
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning dynamic coordination bonds in polar hybrid crystals for a high-temperature ferroelastic transition†
Yao-Bin
Li
a,
Xiao-Xian
Chen
a,
Wei-Jian
Xu
 b,
Ya-Ping
Gong
a,
Hui
Ye
a,
Zhi-Shuo
Wang
a and
Wei-Xiong
Zhang
*a
b,
Ya-Ping
Gong
a,
Hui
Ye
a,
Zhi-Shuo
Wang
a and
Wei-Xiong
Zhang
*a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, IGCME, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: zhangwx6@mail.sysu.edu.cn
bDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, 3810-193 Aveiro, Portugal
First published on 30th January 2024
Abstract
Ferroelastic materials have gained widespread attention as promising candidates for mechanical switches, shape memory, and information processing. Their phase-transition mechanisms usually originate from conventional order–disorder and/or displacive types, while those involving dynamic coordination bonds are still scarce. Herein, based on a strategic molecular design of organic cations, we report three new polar hybrid crystals with a generic formula of AA′RbBiCl6 (A = A′ = Me3SO+ for 1; A = Me3SO+ and A′ = Me4N+ for 2; A = A′ = Me3NNH2+ for 3). Their A-site cations link to the [RbBiCl6]n2n− inorganic framework with lon topology through Rb–O/N coordination bonds, while their significantly different interactions between A′-site cations and inorganic frameworks provide distinct phase-transition behaviour. In detail, the strongly coordinative A′-site Me3SO+ cations prevent 1 from a structural phase transition, while coordinatively free A′-site Me4N+ cations trigger a conventional order–disorder ferroelastic transition at 247 K in 2, accompanied by a latent heat of 0.63 J g−1 and a usual “high → low” second-harmonic-generation (SHG) switch. Interestingly, the A′-site Me3NNH2+ cations in 3 reveal unusual dynamic coordination bonds, driving a high-temperature ferroelastic transition at 369 K with a large latent heat of 18.34 J g−1 and an unusual “low → high” SHG-switching behaviour. This work provides an effective molecular assembly strategy to establish dynamic coordination bonds in a new type of host–guest model and opens an avenue for designing advanced ferroelastic multifunctional materials.
Introduction
Ferroelastics, crucial ferroic materials known for their stress–strain behaviour, play a pivotal role in various applications, including mechanical switches, shape memory, and actuator.1–5 Among them, research on organic-inorganic hybrid crystals exhibiting ferroelastic phase transitions has thrived over the past decade, benefiting from their structural diversity, eco-friendly synthesis, and mechanical flexibility.6–15 Currently, the majority of documented ferroelastic phase transitions in hybrid crystals can be classified as mechanisms involving order–disorder and/or displacive type mechanisms.16–27 For instance, (homopiperazine-1,4-diium)[K(BF4)3] exhibits a ferroelastic phase transition above room temperature, which is attributed to synergic order–disorder transitions of organic cations and BF4− bridges.19 Besides, an unconventional type of structural phase transition, termed the bond-switching phase transition, has gained significant attention.28–31 Unlike order–disorder and displacive ones, bond-switching phase transitions, characterized by reversible generation/cleavage of coordination bonds, induce dramatic changes in the crystal structure crossing different crystal systems, thereby facilitating the triggering of ferroelastic transitions.32,33 Understanding the mechanism behind such unconventional bond switching is essential for the design of novel ferroic materials. However, the relevant research is in its infant stages, as there are very few examples of hybrid crystals with dynamic (switchable or changeable) coordination bonds.We previously reported two cyanide-bridged hybrid crystals, (Me3NOH)[KFe(CN)6] and (Me3NNH2)2[Co(CN)6Na(H2O)], both of which show bond-switching transitions.28,33 We noticed that the switchable coordination bonds should be highly associated with delicate host–guest interactions, which are significantly related to the selection of the host–guest model, metal ions, and organic cations with coordination groups. In this context, besides the few known examples of bond-switching transitions in cyanide-bridged hybrid systems, it is a prerequisite to broaden the scope of host–guest systems for comprehensively understanding and exploring bond-switching transitions.
Recently, our attention was captivated by some bimetal halides with non-perovskite structures,34–36 such as (R-3-hydroxypyrrolidinium)2RbBiBr6 and (3-hydroxypyrrolidinium)2RbBiBr6.34,35 They share the same formula of A2BB′X6, where A is an organic cations, X is a halogen anion, and B and B′ are trivalent and monovalent metal cations, respectively, and adopt a non-perovskite three-dimensional (3D) structure consisting of a [BB′X6]n2n− inorganic framework with a fascinating 4-connected lon topology created through alternating connected face- and corner-sharing Rb/Bi-centred polyhedra, resembling the 4H BaRuO3 structure.37,38 From a microscopic perspective, the interaction of the organic components with an inorganic framework through Rb–O coordination bonds provides a significant clue for designing bond-switching transitions. However, it is likely due to their larger size that the employed cyclic organic cations (such as R-3-hydroxypyrrolidinium and 3-hydroxypyrrolidinium) are hard to rotate in the relatively smaller pores but are strongly connected to the inorganic framework with coordination bonds, and thus their structural phase-transition mechanisms are not involved in dynamic coordination bonds.
Herein, we introduce some globular organic cations with coordination capacity to design new hybrid crystals based on the strategy of coordinate unit-transmutation from a 4H BaRuO3 structure and demonstrate how to achieve an unconventional transition with dynamic coordination bonds. Three new hybrid crystals with a formula of AA′RbBiCl6 (A = A′ = Me3SO+ for 1; A = Me3SO+ and A′ = Me4N+ for 2; A = A′ = Me3NNH2+ for 3) were successfully synthesized, in which all A-site cations coordinatively link to Rb+ ions whereas the different coordination interactions of A′-site cations endow 1–3 with distinct phase-transition behaviours (Scheme 1). In detail, 1 remains phase stable as its A′-site Me3SO+ cations form strong Rb–O coordination bonds. 2 undergoes a phase transition at 247 K, mainly induced by the order–disorder transition of coordinatively free A′-site Me4N+ cations, suggesting that smaller-sized organic cations that are reasonably substituted drive the dynamics changes of the coordination bond. By contrast, 3 undergoes a phase transition with a large latent heat at 369 K as its A′-site Me3NNH2+ cations reveal unusual dynamic coordination bonds, i.e., changing from a coordinative and order state at a room-temperature phase to a much weaker coordinative and disorder state at a high-temperature phase. Detailed structural phase-transition analyses were conducted by using thermal methods, dielectric measurements, and variable-temperature single-crystal structures, highlighting the role of organic cations in promoting bond-switching transitions. Noteworthily, 2 and 3 exhibit distinct heat-driven second-harmonic-generation (SHG) behaviour due to vastly different changes of coordination interactions, i.e. a usual “high → low” SHG-switching behaviour in 2 and an unusual “low → high” SHG-switching behaviour in 3.
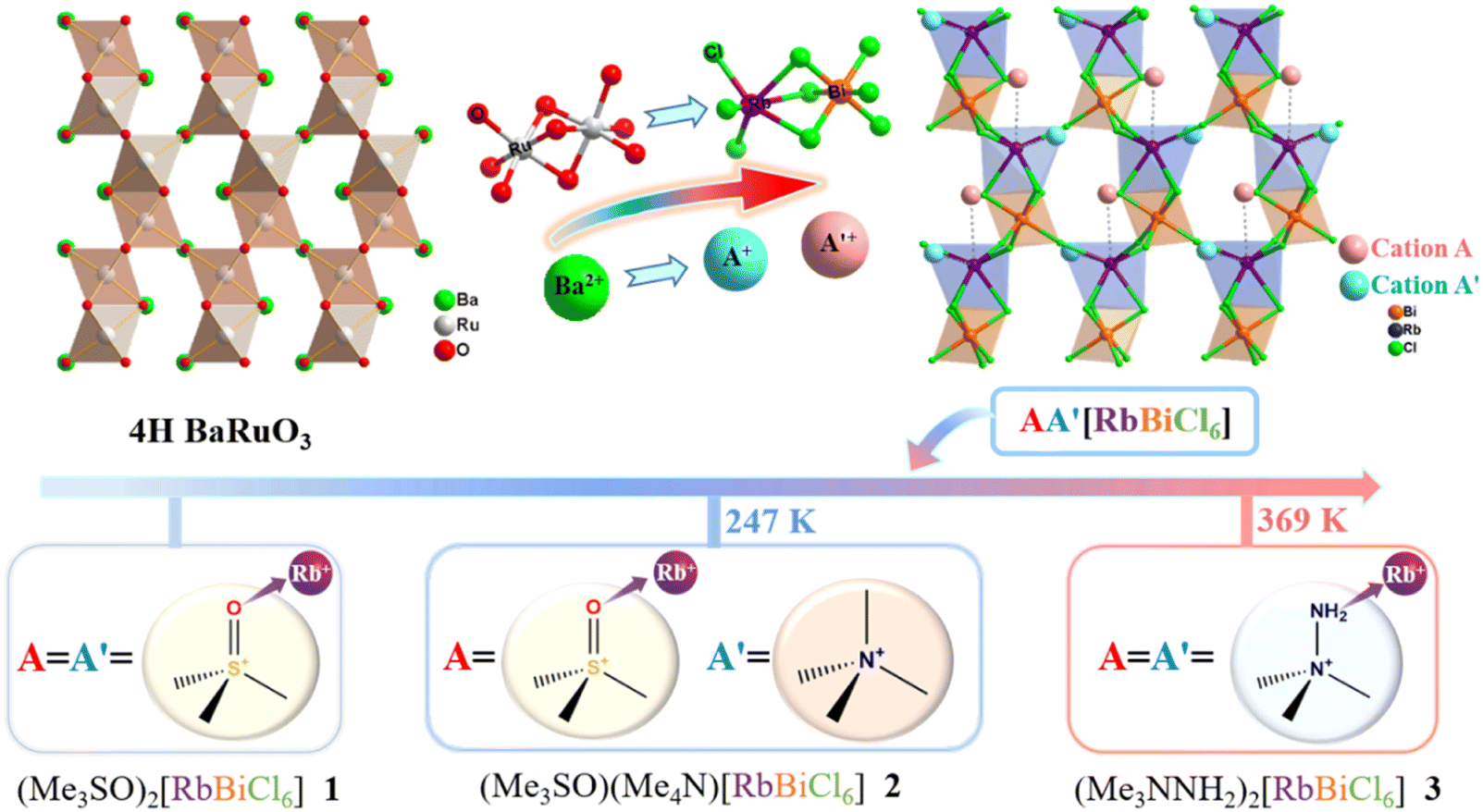 | ||
| Scheme 1 Three new polar hybrid crystals obtained by modifying the 4H BaRuO3 structure and a strategic molecular design of organic cations, which adopt a generic formula of AA′[RbBiCl6], exhibiting different phase-transition behaviours due to different coordination interactions of A′-site organic cations. | ||
Experimental
Materials and syntheses
All chemicals were commercially available and used without further purification. The crystals of the three compounds were prepared by the slow evaporation of the hydrochloric acid solutions of RbCl, BiCl3, and corresponding organic ammonium salts in a stoichiometric ratio. The yield was 82% for 1, 75% for 2 and 72% for 3, respectively, based on BiCl3. The purities of samples were verified using powder X-ray diffraction (PXRD) patterns (Fig. S1–S3†).Instrumentation
PXRD patterns were obtained using a Bruker D8 ADVANCE X-ray powder diffractometer (Cu Kα, λ = 1.54184 Å). Using 5 mg powder samples, thermogravimetric analysis (TGA) was carried out on a TG 209 F3 Tarsus system with a heating rate of 10 K min−1 under a nitrogen atmosphere. Differential scanning calorimetry (DSC) measurements were performed on a TA DSC Q2000 instrument by using 8 mg powder samples. Using powder-pressed samples (plate samples with an area of 113 mm2 and a thickness of 0.3 mm), the temperature-dependent dielectric measurement was performed on a TH2838 impedance analyser at frequencies from 10 kHz to 1 MHz, with an applied voltage of 1.0 V. The SHG signal was measured using an XPL1064-200 instrument at a heating/cooling rate of 5 K min−1. The observation of ferroelastic domains was performed by using an OLYMPUS BX41 polarizing microscope at a heating/cooling rate of 3 K min−1.Single-crystal X-ray diffraction data were collected on a SuperNova single-crystal diffractometer by using Mo Ka (λ = 0.71073 Å) radiation. Absorption corrections were applied by using the multi-scan program CrysAlisPro. All structures were solved by direct methods and refined by the full-matrix least-squares technique with the SHELX program package on Olex2.39,40 Anisotropic thermal parameters were applied to all non-hydrogen atoms. The hydrogen atoms were generated geometrically. Detailed crystallographic data and structural refinement parameters are listed in Tables S1 and S2.†
Results and discussion
Thermal analyses
Thermogravimetric analysis indicated that the three compounds exhibit thermal stability up to 490 K (1), 510 K (2), and 540 K (3), respectively. DSC measurements were conducted to analyze the phase transition behaviours. As shown in Fig. 1, the DSC curves for 1 do not exhibit any endothermic or exothermic peaks, indicating the absence of a structural phase transition within the measured temperature range. In contrast, distinct heat anomalies were observed during heating and cooling processes for 2 and 3. In detail, we observed a pair of endothermic/exothermic peaks at 247/245 K during the heating/cooling runs for 2, indicating a reversible phase transition, while 3 exhibited a pair of prominent thermal anomalies at 369/364 K, revealing an above-room-temperature phase transition. The total latent heat values of 2 and 3 in the heating process are estimated to be 0.63 and 18.34 J g−1, respectively. Accordingly, their total entropy changes (ΔS) in the heating process are estimated to be 1.71 and 32.68 J mol−1 K−1, respectively. In comparison, 3 reveals a greater thermal stability, a higher phase-transition temperature, a larger latent heat, and a larger entropy change than 2, implying relatively dramatic changes during the phase transition in 3. For convenience, the phases above phase-transition temperature are labeled as the high-temperature phases (HTPs) and the phases below phase-transition temperature as the low-temperature phases (LTPs).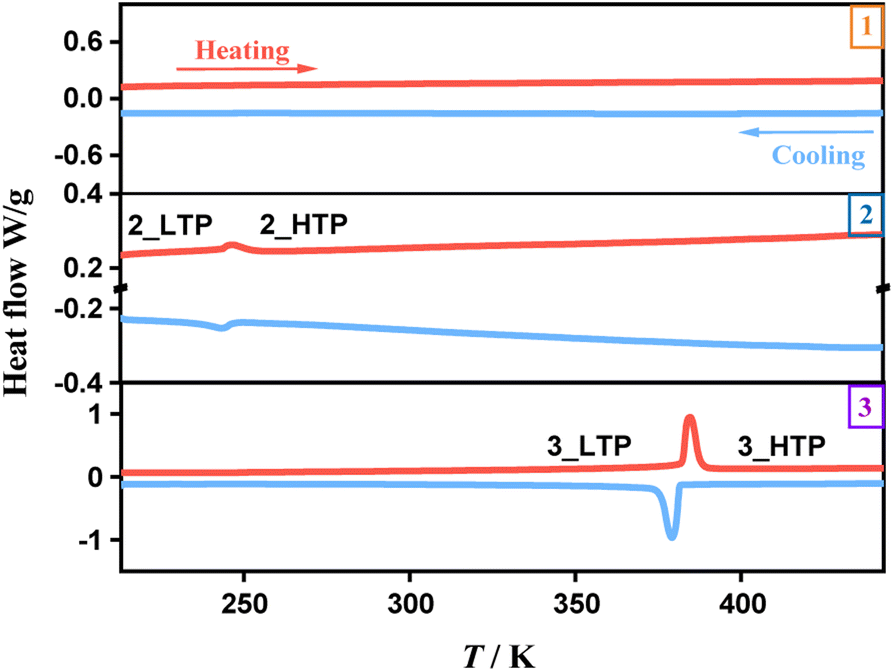 | ||
| Fig. 1 DSC curves in a heating–cooling run for 1, 2, and 3. | ||
Crystal structures
The crystal structures of the three compounds were determined by using variable-temperature single-crystal X-ray diffraction to investigate their phase transition mechanisms. 1 adopts a 3D structure formed by ordered Me3SO+ cations and an [RbBiCl6]n2n− anionic framework with a polar space group of P1121 at 210 K. As shown in Fig. 2a, each Bi3+ ion is surrounded by six Cl− ions to form a BiCl6 octahedron, while each Rb+ ion is located in a distinct RbCl6O2 dodecahedron and coordinated by six Cl− ions and two axially occupied O atoms from two organic cations, namely, the guest cations link to the host inorganic skeleton by Rb–O coordination bonds. Notably, the structure of 1 is distinctive to conventional 3D hybrid double perovskites, such as (CH3NH3)2KGdCl6 and (CH3NH3)2AgBiBr6, where both metal halogen octahedra are interconnected through corner-sharing mode.41,42 It features a unique 3D network where the BiCl6 octahedron and RbCl6O2 dodecahedron are alternately linked through a combination of corner-sharing and face-sharing. Such a 3D inorganic framework can be described as a 4-connected lon topology with a Schlafli symbol of 66, in which both the Rb and Bi centres are the 66 nodes. Furthermore, the inorganic BiCl6 octahedron exhibits a slightly distorted configuration, as evidenced by the unequal Bi–Cl lengths (Table S3†). The RbCl6O2 dodecahedron shows more pronounced distortion due to the presence of Rb–O coordination bonds. Specifically, the Rb–O coordination bond lengths (2.782 and 2.995 Å) are significantly shorter than the Rb–Cl bond lengths (ranging from 3.346–3.448 Å).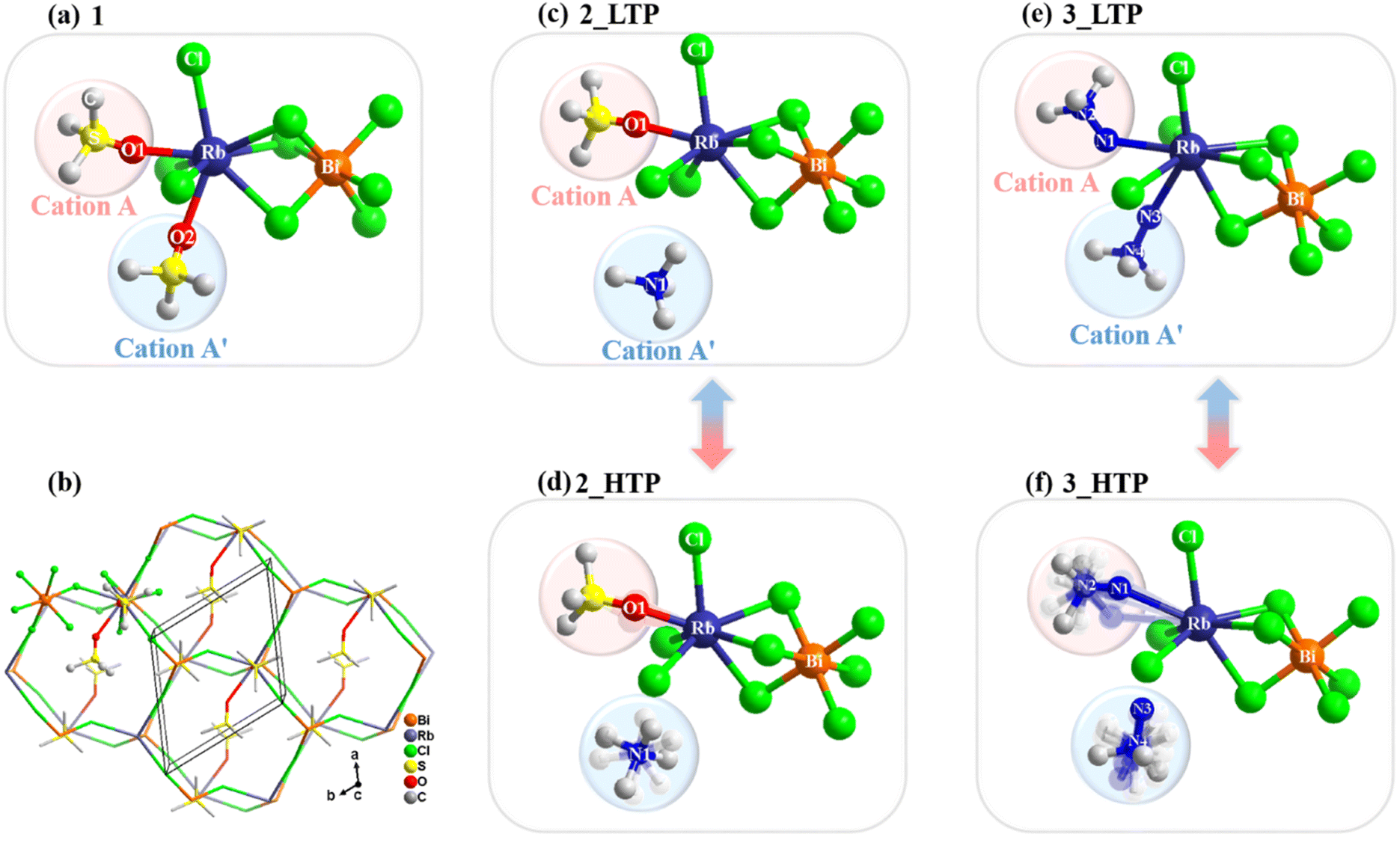 | ||
| Fig. 2 The basic unit of the framework in 1 (a), 2_LTP (c), 2_HTP (d), 3_LTP (e), and 3_HTP (f). (b) Perspective view of the packing structure along the c-axis of 1. H atoms of the organic cations were omitted for clarity. | ||
The crystal structure of the 2_LTP closely resembles that of 1, adopting a 3D lon topology structure and crystallizing in monoclinic space group P1121. As shown in Fig. 2c, each Bi3+ ion forms coordination with six Cl− ions, creating a BiCl6 octahedron. Each Rb+ ion connects with six Cl− ions and one Me3SO+ ion, while Me4N+ cations occupy the void of the 3D inorganic framework formed by the alternately connected BiCl6 octahedron and RbCl6O decahedron. Likewise, the collective arrangement of the organic components and inorganic framework results in polarity along the c axis.
Upon heating to above 247 K (2_HTP), the crystal structure transformed into hexagonal space group P63. As shown in Fig. 2d, Me3SO+ cations have slight swing while Me4N+ cations become highly disordered as they can freely rotate in the voids of the 3D framework. Consequently, the phase transition mechanism of 2 can be classified as an order–disorder type. During the phase transition from P63 to P1121, the number of symmetry operations decreased from 6 (E, 2C6, 2C3, and C2) to 2 (E and C2), indicating that 2 belongs to a ferroelastic transition (6F2) according to Aizu.43
Similarly, the 3_LTP features a 3D [RbBiCl6]n2n− anionic inorganic framework with lon topology and well-ordered organic cations. As shown in Fig. 2e, each Bi3+ coordinates with six Cl− ions, with Bi–Cl bond lengths ranging from 2.685–2.736 Å. The Rb+ ion exhibits a dual coordination environment, involving six Cl− ions (Rb–Cl bond lengths of 3.258–3.524 Å) and two axially occupied –NH2 groups from two Me3NNH2+ cations. Both weak Rb–N coordination interactions (3.217–3.570 Å) and N–H⋯Cl hydrogen bonds lead to an orthorhombic Pna21 space group for the 3_LTP, distinguishing between 1 and 2. The pronounced structural distortion in the RbCl6N2 dodecahedron of 3, akin to that observed in the RbCl6O2 polyhedron in 1, primarily arises from the unequal bond lengths in the Rb–Cl and Rb–N bonds.
Upon heating, the structure transforms from the orthorhombic Pna21 phase (3_LTP) into polar hexagonal space group P63mc (3_HTP). In the 3_HTP, both Me3NNH2+ organic cations become 6-fold disordered, while half of them no longer remain connected to Rb+ ions through Rb–N coordination interactions. Meanwhile, the BiCl6 octahedron shows a more pronounced distortion than that in the 3_LTP (Bi–Cl bond lengths, 2.681–2.721 Å). Therefore, it is reasonable to conclude that during the transition from the 3_LTP to the 3_HTP, the dynamical motions of organic cations drive the Rb–N coordination bonds to experience a bond-switching behaviour from a bonded state to a broken state. Such a unique phase-transition mechanism stands in stark contrast to the mechanisms observed in previously reported metal halides. Moreover, such bond-switching behaviour in 3 leads to a larger entropy change compared to that in 2, which could provide a significant clue for exploration of barocaloric materials.33,44 The space group changes from P63mc in the 3_HTP (with symmetric elements: E, 2C6, 2C3, C2, 3σv, and 3σd) to Pna21 in the 3_LTP (with symmetric elements: E, C2, and 2σv), following the symmetry breaking principle with an Aizu notation 6mmFmm2, indicating a possible ferroelastic transition.
Temperature-dependent SHG signal and dielectric responses
The structural phase transitions in 2 and 3 were further verified by variable-temperature SHG and dielectric measurements. Fig. 3a and c depict the temperature-dependent SHG signal of powder samples for 2 and 3, showing their SHG-activating nature in all phases, consistent with non-centrosymmetric structures as revealed by single-crystal X-ray diffraction. The transition temperatures of the SHG signal are coincident with the corresponding peak temperatures observed in DSC curves, testifying their structural phase transitions. For 2, a noticeable SHG signal (0.42 times of potassium dihydrogen phosphate, KDP) was observed at the 2_LTP, and gradually decreased to 0.12 times of KDP for the 2_HTP, exhibiting a common “high → low” SHG-switching behaviour. In contrast, for 3, the SHG signal at the 3_LTP was about 0.22 times of KDP and increased to 0.46 times of KDP at the 3_HTP, exhibiting an unusual “low → high” SHG-switching behaviour. This phenomenon differs from most of the known SHG switches, which reveal an “on → off” or “high → low” SHG switch upon heating,45–47 such as NH4[(CH3)4N]SO4·H2O and [(R)-3-hydroxy-pyrrolidinium]5Sb4Br17.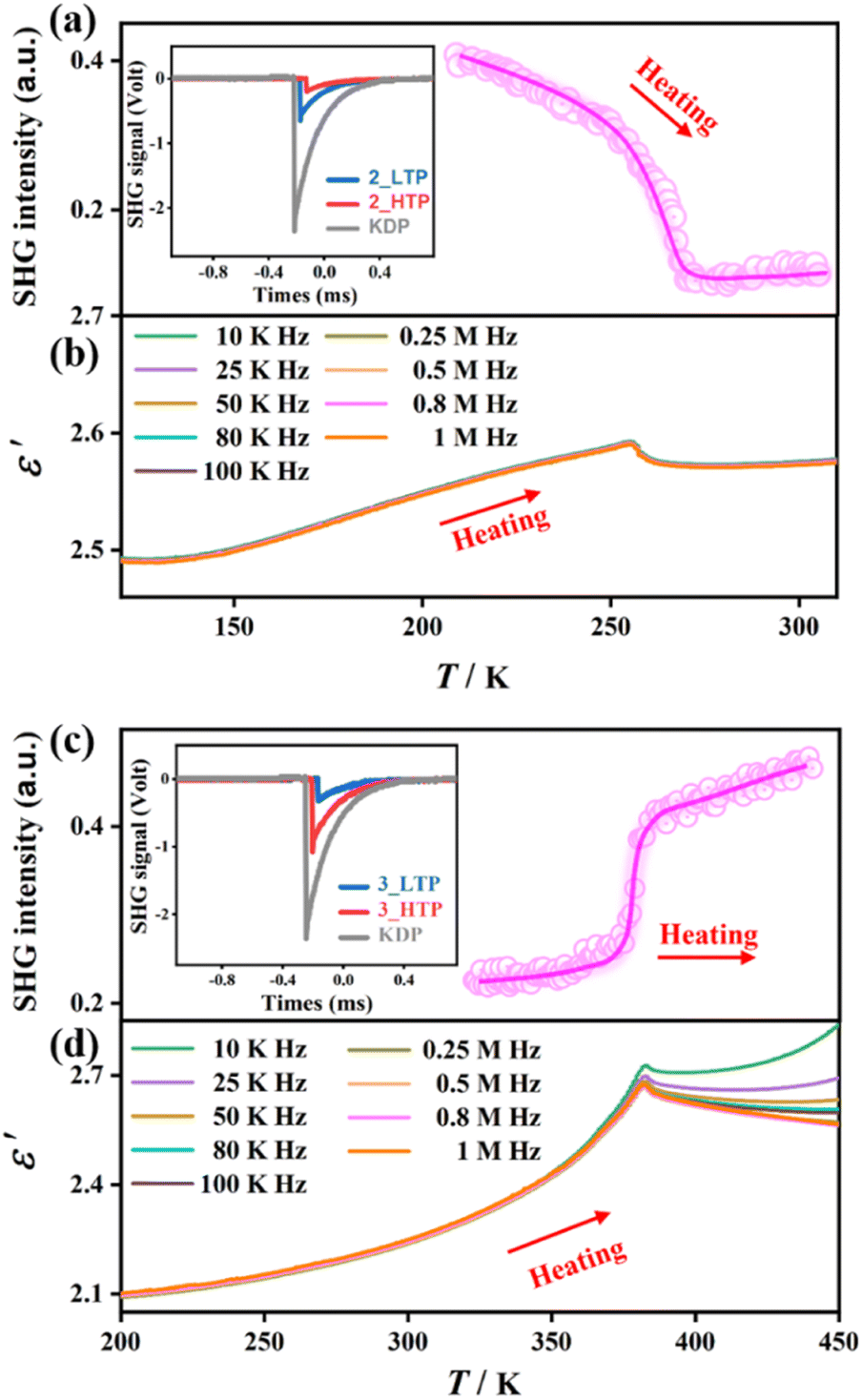 | ||
| Fig. 3 Temperature-dependent SHG intensity and the real part of complex dielectric permittivity for 2 (a) and (b) and 3 (c) and (d). | ||
The different SHG behaviours between 2 and 3 indicate a direct correlation between the observed SHG variations and their unique structural changes, particularly related to the distortion of inorganic octahedra and the order–disorder transition of organic cations. To understand structure–property relationship, we quantitatively estimated the distortion of the octahedra (Δd) using the following equation:
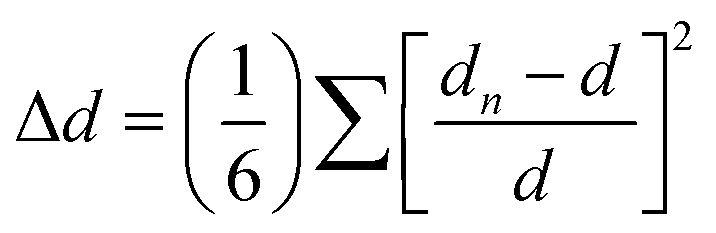
The variable-temperature dielectric constants were measured on the pressed polycrystalline samples for 2 and 3. Upon heating, as illustrated in Fig. 3b and d, prominent dielectric anomalies are observed near the phase-transition temperature, consistent with the results of DSC measurement. Such dielectric anomalies recur during both the heating and cooling processes, demonstrating the reversible nature of phase transitions. Specifically, upon heating, 2 exhibits a slight dielectric anomaly, with the real part of dielectric constants (ε′) at a frequency of 1 MHz varying from 2.49 at 120 K to 2.57 at 290 K. In contrast, 3 exhibits relatively noticeable dielectric anomalies around the phase-transition temperature, with the ε′ value varying from 2.1 at 200 K to 2.6 at 440 K. The distinct dielectric responses should be attributed to the order–disorder transition of the non-polar Me4N+ cation in 2 and the polar Me3NNH2+ cation in 3.50
Temperature-dependent lattice parameters
The temperature-dependent lattice parameters were obtained via Pawley refinements on the variable-temperature PXRD patterns in the temperature range of 193–303 K for 2 and 333–413 K for 3 (Tables S6 and S7†), and the thermal expansion coefficients were calculated using the PASCal program (Table S10†).51 The volume expansion coefficient decreases from +240 × 10−6 K−1 in the 2_LTP to +140 × 10−6 K−1 in the 2_HTP and decreases from +260 × 10−6 K−1 in the 3_LTP to +185 × 10−6 K−1 in the 3_HTP. As shown in Fig. 4a, upon heating, an increase of the crystallographic axes by 0.13–0.97% and a slight increase in cell volume by ca. 0.39% at 245 K were observed during the transition from the 2_LTP to the 2_HTP. In contrast, during the transition from the 3_LTP to the 3_HTP at 369 K, a significant decrease in the a-axis length by 2.49% was observed. Meanwhile, a substantial increase was observed in the b-axis lengths by 3.08%, while an increase in c-axis lengths by 0.63% and a slight increase in cell volume by 1.15% were detected. Such large changes on the a-axis and b-axis lengths in 3 are in accord with the larger entropy changes and dielectric anomalies arising from the dynamic coordination bonds.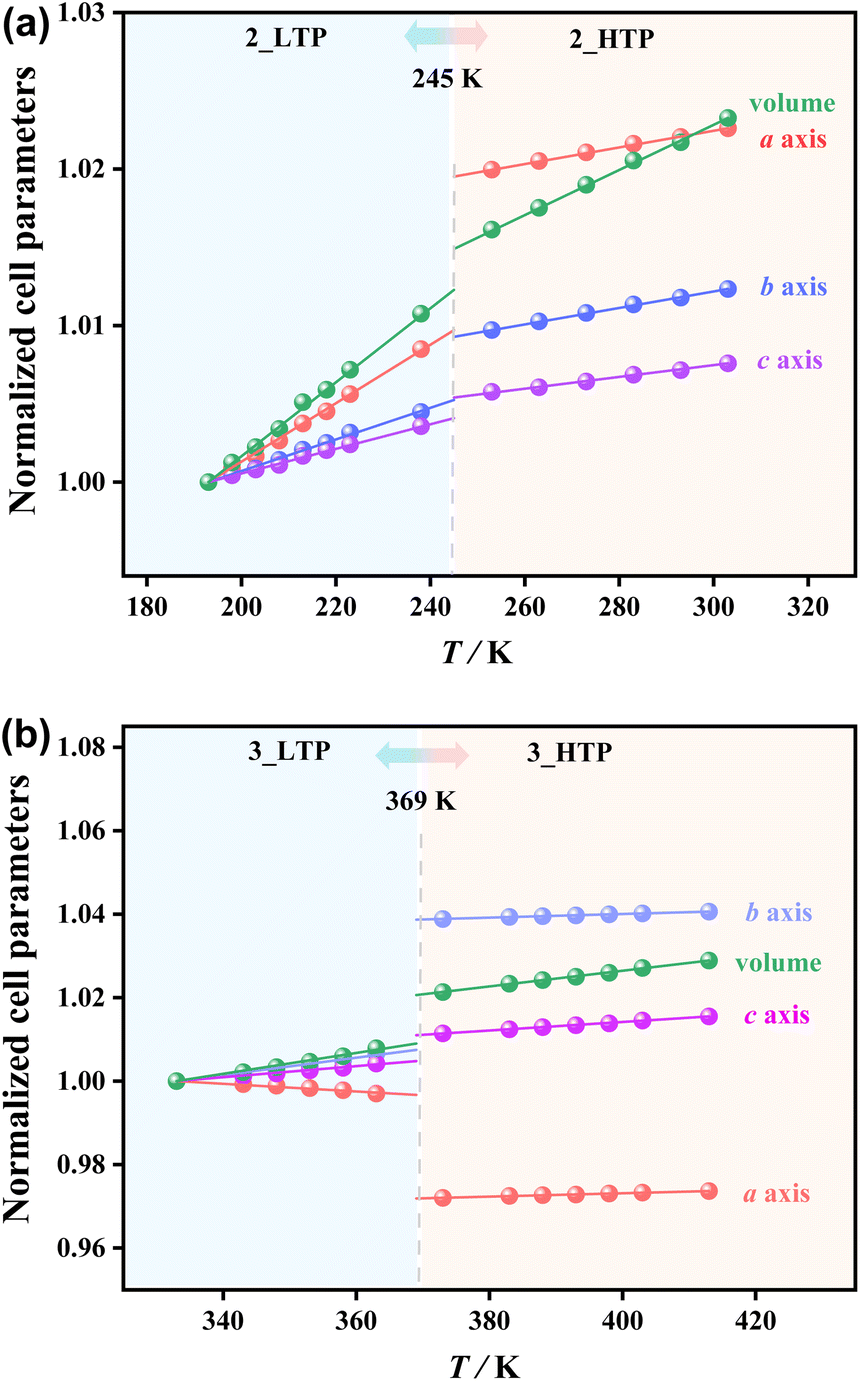 | ||
| Fig. 4 Temperature-dependent normalized cell parameters for 2 (a) and 3 (b). | ||
Ferroelastic phase transitions and spontaneous strains
To further verify the ferroelastic phase transition for 2 and 3, the evolution of domains on an as-grown single crystal was inspected by thermal stimulus in polarization microscopy (Fig. S12 and S13†). As shown in Fig. 5a, no observable domain structures appeared at the 2_HTP (the paraelastic phase). As the sample cooled to 240 K (the ferroelastic phase), the complex but fuzzy domains emerged, which faded away upon reheating to the paraelastic phase. Moreover, using the cell parameters at 245 K deduced by linear fitting variable-temperature cell parameters (Table S8†), the total spontaneous strain of 0.0167 was estimated for 2, and the fuzzy ferroelastic domains might be ascribed to the small spontaneous strain. As mentioned above in structure analysis, 3 exhibits a one-step ferroelastic transition with an Aizu notation of 6mmFmm2 accompanied by a total spontaneous strain of 0.0399 from the cell parameters at 369 K deduced by linear fitting variable-temperature cell parameters (Table S9†). These spontaneous strain values are among the intermediate levels in the reported hybrid ferroelastics.6–15 It should be emphasized that the greater spontaneous strain in 3 compared to 2 is closely associated with more drastic structural changes. As shown in Fig. 5b, a multi-domain structure was absent at the 3_LTP, with the mono-domain state remaining persistent through the 3_HTP. Upon cooling the sample to 360 K (the ferroelastic phase), evident striped domains emerged, disappearing when heated back to 375 K. | ||
| Fig. 5 Polarized microscopy observations at different temperatures for 2 (a) and 3 (b). | ||
Hirshfeld surface analyses
These three compounds share a similar host inorganic framework but differ in their guest organic cations, implying that their distinct phase-transition behaviours primarily stem from varying host–guest intermolecular interactions. Therefore, a comprehensive investigation into the intermolecular interactions among the three compounds was performed by Hirshfeld surface analyses.52 The surfaces are depicted using a colour scheme of red, white, and blue indicating the shorter contacts, the van der Waals separation, and longer contacts, respectively. As shown in Fig. 6a, d and g, the red areas of the surfaces for guest cations originate from the H⋯Cl and O/N⋯Rb contacts in 1, 2, and 3, respectively, revealing that the predominant host–guest interactions between the inorganic skeleton and organic cations are hydrogen bonding interactions and Rb–O/N coordination bonds. Among them, A-site cations always coordinatively link to Rb+ ions whereas the A′-site cations adopt different coordination modes; thus A-site cations from the three compounds exhibit the same host–guest interactions, while their A′-site cations show markedly different host–guest interactions. In decomposed fingerprint plots (Fig. 6c, f and i), approximately 3.3%, 0%, and 1.9% of the Hirshfeld surface area of A′-site cations were associated with Rb–O/N coordination bonds in 1, 2, and 3, respectively. Meanwhile, the smallest (di, de) pairs from A′-site cations indicated that the strongest O/N⋯Rb short contacts are (1.36 Å, 1.64 Å) for 1 and (1.61 Å, 1.91 Å) for 3. Specifically, the interactions associated with the Rb–O coordination bond in 1 appear to be stronger than that of the Rb–N coordination bond in 3. This observation suggests that the Rb–O coordination bond formed by Me3SO+ cations is more resilient and less prone to breakage compared to the Rb–N coordination bond from Me3NNH2+ cations. Consequently, 1 does not undergo a bond-switching phase transition before its decomposition, whereas 3 undergoes an above-room-temperature phase transition due to the changeable/dynamic Rb–N coordination bonds.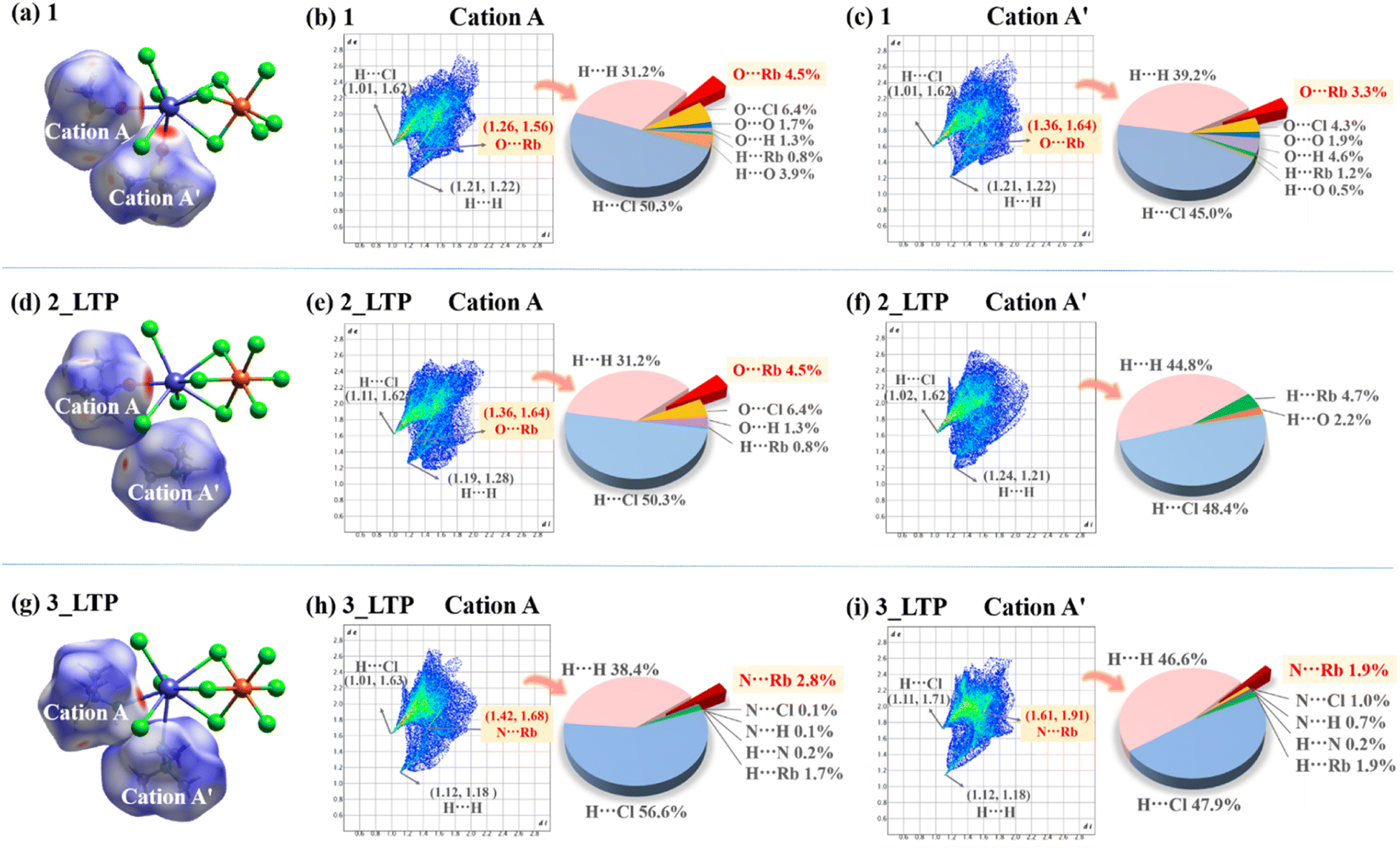 | ||
| Fig. 6 The Hirshfeld surfaces of 1 (a), 2_LTP (d), and 3_LTP (g). The fingerprint plots of guest cations for 1 (b) and (c), 2_LTP (e) and (f), and 3_LTP (h) and (i). | ||
For 2, coordinatively free A′-site Me4N+ cations primarily engaged in weak C–H⋯Cl host–guest interactions and H⋯H guest–guest interactions, enabling it to readily undergo an order–disorder phase transition at a relatively low temperature of 247 K. In contrast, strong guest–guest interactions are observed in 3, evidenced by the smallest H⋯H short contact (1.12 Å, 1.18 Å) and the largest Hirshfeld surface area of H⋯H short contact. As a result, the intensified repulsive interactions among the adjacent guest cations, together with the Rb–N coordination interactions from A′-site cations in 3, lead to a ferroelastic transition occurring at above room temperature.
In short, it is reasonable to deduce that the subtle changes in guest molecules influence the host–guest and guest–guest interactions, fostering competitive yet balanced intermolecular dynamics. These modifications enable the creation of multiple inter-convertible crystalline states, governed by specific activation energy barriers, thus leading to distinct phase-transition behaviours. This understanding is crucial for designing and comprehending unique structural phase transitions, particularly bond-switching transitions.
Conclusions
In summary, we demonstrated a strategic molecular design of organic cations in a new kind of host–guest model with an lon topologic inorganic framework, by presenting three new polar hybrid crystals, i.e., 1–3, with a generic formula of AA′RbBiCl6. Their A-site cations link to Rb+ ions via strong Rb–O/N coordination bonds, while their A′-site cations reveal significantly different interactions that drive distinct phase-transition behaviours. A′-site Me3SO+ cations form strong Rb–O coordination bonds, preventing 1 from triggering a structural phase transition, whereas coordinatively free A′-site Me4N+ cations induce an order–disorder ferroelastic transition at 247 K and a usual heating-induced “high → low” SHG switch in 2. Remarkably, A′-site Me3NNH2+ cations form unusual dynamic coordination bonds in 3, driving a high-temperature ferroelastic phase transition accompanied by the larger entropy change and an unusual heating-induced “low → high” SHG switch. Such systematic investigations into phase transitions not only deepen the understanding of exceptional bond-switching transitions but also hold significant promise for guiding the development of functional hybrid materials.Author contributions
WX Zhang and YB Li conceived the idea, designed the experiments, and co-wrote the manuscript. YB Li performed the synthesis experiments, DSC, dielectric, SHG, crystal structural analysis, ferroelastic domain observations, and the Hirshfeld surface analyses. XX Chen and WJ Xu assisted in crystal structural analyses. YP Gong and H Ye assisted in the Pawley refinements. ZS Wang offered advice on experimental design.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (22071273 and 21821003), Fundamental Research Funds for the Central Universities (Sun Yat-Sen University, 23lgzy001).Notes and references
- Y. Hu, L. You, B. Xu, T. Li, S. A. Morris, Y. Li, Y. Zhang, X. Wang, P. S. Lee and H. J. Fan, Nat. Mater., 2021, 20, 612–617 CrossRef CAS PubMed.
- E. K. Salje, Annu. Rev. Mater. Res., 2012, 42, 265–283 CrossRef CAS.
- M. Liu, B. M. Howe, L. Grazulis, K. Mahalingam, T. Nan, N. X. Sun and G. J. Brown, Adv. Mater., 2013, 25, 4886–4892 CrossRef CAS PubMed.
- S. Baek, H. Jang, C. Folkman, Y. Li, B. Winchester, J. Zhang, Q. He, Y. Chu, C. Nelson and M. Rzchowski, Nat. Mater., 2010, 9, 309–314 CrossRef CAS PubMed.
- Y. Zhang, M.-G. Han, J. A. Garlow, Y. Tan, F. Xue, L.-Q. Chen, P. Munroe, N. Valanoor and Y. Zhu, Nano Lett., 2019, 19, 5319–5326 CrossRef CAS PubMed.
- E. Strelcov, Q. Dong, T. Li, J. Chae, Y. Shao, Y. Deng, A. Gruverman, J. Huang and A. Centrone, Sci. Adv., 2017, 3, e1602165 CrossRef PubMed.
- B.-Q. Zhao, X.-X. Chen, H. Ye, Y.-P. Gong, J. Wang, L. Ye and W.-X. Zhang, Chem. Sci., 2023, 14, 5965–5973 RSC.
- R. G. Xiong, S. Q. Lu, Z. X. Zhang, H. Cheng, P. F. Li and W. Q. Liao, Angew. Chem., Int. Ed., 2020, 132, 9661–9665 CrossRef.
- M. M. Lun, C. Y. Su, J. Li, Q. Q. Jia, H. F. Lu, D. W. Fu, Y. Zhang and Z. X. Zhang, Small, 2023, 19, 2303127 CrossRef CAS PubMed.
- Z. Yue, F. Wu, X. Li, Y. Liu, J. Luo and X. Liu, Sci. China Mater., 2023, 66, 3977–3983 CrossRef CAS.
- Q. Liu, H. Peng, J.-C. Qi, Y.-Z. Lu, S.-J. Yang and W.-Q. Liao, Chem. Commun., 2023, 59, 1793–1796 RSC.
- D.-X. Liu, Z.-H. Yu, X.-X. Chen, W.-X. Zhang and X.-M. Chen, Chin. Chem. Lett., 2023, 34, 107310 CrossRef CAS.
- Y.-P. Gong, X.-X. Chen, G.-Z. Huang, W.-X. Zhang and X.-M. Chen, J. Mater. Chem. C, 2022, 10, 5482–5488 RSC.
- X.-X. Chen, D.-X. Liu, Y.-P. Gong, S.-S. Wang, W.-X. Zhang and X.-M. Chen, Inorg. Chem., 2022, 61, 2219–2226 CrossRef CAS PubMed.
- D.-X. Liu, X.-X. Chen, Z.-M. Ye, W.-X. Zhang and X.-M. Chen, Sci. China Mater., 2022, 65, 263–267 CrossRef CAS.
- W. Luo, N. Wang, H.-K. Li, Z.-J. Xu, Y. Feng, X.-b. Fu, C. Shi, H.-Y. Ye and L.-P. Miao, Inorg. Chem. Front., 2023, 10, 5082–5088 RSC.
- H. Ye, X.-X. Chen, D.-X. Liu, B.-Q. Zhao, Y.-B. Li, Y. Zeng, W.-X. Zhang and X.-M. Chen, Chem. Sci., 2022, 13, 14124–14131 RSC.
- Y. Xu, K. Xu, L. He, T.-J. Yin, J. Mu, J.-T. Men, W. Zhang and Q. Ye, Inorg. Chem., 2023, 62, 1279–1285 CrossRef CAS PubMed.
- X.-X. Chen, X.-Y. Zhang, D.-X. Liu, R.-K. Huang, S.-S. Wang, L.-Q. Xiong, W.-X. Zhang and X.-M. Chen, Chem. Sci., 2021, 12, 8713–8721 RSC.
- H. Zhang, Q.-L. Li, Y.-H. Tan, Y.-Z. Tang, X.-W. Fan, J.-L. Luo, F.-X. Wang and M.-Y. Wan, Inorg. Chem., 2023, 62, 10847–10853 CrossRef CAS PubMed.
- Q.-R. Meng, W.-J. Xu, W.-H. Hu, H. Ye, X.-X. Chen, W. Yuan, W.-X. Zhang and X.-M. Chen, Chem. Commun., 2021, 57, 6292–6295 RSC.
- W. Yuan, Y. Zeng, Y.-Y. Tan, J.-H. Zhou, W.-J. Xu, W.-X. Zhang and X.-M. Chen, Chem. Commun., 2019, 55, 8983–8986 RSC.
- Y. Gong, X. Chen, B. Zhao, J. Wang, W. Zhang and X. Chen, Chin. Chem. Lett., 2023, 34, 108282 CrossRef CAS.
- W.-J. Xu, P. Zelenovskii, A. Tselev, L. Verissimo, K. Romanyuk, W. Yuan, W.-X. Zhang, A. Kholkin and J. Rocha, Chem. Commun., 2023, 59, 11264–11267 RSC.
- T. J. Yin, K. Xu, L. He, X. Meng, Y. Xu, J. T. Men, J. Mu, Q. Ye and P. P. Shi, Chem.–Eur. J., 2023, 29, e202203606 CrossRef CAS PubMed.
- G.-H. Ni, Y.-X. Sun, C.-X. Ji, Y.-W. Jin, M.-Y. Liu, J.-P. Zhao and F.-C. Liu, Cryst. Growth Des., 2022, 22, 3428–3434 CrossRef CAS.
- D. Fu, Z. Hou, Y. He, J.-C. Liu, H.-P. Lv and Y.-Y. Tang, Chem. Mater., 2022, 34, 3518–3524 CrossRef CAS.
- W.-J. Xu, P.-F. Li, Y.-Y. Tang, W.-X. Zhang, R.-G. Xiong and X.-M. Chen, J. Am. Chem. Soc., 2017, 139, 6369–6375 CrossRef CAS PubMed.
- H.-Y. Zhang, N. Zhang, Y. Zhang, H.-H. Jiang, Y.-L. Zeng, S.-Y. Tang, P.-F. Li, Y.-Y. Tang and R.-G. Xiong, Phys. Rev. Lett., 2023, 130, 176802 CrossRef CAS PubMed.
- W.-J. Xu, K. Romanyuk, Y. Zeng, A. Ushakov, V. Shur, A. Tselev, W.-X. Zhang, X.-M. Chen, A. Kholkin and J. Rocha, J. Mater. Chem. C, 2021, 9, 10741–10748 RSC.
- R. Li, T. Zhu, Z. k. Zhu, J. Wu, Y. Geng and J. Luo, Small, 2023, 2306825 CrossRef PubMed.
- X.-G. Chen, Z.-X. Zhang, Y.-L. Zeng, S.-Y. Tang and R.-G. Xiong, Chem. Commun., 2022, 58, 3059–3062 RSC.
- W.-J. Xu, Y. Zeng, W. Yuan, W.-X. Zhang and X.-M. Chen, Chem. Commun., 2020, 56, 10054–10057 RSC.
- L. He, K. Xu, P.-P. Shi, Z.-B. Liu, W. Zhang and Q. Ye, Sci. China Mater., 2022, 65, 2879–2883 CrossRef.
- L. He, P.-P. Shi, L. Zhou, Z.-B. Liu, W. Zhang and Q. Ye, Chem. Mater., 2021, 33, 6233–6239 CrossRef CAS.
- J. Zhou, P. Xie, C. Wang, T. Bian, J. Chen, Y. Liu, Z. Guo, C. Chen, X. Pan, M. Luo, J. Yin and L. Mao, Angew. Chem., Int. Ed., 2023, 62, e202307646 CrossRef CAS PubMed.
- H. Seung-Tae and A. W. Sleight, J. Solid State Chem., 1997, 128, 251–255 CrossRef.
- C. Felser and R. Cava, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 10005 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- F. Wei, Z. Deng, S. Sun, F. Zhang, D. M. Evans, G. Kieslich, S. Tominaka, M. A. Carpenter, J. Zhang and P. D. Bristowe, Chem. Mater., 2017, 29, 1089–1094 CrossRef CAS.
- Z. Deng, F. Wei, F. Brivio, Y. Wu, S. Sun, P. D. Bristowe and A. K. Cheetham, J. Phys. Chem. Lett., 2017, 8, 5015–5020 CrossRef CAS PubMed.
- K. Aizu, A. Kumada and H. Yumoto, J. Phys. Soc. Jpn., 1969, 27, 511 CrossRef CAS.
- B. Li, Y. Kawakita, S. Ohira-Kawamura, T. Sugahara, H. Wang, J. Wang, Y. Chen, S. I. Kawaguchi, S. Kawaguchi, K. Ohara, K. Li, D. Yu, R. Mole, T. Hattori, T. Kikuchi, S.-I. Yano, Z. Zhang, Z. Zhang, W. Ren, S. Lin, O. Sakata, K. Nakajima and Z. Zhang, Nature, 2019, 567, 506–510 CrossRef CAS PubMed.
- S. Han, L. Li, C. Ji, X. Liu, G.-E. Wang, G. Xu, Z. Sun and J. Luo, J. Am. Chem. Soc., 2023, 145, 12853–12860 CrossRef CAS PubMed.
- H.-Y. Shen, L. He, P.-P. Shi and Q. Ye, J. Mater. Chem. C, 2021, 9, 4338–4343 RSC.
- S. Liu, Z. Sun, C. Ji, L. Li, S. Zhao and J. Luo, Chem. Commun., 2017, 53, 7669–7672 RSC.
- K. Robinson, G. Gibbs and P. Ribbe, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
- W.-J. Xu, S.-L. Chen, Z.-T. Hu, R.-B. Lin, Y.-J. Su, W.-X. Zhang and X.-M. Chen, Dalton Trans., 2016, 45, 4224–4229 RSC.
- M. J. Cliffe and A. L. Goodwin, J. Appl. Crystallogr., 2012, 45, 1321–1329 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2313516–2313519 and 2313598. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06702e |
| This journal is © The Royal Society of Chemistry 2024 |