
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Terminal dysprosium and holmium organoimides†
Theresa E.
Rieser
,
Dorothea
Schädle
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 22nd January 2024
Abstract
Terminal rare-earth-metal imide complexes TptBu,MeLn(NC6H3iPr2-2,6)(dmap) of the mid-late rare-earth elements dysprosium and holmium were synthesized via double methane elimination of Lewis acid stabilized dialkyl precursors TptBu,MeLnMe(GaMe4) with primary aniline derivative H2NC6H3iPr2-2,6 (H2NAriPr). Exploiting the weaker Ln–CH3⋯[GaMe3] interaction compared to the aluminium congener, addition of the aniline derivative leads to the mixed methyl/anilido species TptBu,MeLnMe(HNAriPr) which readily eliminate methane after being exposed to the Lewis base DMAP (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N,N-dimethyl-4-aminopyridine). Under the same conditions, [AlMe3]-stabilized dimethyl rare-earth-metal complexes transform immediately to Lewis acid bridged imides TptBu,MeLn(μ2-NC6H3Me2-2,6)(μ2-Me)AlMe2 (Ln = Dy, Ho). DMAP/THF donor exchange is accomplished by treatment of TptBu,MeLn(NC6H3iPr2-2,6)(dmap) with 9-BBN in THF while the terminal imides readily insert carbon dioxide to afford carbamate complexes.
N,N-dimethyl-4-aminopyridine). Under the same conditions, [AlMe3]-stabilized dimethyl rare-earth-metal complexes transform immediately to Lewis acid bridged imides TptBu,MeLn(μ2-NC6H3Me2-2,6)(μ2-Me)AlMe2 (Ln = Dy, Ho). DMAP/THF donor exchange is accomplished by treatment of TptBu,MeLn(NC6H3iPr2-2,6)(dmap) with 9-BBN in THF while the terminal imides readily insert carbon dioxide to afford carbamate complexes.
Introduction
Recent years have witnessed major progress in the field of rare-earth-metal (Ln) complexes with multiply bonded (dianionic) main-group ligands, most notably (organo)imide chemistry.1–3 Upon closer inspection this is not all that surprising, since [LnNR] moieties display a favourable Pearson hard/hard match and enhanced steric/electronic variability through the imido substituent R,4 compared to other fragments such as [LnO],5 [LnPR],6 or [Ln=CR2].7 Of particular interest have been the synthesis of terminal rare-earth-metal imides8–15 and a fundamental understanding of the Ln–imido bonding as well as reactivity,16 and in particular small-molecule-activation scenarios.17 Closely based on Chen's synthesis protocol of the first terminal scandium imide complex L1Sc(NAriPr)(dmap) (AriPr = C6H3iPr2-2,6; L1 = [AriPrNC(Me)CHC(Me)N(CH2)2NMe2]),8 a donor-promoted intramolecular methane/tetramethylsilane elimination from mixed methyl(neosilyl)/primary amido precursors emerged as the most efficient approach for accessing terminal Ln(III) imides (Scheme 1/A).8–13 Meanwhile, another three strategies have been successfully pursued for the early larger rare-earth metals. While anionic terminal ceric imides could be obtained by the deprotonation of a neutral primary amide complex with alkali-metal silylamides (Scheme 1/B),14 a [LnCH2(GaMe3)2] → [LnNR(thf)2] transformation proved feasible for generating the open-shell terminal imides TptBu,MeLn(NAriPr)(thf)2 (Ln = Ce, Nd, Sm) (Scheme 1/C).15 More recently, an anionic cerium(IV) terminal imide was accessed by a two-electron oxidation of a “Ce(II)” complex supported by a tripodal tris(amido)arene ligand using azide N3ArCF3 (ArCF3 = C6H3(CF3)2-3,5) (Scheme 1/D).18 Less surprising, terminal imides of the extremely large divalent rare-earth-metal centres have remained elusive.19
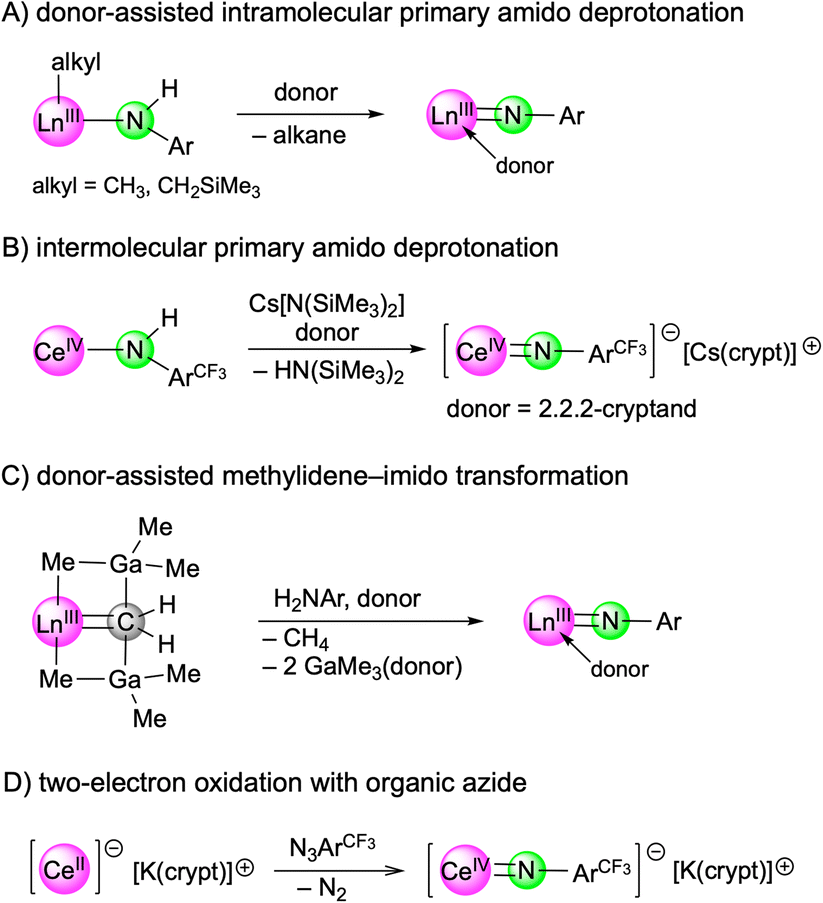 | ||
| Scheme 1 Synthesis strategies for terminal rare-earth-metal imides. | ||
Common features of all terminal rare-earth-metal imides, reported so far, are that the imido ligand is derived from a substituted aniline, H2NArR (R = iPr, Me, CF3), and an indispensable kinetic stabilization by use of sterically demanding ancillary co-ligands. Aliphatic amines, benzylic amines, and silylamines engage in imido ligand formation as well but have been detected only as metal-bridging and Lewis acid stabilized versions.3 Successfully applied ancillaries include β-diketiminato (nacnac),8,9,11 phosphazene,10 TriNox,14 and multidentate pyrazolato ligands.12,13,15 We found that especially the bulky monoanionic scorpionato ligand hydrotris(3-tert-butyl-5-methylpyrazolyl)borato (TptBu,Me) provides a useful scaffold for stabilizing terminal imides as well as phosphinidenes.6c,12,15 However, like for all terminal Ln(III) imides, stabilization of the highly polarized LnN bond, which predominantly consists of non-directional ionic interactions, is still challenging as it readily reacts with solvent molecules or the ancillary ligand of the aspired complexes.2,3 DFT calculations performed on the yttrium compound TptBu,MeY(NC6H3Me2-2,6)(dmap) confirmed a large ionic character of the Y–N(imido) bond but also a significant covalent bonding pattern with one σ-type and two π-type interactions.6c
Given the feasibility of terminal imides of the early open-shell cations Ce(III) (f1), Nd(III) (f3), and Sm(III) (f5),15 we herein envisaged the synthesis of those of the mid-late open-shell cations Dy(III) (f9) and Ho(III) (f10). These metal centres exhibit ionic radii similar to Y(III) but would (if at all) contribute to a distinct covalent bonding. On the other hand, their much higher molar mass might promote the crystallization behaviour and hence, the single-crystal X-ray structure diffraction (SCXRD) analysis of the targeted complexes. We also examined the reactivity of the first terminal, trivalent dysprosium and holmium imide complexes.
Results and discussion
Selection of precursors
Our original approach toward the terminal yttrium imide TptBu,MeY(NC6H3Me2-2,6)(dmap) involved the mixed methyl/tetramethylgallato complex TptBu,MeYMe(GaMe4) as a suitable precursor.12 There, the ease of GaMe3 displacement proved to be crucial for the successful synthesis. Consequently, we chose the trimethylgallium-stabilized dialkyl complexes TptBu,MeLnMe(GaMe4) (1-LnGa; Ln = Y,12 Dy,6c Ho) as precursors for the present study. Like their aluminium congeners,20 complexes 1-LnGa are available in moderate yield via protonolysis of the homoleptic gallates Ln(GaMe4)3 (ref. 21) with H[TptBu,Me] (ref. 22) and precipitation from toluene or n-hexane solution (Scheme 2; for detailed metrics of Ln(GaMe4)3 (Dy, Ho) and 1-HoGa; see the ESI, Fig. S1–S3†). The solid-state structure of the bimetallic compounds TptBu,MeLnMe(μ2-MeEMe3) (1-LnE, E = Al, Ga), depicting one terminal methyl group and an almost linear Ln–Me–E linkage, is not reflected in the solution NMR spectra, which reveal highly fluxional methyl groups at ambient temperature, an even higher mobility in case of the gallium derivatives. The isostructural 1-LnGa show Ln–C(Me) (Dy: 2.389(3) Å,6c Y: 2.385(3) Å,12 Ho: 2.356(5) Å) and Ln–C(MeGa) distances (Dy: 2.736(2) Å,6c Y: 2.688(2) Å,12 Ho: 2.652(4) Å) in accordance with the distinct Ln(III) radii.23 Unexpectedly, the solid-state structures of the formally five-coordinate 1-LnGa of the similar sized yttrium and holmium differ in the hapticity of the TptBu,Me ligand and in the Ln1–C26–Ga1 angle, which is more linear for the holmium derivative (174.9(2) vs. 163.3(1)°). Packing effects of co-crystallizing toluene in 1-HoGa have presumably a major impact on the coordination of the GaMe4 moiety and might also cause the bending of one pyrazolyl moiety toward the rare-earth metal centre. The pyrazolyl nitrogen atoms exhibit Ho–N interatomic distances ranging from 2.334(3) to 2.376(3) Å with an additional close contact to the tilted pyrazolato ligand (Ho⋯N5, 2.859(3) Å, see Fig. S3†). The proposed mechanism for the formation of 1-LnGa includes the preformation of [TptBu,MeLn(GaMe4)2] under release of methane and trimethylgallium, and the elimination of a second molecule GaMe3 sterically induced by the bulky TptBu,Me ligand. | ||
| Scheme 2 Formation of TptBu,MeLn(NAriPr)(dmap) (4-Ln: Ln = Y, Dy, Ho) via reaction of TptBu,MeLnMe(GaMe4)] (1-LnGa) with primary aniline H2NAriPr to afford mixed methyl/amido complexes TptBu,MeLnMe(HNAriPr) (2-Ln), and subsequent addition of DMAP. Use of 1-adamantylamine led to bis(amido) complex [TptBu,MeHo(HNAd)2] (3-Ho). Nucleophilic attack of acetonitrile by Dy–CH3 affords 7-Dy. | ||
The primary aniline H2NAriPr (AriPr = C6H3iPr2-2,6) has proven a privileged imido ligand precursor in rare-earth-metal chemistry.1–3 The molecule not only features the right balance of adequately acidic protons, but also a sufficient steric protection with bulky substituents in the positions 2 and 6 at the aryl group. The reaction of bis(alkyl) 1-LnGa with H2NAriPr to form the mixed methyl/primary amido complexes TptBu,MeLnMe(HNAriPr) 2-Ln (Ln = Y, Dy, Ho) is clearly visible by the elimination of methane and displacement of trimethylgallium (Scheme 2).
Complexes 2-Dy and 2-Ho are isostructural but crystallize in different space groups (2-Dy: P21/c; 2-Ho: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; for the crystal structures and detailed metrics, see the ESI, Fig. S4/S5†). Both compounds are insoluble in n-hexane, but dissolve in aromatic solvents such as toluene. The ancillary ligand coordinates in a κ3 fashion (N, N′, N′′) with considerably varying Ln–Npz distances (2-Dy: 2.3752(17)–2.5163(18) Å; 2-Ho: 2.369(2)–2.536(2) Å). The Ln–Namido (2-Dy: 2.212(2) Å; 2-Ho: 2.222(2) Å) and the Ln–CH3 (2-Dy: 2.436(2) Å; 2-Ho: 2.427(3) Å) interatomic distances lie in the range of the similar mixed methyl/amido complexes TptBu,MeLuMe(HNArR) (ArR = ArMe2 = C6H3Me2-2,6: Lu–Namido 2.189(2) Å, Lu–CH3 2.369(2) Å; ArR = ArCF3 = C6H3(CF3)2-3,5: Lu–Namido 2.215(1) Å, Lu–CH3 2.360(1) Å).12 The Ln–Namido–Cipso angle spans a wide range from 142.5(1)° (Ln = Lu, ArR = ArCF3),12 153.8(1)° (Ln = Lu, ArR = ArMe2)12 over 155.84(16)° (Ln = Dy, ArR = AriPr) to 160.28(19)° (Ln = Ho, ArR = AriPr), dependent on the Ln(III) centre and the substitution pattern of the amido ligand. As the paramagnetic nature of dysprosium and holmium impedes conclusive interpretations of their NMR spectra, the yttrium congener 2-Y was accessed from 1-YGa and H2NAriPr. The 1H NMR spectrum of 2-Y shows a sharp singlet at 0.46 ppm for the methyl group and a broader singlet at 4.87 ppm for the proton of the amido ligand.
; for the crystal structures and detailed metrics, see the ESI, Fig. S4/S5†). Both compounds are insoluble in n-hexane, but dissolve in aromatic solvents such as toluene. The ancillary ligand coordinates in a κ3 fashion (N, N′, N′′) with considerably varying Ln–Npz distances (2-Dy: 2.3752(17)–2.5163(18) Å; 2-Ho: 2.369(2)–2.536(2) Å). The Ln–Namido (2-Dy: 2.212(2) Å; 2-Ho: 2.222(2) Å) and the Ln–CH3 (2-Dy: 2.436(2) Å; 2-Ho: 2.427(3) Å) interatomic distances lie in the range of the similar mixed methyl/amido complexes TptBu,MeLuMe(HNArR) (ArR = ArMe2 = C6H3Me2-2,6: Lu–Namido 2.189(2) Å, Lu–CH3 2.369(2) Å; ArR = ArCF3 = C6H3(CF3)2-3,5: Lu–Namido 2.215(1) Å, Lu–CH3 2.360(1) Å).12 The Ln–Namido–Cipso angle spans a wide range from 142.5(1)° (Ln = Lu, ArR = ArCF3),12 153.8(1)° (Ln = Lu, ArR = ArMe2)12 over 155.84(16)° (Ln = Dy, ArR = AriPr) to 160.28(19)° (Ln = Ho, ArR = AriPr), dependent on the Ln(III) centre and the substitution pattern of the amido ligand. As the paramagnetic nature of dysprosium and holmium impedes conclusive interpretations of their NMR spectra, the yttrium congener 2-Y was accessed from 1-YGa and H2NAriPr. The 1H NMR spectrum of 2-Y shows a sharp singlet at 0.46 ppm for the methyl group and a broader singlet at 4.87 ppm for the proton of the amido ligand.
Noteworthy, the reaction of 1-HoGa with 1-adamantylamine in a 0.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio gave the bis(amido) holmium complex TptBu,MeHo(HNAd)2 (3-Ho, Ad = adamantyl), even though the primary amine was added in deficit. Apparently, the single deprotonation is favored over the second deprotonation, as the less Brønsted acidic second proton is less prone to be abstracted than the first proton of a second primary aniline. According to the present synthesis protocol, so far only sufficiently acidic aniline derivatives lead to the successful isolation of terminal rare-earth-metal imides, as other, less bulky and less electronically advantageous amines only form bis(amido) complexes,3,6c or in case 1-LnAl are employed result in trimethylaluminium-stabilized imide species.20a Like its precursors, complex 3-Ho is insoluble in aliphatic solvents, but readily dissolves in toluene and THF. The crystal structure of 3-Ho (triclinic P space group) shows the expected κ3 fashion (N, N′, N′′) of the ancillary ligand with Ho–Npz distances in the range of 2.393(3)–2.610(3) Å (Fig. 1).
:1 ratio gave the bis(amido) holmium complex TptBu,MeHo(HNAd)2 (3-Ho, Ad = adamantyl), even though the primary amine was added in deficit. Apparently, the single deprotonation is favored over the second deprotonation, as the less Brønsted acidic second proton is less prone to be abstracted than the first proton of a second primary aniline. According to the present synthesis protocol, so far only sufficiently acidic aniline derivatives lead to the successful isolation of terminal rare-earth-metal imides, as other, less bulky and less electronically advantageous amines only form bis(amido) complexes,3,6c or in case 1-LnAl are employed result in trimethylaluminium-stabilized imide species.20a Like its precursors, complex 3-Ho is insoluble in aliphatic solvents, but readily dissolves in toluene and THF. The crystal structure of 3-Ho (triclinic P space group) shows the expected κ3 fashion (N, N′, N′′) of the ancillary ligand with Ho–Npz distances in the range of 2.393(3)–2.610(3) Å (Fig. 1).
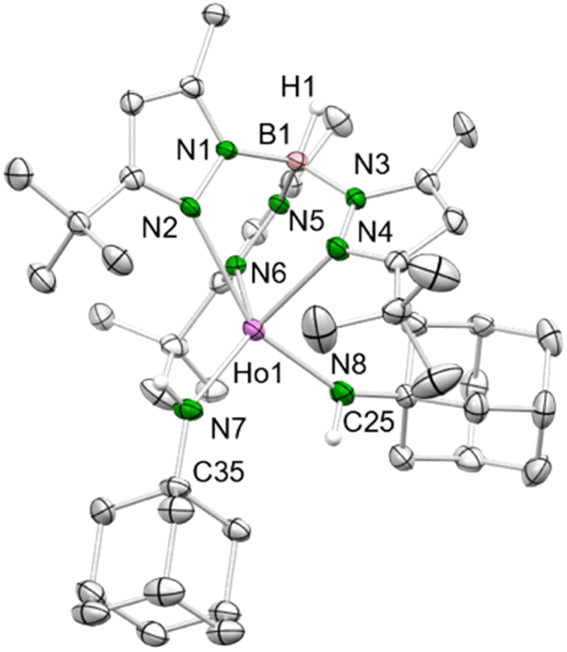 | ||
| Fig. 1 Crystal structure of 3-Ho. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Solvent molecules and hydrogen atoms except for those of B–H and N–H are omitted for clarity. Selected interatomic distances (Å) and angles (°): Ho1–N7 2.172(3), Ho1–N8 2.170(3); Ho1–N7–C35 144.0(3), Ho1–N8–C25 148.5(3) (for further metrics, see ESI†). | ||
The Ln–Namido distances (ø 2.171 Å) are longer compared to the Ln–Nimido interactions in TptBu,MeHo(μ2-NAd)AlMe3 (2.087(2) Å),20a but slightly shorter compared to the mixed methyl/amido complexes 2-Ln (Ln = Dy, Ho) described beforehand. This reflects a better electron donation of two nitrogen atoms to the metal centre, despite increased steric bulk. However, the angles Ho1–N7–C35 (144.0(3)°) and Ho1–N8–C25 (148.5(3)°) are significantly more bent compared to the methyl/anilido complex 2-Ho (160.28(19)°).
Ln(III) imide synthesis
Treatment of the mixed methyl/anilido complexes 2-Ln with the Lewis base DMAP led to the isolation of the targeted terminal lanthanide imide complexes TptBu,MeLn(NAriPr)(dmap) (4-Ln: Ln = Y, Dy, Ho). DMAP was used previously for the synthesis of terminal rare-earth-metal imide complexes,8,10–12 exploiting its strong donor capacity (versus e.g., THF) to induce the elimination of methane via inner-sphere deprotonation of the amido species. We assume, that the preceding coordination of DMAP to the metal centre affects the geometry of its coordination sphere in a way that the methyl group and the amido proton come into close proximity and finally evolve methane, being a very potent leaving group. The 1H NMR spectrum of 4-Y evidences the deprotonation of the amido functionality via the methyl ligand since both the N–H and methyl signal disappeared (see Fig. S21†). Complexes 4-Ln are insoluble in non-polar solvents (e.g. n-hexane), but easily dissolve in aromatic or polar solvents like toluene and THF.While the yttrium complex 4-Y could not be obtained in very pure form and gave only poor crystal quality (connectivity structure only, see ESI Fig. S7†), the dysprosium and holmium congeners displayed good crystallization and diffraction behaviours (isotypic, monoclinic space group C2/c, Fig. 2 and S8/9†). The metal centres are pentacoordinate with the ancillary TptBu,Me ligand coordinating in the familiar κ3 fashion (N, N′, N′′). The Ln–Npz distances typical of scorpinate ligands show two shorter bonds (4-Dy: 2.450(3)/2.452(3) Å, 4-Ho: 2.426(4)/2.436(3) Å) and one longer (4-Dy: 2.517(3) Å, 4-Ho: 2.492(4) Å). The Ln–Nimido distances are in line with those of other terminal imides, considering the changes in the ionic radii (Table 1). Except for the anionic ceric complex [(TriNOx)Ce(NArCF3)[Cs(2.2.2-cryptand)] (157.3(4)°),14a the Ln–Nimido–Cipso angles of all trivalent terminal imides are larger than 165° and almost linear (4-Dy: 166.0(2)°; 4-Ho: 166.7(3)°; Table 1). Exchange of the donor ligand dmap for thf seems to entail a shortening of the Ln–Nimido bond but doesn't appear to affect the Ln–Nimido–Cipso angle.
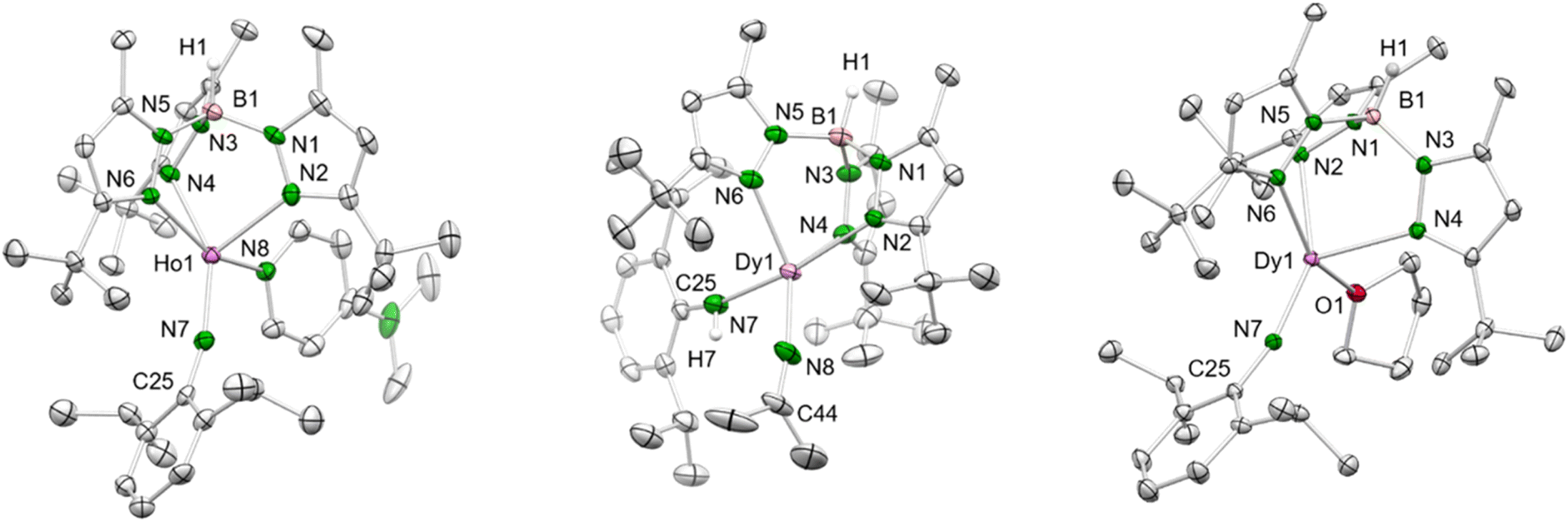 | ||
| Fig. 2 Left: Crystal structure of 4-Ho. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Solvent molecules, and hydrogen atoms except for that of B–H are omitted for clarity. For selected interatomic distances and angles, see Table 1 and ESI.† Middle: Crystal structure of 7-Dy. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Solvent molecules and hydrogen atoms except for those of B–H and N–H are omitted for clarity. Selected interatomic distances (Å) and angles (°): Dy1–N7 2.219(3), Dy1–N8 2.148(3); Dy1–N7–C25 156.3(3), Dy1–N8–C44 165.3(3) (for further metrics, see ESI†). Right: Crystal structure of 8-Dy. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Only one molecule of the asymmetric unit is shown. Solvent molecules, and hydrogen atoms except for that of B–H are omitted for clarity. For selected interatomic distances and angles, see Table 1 and ESI.† | ||
| Compound | LnNimido/Å |
Ln–do/Å | Ln–Nimido–Cipso/deg | IRd/Å (ref. 23) | CN | Ref. |
|---|---|---|---|---|---|---|
| a Two molecules in the asymmetric unit. b nacnacR1 = [AriPrNC(Me)CHC(Me)N(CH2)2NMe2]. c nacnacR2 = [AriPrNC(Me)CHC(Me)N(CH2)2N(CH2)2NMe2]. d Effective ionic radii. | ||||||
| TptBu,MeLu(NArCF3)(dmap) | 1.993(5) | 2.377(5) | 175.8(5) | 0.861 | 5 | 12 |
| TptBu,MeY(NArMe2)(dmap) | 2.024(4) | 2.426(4) | 173.6(4) | 0.900 | 5 | 12 |
| TptBu,MeHo(NAriPr)(dmap) (4-Ho) | 2.012(4) | 2.429(4) | 166.7(3) | 0.901 | 5 | This work |
| TptBu,MeDy(NAriPr)(dmap) (4-Dy) | 2.017(3) | 2.450(3) | 166.0(2) | 0.912 | 5 | This work |
| TptBu,MeDy(NAriPr)(thf) (8-Dy)a | 2.008(3)/2.004(4) | 2.397(3)/2.392(3) | 166.9(3)/165.3(3) | 0.912 | 5 | This work |
| TptBu,MeSm(NAriPr)(thf)2 | 2.067(5) | 2.527(5)/2.560(4) | 169.3(5) | 0.958 | 6 | 15 |
| TptBu,MeNd(NAriPr)(thf)2 | 2.076(4) | 2.557(4)/2.594(3) | 169.2(4) | 0.983 | 6 | 15 |
| TptBu,MeNd(NAriPr)(thf)a | 2.036(7)/2.047(7) | 2.517(6)/2.511(6) | 165.8(4)/163.2(6) | 0.983 | 5 | 15 |
| TptBu,MeCe(NAriPr)(thf)2 | 2.101(5) | 2.599(3)/2.628(3) | 171.3(3) | 1.01 | 6 | 15 |
| (nacnacR1)Sc(NAriPr)(dmap)b | 1.881(8) | 2.271(5) | 169.6(5) | 0.745 | 5 | 8 |
| (nacnacR1)Sc(NAriPr)(thf)b | 1.852(4) | 2.251(3) | 168.6(3) | 0.745 | 5 | 11 |
| (nacnacR2)Sc(NAriPr)(dmap)c | 1.8591(18) | 2.369(2) | 167.90(17) | 0.745 | 5 | 9 |
| [(PhN = Ph2P)2N]Sc(NAriPr)(dmap)2 | 1.853(3) | 2.379(3)/2.326(3) | 168.8(3) | 0.745 | 6 | 10 |
| (BPz2Py3)Sc(NAriPr) | 1.877(3) | — | 173.1(3) | 0.745 | 6 | 13 |
| [(TriNOx)Ce(NArCF3)][Cs(2.2.2-cryptand)] | 2.077(3) | — | 157.3(4) | 0.87 | 8 | 14a |
| [(AdTPBN3)Ce(NArCF3)][K(2.2.2-cryptand)] | 2.0742(4) | — | 176.3(4) | <0.87 | 4 | 18 |
In contrast, and as pointed out previously, the reaction of the primary aniline H2NArMe2 (ArMe2 = C6H3Me2-2,6) with the aluminium congeners TptBu,MeLnMe(AlMe4) (1-LnAl) implies the formation of trimethylaluminium-stabilized imide complexes.12 These complexes cannot be converted into unsupported terminal rare-earth-metal imide complexes by applying Lewis bases such as 1,4-dioxane, pyridine, DMAP, or TMEDA (N,N,N′,N′-tetramethylethylenediamine). Complex 1-DyAl is accessible from Dy(AlMe4)3 (ref. 24) and H[TptBu,Me],22 in analogy to the yttrium and holmium complexes reported previously.20 In order to probe the effect of the substituents on the aniline, compounds 1-LnAl (Ln = Dy, Ho) were reacted with H2NAriPr, H2NArMe2, and H2NArMe3 (ArMe3 = C6H2Me3-2,4,6) (Scheme 3).
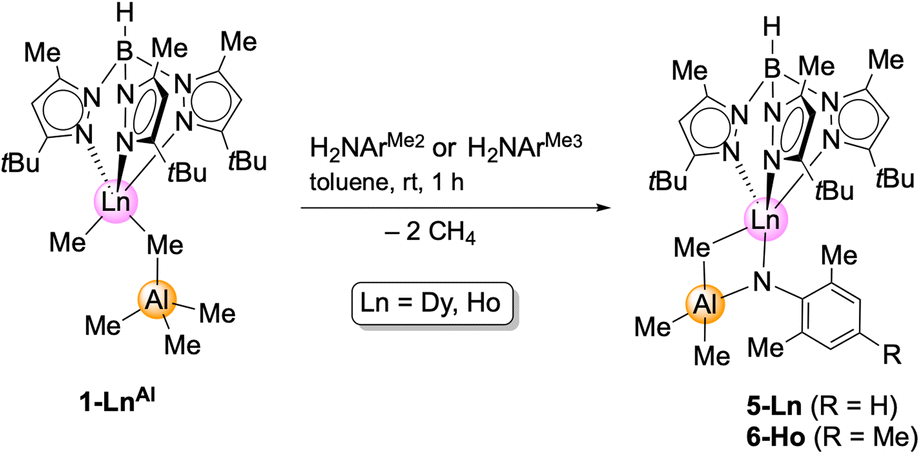 | ||
| Scheme 3 Synthesis of Lewis acid stabilized, bimetallic imides TptBu,MeLn(NArMe2)(μ2-MeAlMe3) (5-Ln; Ln = Dy, Ho) and TptBu,MeHo(NArMe3)(μ2-MeAlMe3). | ||
Contrary to the reaction with the weaker coordinated trimethylgallium in complexes 1-LnGa, the 1-LnAl/H2NAriPr reaction was inconclusive. Like in the case of yttrium,12 the sterically less demanding anilines gave the trimethylaluminium-stabilized imides TptBu,MeLn(μ2-NArMe2)AlMe3 (5-Ln: Ln = Dy, Ho) and TptBu,MeHo(μ2-NArMe3)AlMe3 (6-Ln). The coordinated trimethylaluminium is not removable, neither under vacuum, nor with Lewis bases (e.g., DMAP, THF). Presumably, after the first methane elimination and the coordination of the primary amido functionality to the metal centre, one of the methyl groups at the [μ2-MeAlMe3] unit abstracts the second amido proton via release of another molecule of methane, resulting in 5-Ln. Compounds 5-Ln are insoluble in aliphatic solvents, but dissolve in aromatic and polar solvents. The isostructural complexes crystallize in different space groups (5-Dy: monoclinic, P21/n; 5-Ho: triclinic, P; 6-Ho: monoclinic, Cc; Fig. 3, S10 and S11†).
 | ||
| Fig. 3 Crystal structure of 5-Ho. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Solvent molecules, and hydrogen atoms except for that of B–H are omitted for clarity. Selected interatomic distances (Å) and angles (°): Ho1–N7 2.116(4), Ho1–C25 2.549(5), Al1–C25 2.105(6); Ho1–N7–C28 146.7(4) (for isostructural 5-Dy and 6-Ho and further metrics, see ESI†). | ||
The bulky TptBu,Me ligand coordinates again in the κ3 fashion (N, N′, N′′) with interatomic Ln–N distances in the range of 2.423(2)–2.492(2) Å (5-Dy), 2.407(4)–2.437(4) Å (5-Ho) and 2.394(3)–2.454(3) Å (6-Ho). The central metal ion is pentacoordinate and the Ln–Nimido–Cipso angle is strongly bent (5-Dy: 146.68(17)°; 5-Ho: 146.7(4)°; 6-Ho: 151.0(2)°), which is due to the interaction with the Lewis acid trimethylaluminium. Hence, the electronic situation differs considerably compared to the terminal rare-earth-metal imide complexes 4-Ln (Ln = Dy, Ho) as electron density of the imido nitrogen is shifted to the empty p orbitals of the aluminium ion. This is also reflected in the Ln–Nimido bonds of 5-Ln and 6-Ho which are elongated by ca. 0.1 Å (5-Dy: 2.129(2) Å; 5-Ho: 2.116(4) Å; 6-Ho: 2.116(4) Å), matching those in TptBu,MeY(μ2-NArMe)AlHMe2 (Y–Nimido: 2.133(2) Å; Y–Nimido–Cipso: 145.1(2)°).25 Further structural comparison with the similar holmium imides TptBu,MeHo(μ2-NR)AlMe3 (R = tBu, adamantyl) clearly indicate a more pronounced Ln–Nimido interaction for the latter (Ho–Nimido: 2.083(2), 2.087(2) Å; Ho–Nimido–Cipso: 140.4(2), 140.2(1)°).20a Other Lewis acid supported monomeric rare-earth-metal imide complexes include Mindiola's (PNP)Sc(μ2-NAriPr)(μ2-Me)AlMe2 [PNPN(2-P-(CHMe2)2-4-methylphenyl)2] (ref. 26) or TptBu,MeLn(NAriPr)(MMe3) (Ln = Ce, Nd, Sm; M = Al, Ga) from our group.6c
Probing donors other than DMAP
N,N-Dimethyl-4-aminopyridine (DMAP) emerged as a most valuable donor for forcing the elimination of methane via inner-sphere deprotonation of the primary amido species in 2-Ln-type complexes. In order to assess the impact of the donor molecule on the Ln–Nimido bonding, the implementation of other donor ligands was examined. In general, donor ligands might be introduced according to Scheme 1 promoting methane elimination (route A) or via post-imide-synthesis exchange (Scheme 4). Since imido ligand formation according to route A could not be achieved with ethereal donors such as OEt2 or THF, complex 2-Dy was treated with N-donors TMEDA (tetramethylethylenediamine), TMPDA (tetramethylpropane-1,3-diamine) and acetonitrile. While the potentially bidentate diamines did not undergo any reaction yielding only in the isolation of the starting compounds, acetonitrile formed the insertion complex TptBu,MeDy[NC(Me)2](HNAriPr) (7-Dy, Scheme 2). It was previously shown that acetonitrile does react with rare-earth-metal alkyls either via C–H-bond activation/deprotonation or insertion.27–29 While the product of the deprotonation reaction (C5Me5)2La[CH(SiMe3)2]/CH3CN was structurally characterized as [(C5Me5)2La(μ-CH2CN)]2,27 the insertion product of the reaction (C5Me5)2ScCH3/CH3CN was only spectroscopically analyzed as (C5Me5)2Sc[NC(Me)2].28 The SCXRD study of five-coordinate 7-Dy features distinct bonding behaviour of the primary amido and the dimethyliminato ligand (Dy–N: 2.219(3) versus 2.148(3) Å; Dy–N–C: 156.3(3) versus 165.3(3)°) (Fig. 2). Thus, the dimethyliminato coordination compares to that of imidazolin-2-iminato complexes like LY(CH2SiMe3)2(thf)2 (L = 1,3-bis(2,6-diisopropylphenyl)imidazoline-2-iminato; Y–N, 2.1255(13) Å, Y–N–C, 176.85(12)° or LYCl2(thf)3 Y–N, 2.1278(18) Å, Y–N–C, 174.35(16)°) supposedly featuring very short Ln–Niminato bonds.30,31 | ||
| Scheme 4 Reactivity of TptBu,MeDy(NAriPr)(dmap) (4-Dy) (Ln = Y, Dy) towards Lewis acid 9-BBN in THF and toluene. | ||
The post-synthesis exchange approach (route 2, Scheme 4) was probed with 4-Dy and the strong Lewis acid 9-borabicyclo[3.3.1]nonane (9-BBN) in THF.11 Accordingly, the equimolar reaction led to the displacement of DMAP and coordination of one THF molecule to the dysprosium centre in TptBu,MeDy(NAriPr)(thf) (8-Dy, Scheme 4). Like the dmap adduct, 8-Dy is soluble in toluene and THF, but insoluble in aliphatic solvents. It crystallized in the orthorhombic space group Pna21 and shows the same κ3 coordination of the TptBu,Me ligand as complex 4-Dy before (Dy–Npz: 2.465(4)–2.510(4)/2.450(3)–2.539(4) Å) (Fig. 2). As the coordination number did not change, the angle of the imido functionality stayed nearly the same (Dy–Nimido–Cipso: 165.2(3)/166.9(3)°) as detected for 4-Dy. However, the Dy–Nimido distance of 2.004(4)/2.008(3) Å) in 8-Dy appears to be slightly shorter compared to 4-Dy (Table 1) which can be attributed to the weaker donor properties of the thf ligand.
The 9-BBN-promoted donor exchange was previously introduced by Chen, revealing that prior activation of L1Sc(NAriPr)(dmap) (L1 = [AriPrNC(Me)CHC(Me)N(CH2)2NMe2]) with 9-BBN led to abstraction of the donor molecule DMAP.11 The emerging donor-free imide intermediate [L1Sc(NAriPr)] could be trapped with THF to afford L1Sc(NAriPr)(thf) featuring also a shorter Sc–Nimido bond than the DMAP adduct (Table 1: 1.852(4) versus 1.881(8) Å). It was also mentioned that the direct synthesis of the THF adduct L1Sc(NAriPr)(thf) is not possible by thermolysis of the mixed methyl/amido scandium complex in THF, which is the same case for 8-Dy as well.2 In contrast, terminal imides TptBu,MeLn(NAriPr)(thf)2 of the larger rare-earth metals can be obtained directly according to the donor(THF)-assisted methylidene → imido transformation (Scheme 1 and Table 1).6c
Treatment of 4-Dy with excess of 9-BBN in toluene gave the mixed primary amido/hydroborato complex 9-Dy (Scheme 4 and Fig. 4). Again, this is in line with the observation made by Chen with the system L2Sc(NAriPr)/9-BBN (L2 = [AriPrNC(Me)CHC(Me)N(CH2)2N(CH2)2NMe2]).32 Correspondingly, it can be hypothesized that initially the strong Lewis acid 9-BBN displaces all of the coordinated DMAP, rendering a highly reactive donor-free terminal imide [TptBu,MeDy(NAriPr)] (Scheme 4, intermediate I1, lower trace). Subsequent 1,2-addition of a tBu methyl group (C–H-bond activation) across the highly reactive Dy–Nimido bond of transient species I1 reforms the primary amido ligand along with a 5-membered metallacycle in I2. Then, the highly nucleophilic alkyl attached to the dysprosium attacks a second molecule of 9-BBN to afford the alkylhydroborato moiety. The resulting Dy–Namido–Cipso angle (143.4(2)°) and the Dy–Namido distance (2.237(3) Å) of 9-Dy are comparable to the dysprosium amide complexes discussed beforehand. The Dy–B distance of 2.703(4) Å is in the range of the Y–B distances in [(Me3Si)2NC(NiPr)2]Y[μ-H(μ-Et)2BEt]2(thf)2 (2.658(4) and 2.671(4) Å)33 and (C5Me5)2YH(9-BBN) (2.767(6) Å),34 but longer than those observed in (tBu4Carb)Dy(BH4)2(thf) (2.473(2) and 2.487(2) Å).35
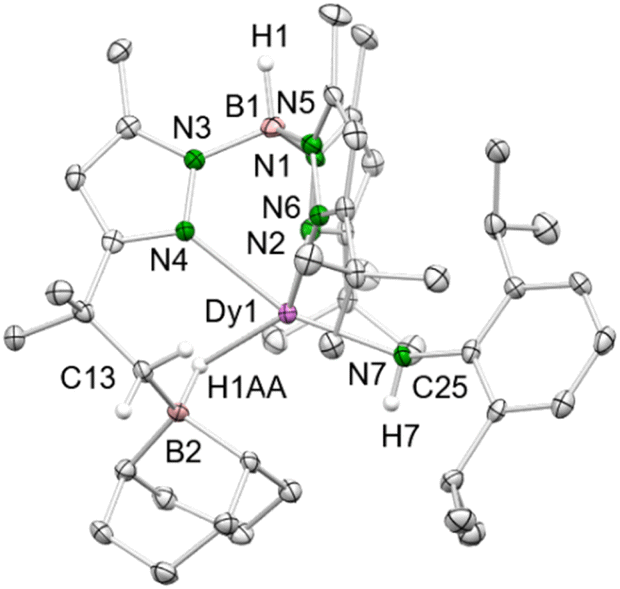 | ||
| Fig. 4 Crystal structure of 9-Dy. All atoms are represented by atomic displacement ellipsoids set at 50% probability. Solvent molecules and hydrogen atoms except for those of B–H and N–H are omitted for clarity. Selected bond distances (Å) and angels (°) for 9-Dy: Dy1–N7 2.237(3), Dy 1–N2 2.452(3), Dy 1–N4 2.405(3), Dy 1–N6 2.461(3), Dy 1–B2 2.703(4); Dy1–H1AA 2.17(3), B2–H1AA 1.23(3); Dy1–N7–C35 143.4(2), N7–Dy1–B2 117.06(11), N7–Dy1–N4 161.07(10), N7–Dy1–N2 93.18(10), N7–Dy1–N6 92.04(10). | ||
In Chen's system [L2Sc(NAriPr)]/9-BBN the respective C(sp3)–H bond borylation took place at the [–CH2NMe2] side arm of the ancillary nacnac ligand (Sc–Hhydrido, 2.01(3) Å).32 Chen also reported on the reaction of L1Sc(NAriPr)(dmap) with three equivalents of 9-BBN which led to mixed boroamido/hydroborato complex L1Sc[NAriPr(9-BBN–H)](9-BBN + H) (Sc–Hhydrido, 1.89 Å and 1.20 Å).11
Preliminary studies on the reactivity of terminal imide complex 4-Dy towards carbon dioxide (1 bar) in toluene at ambient temperature clearly indicate CO2 insertion into the Dy–Nimido bond and, hence, carbamate formation in product 10-Dy. The DRIFT spectrum of 10-Dy revealed typical carbonyl vibrations at 1701 cm−1 and 1641 cm−1 assigned to asymmetric and symmetric CO stretching vibrations (Fig. S17†). Moreover, the B–H vibration of 4-Dy at 2557 cm−1, typical for the terminal B–H stretch of the TptBu,Me ligand when coordinated in a tridentate fashion,36 changed to 2431 cm−1 in 10-Dy. Such a dramatic change of the position of the B–H band can be ascribed to a κ3 → κ2 coordination switch of the TptBu,Me ligand, likely involving a Dy⋯H–B interaction.37 Unfortunately, crystals of 10-Dy suitable for SCXRD analysis could not be obtained, but 10-Dy can be tentatively assigned as the dicarboxylate species [TptBu,MeDy{(O2C)2NAriPr}] on the basis of the double CO2-insertion chemistry of the terminal scandium imides L2Sc(NAriPr) (A, Fig. 5)17a and (BPz2Py3)Sc(NAriPr) (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1685 and 1632 cm−1).13 For further comparison, mono insertion of carbon dioxide was observed for the Lu3(μ3-NPh) moiety of the trinuclear cluster [L3Lu3(μ2-Me)3(μ3-Me)(μ3-NPh)] (L3 = PhC(NC6H3iPr2-2,6; C, Fig. 5)17d and the alkali-metal stabilized Ce(IV)–Nimido bond of [(TriNOx)Ce(NArCF3)][Cs(2.2.2-cryptand)] (B, Fig. 5).38
= 1685 and 1632 cm−1).13 For further comparison, mono insertion of carbon dioxide was observed for the Lu3(μ3-NPh) moiety of the trinuclear cluster [L3Lu3(μ2-Me)3(μ3-Me)(μ3-NPh)] (L3 = PhC(NC6H3iPr2-2,6; C, Fig. 5)17d and the alkali-metal stabilized Ce(IV)–Nimido bond of [(TriNOx)Ce(NArCF3)][Cs(2.2.2-cryptand)] (B, Fig. 5).38
 | ||
| Fig. 5 Structurally authenticated CO2-insertion products A,17aB,38 and C (ref. 17d) emerged from rare-earth-metal imides. | ||
Conclusion
The successful application of the donor-assisted intramolecular primary amido deprotonation protocol towards terminal imides of the rare-earth metals yttrium, dysprosium, and holmium depends on two crucial factors: first, the kinetic stabilization of the targeted imide via appropriate steric shielding of the ancillary ligand, herein supplied by the bulky TptBuMe scorpionate ligand; second, sufficient Brønsted acidity and steric demand of the employed primary amine, herein provided by H2NAriPr (AriPr = C6H3iPr2-2,6). With these key factors in mind, it was possible to generate a series of terminal rare-earth-metal imides TptBu,MeLn(NAriPr)(dmap) for mid-sized to small rare-earth metals. Their reactivity parallels that of terminal scandium imides, as revealed by treatment with the strong Lewis acid 9-borabicyclo[3.3.1]nonane (9-BBN) and the heteroallene CO2. These reactions revealed effective DMAP/THF donor exchange and CO2 insertion into the Ln–Nimido bond (carbamate formation). The reactivity studies also include the isolation and structural characterization of the bis(amido) species TptBu,MeHo(NAd)2 (Ad = adamantyl) and dimethyliminate complex TptBu,MeDy[NC(Me)2](HNAriPr).Data availability
Experimental, spectroscopic and structural data supporting this article have been uploaded as part of the ESI. Crystallographic data for all compounds have been deposited at the CCDC under 2312212–2312226, and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Author contributions
TR, synthesis and characterization of compounds, writing original draft; DS, synthesis and characterization of compound 1-HoGa, editing original draft; CM-M, crystallography, editing original draft; RA, conceptualization, supervision, writing and project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Felix Kracht for assistance with recording NMR and IR spectra.Notes and references
- O. T. Summerscales and J. C. Gordon, Complexes containing multiple bonding interactions between lanthanoid elements and main-group fragments, RSC Adv., 2013, 3, 66–6692 RSC.
- E. Lu, J. Chu and Y. Chen, Scandium Terminal Imido Chemistry, Acc. Chem. Res., 2018, 51, 557–566 CrossRef CAS PubMed.
- D. Schädle and R. Anwander, Rare-earth metal and actinide organoimide chemistry, Chem. Soc. Rev., 2019, 48, 5752–5805 RSC.
- R. Anwander, “Self-Assembly” in Organolanthanide Chemistry: Formation of Rings and Clusters, Angew. Chem., Int. Ed., 1998, 37, 599–602 CrossRef CAS PubMed.
- (a) J. Scott, H. Fan, B. F. Wicker, A. R. Fout, M.-H. Baik and D. J. Mindiola, Lewis Acid Stabilized Methylidene and Oxoscandium Complexes, J. Am. Chem. Soc., 2008, 130, 14438–14439 CrossRef CAS PubMed; (b) Y.-M. So, G.-C. Wang, Y. Li, H. H.-Y. Sung, I. D. Williams, Z. Lin and W.-H. Leung, A Tetravalent Cerium Complex Containing a Ce=O Bond, Angew. Chem., Int. Ed., 2014, 53, 1626–1629 CrossRef CAS PubMed; (c) P. L. Damon, G. Wu, N. Kaltsoyannis and T. W. Hayton, Formation of a Ce(IV) Oxo Complex via Inner Sphere Nitrate Reduction, J. Am. Chem. Soc., 2016, 138, 12743–12746 CrossRef CAS PubMed; (d) M. K. Assefa, G. Wu and T. W. Hayton, Synthesis of a terminal Ce(IV) oxo complex by photolysis of a Ce(III) nitrate complex, Chem. Sci., 2017, 8, 7873–7878 RSC.
- (a) B. F. Wicker, J. Scott, J. G. Andino, X. Gao, H. Park, M. Pink and D. J. Mindiola, Phosphinidene Complexes of Scandium: Powerful PAr Group-Transfer Vehicles to Organic and Inorganic Substrates, J. Am. Chem. Soc., 2010, 132, 3691–3693 CrossRef CAS PubMed; (b) B. Feng, L. Xiang, A. Carpentier, L. Maron, X. Leng and Y. Chen, Scandium-Terminal Boronylphosphinidene Complex, J. Am. Chem. Soc., 2021, 143, 2705–2709 CrossRef CAS PubMed; (c) T. E. Rieser, P. Wetzel, P. Sirsch, C. Maichle-Mössmer and R. Anwander, A Terminal Yttrium Phosphinidene, J. Am. Chem. Soc., 2023, 145, 4102–4113 CrossRef PubMed.
- J. Kratsch and P. W. Roesky, Rare-Earth-Metal Methylidene Complexes, Angew. Chem., Int. Ed., 2014, 53, 376–383 CrossRef CAS PubMed.
- E. Lu, Y. Li and Y. Chen, A scandium terminal imido complex: synthesis, structure and DFT studies, Chem. Commun., 2010, 46, 4469–4471 RSC.
- E. Lu, J. Chu, Y. Chen, M. V. Borzov and G. Li, Scandium terminal imido complex induced C–H bond selenation and formation of an Sc–Se bond, Chem. Commun., 2011, 47, 743–745 RSC.
- W. Rong, J. Cheng, Z. Mou, H. Xie and D. Cui, Facile Preparation of a Scandium Terminal Imido Complex Supported by a Phosphazene Ligand, Organometallics, 2013, 32, 5523–5529 CrossRef CAS.
- J. Chu, X. Han, C. E. Kefalidis, J. Zhou, L. Maron, X. Leng and Y. Chen, Lewis Acid Triggered Reactivity of a Lewis Base Stabilized Scandium-Terminal Imido Complex: C–H Bond Activation, Cycloaddition, and Dehydrofluorination, J. Am. Chem. Soc., 2014, 136, 10894–10897 CrossRef CAS PubMed.
- D. Schädle, M. Meermann-Zimmermann, C. Schädle, C. Maichle-Mössmer and R. Anwander, Rare-Earth Metal Complexes with Terminal Imido Ligands, Eur. J. Inorg. Chem., 2015, 1334–1339 CrossRef.
- E. A. Patrick, Y. Yang, W. E. Piers, L. Maron and B. S. Gelfand, A monoanionic pentadentate ligand platform for scandium–pnictogen multiple bonds, Chem. Commun., 2021, 57, 8640–8643 RSC.
- (a) L. A. Solola, A. V. Zabula, W. L. Dorfner, B. C. Manor, P. J. Carroll and E. J. Schelter, Cerium(IV) Imido Complexes: Structural, Computational, and Reactivity Studies, J. Am. Chem. Soc., 2017, 139, 2435–2442 CrossRef CAS PubMed; (b) T. Cheisson, L. A. Solola, M. R. Gau, P. J. Carroll and E. J. Schelter, Silyl Transfer Pathway to a Ce(IV) Imido Complex, Organometallics, 2018, 37, 4332–4335 CrossRef CAS.
- T. E. Rieser, R. Thim-Spöring, D. Schädle, P. Sirsch, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Open-Shell Early Lanthanide Terminal Imides, J. Am. Chem. Soc., 2022, 144, 4102–4113 CrossRef CAS PubMed.
- (a) J. C. Gordon, G. R. Giesbrecht, D. L. Clark, P. J. Hay, D. W. Keogh, R. Poli, B. L. Scott and J. G. Watkin, The First Example of a μ2-Imido Functionality Bound to a Lanthanide Metal Center: X-ray Crystal Structure and DFT Study of [(μ-ArN)Sm(μ-NHAr)(μ-Me)AlMe2]2 (Ar = 2,6-iPr2C6H3), Organometallics, 2002, 21, 4726–4734 CrossRef CAS; (b) T. Cheisson, K. D. Kersey, N. Mahieu, A. McSkimming, M. R. Gau, P. J. Carroll and E. J. Schelter, Multiple Bonding in Lanthanides and Actinides: Direct Comparison of Covalency in Thorium(IV)- and Cerium(IV)-Imido Complexes, J. Am. Chem. Soc., 2019, 141, 9185–9190 CrossRef CAS PubMed.
- (a) J. Chu, E. Lu, Z. Liu, Y. Chen, X. Leng and H. Song, Reactivity of a Scandium Terminal Imido Complex towards Unsaturated Substrates, Angew. Chem., Int. Ed., 2011, 50, 7677–7680 CrossRef CAS PubMed; (b) Z. Jian, W. Rong, Z. Mou, Y. Pan, H. Xie and D. Cui, Intramolecular C–H bond activation induced by a scandium terminal imido complex, Chem. Commun., 2012, 48, 7515–7518 Search PubMed; (c) J. Chu, E. Lu, Y. Chen and X. Leng, Reversible Addition of the Si-H Bond of Phenylsilane to the Sc=N Bond of a Scandium terminal Imido Complex, Organometallics, 2013, 32, 1137–1140 CrossRef CAS; (d) J. Hong, L. Zhang, K. Wang, Y. Zhang, L. Weng and X. Zhou, Methylidene Rare-Earth-Metal Complexes Mediated Transformations of C=N, N=N and N–H Bonds: New Routes to Imido Rare-Earth-Metal Clusters, Chem.–Eur. J., 2013, 19, 7865–7873 CrossRef CAS PubMed.
- Y. Wang, J. Liang, C. Deng, R. Sun, P.-X. Fu, B. W- Wang, S. Gao and W. Huang, Two-Electron Oxidations at a Single Cerium Center, J. Am. Chem. Soc., 2023, 145, 22466–22474 CrossRef CAS PubMed.
- M. Katzenmayer, F. Kracht, C. Maichle-Mössmer and R. Anwander, Potential precursors for terminal ytterbium(II) imide complexes bearing the tris(3-tert-butyl-5-methylpyrazolyl)hydroborato ligand, Dalton Trans., 2023, 72, 6273–6283 RSC.
- (a) D. Schädle, C. Maichle-Mössmer, C. Schädle and R. Anwander, Rare-Earth-Metal Methyl, Amide, and Imide Complexes Supported by a Superbulky Scorpionate Ligand, Chem.–Eur. J., 2015, 21, 662–670 CrossRef PubMed; (b) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, A Rare-Earth Metal Variant of the Tebbe Reagent, Angew. Chem., Int. Ed., 2008, 47, 9560–9564 CrossRef PubMed; (c) M. Zimmermann, J. Takats, G. Kiel, K. W. Törnroos and R. Anwander, Ln(III) methyl and methylidene complexes stabilized by a bulky hydrotris(pyrazolyl)borate ligand, Chem. Commun., 2008, 612–614 RSC.
- (a) W. J. Evans, R. Anwander, R. J. Doedens and J. W. Ziller, The Use of Heterometallic Bridging Moieties To Generate Tractable Lanthanide Complexes of Small Ligands, Angew. Chem. Int. Ed. Engl., 1994, 33, 1641–1644 CrossRef; (b) H. M. Dietrich, G. Raudaschl-Sieber and R. Anwander, Trimethylyttrium and Trimethyllutetium, Angew. Chem., Int. Ed., 2005, 44, 5303–5306 CrossRef CAS PubMed.
- K. O. Saliu, J. Chen, R. McDonald and J. Takats, Acid form of Trofimenko’s scorpionates, H(TpR,R’); comments on the synthesis and solid-state structure of H(TptBu,Me), Aust. J. Chem., 2022, 75, 566–570 CrossRef CAS.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- S. N. König, N. F. Chilton, C. Maichle-Mössmer, E. M. Pineda, T. Pugh, R. Anwander and R. A. Layfield, Fast magnetic relaxation in an octahedral dysprosium tetramethyl-aluminate complex, Dalton Trans., 2014, 43, 3035–3038 RSC.
- C. Schädle, D. Schädle, K. Eichele and R. Anwander, Methylaluminum-Supported Rare-Earth-Metal Dihydrides, Angew. Chem., Int. Ed., 2013, 52, 13238–13242 CrossRef PubMed.
- J. Scott, F. Basuli, A. R. Fout, J. C. Huffman and D. J. Mindiola, Evidence for the Existence of a Terminal Imidoscandium Compound: Intramolecular C–H Activation and Complexation Reactions with the Transient Sc=NAr Species, Angew. Chem., Int. Ed., 2008, 47, 8502–8505 CrossRef CAS PubMed.
- (a) H. J. Heeres, A. Meetsma and J. H. Teuben, CH Activation of Acetonitrile by Alkyl Compounds of the Early Lanthanoids: Dimeric Cyanomethyl-Lanthanoid Complexes with CH2CN Bridges, Angew. Chem., Int. Ed., 1990, 29, 420–422 CrossRef; (b) R. Duchateau, C. T. van Wee and J. H. Teuben, Insertion and C–H Bond Activation of Unsaturated Substrates by Bis(benzamidinato)yttrium Alkyl, [PhC(NSiMe3)2]2YR (R = CH2Ph THF, CH(SiMe3)2, and Hydrido, {[PhC(NSiMe3)2]2Y(μ-H)}2, Compounds, Organometallics, 1996, 15, 2291–2302 CrossRef CAS.
- J. E. Bercaw, D. L. Davies and P. T. Wolczanski, Reactions of Alkyl and Hydride Derivatives of Permethylscandocene and -zirconocene with Nitriles and Amines. Catalytic Hydrogenation of tert-Butyl Cyanide with Permethylscandocene Hydride, Organometallics, 1985, 5, 443–450 CrossRef.
- W.-Y. Yeh, C.-S. Ting, S.-M. Peng and G.-H. Lee, Reduction of Acetonitrile Ligand on W(PhCCPh)3(NCMe) and W(η4-C4Ph4)(PhCCPH)2(NCMe): Crystal Structure of W(PCCPh)3(NH=C(Me)2), Organometallics, 1995, 14, 1417–1422 CrossRef CAS.
- A. G. Trambitas, T. K. Panda, J. Jenter, P. W. Roesky, C. Daniluic, C. G. Hrib, P. Jones and M. Tamm, Rare-Earth Metal Alkyl, Amido, and Cyclopentadienyl Complexes Supported by Imidazolin-2-iminato Ligands: Synthesis, Structural Characterization, and Catalytic Application, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed.
- T. K. Panda, S. Randoll, C. G. Hrib, P. Jones, T. Bannenberg and M. Tamm, Syntheses and structures of mononuclear lutetium imido complexes with very short Lu–N bonds, Chem. Commun., 2007, 5007–5009 RSC.
- J. Chu, C. Wang, L. Xiang, X. Leng and Y. Chen, Reactivity of Scandium terminal Imido Complex toward Boranes: C(sp3)–H Bond Borylation and B–O Bond Cleavage, Organometallics, 2017, 36, 4620–4625 CrossRef CAS.
- D. M. Lyubov, G. K. Fukin and A. A. Trifonov, N,N’-Diisopropyl-N``-bis(trimethylsilyl)guanidinate Ligand as a Supporting Coordination Environment in Yttrium Chemistry. Synthesis, Structure, and Properties of Complexes [(Me3Si)2NC(Ni-Pr)2]YCl2(THF)2, [(Me3Si)2NC(Ni-Pr)2]Y(CH2SiMe3)2(THF)2, and [(Me3Si)2NC(Ni-Pr)2]Y[μ-H](μ-Et)2BEt]2(THF)2, Inorg. Chem., 2007, 46, 11450–11456 CrossRef CAS PubMed.
- W. J. Evans, S. E. Lorenz and J. W. Ziller, Yttrium metallocene borane chemistry: isolation of 9-BBN substitution and coordination complexes in a single crystal, {(C5Me5)2Y[η3-C3H4(BC8H14)]} and {(C5Me5)2Y(μ-H)2(BC8H14)}, Chem. Commun., 2007, 4662–4664 RSC.
- J. Long, A. N. Selikhov, N. Y. Radkova, A. V. Cherkasov, Y. Guari, J. Larionova and A. A. Trifonov, Synthesis, Structures and Magnetic Properties of two Heteroleptic Dy3+ Borohydride Complexes, Eur. J. Inorg. Chem., 2021, 3008–3012 CrossRef CAS.
- X. W. Zhang, G. H. Maunder, S. Gießmann, R. MacDonald, M. J. Ferguson, A. H. Bond, R. D. Rogers, A. Sella and J. Takats, Stable heteroleptic complexes of divalent lanthanides with bulky pyrazolylborate ligands – iodides, hydrocarbyls and triethylborohydrides, Dalton Trans., 2011, 40, 195–210 RSC.
- (a) D. L. Reger, J. A. Lindemann and L. Lebioda, Synthesis, X-ray Crystal Structure, and Multinuclear NMR Study of the Dynamic Behavior of Tris[dihydridobis(1-pyrazolyl)borato]yttrium(III): A Molecule with Three Three-Center, Two-Electron Bonds, Inorg. Chem., 1988, 27, 1890–1896 CrossRef CAS; (b) M. Akita, K. Ohta, Y. Takahashi, S. Hikichi and Y. Moro-oka, Synthesis and Structure Determination of Rh–diene Complexes with the Hydridotris(3,5-diisopropylpyrazolyl)borate ligand, TpiPrRh(diene) (diene = cod, nbd): Dependence of the v(B–H) Values on the Hapticity of the TpiPr Ligand (κ2 vs. κ3), Organometallics, 1997, 16, 4121–4128 CrossRef CAS; (c) X. Zhang, R. MacDonald and J. Takats, Synthesis and structure of the first bis-hydridotris(3-tBu-5-Mepyrazolyl)borate complexes, Ln(TptBu,Me)2 (Ln = Sm, Yb): fluxionality, bonding mode exchange and B-H-Ln bridge bonding, New J. Chem., 1995, 19, 573–585 CAS.
- E. N. Lapsheva, T. Cheisson, C. Álvarez Lamsfus, P. J. Carroll, M. R. Gau, L. Maron and E. J. Schelter, Reactvity of Ce(IV) imido compounds with heteroallenes, Chem. Commun., 2020, 56, 4781–4784 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2312212–2312226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc06584g |
| This journal is © The Royal Society of Chemistry 2024 |