
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping an abnormal high-Na-content P2-type layered oxide cathode with near-zero-strain for high-performance sodium-ion batteries †
Hai-Yan
Hu‡
ab,
Jia-Yang
Li‡
b,
Yi-Feng
Liu
b,
Yan-Fang
Zhu
*ab,
Hong-Wei
Li
b,
Xin-Bei
Jia
ab,
Zhuang-Chun
Jian
ab,
Han-Xiao
Liu
ab,
Ling-Yi
Kong
ab,
Zhi-Qi
Li
ab,
Hang-Hang
Dong
b,
Meng-Ke
Zhang
d,
Lang
Qiu
d,
Jing-Qiang
Wang
 b,
Shuang-Qiang
Chen
ab,
Xiong-Wei
Wu
c,
Xiao-Dong
Guo
d and
Yao
Xiao
*ab
b,
Shuang-Qiang
Chen
ab,
Xiong-Wei
Wu
c,
Xiao-Dong
Guo
d and
Yao
Xiao
*ab
aInstitute for Carbon Neutralization, College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325035, P. R. China. E-mail: yanfangzhu@wzu.edu.cn; xiaoyao@wzu.edu.cn
bWenzhou Key Laboratory of Sodium-Ion Batteries, Wenzhou University Technology Innovation Institute for Carbon Neutralization, Wenzhou, 325035, P. R. China
cSchool of Chemistry and Materials Science, Hunan Agricultural University, Changsha, 410128, P. R. China
dCollege of Chemical Engineering, Sichuan University, Chengdu, 610065, P. R. China
First published on 28th February 2024
Abstract
Layered transition metal oxides (NaxTMO2) possess attractive features such as large specific capacity, high ionic conductivity, and a scalable synthesis process, making them a promising cathode candidate for sodium-ion batteries (SIBs). However, NaxTMO2 suffer from multiple phase transitions and Na+/vacancy ordering upon Na+ insertion/extraction, which is detrimental to their electrochemical performance. Herein, we developed a novel cathode material that exhibits an abnormal P2-type structure at a stoichiometric content of Na up to 1. The cathode material delivers a reversible capacity of 108 mA h g−1 at 0.2C and 97 mA h g−1 at 2C, retaining a capacity retention of 76.15% after 200 cycles within 2.0–4.3 V. In situ diffraction studies demonstrated that this material exhibits an absolute solid-solution reaction with a low volume change of 0.8% during cycling. This near-zero-strain characteristic enables a highly stabilized crystal structure for Na+ storage, contributing to a significant improvement in battery performance. Overall, this work presents a simple yet effective approach to realizing high Na content in P2-type layered oxides, offering new opportunities for high-performance SIB cathode materials.
Introduction
The utilization of renewable energy resources, such as solar or wind is becoming increasingly crucial to alleviate energy crises and environmental pollution. However, their intermittent characteristics need to be addressed by developing advanced large-scale energy storage systems (EESs).1,2 Rechargeable lithium-ion batteries (LIBs) have been successfully commercialized, supplying power for 3C electronic products and electric vehicles. However, due to the high cost of LIBs resulting from limited lithium resources, their application in next-generation EESs is not considered an economically viable option. Consequently, sodium-ion batteries (SIBs), which share similar working mechanisms and cell configurations to LIBs but utilize abundant and low-cost sodium resources, present a potential solution for EESs.3–8To improve the comprehensive performance of SIBs, substantial efforts have been dedicated to key materials, especially cathode materials. There are various categories of cathode materials, including layered oxides, Prussian blue analogs, polyanionic compounds and organic frameworks.9–12 Among these cathode candidates, layered transition metal oxides (NaxTMO2, where 0 < x < 1 and TM represents a 3d transition metal) have garnered considerable interest due to their excellent ionic conductive ability, high theoretical capacity and environmental benignity.13–16 The development of NaxTMO2 resembles that of LIB cathode materials, such as ternary materials (Li[NixCoyMnz]O2). Most of the modification strategies and manufacturing technologies of LIB cathode materials can be adapted for NaxTMO2, making them more feasible for industrial production compared to other SIB cathode materials.17–19 In general, NaxTMO2 can be categorized as O3-, O2-, P3- and P2-type. The letters “O” and “P” refer to the occupation of Na+ at octahedral and prismatic sites, while the numbers “2” and “3” signify the oxygen stacking sequence.20–23 Determined by their chemical composition and synthesis conditions, NaxTMO2 exhibit a range of structural features and electrochemical intercalation chemistry. O3-NaxTMO2 (0.8 ≤ x ≤ 1) deliver a high Na storage capacity due to the Na-rich composition of the host structure, but their narrow octahedral diffusion channels are unfavorable for Na+ kinetics.24–26 Furthermore, O3-NaxTMO2 generally undergo complex irreversible phase transitions during charging/discharging, such as O3-O3′-P3-P3′-P3′′, making them difficult to achieve good cycle performance. In contrast, P2-NaxTMO2 (x ≤ 0.7) exhibit better rate capability owing to their more open prismatic transport and lower energy barrier for Na+ migration, but lower sodium content in the structure is extremely unfavorable for obtaining a decent capacity.27–32 Additionally, once charged to high voltages, transition metal layers tend to slide and initiate Na+/vacancy ordering arrangements as well as phase transitions from P2 to O2 or OP4 since little sodium remains in the primary structure.33–35 Frequent phase transitions during cycling lead to continuous volume variations and huge lattice strain accumulation, resulting in rapid structural degradation.36–40 Therefore, it remains uncertain to achieve a high sodium content in P2-NaxTMO2 to suppress Na+/vacancy ordering and phase transition, thus realizing an absolute solid-solution reaction mechanism for Na+ storage performance.
Inspired by the classical cathode material LiNi1/3Co1/3Mn1/3O2 for LIBs, an analogous composition of NaNi1/3Co1/3Mn1/3O2 (denoted as NaNCM) was synthesized by thermal polymerization combined with high-temperature calcination and applied as a cathode material for SIBs. The NaNCM cathode material was well indexed to a composite structure with P2 and P3 phases, and its formation process concerning detailed dynamic structural evolution was revealed by in situ high-energy X-ray diffraction. However, in situ charge and discharge XRD indicated that P2/P3-NaNCM underwent a complex phase evolution process of P2/P3-P2/P3′′-P2/P3-P2′/P3′′ during cycling, resulting in rapid capacity decay and poor rate performance. In addition to this, electrochemically inactive Mg2+ was carefully incorporated into P2/P3-NaNCM, and it was found that the P2/P3 composite structure was transformed into a pure P2 single-phase structure, while the Na content remained at a high stoichiometric value of 1. The high Na content in the obtained P2-NaNi2/9Mg1/9Co1/3Mn1/3O2 (P2-NaNMCM) could provide sufficient reservoir for Na (de)intercalation without compromising specific capacity, while maintaining a P2-type structure for fast Na+ diffusion. As a result, P2-NaNMCM exhibited superior rate capability (89.8% capacity retention at 2C vs. 0.2C) and cycle stability (76.15% capacity retention at 1C for 200 cycles) over a wide voltage range of 2.0–4.3 V. The improved performance could be attributed to its high Na content and P2 structure which lead to a phase-transition-free process except for only 0.8% volume variation, thus resulting in high structural stability. These findings afford a promising strategy for achieving high Na content in P2-type layered cathode materials to fabricate high-performance SIBs.
Results and discussion
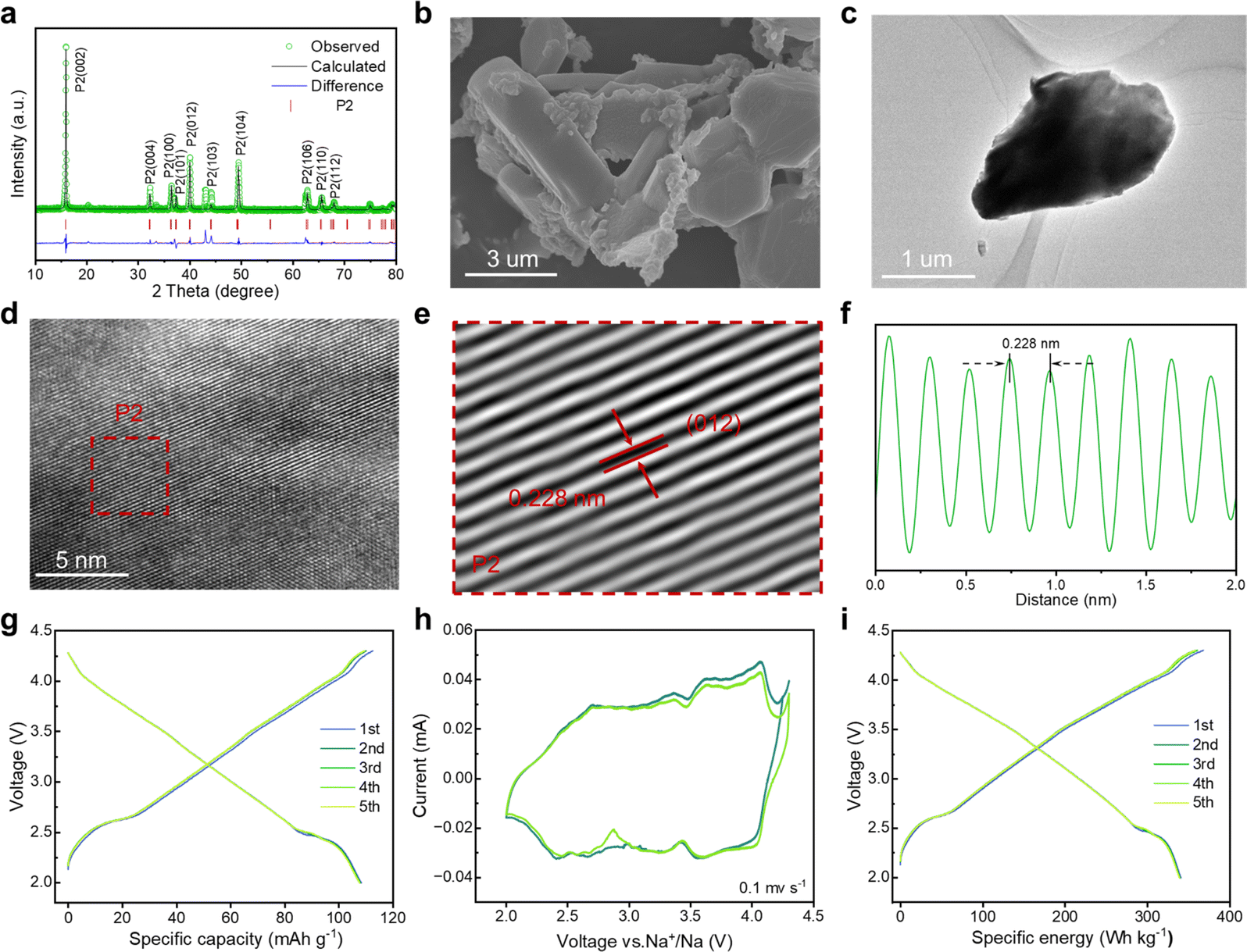
To gain in-depth structural information of the as-synthesized NaNi1/3Co1/3Mn1/3O2 (NaNCM) cathode material, in situ high-energy X-ray diffraction (HE-XRD) was carried out to investigate its formation process. The final product went through three stages: amorphous, P3, and P2/P3, as shown in Fig. 1a and S1.† When the temperature is below 300 °C, amorphous peaks can be observed. As the temperature increases, (003), (006), (012), (104), (015), (107), and (108) characteristic diffraction peaks gradually appear, which are ascribed to the P3 phase.41–43 When the temperature is up to 610 °C, (002), (004), (100), (012), (103), (106), and (110) characteristic peaks of the P2 phase emerge.44 The detailed structural evolution followed by changing temperature is displayed in Fig. 1b and S2 (ESI†). The crystal structure of NaNCM was studied by powder XRD with Rietveld refinement (Fig. 1c), which could be ascribed to the hexagonal P2 phase with a P63/mmc space group and the hexagonal P3 phase with an R3m space group, indicating the successful fabrication of the P2/P3 composite structure.45–47 The specific structural information and lattice parameters of the P2 phase (a = 2.844(2) Å, c = 11.070(6) Å, and V = 77.561(2) Å3) and P3 phase (a = 2.868(2) Å, c = 16.514(6) Å, and V = 117.658(1) Å3) are available in Table S1 (ESI†), which shows a mass ratio of about 3 to 7. The low Rp and Rwp values also illustrate a good agreement between the experimental and calculated patterns, making the calculated results of the crystallographic parameters from the Rietveld refinement more convincing. The morphology of P2/P3-NaNCM was observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1d, e and S3 (ESI†), P2/P3-NaNCM is chunk-like and uniformly dispersed. The elaborate crystalline facet information was detected by high-resolution TEM (HR-TEM) in Fig. 1f. The interplanar lattice distances of 0.235 nm and 0.228 nm are assigned separately to P3 (012) and P2 (012) as shown in Fig. 1g, h and S4 (ESI†).48–50 The HR-TEM images correspond to the XRD results and together confirm the presence of a combination of P2 and P3 structures in NaNCM. The distributed elements in P2/P3-NaNCM were analyzed using energy dispersive spectroscopy (EDS), which showed that Na, Ni, Co and Mn were uniformly distributed in the material (Fig. 1i and S5, ESI†). Besides, inductively coupled plasma mass spectrometry (ICP-MS) was used to analyze the chemical composition of P2/P3-NaNCM, as listed in Table S2.† Based on the results of the advanced techniques described above, it can be proven that P2 and P3 composite structures were indeed obtained. | ||
| Fig. 1 Structure characterization of the P2/P3-NaNCM cathode material. (a) In situ high-energy XRD patterns of the as-synthesized precursor of the P2/P3-NaNCM cathode material during calcination at different temperatures and (b) corresponding contour maps (bird's eye view) showing the evolution of the main characteristic diffraction peaks. (c) Rietveld refinement of the XRD pattern. (d) SEM image. (e) TEM image. (f) HR-TEM image. (g and h) Enlarged HR-TEM images of P3 and P2 phases, respectively. (i) EDS mapping. | ||
The electrochemical performance of P2/P3-NaNCM was investigated within 2.0–4.3 V. The galvanostatic charge–discharge (GCD) curve of P2/P3-NaNCM (Fig. 2a) exhibits multiple voltage plateaus corresponding to its cyclic voltammetry (CV) curve as shown in Fig. 2b, which also reflected complex redox reactions and structural rearrangements. It is worth noting that P2/P3-NaNCM could deliver a reversible specific capacity of 125.1 mA h g−1 equivalent to 386.1 W h kg−1 at 0.2C (Fig. 2c), which makes it a possible commercial cathode candidate for SIBs. To further illustrate the dynamic phase transition of P2/P3-NaNCM, the structural changes during Na+ (de)intercalation were observed by in situ charge–discharge XRD within 2.0–4.3 V. Pristine P2/P3-NaNCM can be distinguished based on the characteristic peaks of both the P2 and P3 phases. The P2 phase is characterized by distinctive peaks at (002), (004), (100), (012), (103), and (104), while the P3 phase is recognized by a characteristic peak at (015).51 These specific peak positions identify the presence of P2/P3-NaNCM and distinguish it from Al foil. Fig. 2d demonstrates that the P2/P3-NaNCM cathode undergoes irreversible multiphase transitions. With the extraction of Na+, the (002) and (004) peaks associated with the c parameter consistently continue to shift to low angles, indicating an increased interlayer distance caused by repulsion between adjacent oxygen layers. In contrast, the (100) and (012) peaks related to a/b parameters showed opposite behaviors, which could be ascribed to the reduction of TM–O distance as a result of charge compensation.52,53 In the subsequent extraction process, the (003)/(104) peaks of P3′′ appeared, while the (015) peak of P3 disappeared, indicating a phase transition from P3 to P3′′. Upon the embedding of Na+, the (015) peak of P3 reappeared, accompanied by the disappearance of the (003)/(104) peaks of P3′′, indicating a phase transition from P3′′ back to P3. Additionally, during the subsequent embedding process, the (002) peaks of P2′ appeared, indicating the presence of a phase transition from the P2 to the P2′ phase. These observations were further supported by the intensity contour maps shown in Fig. 2e and S6 (ESI†). The variation in specific cell parameters (a/b, c and V) of P2/P3-NaNCM was obtained by refining the in situ XRD results at different charge/discharge states as shown in Fig. 2f. The a/b and c parameters were varied in opposite ways upon Na+ insertion/extraction, with maximum changes of 2.4% and 4.6% for the P2 phase (4.5% and 4.5% for the P3 phase), respectively. The change in the V parameter was calculated to be 1.3% for the P2 phase (4.5% for the P3 phase), indicating that the structural reversibility of the P2 phase is relatively higher than that of the P3 phase.54–56Fig. 2g reveals the changes in lattice strain and stress of P2/P3-NaNCM during the initial cycling, showing that lattice strain increases with the extraction of Na+ and reaches a maximum (ΔS = 0.04 GPa) at the end of charging, while it decreases with the insertion of Na+ and returns to the initial value at the end of discharging. The entire structural evolution process is illustrated in Fig. 2h, revealing a complex irreversible phase transition (P2/P3-P2/P3′′–P2/P3-P2′/P3′′) in the voltage range of 2.0–4.3 V. Furthermore, in situ XRD measurements conducted at different charge/discharge states confirmed the structural evolution (Fig. S7, ESI†). The observed structural changes reflect complex electrochemical behaviors that may cause severe capacity degradation, thus hampering the long-term cycling performance of P2/P3-NaNCM.
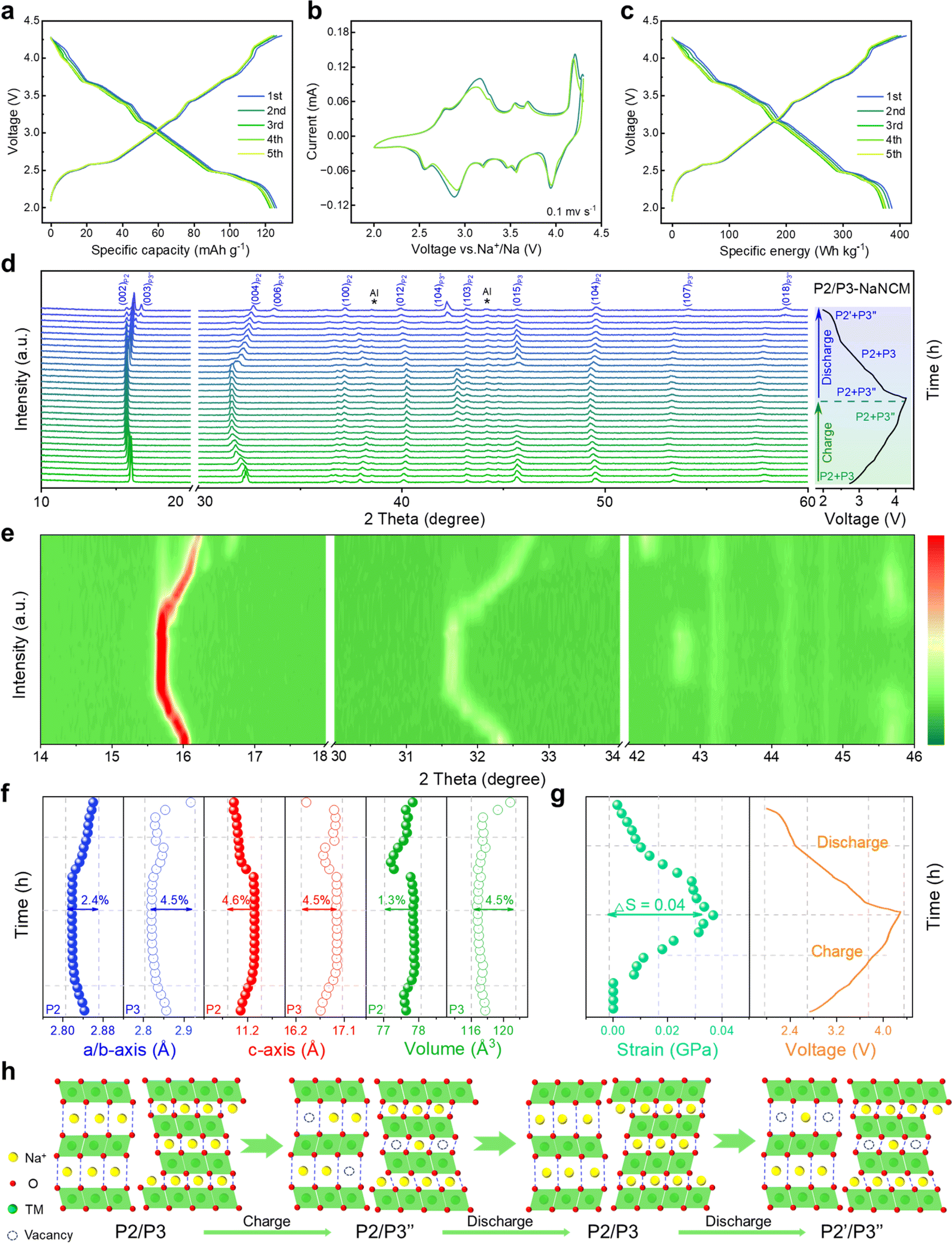 | ||
| Fig. 2 Electrochemical performance of the P2/P3-NaNCM cathode material in a half-cell system. (a) GCD versus specific capacity at 0.2C for different cycles. (b) CV curves at 0.1 mV s−1. (c) GCD versus specific energy at 0.2C for different cycles. (d) In situ charge and discharge XRD patterns, and (e) corresponding evolution of the main characteristic diffraction peaks. (f) Change in lattice parameters, and (g) change in strain during the initial cycle. (h) Schematic illustration of the crystal structural evolution process during the first charging/discharging at 0.1C. | ||
Mitigating multiphase transitions is crucial for enhancing comprehensive electrochemical performance for cathode materials. Therefore, a Mg substitution strategy was adapted to endow P2/P3-NaNCM with a stabilized structure for absolute solid-solution reactions. By partially replacing 1/9 Ni with Mg in P2/P3-NaNCM, a new cathode material (NaNMCM) with a pure P2 structure is generated, which is identified by XRD. The Rietveld refinement results of P2-NaNMCM are shown in Fig. 3a, and lattice parameters (a = 2.846(7) Å, c = 11.091(3) Å, and V = 77.841(2) Å3) are available in Table S3 (ESI†). The results show that Mg substitution leads to a structural transformation from the composite P2/P3 structure to a pure P2 structure (Fig S8, ESI†), while the initial sodium content remains at 1. It is noteworthy that such a high Na content in the P2-type structure has not been reported so far.57 SEM and TEM images show that the P2-NaNMCM material still exhibits a chunk-like morphology with a smooth surface after Mg incorporation (Fig. 3b, c and S9, ESI†). The HR-TEM images and line profile of P2-NaNMCM are separately shown in Fig. 3d–f and S10 (ESI†), and the spacing between the neighboring transition metal atoms was observed to be 0.228 nm by the fast Fourier transform corresponding to the (012) crystal plane of the P2 phase.58 The EDS and ICP-MS results also confirmed that element Mg has been successfully incorporated into the P2-NaNMCM cathode material (Fig. S11, S12 and Table S4, ESI†).
 | ||
| Fig. 3 Structure characterization of the P2-NaNMCM cathode material. (a) Rietveld refinement of the XRD pattern. (b) SEM image. (c) TEM image. (d) HR-TEM image. (e) Enlarged HR-TEM image of the P2 phase. (f) The line profile from the selected area. Electrochemical performance of the P2-NaNMCM cathode material in the half-cell system. (g) GCD versus specific capacity at 0.2C for different cycles. (h) CV curves at 0.1 mV s−1. (i) GCD versus specific energy at 0.2C for different cycles. | ||
To investigate the effect of Mg introduction on electrochemical performance of P2-NaNMCM, GCD and CV tests were performed as shown in Fig. 3g and h, respectively. It is clear that multiple plateaus in the GCD curve of P2-NaNMCM disappear, corresponding to its relatively smooth CV curve, thus showing a complete solid-solution reaction behavior. Although a portion of the active Ni element involved in redox reactions has been replaced, P2-NaNMCM still displays a high specific capacity of 108.0 mA h g−1 and energy density 339.9 W h kg−1 at 0.2C (Fig. 3i) and maintains a very good consistency in the first five cycles. The substantial increase in reversibility further supports the conclusion that the substitution of Mg not only achieves structural modulation from the P2/P3 composite structure to a pure P2 structure but also plays a crucial role in enhancing electrochemical properties.
The comparison of the electrochemical performance of P2/P3-NaNCM and P2-NaNMCM cathode materials was assessed in the voltage window of 2.0–4.3 V vs. Na/Na+ in a half-cell system. The CV curves in Fig. 4a at different sweep speeds are highly consistent with the GCD curves in Fig. 4b, which proves that the P2/P3-NaNCM cathode material experiences complex phase transitions in agreement with the in situ XRD results. The rate performance of P2/P3-NaNCM is revealed in Fig. 4c, showing a rapid capacity degradation without Mg substitution. As shown in Fig. 4d and e, a stark contrast can be seen in that P2-NaNMCM exhibits smoother GCD and CV curves, which implies a more simple and reversible capacity contribution process. Benefiting from the large spacing of prismatic sites in the P2 structure for fast Na+ diffusion, the P2-NaNMCM cathode shows excellent rate performance (Fig. 4f). Even tested at 2C, a capacity retention of 89.8% (vs. 0.2C) is obtained for P2-NaNMCM, which is higher than that of P2/P3-NaNCM. The comparison of capacity retention at different rates corresponding to 0.2C is shown in Fig. S13 (ESI†), and detailed charging and discharging specific capacities are recorded in Table S5 (ESI†). The calculated specific energy density of P2-NaNMCM electrodes is approaching 301.5 W h kg−1, while that of P2/P3-NaNCM electrodes is estimated to be only 269.7 W h kg−1 at 2C (Fig. S14 and Table S6, ESI†). The cycle performance in Fig. 4g revealed that P2-NaNMCM possesses higher stability than P2/P3-NaNCM, showing a capacity retention of 76.15% after 200 cycles. The capacity retention of P2/P3-NaNCM is slightly higher at the beginning of the cycling process; this may be due to some irreversible capacity stimulated by side reactions at the interface between the cathode material and the electrolyte (Fig. S15, ESI†).59,60 At the same time, complex phase transitions lead to large volume changes and lattice stresses, which destabilize the cathode/electrolyte interface and generate more side reactions.61 The accumulation of unfavorable side reactions can further hinder Na+ transport and trigger structural collapse of the material from the surface to the interior, leading a poor cycling stability.
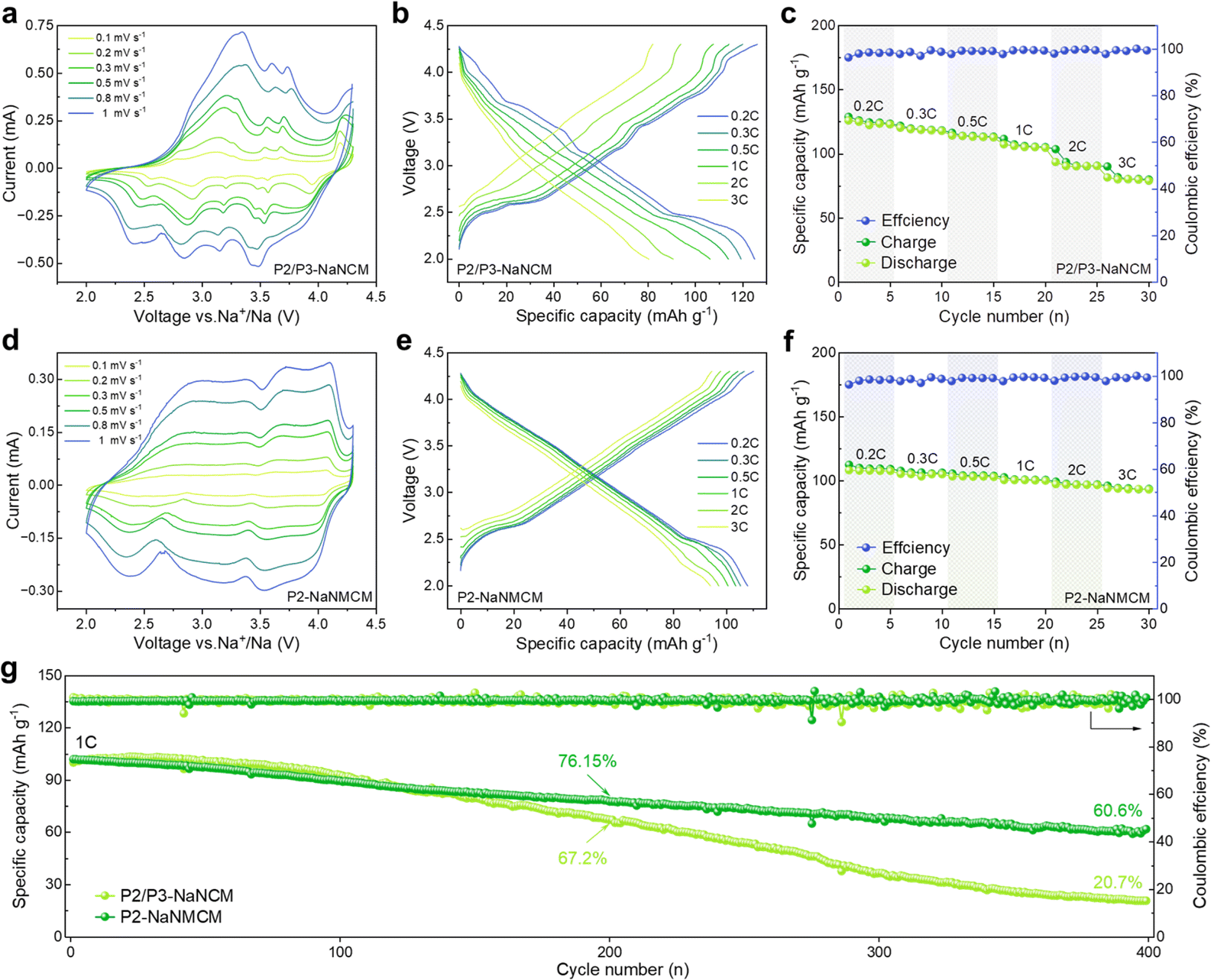 | ||
| Fig. 4 Comparison of electrochemical properties of P2/P3-NaNCM and P2-NaNMCM cathode materials in the half-cell system. (a and d) CV curves at different scan rates. (b and e) GCD versus specific capacity. (c and f) Rate performance at different rates. (g) Cycling performance at 1C after rate tests. | ||
Since the electrochemical properties of P2-NaNMCM are superior to those of P2/P3-NaNCM, in situ XRD measurements were performed to better understand the enhancement mechanism as shown in Fig. 5a, and the evolution of the different peaks during the initial cycling is shown in Fig. 5b, S16 and S17 (ESI†) as contour plots. The charging process causes the (002) and (004) diffraction peaks to gradually move towards lower angles, expanding the interlayer distance along the c-axis, which is attributed to an increase in electrostatic attraction between adjacent oxygen layers. Simultaneously, the (100) and (012) diffraction peaks are shifted to a higher angle, which suggests that the ionic radii of the high-valent transition metal ions formed during charging become smaller, leading to the contraction of the a–b plane. Then, all of these peaks return to their original positions during discharging and no new peaks can be detected, suggesting an absolute solid-solution reaction over a wide voltage range of 2.0–4.3 V. The variation in cell parameters of P2-NaNMCM is shown in Fig. 5c, and the change in a/b, c and V parameters is separately calculated only to be 1.6%, 3.7% and 0.8%, which are lower than those of P2/P3-NaNCM. This ultra-low volume change of P2-NaNMCM within 2.0–4.3 V can be characterized as a “zero strain” state (ΔV < 1%),62,63 which is smaller than that of previously reported cathode materials including Na0.76Ca0.05[Ni0.23□0.08Mn0.69]O2 (4.1%),64 Na0.75Ca0.04-[Li0.1Ni0.2Mn0.67]O2 (1.7%),65 and Na0.85Li0.12Ni0.22Mn0.66 O2 (1.7%).66 The change in lattice strain of P2-NaNMCM is displayed in Fig. 5d, and it is worth noting that a strain value of about 0.03 GPa exists at the initial stage of charging, which could be ascribed to its high Na content inducing strong Na+–Na+ electrostatic repulsions within Na layers. The calculated strain change (ΔS = 0.027 GPa) of P2-NaNMCM is also lower than that of P2/P3-NaNCM, which further confirms its near-zero-strain characteristics. The dynamic crystal structure evolution of P2-NaNMCM is illustrated in Fig. 5e. Since a large amount of Na+ can be retained in the host structure, serving as a pillar to suppress the gliding of transition metal layers, its P2 structure is well preserved without any phase transition except for a negligible volume variation during the entire charging and discharging process. These results show that rational elemental substitution with Mg can induce the formation of abnormal P2 structures with high Na content, achieving desired electrochemical performance.
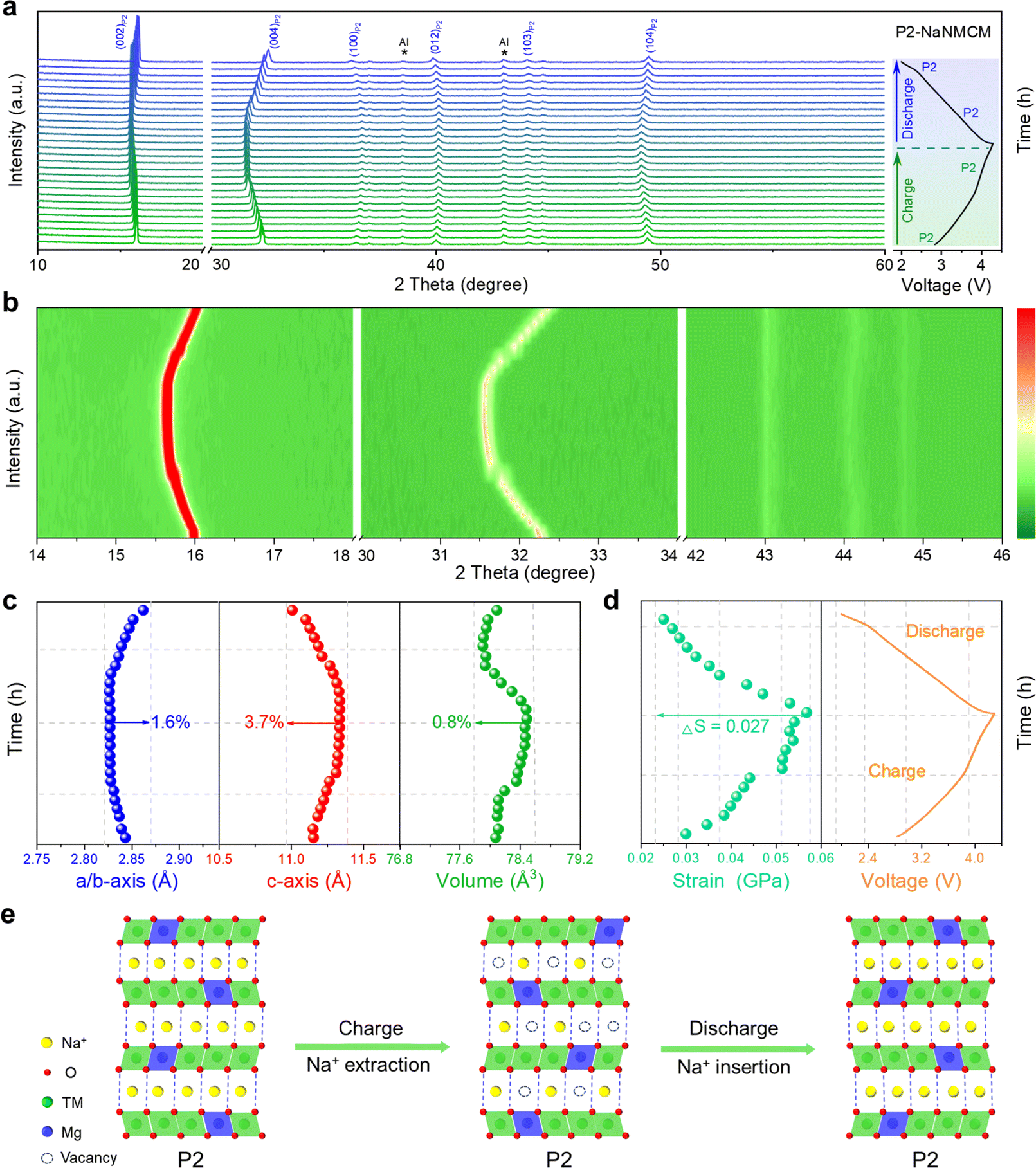 | ||
| Fig. 5 (a) In situ charge and discharge XRD pattern, and (b) corresponding evolution of the main characteristic diffraction peaks. (c) Change in lattice parameters, and (d) change in strain during the initial cycle. (e) Schematic illustration of the crystal structural evolution process of the P2-NaNMCM cathode material during the first charging/discharging at 0.1C. | ||
Conclusions
In summary, we have developed a high-Na-content P2-NaNi2/9Mg1/9Co1/3Mn1/3O2 (P2-NaNMCM) cathode material by partial replacement of 1/9 Ni with Mg. The substitution of Mg induces a unique structural transformation from a composite P2/P3 structure to a pure P2 structure at a high stoichiometric content of Na up to 1. Such a high Na content in the P2 structure can not only enhance rate performance by providing sufficient prismatic channels for rapid Na+ migration, but also improve structural stability via allowing more Na+ to reside in the host structure when charging to high cut-off voltages, which is conducive to suppressing Na+/vacancy ordering and phase transition. In situ charge and discharge XRD also indicated that the P2-NaNMCM cathode material experiences an absolute solid-solution reaction with near-zero-strain upon Na+ extraction and insertion, resulting in better electrochemical performance. The outcomes demonstrated in this work based on a general strategy could provide valuable insights into the development of high-performance P2-type layered oxide cathode materials for SIBs.Data availability
Essential data are fully provided in the main text and ESI.†Author contributions
Hai-Yan Hu and Jia-Yang Li: conceptualization, data curation, writing-original draft. Yi-Feng Liu: methodology, data curation. Yan-Fang Zhu: project administration. Hong-Wei Li: software. Xin-Bei Jia, Zhuang-Chun Jian, Han-Xiao Liu, Ling-Yi Kong, and Zhi-Qi Li: investigation, visualization. Hang-Hang Dong, Meng-Ke Zhang, Lang Qiu, and Jing-Qiang Wang: formal analysis. Shuang-Qiang Chen, Xiong-Wei Wu, and Xiao-Dong Guo: resources. Yao Xiao: supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52202284), Zhejiang Provincial Natural Science Foundation (LQ23E020002), Wenzhou Key Scientific and Technological Innovation Research Projects (ZG2023053), Wenzhou Natural Science Foundation (G20220019 and G20220021), Cooperation between Industry and Education Project of Ministry of Education (220601318235513), China Scholarship Council (202106370062), and Doctoral Innovation Foundation of Wenzhou University (3162023001001).References
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC
.
- Q. Y. Zhao, G. Y. Yin, Y. F. Liu, R. R. Tang, X. W. Wu and X. X. Zeng, Carbon Neutralization, 2022, 2, 90–114 CrossRef
- Y. Su, B. Johannessen, S. Zhang, Z. Chen, Q. Gu, G. Li, H. Yan, J. Y. Li, H. Y. Hu, Y. F. Zhu, S. Xu, H. Liu, S. Dou and Y. Xiao, Adv. Mater., 2023, 35, 2305149 CrossRef CAS PubMed
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y. S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS
- Y. F. Zhu, Y. Xiao, S. X. Dou, Y. M. Kang and S. L. Chou, eScience, 2021, 1, 13–27 CrossRef
- W. Zhang, Y. Wu, Y. Dai, Z. Xu, L. He, Z. Li, S. Li, R. Chen, X. Gao, W. Zong, F. Guo, J. Zhu, H. Dong, J. Li, C. Ye, S. Li, F. Wu, Z. Zhang, G. He, Y. Lai and I. P. Parkin, Chem. Sci., 2023, 14, 8662–8671 RSC
- H. Ren, Y. Li, Q. Ni, Y. Bai, H. Zhao and C. Wu, Adv. Mater., 2022, 34, 2106171 CrossRef CAS PubMed
- K. Wang, H. Zhuo, J. Wang, F. Poon, X. Sun and B. Xiao, Adv. Funct. Mater., 2023, 33, 2212607 CrossRef CAS
- J. Li, H. Hu, J. Wang and Y. Xiao, Carbon Neutralization, 2022, 1, 96–116 CrossRef
- H. Zhang, Y. Gao, X. Liu, L. Zhou, J. Li, Y. Xiao, J. Peng, J. Wang and S. L. Chou, Adv. Energy Mater., 2023, 13, 2300149 CrossRef CAS
- H. R. Yao, L. Zheng, S. Xin and Y. G. Guo, Sci. China: Chem., 2022, 65, 1076–1087 CrossRef CAS
- Q. Y. Zhao, J. Y. Li, M. J. Chen, H. Wang, Y. T. Xu, X. F. Wang, X. Ma, Q. Wu, X. Wu and X. X. Zeng, Nano Lett., 2022, 22, 9685–9692 CrossRef CAS PubMed
- H. Xu, Q. Yan, W. Yao, C. S. Lee and Y. Tang, Small Struct., 2022, 3, 2100217 CrossRef CAS
- C. Shi, L. Wang, X. Chen, J. Li, S. Wang, J. Wang and H. Jin, Nanoscale Horiz., 2022, 7, 338–351 RSC
- W. Zuo and Y. Yang, Acc. Mater. Res., 2022, 3, 709–720 CrossRef CAS
- C. Peng, X. Xu, F. Li, L. Xi, J. Zeng, X. Song, X. Wan, J. Zhao and J. Liu, Small Struct., 2023, 4, 2300150 CrossRef CAS
- R. Zhang, S. Yang, H. Li, T. Zhai and H. Li, InfoMat, 2022, 4, e12305 CrossRef CAS
- L. Qiu, M. Zhang, Y. Song, Z. Wu, H. Zhang, W. Hua, Y. Sun, Q. Kong, W. Feng, K. Wang, Y. Xiao and X. Guo, Angew. Chem., Int. Ed., 2023, 62, e202300209 CrossRef CAS PubMed
- H. Yang, R. M. Gao, X. D. Zhang, J. Y. Liang, X. H. Meng, Z. Y. Lu, F. F. Cao and H. Ye, Adv. Mater., 2022, 34, e2204835 CrossRef PubMed
- Y. F. Liu, K. Han, D. N. Peng, L. Y. Kong, Y. Su, H. W. Li, H. Y. Hu, J. Y. Li, H. R. Wang, Z. Q. Fu, Q. Ma, Y. F. Zhu, R. R. Tang, S. L. Chou, Y. Xiao and X. W. Wu, InfoMat, 2023, 5, e12422 CrossRef CAS
- Y. Xiao, Y. F. Zhu, L. Li, P. F. Wang, W. Zhang, C. Li, S. X. Dou and S. L. Chou, Cell Rep. Phys. Sci., 2021, 2, 100547 CrossRef CAS
- W. Zuo, X. Liu, J. Qiu, D. Zhang, Z. Xiao, J. Xie, F. Ren, J. Wang, Y. Li, G. F. Ortiz, W. Wen, S. Wu, M. S. Wang, R. Fu and Y. Yang, Nat. Commun., 2021, 12, 4903 CrossRef CAS PubMed
- W. Zuo, J. Qiu, X. Liu, F. Ren, H. Liu, H. He, C. Luo, J. Li, G. F. Ortiz, H. Duan, J. Liu, M. S. Wang, Y. Li, R. Fu and Y. Yang, Nat. Commun., 2020, 11, 3544 CrossRef CAS PubMed
- H. Y. Hu, H. Wang, Y. F. Zhu, J. Y. Li, Y. Liu, J. Wang, H. X. Liu, X. B. Jia, H. Li, Y. Su, Y. Gao, S. Chen, X. Wu, S. X. Dou, S. Chou and Y. Xiao, ACS Nano, 2023, 17, 15871–15882 CrossRef CAS PubMed
- Y. Niu, Z. Hu, B. Zhang, D. Xiao, H. Mao, L. Zhou, F. Ding, Y. Liu, Y. Yang, J. Xu, W. Yin, N. Zhang, Z. Li, X. Yu, H. Hu, Y. Lu, X. Rong, J. Li and Y. S. Hu, Adv. Energy Mater., 2023, 13, 2300746 CrossRef CAS
- T. Yuan, S. Li, Y. Sun, J. H. Wang, A. J. Chen, Q. Zheng, Y. Zhang, L. Chen, G. Nam, H. Che, J. Yang, S. Zheng, Z. F. Ma and M. Liu, ACS Nano, 2022, 16, 18058–18070 CrossRef CAS PubMed
- J. Y. Li, H. Y. Hu, L. F. Zhou, H. W. Li, Y. J. Lei, W. H. Lai, Y. M. Fan, Y. F. Zhu, G. Peleckis, S. Q. Chen, W. K. Pang, J. Peng, J. Z. Wang, S. X. Dou, S. L. Chou and Y. Xiao, Adv. Funct. Mater., 2023, 33, 2213215 CrossRef CAS
- L. Yu, H. Dong, Y. X. Chang, Z. Cheng, K. Xu, Y. H. Feng, D. Si, X. Zhu, M. Liu, B. Xiao, P. F. Wang and S. Xu, Sci. China: Chem., 2022, 65, 2005–2014 CrossRef CAS
- F. Fu, X. Liu, X. Fu, H. Chen, L. Huang, J. Fan, J. Le, Q. Wang, W. Yang, Y. Ren, K. Amine, S. G. Sun and G. L. Xu, Nat. Commun., 2022, 13, 2826 CrossRef CAS PubMed
- T. Y. Yu, H. H. Ryu, G. Han and Y. K. Sun, Adv. Energy Mater., 2020, 10, 2001609 CrossRef CAS
- F. Ding, H. Wang, Q. Zhang, L. Zheng, H. Guo, P. Yu, N. Zhang, Q. Guo, F. Xie, R. Dang, X. Rong, Y. Lu, R. Xiao, L. Chen and Y. S. Hu, J. Am. Chem. Soc., 2023, 145, 13592–13602 CrossRef CAS PubMed
- H. R. Yao, X. G. Yuan, X. D. Zhang, Y. J. Guo, L. Zheng, H. Ye, Y. X. Yin, J. Li, Y. Chen, Y. Huang, Z. Huang and Y. G. Guo, Energy Storage Mater., 2023, 54, 661–667 CrossRef
- G. Shao, W. Kong, Y. Yu, J. Zhang, W. Yang, J. Yang, Y. Li and X. Liu, Inorg. Chem., 2023, 62, 9314–9323 CrossRef CAS PubMed
- T. Yang, Y. Huang, J. Zhang, H. Zhu, J. Ren, T. Li, L. C. Gallington, S. Lan, L. Yang and Q. Liu, J. Energy Chem., 2022, 73, 542–548 CrossRef CAS
- Y. Wang, X. Zhao, J. Jin, Q. Shen, Y. Hu, X. Song, H. Li, X. Qu, L. Jiao and Y. Liu, J. Am. Chem. Soc., 2023, 145, 22708–22719 CrossRef CAS PubMed
- H. Y. Hu, Y. F. Zhu, Y. Xiao, S. Li, J. Y. Li, Z. Q. Hao, J. H. Zhao and S. L. Chou, Adv. Energy Mater., 2022, 12, 2201511 CrossRef CAS
- P. F. Wang, H. R. Yao, X. Y. Liu, Y. X. Yin, J. N. Zhang, Y. Wen, X. Yu, L. Gu and Y. G. Guo, Sci. Adv., 2018, 4, eaar6018 CrossRef PubMed
- Z. Wu, Y. Ni, S. Tan, E. Hu, L. He, J. Liu, M. Hou, P. Jiao, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2023, 145, 9596–9606 CrossRef CAS PubMed
- J. Xia, H. Zhao, W. K. Pang, Z. Yin, B. Zhou, G. He, Z. Guo and Y. Du, Chem. Sci., 2018, 9, 3421–3425 RSC
- L. Yang, X. Li, J. Liu, S. Xiong, X. Ma, P. Liu, J. Bai, W. Xu, Y. Tang, Y. Y. Hu, M. Liu and H. Chen, J. Am. Chem. Soc., 2019, 141, 6680–6689 CrossRef CAS PubMed
- P. Hou, J. Yin, X. Lu, J. Li, Y. Zhao and X. Xu, Nanoscale, 2018, 10, 6671–6677 RSC
- X. Chen, X. Zhou, M. Hu, J. Liang, D. Wu, J. Wei and Z. Zhou, J. Mater. Chem. A, 2015, 3, 20708–20714 RSC
- Y. N. Zhou, P. F. Wang, X. D. Zhang, L. B. Huang, W. P. Wang, Y. X. Yin, S. Xu and Y. G. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 24184–24191 CrossRef CAS PubMed
- Y. Xiao, Y. F. Liu, H. W. Li, J. Y. Li, J. Q. Wang, H. Y. Hu, Y. Su, Z. C. Jian, H. R. Yao, S. Q. Chen, X. X. Zeng, X. W. Wu, J. Z. Wang, Y. F. Zhu, S. X. Dou and S. L. Chou, InfoMat, 2023, 5, e12475 CrossRef CAS
- Y. N. Zhou, P. F. Wang, Y. B. Niu, Q. H. Li, X. Q. Yu, Y. X. Yin, S. L. Xu and Y. G. Guo, Nano Energy, 2019, 55, 143–150 CrossRef CAS
- N. Jiang, Q. Liu, J. Wang, W. Yang, W. Ma, L. Zhang, Z. Peng and Z. Zhang, Small, 2021, 17, e2007103 CrossRef PubMed
- Y. Su, N. N. Zhang, J. Y. Li, Y. Liu, H. Y. Hu, J. Wang, H. Li, L. Y. Kong, X. B. Jia, Y. F. Zhu, S. Chen, J. Z. Wang, S. X. Dou, S. Chou and Y. Xiao, ACS Appl. Mater. Interfaces, 2023, 15, 44839–44847 CrossRef CAS PubMed
- Z. Cheng, X. Y. Fan, L. Yu, W. Hua, Y. J. Guo, Y. H. Feng, F. D. Ji, M. Liu, Y. X. Yin, X. Han, Y. G. Guo and P. F. Wang, Angew. Chem., Int. Ed., 2022, 61, e202117728 CrossRef CAS PubMed
- Z. Yan, L. Tang, Y. Huang, W. Hua, Y. Wang, R. Liu, Q. Gu, S. Indris, S. L. Chou, Y. Huang, M. Wu and S. X. Dou, Angew. Chem., Int. Ed., 2019, 58, 1412–1416 CrossRef CAS PubMed
- L. Zhang, J. Wang, G. Schuck, F. Xi, L. Du, M. Winter, G. Schumacher and J. Li, Small Methods, 2020, 4, 2000422 CrossRef CAS
- S. J. Park, J. Lee, G. H. Yoon, C. Koo, S. H. Lee, S. Koo, D. Kwon, S. H. Song, T. Y. Jeon, H. Baik, H. Kim, D. Kim and S. H. Yu, Energy Storage Mater., 2022, 53, 340–351 CrossRef
- H. Fang, H. Ji, J. Zhai, C. Wang, C. Zhu, G. Chen, M. Chu, T. Zhang, Z. Ma, W. Zhao, W. Ji and Y. Xiao, Small, 2023, 19, 2301360 CrossRef CAS PubMed
- S. Yuan, L. Yu, G. Qian, Y. Xie, P. Guo, G. Cui, J. Ma, X. Ren, Z. Xu, S. J. Lee, J. S. Lee, Y. Liu, Y. Ren, L. Li, G. Tan and X. Liao, Nano Lett., 2023, 23, 1743–1751 CrossRef CAS PubMed
- S. Chu, C. Zhang, H. Xu, S. Guo, P. Wang and H. Zhou, Angew. Chem., Int. Ed., 2021, 60, 13366–13371 CrossRef CAS PubMed
- Y. Wang, L. Wang, H. Zhu, J. Chu, Y. J. Fang, L. Wu, L. Huang, Y. Ren, C. J. Sun, Q. Liu, X. P. Ai, H. X. Yang and Y. L. Cao, Adv. Funct. Mater., 2020, 30, 1910327 CrossRef CAS
- Y. Cao, Q. Zhang, Y. Wei, Y. Guo, Z. Zhang, W. Huang, K. Yang, W. Chen, T. Zhai, H. Li and Y. Cui, Adv. Funct. Mater., 2019, 30, 1907023 CrossRef
- C. Zhao, Z. Yao, Q. Wang, H. Li, J. Wang, M. Liu, S. Ganapathy, Y. Lu, J. Cabana, B. Li, X. Bai, A. Aspuru-Guzik, M. Wagemaker, L. Chen and Y. S. Hu, J. Am. Chem. Soc., 2020, 142, 5742–5750 CrossRef CAS PubMed
- Y. Xiao, H. R. Wang, H. Y. Hu, Y. F. Zhu, S. Li, J. Y. Li, X. W. Wu and S. L. Chou, Adv. Mater., 2022, 34, 2202695 CrossRef CAS PubMed
- Y. F. Liu, H. R. Wang, J. Y. Li, M. J. Chen, H. Chen, B. Y. Lu, Q. Ma, X. W. Wu and X. X. Zeng, Appl. Surf. Sci., 2021, 541, 148294 CrossRef CAS
- J. Zhang, J. Gai, K. Song and W. Chen, Cell Rep. Phys. Sci., 2022, 3, 100868 CrossRef CAS
- N. Li, S. Wang, E. Zhao, W. Yin, Z. Zhang, K. Wu, J. Xu, Y. Kuroiwa, Z. Hu, F. Wang, J. Zhao and X. Xiao, J. Energy Chem., 2022, 68, 564–571 CrossRef CAS
- R. Zhang, C. Wang, P. Zou, R. Lin, L. Ma, L. Yin, T. Li, W. Xu, H. Jia, Q. Li, S. Sainio, K. Kisslinger, S. E. Trask, S. N. Ehrlich, Y. Yang, A. M. Kiss, M. Ge, B. J. Polzin, S. J. Lee, W. Xu, Y. Ren and H. L. Xin, Nature, 2022, 610, 67–73 CrossRef CAS PubMed
- X. Yang, S. Wang, H. Li, J. Peng, W. J. Zeng, H. J. Tsai, S. F. Hung, S. Indris, F. Li and W. Hua, ACS Nano, 2023, 17, 18616–18628 CrossRef CAS PubMed
- Q. Shen, Y. Liu, X. Zhao, J. Jin, Y. Wang, S. Li, P. Li, X. Qu and L. Jiao, Adv. Funct. Mater., 2021, 31, 2106923 CrossRef CAS
- J. Jin, Y. Liu, Q. Shen, X. Zhao, J. Zhang, Y. Song, T. Li, X. Xing and J. Chen, Adv. Funct. Mater., 2022, 32, 2203424 CrossRef CAS
- T. Jin, P. F. Wang, Q. C. Wang, K. Zhu, T. Deng, J. Zhang, W. Zhang, X. Q. Yang, L. Jiao and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 14511–14516 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, crystallography data and additional figures. See DOI: https://doi.org/10.1039/d3sc06878a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |