DOI:
10.1039/D3SC06618E
(Edge Article)
Chem. Sci., 2024,
15, 3552-3561
Dual role of nitroarenes as electrophiles and arylamine surrogates in Buchwald–Hartwig-type coupling for C–N bond construction†
Received
9th December 2023
, Accepted 24th January 2024
First published on 25th January 2024
Abstract
One of the most widely utilized methods for the construction of C(sp2)–N bonds is the transition-metal-catalyzed cross-coupling of aryl halides/boronic acids with amines, known as Ullmann condensation, Buchwald–Hartwig amination, and Chan–Lam coupling. However, aryl halides/boronic acids often require multi-step preparation while generating a large amount of corrosive and toxic waste, making the reaction less attractive. Herein, we present an unprecedented method for the C(sp2)–N formation via Buchwald–Hartwig-type reactions using synthetically upstream nitroarenes as the sole starting materials, thus eliminating the need for arylhalides and pre-formed arylamines. A diverse range of symmetrical di- and triarylamines were obtained in a single step from nitroarenes, and more importantly, various unsymmetrical di- and triarylamines were also highly selectively synthesized in a one-pot/two-step process. Furthermore, the success of the scale-up experiments, the late-stage functionalization of a drug intermediate, and the rapid preparation of hole-transporting material TCTA showcased the utility and practicality of this protocol in synthetic chemistry. Mechanistic studies indicate that this transformation may proceed via an arylamine intermediate generated in situ from the reduction of nitroarenes, which is followed by a denitrative Buchwald–Hartwig-type reaction with another nitroarene to form a C–N bond.
Introduction
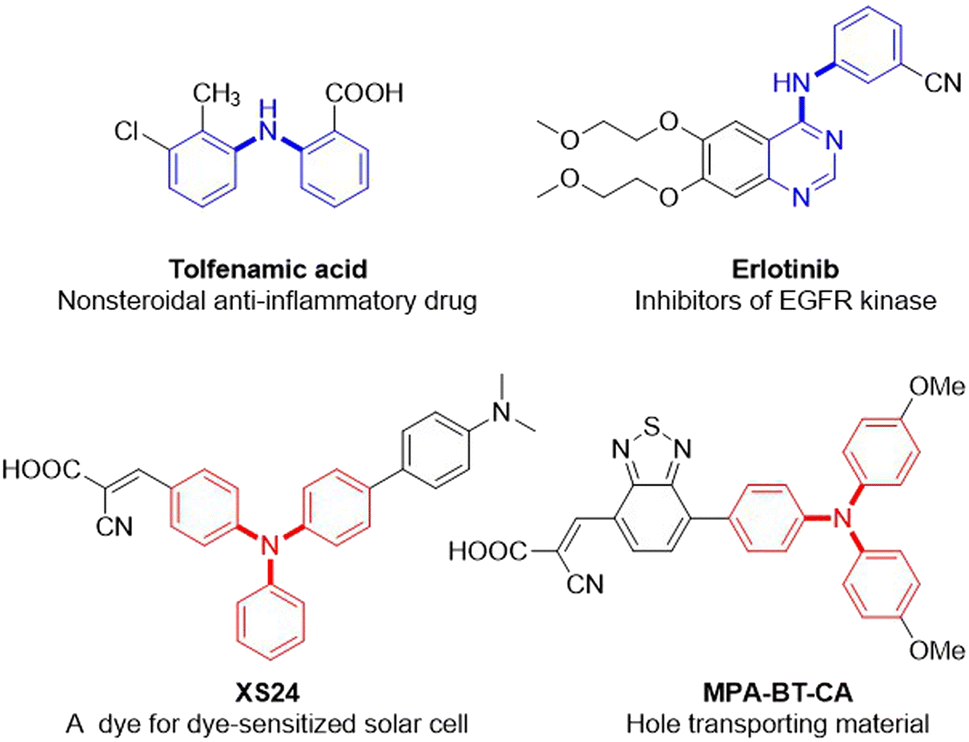
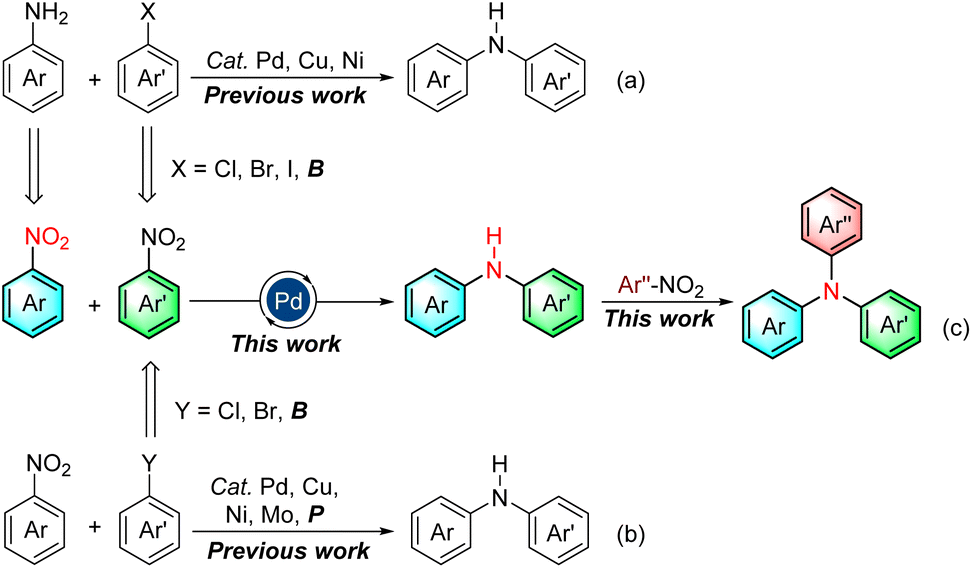
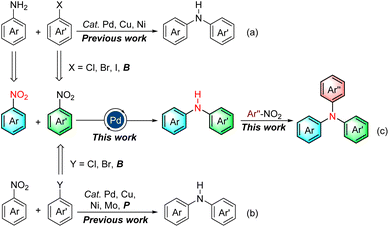
Due to the ubiquitous nature of the C(sp2)–N bond in pharmaceuticals,1 agrochemicals,2 dyes,3 electronic materials,4 polymers,5 and other industrial chemicals (Fig. 1),6 the construction of the C(sp2)–N bond is a significant research topic in both academic research and industrial processes. Consequently, considerable efforts have been devoted to its construction, and diverse means have been developed.7–13 Among them, the transition-metal-catalyzed C–N cross-coupling reaction is one of the most widely utilized means for the construction of C(sp2)–N bonds, including Ullmann condensation,7 Buchwald–Hartwig amination,8 and Chan–Lam coupling.9 Notably, the Buchwald–Hartwig amination is ranked among the top 20 most commonly used reactions in medicinal chemistry owing to its broad substrate scope, high reproducibility, and scalability.14 This strategy for C(sp2)–NAr bond formation is frequently through the transition-metal-catalyzed cross-coupling of aryl halides or aryl borons with arylamines, which are usually prepared in advance by the reduction of nitroarenes (Scheme 1a). Using nitroarenes instead of arylamines is attractive as it avoids pre-synthesizing and isolating anilines, saving time and cost. In addition, nitroarenes are among the most fundamental chemical feedstocks, which can be conveniently synthesized through the nitration of arenes. Toward this end, the groups of Baran15 and Hu16 have elegantly disclosed that nitroarenes are ideal surrogates of arylamines in organic synthesis, respectively. Recently, a PIII/PV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O catalyzed reductive C–N coupling of nitroarenes with arylboronic acids and a Mo-catalyzed reductive amination of nitroarenes with arylboronic acids have been successfully uncovered by Radosevich17 and Sanz groups,18 respectively. In 2021, Driver and co-workers reported an innovative copper-catalyzed cross-coupling of nitroarenes with aryl boronic acids to construct diarylamines via a nitrosoarene intermediate utilizing phenylsilane as a terminal reductant.19 Xue later made a notable discovery of a nickel-catalyzed C–N coupling of aryl halides with nitroarenes via a nitrosoarene intermediate facilitated by photo-assisted processes.20 Very recently, Weix and co-workers have impressively demonstrated a reductive arylation of nitroarenes with chloroarenes through an azoarene intermediate using manganese powder as a reductant.21 Despite these remarkable advances,15–22 requiring aryl halides or aryl boronic acids limits their broad applications in C(sp2)–N bond formation (Scheme 1b).
O catalyzed reductive C–N coupling of nitroarenes with arylboronic acids and a Mo-catalyzed reductive amination of nitroarenes with arylboronic acids have been successfully uncovered by Radosevich17 and Sanz groups,18 respectively. In 2021, Driver and co-workers reported an innovative copper-catalyzed cross-coupling of nitroarenes with aryl boronic acids to construct diarylamines via a nitrosoarene intermediate utilizing phenylsilane as a terminal reductant.19 Xue later made a notable discovery of a nickel-catalyzed C–N coupling of aryl halides with nitroarenes via a nitrosoarene intermediate facilitated by photo-assisted processes.20 Very recently, Weix and co-workers have impressively demonstrated a reductive arylation of nitroarenes with chloroarenes through an azoarene intermediate using manganese powder as a reductant.21 Despite these remarkable advances,15–22 requiring aryl halides or aryl boronic acids limits their broad applications in C(sp2)–N bond formation (Scheme 1b).
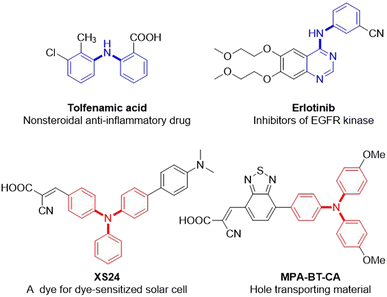 |
| | Fig. 1 Selected examples of the C(sp2)–N bond in pharmaceuticals, dyes, and electronic materials. | |
 |
| | Scheme 1 Comparison of various starting materials and strategies for the C(sp2)–N bond formation via cross-coupling methods. | |
Aryl halides, usually prepared from nitroarenes through reduction, diazotization, and halogenation processes, frequently serve as electrophiles in Buchwald–Hartwig amination. Meanwhile, aryl boronic acids are commonly prepared from these aryl halides. However, the additional steps necessary to pre-synthesize aryl halides/boronic acids from nitroarenes and the large amount of toxic halide waste generated during the preparation and cross-coupling process make them less attractive. In sharp contrast, the direct utilization of synthetically upstream nitroarenes as the electrophiles would significantly enhance the step- and atom-economy of the C(sp2)–N coupling and circumvents many problems associated with the preparation and usage of aryl halides, including tedious synthesis, heavy pollution, and high cost. Indeed, the realm of denitrative coupling reactions has witnessed significant achievements, spearheaded by Nakao,23 Yamaguchi,24 You,25 and others,26 subsequent to the seminal work on the denitrative Suzuki–Miyaura reaction documented by Nakao.23a However, the construction of C–N bonds employing nitroarenes as both electrophiles and arylamine surrogates remains a highly formidable challenge, primarily due to the following reasons: (a) the presence of numerous potential reduction intermediates of nitroarenes, including nitrosoarene, azoarene, azoxyarene, 1,2-arylhydrazine, and N-arylhydroxylamine. These diverse intermediates complicate the reaction pathway. (b) The existence of multiple possible competing processes, such as denitrative hydrogenation of nitroarenes, reductive amination to form triarylamines, and reductive homo- or cross-coupling of nitroarenes. These side reactions can hinder the desired C–N bond formation. (c) The fundamental aspect of ensuring compatibility between the reduction procedure of nitroarenes and the denitrative Buchwald–Hartwig-type process. This requirement adds an additional layer of complexity to the overall reaction setup. Given the synthetic importance of the C(sp2)–N bonds, addressing these challenges represents a significant frontier in the development of efficient, reliable, and step-economic methodologies for constructing C–N bonds. Herein, we wish to disclose an efficient and straightforward protocol for C(sp2)–N bond formation via Buchwald–Hartwig-type amination using synthetically upstream nitroarenes as the sole starting materials, thus eliminating the need for pre-synthesizing arylhalides and isolating arylamines (Scheme 1c).
Results and discussion
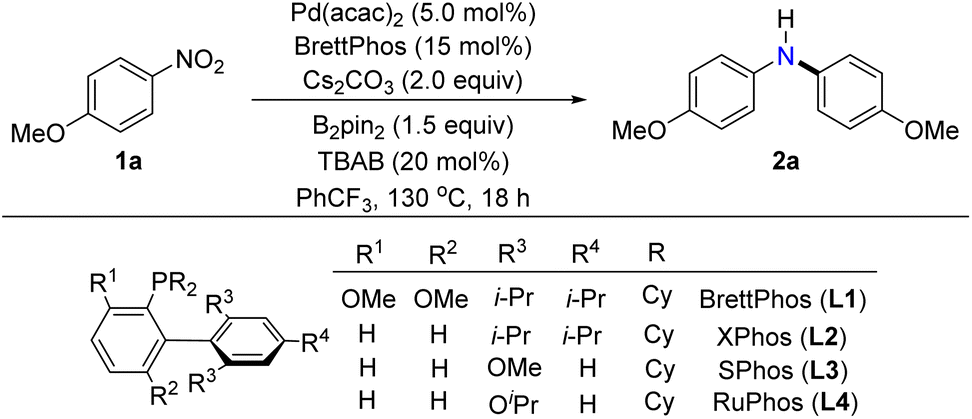
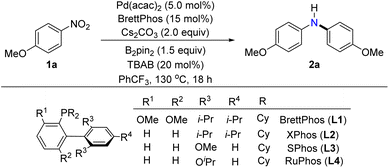
To verify the feasibility of a palladium-catalyzed reductive arylation of dual nitroarenes, we initiated our investigations by evaluating the reaction of the 4-nitroanisole (1a) in the presence of the palladium catalyst, phosphine ligand, reductant, and base at 130 °C for 18 hours (Table 1, please see the ESI† for details). A key step of our hypothesis strategy was the in situ generation of p-anisidine from 4-nitroanisole by reduction. The choice of reductant was found to be extremely crucial to the reduction of 4-nitroanisole and the consequent formation of the desired diarylamine (2a). We first examined the PhSiH3, PHMS, and B2(OH)4, commonly utilized reductants in the reduction of nitroarenes; all the reactions furnished p-anisidine instead of the desired diarylamine (entry 2). These results implied that the denitrative Buchwald–Hartwig-type process was incompatible with these strong reductants. Similarly, employing B2cat2 or Ph3P as reductants did not result in the production of the target products, as the starting materials remained unchanged (entry 3). Furthermore, other reductants, such as Mn and Fe, promoted the generation of a complex mixture containing triarylamine. When propan-2-ol was utilized as a reductant, the 4-nitroanisole predominantly underwent the competing denitrative hydrogenation of nitroarenes, leading to the formation of anisole (please see ESI, Table S1,† not shown in Table 1). To our delight, using B2nep2 or HSiEt3 as reductants gave the target product, albeit with modest yields, and a portion of the starting materials remained unconverted (entry 4). Investigation of HBpin revealed the formation of both diarylamines and triarylamines (entry 5). These outcomes indicated low efficiency of the reduction process. Fortunately, persistence and systematic investigation led us to discover the optimal reducing reagent: B2pin2. This mild, stable, and cost-effective reductant proved to be the key to achieving the desired transformation.27 The subsequent investigation revealed that the amount of the reductant significantly influenced the yield of the desired product. It was found that an excessive or insufficient amount of the reductant B2pin2 resulted in a decrease in the production of the target product. Controlled experiments revealed that when B2pin2 was used at an equivalent of 1.5, the highest yield of the target product at 83% was obtained. However, reducing the amount of B2pin2 to an equivalent of 1.0 decreased the yield of the target product to 36%, whereas increasing the reductant amount to an equivalent of 2.0 yielded a product with 61% yield (please see ESI, Table S1,† entries 12–14, not shown in Table 1). Furthermore, the use of different ligands, such as RuPhos (monodentate phosphine ligand), XantPhos (bidentate phosphine ligand), and IPr·HCl (N-heterocyclic carbene ligand), resulted in inferior reaction yields or complete inhibition of the reaction (entry 6 and ESI, Table S2†). Subsequently, various Pd(0) and Pd(II) catalysts, such as
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
Deviation from ‘standard conditions’ |
Yield 2ab (%) |
|
Standard reaction conditions: 4-nitroanisole 1a (0.30 mmol), Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%), Cs2CO3 (0.60 mmol), B2pin2 (0.45 mmol), and TBAB (20 mol%) in PhCF3 (1.5 mL) at 130 °C for 18 hours.
Isolated yield.
p-Anisidine was formed.
Most all of 4-nitroanisole remained.
A portion of 4-nitroanisole remained.
Diarylamine and triarylamine were formed.
|
| 1 |
None |
83 |
| 2c |
PhSiH3, PHMS, or B2(OH)4 instead of B2pin2 |
0 |
| 3d |
B2cat2, Ph3P, Fe, or Mn instead of B2pin2 |
0 |
| 4e |
B2nep2 or HSiEt3 instead of B2pin2 |
19–41 |
| 5f |
HBpin instead of B2pin2 |
55 |
| 6 |
L2, L3 or L4 instead of L1 |
0–46 |
| 7 |
Pd(tBu3P)2, Pd(OAc)2, or [Pd(allyl)Cl]2 instead of Pd(acac)2 |
0–50 |
| 8 |
K2CO3, t-BuOK, CsOH·H2O, or K3PO4 instead of Cs2CO3 |
29–57 |
| 9 |
THF, DMF, toluene, or n-heptane instead of PhCF3 |
0–55 |
| 10 |
TBAI, TBAA, or TBAC instead of TBAB |
66–75 |
Pd(tBu3P)2, Pd(acac)2, Pd(OAc)2, and [Pd(allyl)Cl]2 were examined, and the Pd(acac)2 exhibited the highest catalytic activity for this transformation (entry 7). Further investigation on the effect of the base showed that Cs2CO3 was the best one for the coupling, while K3PO4 and K2CO3 afforded the target products with diminished yields (entry 8). Replacing solvent PhCF3 with THF, DMF, toluene, or n-heptane led to sharply decreased yields of the desired products (entry 9). With tetrabutylammonium bromide (TBAB) absent or replaced by other quaternary ammonium salts such as tetrabutylammonium iodide (TBAI), tetrabutylammonium acetate (TBAA), and tetrabutylammonium chloride (TBAC), a decrease of the yield was noted (entry 10). After systematic studies, we found that employing Pd(acac)2 (5 mol%), BrettPhos (15 mol%), Cs2CO3 (2.0 equiv), B2pin2 (1.5 equiv), and TBAB (20 mol%) with PhCF3 as solvent, the target diarylamine (2a) was isolated in 83% yield (entry 1).
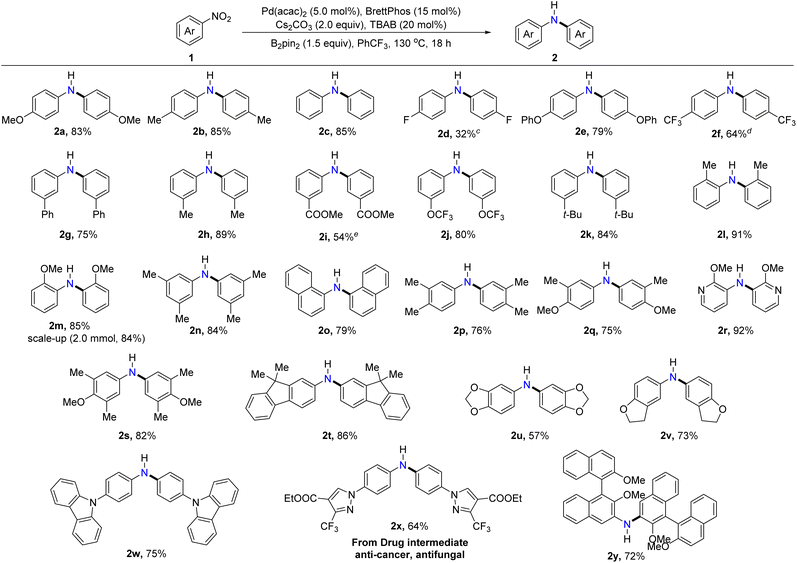
Assessment of reaction scope for synthesizing symmetrical diarylamines
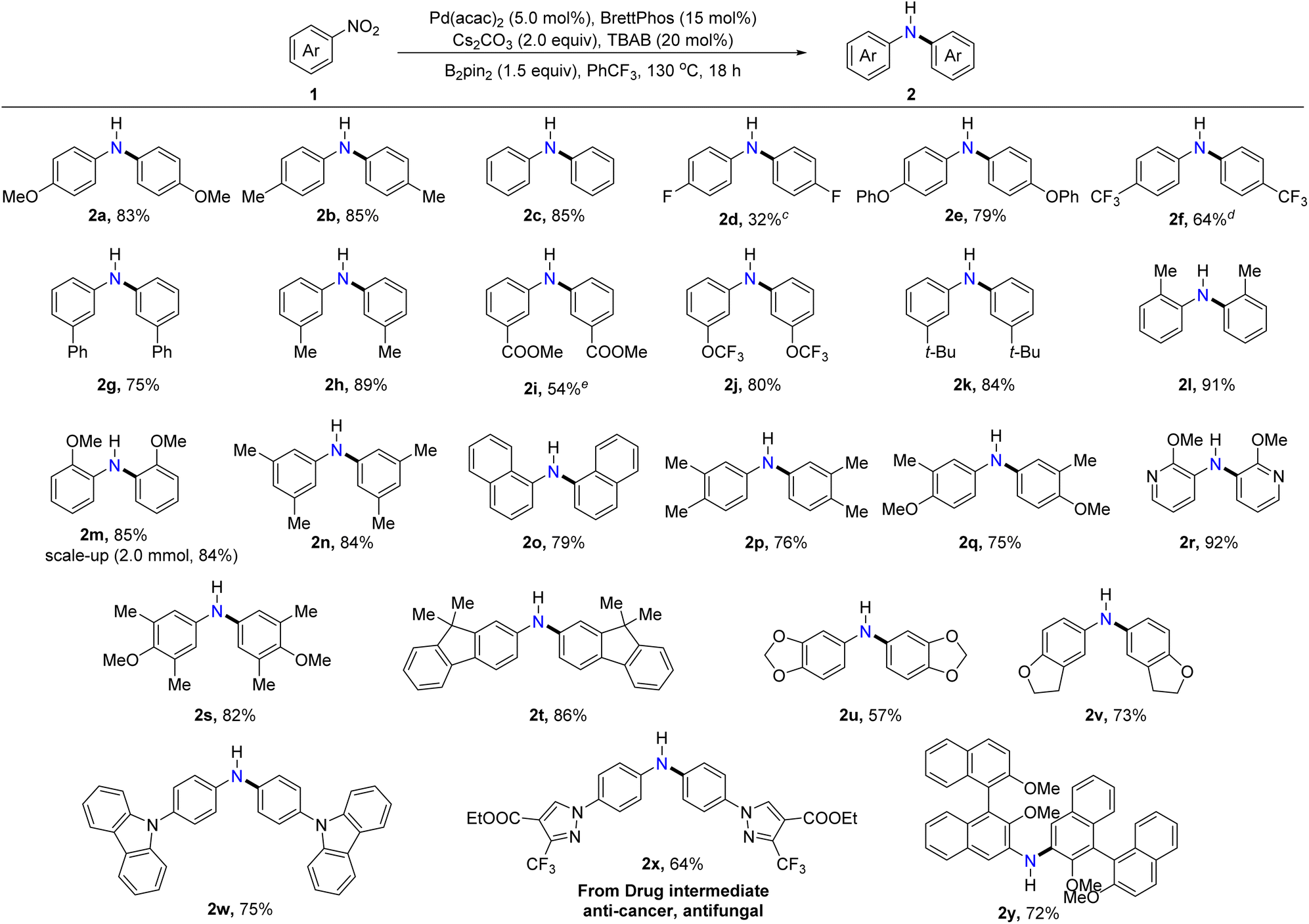
With the optimized reaction conditions in hand, we set out to explore the generality of this protocol for the formation of symmetrical diarylamines and the scope of nitroarenes. As illustrated in Table 2, nitroarenes bearing electron-donating substituents, including methyl (2b, 2h, 2l, and 2n), methoxy (2a and 2m), tertiary butyl (2k), phenoxy (2e), and phenyl (2g) substituents underwent the desired reductive coupling to form the expected products in moderate to excellent yields (75–91%). Moreover, nitroarenes with electron-withdrawing substituents such as fluorine (2d), trifluoromethyl (2f), ester (2i), and trifluoromethoxy (2j) were all converted into the target products, albeit with slightly diminished yields. It was noteworthy that the nitroarenes with ortho-substituted methyl (2l) or methoxy (2m) highly efficiently afforded the desired diarylamines, indicating the high compatibility with the steric hindrance of substrates in this protocol. Encouragingly, the reaction scale could be increased to 2.0 mmol without a significant decrease in yield (2m). Gratifyingly, disubstituted and trisubstituted nitroarenes also worked successfully under the developed catalytic systems, showcasing the wide substrate scope of nitroarenes in the protocol (2n, 2p, 2q, and 2s). Furthermore, π-extended nitroarenes such as 1-nitronaphthalene and 9,9-dimethyl-2-nitro-9H-fluorene proceeded efficiently and yielded the corresponding diarylamines 2o and 2t in 79% and 86% yields, respectively. Importantly, various nitroheteroarenes bearing pyridine, benzo[d][1,3]dioxole, 2,3-dihydrobenzofuran, and carbazole motifs could participate in the reaction to furnish the desired products 2r, 2u, 2v, and 2w in 92%, 57%, 73%, and 75% yields, respectively. When an anti-cancer and antifungal drug intermediate was subjected to the current optimized conditions, the corresponding diarylamine product (2x) was formed in 64% yield, demonstrating the synthetic utility and practicality of this protocol. Interestingly, 2,2′-dimethoxy-3-nitro-1,1′-binaphthalene, a sterically encumbered nitroarene, was also compatible with this C–N coupling and provided the desired diarylamine (2y) in 72% yield.
Table 2 Substrate scope of nitroarenes to form symmetrical diarylaminesa,b
Reaction conditions A: nitroarenes (0.30 mmol), Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%), TBAB (20 mol%), B2pin2 (0.45 mmol) and Cs2CO3 (0.60 mmol) in PhCF3 (1.5 mL) at 130 oC for 18 hours.
Isolated yield.
Cs2CO3 (1.0 equiv.) without TBAB at 150 °C for 24 hours.
CsOH·H2O in dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PhCF3 = 1:3 at 150 °C for 24 hours.
Cs2CO3 (1.0 equiv.) was used at 150 °C for 24 hours. :PhCF3 = 1:3 at 150 °C for 24 hours.
Cs2CO3 (1.0 equiv.) was used at 150 °C for 24 hours.
|
|
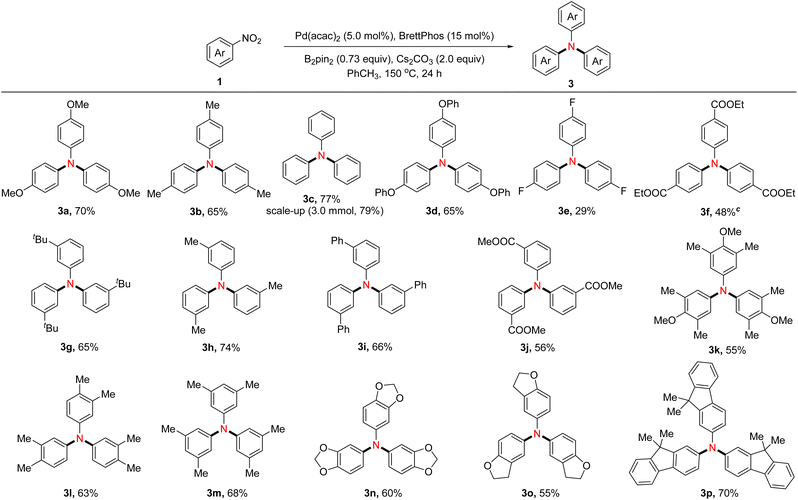
While exploring the scope and limitations of this transformation, we were delighted to discover that reducing the amounts of reductant and elevating the reaction temperature were beneficial for affording triarylamines. Subsequently, the effects of different bases, solvents, and the amount of B2pin2 were explored, and the yield of the target triarylamine increased to 70% when the reaction was performed under the following conditions: B2pin2 (0.73 equiv), Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%), and Cs2CO3 (2.0 equiv.) in toluene at 150 °C for 24 hours (please see ESI, Table S8†).
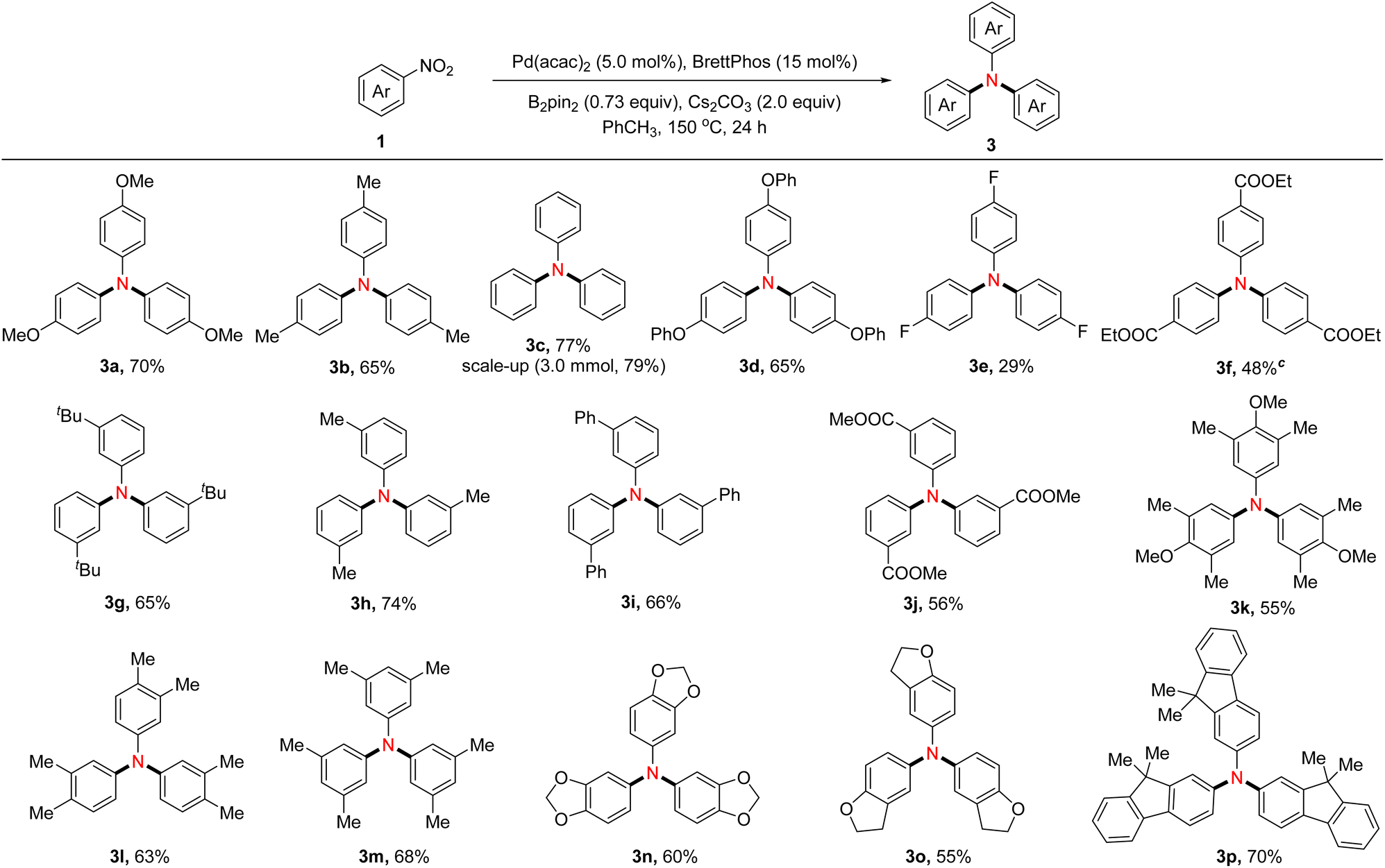
Assessment of reaction scope for synthesizing symmetrical triarylamines
Under the optimized reaction conditions, the scope of nitroarenes for symmetrical triarylamine synthesis was investigated (Table 3). In general, the substrates containing electron-donating groups such as methyl (3b, 3h, 3l, and 3m), tertiary butyl (3g), and phenoxy (3d) were well tolerated, providing the target products with moderate yields. Nitroarenes bearing electron-withdrawing groups such as ester (3f and 3j) proceeded uneventfully, while fluorine-substituted nitroarene (3e) gave the desired product with a lower yield. Of note, nitroarenes possessing a heterocycle, such as benzo[d][1,3]dioxole (3n) and 2,3-dihydrobenzofuran (3o), reacted under standard conditions to form the expected products smoothly. In contrast, 1-methyl-2-nitrobenzene was found to be an ineffective substrate under the optimized conditions, indicating that the steric hindrance significantly affects the conversion of nitroarenes to triarylamines (not shown in Table 3). The synthetic utility was illustrated with a 3.0 mmol scale synthesis of 3c in 79% yield.
Table 3 Substrate scope of nitroarenes to form symmetrical triarylaminesa,b
|
Reaction conditions B: nitroarenes 1 (0.30 mmol), Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%), B2pin2 (0.22 mmol) and Cs2CO3 (0.60 mmol) in PhCH3 (1.0 mL) at 150 oC for 24 hours;
Isolated yield.
PhCF3 instead of PhCH3.
|
|
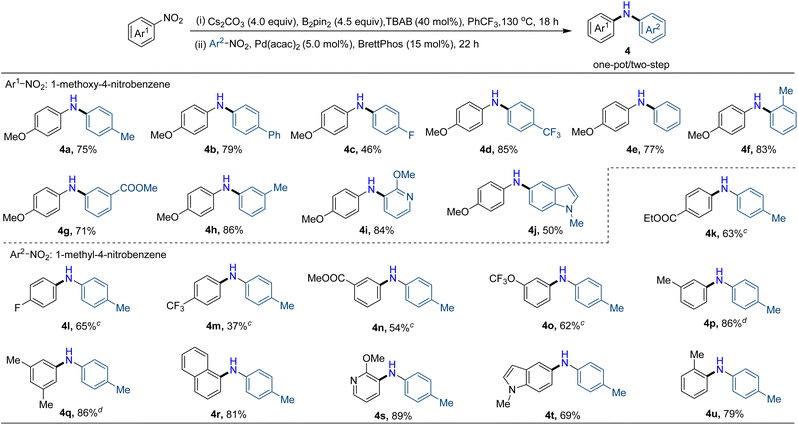
Assessment of reaction scope for synthesizing unsymmetrical diarylamines
Motivated by these results, we next turned our attention to the synthesis of unsymmetrical arylated amines, which are more frequently encountered in pharmaceuticals and functional materials. We first studied the synthesis of unsymmetrical diarylamines, with the main challenge being to avoid the formation of symmetrical diarylamines, which result from the homocoupling of two different nitroarenes for their respective reductive arylation. We realized that it might be accomplished in a one-pot/two-step process by using B2pin2 to reduce the initially introduced nitroarene to arylamine, followed by Buchwald–Hartwig-type coupling with another nitroarene, diminishing the formation of symmetrical diarylamines. Excitingly, after slightly adjusting the conditions, the desired unsymmetrical diarylamine (4a) was obtained in 75% yield using the two-step/one-pot method. Subsequently, the scope of two nitroarenes to form unsymmetrical diarylamines was examined. As shown in Table 4, the one-pot/two-step synthesis of unsymmetrical diarylamines exhibited good functional group compatibility with both reaction partners. Generally, nitroarenes Ar2 possessing electron-donating groups such as methyl (4a, 4f, and 4h) and phenyl (4b) as well as electron-withdrawing groups such as fluorine (4c), trifluoromethyl (4d), and ester (4g) were amenable in this transformation, affording the target unsymmetrical diarylamines in modest to good yields. To our satisfaction, the nitroheteroarenes with pyridine or indole scaffolds were also applicable in the reaction, leading to the expected products (4i) and (4j) in 84% and 50% yields, respectively. We next investigated the scope of another nitroarene Ar1. With a 10% catalyst, 30% ligand, and elevated temperature, the reaction exhibited notable tolerance towards nitroarenes Ar1, including electron-withdrawing groups (4k–4o) and electron-donating groups (4p, 4q, and 4u) irrespective of the position of the substituent groups on the aryl ring. Notably, the nitroarene with sterically demanding ortho-substituted methyl was also a suitable substrate for the reaction, and the expected products (4f and 4u) were obtained with 83% and 79% yields, respectively.
Table 4 Selective one-pot/two-step construction of unsymmetrical diarylaminesa,b
|
Reaction conditions: nitroarenes Ar1 (0.15 mmol), B2pin2 (0.45 mmol), Cs2CO3 (0.40 mmol), and TBAB (40 mol%) in PhCF3 (1.5 mL) at 130 oC for 18 hours. After the first step reaction, without any extra work-up, Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%) and nitroarenes Ar2 (0.10 mmol) were added in situ to react at 130 °C for 22 hours;
Isolated yield.
Pd(acac)2 (10 mol%) and BrettPhos (30 mol%) at 150 °C.
Pd(acac)2 (10 mol%) and BrettPhos (30 mol%).
|
|
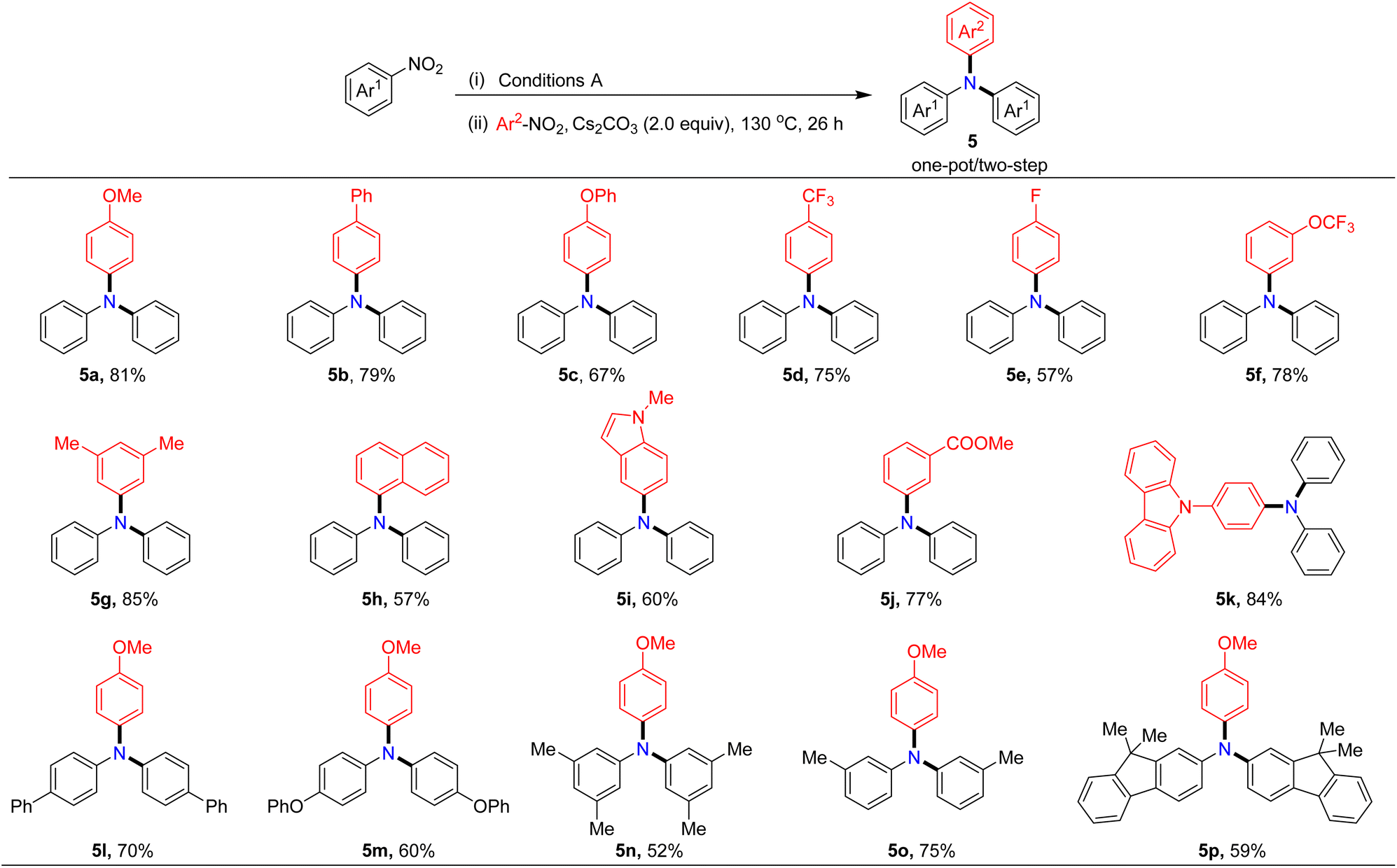
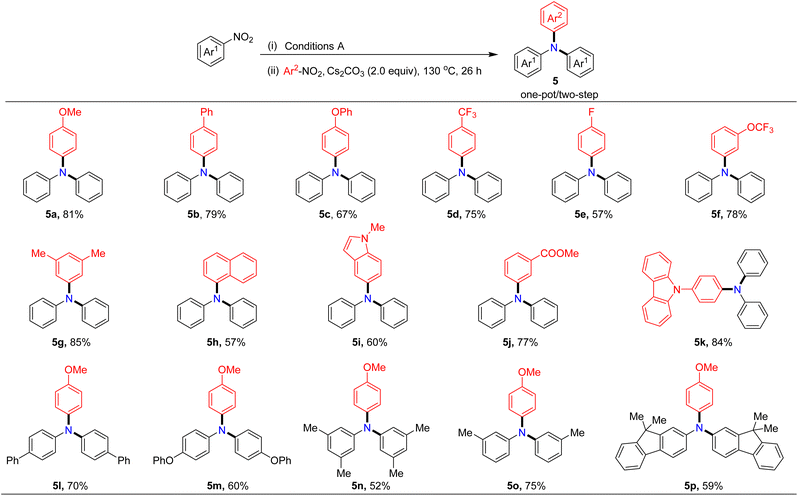
Assessment of reaction scope for synthesizing unsymmetrical triarylamines
Considering the importance of unsymmetrical triarylamines despite the challenge of their synthesis, we next explored the application of our protocol to their preparation. After a series of studies, we found that the unsymmetrical triarylamine 5a could be isolated in 81% yield when two additional equivalents of Cs2CO3 and a second nitroarene were added in situ to react for 26 hours after the generation of symmetrical diarylamines in one-pot. It is noteworthy to mention that the Pd/BrettPhos still retained high catalytic activity in the second step reaction. The reaction scope of unsymmetrical triarylamine synthesis was examined next. As illustrated in Table 5, the catalytic system was compatible with both electron-donating (methoxy, phenyl, phenoxy, and methyl) and electron-withdrawing substituents (trifluoromethyl, trifluoromethoxy, fluorine, and ester) at either the para- or meta-position on the aryl ring of nitroarenes, and the target unsymmetrical triarylamines were formed in 52–85% yields with high selectivity. Remarkably, nitroheteroarenes containing indole or carbazole skeletons were competent substrates, giving the desired products in 60% and 84% yields, respectively (5i and 5k). 1-Nitronaphthalene, a π-extended nitroarene, was also found to be a viable partner, generating the corresponding triarylamine in 57% yield (5h). Moreover, various symmetrical diarylamines generated in situ could also be coupled with 4-nitroanisole in one-pot to give the corresponding triarylamines in moderate to good yields (5l–5p).
Table 5 Selective one-pot/two-step construction of the unsymmetrical triarylaminesa,b
|
Reaction conditions: nitroarenes Ar1 (0.20 mmol), Pd(acac)2 (5.0 mol%), BrettPhos (15 mol%), TBAB (20 mol%), B2pin2 (0.3 mmol) and Cs2CO3 (0.4 mmol) in PhCF3 (1.0 mL) at 130 °C for 18 hours under N2. After the first step reaction, without any additional work-up, Cs2CO3 (0.4 mmol) and nitroarenes Ar2 (0.15 mmol) were added in situ to react at 130 °C for 26 hours.
Isolated yield.
|
|
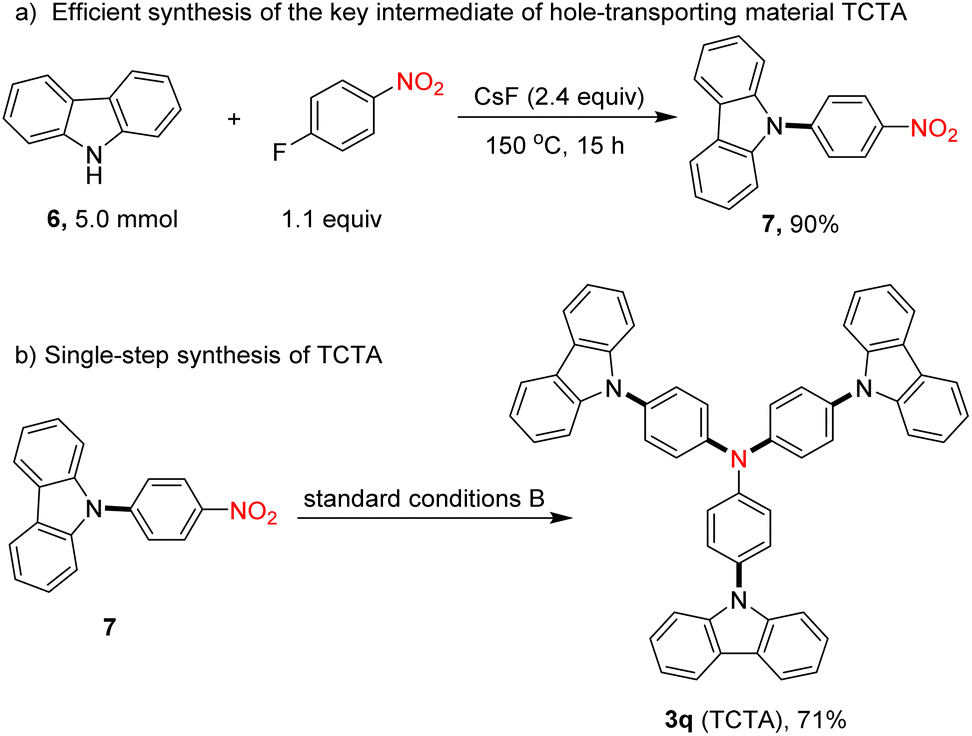
Exploration of synthetic applications
To illustrate the synthetic applications of this protocol, we also conducted the rapid synthesis of a functional material using this transformation as the critical step. After nucleophilic substitution of carbazole (6) with 1-fluoro-4-nitrobenzene, 9-(4-nitrophenyl)-9H-carbazole (7) was afforded in excellent yield (Scheme 2a).28 Then, using our protocol, TCTA (3q), a valuable hole-transporting material,29 was readily obtained in 71% isolated yield (Scheme 2b).
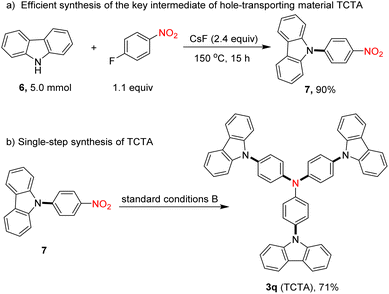 |
| | Scheme 2 The application in the synthesis of hole-transporting material TCTA. | |
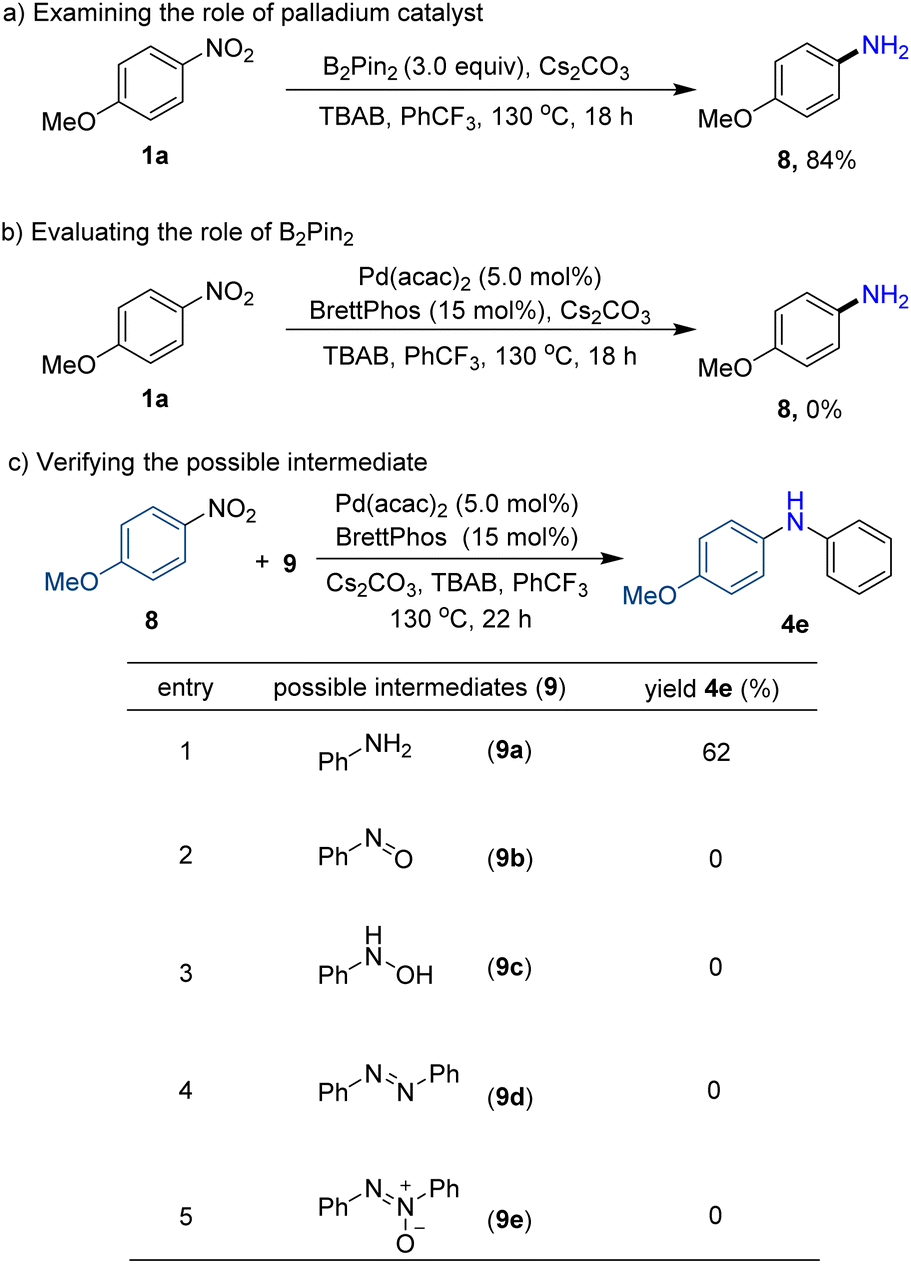
Mechanistic studies
In order to elucidate the reaction mechanism, preliminary mechanistic studies have been performed (Scheme 3). As expected, the reductive product p-anisidine (8) could be isolated in 84% yield without diarylamine formation when the palladium catalyst and ligand were excluded, implying that arylamine may be a possible intermediate (Scheme 3a). Furthermore, neither the desired product diarylamine nor reductive product p-anisidine (8) was observed without reductant agent B2pin2, indicating the crucial role of B2pin2 in reducing nitroarenes (Scheme 3b). In addition, the reaction of possible intermediates such as aniline (9a), nitrosobenzene (9b), N-phenylhydroxylamine (9c), azobenzene (9d), and azoxybenzene (9e) with nitroarenes was tested in the absence of reductant B2pin2. As a result, only using aniline, the desired diarylamine product (4e) was obtained with a yield of 62% (Scheme 3c, entries 1–5). The results further confirmed that arylamine may be a key intermediate in our protocol.
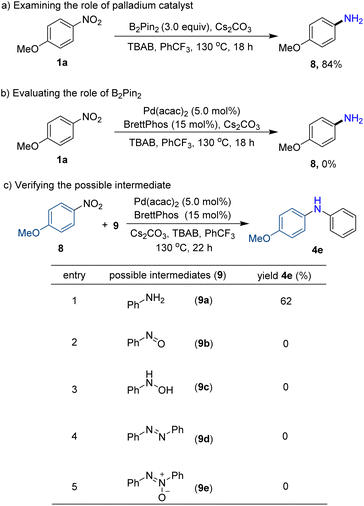 |
| | Scheme 3 Mechanistic studies. | |
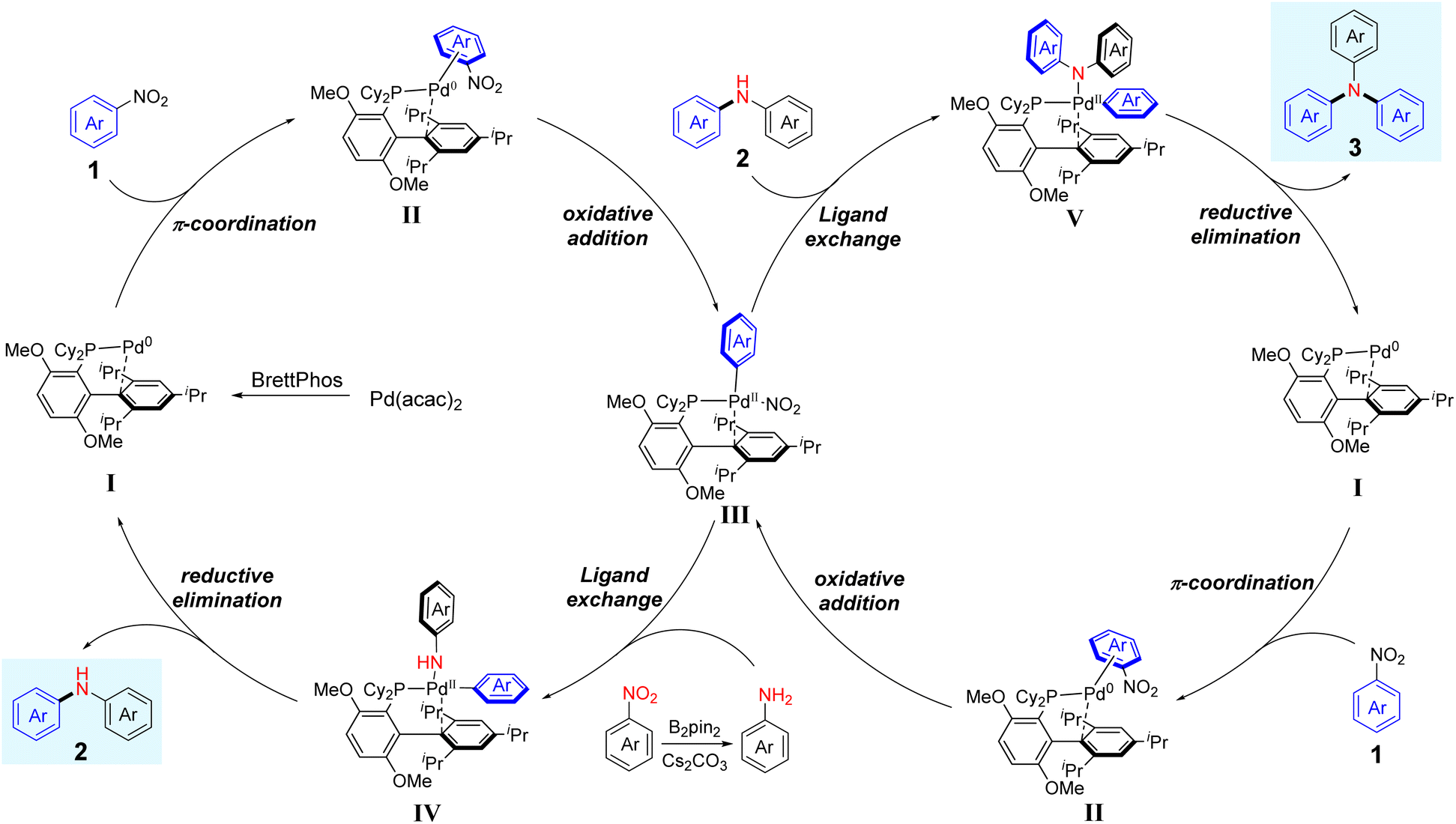
Based on previous reports23a,27 and our present studies, a plausible catalytic cycle for the reductive arylation of nitroarenes is described in Scheme 4. Initially, the η2-arene-palladium(0) complex II is formed by π-coordination of nitroarenes with the in situ generated palladium(0) complex, which is derived from the reaction of BrettPhos with Pa(acac)2. Subsequently, the oxidative addition of an Ar–NO2 bond to the Pd centre affords an Ar–Pd(II)–NO2 complex III. Next, intermediate III undergoes ligand exchange with arylamine, which is generated from the reduction of nitroarene by B2pin2, forming intermediate IV. Finally, reductive elimination by intermediate IV delivers diarylamine 2 and regenerates the Pd(0) catalyst I. Concerning the generation of triarylamine, diarylamine 2, acting as a nucleophile, undergoes ligand exchange with intermediate III, which comes from the same processes as aforementioned, furnishing the intermediate V. Following reductive elimination of intermediate V provides the triarylamine 3 and extrudes the active catalyst I to complete the catalytic cycle.
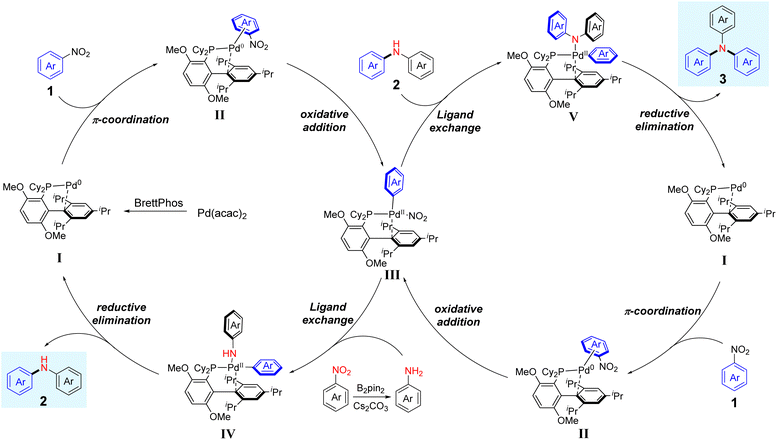 |
| | Scheme 4 A plausible mechanism for the reductive and denitrative C–N coupling of nitroarenes. | |
Conclusions
In summary, we have developed an efficient and straightforward method for the C(sp2)–N formation via Pd-catalyzed reductive and denitrative amination of nitroarenes. This protocol allows the inexpensive and readily available nitroarenes as both electrophiles and amine sources, thereby circumventing the necessity for isolation and purification of arylamines and the employment of halides. Consequently, it provides a highly step-economic strategy for highly selective construction of symmetrical and unsymmetrical di- and triarylamines. The reaction demonstrates compatibility with a broad range of functional groups and nitroheteroarene substrates using commercially available B2Pin2 as a reductant. Preliminary mechanistic investigations support that the reaction involves the formation of an arylamine intermediate, which is subsequently captured by the arylpalladium species, derived from the oxidation addition of a CAr–nitro bond to the palladium complex, leading to the production of diarylamine. The obtained diarylamine then undergoes denitrative Buchwald–Hartwig-type coupling with the subsequently introduced nitroarene to generate the triarylamine in a one-pot process.
Data availability
The data supporting this article have been included in ESI.†
Author contributions
L. Yu: supervision, conceptualization, writing – review and editing. W. Duan: review and editing. C.-J. Li: review and editing. Z. Lei: methodology, investigation and writing – original draft. J. Yao: investigation and review. Y. Xiao: partial substrates expansion. W. H. Liu: review and editing. All authors discussed the experimental results and commented on the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the contributions of Mr Jinshuo Wang to this manuscript. We gratefully acknowledge the financial support from the Guangxi Science and Talent Special Project under Grant No. AD23026131, the Guangxi Natural Science Foundation under Grant No. 2021GXNSFBA220027, and the Guangxi University (Start-up Grant No. A3040051017).
Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
-
B. Amini and S. Lowenkron, Aniline and its Derivatives: Kirk-Othmer Encyclopedia of Chemical Technolog, Wiley, Hoboken, 2003, pp. 783–809 Search PubMed.
- H. Zhang and C. Liu, Dyes Pigm., 2017, 143, 143–150 CrossRef CAS.
-
(a) D. Meng, J. Xue, Y. Zhao, E. Zhang, R. Zheng and Y. Yang, Chem. Rev., 2022, 122, 14954–14986 CrossRef CAS PubMed;
(b) K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS PubMed.
- H. Gao, L. Xue, S. Xin and J. B. Goodenough, Angew. Chem., Int. Ed., 2018, 57, 5449–5453 CrossRef CAS PubMed.
-
(a)
Z. Rappoport, The Chemistry of Anilines, WileyVCH, Weinheim, 2007 CrossRef;
(b)
A. Ricci, Amino Group Chemistry: From Synthesis to the Life Sciences, WileyVCH, Weinheim, 2008 Search PubMed.
- Selected reviews and examples on Ullmann condensation:
(a) S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed;
(b) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC;
(c) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449 CrossRef CAS PubMed;
(d) S. Li, X. Huang, Y. Gao and J. Jin, Org. Lett., 2022, 24, 5817–5824 CrossRef CAS PubMed.
- Selected reviews on Buchwald–Hartwig amination:
(a) R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed;
(b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed;
(c) J. F. HARTWIG, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS PubMed;
(d) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed;
(e) P. A. Forero-Cortés and A. M. Haydl, Org. Process Res. Dev., 2019, 23, 1478–1483 CrossRef;
(f) D. S. Surry and S. L. Buchwald, Chem. Sci., 2011, 2, 27–50 RSC;
(g) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- Selected review and examples on Chan–Lam coupling:
(a) J. Q. Chen, J. H. Li and Z. B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS;
(b) D. M. T. Chan, K. L. Monaco, R.-P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933–2936 CrossRef CAS;
(c) P. Y. S. Lam, C. G. Clarkt, S. Saubernt, J. Adamst, M. P. Winters, D. M. T. Chan and A. Combst, Tetrahedron Lett., 1998, 39, 2941–2944 CrossRef CAS.
- Selected reviews and examples on C–H amination:
(a) J.-Q. Yu and Z.-J. Shi, C-H Activation, Top. Curr. Chem., 2010, 292, 347–378 CrossRef;
(b) J. Jiao, K. Murakami and K. Itami, ACS Catal., 2015, 6, 610–633 CrossRef;
(c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed;
(d) C. Du, P.-X. Li, X. Zhu, J.-N. Han, J.-L. Niu and M.-P. Song, ACS Catal., 2017, 7, 2810–2814 CrossRef CAS.
- Selected reviews and examples on dehydrogenative aromatization ways:
(a) Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed;
(b) Z. Chen, H. Zeng, S. A. Girard, F. Wang, N. Chen and C. J. Li, Angew. Chem., Int. Ed., 2015, 54, 14487–14491 CrossRef CAS PubMed;
(c) S. A. Girard, X. Hu, T. Knauber, M.-O. Simon, G.-J. Deng, C.-J. Li and F. Zhou, Org. Lett., 2012, 14, 5606–5609 CrossRef CAS PubMed;
(d) K. Taniguchi, X. Jin, K. Yamaguchi, K. Nozaki and N. Mizuno, Chem. Sci., 2017, 8, 2131–2142 RSC;
(e) S. Takayama, T. Yatabe, Y. Koizumi, X. Jin, K. Nozaki, N. Mizuno and K. Yamaguchi, Chem. Sci., 2020, 11, 4074–4084 RSC.
- Selected book and example on “classical” nitration and reduction procedures:
(a)
U. Scholz, Evolution of Transition Metal-Catalyzed Amination Reactions: the Industrial Approach, in Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley-VCH, 2008 Search PubMed;
(b) R. S. Downing, P. J. Kunkeler and H. v. Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS.
- Other methods:
(a)
A. Ricci, Modern Amination Methods, WileyVCH, Weinheim, 2000 CrossRef;
(b) J. S. K. Clark, M. J. Ferguson, R. McDonald and M. Stradiotto, Angew. Chem., Int. Ed., 2019, 58, 6391–6395 CrossRef CAS PubMed;
(c) R. T. McGuire, J. F. J. Paffile, Y. Zhou and M. Stradiotto, ACS Catal., 2019, 9, 9292–9297 CrossRef CAS;
(d) A. Ribaucourt and J. Cossy, ACS Catal., 2020, 10, 10127–10148 CrossRef CAS;
(e) K. M. Morrison, C. S. Yeung and M. Stradiotto, Angew. Chem., Int. Ed., 2023, 62, e202300686 CrossRef CAS PubMed;
(f) L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed;
(g) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed;
(h) K. Wang, Z. H. Deng, S. J. Xie, D. D. Zhai, H. Y. Fang and Z. J. Shi, Nat. Commun., 2021, 12, 248 CrossRef CAS PubMed;
(i) G. Song, D.-Z. Nong, Q. Li, Y. Yan, G. Li, J. Fan, W. Zhang, R. Cao, C. Wang, J. Xiao and D. Xue, ACS Catal., 2022, 12, 15590–15599 CrossRef CAS.
- D. G. Brown and J. Boström, J. Med. Chem., 2015, 59, 4443–4458 CrossRef PubMed.
- J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. L. Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed.
-
(a) C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494 CrossRef PubMed;
(b) C. W. Cheung, J.-A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789–6792 CrossRef CAS PubMed;
(c) C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef PubMed;
(d) M. L. Ploeger, A. Darù, J. N. Harvey and X. Hu, ACS Catal., 2020, 10, 2845–2854 CrossRef CAS;
(e) C. W. Cheung, M. L. Ploeger and X. Hu, Chem. Sci., 2018, 9, 655–659 RSC.
-
(a) T. V. Nykaza, J. C. Cooper, G. Li, N. Mahieu, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 15200–15205 CrossRef CAS PubMed;
(b) G. Li, T. V. Nykaza, J. C. Cooper, A. Ramirez, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 6786–6799 CrossRef CAS PubMed.
- S. Suárez-Pantiga, R. Hernández-Ruiz, C. Virumbrales, M. R. Pedrosa and R. Sanz, Angew. Chem., Int. Ed., 2019, 58, 2129–2133 CrossRef PubMed.
- X. Guan, H. Zhu and T. G. Driver, ACS Catal., 2021, 11, 12417–12422 CrossRef CAS PubMed.
- G. Li, L. Yang, J. J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS PubMed.
- B. D. Akana-Schneider and D. J. Weix, J. Am. Chem. Soc., 2023, 145, 16150–16159 CrossRef CAS PubMed.
- T. Cao, Y.-P. Luo, L. Cheng, J.-L. Zhao, Q.-S. Jia, S. Zhang and X.-W. Liu, Eur. J. Org Chem., 2023, 26, e202300494 CrossRef CAS.
-
(a) M. R. Yadav, M. Nagaoka, M. Kashihara, R. L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423–9426 CrossRef CAS PubMed;
(b) F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 13307–13309 CrossRef CAS PubMed;
(c) M. Kashihara, M. R. Yadav and Y. Nakao, Org. Lett., 2018, 20, 1655–1658 CrossRef CAS PubMed.
-
(a) K. Muto, T. Okita and J. Yamaguchi, ACS Catal., 2020, 10, 9856–9871 CrossRef CAS;
(b) K. K. Asahara, T. Okita, A. N. Saito, K. Muto, Y. Nakao and J. Yamaguchi, Org. Lett., 2019, 21, 4721–4724 CrossRef CAS PubMed;
(c) T. Okita, K. K. Asahara, K. Muto and J. Yamaguchi, Org. Lett., 2020, 22, 3205–3208 CrossRef CAS PubMed.
-
(a) B. Feng, Y. Yang and J. You, Chem. Sci., 2020, 11, 6031–6035 RSC;
(b) B. Feng, Y. Yang and J. You, Chem. Commun., 2020, 56, 790–793 RSC;
(c) F. Zhou, F. Zhou, R. Su, Y. Yang and J. You, Chem. Sci., 2020, 11, 7424–7428 RSC.
-
(a) K. Chen, W. Chen, X. Yi, W. Chen, M. Liu and H. Wu, Chem. Commun., 2019, 55, 9287–9290 RSC;
(b) W. Chen, K. Chen, W. Chen, M. Liu and H. Wu, ACS Catal., 2019, 9, 8110–8115 CrossRef CAS;
(c) L. Feng, J. Yao, L. Yu and W. Duan, Palladium-Catalyzed Denitrative N-Arylation of Nitroarenes with Pyrroles, Indoles, and Carbazoles, Org. Chem. Front., 2022, 9, 2351–2356 RSC;
(d) J. Yao, L. Yu, W. Duan and C.-J. Li, Palladium-Catalyzed C–Si Bond Formation via Denitrative Cross-Coupling of Nitroarenes with Hexamethyldisilane, Org. Chem. Front., 2023, 10, 524–530 RSC.
-
(a) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed;
(b) H. Lu, Z. Geng, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2016, 18, 2774–2776 CrossRef CAS PubMed;
(c) K. Yang, F. Zhou, Z. Kuang, G. Gao, T. G. Driver and Q. Song, Org. Lett., 2016, 18, 4088–4091 CrossRef CAS PubMed;
(d) M. Rauser, C. Ascheberg and M. Niggemann, Angew. Chem., Int. Ed., 2017, 56, 11570–11574 CrossRef CAS PubMed.
- G.-S. Liou, S.-H. Hsiao and H.-W. Chen, J. Mater. Chem., 2006, 16, 1831–1842 RSC.
- Y. Kuwabara, H. Ogawa, H. Inada, N. Noma and Y. Shirota, Adv. Mater., 1994, 6, 677–679 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Lin
Yu
c,
Lin
Yu