
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemo-selective Stille-type coupling of acyl-chlorides upon phosphine-borane Au(I) catalysis†
Nereida
Hidalgo
 ab,
Arnaud
Le Gac
a,
Sonia
Mallet-Ladeira
c,
Ghenwa
Bouhadir
*a and
Didier
Bourissou
*a
ab,
Arnaud
Le Gac
a,
Sonia
Mallet-Ladeira
c,
Ghenwa
Bouhadir
*a and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: didier.bourissou@univ-tlse3.fr
bDepartamento de Química Inorgánica, Universidad de Sevilla, 41071 Sevilla, Spain
cInstitut de Chimie de Toulouse (FR 2599), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France
First published on 1st March 2024
Abstract
Phosphine-boranes do not promote oxidative addition of acyl chlorides to gold, but the phosphine-borane gold triflimide complex [iPr2P(o-C6H4)BCy2]AuNTf2 was found to catalyze the coupling of acyl chlorides and aryl stannanes. The reaction involves aryl/chloride-bridged dinuclear gold(I) complexes as key intermediates, as substantiated by spectroscopic and crystallographic analyses. Similar to Pd(0)/Pd(II)-catalyzed Stille coupling with phosphine-borane ligands, the gold-catalyzed variant shows complete chemoselectivity for acyl chlorides over aryl iodides and bromides, enabling straightforward access to halogenated aryl ketones.
Introduction
Over the past 15 years, phosphine-boranes have emerged as versatile and powerful ligands. The presence of a Lewis moiety in the coordination sphere of transition metals, either directly bonded to the metal or pendant, has been shown to impart peculiar reactivity and to open new catalytic paths.1 In this context, we have shown that phosphine-boranes enable chemoselective activation and catalytic Stille coupling of acid chlorides at palladium.2Recently, we have started to explore the impact of phosphine-boranes on gold chemistry and discovered some unique features, namely facile organic group transfer between boron and gold(I)3 and Lewis acid-assisted C(sp3)–C(sp3) coupling at gold(III).4 Here, we investigated the possibility of activating acyl chlorides at gold thanks to the use of phosphine-borane ligands. This work was stimulated by two recent reports. First, Peters et al. achieved Stille coupling under gold catalysis for the first and only time so far. Gold nanoparticles supported on active carbon were shown to efficiently promote the coupling of allyl stannanes and alkyl bromides.5 Second, Gabbaï et al. demonstrated the ability of Lewis acids to trigger Au(I) to Au(III) oxidation using a phosphine–xanthylium ambiphilic ligand.6 As detailed hereafter, we found that phosphine-boranes do not emulate oxidation addition of acyl chlorides to gold, but we discovered that Stille-type coupling is possible under catalytic conditions and with complete chemoselectivity for acyl chlorides over aryl iodides and bromides. Aryl-bridged dinuclear gold(I) complexes with pendant borane moieties have been spectroscopically and structurally authenticated. They are proposed to be key intermediates for acyl chloride activation and aryl transfer.
Results & discussion
The phosphine-borane iPr2P(o-C6H4)BCy2 (referred to as PBCy2) was chosen for this study. It is easily accessible and rather stable. The Lewis acidity and steric hindrance of the borane moiety are intermediate compared to other phosphine-boranes. The (PBCy2)AuCl complex I was prepared following the reported procedure.7 Treatment with AgNTf2 then readily afforded the corresponding (PBCy2)AuNTf2 complex II (Scheme 1).8 For the sake of completeness, the (PBCy2)AuPh complex III, a possible intermediate in Stille coupling, was also targeted. It was synthesized by reacting I or II with half an equivalent of ZnPh2 (or one equivalent of PhZnCl). All complexes were characterized by multi-nuclear NMR spectroscopy and mass spectrometry. Single crystals of complex III were also analyzed by X-ray diffraction. The complex adopts T-shaped geometry with weak Au → B interaction. The Au⋯B distance (3.086(2) Å, r 1.40)9 and sum of CBC bond angles (Σ(CBC) 358.38°) are similar to those found in I (2.903(5) Å, r 1.32, 358.6°).7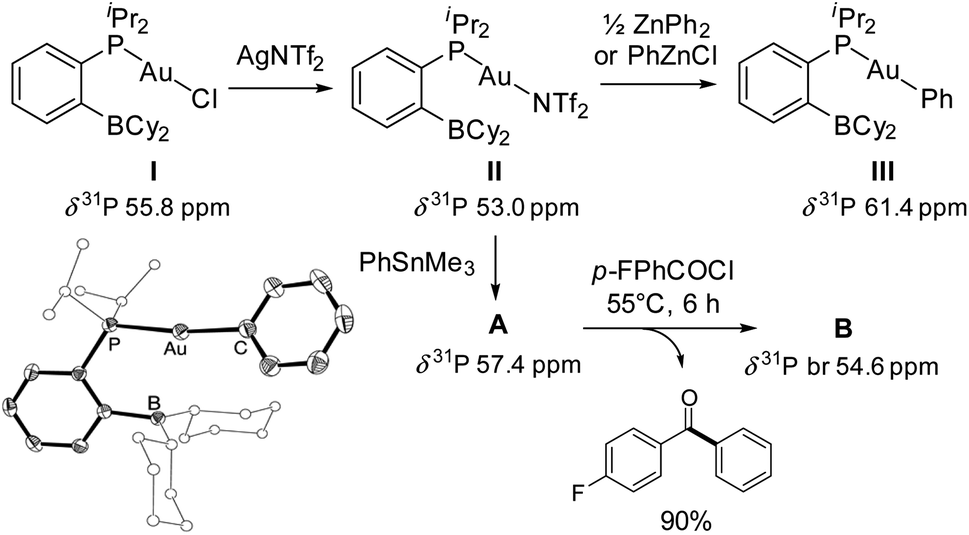 | ||
| Scheme 1 Synthesis of the (PBCy2) gold complexes II and III. Molecular structure of III (bottom left) with ellipsoids shown at 50% probability and H atoms omitted, and Cy/iPr substituents at boron/phosphorus simplified. Stoichiometric reactions with pF-PhCOCl and PhSnMe3. | ||
The PBCy2 gold complexes were then reacted with p-fluoro benzoyl chloride pF-PhCOCl and phenyltrimethyl stannane PhSnMe3 as benchmark substrates. With the acyl chloride alone, no reaction was observed with the chloro and triflimide complexes, as well as the phenyl complex, even upon heating at 65 °C for hours. Apparently, oxidative addition of acyl chloride to gold is not possible whatever the X coligand at gold (Cl, NTf2 or Ph) and despite the presence of the borane moiety.10 The (PBCy2)AuCl complex I also proved inert towards the stannane, but the triflimide complex II readily reacted with PhSnMe3 over 15 minutes at 25 °C to give a new complex A (Scheme 1), as indicated by the appearance of a new 31P NMR signal at δ 57.4 ppm (versus 53.0 ppm for II). This is not the (PBCy2)AuPh complex III whose 31P NMR signal appears at δ 61.4 ppm. Interestingly, addition of the acyl chloride pF-PhCOCl to complex A led after 6 hours of heating at 55 °C to the Stille coupling product, i.e. the pF-PhCO–Ph ketone (in 90% yield, as determined by 19F NMR spectroscopy with C6F6 as an internal standard), along with a new (PBCy2)Au complex B (associated with a broad 31P NMR signal at δ 54.6 ppm).
Consistently, reacting the triflimide complex II with one equivalent of pF-PhCOCl and PhSnMe3 afforded ketone coupling in 78% yield after 5 hours at 55 °C, and gratifyingly, the reaction also works under catalytic conditions (Scheme 2). Using 5 mol% of complex II, the pF-PhCO–Ph ketone was obtained in 89% yield after 12 hours at 55 °C. No difference was observed when the gold complex was generated in situ, from (PBCy2)AuCl and AgNTf2 (83% yield in ketone after 12 hours at 55 °C with 5 mol% catalyst). Conversely, the chloride complex (PBCy2)AuCl alone showed no activity, as did the diphosphine-borane [iPr2P(o-C6H4)]2BPh AuCl and AuNTf2 complexes.11 Of note, a mercury test performed with the (PBCy2)AuNTf2 catalyst showed no drop in efficiency of the Stille coupling, suggesting that homogeneous not nanoparticle catalysis is operating. A control experiment without gold was also performed and no coupling product was obtained with the phosphine-borane PBCy2 alone.
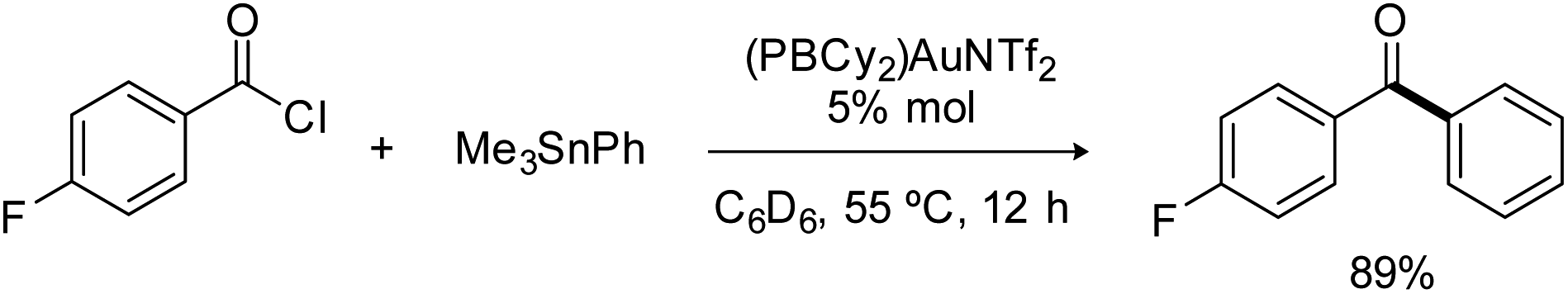 | ||
| Scheme 2 Stille coupling catalyzed by the PBCy2 triflimide gold complex II. | ||
The scope of the Stille coupling catalyzed by the (PBCy2)AuNTf2 complex II was then investigated. Besides pF-PhCOCl, four benzoyl chlorides were selected to test the compatibility of electron-donating and withdrawing groups and of substitution in ortho, meta and para positions (Scheme 3). In all cases, upon reaction with PhSnMe3, the ketone coupling product was obtained in good to high yield. The best result was obtained with pMeO-PhCOCl, 90% yield within 1 hour at room temperature, while pF3C-PhCOCl required prolonged heating at 70 °C and afforded only 50% of the ketone. Aliphatic acid chlorides are also efficiently coupled, including those featuring sterically demanding isopropyl, cyclohexyl and tert-butyl substituents. As for the stannane partner, the coupling of MesSnMe3 with pF-PhCOCl worked nicely, showing again good tolerance to steric hindrance.
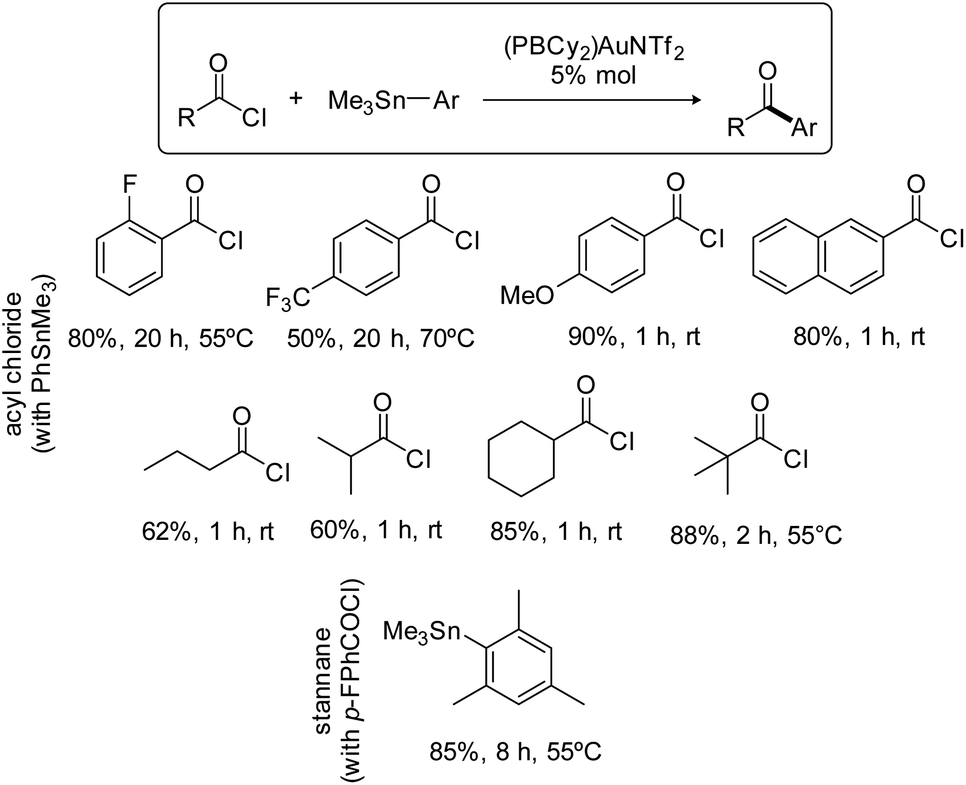 | ||
| Scheme 3 Scope of the Stille coupling catalyzed by the PBCy2 triflimide gold complex II. Yields in ketone, as determined by 19F or 1H NMR spectroscopy with internal standard, C6F6 or ClCH2CH2Cl. | ||
Having shown that phosphine-borane ligands impart complete chemoselectivity for acyl chlorides in the Pd-catalyzed Stille coupling,2 we then investigated the functional group compatibility of the phosphine-borane gold catalyzed transformation towards common aryl electrophiles (Scheme 4). First, competitive experiments were carried out using 1 equivalent of pF-PhCOCl acyl chloride and 1 equivalent of p-tolyl bromide, iodide or triflate. In all cases, the ArX (X = Br, I, OTf) additive remained intact and the yield of the ketone coupling product was not significantly compromised. Then, the coupling of bifunctional substrates was attempted. The pBr-PhCOCl and pI-PhCOCl acyl chlorides reacted selectively with PhSnMe3 to afford the corresponding p-halogenated dibenzophenones in 75–85% yield. In the same way, the coupling of the pI-PhSnMe3 stannane with pF-PhCOCl proceeded cleanly and efficiently to give the corresponding p-dihalogenated benzophenone (80% yield). Thus, the phosphine-borane gold complex II displays high selectivity for acyl chlorides and tolerates C(sp2)–X bonds in both partners of the transformation, enabling straightforward access to functionalized ketones.
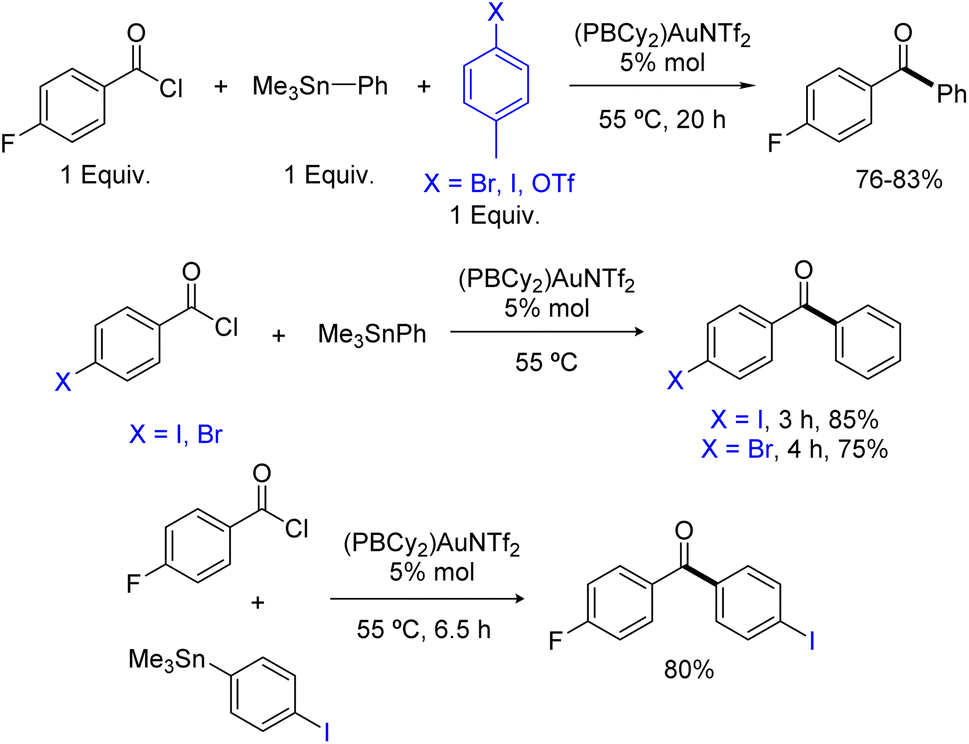 | ||
| Scheme 4 Chemoselectivity of the Stille coupling catalyzed by the PBCy2 triflimide gold complex II. Yields in ketone, as determined by 19F or 1H NMR spectroscopy with internal standard, C6F6 or ClCH2CH2Cl. | ||
Efforts were then made to decipher how the catalytic transformation works. To this end, we first aimed at isolating and characterizing the complexes A and B formed in our initial stoichiometric studies and supposed to be key intermediates. Complex A was most efficiently prepared by reacting the triflimide complex II with half an equivalent of PhSnMe3 in dichloromethane. It was isolated in 61% yield and unambiguously authenticated as the geminal digold(I) aryl complex (μ-Ph)[Au(PBCy2)]2NTf2 by single-crystal X-ray diffraction analysis (Scheme 5a). Since the seminal contribution of Schmidbaur et al. in 2003,12 such aryl-bridged dinuclear gold complexes have attracted considerable attention13 and been identified as off-cycle resting states in some catalytic transformations.14 The solid-state structure of complex A shows a W-shape conformation with an eclipsed arrangement of the PBCy2 ligands, which probably minimizes steric constraints. The Au⋯Au distance (2.795(1) Å) and Au–C–Au bond angle (79.9(3)°) fall in the middle range of those found in related complexes (2.70–2.88 Å and 76.6–85.7°),15 indicating strong aurophilic interaction. The Au⋯B distances (3.17(1) and 3.18(1) Å) are slightly elongated compared to that found in the (PBCy2)AuPh complex III (3.086(2) Å). Complex A was also characterized by multi-nuclear NMR spectroscopy and mass spectrometry. Worthy of note is the 13C NMR signal for the carbon atom of the phenyl ring bridging the two gold centers. While this signal has proved so far undetectable,12,16 it could be easily observed and authenticated for complex A. It appears as a triplet at δ 145.7 ppm (2JCP 49 Hz), shielded by about 33 ppm compared with the monometallic (PBCy2)AuPh complex III (δ 178.9 ppm, 2JCP 113 Hz), in line with a Wheland-type structure.17 The stability and catalytic activity of complex A were then investigated. It proved thermally robust (no sign of decomposition was detected by NMR spectroscopy after 2 days at 55 °C), gave the same results as complex II in the catalytic coupling of pF-PhCOCl or tBuCOCl and Me3SnPh (87% yield after 20 h at 55 °C and 70% yield after 1 h 30 at 55 °C, respectively) and did not interfere with C(sp2)–I bonds (as substantiated by the coupling of pF-PhCOCl and PhSnMe3 in the presence of p-tol-I as well as the coupling of pF-PhCOCl and p-IPhSnMe3).18
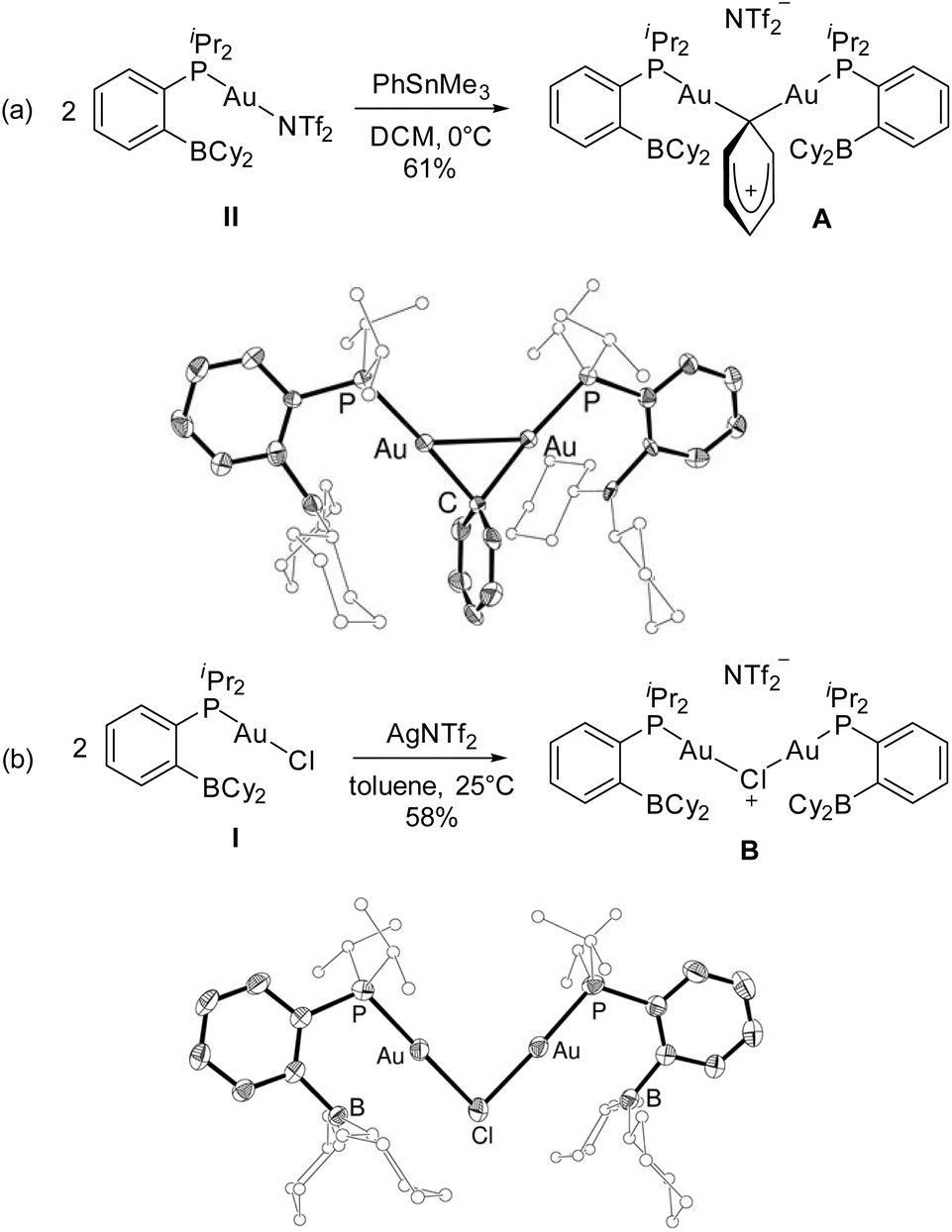 | ||
| Scheme 5 Synthesis and molecular structures of the PB-ligated dinuclear Au(I) complexes A (a) and B (b). Ellipsoids are drawn at 50% probability. For clarity, the counter-anion and hydrogen atoms are omitted, the iPr and Cy substituents are simplified, and only one molecule of the unit cell is shown for A. | ||
Complex B was formed along with the Stille coupling product upon the reaction of complex A with pF-PhCOCl (Scheme 1). Given the structure of A, we surmised the related chloro-bridged digold(I) structure for complex B. Independent synthesis upon reacting the chloro complex I with half-an-equivalent of AgNTf2 confirmed this hypothesis. The resulting complex showed spectroscopic features identical to those of B and it was unequivocally identified as (μ-Cl)[Au(PBCy2)]2,NTf2 by single-crystal X-ray diffraction (Scheme 5b). Like A, complex B adopts a W-shape eclipsed conformation in the solid state, but the Au⋯Au distance (3.2846(7) Å) and bridging Au–Cl–Au bond angle (88.39(3)°) are substantially larger. The Au⋯B distances (2.946(4)/2.988(4) Å) are similar to that found in the (PBCy2)AuCl complex I (2.903(5) Å). Chloro-bridged dinuclear Au(I) complexes are well-known.13 Their propensity to further aggregate and form clusters sustained by Au⋯Au aurophilic interactions has been recognized early on.19 They are also common by-products in the activation of LAuCl precatalysts with silver salts, often displaying reduced activity if not completely inert.20 To assess the possibility of catalytic turnover from complex B, it was reacted with Me3SnPh and pleasingly, the phenyl-bridged complex A was quantitatively reformed within 20 minutes at 25 °C.
To gain further mechanistic insights, kinetic investigations were performed on the coupling reaction between pF-PhCOCl and Me3SnPh with different concentrations of (PBCy2)AuNTf2II.18 Using the initial rate method, the partial order in the mononuclear gold(I) complex was found to be 0.5, in line with a dinuclear mechanism (see below). In addition, two catalytic reactions were monitored by 31P NMR spectroscopy.18 In both cases, the aryl-bridged digold(I) complex A was found to be very major species, with a minor (and increasing over the course of the reaction) amount of chloro-bridged digold(I) complex B. Complex A thus stands as the resting state of the catalytic cycle.
Based on all these experimental observations, we propose the catalytic cycle displayed in Fig. 1 to account for the chemoselective Stille coupling with the phosphine-borane gold(I) complex II. It does not proceed by oxidative addition of the acyl chloride to gold and Au(I)/Au(III) redox cycling,21,22 but rather operates at a constant Au(I) redox state with aryl-bridged dinuclear complexes such as A as key intermediates.
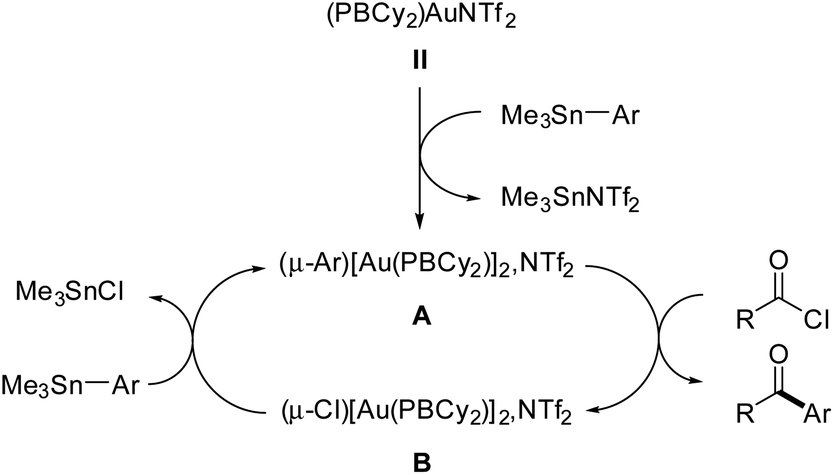 | ||
| Fig. 1 Catalytic cycle proposed to account for the Stille coupling of acyl chlorides with organostannanes with phosphine-borane gold(I) complexes. | ||
To assess the role of the borane moiety, Gagosz's complex (Ph3P)AuNTf2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 was tested in the catalytic coupling of pF-PhCOCl and PhSnMe3 under similar conditions. The coupling product was obtained in only 3% yield. The C–C bond formation is likely to occur via a Friedel–Crafts-type mechanism, although it is difficult to ascertain precisely in the absence of detectable intermediates besides the dinuclear gold(I) complexes A and B. At this stage, we surmise that the pendant borane moieties of complex A activate the acyl chloride while gold transfers the μ-aryl bridged group. It is worth mentioning that Friedel–Crafts coupling of acyl chlorides and aryl stannanes is known with strong Lewis acids such as AlCl3.24 Of note, no coupling was observed combining the aryl-bridged dinuclear gold complex (μ-MeOPh)[Au(PPh3)]2NTf216a with exogeneous boranes (BPh3 or BMes3),18 demonstrating the critical role of the phosphine-borane ligand. To further support the contribution of the borane moiety, the accessibility and Lewis acidity of boron were reduced. The cationic gold complexes (PB)AuNTf2 deriving from PBMes2 and PBpin phosphine-boranes25 showed significantly lower activity in the catalytic coupling of pF-PhCOCl and PhSnMe3 than that derived from PBCy2. The coupling product was obtained in only 62 and 23% yields after 20 hours at 55 °C with PBMes2 and PBpin, compared to 89% within 12 h with PBCy2.
23 was tested in the catalytic coupling of pF-PhCOCl and PhSnMe3 under similar conditions. The coupling product was obtained in only 3% yield. The C–C bond formation is likely to occur via a Friedel–Crafts-type mechanism, although it is difficult to ascertain precisely in the absence of detectable intermediates besides the dinuclear gold(I) complexes A and B. At this stage, we surmise that the pendant borane moieties of complex A activate the acyl chloride while gold transfers the μ-aryl bridged group. It is worth mentioning that Friedel–Crafts coupling of acyl chlorides and aryl stannanes is known with strong Lewis acids such as AlCl3.24 Of note, no coupling was observed combining the aryl-bridged dinuclear gold complex (μ-MeOPh)[Au(PPh3)]2NTf216a with exogeneous boranes (BPh3 or BMes3),18 demonstrating the critical role of the phosphine-borane ligand. To further support the contribution of the borane moiety, the accessibility and Lewis acidity of boron were reduced. The cationic gold complexes (PB)AuNTf2 deriving from PBMes2 and PBpin phosphine-boranes25 showed significantly lower activity in the catalytic coupling of pF-PhCOCl and PhSnMe3 than that derived from PBCy2. The coupling product was obtained in only 62 and 23% yields after 20 hours at 55 °C with PBMes2 and PBpin, compared to 89% within 12 h with PBCy2.
Conclusion
In summary, the phosphine-borane gold triflimide complex II was found to efficiently promote the Stille coupling of acyl chlorides and aryl stannanes with complete chemoselectivity over aryl iodides and bromides. The reaction involves an aryl-bridged dinuclear gold(I) complex as the key intermediate, as supported by NMR spectroscopy and X-ray diffraction.Over the past 5 years, ligand design has been shown to dramatically impact the reactivity profile of gold complexes, emulating new reactivity paths and enabling new catalytic transformations. Chelating P^P and hemilabile P^N ligands has in particular opened the way to a variety of Au(I)/Au(III) redox cross-coupling. The results reported here demonstrate that phosphine-boranes, prototype ambiphilic ligands, may also trigger unusual reactivity at gold and expand the scope of gold-catalyzed transformations. Future studies are planned to explore further the impact of Lewis acid moieties on the reactivity and catalytic activity of gold complexes. Special interest will also be devoted to the ability of ambiphilic ligands to drive chemoselectivity upon C–X bond activation at transition metals.
Data availability
All experimental and characterization data in this manuscript are available in the ESI.† Crystallographic data for complexes II, A and B have been deposited at the CCDC and assigned the numbers 2298251, 2298252 and 2298253, respectively.Author contributions
N. Hidalgo and A. Le Gac performed the experimental work and spectroscopic analyses. S. M.-L. carried out the X-ray diffraction analyses. All authors analyzed the data and contributed to the manuscript preparation. G. B. and D. B. conceived and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Centre National de la Recherche Scientifique (CNRS), the Université Paul Sabatier (UPS), the Agence Nationale de la Recherche (project ANR-CE07-0001-02) and the Région Occitanie (PhD fellowship to A. L. G.) are acknowledged for financial support of this work. N. H. thanks the University of Seville, Spanish Ministry of Universities and European Union (Margarita Salas grant funded by the European Union-Next Generation EU).Notes and references
- For selected reviews, see:
(a) A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC
; (b) H. Kameo and H. Nakazawa, Chem.–Asian J., 2013, 8, 1720 CrossRef CAS PubMed
- M. Boudjelel, O. Sadek, S. Mallet-Ladeira, Y. García-Rodeja, E. D. Sosa Carrizo, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2021, 11, 3822 CrossRef CAS
-
(a) C. A. Theulier, Y. García-Rodeja, N. Saffon-Merceron, K. Miqueu, G. Bouhadir and D. Bourissou, Chem. Commun., 2021, 57, 347 RSC
- C. A. Theulier, Y. García-Rodeja, K. Miqueu, G. Bouhadir and D. Bourissou, J. Am. Chem. Soc., 2023, 145, 10800 CrossRef CAS PubMed
- J. Holz, C. Pfeffer, H. Zuo, D. Beierlein, G. Richter, E. Klemm and R. Peters, Angew. Chem., Int. Ed., 2019, 58, 10330 CrossRef CAS PubMed
- L. C. Wilkins, Y. Kim, E. D. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266 CrossRef CAS PubMed
- S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed
- For gold(I) triflimide complexes deriving from vicinal phosphine-boranes, see: A. Ueno, K. Watanabe, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2019, 55, 4367 RSC
- To compare M → Z complexes, taking into account the size of the involved atoms, we commonly refer to the ratio r between the MZ distance and the sum of the covalent radii of M and Z. See ref. 1a.
- For a recent example of Au(I) to Au(III) oxidation assisted by the Lewis acid moiety of an ambiphilic ligand, see ref. 6.
-
(a) M. Sircoglou, S. Bontemps, M. Mercy, N. Saffon, M. Takahashi, G. Bouhadir, L. Maron and D. Bourissou, Angew. Chem., Int. Ed., 2007, 56, 8583 CrossRef PubMed
- R.-Y. Liau, A. Schier and H. Schmidbaur, Organometallics, 2003, 22, 4922 CrossRef
- T. A. C. A. Bayrakdar, T. Scattolin, X. Ma and S. P. Nolan, Chem. Soc. Rev., 2020, 49, 7044 RSC
-
(a) J. Preindl, K. Jouvin, D. Laurich, G. Seidel and A. Fürstner, Chem.–Eur. J., 2016, 22, 237 CrossRef CAS PubMed
- According to a Cambridge database search, slightly over 30 related (hetero)aryl-bridged dinuclear gold(I) complexes have been characterized by X-ray diffraction analysis. The mean values of Au⋯Au distances and Au–C–Au bond angles are 2.775 Å and 80.73°, respectively.
-
(a) D. Weber, T. D. Jones, L. L. Adduci and M. R. Gagné, Angew. Chem., Int. Ed., 2012, 51, 2452 CrossRef CAS PubMed
- A mono-metallic Wheland-type complex has been recently authenticated spectroscopically, see: M. S. M. Holmsen, C. Blons, A. Amgoune, M. Regnacq, D. Lesage, E. D. Sosa Carrizo, P. Lavedan, Y. Gimbert, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2022, 144, 22722 CrossRef CAS PubMed
- See the ESI.†.
-
(a) A. Hamel, N. W. Mitzel and H. Schmidbaur, J. Am. Chem. Soc., 2001, 123, 5106 CrossRef CAS PubMed
-
(a) A. Homms, I. Escofet and A. M. Echavarren, Org. Lett., 2013, 15, 5782 CrossRef PubMed
- For recent reviews dealing with Au(I)/Au(III) (photo)catalysis, see:
(a) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed
- We believe that this scenario is very unlikely for several reasons: (i) none of the features we found crucial to enable oxidative addition to gold are present in complexes II and A, (ii) the ensuing Au(III) species is likely to lie very high in energy (the phosphine-borane ligand cannot provide stabilization by chelation), and (iii) activation of the acyl chloride by the borane moiety (via O → B interaction) is not expected to enable oxidative addition to gold by itself.
- N. Mézailles, L. Ricard and F. Gagosz, Org. Lett., 2005, 7, 4133 CrossRef PubMed
-
(a) M. J. L. Feigo, G. F. Silbestri, A. B. Chopa and M. T. Lockhart, J. Org. Chem., 2011, 76, 1707 CrossRef PubMed
-
(a) M. W. P. Bebbington, S. Bontemps, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2007, 46, 3333 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2298251, 2298252 and 2298253. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06193k |
| This journal is © The Royal Society of Chemistry 2024 |