
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise synthesis of BN embedded perylene diimide oligomers for fast-charging and long-life potassium–organic batteries†
Guangwei
Shao‡
ab,
Hang
Liu‡
ab,
Li
Chen
a,
Mingliang
Wu
a,
Dongxue
Wang
 *ab,
Di
Wu
*ab and
Jianlong
Xia
*abc
*ab,
Di
Wu
*ab and
Jianlong
Xia
*abc
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Center of Smart Materials and Devices, Wuhan University of Technology, Wuhan 430070, China. E-mail: chemwd@whut.edu.cn
bSchool of Chemistry, Chemical Engineering and Life Science, Wuhan University of Technology, Wuhan 430070, China. E-mail: wangdongxue@whut.edu.cn
cInternational School of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China. E-mail: jlxia@whut.edu.cn
First published on 23rd January 2024
Abstract
Replacing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond with an isoelectronic BN unit is an effective strategy to tune the optoelectronic properties of polycyclic aromatic hydrocarbons (PAHs). However, precise control of the BN orientations in large PAH systems is still a synthetic challenge. Herein, we demonstrate a facile approach for the synthesis of BN embedded perylene diimide (PDI) nanoribbons, and the polarization orientations of the BN unit were precisely regulated in the two PDI trimers. These BN doped PDI oligomers show great potential as organic cathodes for potassium-ion batteries (PIBs). In particular, trans-PTCDI3BN exhibits great improvement in voltage potential, reversible capacities (ca. 130 mA h g−1), superior rate performance (19 s to 69% of the maximum capacity) and ultralong cyclic stability (nearly no capacity decay over 30
C bond with an isoelectronic BN unit is an effective strategy to tune the optoelectronic properties of polycyclic aromatic hydrocarbons (PAHs). However, precise control of the BN orientations in large PAH systems is still a synthetic challenge. Herein, we demonstrate a facile approach for the synthesis of BN embedded perylene diimide (PDI) nanoribbons, and the polarization orientations of the BN unit were precisely regulated in the two PDI trimers. These BN doped PDI oligomers show great potential as organic cathodes for potassium-ion batteries (PIBs). In particular, trans-PTCDI3BN exhibits great improvement in voltage potential, reversible capacities (ca. 130 mA h g−1), superior rate performance (19 s to 69% of the maximum capacity) and ultralong cyclic stability (nearly no capacity decay over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles), which are among those of state-of-the-art organic-based cathodes. Our synthetic approach stands as an effective way to access large PAHs with precisely controlled BN orientations, and the BN doping strategy provides useful insight into the development of organic electrode materials for secondary batteries.
000 cycles), which are among those of state-of-the-art organic-based cathodes. Our synthetic approach stands as an effective way to access large PAHs with precisely controlled BN orientations, and the BN doping strategy provides useful insight into the development of organic electrode materials for secondary batteries.
Introduction
Perylene diimide (PDI) is a versatile building block for the construction of novel functional materials for organic electronics.1–7 In the past few decades, numerous tailor-made PDI derivatives have been developed for application in electronic devices,8–13 sensors,14–17 and energy storage.18–21 Due to the presence of multiple redox-active carbonyl groups, as well as the merits including but not limited to flexible structure design, low-cost production, excellent stability and environmental friendliness, PDI derivatives have been widely used in almost all kinds of rechargeable ion batteries.22–25 In terms of material diversity and tunable redox properties, PDI-based electrodes show great potential for application in potassium-ion batteries (PIBs),26 which have recently been proposed as an alternative to lithium-ion batteries owing to the earth-abundant and widely distributed potassium resources.27–30 One of the main challenges in the development of PDI-based electrodes is the low electron conductivity of organic materials. Continuous efforts have been devoted to improving the performance of PDI-based energy storage devices;28,31–36 the construction of oligomers and/or hybrids with extended π-conjugation and an increased number of redox-active sites has been proved to be a robust strategy to obtain high-performance PDI-based electrodes.22 The enlarged π conjugation can significantly improve the electron conductivity, and the definite chemical structures of PDI oligomers/hybrids can avoid batch-to-batch variations and reduce the use of redox-inactive linkers and side chains in comparison with their polymer counterparts.37,38 Very recently, Nuckolls and co-workers demonstrated the iterative synthesis of helical PDI nanoribbons; the longest oligomer with six PDI units displayed remarkable cathode performance in lithium batteries, with both high electrical conductivity and high ionic diffusivity.39 However, the synthetic route toward oligomers with increasing length is somewhat cumbersome and expensive, which hinders further application of this PDI hexamer.Heteroatom doping is a powerful strategy for tuning the electronic properties of carbon-based materials toward improved electrochemical storage performance; the boron (B) and nitrogen (N) co-doping approach has achieved great success in the development of carbonaceous electrodes.40–42 And it has also been well established that the substitution of CC bonds with BN units endows the resultant BN embedded polycyclic aromatic hydrocarbons (PAHs) with similar structural features but with divergent optoelectronic properties.43–50 We speculated that the use of the B and N co-doping strategy would provide an alternative way for tuning the electrochemical properties of PDI oligomers, besides extending the π conjugation through increasing PDI units with tremendous synthetic efforts. Although the synthetic routes of BN doped PAHs have been well documented, a particular challenge is the precise control of the polarization orientations of BN units in BN-embedded PDI derivatives.48
Herein, to prove our hypothesis, a BN bridged PDI dimer PDI2BN and a pair of trimers trans- and cis-PDI3BN with different BN orientations were designed and precisely synthesized. It is worth noting that the direct bromination of the commercially available PDI monomer usually yields a regioisomeric mixture of 1,7- and 1,6-dibrominated PDI precursors. Although the regioisomeric mixtures of dibrominated PDI intermediates were elaborately used to reduce synthetic efforts in Nuckolls' iterative synthesis of helical PDI nanoribbons,39 the use of a mixture of 1,7- and 1,6-dibrominated PDI as a starting material gives a mixture of trans- and cis-PDI3BN, which could not be separated by column chromatography and crystallization methods. By using a synthetic strategy recently developed in our lab, the regiochemically pure 1,6-di(substituted) PDI precursor can be easily accessed.51 With these key building blocks in hand, trans- and cis-PDI3BN can be separately synthesized. In comparison with the corresponding vinylene bridged counterparts, the photophysical and electronic properties of the PDI oligomers were pronouncedly tuned by the introduction of BN units. The development of PDI-based PIBs lags behind that of other counterparts in terms of understanding the structure–property–performance relationships.52 The first example of a PDI derivative (3,4,9,10-perylene-tetracarboxylic dianhydride) based PIB cathode with a decent reversible capacity was reported in 2015; however, the potassium storage suffered from poor cyclability.53 To investigate the electrochemical storage performance of these BN co-doped PDI oligomers, the dealkylated oligomers PTCDI2BN, trans- and cis-PTCDI3BN (Scheme 1) were prepared, and they all show more superior performance than the vinylene bridged PDI dimer (PTCDI2) when employed as the cathode in potassium batteries. The rate performance and cycle stability of the two trimers are among the top class of carbonyl-based potassium batteries, while trans-PTCDI3BN shows certain superiority compared with the cis-isomer, illustrating the vital role of BN orientation in the performance of organic batteries (Scheme 1).
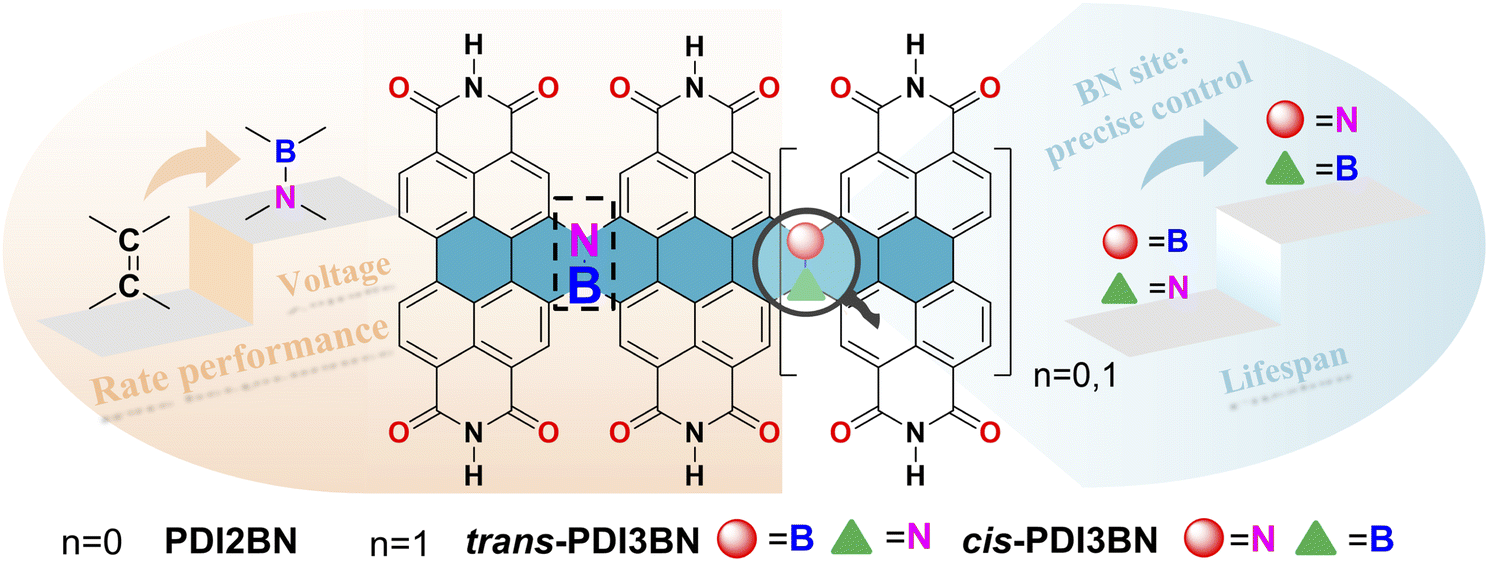 | ||
| Scheme 1 The design strategy of BN embedded PDI oligomers for high performance PIBs; the structure highlights the effect of BN orientation and the length of conjugation. | ||
Results and discussion
As shown in Scheme 2, the synthesis of PDI2BN commences with the preparation of PDI2NH by reacting 1-Br-PDI 154,55 with 1-NH2-PDI 256,57via a Buchwald–Hartwig coupling reaction.58 Subsequently, borylation of PDI2NH by treatment with BCl3, Et3N affords PDI2BN in a yield of 76%. The trimers trans- and cis-PDI3BN were synthesized based on the same sequence using the regiochemically pure 1,7- and 1,6-di(substituted) PDI precursors 3 and 4 as staring materials, respectively (Scheme 2). The corresponding dealkylated oligomers PTCDI2BN, trans- and cis-PTCDI3BN are prepared by vacuum thermolysis at 400 °C (Scheme S7–S9†).39 The weight loss percentage of these PTCDI oligomers was approximately 48% at 400 °C in thermogravimetric analysis (Fig. S21†), which aligned with the soluble alkyl chain content. And as shown in Fig. S22,† vibrational modes of alkyl chains disappeared in the PTCDI oligomers, accompanied by the appearance of new signals identified as those of N–H stretching vibration. These results indicated that PTCDI oligomers were successfully synthesized. The vinylene bridged PDI dimer PDI2 and PTCDI2 (Scheme S10†) were synthesized for comparison following the method described in the literature.28,59,60 The chemical structures of all new compounds were well characterized by 1H NMR, 13C NMR and HRMS and the synthetic details are illustrated in the ESI.†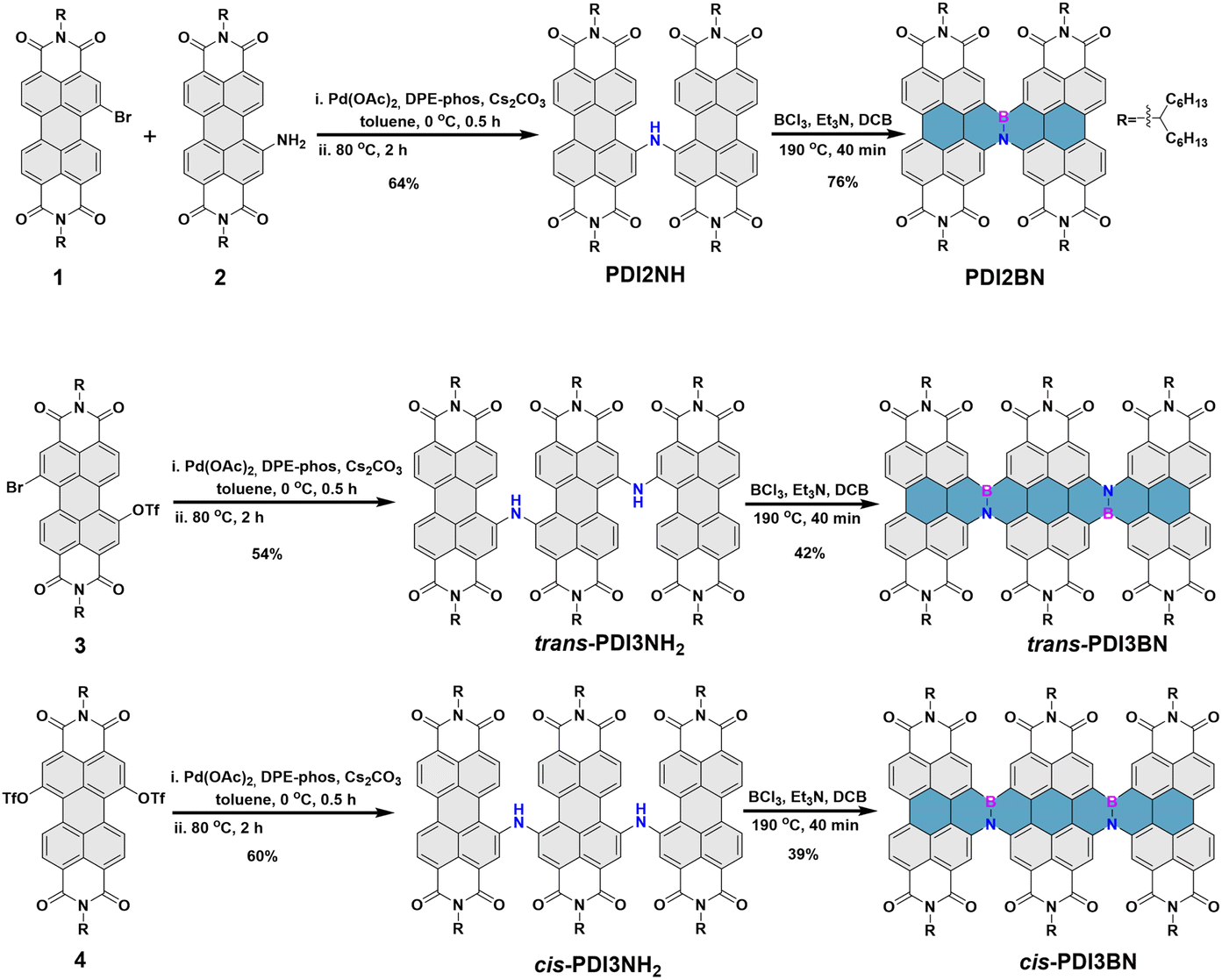 | ||
| Scheme 2 The synthetic routes of (a) PDI2BN, (b) trans-PDI3BN and (c) cis-PDI3BN. | ||
The photophysical properties of these BN embedded oligomers were examined by using the ultraviolet-visible (UV-vis) absorption spectra and fluorescence spectra (Fig. 1 and Table 1). As shown in Fig. 1a, on comparing with that of PDI2, the absorption band of PDI2BN in the 350–425 nm range is almost vanished, which is the characteristic absorption of vinylene bridged PDI2.60–62 Similarly, the characteristic absorption of trans- and cis-PDI3BN also disappeared in comparison with vinylene bridged PDI3 (Fig. S1†).60trans- and cis-PDI3BN showcase a bathochromic-shift of around 20 nm and increased absorption coefficient compared to that of PDI2BN, demonstrating the effect of extended conjugation. Furthermore, the different BN orientations in PDI3BN lead to distinct fine structures in the absorption spectra, and the cis-isomer shows an absorption peak and shoulder at 385 and 609 nm, which may be ascribed to the different dipole–dipole interactions of BN units.48,49 All the three BN embedded PDI oligomers exhibit slightly red-shifted fluorescence compared to vinylene bridged PDI2 (Fig. 1b) with decreases in fluorescence quantum yields (Table S1†), which are consistent with that of the reported BN embedded PDI.49 The redox properties of BN embedded oligomers were examined by cyclic voltammetry (CV). All compounds exhibit multiple reduction waves at a given potential (Fig. S2†), which is similar to the reported B–N embedded PDI.49 The overlapped CV curves of the five cycles of scan and intensified redox peaks with the increase of scanning rates were also observed, demonstrating the splendid reversibility of these oligomers (Fig. S2†).63–65 The BN doping exerts meagre influence on the energy levels of PDI2BN in comparison to PDI2 (Table 1). In the case of trans-PDI3BN and cis-PDI3BN, the orientations of BN units obviously adjust the highest occupied molecular orbital (HOMO) energy level, while the impact on the lowest unoccupied molecular orbital (LUMO) energy level is negligible (Table 1).49,50
 | ||
| Fig. 1 (a) UV-vis absorption spectra of PDI2, PDI2BN, trans-PDI3BN and cis-PDI3BN in chloroform (5 × 10−6 M); (b) normalized fluorescence spectra of PDI2, PDI2BN, trans- and cis-PDI3BN in chloroform (5 × 10−6 M). | ||
| Compounds | λ abs (nm) | ε × 10−5 (M−1 cm−1) | E LUMO (eV) | E HOMO (eV) | E g (eV) |
|---|---|---|---|---|---|
| a λ abs maxima were measured in the CHCl3 solutions of a concentration of 5.0 × 10−6 M. b The ELUMO was calculated by using the following equation: ELUMO = −(Eonesetred − EFc + 4.8) eV, where the first reduction potential was measured by CV and EFc was the half-wave potential of ferrocene. The EHOMO was calculated by using the following equation: EHOMO = −Eg + ELUMO eV. c The optical bandgap was estimated from the onset positions of their absorption spectra and calculated by using the equation: Eg = 1240/λonset. | |||||
| PDI2 | 547 | 1.04 | −3.78 | −5.97 | 2.19 |
| PDI2BN | 482 | 0.82 | −3.75 | −5.93 | 2.18 |
| trans-PDI3BN | 489 | 1.41 | −3.79 | −5.90 | 2.11 |
| cis-PDI3BN | 486 | 1.42 | −3.77 | −5.72 | 1.95 |
Due to the notable redox characteristics of these BN bridged PDI oligomers, they were tested as electrode materials in PIBs. The energy storage performances of these PTCDI oligomers were systematically measured in coin cells with potassium metal as the counter electrode and 1 M KPF6 in ethylene glycol dimethyl ether (DME) as the electrolyte. As shown in Fig. 2a, the cyclic voltammograms (CVs) of the initial three cycles for PTCDI2BN are nearly superimposed on each other, while PTCDI2 shows two irreversible reduction peaks during the initial cathodic scan, implying an irreversible structural transformation.28 The significant enhancement in reversibility may be related to the changed electron distribution of the carbon plane induced by the heteroelectronic structure afforded by BN doping.66 Besides, both cis- and trans-PTCDI3BN display similar redox behaviors, but obviously extended CV area when compared with PTCDI2BN, indicative of their enhanced potassium storage capability. Correspondingly, the galvanostatic charge/discharge (GCD) profiles (Fig. 2b) for all samples are well-consistent with the CV results, i.e., PTCDI2BN, trans- and cis-PTCDI3BN show similar GCD curves, and both trimers can deliver higher reversible capacities of ca. 140 mA h g−1 compared to dimers (ca. 90 mA h g−1). This should highlight that such a high specific capacity is at the forefront of all carbonyl cathodes for PIBs.27,31,35,53,67–69 These additional capacities may be rooted in heteroatom substituents in the carbon plane, which changes the surface-chemical properties and enhances the electrochemical adsorption of K+-ions, further leading to an improved capacity.41,70 Besides, BN embedded PTCDI2BN also shows a ca. 0.2 V higher plateau than PTCDI2 (Fig. 2c and S23†), which can potentially increase the operating voltage of these BN derivative-based full PIBs. It has been shown that a higher redox potential is normally obtained by introducing inactive electron-withdrawing groups, which obviously damages the reversible capacities.71 Moreover, the balance between stabilizing redox-potential and increasing π-conjugation is always a dilemma since the average voltage would inevitably decrease with an increased π-conjugation degree.71–74 This dilemma can be effectively circumvented through precise regulation of the polarization orientations of BN units in trans- and cis-PTCDI3BN. As displayed in Fig. S23,† a rather low voltage-loss of ca. 2.79% is obtained in trans-PTCDI3BN (2.09 V vs. 2.15 V for PTCDI2BN), and a much higher one of ca. 12.56% occurred in cis-PTCDI3BN (1.88 V vs. 2.15 V for PTCDI2BN). More particularly, both trans- and cis-PTCDI3BN demonstrated remarkable smaller overpotentials of ca. 0.02 V than PTCDI2BN (Fig. 2d and S24†), implying their better kinetic properties. In summary, BN co-doped PDI oligomers show unique superiorities as follows: (1) heteroelectronic BN linkers can change the electron distributions of the carbon plane, contributing to remarkable improvement in redox potentials of PTCDI derivatives; (2) an increased number of BN linkers offer additional K+ storage sites, and improve the charge diffusion capability in bulk BN co-doped PTCDI trimers; (3) precise control of BN units in PDI trimers as trans-PTCDI3BN can effectively reduce voltage loss even with increased π-conjugation. Fig. 2e demonstrates that the charge transfer resistance (Rct) is significantly reduced with the increase of BN linkers (Fig. S25†), and trans-PTCDI3BN displayed the smallest Rct value, reflecting its fastest reaction kinetics. Detailed fitted Rct values are presented in Table S2.† Furthermore, a series of rectangle-like CV curves with recognized redox peaks (Fig. S26†) are observed at various scan rates from 0.5 to 10 mV s−1, suggestive of co-managed Faradaic and non-Faradaic behaviors during repeated K+ release/storage in these four PTCDI oligomers. According to the power law between measured current (i) and sweep rates (v) shown in eqn S1 and S2,†PTCDI2BN, trans- and cis-PTCDI3BN demonstrate a higher b-value compared to PTCDI2, implying that BN co-doping is beneficial to increase the contribution from pseudocapacitance (Fig. 2f and S27†).
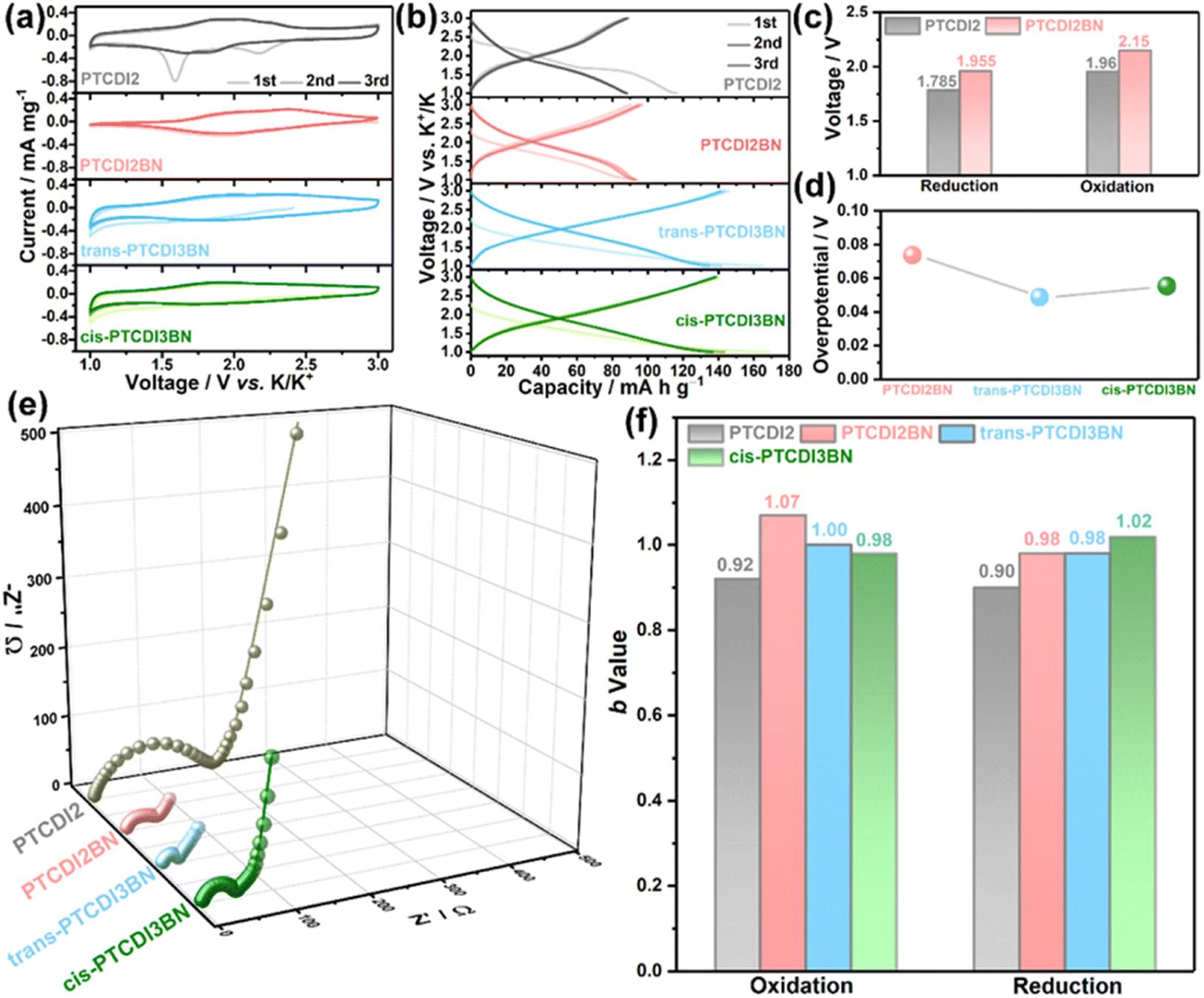 | ||
| Fig. 2 (a) CV curves at a scan rate of 2 mV s−1 and (b) GCD splines at 500 mA g−1 during the initial three cycles for PTCDI oligomers. (c) The average oxidation and reduction potential comparison picture of PTCDI2 and PTCDI2BN. (d) Overpotential image of BN embedded PTCDI derivatives. (e) Nyquist plots and its simulation lines at the third cycle and (f) the histogram for the b-value of PTCDI oligomers. | ||
Encouraged by the improvement in K+ storage and transfer capability through BN co-doping, we further assessed their rate performance. As shown in Fig. 3a, all the BN doped PTCDI oligomers displayed apparent superiority in high-rate capability compared to PTCDI2, and both trans- and cis-PTCDI3BN exhibit enhanced reversible capacities and capacity retention at various current densities. Specifically, trans-PTCDI3BN exhibits a capacity response of ca. 135.2 mA h g−1 at a current density of 0.5 A g−1, and 69% of it can be retained even at a high rate of 20 A g−1, suggestive of its excellent structural stability. Moreover, GCD curves collected at various current densities (Fig. 3b) reveal the electrode polarization order to be trans-PTCDI3BN < cis-PTCDI3BN < PTCDI2BN < PTCDI2, which is in accordance with EIS results and indicates the outstanding high-rate endurance of both trans- and cis-PTCDI3BN again. Furthermore, all three BN embedded PTCDI oligomers outperform most of the state-of-the-art carbonyl cathodes for PIBs (Fig. 3c).28,33,35,36,38,53,75–82 Besides boosting the rate performance, the extended conjugated structure of PTCDI3BN also contributes to remarkable improvement in cyclic durability, which was evaluated at high current densities of 10 A g−1 and 20 A g−1, respectively (Fig. 3d and e). Moreover, trans-PTCDI3BN exhibits the best cyclability among these four PTCDI oligomers, followed by by cis-PTCDI3BN. In detail, there is nearly no capacity fading in the trans-PTCDI3BN cathode even after 13000 cycles at 10 A g−1 and 30000 cycles at 20 A g−1. Furthermore, the cis-PTCDI3BN cathode exhibits a high reversible capacity of 78.4 mA h g−1 over 9000 cycles at 10 A g−1, and retained 91% of its initial value. Even after 20000 cycles, a capacity retention of 85% can still be achieved. It should be highlighted that such an ultra-long lifespan over 30000 cycles with a capacity retention of nearly 100% is among the best ones in state-of-the-art potassium storage.
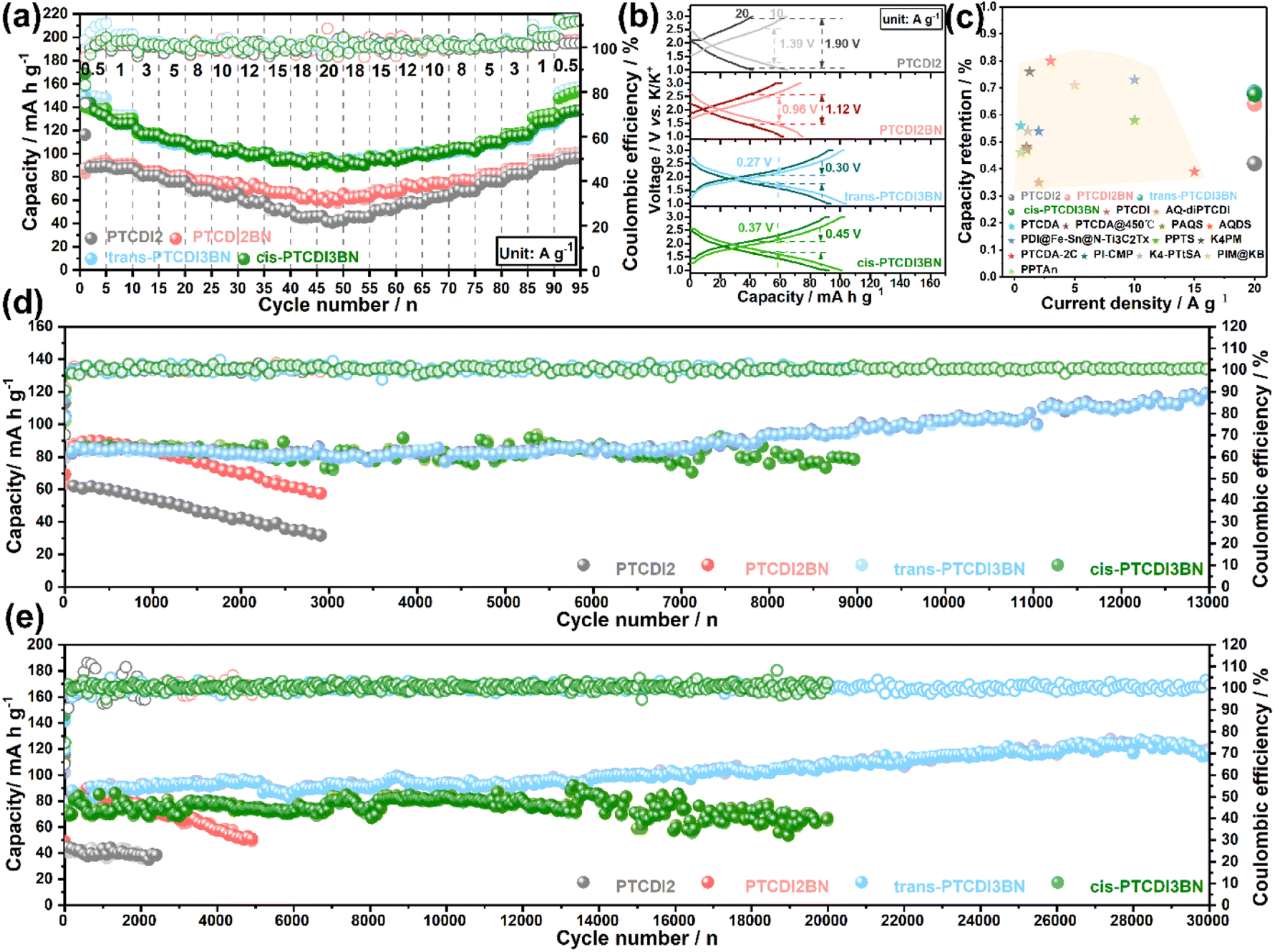 | ||
| Fig. 3 (a) The cycle performance at different current densities and (b) ohmic polarization at the selected current density for PTCDI oligomers. (c) Comparison of the capacity retention among various reported OEMs. Cycle stability of PTCDI oligomers at (d) 10 A g−1 and (e) 20 A g−1. | ||
Conclusions
In conclusion, a two-step synthetic route toward boron and nitrogen co-doped PDI oligomers with precisely controlled BN orientations was developed. The optoelectronic properties of the obtained PDI2BN and trans- and cis-PDI3BN were investigated. The introduction of BN units causes pronounced changes in the absorption and emission characteristics as compared with those of the vinylene bridged PDI oligomers. The difference in LUMO energy levels of trans- and cis-PDI3BN reveals the influence of BN orientations. Furthermore, the successful application of dealkylated BN embedded PDI oligomers in PIBs strongly verifies the superiority of the B, N co-doping design strategy. Potassium batteries using PTCDI2BN and trans- and cis-PTCDI3BN as cathodes show excellent rate performance and cycle stability. Among them, trans-PTCDI3BN shows remarkable superiority with ultrafast-charging capacity and a capacity retention of 100% over 30000 cycles, which displays great potential for practical application in PIBs. The synthetic route developed here offers an applicable method to access BN embedded PDI derivatives, and the B, N co-doping strategy provides an effective way for tuning the electrochemical performance of organic electrodes for potassium batteries with excellent rate performance and cycle stability.
Data availability
All experimental details and data supporting the findings of this study are available within the paper and its ESI.† And original data can be obtained by contacting the corresponding authors.Author contributions
G. Shao and H. Liu conducted the experiments and finished the manuscript. L. Chen and M. Wu provided assistance for the investigation. D. Wang, D. Wu. and J. Xia conducted the project administration, and writing as well as editing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 21801201, 51773160, 21975194, 22209127 and 22175134) and Natural Science Foundation of Hubei Province (No. 2023AFA014).Notes and references
- F. Würthner, C. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2015, 116, 962–1052 CrossRef.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513–1528 RSC.
- E. Krieg, A. Niazov-Elkan, E. Cohen, Y. Tsarfati and B. Rybtchinski, Acc. Chem. Res., 2019, 52, 2634–2646 CrossRef PubMed.
- Z. Yang and X. Chen, Acc. Chem. Res., 2019, 52, 1245–1254 CrossRef PubMed.
- C. Schaack, A. Evans, F. Ng, M. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2021, 144, 42–51 CrossRef PubMed.
- W. Jiang and Z. Wang, J. Am. Chem. Soc., 2022, 144, 14976–14991 CrossRef PubMed.
- G. Zhang, J. Zhao, P. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef.
- P. Cheng, X. Zhao and X. Zhan, Acc. Mater. Res., 2022, 3, 309–318 CrossRef.
- X. Zhan, A. Facchetti, S. Barlow, T. Marks, M. Ratner, M. Wasielewski and S. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef.
- B. Zhang, G. Lyu, E. Kelly and R. Evans, Adv. Sci., 2022, 9, 2201160 CrossRef.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef.
- J. Anthony, A. Facchetti, M. Heeney, S. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef.
- V. Praveen, B. Vedhanarayanan, A. Mal, R. Mishra and A. Ajayaghosh, Acc. Chem. Res., 2020, 53, 496–507 CrossRef.
- P. Singh, A. Hirsch and S. Kumar, TrAC, Trends Anal. Chem., 2021, 138, 116237 CrossRef.
- Z. Wang, T. Liu, H. Peng and Y. Fang, J. Phys. Chem. B, 2023, 127, 828–837 CrossRef.
- S. Chen, M. Zhou, L. Zhu, X. Yang and L. Zang, Chemosensors, 2023, 11, 293 CrossRef.
- M. Wu and Z. Zhou, Interdiscip. Mater., 2023, 2, 231–259 CrossRef.
- J. Russell, V. Posey, J. Gray, R. May, D. Reed, H. Zhang, L. Marbella, M. Steigerwald, Y. Yang, X. Roy, C. Nuckolls and S. Peurifoy, Nat. Mater., 2021, 20, 1136–1141 CrossRef.
- L. Zhu, G. Ding, L. Xie, X. Cao, J. Liu, X. Lei and J. Ma, Chem. Mater., 2019, 31, 8582–8612 CrossRef.
- H. g. Wang and X. b. Zhang, Chem. - Eur. J., 2018, 24, 18235–18245 CrossRef.
- Y. Yu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142 CrossRef.
- J. Kim, Y. Kim, J. Yoo, C. Kwon, Y. Ko and K. kang, Nat. Rev. Chem, 2023, 8, 54–70 Search PubMed.
- T. Schon, B. McAllister, P. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef.
- Y. Feng, Y. Lv, H. Fu, M. Parekh, A. M. Rao, H. Wang, X. Tai, X. Yi, Y. Lin, J. Zhou and B. Lu, Natl. Sci. Rev., 2023, 10, nwad118 CrossRef.
- H. Liu, M. Cheng, Z. Tian, L. Cui, D. Wu, D. Wang, L. Zhou and J. Xia, Adv. Funct. Mater., 2023, 2306424 CrossRef.
- D. Wang, X. You, M. Wu, H. Huang, L. Chen, D. Wu and J. Xia, ACS Appl. Mater. Interfaces, 2021, 13, 16396–16406 CrossRef.
- H. Wang, F. Liu, R. Yu and J. Wu, Interdiscip. Mater., 2022, 1, 196–212 CrossRef.
- Y. Huang, Interdiscip. Mater., 2022, 1, 323–329 CrossRef.
- L. Fan, R. Ma, J. Wang, H. Yang and B. Lu, Adv. Mater., 2018, 30, 1805486 CrossRef.
- Z. Zhang, Y. Zhu, M. Yu, Y. Jiao and Y. Huang, Nat. Commun., 2022, 13, 6489 CrossRef.
- Z. Tong, S. Tian, H. Wang, D. Shen, R. Yang and C. Lee, Adv. Funct. Mater., 2019, 30, 1907656 CrossRef.
- J. Ge, X. Yi, L. Fan and B. Lu, J. Energy Chem., 2021, 57, 28–33 CrossRef.
- J. Zheng, X. Liu, W. Li, W. Li, X. Feng and W. Chen, Nano Res., 2023, 16, 9538–9545 CrossRef.
- P. Han, F. Liu, Y. Zhang, Y. Wang, G. Qin, L. Hou and C. Yuan, Angew. Chem., Int. Ed., 2021, 60, 23596–23601 CrossRef.
- M. Bhosale, S. Chae, J. Kim and J. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC.
- Y. Hu, W. Tang, Q. Yu, X. Wang, W. Liu, J. Hu and C. Fan, Adv. Funct. Mater., 2020, 30, 2000675 CrossRef.
- Z. Jin, Q. Cheng, S. Bao, R. Zhang, A. M. Evans, F. Ng, Y. Xu, M. Steigerwald, A. McDermott, Y. Yang and C. Nuckolls, J. Am. Chem. Soc., 2022, 144, 13973−13980 Search PubMed.
- C. Sathish, G. Kothandam, P. Selvarajan, Z. Lei, J. Lee, J. Qu, A. Al-Muhtaseb, X. Yu, M. Breese, R. Zheng, J. Yi and A. Vinu, Adv. Sci., 2022, 9, 2105603 CrossRef.
- X. Lian, J. Zhou, Y. You, Z. Tian, Y. Yi, J. Choi, M. Rümmeli and J. Sun, Adv. Funct. Mater., 2022, 32, 2109969 CrossRef.
- M. Wang, Y. Yang, Z. Yang, L. Gu, Q. Chen and Y. Yu, Adv. Sci., 2017, 4, 1600468 CrossRef PubMed.
- X. Chen, D. Tan and D. Yang, J. Mater. Chem. C, 2022, 10, 13499–13532 RSC.
- X. Wang, J. Wang and J. Pei, Chem. - Eur. J., 2014, 21, 3528–3539 CrossRef PubMed.
- J. Wang and J. Pei, Chin. Chem. Lett., 2016, 27, 1139–1146 CrossRef.
- H. Helten, Chem. - Eur. J., 2016, 22, 12972–12982 CrossRef.
- T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef.
- P. Zhang, J. Zeng, F. Zhuang, K. Zhao, Z. Sun, Z. Yao, Y. Lu, X. Wang, J. Wang and J. Pei, Angew. Chem., Int. Ed., 2021, 60, 23313–23319 CrossRef.
- K. Zhao, Z. Yao, Z. Wang, J. Zeng, L. Ding, M. Xiong, J. Wang and J. Pei, J. Am. Chem. Soc., 2022, 144, 3091–3098 CrossRef.
- Y. Chen, W. Chen, Y. Qiao, X. Lu and G. Zhou, Angew. Chem., Int. Ed., 2020, 59, 7122–7130 CrossRef PubMed.
- G. Shao, M. Wu, X. Wang, J. Zhao, X. You, D. Wu and J. Xia, J. Org. Chem., 2022, 87, 14825–14832 CrossRef PubMed.
- L. Li, Y. Yin, J. Hei, X. Wan, M. Li and Y. Cui, Small, 2021, 17, 2005752 CrossRef PubMed.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef.
- Z. Luo, F. Wu, T. Zhang, X. Zeng, Y. Xiao, T. Liu, C. Zhong, X. Lu, L. Zhu, S. Yang and C. Yang, Angew. Chem., Int. Ed., 2019, 58, 8520–8525 CrossRef PubMed.
- Z. Luo, K. Wu, Y. Zhao, B. Qiu, Y. Li and C. Yang, Dyes Pigm., 2019, 163, 356–362 CrossRef.
- A. Goujon, L. Rocard, T. Cauchy and P. Hudhomme, J. Org. Chem., 2020, 85, 7218–7224 CrossRef.
- X. Wang, F. Zhuang, X. Zhou, D. Yang, J. Wang and J. Pei, J. Mater. Chem. C, 2014, 2, 8152–8161 RSC.
- Y. Li, C. Wang, L. Cheng, S. Di Motta, F. Negri and Z. Wang, Org. Lett., 2012, 14, 5278–5281 CrossRef.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122–8130 CrossRef PubMed.
- Y. Zhong, M. Trinh, R. Chen, W. Wang, P. Khlyabich, B. Kumar, Q. Xu, C. Nam, M. Sfeir, C. Black, M. Steigerwald, Y. Loo, S. Xiao, F. Ng, X. Zhu and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 15215–15221 CrossRef PubMed.
- M. Wu, J. Yi, L. Chen, G. He, F. Chen, M. Sfeir and J. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 27894–27901 CrossRef PubMed.
- J. Tan, G. Zhang, C. Ge, J. Liu, L. Zhou, C. Liu, X. Gao, A. Narita, Y. Zou and Y. Hu, Org. Lett., 2022, 24, 2414–2419 CrossRef.
- S. Zhao, Z. Bian, Z. Liu, Y. Wang, F. Cui, H. g. Wang and G. Zhu, Adv. Funct. Mater., 2022, 32, 2204539 CrossRef.
- X. Li, W. Liu, Y. Wang, L. Lv, H. Feng, W. Huang, Y. Sun, W. Xiong and H. Zheng, Chem. Eng. J., 2023, 473, 145310 CrossRef.
- D. Chao, C. Zhu, P. Yang, X. Xia, J. Liu, J. Wang, X. Fan, S. V. Savilov, J. Lin, H. Fan and Z. Shen, Nat. Commun., 2016, 7, 1–8 Search PubMed.
- S. Li, J. Cao, T. Wang, L. Wang, H. Deng, Q. Zhang, Y. Cheng, J. Zhu and B. Lu, Chem. Eng. J., 2022, 431, 133215 CrossRef.
- J. Zhao, J. Yang, P. Sun and Y. Xu, Electrochem. Commun., 2018, 86, 34–37 CrossRef.
- A. Yu, Q. Pan, M. Zhang, D. Xie and Y. Tang, Adv. Funct. Mater., 2020, 30, 2001440 CrossRef.
- X. Chen, P. Ye, H. Wang, H. Huang, Y. Zhong and Y. Hu, Adv. Funct. Mater., 2023, 33, 2212915 CrossRef.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 Search PubMed.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef.
- Z. Tie and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303 CrossRef PubMed.
- B. Li, J. Zhao, Z. Zhang, C. Zhao, P. Sun, P. Bai, J. Yang, Z. Zhou and Y. Xu, Adv. Funct. Mater., 2018, 29, 1807137 CrossRef.
- Q. Pan, Y. Zheng, Z. Tong, L. Shi and Y. Tang, Angew. Chem., Int. Ed., 2021, 60, 11835–11840 CrossRef.
- J. Wang, X. Liu, H. Jia, P. Apostol, X. Guo, F. Lucaccioni, X. Zhang, Q. Zhu, C. Morari, J.-F. Gohy and A. Vlad, ACS Energy Lett., 2022, 7, 668–674 CrossRef.
- Y. Hu, Y. Gao, L. Fan, Y. Zhang, B. Wang, Z. Qin, J. Zhou and B. Lu, Adv. Energy Mater., 2020, 10, 2002780 CrossRef.
- B. Tian, J. Zheng, C. Zhao, C. Liu, C. Su, W. Tang, X. Li and G.-H. Ning, J. Mater. Chem. A, 2019, 7, 9997–10003 RSC.
- C. Zhang, Y. Xu, K. He, Y. Dong, H. Zhao, L. Medenbach, Y. Wu, A. Balducci, T. Hannappel and Y. Lei, Small, 2020, 16, 2002953 CrossRef.
- L. Fan, Q. Liu, Z. Xu and B. Lu, ACS Energy Lett., 2017, 2, 1614–1620 CrossRef.
- M. Tang, Y. Wu, Y. Chen, C. Jiang, S. Zhu, S. Zhuo and C. Wang, J. Mater. Chem. A, 2019, 7, 486–492 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06331c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |