabilised 94 and free bisNHCP (Scheme 25).118
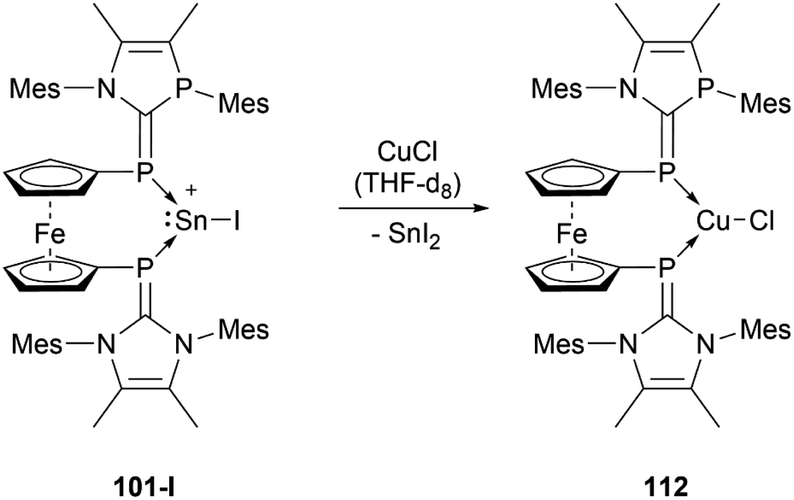 | ||
| Scheme 24 Transmetalation reaction of stannyliumylidene 101-I. | ||
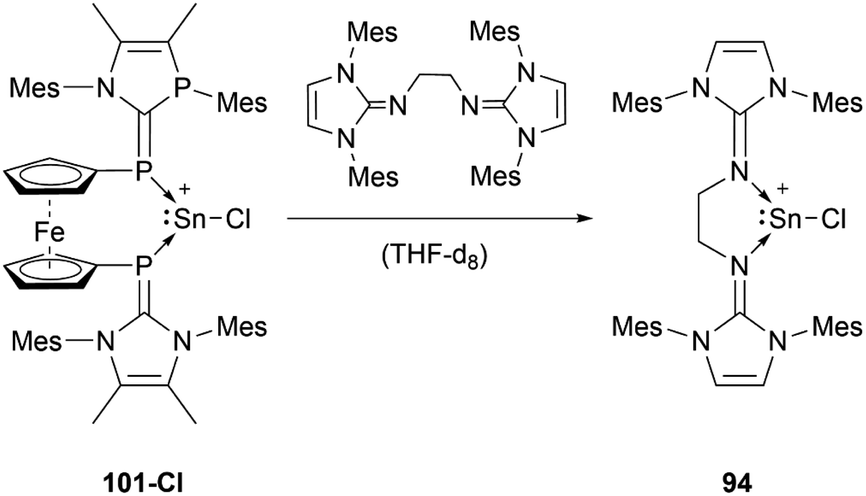 | ||
| Scheme 25 Sn(II) transfer reaction between bisNHCP and bisNHI. | ||
To date, to the best of our knowledge, except for the dinuclear tin complex 95 that was found to catalyze the hydroboration of aldehydes and ketones, no other examples of catalytic transformations that utilize stannyliumylidenes have been reported.115
Plumbyliumylidenes and catalytic reactions
Compared to lighter congeners, the chemistry of the heaviest plumbyliumylidenes [R–Pb:]+ remains underdeveloped. This can be attributed to the bad reputation of lead due to its toxicity. Like the lighter congeners, the first example of isolable plumbyliumylidene emerged from Jutzi's group. Accordingly, in 1989, Jutzi and Nöth reported 113, a plumbyliumylidene stabilised by a pentamethylcyclopentadienyl ligand (Fig. 6).30 Since then, several examples of isolable plumbyliumylidenes stabilised by bulky and strongly donating substituents have been reported. Power and co-workers succeeded in isolating the plumbyliumylidene 114 with a terphenyl group in 2004.123 Subsequent to this, Fulton and co-workers reported the plumbyliumylidenes 115 that are stabilised by a β-diketiminate ligand.120 In 2006, Godwin et al. obtained the three-coordinated N2S(alkylthiolate) stabilised plumbyliumylidene ion 116.124 In addition, the coordination of platinum complex Pt(PCy3)2 (Cy = cyclohexyl) can stabilise the plumbyliumylidene 117.66 The carbazole-based bulky substituent that has been utilised in Si, Ge, and Sn analogues was also used to isolate the corresponding plumbyliumylidene 118.109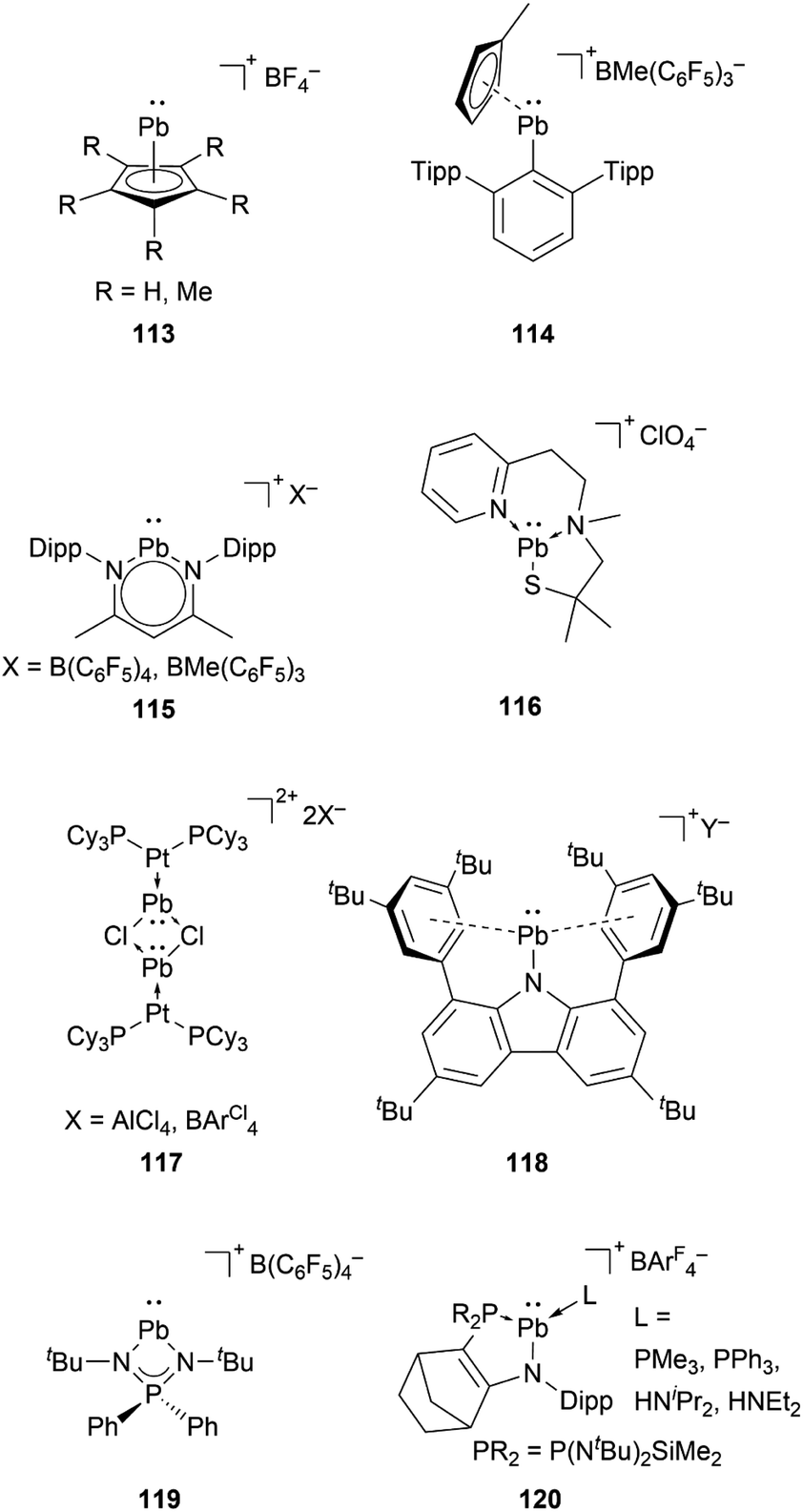 | ||
| Fig. 6 Reported plumbyliumylidene ions (Cy = cyclohexyl; Y = B(C6F5)4 or Al(OC4F9)4). | ||
Recently in 2022, Nakata and co-workers prepared the plumbyliumylidene 119 supported by an N,N′-di-tert-butyl-iminophosphonamide ligand. 119 is one of the two examples of a cationic low-coordination lead compound capable of acting as a catalyst.51 In the proposed mechanism, which was also supported by quantum chemical calculation, the Lewis acidic 119 captures a carbonyl compound (benzophenone or benzaldehyde) to furnish the plumbyliumylidene–benzophenone complex Int-121-[B(C6F5)4]. This reactive intermediate can further react with an HBpin (HBpin = 4,4,5,5-tetramethyl-1,3,2-dioxaborolane) to form a four-membered intermediate Int-123. Subsequent reductive elimination of a boronate ester 124 regenerates the plumbyliumylidene 119. The catalytic cycle proceeds at room temperature with a low for a main-group complex catalyst loading of 0.1 mol% (Scheme 26).
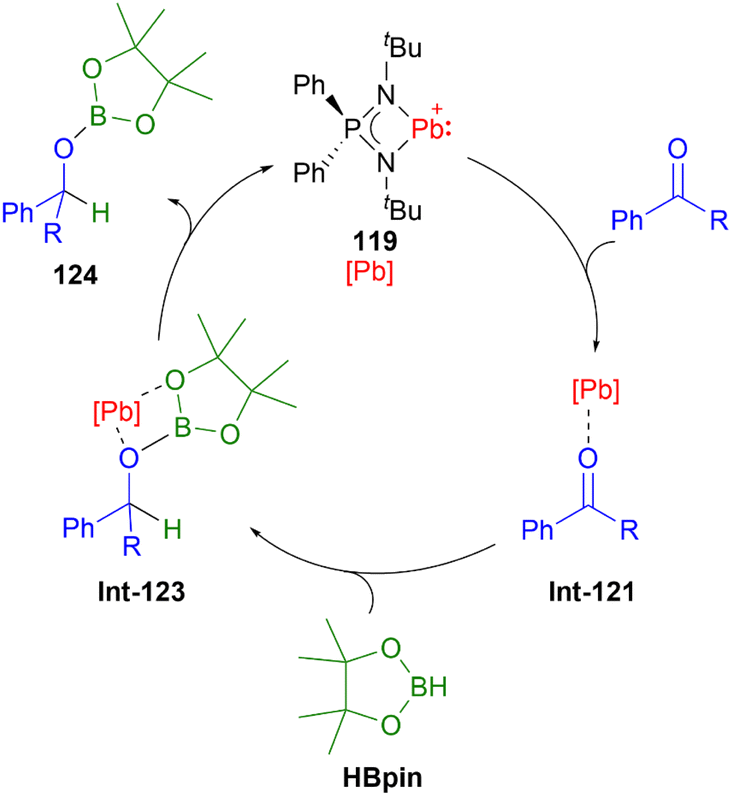 | ||
| Scheme 26 Plumbyliumylidene-catalysed hydroboration of benzaldehyde (R = H) and benzophenone (R = Ph). | ||
At around the same time, the group of Kato obtained the phosphine or amine stabilised plumbyliumylidene 120, which readily reacts with phenylacetylene to the corresponding cationic vinylplumbylene 125via alkyne insertion into the Pb–L bond (Scheme 27).125
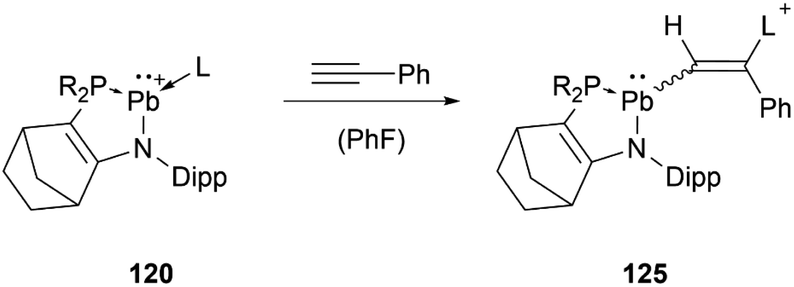 | ||
| Scheme 27 Reaction of 120 with phenylacetylene (R2P = P(NtBu)2SiMe2, L = PMe3, PPh3, HNiPr2, HNEt2). | ||
In the case of amine stabilisation, plumbyliumylidene 120 can catalyse the hydroamination of phenylacetylene to give the corresponding enamine 126 in a regioselective manner (Scheme 28). The yield improves drastically, when using HNEt2 instead of HNiPr2. The faster attack of amine on the activated acetylene bound to the cationic plumbylene prevents side reactions, which can deactivate the catalyst. Further reaction of 126 with Pb-activated phenylacetylene can lead to the formation of the corresponding diene 127. The use of an excess of amine can improve the selectivity of the reaction to enamine 126.125
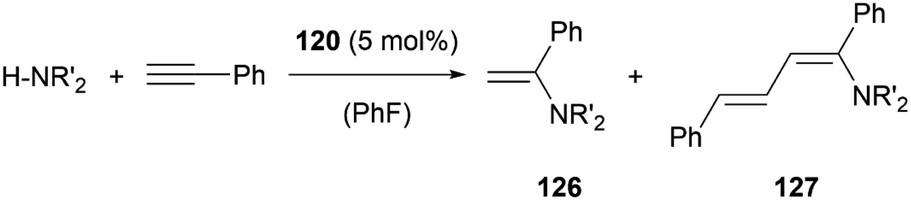 | ||
| Scheme 28 Catalytic hydroamination of phenylacetylene (R′ = iPr, Et). | ||
Conclusions
Pioneering work by Jutzi and co-workers set the stage for the isolation and stabilisation of the unique E(II) cations (E = Si, Ge, Sn, Pb), which merge the electrophilicity of tetrylium cations with the ambiphilic character of tetrylenes.29,30 The subsequent decades witnessed the isolation of various stable tetryliumylidene ions with different ligand stabilisation strategies. The following studies demonstrated promising reactivity of tetryliumylidene ions in various synthetic transformations. These developments expanded our understanding of tetryliumylidene capabilities and paved the way for their application in small molecule activation and catalysis. Despite these achievements, the chemistry of tetryliumylidenes remains largely underexplored. We hope that this review will highlight the topic and stimulate further research aimed at developing more tetryliumylidene-based systems capable of facilitating unique chemical transformations and fostering novel catalytic applications.Author contributions
All authors discussed the concepts and contributed to the final manuscript. S. S. and S. F. performed literature reviews and wrote the original draft. A. K. carried out the quantum chemical calculations. A. K. and S. I. conceived the topic, strategised on content and information organisation, and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the Alexander von Humboldt Foundation for a Research Fellowship (S.F.) and the European Research Council (ALLOWE 101001591).Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed.
- M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed.
- C. Weetman, in Encyclopedia of Inorganic and Bioinorganic Chemistry, ed. R. A. Scott, John Wiley & Sons, Hoboken, New Jersey, 2021, vol. 2021, pp. 1–27 Search PubMed.
- S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 486–501 CrossRef CAS.
- B. Blom and M. Driess, in Functional Molecular Silicon Compounds II: Low Oxidation States, ed. D. Scheschkewitz, Springer International Publishing, Cham, 2014, pp. 85–123, DOI:10.1007/430_2013_95.
- S. Raoufmoghaddam, Y.-P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS.
- S. K. Mandal and H. W. Roesky, Acc. Chem. Res., 2012, 45, 298–307 CrossRef CAS PubMed.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- J. Zheng, Z. H. Li and H. Wang, Chem. Sci., 2018, 9, 1433–1438 RSC.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- H. F. T. Klare, L. Albers, L. Süsse, S. Keess, T. Müller and M. Oestreich, Chem. Rev., 2021, 121, 5889–5985 CrossRef CAS PubMed.
- T. Müller, in Organogermanium Compounds: Theory, Experiment, and Applications, ed. V. Y. Lee, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2023, pp. 299–338, DOI:10.1002/9781119613466.ch6.
- J. B. Lambert, in Tin Chemistry: Fundamentals, Frontiers, and Applications, ed. A. G. Davies, M. Gielen, K. H. Pannell and E. R. T. Tiekink, John Wiley & Sons, Chichester, 2008, pp. 152–159 Search PubMed.
- M. Janssen, S. Mebs and J. Beckmann, Chem. Commun., 2023, 59, 7267–7270 RSC.
- V. S. V. S. N. Swamy, S. Pal, S. Khan and S. S. Sen, Dalton Trans., 2015, 44, 12903–12923 RSC.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. Kaupp, in The Chemical Bond, ed. G. Frenking and S. Shaik, 2014, pp. 1–24, DOI:10.1002/9783527664658.ch1.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis, and F. Weinhold, NBO 7.0, Madison, 2018 Search PubMed.
- P. P. Gaspar, X. Liu, D. Ivanova, D. Read, J. S. Prell, and M. L. Gross, in Modern Aspects of Main Group Chemistry, American Chemical Society, 2005, ch. 4, vol. 917, pp. 52–65 Search PubMed.
- G. Bertrand, Science, 2004, 305, 783–785 CrossRef CAS PubMed.
- P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y.-H. Tsay, Chem. Ber., 1980, 113, 757–769 CrossRef CAS.
- P. Jutzi, R. Dickbreder and H. Nöth, Chem. Ber., 1989, 122, 865–870 CrossRef CAS.
- R. Jambor and M. Novák, Eur. J. Inorg. Chem., 2023, 26, e202300505 CrossRef CAS.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 CrossRef CAS.
- M. Stender, A. D. Phillips and P. P. Power, Inorg. Chem., 2001, 40, 5314–5315 CrossRef CAS PubMed.
- H. V. R. Dias and Z. Wang, J. Am. Chem. Soc., 1997, 119, 4650–4655 CrossRef CAS.
- T. Probst, O. Steigelmann, J. Riede and H. Schmidbaur, Angew. Chem., Int. Ed., 1990, 29, 1397–1398 CrossRef.
- P. A. Rupar, V. N. Staroverov, P. J. Ragogna and K. M. Baines, J. Am. Chem. Soc., 2007, 129, 15138–15139 CrossRef CAS PubMed.
- Y. Xiong, T. Szilvási, S. Yao, G. Tan and M. Driess, J. Am. Chem. Soc., 2014, 136, 11300–11303 CrossRef CAS PubMed.
- A. P. Singh, H. W. Roesky, E. Carl, D. Stalke, J.-P. Demers and A. Lange, J. Am. Chem. Soc., 2012, 134, 4998–5003 CrossRef CAS PubMed.
- A. Schäfer, F. Winter, W. Saak, D. Haase, R. Pöttgen and T. Müller, Chem.–Eur. J., 2011, 17, 10979–10984 CrossRef PubMed.
- P. Jutzi, A. Mix, B. Rummel, W. W. Schoeller, B. Neumann and H.-G. Stammler, Science, 2004, 305, 849–851 CrossRef CAS PubMed.
- Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147–7150 CrossRef CAS.
- Y. Xiong, S. Yao, G. Tan, S. Inoue and M. Driess, J. Am. Chem. Soc., 2013, 135, 5004–5007 CrossRef CAS PubMed.
- A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS.
- S. U. Ahmad, T. Szilvási, E. Irran and S. Inoue, J. Am. Chem. Soc., 2015, 137, 5828–5836 CrossRef CAS PubMed.
- D. Sarkar, D. Wendel, S. U. Ahmad, T. Szilvási, A. Pöthig and S. Inoue, Dalton Trans., 2017, 46, 16014–16018 RSC.
- A. Porzelt, J. I. Schweizer, R. Baierl, P. J. Altmann, M. C. Holthausen and S. Inoue, Inorganics, 2018, 6, 54 CrossRef.
- D. Sarkar, V. Nesterov, T. Szilvási, P. J. Altmann and S. Inoue, Chem.–Eur. J., 2019, 25, 1198–1202 CrossRef CAS PubMed.
- E. Fritz-Langhals, Org. Process Res. Dev., 2019, 23, 2369–2377 CrossRef CAS.
- S. Sinhababu, D. Singh, M. K. Sharma, R. K. Siwatch, P. Mahawar and S. Nagendran, Dalton Trans., 2019, 48, 4094–4100 RSC.
- D. Sarkar, S. Dutta, C. Weetman, E. Schubert, D. Koley and S. Inoue, Chem.–Eur. J., 2021, 27, 13072–13078 CrossRef CAS.
- K. Nakaya, S. Takahashi, A. Ishii and N. Nakata, Inorg. Chem., 2022, 61, 15510–15519 CrossRef CAS PubMed.
- B.-X. Leong, J. Lee, Y. Li, M.-C. Yang, C.-K. Siu, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2019, 141, 17629–17636 CrossRef CAS.
- B.-X. Leong, Y.-C. Teo, C. Condamines, M.-C. Yang, M.-D. Su and C.-W. So, ACS Catal., 2020, 10, 14824–14833 CrossRef CAS.
- Y.-C. Teo, D. Loh, B.-X. Leong, Z.-F. Zhang, M.-D. Su and C.-W. So, Inorg. Chem., 2023, 62, 16867–16873 CrossRef CAS PubMed.
- N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958–17968 CrossRef CAS PubMed.
- H.-X. Yeong, Y. Li and C.-W. So, Organometallics, 2014, 33, 3646–3648 CrossRef CAS.
- P. Ghana, M. I. Arz, G. Schnakenburg, M. Straßmann and A. C. Filippou, Organometallics, 2018, 37, 772–780 CrossRef CAS.
- P. Frisch and S. Inoue, Dalton Trans., 2020, 49, 6176–6182 RSC.
- C. Seow, M. L. B. Ismail, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Organometallics, 2018, 37, 1368–1372 CrossRef CAS.
- R. Jambor, B. Kašná, S. G. Koller, C. Strohmann, M. Schürmann and K. Jurkschat, Eur. J. Inorg. Chem., 2010, 2010, 902–908 CrossRef.
- R. Dostálová, L. Dostál, A. Růžička and R. Jambor, Organometallics, 2011, 30, 2405–2410 CrossRef.
- J. Martincová, L. Dostál, S. Herres-Pawlis, A. Růžička and R. Jambor, Chem.–Eur. J., 2011, 17, 7423–7427 CrossRef PubMed.
- P. M. Keil and T. J. Hadlington, Chem. Commun., 2022, 58, 3011–3014 RSC.
- P. M. Keil, A. Soyemi, K. Weisser, T. Szilvási, C. Limberg and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202218141 CrossRef CAS PubMed.
- A. Schulz, T. L. Kalkuhl, P. M. Keil and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202305996 CrossRef CAS PubMed.
- H. Braunschweig, M. A. Celik, R. D. Dewhurst, M. Heid, F. Hupp and S. S. Sen, Chem. Sci., 2015, 6, 425–435 RSC.
- P. Jutzi, A. Mix, B. Neumann, B. Rummel and H.-G. Stammler, Chem. Commun., 2006, 3519–3521 RSC.
- A. Hinz, Angew. Chem., Int. Ed., 2020, 59, 19065–19069 CrossRef CAS PubMed.
- M. Driess, S. Yao, M. Brym and C. van Wüllen, Angew. Chem., Int. Ed., 2006, 45, 6730–6733 CrossRef CAS PubMed.
- D. C. H. Do, A. V. Protchenko, M. Á. Fuentes, J. Hicks, P. Vasko and S. Aldridge, Chem. Commun., 2020, 56, 4684–4687 RSC.
- S. Takahashi, M. Frutos, A. Baceiredo, D. Madec, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2022, 61, e202208202 CrossRef CAS PubMed.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974–6978 CrossRef CAS.
- T. Agou, N. Hayakawa, T. Sasamori, T. Matsuo, D. Hashizume and N. Tokitoh, Chem.–Eur. J., 2014, 20, 9246–9249 CrossRef CAS.
- S. U. Ahmad, T. Szilvási and S. Inoue, Chem. Commun., 2014, 50, 12619–12622 RSC.
- Y. Li, Y.-C. Chan, Y. Li, I. Purushothaman, S. De, P. Parameswaran and C.-W. So, Inorg. Chem., 2016, 55, 9091–9098 CrossRef CAS PubMed.
- Y. Li, Y.-C. Chan, B.-X. Leong, Y. Li, E. Richards, I. Purushothaman, S. De, P. Parameswaran and C.-W. So, Angew. Chem., Int. Ed., 2017, 56, 7573–7578 CrossRef CAS.
- N. Hayakawa, K. Sadamori, S. Mizutani, T. Agou, T. Sugahara, T. Sasamori, N. Tokitoh, D. Hashizume and T. Matsuo, Inorganics, 2018, 6, 30 CrossRef.
- P. Frisch and S. Inoue, Dalton Trans., 2019, 48, 10403–10406 RSC.
- F. Hanusch, D. Munz, J. Sutter, K. Meyer and S. Inoue, Angew. Chem., Int. Ed., 2021, 60, 23274–23280 CrossRef CAS PubMed.
- S. V. Hirmer, F. S. Tschernuth, F. Hanusch, R. Baierl, M. Muhr and S. Inoue, Mendeleev Commun., 2022, 32, 16–18 CrossRef CAS.
- Y. Xiong, S. Yao, S. Inoue, E. Irran and M. Driess, Angew. Chem., Int. Ed., 2012, 51, 10074–10077 CrossRef CAS PubMed.
- H.-X. Yeong, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, Chem.–Eur. J., 2013, 19, 11786–11790 CrossRef CAS PubMed.
- R. Nougué, S. Takahashi, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Angew. Chem., Int. Ed., 2023, 62, e202215394 CrossRef PubMed.
- S. Stigler, M. Park, A. Porzelt, A. Kostenko, D. Henschel and S. Inoue, Organometallics, 2022, 41, 2088–2094 CrossRef CAS.
- P. Frisch and S. Inoue, Chem. Commun., 2018, 54, 13658–13661 RSC.
- P. Frisch, T. Szilvási, A. Porzelt and S. Inoue, Inorg. Chem., 2019, 58, 14931–14937 CrossRef CAS PubMed.
- P. Frisch, T. Szilvási and S. Inoue, Chem.–Eur. J., 2020, 26, 6271–6278 CrossRef CAS PubMed.
- R. Nougué, S. Takahashi, A. Dajnak, E. Maerten, A. Baceiredo, N. Saffon-Merceron, V. Branchadell and T. Kato, Chem.–Eur. J., 2022, 28, e202202037 CrossRef.
- D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS PubMed.
- B. Su, R. Ganguly, Y. Li and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 13106–13109 CrossRef CAS PubMed.
- B. Su, R. Ganguly, Y. Li and R. Kinjo, Chem. Commun., 2016, 52, 613–616 RSC.
- M. T. Nguyen, D. Gusev, A. Dmitrienko, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, J. Am. Chem. Soc., 2020, 142, 5852–5861 CrossRef CAS.
- D. Paul, F. Heins, S. Krupski, A. Hepp, C. G. Daniliuc, K. Klahr, J. Neugebauer, F. Glorius and F. E. Hahn, Organometallics, 2017, 36, 1001–1008 CrossRef CAS.
- Y.-P. Zhou, M. Karni, S. Yao, Y. Apeloig and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 15096–15099 CrossRef CAS PubMed.
- A. Rit, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2016, 55, 378–382 CrossRef CAS PubMed.
- R. J. Mangan, A. R. Davies, J. Hicks, C. P. Sindlinger, A. L. Thompson and S. Aldridge, Polyhedron, 2021, 196, 115006 CrossRef CAS.
- M. M. D. Roy, P. A. Lummis, M. J. Ferguson, R. McDonald and E. Rivard, Chem.–Eur. J., 2017, 23, 11249–11252 CrossRef CAS PubMed.
- T. Ochiai, D. Franz, X.-N. Wu and S. Inoue, Dalton Trans., 2015, 44, 10952–10956 RSC.
- T. Ochiai and S. Inoue, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 624–627 CrossRef CAS.
- T. Ochiai, T. Szilvási, D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2016, 55, 11619–11624 CrossRef CAS PubMed.
- F. S. Tschernuth, F. Hanusch, T. Szilvási and S. Inoue, Organometallics, 2020, 39, 4265–4272 CrossRef CAS.
- M. Majumdar, R. K. Raut, P. Sahoo and V. Kumar, Chem. Commun., 2018, 54, 10839–10842 RSC.
- P. Sahoo, R. K. Raut, D. Maurya, V. Kumar, P. Rani, R. G. Gonnade and M. Majumdar, Dalton Trans., 2019, 48, 7344–7351 RSC.
- Y. Su, Y. Li, R. Ganguly and R. Kinjo, Eur. J. Inorg. Chem., 2018, 2228–2231 CrossRef CAS.
- Y. Xiong, S. Yao, S. Inoue, A. Berkefeld and M. Driess, Chem. Commun., 2012, 48, 12198–12200 RSC.
- M. Bouška, L. Dostál, A. Růžička and R. Jambor, Organometallics, 2013, 32, 1995–1999 CrossRef.
- D. Maurya, J. Karmakar, P. Sahoo, R. K. Raut and M. Majumdar, Inorg. Chim. Acta, 2020, 503, 119380 CrossRef CAS.
- A. Schäfer, W. Saak, D. Haase and T. Müller, Chem.–Eur. J., 2009, 15, 3945–3950 CrossRef PubMed.
- A. Hinz, Chem.–Eur. J., 2019, 25, 3267–3271 CrossRef CAS PubMed.
- S. Khan, G. Gopakumar, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2013, 52, 5644–5647 CrossRef CAS.
- T. J. Hadlington, A. Kostenko and M. Driess, Chem.–Eur. J., 2021, 27, 2476–2482 CrossRef CAS PubMed.
- D. Sarkar, C. Weetman, S. Dutta, E. Schubert, C. Jandl, D. Koley and S. Inoue, J. Am. Chem. Soc., 2020, 142, 15403–15411 CrossRef CAS PubMed.
- R. S. P. Turbervill and J. M. Goicoechea, Aust. J. Chem., 2013, 66, 1131–1137 CrossRef CAS.
- T. Ochiai, D. Franz, E. Irran and S. Inoue, Chem.–Eur. J., 2015, 21, 6704–6707 CrossRef CAS PubMed.
- X.-X. Zhao, J. A. Kelly, A. Kostenko, S. Fujimori and S. Inoue, Z. Anorg. Allg. Chem., 2022, 648, e202200220 CrossRef CAS.
- R. K. Raut and M. Majumdar, J. Organomet. Chem., 2019, 887, 18–23 CrossRef CAS.
- I. Objartel, H. Ott and D. Stalke, Z. Anorg. Allg. Chem., 2008, 634, 2373–2379 CrossRef CAS.
- R. Baierl, A. Kostenko, F. Hanusch and S. Inoue, Dalton Trans., 2021, 50, 14842–14848 RSC.
- H. V. R. Dias and W. Jin, J. Am. Chem. Soc., 1996, 118, 9123–9126 CrossRef CAS.
- M. J. Taylor, A. J. Saunders, M. P. Coles and J. R. Fulton, Organometallics, 2011, 30, 1334–1339 CrossRef CAS.
- H. Arii, M. Matsuo, F. Nakadate, K. Mochida and T. Kawashima, Dalton Trans., 2012, 41, 11195–11200 RSC.
- M. Wagner, T. Zöller, W. Hiller, M. H. Prosenc and K. Jurkschat, Chem.–Eur. J., 2013, 19, 9463–9467 CrossRef CAS PubMed.
- S. Hino, M. Brynda, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2004, 43, 2655–2658 CrossRef CAS PubMed.
- R. J. Andersen, R. C. diTargiani, R. D. Hancock, C. L. Stern, D. P. Goldberg and H. A. Godwin, Inorg. Chem., 2006, 45, 6574–6576 CrossRef CAS PubMed.
- A. Chandran, J. M. Léon Baeza, V. Timofeeva, R. Nougué, S. Takahashi, R. Ohno, A. Baceiredo, R. S. Rojas Guerrero, M. Syroeshkin, T. Matsuo, N. Saffon-Merceron and T. Kato, Inorg. Chem., 2022, 61, 16156–16162 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |