
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational electrocatalysis beyond conventional hydrogen electrode model: CO2 reduction to C2 species on copper facilitated by dynamically formed solvent halide ions at the solid–liquid interface†
Xin
Mao
a,
Tianwei
He
b,
Gurpreet
Kour
a,
Hanqing
Yin
a,
Chongyi
Ling
 *c,
Guoping
Gao
*d,
Yonggang
Jin
e,
Qingju
Liu
b,
Anthony P.
O'Mullane
a and
Aijun
Du
*a
*c,
Guoping
Gao
*d,
Yonggang
Jin
e,
Qingju
Liu
b,
Anthony P.
O'Mullane
a and
Aijun
Du
*a
aSchool of Chemistry and Physics, Centre for Material Science, Faculty of Science, Queensland University of Technology, Gardens Point Campus, Brisbane, QLD 4001, Australia. E-mail: aijun.du@qut.edu.au
bYunnan Key Laboratory for Micro/Nano Materials & Technology, National Center for International Research on Photoelectric and Energy Materials, School of Materials and Energy, Yunnan University, Kunming 650091, China
cSchool of Physics, Southeast University, Nanjing, 211189, China. E-mail: lingchy@seu.edu.cn
dMOE Key Lab for Nonequilibrium Synthesis and Modulation of Condensed Matter, School of Physics, Xi'an Jiaotong University, Xi'an, 710049, China. E-mail: guopinggao@xjtu.edu.cn
eCSIRO Mineral Resources, 1 Technology Court, Pullenvale, QLD 4069, Australia
First published on 25th January 2024
Abstract
The reduction of CO2 into value-added chemicals and fuels has been actively studied as a promising strategy for mitigating carbon dioxide emissions. However, the dilemma for the experimentalist in choosing an appropriate reaction medium and neglecting the effect of solvent ions when using a simple thermochemical model, normally leads to the disagreement between experimental observations and theoretical calculations. In this work, by considering the effects of both the anion and cation, a more realistic CO2 reduction environment at the solid–liquid interface between copper and solvent ions has been systematically studied by using ab initio molecular dynamics and density functional theory. We revealed that the co-occurrence of alkali ions (K+) and halide ions (F−, Cl−, Br−, and I−) in the electric double layer (EDL) can enhance the adsorption of CO2 by more than 0.45 eV compared to that in pure water, and the calculated energy barrier for CO–CO coupling also decreases 0.32 eV in the presence of I ion on a negatively charged copper electrode. The hydrated ions can modulate the distribution of the charge near the solid–liquid interface, which significantly promotes CO2 reduction and meanwhile impedes the hydrogen evolution reaction. Therefore, our work unveils the significant role of halide ions at the electrode–electrolyte interface for promoting CO2 reduction on copper electrode.
Introduction
The excessive emission of CO2 into the atmosphere has given rise to severe environmental issues, such as global warming and ocean acidification.1,2 One promising way to establish a carbon-neutral economy is to transform CO2 into some value-added chemicals and fuels via the electrochemical reduction of CO2 (CO2RR).3–11 Copper is reported as the most promising catalytic materials to achieve this process, and significant yields (70% current efficiency) of methane, ethane, methanol, ethanol, and even some C3 products can be obtained at an applied potential of −1.0 V vs. RHE over a copper electrode.4 The catalytic activity for CO2 reduction on copper can be significantly affected by the exposed surface, grain boundaries, and more importantly the reaction medium.12–17The reaction medium around an electrode is usually made of several layers. The layer that is the closest to the electrode surface and normally referred to as the inner Helmholtz or Stern layer, contains water dipoles, and sometimes other species, like cations, and anions, which are specifically adsorbed on the electrode surface, while some ions move and diffuse in the solution at a distance from the electrode, i.e. the outer Helmholtz layer.16,18 The microscopic structure of this type of solid–liquid interface will have a strong impact on CO2 reduction, but how these interface solvent ions affect the activity and selectivity of the CO2 reduction process is still under debate.19 For example, Singh et al.20 proposed that the presence of solvent ions can buffer the interfacial pH, while Chen et al.21,22 suggested that the ions can stabilize the hydrocarbon intermediates through electrostatic interaction. Waegele et al.23 postulated that the solvent ions can modify the local electric field at the electrode surface. All of these explanations lead to a dilemma for the experimentalist to select a suitable reaction medium to catalyze the reduction of CO2.
Recently, Koper's group19 revealed that the CO2 reduction process cannot take place in the absence of solvent metal cations in the electric double layer. Huang's group24 and Hu's group18 reported that the hydrated K+ near the electrode surface of a copper electrode in strong acid media (pH < 1) can catalyze CO2 to multi carbon products with 50% conversion efficiency at a current density of 1.20 A cm−2. There have been numerous intriguing experimental and computational studies investigating the impact of cations at the solid–liquid interface. In the case of halide ions, several research groups have highlighted their significant role in CO2 reduction.25–30 However, there has been limited research on elucidating the potential mechanisms originating from the presence of solvent halide ions.
To further develop novel catalysts enabling a smaller overpotential, high catalytic activity and selectivity, an in-depth understanding of the underlying CO2 reduction mechanism at the liquid–solid interface is urgently required to make CO2 reduction more commercially viable.31,32 Besides, machine-learning accelerated catalyst screening has also becoming another promising strategy for CO2 reduction on copper based materials in recent years.33,34
To date, almost all computational works have been done using a simple thermochemical model (TCM) based on the computational hydrogen electrode (CHE).4,35 The CHE model has demonstrated success in predicting the overpotential for some simple chemical reactions such as the hydrogen evolution reaction (HER) as well as the oxygen evolution and reduction reactions (OER/ORR). However, detailed reaction process cannot be reproduced when dealing with more complex CO2 reduction process.35 Moreover, electrochemical reactions in reality occur under constant electrode potentials, which is normally neglected in computational studies. Therefore, a more complex model for studying the realistic reaction processes at the solid–liquid interface by considering the negatively charged metal surface and electrode potential36–41 is highly desirable.
In this work, by using an ab initio molecular dynamics (AIMD) method, several consecutive simulations have been conducted to investigate the role of the reaction medium on CO2 reduction over a negatively charged copper surface. The simulations include studies of the initial process of getting stable electric double layers on the copper surface; the insertion of both halide ion and alkali ion into the system to obtain the local charge distribution near the solid–liquid interface; and adding CO2 into the reaction medium to search for the most probable reaction mechanism. We found that hydrated halide and alkali shells can be formed in the electric double layer. The hydrated ions can significantly modify the distribution of the charge in the electric double layers and substantially enhance the short-range electrostatic attraction compared to that in a pure water solvent. Further DFT calculations indicate that halide ions transfer electrons to the lowest unoccupied molecular orbital (LUMO) of CO2, which facilitates the activation of the inert C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond. The I-assisted Cu–H2O catalyst demonstrates the efficient reduction of CO2 to CH4 and C2H4 with energy barriers of 0.80 eV and 0.66 eV, respectively, which are much lower than that of Cu–H2O in the absence of halide ions (1.21 eV, and 0.98 eV). Therefore, in this work, by utilizing the well-adopted methodology of work function tuning by introducing extra electrons, with sufficiently thick explicit copper–water interface and a water–air interfaces, the mechanism of the catalytic CO2 reduction in an aqueous environment over copper electrode and the corresponding reaction kinetics are systematically investigated by a combined AIMD and DFT approach.
O double bond. The I-assisted Cu–H2O catalyst demonstrates the efficient reduction of CO2 to CH4 and C2H4 with energy barriers of 0.80 eV and 0.66 eV, respectively, which are much lower than that of Cu–H2O in the absence of halide ions (1.21 eV, and 0.98 eV). Therefore, in this work, by utilizing the well-adopted methodology of work function tuning by introducing extra electrons, with sufficiently thick explicit copper–water interface and a water–air interfaces, the mechanism of the catalytic CO2 reduction in an aqueous environment over copper electrode and the corresponding reaction kinetics are systematically investigated by a combined AIMD and DFT approach.
Computational methodologies
All calculations were performed by using the density functional theory (DFT) method as implemented in the Vienna Ab initio Simulation Package (VASP). The Perdew–Burke–Ernzerhof (PBE) functional of the generalized gradient approximation (GGA) was used for the calculation of electron exchange–correlation with the projector augmented wave (PAW) method.42–44 Spin-polarization was also included through the calculations, and the cut-off energy of 500 eV for plain-wave basis sets was adopted. The convergence threshold was set to 10−5 eV, and 5 × 10−3 eV Å−1 for energy and force, respectively. For ab initio molecular dynamics simulations (AIMD), the optimization was considered to be converged when energy change between two steps was smaller than 10−4 eV. The Brillouin-zone integrations were approximated by using the k-point sampling of the Monkhorst–Pack scheme. The weak interaction was treated by DFT + D3 method using an empirical correction in Grimme's scheme.45 The minimum energy pathway for each elementary step was determined by using a climbing image nudged elastic band method (CI-NEB), and the transferred hydrogen atom was from the surrounding water molecule.46 The electron distribution was calculated by Bader charge analysis. The total z-direction length is 40 Å, including 5-layered copper slab, six explicit water layers about 20 Å, and another 15 Å for the vacuum space layer, which is thick enough to simulate the electric double layer over copper electrode. For the calculation of work function, an extra 10 Å vacuum layer was added to the z-direction.A six layer of water molecules that well matches the Cu(110) surface as shown in the previous work47 was put on top of the Cu surface with a vacuum space of 15 Å. The initial water molecules in each layer are in the shape of ice-like hexagonal configurations. After introducing negative charges on the copper surface, the water molecules are all restructured into the most stable configuration.
Ab initio molecular dynamics simulations (AIMD) were carried out to evaluate the thermodynamic stability and simulate the realistic reaction process of in total 30 ps near the solid–liquid interface at a temperature of 300 K on a negatively charged copper surface under a canonical ensemble (NVT) with each time step of 1 fs. The first 10 ps is for the initial structure relaxation, and the period of 10 ps to 20 ps is for the optimization to get the stable position for alkali and halide ions, and the last 10 ps is for the investigation of the effect of hydrated ions on the adsorbed CO2 molecules. To verify the sufficient simulation time to reach the well-equilibrated model, the radial distribution functions of O–O and O–H has been calculated as shown in Fig. S1,† which is in good agreement with previous work.48 We add different numbers of extra electrons and calculate the corresponding absolute Fermi levels with respect to the electrostatic potential in the region far from copper slab and explicit water molecules. By interpolating the extra electrons and the work function relation, we can get the exact extra electrons for the target electrode potential of −1.0 V vs. RHE.36,37
Results and discussion
To simulate CO2 reduction in a reaction medium on copper surface, we carried out ab initio molecular dynamics (AIMD) on Cu–water interface. Six layers of explicit water molecules are adsorbed on the top of copper surface (Cu–H2O) in electric double layer with ice-like hexagonal initial configurations to mimic the effect of explicit solvent.47 A vacuum space of 15 Å between two slabs is used to avoid image interaction, and the total model length in z-direction is set to be 40 Å, containing 10 Å of copper–water interface, and 10 Å of water–air interface. Then, extra electrons have been introduced into the model to tune the work function and electrode potential to better understand each reaction process and infer what really happens under a negatively charged copper surface. Normally, the hydrogen bond relaxation in liquid water and copper–water interface requires about 10 ps to reach a well-equilibrated system, thus the first 10 ps of AIMD is conducted to get the stable initial structure. To verify the sufficient dynamic time scale, the radial distribution functions for O–O and O–H bonds have been calculated as shown in Fig. S1,† which is in full agreement with previous results.48 After 10 ps of AIMD simulation, the oriented interfacial water molecules present a configuration of H-pointing towards surface due to the negatively charged copper surface, and the temperature and the total energy of the studied system also changed slightly until 10 ps as shown in Fig. 1a.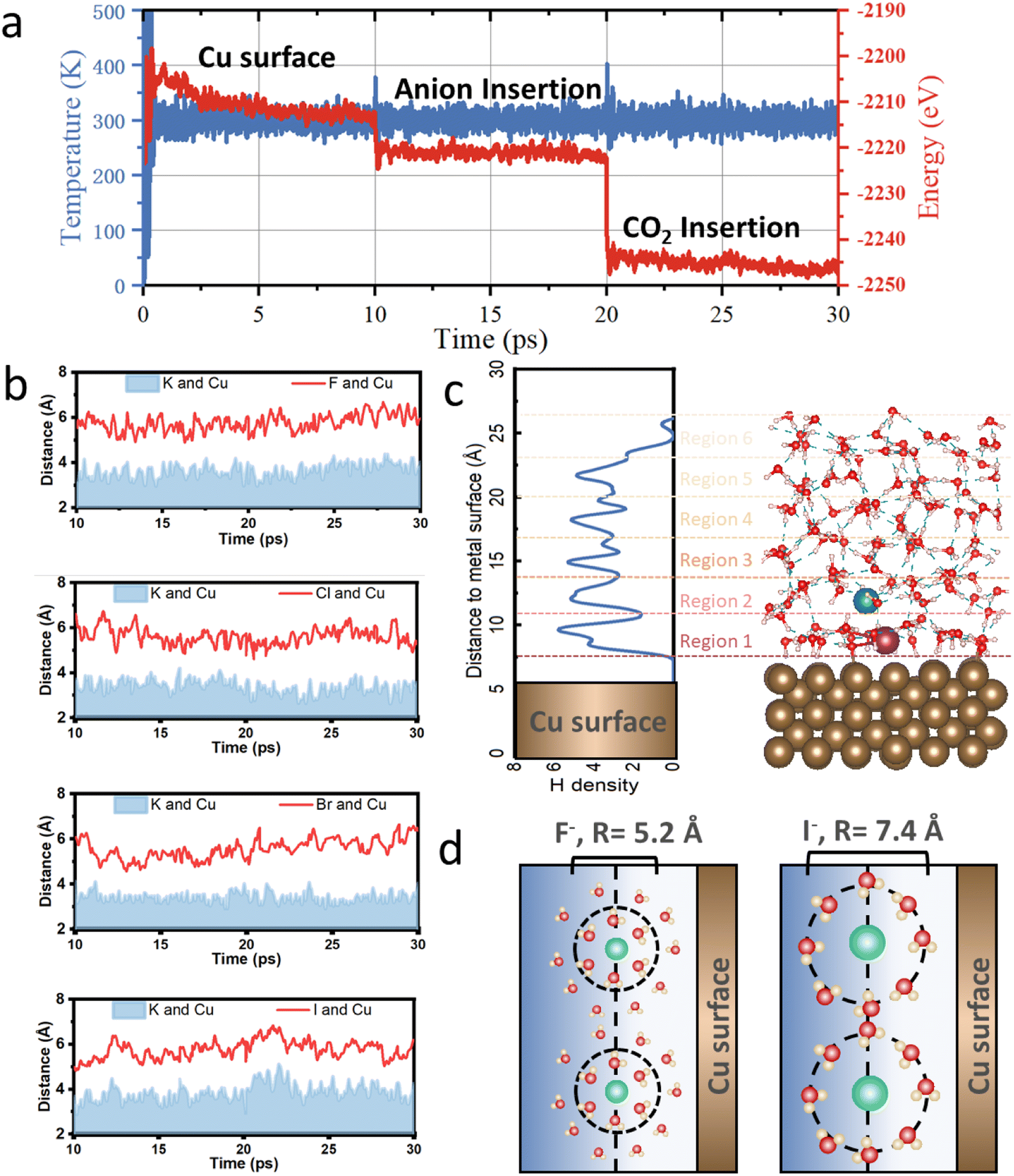 | ||
| Fig. 1 (a) Variation of temperature and total energy for I-assisted Cu–H2O catalyst, and the simulation time is 30 ps at a temperature of 300 K under a canonical ensemble (NVT) with each time step of 1 fs. (b) The distance to copper surface for halide and cation ions in Cu–H2O catalyst. (c) Schematic figure for the model, including Cu slab, 10 Å of copper–water interface layer and 10 Å of water–air interface, and vacuum layer is not shown here in the picture. The purple and green balls refer to cation and anion ion. (d) The schematic diagram of the formed F− hydration shell and I− hydration shell with their hydration diameters labeled in the picture. | ||
Then another 10 ps simulation has been performed to get the stable cation and anion position. K+ shows a closer distance between copper atoms than other halide ions (Fig. 1b), and both ions can form a hydration shell in electric double layer. For different halide ions, a different size of hydration shell can be formed as shown in Fig. 1d. Due to a relatively smaller radius and higher electronegativity, F-assisted Cu–H2O produces a strong ion–solvent interaction with surrounding water molecules, and the obtained average hydration diameter is 5.2 Å, which is calculated from the first shell of the radial distribution functions of anion-oxygen. The calculated hydration diameter increases with the decreases of electronegativity, which are 6.4 Å, 6.8 Å, and 7.4 Å for Cl, Br, and I on a copper electrode, respectively. Moreover, the coordination number of different halide ion with surrounding water molecules have also been calculated, and they are 6.0, 6.4, 6.5, and 7.0 for F, Cl, Br, and I-assisted Cu–H2O catalysts, respectively, which indicates a relatively harder hydrated ion shell can be formed for F-assisted Cu–H2O. These computational findings are consistent with experimental observations, further demonstrating the reliability of our model and simulation approach.16 As for the alkali K+ ion, it can move to the copper surface and is partially covered with water molecules as observed in experiment. And the simulated average coordination number of the K+ ion with water molecules is 6.0.49 Fig. S2† shows a plot of the charge difference between ions and the copper surface, and demonstrates that the charge is transferred from the copper surface to the hydrated ions. Thus, the insertion of the ions can form a local charge near the solid–liquid interface, and the produced charge distribution is believed to have a promotional effect on the subsequent adsorption and activation of inert CO2 molecules.
Fig. 2a presents the calculated anion-oxygen radial distance function (RDF), which can show how halide ions form the anion hydration shell at the solid–liquid interface. The RDF values for halide ions increases from F (2.6 Å), Cl (3.0 Å), Br (3.2 Å) to I-assisted Cu–H2O (3.5 Å), which is in good agreement with previous experimental results.49 Generally, the larger RDF values suggest a softer hydration shell formed around the halide ions, resulting in a higher ion concentration at the copper surface and thus stronger binding of CO2 molecules. However, ions with a smaller RDF, such as F-assisted Cu–H2O, the produced hydration shell will be hard, thus resulting in a poorer bonding with CO2 molecules.
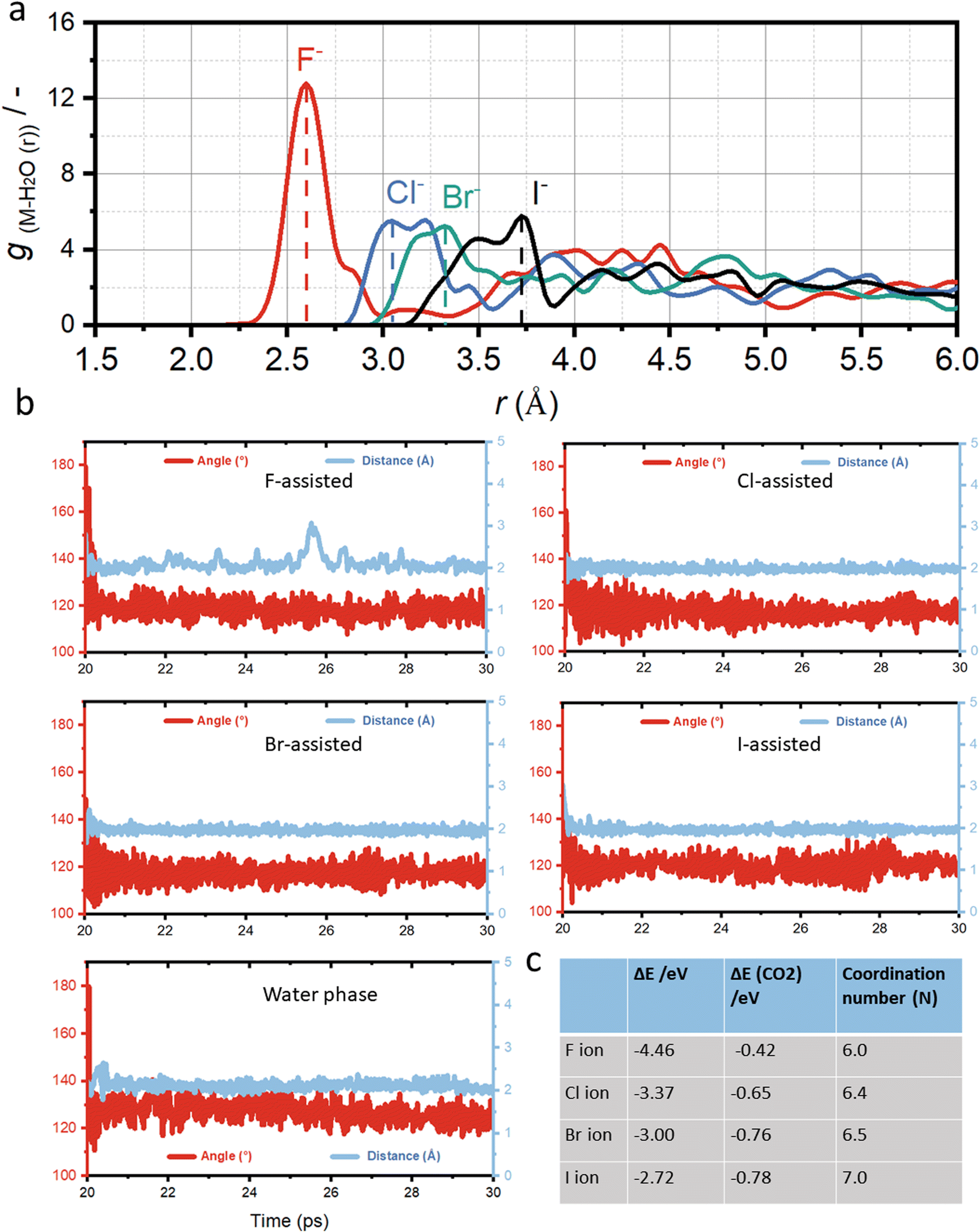 | ||
| Fig. 2 (a) The calculated radial distribution functions for four different halide ions in the Cu–H2O catalyst. (b) Simulated O–C–O bond angle and its distance to copper surface for five different catalysts, including F-assisted Cu–H2O, Cl-assisted Cu–H2O, Br-assisted Cu–H2O, and I-assisted Cu–H2O catalyst, red line indicates the O–C–O bond angle against simulation time, and blue line indicates the distance to copper against simulation time. (c) Formation energies and coordination numbers of different halide ions in the Cu–H2O catalyst, and the binding energies of CO2 molecules. | ||
Fig. 2b shows the AIMD simulation of Cu–H2O in the presence of different halide ions after placing CO2 molecules into the copper surface over a period of 20 ps to 30 ps simulation scale. Initially, a linear CO2 molecule was placed into the system at a distance of 2.0 Å to the copper surface. Under the constant potential model, the O–C–O bond angle can be reduced from 180° to 120°, with significant elongation of the CO bond length, indicating the full capture and potential activation of the CO2 molecule on the copper surface. The final bonding configuration of CO2 on the copper surface, involves carbon and one oxygen attaching to copper, while the second oxygen is oriented towards neighboring water molecules. However, in the presence of halide ions in the solvent, the adsorption binding energy of CO2 molecule is much stronger compared to the water phase as shown in Fig. 2c. The adsorption energy is −0.42 eV for F-assisted Cu–H2O catalyst, and the value reduces with the decrease of electronegativity of halide ions from F− to I−.
It has been reported that the initial stage of CO2 reduction requires a voltage of about −1.90 V vs. SHE due to the high energy needed to restructure the stable linear OCO double bonds, such as bending them into a radical anion form thereby forming chemical bonds with the catalyst. However, the hydrated halide ion in the electric double layer of the Cu–H2O catalyst is believed to have a promotional effect for this process as shown in Fig. 3a. Unlike the conventionally accepted belief that copper transfers fully occupied 3d electrons to the LUMO orbital of CO2, the CO2 activation process is mainly attributed to the halide ions in the solvent.50 CO2 has a linear triatomic configuration with two equivalent C–O double bonds, and the molecular orbital energy level of CO2 is (K)(K)(K) (3σg)2(2σu)2(4σg)2(3σu)2(1πu)4(1πg)4(2πu)0. The formation of a CO2− radical anion needs to accept one electron from the external system, which is then transferred into the LUMO (2πu) of the CO2 molecules. During this process, the linear CO2 molecule can be activated and bent. As shown in Fig. S3,† electrons from the fully occupied p orbitals of halide ions can flow to the 2πu orbital of the CO2 molecules, and the calculated charge decreases from 1.0 e to 0.30 e for the I− ion, which indicates that 0.70 e is transferred to the CO2 molecule. Consequently, CO2 is now negatively charged accompanied by a structural transition from a linear to bent form.
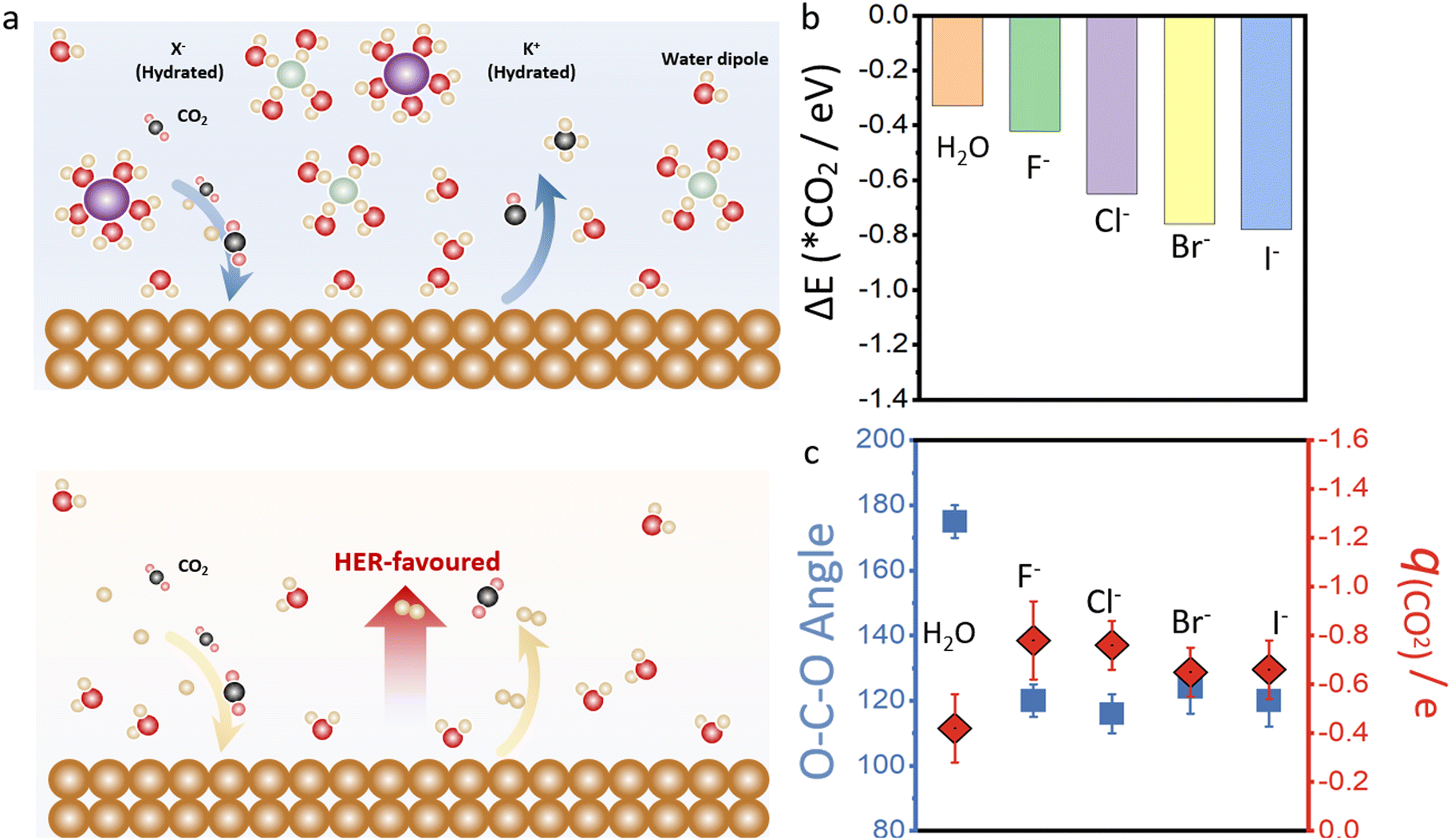 | ||
| Fig. 3 (a) The schematic diagram for CO2 reduction to C1, and C2 products on the surface of Cu–H2O, with and without ions in the electric double layer. The yellow, red, white, purple, and green balls represent Cu, O, H, K, and halide ions, respectively. (b) Binding energies of CO2 molecules on Cu–H2O catalyst in the presence and absence of halide ions. (c) The calculated bond angle for O–C–O and charge for CO2 molecules on Cu–H2O catalyst in the presence and absence of halide ions. | ||
Fig. 4 shows the reaction pathways for the reduction of CO2 to CH3OH and CH4 molecules at a applied potential of −1.0 V vs. RHE on I-assisted Cu–H2O catalyst to determine the free energy change and reaction barriers. For comparison, free energy change diagram for pure Cu–H2O without I ion has also been calculated and shown in Fig. S4 in ESI.† As discussed above, in the presence of I ion, the adsorption of CO2 molecules becomes more favorable than pure water solvent. After that, the first proton–electron transfer step is the activation of CO2 to form COOH or HCOO species, and the obtained energy barriers are 0.56 eV, and 0.83 eV, respectively. Whereas these values become much more unfavorable for Cu–H2O without I assisted, and they are 0.68 eV, and 1.21 eV for the formation of COOH and HCOO species, respectively. The second proton–electron transfer step is the protonation of COOH to CO species with an energy barrier of 0.44 eV for I-assisted Cu–H2O, and 0.56 eV for Cu–H2O without I ion. The third proton–electron transfer step may either lead to *CHO or *COH by attacking *CO species through C side or O side. The obtained energy barriers are 0.80 eV and 1.41 eV, indicative of the difficulty in producing *COH on the surface for I-assisted Cu–H2O. Then *CHOH species can be formed on the copper surface with an energy barrier of only 0.09 eV, proving the fast kinetic process. For *COH species, the subsequent product can be *C species, however, the calculated energy barrier is as high as 1.26 eV, which is difficult to take place. Then, *CH species can be formed with a low barrier of 0.49 eV. After that, three hydrogen atoms can continue attacking *CH species until the formation of CH4 molecule. For the formation of CH3OH, the calculated energy barrier is much higher, reaching 0.79 eV. The figure shows that the rate-limiting step (RLS) is the formation of *CHO species for Cu–H2O both with and without I-assisted, and the calculated energy barriers are 0.80 eV and 1.21 eV, respectively.
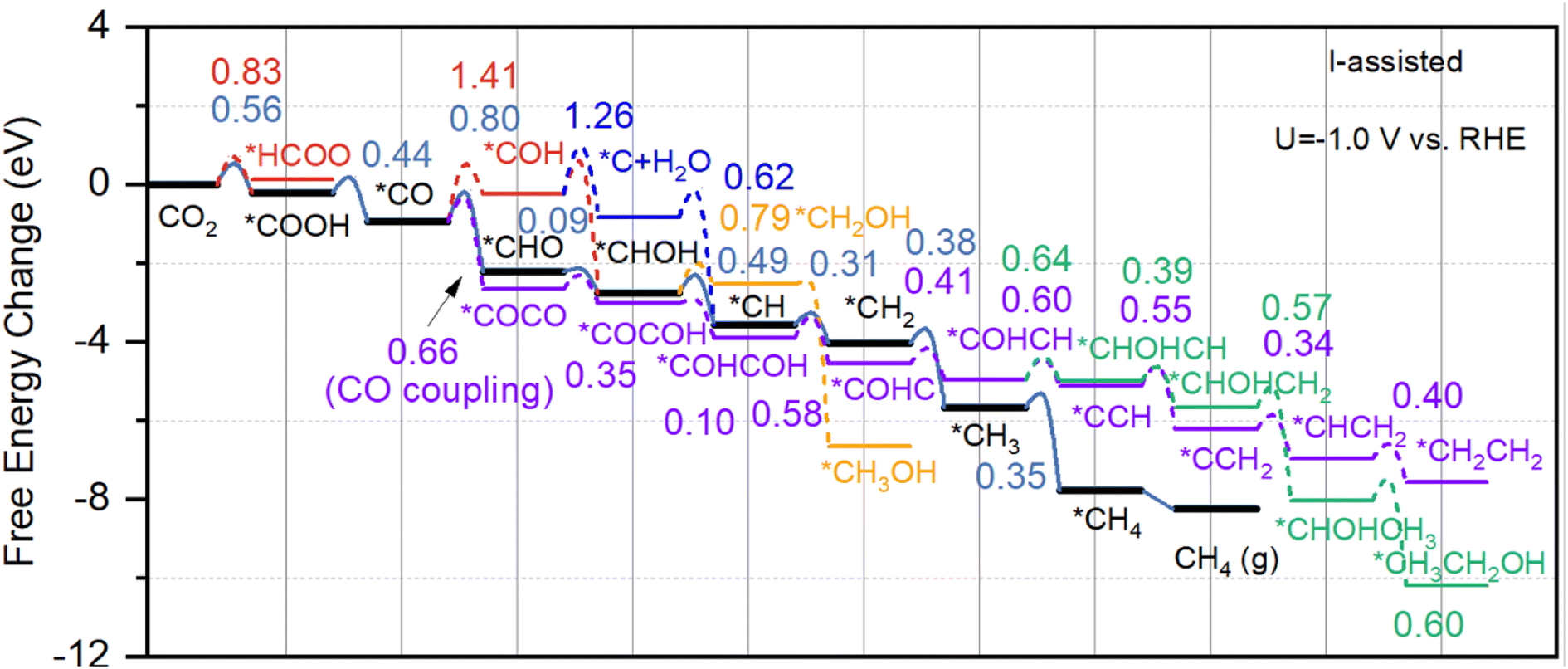 | ||
| Fig. 4 The calculated Gibbs free energy change diagrams with energy barriers for the formation of CH3OH (orange line), CH4 (black line), C2H5OH (green line), and C2H4 (purple line), and some important reaction pathway to produce *HCOO (red line) and *C species (blue line) on the surface of I-assisted Cu–H2O. | ||
In the above, we have described all the possible proton–electron transfer steps for CO2 reduction to C1 products. However, the CO–CO coupling process may also take place along the reaction pathway. DFT-calculated results show that the obtained energy barrier is only 0.66 eV for CO–CO coupling for I-assisted Cu–H2O. In contrast, the calculated energy barrier is as high as 0.98 eV for Cu–H2O in the absence of I ion. The purple and green lines in Fig. 4 indicate the formation of C2H4 and CH3CH2OH molecules on the surface of Cu–H2O with the presence of I ion. For comparison, the free energy landscape for the formation of C2H4 and CH3CH2OH are also given in Fig. S4 in ESI.† After the formation of *COCO species on the active site, then *COCOH species can be formed with energy barrier of 0.35 eV. Then another proton–electron pair can attack *OH species with one water molecule released to the system, and the calculated energy barrier is 0.10 eV, indicating the fast kinetic process at the room temperature. 0.58 eV and 0.41 eV energy barriers are calculated for the formation of *COHC and *COHCH species. After that, *COHCH can either produce *CHOHCH species for the produce of ethanol or *CCH for the produce of ethylene. For the formation of *CHOHCH species, the calculated energy barrier is 0.64 eV, which is slightly higher than the formation of *CCH (0.60 eV). Then, *CCH species can react with another three consecutive proton–electron pairs to produce ethylene. Whereas, after the formation of *CHOHCH species, another proton–electron pair can attack carbon side to produce *CHOHCH2 species with an energy barrier of 0.57 eV. The final step for the formation of ethanol is *CHOHCH3 species reacts with another proton–electron pair with an energy barrier of 0.60 eV. For I-assisted Cu–H2O catalyst, the RLS is the CO–CO coupling process, and the obtained energy barrier is only 0.66 eV, whereas this value is as high as 0.98 eV for the pure Cu–H2O in the absence of I ion. Moreover, the current density for the formation of C1 and C2 products on Cu–H2O with and without I ions at the electric double layer has also been calculated by using micro-kinetic calculation according to previous work,51 and the calculation details can be found in ESI.† The calculated results suggest that the current density of about 0.158 mA cm−2 and 35.43 mA cm−2 for methane and ethylene production, which is much higher than pure water. Therefore, the introduction of anion at the solid–liquid interface can significantly reduce the reaction barrier and enhance the reaction rate compared to the pure Cu–H2O catalyst.
It is worth noting that in the presence of I ion in the electric double layer of Cu–H2O, the water dissociation barrier is 0.76 eV to produce hydrogen proton and OH species. However, the activation of CO2 to produce *COOH is only 0.56 eV, indicating the consumption of H proton is faster than H production and hydrogen evolution process can also be suppressed during this process. Thus, high OH amount can be produced near the solid–liquid interface over copper electrode, leading to a high pH value in electric double layer than bulk phase. This can also be proved by the adsorption of OH ion in six different water layers of I-assisted Cu–H2O, and we found the calculated adsorption energy for the OH species in the first layer is much stronger than in the sixth layer, also proving the high pH value near copper surface. Besides, considering the initial bulk pH in the near-neutral KI solution is about 7.0. We can find that pH reduces with more CO2 captured in the system, however, the more CO2 capture will lower the profit of CO2 reduction, and the formation of CO32− directly reflects the OH− concentration in the vicinity of the cathode which can be possibly utilized to determine the pH value near the cathode surface. When CO2 partial pressure is 1.0 atm, the obtained pH is about 6.8 as shown in Fig. S5,† which is in good agreement with the experimental measured pH of 6.6 for the electrolyte after operating at −1.20 V vs. RHE for 30 min.52
Conclusions
In this work, a realistic model containing hydrated halide and K+ ions with sufficient thick explicit water solvent layer have been employed to simulate the reaction of CO2 near the solid–liquid interface by using AIMD and DFT calculations. Due to the smaller activation barrier for the first hydrogenation step of CO2 reduction compared to the water dissociation process, the HER process can be suppressed on the copper surface. The calculated highest energy barrier for the formation of CH4 is 0.80 eV for I-assisted Cu–H2O, and this value significantly enhanced to 1.21 eV in the absence of I ion. Further DFT simulations indicate that halide ions transfer electrons to the LUMO of CO2 which activates the inert OCO double bond, leading to high catalytic activity for the reduction of CO2 to C2H4, with an energy barrier of 0.66 eV for I-assisted Cu–H2O catalyst, which is much lower than that of Cu–H2O without any halide ions.Data availability
The data supporting this article have been included in ESI.†Author contributions
A. D. conceived and supervised the research. X. M., T. H., and G. K. performed the DFT and MD simulations. C. L., G. G., Y. J., Q. L., and A. O. provided some suggestions. X. M. wrote and revised the manuscript. A. D., A. O. and H. Y. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the generous grants of high-performance computer resources provided by the NCI National Facility and The Pawsey Supercomputing Centre through the National Computational Merit Allocation Scheme supported by the Australian Government and the Government of Western Australia. A. D. greatly appreciates financial support by the Australian Research Council under the Discovery Project (DP 210100721).References
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. Stephens, K. Chan and C. Hahn, Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte, Chem. Rev., 2019, 119(12), 7610–7672 CrossRef CAS PubMed.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Fundamentals and challenges of electrochemical CO2 reduction using two-dimensional materials, Chem, 2017, 3(4), 560–587 CAS.
- X. Lim, How to make the most of carbon dioxide, Nature, 2015, 526(7575), 628–631 CrossRef CAS PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, Calculations of product selectivity in electrochemical CO2 reduction, ACS Catal., 2018, 8(6), 5240–5249 CrossRef CAS.
- K. Schouten, Y. Kwon, C. Van Der Ham, Z. Qin and M. Koper, A new mechanism for the selectivity to C1 and C2 species in the electrochemical reduction of carbon dioxide on copper electrodes, Chem. Sci., 2011, 2(10), 1902–1909 RSC.
- K. J. P. Schouten, Z. Qin, E. Pérez Gallent and M. T. Koper, Two pathways for the formation of ethylene in CO reduction on single-crystal copper electrodes, J. Am. Chem. Soc., 2012, 134(24), 9864–9867 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces, Energy Environ. Sci., 2012, 5(5), 7050–7059 RSC.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces, J. Am. Chem. Soc., 2014, 136(40), 14107–14113 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Opportunities and challenges for a sustainable energy future, Nature, 2012, 488(7411), 294–303 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, What would it take for renewably powered electrosynthesis to displace petrochemical processes?, Science, 2019, 364(6438), eaav3506 CrossRef CAS PubMed.
- H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao and S.-Z. Qiao, C2+ Selectivity for CO2 Electroreduction on Oxidized Cu-Based Catalysts, J. Am. Chem. Soc., 2023, 145(26), 14335–14344 CrossRef CAS PubMed.
- Y. Huang, A. D. Handoko, P. Hirunsit and B. S. Yeo, Electrochemical reduction of CO2 using copper single-crystal surfaces: effects of CO* coverage on the selective formation of ethylene, ACS Catal., 2017, 7(3), 1749–1756 CrossRef CAS.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, Grain-boundary-dependent CO2 electroreduction activity, J. Am. Chem. Soc., 2015, 137(14), 4606–4609 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, Formation of hydrocarbons in the electrochemical reduction of carbon dioxide at a copper electrode in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85(8), 2309–2326 RSC.
- Y. Huang, C. W. Ong and B. S. Yeo, Effects of electrolyte anions on the reduction of carbon dioxide to ethylene and ethanol on copper (100) and (111) surfaces, ChemSusChem, 2018, 11(18), 3299–3306 CrossRef CAS PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Advances and challenges for the electrochemical reduction of CO2 to CO: from fundamentals to industrialization, Angew. Chem., Int. Ed., 2021, 60(38), 20627–20648 CrossRef CAS PubMed.
- A. Xu, N. Govindarajan, G. Kastlunger, S. Vijay and K. Chan, Theories for Electrolyte Effects in CO2 Electroreduction, Acc. Chem. Res., 2022, 55(4), 495–503 CrossRef CAS PubMed.
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Modulating electric field distribution by alkali cations for CO2 electroreduction in strongly acidic medium, Nat. Catal., 2022, 1–9 Search PubMed.
- M. C. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. Koper, Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution, Nat. Catal., 2021, 4(8), 654–662 CrossRef CAS.
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, Hydrolysis of electrolyte cations enhances the electrochemical reduction of CO2 over Ag and Cu, J. Am. Chem. Soc., 2016, 138(39), 13006–13012 CrossRef CAS PubMed.
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, Electric field effects in electrochemical CO2 reduction, ACS Catal., 2016, 6(10), 7133–7139 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, Promoter effects of alkali metal cations on the electrochemical reduction of carbon dioxide, J. Am. Chem. Soc., 2017, 139(32), 11277–11287 CrossRef CAS PubMed.
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, How cations affect the electric double layer and the rates and selectivity of electrocatalytic processes, J. Chem. Phys., 2019, 151(16), 160902 CrossRef PubMed.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, CO2 electrolysis to multicarbon products in strong acid, Science, 2021, 372(6546), 1074–1078 CrossRef CAS PubMed.
- D. Gao, F. Scholten and B. Roldan Cuenya, Improved CO2 electroreduction performance on plasma-activated Cu catalysts via electrolyte design: halide effect, ACS Catal., 2017, 7(8), 5112–5120 CrossRef CAS.
- A. S. Varela, W. Ju, T. Reier and P. Strasser, Tuning the catalytic activity and selectivity of Cu for CO2 electroreduction in the presence of halides, ACS Catal., 2016, 6(4), 2136–2144 CrossRef CAS.
- P. Sebastián-Pascual, A. S. Petersen, A. Bagger, J. Rossmeisl and M. Escudero-Escribano, pH and anion effects on Cu–phosphate interfaces for CO electroreduction, ACS Catal., 2021, 11(3), 1128–1135 CrossRef.
- Z. Zhao, J. Zhang, M. Lei and Y. Lum, Reviewing the impact of halides on electrochemical CO2 reduction, Nano Research Energy, 2023, 2(1), e9120044 CrossRef.
- A. Yoon, J. Poon, P. Grosse, S. W. Chee and B. R. Cuenya, Iodide-mediated Cu catalyst restructuring during CO2 electroreduction, J. Mater. Chem. A, 2022, 10(26), 14041–14050 RSC.
- P. Kuilin, L. Guilin, J. Chongyang, Z. Shaojuan and Z. Xiangping, Research Progress for the Role of Electrolytes in the CO2 Electrochemical Reduction, Chem. J. Chin. Univ., 2022, 43(7), 74–83 Search PubMed.
- F. P. García de Arquer, C.-T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D.-H. Nam, C. Gabardo, A. Seifitokaldani and X. Wang, CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Science, 2020, 367(6478), 661–666 CrossRef PubMed.
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper, Nat. Catal., 2020, 3(6), 478–487 CrossRef CAS.
- X. Li, H. Li, Z. Zhang, J. Q. Shi, Y. Jiao and S.-Z. Qiao, Active-learning accelerated computational screening of A2B@NG catalysts for CO2 electrochemical reduction, Nano Energy, 2023, 115, 108695 CrossRef CAS.
- H. Li, Y. Jiao, K. Davey and S. Z. Qiao, Data-Driven Machine Learning for Understanding Surface Structures of Heterogeneous Catalysts, Angew. Chem., 2023, 135(9), e202216383 CrossRef.
- E. Tayyebi, Y. Abghoui and E. Skulason, Elucidating the mechanism of electrochemical N2 reduction at the Ru (0001) electrode, ACS Catal., 2019, 9(12), 11137–11145 CrossRef CAS.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, Dynamic Stability of Copper Single-Atom Catalysts under Working Conditions, J. Am. Chem. Soc., 2022, 144(37), 17140–17148 CrossRef CAS PubMed.
- X. Zhao and Y. Liu, Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction, J. Am. Chem. Soc., 2021, 143(25), 9423–9428 CrossRef CAS PubMed.
- D. Luan and J. Xiao, Adaptive Electric Fields Embedded Electrochemical Barrier Calculations, J. Phys. Chem. Lett., 2023, 14, 685–693 CrossRef CAS PubMed.
- H. Li, C. Guo, J. Long, X. Fu and J. Xiao, Theoretical understanding of electrocatalysis beyond thermodynamic analysis, Chin. J. Catal., 2022, 43(11), 2746–2756 CrossRef CAS.
- K. Chan and J. K. Nørskov, Electrochemical barriers made simple, J. Phys. Chem. Lett., 2015, 6(14), 2663–2668 CrossRef CAS PubMed.
- K. Chan and J. K. Nørskov, Potential dependence of electrochemical barriers from ab initio calculations, J. Phys. Chem. Lett., 2016, 7(9), 1686–1690 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- M. Forster, R. Raval, A. Hodgson, J. Carrasco and A. Michaelides, c(2× 2) water-hydroxyl layer on Cu (110): a wetting layer stabilized by Bjerrum defects, Phys. Rev. Lett., 2011, 106(4), 046103 CrossRef PubMed.
- J. Wang, G. Román-Pérez, J. M. Soler, E. Artacho and M.-V. Fernández-Serra, Density, structure, and dynamics of water: the effect of van der Waals interactions, J. Chem. Phys., 2011, 134(2), 024516 CrossRef PubMed.
- Y. Marcus, Ionic radii in aqueous solutions, Chem. Rev., 1988, 88(8), 1475–1498 CrossRef CAS.
- T. Chen, J. Hu, K. Wang, K. Wang, G. Gan and J. Shi, Specifically Adsorbed Anions Enhance CO2 Electrochemical Reduction to CO over a Gallium Catalyst in Organic Electrolytes, Energy Fuels, 2021, 35(21), 17784–17790 CrossRef CAS.
- S. Zhou, X. Yang, X. Xu, S. X. Dou, Y. Du and J. Zhao, Boron nitride nanotubes for ammonia synthesis: activation by filling transition metals, J. Am. Chem. Soc., 2019, 142(1), 308–317 CrossRef PubMed.
- X. Zhang, J. Li, Y.-Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, Selective and high current CO2 electro-reduction to multicarbon products in near-neutral KCl electrolytes, J. Am. Chem. Soc., 2021, 143(8), 3245–3255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06471a |
| This journal is © The Royal Society of Chemistry 2024 |