
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolarization of macrophages to an anti-cancer phenotype through in situ uncaging of a TLR 7/8 agonist using bioorthogonal nanozymes†
Xianzhi
Zhang‡
 a,
Yuanchang
Liu‡
a,
Mingdi
Jiang‡
a,
Javier A.
Mas-Rosario
ab,
Stefano
Fedeli
a,
Roberto
Cao-Milan
a,
Liang
Liu
a,
Kyle J.
Winters
a,
Cristina-Maria
Hirschbiegel
a,
Ahmed
Nabawy
a,
Rui
Huang
a,
Michelle E.
Farkas
ab and
Vincent M.
Rotello
*ab
a,
Yuanchang
Liu‡
a,
Mingdi
Jiang‡
a,
Javier A.
Mas-Rosario
ab,
Stefano
Fedeli
a,
Roberto
Cao-Milan
a,
Liang
Liu
a,
Kyle J.
Winters
a,
Cristina-Maria
Hirschbiegel
a,
Ahmed
Nabawy
a,
Rui
Huang
a,
Michelle E.
Farkas
ab and
Vincent M.
Rotello
*ab
aDepartment of Chemistry, University of Massachusetts Amherst, 710 N. Pleasant St., Amherst, MA 01003, USA. E-mail: rotello@chem.umass.edu
bMolecular and Cellular Biology Graduate Program, University of Massachusetts Amherst, 230 Stockbridge Road, Amherst, Massachusetts 01003, USA
First published on 9th January 2024
Abstract
Macrophages are plastic cells of the immune system that can be broadly classified as having pro-inflammatory (M1-like) or anti-inflammatory (M2-like) phenotypes. M2-like macrophages are often associated with cancers and can promote cancer growth and create an immune-suppressive tumor microenvironment. Repolarizing macrophages from M2-like to M1-like phenotype provides a crucial strategy for anticancer immunotherapy. Imiquimod is an FDA-approved small molecule that can polarize macrophages by activating toll-like receptor 7/8 (TLR 7/8) located inside lysosomes. However, the non-specific inflammation that results from the drug has limited its systemic application. To overcome this issue, we report the use of gold nanoparticle-based bioorthogonal nanozymes for the conversion of an inactive, imiquimod-based prodrug to an active compound for macrophage re-education from anti- to pro-inflammatory phenotypes. The nanozymes were delivered to macrophages through endocytosis, where they uncaged pro-imiquimod in situ. The generation of imiquimod resulted in the expression of pro-inflammatory cytokines. The re-educated M1-like macrophages feature enhanced phagocytosis of cancer cells, leading to efficient macrophage-based tumor cell killing.
Introduction
Macrophages are plastic phagocytes that serve as a first-line defense against pathogens including tumors.1–3 Macrophage subtypes exist as a complex spectrum with broadly two major phenotypes: classically-activated M1-like macrophages and alternatively-activated M2-like macrophages.4–6 M1-like macrophages are associated with inflammation by secreting pro-inflammatory cytokines, generating reactive oxygen and nitrogen species, and engulfing antigens to fight viruses, bacteria, or cancers.7 In contrast, M2-like macrophages reduce inflammation, and promote wound healing and angiogenesis.5 Studies have revealed, however, that several cancer types can hijack macrophages, which in the tumor environment facilitate proliferation, angiogenesis, and invasion, and provide defense against anticancer therapies.8–12 These tumor-associated macrophages (TAMs)13 exist primarily in M2-like phenotype and can constitute up to 50% of the tumor mass.14 TAMs provide an immunosuppressive microenvironment surrounding the cancer cells,15 hampering cancer immunotherapies including immune checkpoint blockades16 and chimeric antigen receptor (CAR) T cell therapy.17 Moreover, TAMs can hinder chemotherapy by sacrificing themselves to deplete drugs from cancer cells.11,18–20 Overall, the M2-like TAMs promote cancer growth and pose an obstacle to multiple efficient cancer therapies.In contrast, M1-like macrophages display strong antitumor activity. M1-like macrophages can eliminate tumor cells via direct phagocytosis and secretion of proinflammatory cytokines that induce the apoptosis of cancer cells.21 Moreover, M1-like macrophages can also recruit T cells and B cells to the tumor site to initiate the adaptive immune response.22,23 Therefore, the polarization of TAMs to the M1-like phenotype provides a macrophage-based immunotherapeutic approach to cancer eradication.24
Imiquimod is an FDA-approved small molecule toll-like receptor (TLR) agonist that polarizes macrophages to the M1-like phenotype by activating lysosomal TLR7/8.25–27 Imiquimod is used clinically to treat genital warts28 and basal cell carcinomas.29 The therapeutic use of imiquimod requires topical delivery; systemic administration results in non-specific inflammation including cytokine storms.30 Prodrug strategies can minimize off-target effects via the use of an inert drug precursor (prodrug), which is converted into the active entity following exposure to either an exogenous or endogenous stimulus.31–33 Therefore, converting imiquimod from the inactive precursor can potentially avoid cytokine storms throughout the body. Light,34 enzymes,35 and redox potential36 have been used as stimuli to activate imiquimod prodrugs, however, controlled activation and sustained dosing aspects remain challenging.
Bioorthogonal catalysis via transition metal catalysts (TMCs) offers an approach for prodrug activation that employs chemical reactions inaccessible to natural enzymes.37–45 The in situ activation of prodrugs overcomes the concern of non-specific leakage of the drug through the conventional delivery strategy. Specifically designed catalysts have shown their potential for drug conversion in biomedical applications,46–51 however, the direct use of TMCs can face challenges such as limited solubility,52 low stability,53 and poor biocompatibility54 in biological environments. Surface-engineered nanomaterials can solubilize TMCs and protect them from deactivation, providing a modular system for catalysts.55–58 The loading of TMCs into nanomaterials can be used to fabricate bioorthogonal nanozymes with high catalytic activity for the sustained generation of therapeutics for anticancer,59–69 antimicrobial,70–75 and anti-inflammatory treatments.76
We report here the use of bioorthogonal nanozymes to re-educate tumor-associated macrophages to pro-inflammatory phenotypes through the intracellular uncaging of an imiquimod prodrug. The encapsulation of palladium catalysts into the monolayer of gold nanoparticles yields bioorthogonal nanozymes and avoids non-specific immunogenicity resulting from the hydrophobic catalysts. Significantly, nanozymes are internalized by macrophages via endocytosis, and subsequently activate imiquimod intracellularly, in proximity to the lysosomal TLR7/8 receptors (Fig. 1). The in situ generation of imiquimod facilitates macrophage re-education from an M2-like to the M1-like phenotype, evidenced by significant upregulation of inflammatory biomarkers, resulting in enhanced phagocytosis of cancer cells. M1-like macrophages polarized by this nanozyme-based strategy possess efficient anticancer activity and provide a biocompatible strategy for potential cancer immunotherapy use.
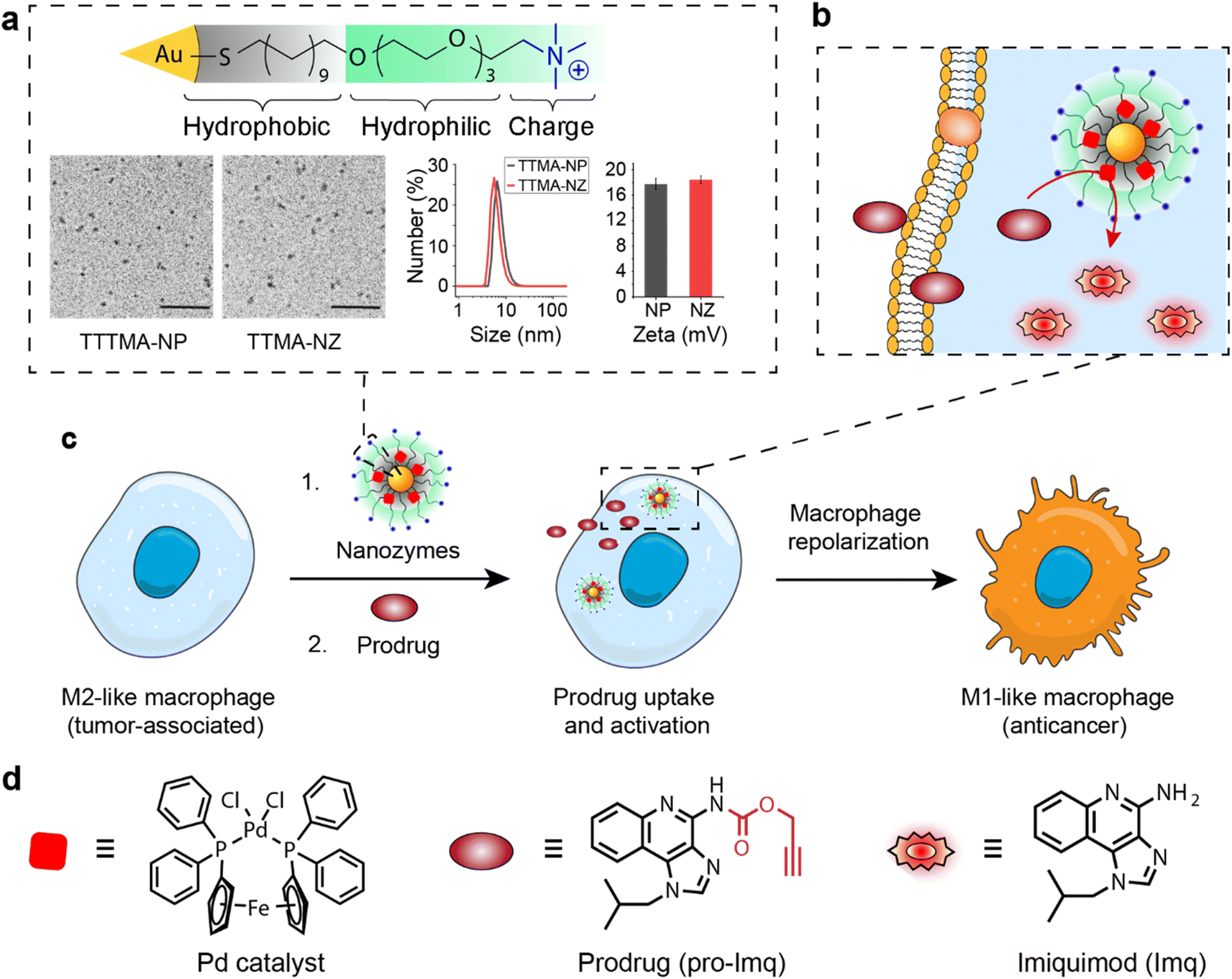 | ||
| Fig. 1 (a) Structure, transmission electron microscopy (TEM) imaging, dynamic light scattering (DLS) measurement, and zeta potential of thioalkyl tetra(ethylene glycol) trimethylammonium nanoparticles and nanozymes (TTMA-NP and TTMA-NZ, respectively). Scale bar = 50 nm. (b) Intracellular activation of the prodrug (pro-Imq) by TTMA-NZ. (c) Schematic representation of macrophage re-education by nanozyme-mediated imiquimod activation. (d) Structure of the palladium catalyst, pro-Imq, and Imq. | ||
Results and discussion
The bioorthogonal nanozyme was comprised of palladium catalysts and 2 nm thioalkyl tetra(ethylene glycol) trimethylammonium ligand (TTMA)-functionalized gold nanoparticles (TTMA-NP). The TTMA ligand consisted of three parts: an aliphatic chain to stabilize the nanoparticle and encapsulate catalysts, an oligo ethylene glycol spacer to enhance water solubility and avoid serum protein denaturation,56,77,78 and a cationic charge to facilitate efficient endocytosis. The TTMA nanozyme (TTMA-NZ) was then generated by encapsulating Pd(dppf)Cl2 catalysts ([1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II)) into the nanoparticle through nanoprecipitation. The hydrophobicity of the ligands kept the palladium catalysts inside the hydrophobic monolayer of nanoparticles for a prolonged period.79 Briefly, Pd catalysts were dissolved in the mixture of acetone and tetrahydrofuran and added dropwise into the nanoparticle solution in water. The mixture was applied to a molecular cut-off filter and washed with water to remove excess catalysts to obtain nanozymes. The resulting nanozyme maintained a similar size and surface charge compared to TTMA-NP after formulation, as indicated by transmission electron microscopy (TEM), dynamic light scattering (DLS), and zeta potential (Fig. 1a). Inductively coupled plasma mass spectrometry (ICP-MS: 106Pd and 197Au) verified the presence of ∼29 Pd molecules per nanozyme (Table S1†).The catalytic activity of nanozymes was evaluated through the activation of a caged fluorophore both in solution and in cell culture. The caged fluorophore was prepared by reacting Rhodamine 110 (Rho) with propargyl chloroformate to obtain the non-fluorescent precursor (pro-Rho).80TTMA-NZ removes the propargyl groups, resulting in the generation of fluorescent Rho (Fig. 2a). The green fluorescence signal increased significantly after adding nanozymes and pro-Rho (Fig. 2b), indicating the decent catalytic activity of nanozymes. The rate (∼2 nM min−1) was calculated by the calibration curve of Rho (Fig. S1†). This activity was further probed in RAW 264.7 murine macrophage cells (M0 macrophages) through intracellular imaging. A cytotoxicity study was first performed to optimize the working concentration of nanozymes. Results showed that 400 nM TTMA-NZ started to have slight toxicity after 48 h treatment, 50 nM nanozyme, however, started to be toxic after 72 h treatment (Fig. S2†). Non-specific immunogenicity of the agents toward macrophages was then determined by evaluating the mRNA expression of the pro-inflammatory biomarker tumor necrosis factor-α (TNF-α) following 24 h treatment with TTMA-NZ (400 nM, containing 11.6 μM Pd catalysts) or the same amount of free catalysts (11.6 μM) using quantitative real-time polymerase chain reaction (qRT-PCR). Compared to the free catalysts, nanozymes are much less immunogenic, presumably due to the shielding of the hydrophobic catalysts from the environment (Fig. 2c). TTMA-NZ was then used for pro-Rho activation in vitro. Confocal microscopy showed the generation of green fluorescence inside the macrophages that were treated with nanozymes and pro-Rho (Fig. 2d). Considering pro-Rho cannot be activated by cellular components, fluorogenesis indicates that nanozymes retain significant activity inside the living cells. Significantly, the green fluorescence co-localized with red fluorescence from the Lysotracker, mirroring the in situ intracellular uncaging of substrates inside the lysosomes.
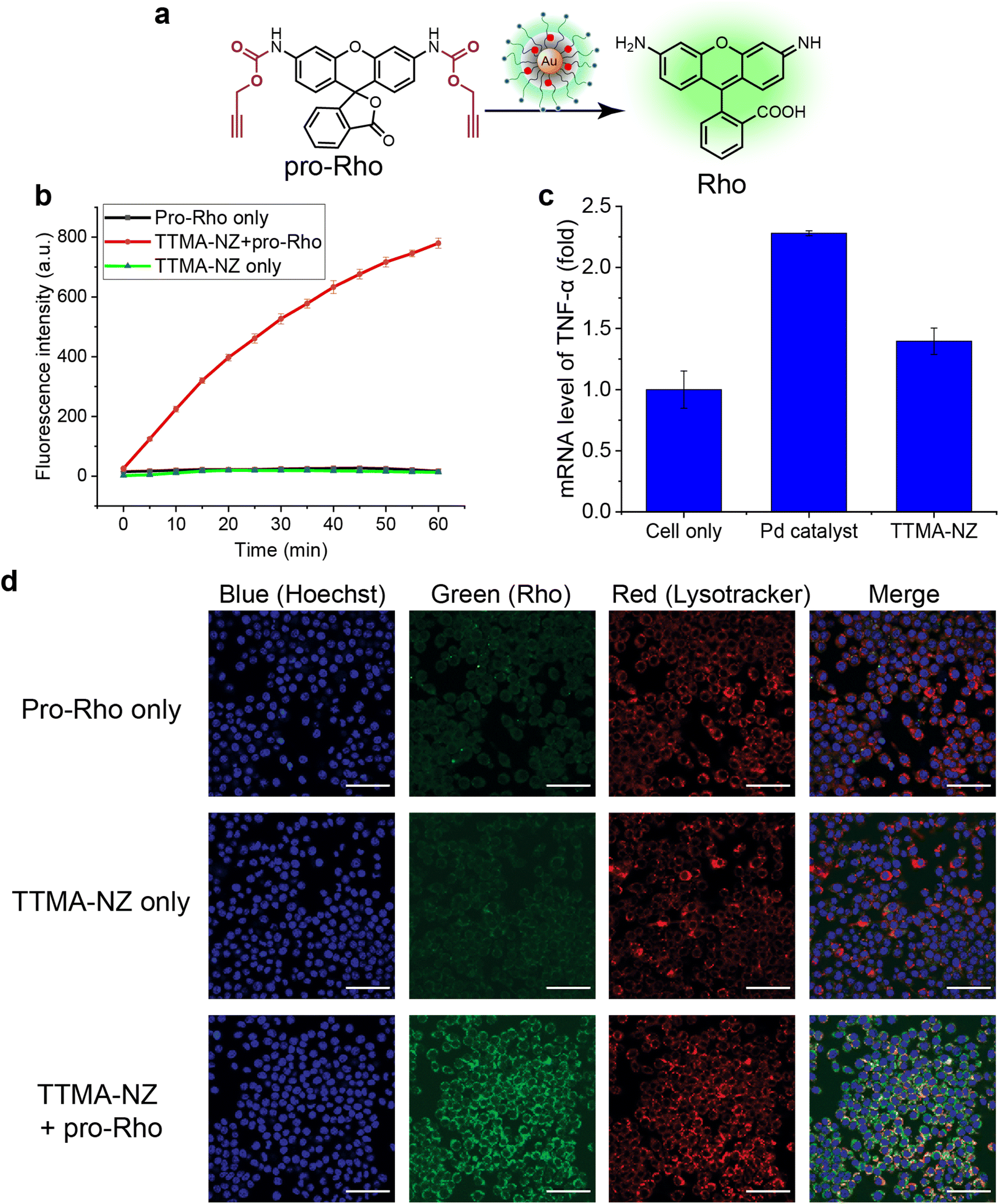 | ||
| Fig. 2 (a) pro-Rho activation by 400 nM TTMA-NZ. (b) Kinetic study of pro-Rho activation by TTMA-NZ in phosphate-buffered saline (PBS) solution at 37 °C. (c) Quantification of TNF-α mRNA levels following 24 h treatment with free catalysts and TTMA-NZ using qRT-PCR. For (b and c), data shown is the average of three replicates; error bars represent standard deviation. (d) Confocal images of RAW 264.7 cells incubated with nanozymes followed by incubation with pro-Rho for 24 h and controls. Nuclei were stained by using Hoechst 33342, and lysosomes were stained with deep red Lysotracker. Scale bar = 50 μm. The colocalization of green and red fluorescence indicates the lysosomal activation of the substrate by TTMA-NZ. | ||
Encouraged by the results obtained from cell imaging, we applied the same caging strategy to generate a prodrug of the pro-inflammatory drug imiquimod (pro-Imq; synthesis and characterization in Fig. S3–S5†). pro-Imq was obtained by blocking the imiquimod (Imq) pharmacophore with a propargyloxycarbonyl group. Compared to Imq, pro-Imq resulted in a more than 50-fold lower expression of the pro-inflammatory cytokine TNF-α (Fig. S6a†) and only 10 μM pro-Imq started to have slight toxicity after 72 h treatment (Fig. S6†). Even though immortalized macrophages mimic the main phenotypical and metabolic features of their primary cell counterparts, they still differ in multiple aspects.81 We therefore, also tested the cytotoxicity of TTMA-NZ and pro-Imq on primary bone marrow-derived macrophages (BMDMs) isolated from C57/B6J mice (Fig. S7†), which demonstrated that TTMA-NZ and pro-Imq showed even lower toxicity toward BMDMs compared to RAW 264.7 cells. Based on the cytotoxicity results, 400 nM TTMA-NZ, 10 μM pro-Imq and 2 μM Imq were used for the following studies.
We next investigated macrophage re-polarization through intracellular uncaging of the pro-Imq using nanozymes. M2-like macrophages were induced by incubating RAW 264.7 cells with interleukin-4 (lL-4) for 24 h. The M2-polarized RAW 264.7 macrophages were then incubated with nanozymes for 24 h, followed by washing with PBS four times to remove the non-internalized nanozymes. pro-Imq was then added to the cells. After 24 h, the supernatant was collected for analysis. The pro-Imq and Imq alone were used as negative and positive controls, respectively. The re-polarization of M2-like macrophages to M1-like macrophages was investigated through analysis of the expression of pro-inflammatory cytokines (TNF-α and interleukin-6 (IL-6)), and nitrite production. Nanozyme and pro-Imq treated RAW 264.7 macrophages expressed significantly higher levels of all three proinflammatory signaling molecules (Fig. 3), comparable to the positive controls. Negative controls (nanozyme and pro-Imq, independently) did not show elevated expression of pro-inflammatory cytokines or nitrite levels. M1-like polarization was further assessed by analyzing the expression of the M1-like macrophage-related surface marker CD80. CD80 serves as a co-stimulatory molecule for T cell activation that initiates cell-based immunotherapy.82 RAW 264.7 macrophages treated with nanozymes and pro-Imq showed a two-fold greater expression of CD80 receptors versus negative control groups, as determined by flow cytometry (Fig. 3d). Together, in situ uncaging of pro-Imq by nanozymes results in the efficient polarization of RAW 264.7 macrophages to M1-like macrophages. In addition, we determined the TNF-α and IL-6 expression of IL-4-induced M2-like BMDMs after treatment with nanozyme and pro-Imq (Fig. S8†). The significantly increased levels of TNF-α and IL-6 indicated that nanozymes uncaging pro-Imqin situ can also induce efficient M2-like to M1-like repolarization of primary macrophages, further demonstrating the immunotherapeutic potential of our system.
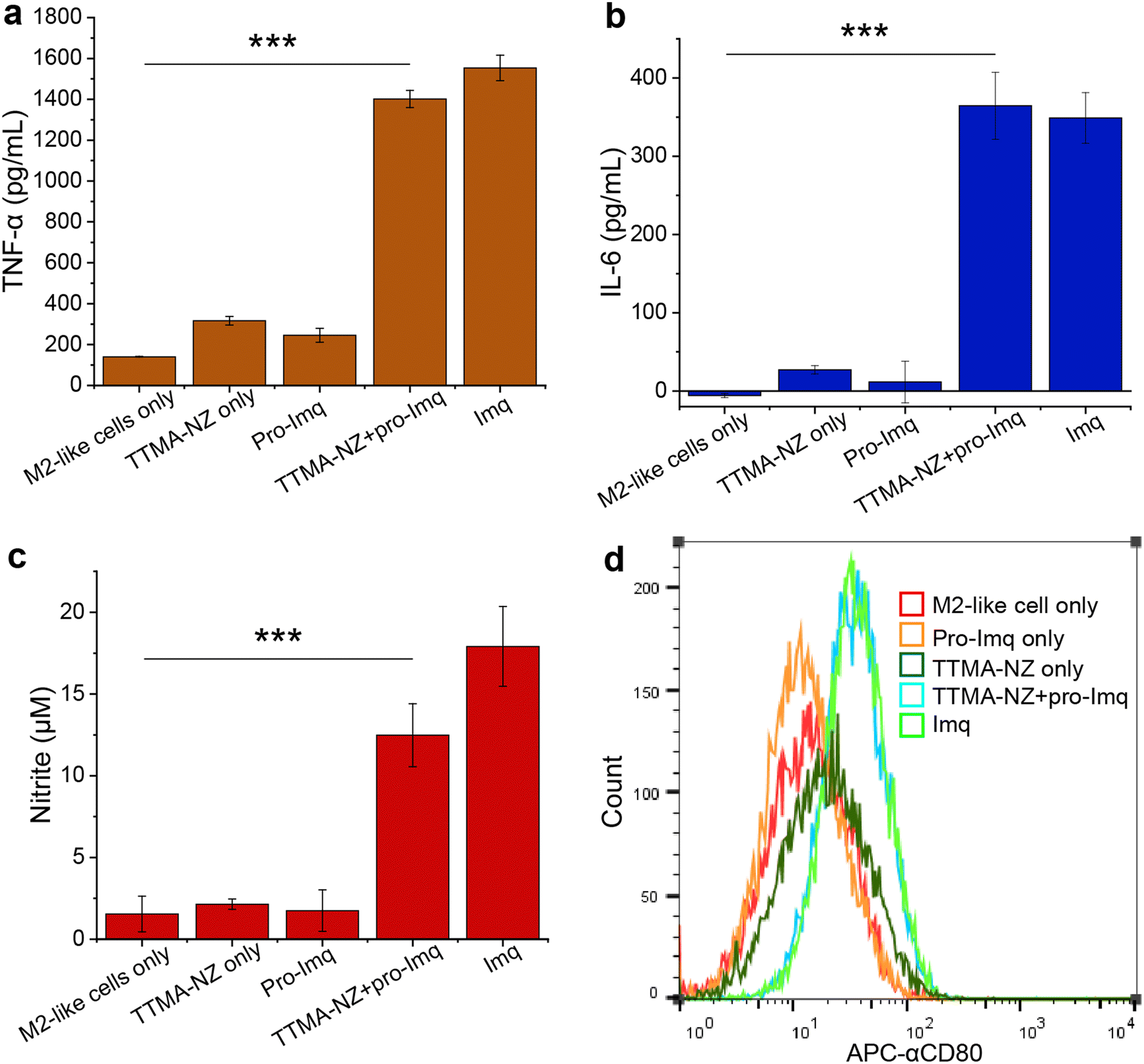 | ||
| Fig. 3 Re-education of M2-like RAW 264.7 cells by nanozyme-mediated pro-Imq activation. Cells were treated with 400 nM nanozymes for 24 h, followed by the addition of 10 μM pro-Imq for another 24 h. Imq (2 μM) was used as a positive control. Quantification of expression of (a) TNF-α and (b) IL-6 of RAW 264.7 macrophages using enzyme-linked immunosorbent assay (ELISA). (c) Nitrite expression in RAW 264.7 macrophages determined by Griess assay. (d) The expression of M1-related surface marker CD80 tested by flow cytometry using APC-labeled CD80 antibody (APC-αCD80). M2-like RAW 264.7 macrophages treated with nanozymes and pro-Imq expressed significantly higher levels of pro-inflammatory biomarkers, showing the re-education of the macrophages to the M1-like phenotype. Data shown is the average of three biological replicates and error bars represent the standard deviation. Statistical significance was determined by a two-tailed Student's t-test. ***: p < 0.001. | ||
A key aspect of macrophage-mediated anticancer activity is their engulfment of cancer cells via phagocytosis;83–85 M2-like macrophages repolarized to the M1-like phenotype have enhanced phagocytosis.86 We tested the phagocytic ability of M1-like macrophages re-polarized by the nanozyme-based treatment against green-fluorescent protein-expressing osteosarcoma U2OS cancer cells (GFP-U2OS cells). The cytotoxicity of Imq itself toward GFP-U2OS cells was first determined (Fig. S9†), with 2 μM Imq exhibiting no toxicity after up to 72 h treatment. For phagocytosis studies, M2-like RAW 264.7 macrophages were pre-treated with nanozymes and pro-Imq subsequently. Afterward, the macrophages were harvested and labeled by phycoerythrin (PE)-conjugated F4/80 antibody. The cells were then washed and incubated with GFP-U2OS cells for 4 h at 37 °C. Phagocytosis was evaluated by tracking the red PE channel and green FITC channel using flow cytometry. RAW 264.7 macrophages treated with NZ and pro-Imq showed a 4-fold increase in phagocytosis of U2OS cells compared to the non-treated controls, reaching the same level as the positive control (Fig. 4 and S10†). This result reflects the efficient recognition and engulfment of cancer cells by the re-educated macrophages using nanozymes.
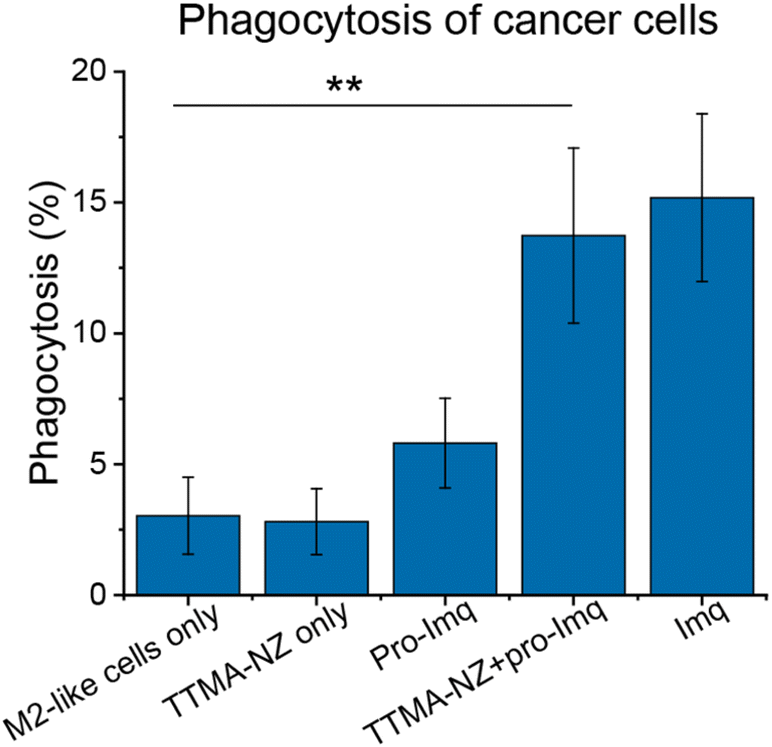 | ||
| Fig. 4 Phagocytosis percentage of M2-like RAW 264.7 macrophages against U2OS cells determined by flow cytometry (Fig. S10†). Cells were treated with 400 nM nanozymes for 24 h followed by the addition of 10 μM pro-Imq for another 24 h. Imq (2 μM) was used as a positive control. Nanozyme-based strategy resulted in elevated phagocytosis by ∼4-fold compared to untreated controls. Data shows the average of three biological replicates and error bars represent the standard deviation. Statistical significance was determined by a two-tailed Student's t-test. **: 0.01 > p > 0.001. | ||
The anti-cancer potential of nanozyme re-educated macrophages was further evaluated through a co-culture model. M2-like RAW 264.7 macrophages were seeded with GFP-U2OS cells in confocal microscopy dishes followed by sequential treatment with nanozymes, pro-Imq and indicated controls. The cells were then incubated for 24 h and imaged using confocal microscopy. GFP fluorescence was used as a surrogate readout for the viability of the cancer cells. Diminished levels of green fluorescence were observed for cells treated with nanozymes and pro-Imq, and Imq alone, which can be attributed to cancer cell death and/or inhibition of proliferation. As expected, nanozyme alone or pro-Imq alone showed no appreciable effects on the cancer cells (Fig. 5).
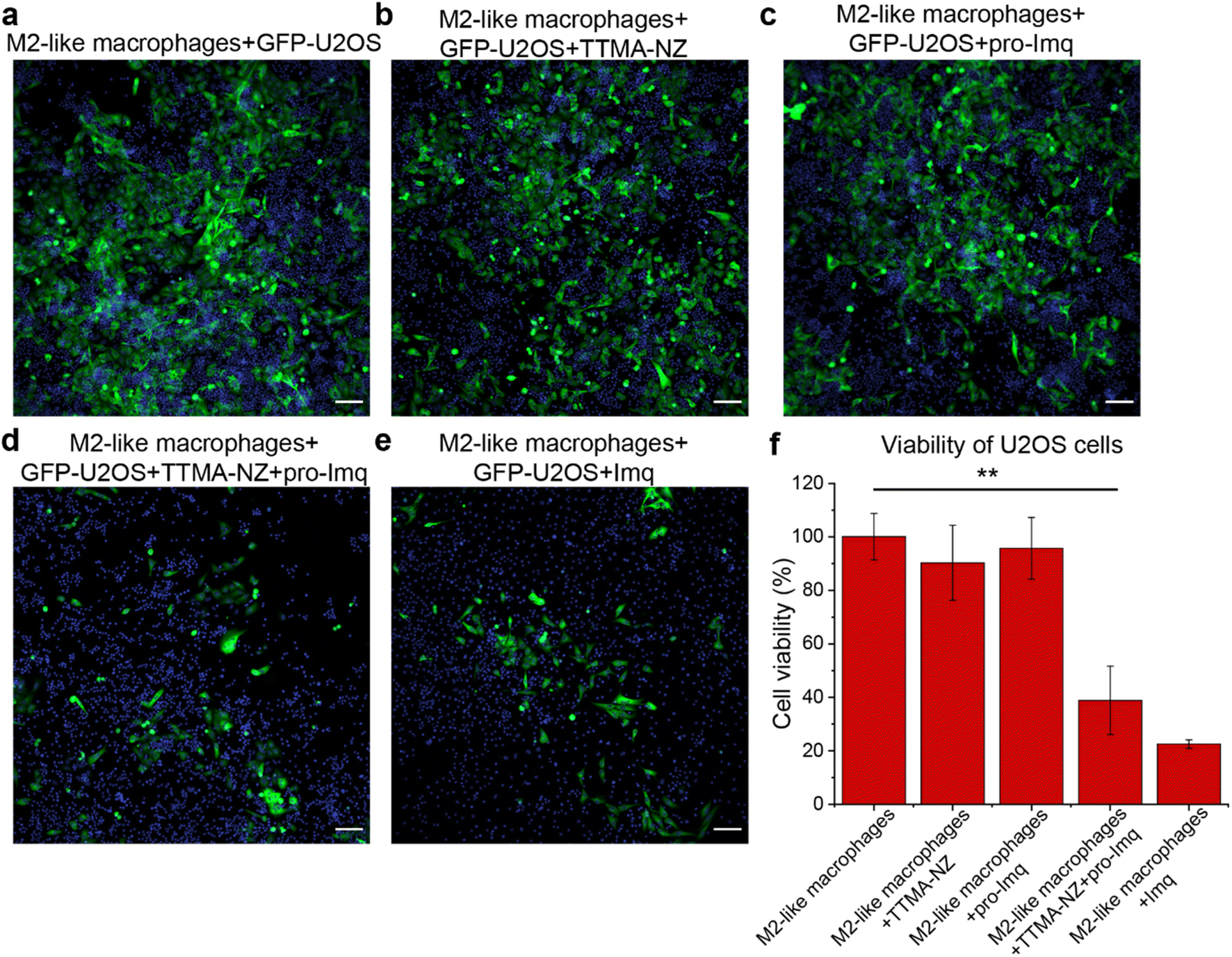 | ||
| Fig. 5 Confocal imaging of M2-like RAW 264.7 macrophages and GFP-U2OS cells (a) without further treatment, and with M2 macrophages treated by (b) pro-Imq, (c) Imq, (d) TTMA-NZ, and (e) TTMA-NZ + pro-Imq subsequently. Where applicable, cells were treated with nanozymes (400 nM) and pro-Imq (10 μM). Imq (2 μM) was used as the positive control. The nucleus was stained by Hoechst 33342. Scale bar = 100 μm. (f) Viability of cancer cells. The result was determined by tracking the intensity of green fluorescence. Data shows the average of triplicates and error bars represent the standard deviation. Statistical significance was determined by a two-tailed Student's t-test. **: 0.01 > p > 0.001. | ||
Conclusion
In summary, we report the polarization of tumor-associated macrophages to an anticancer phenotype by in situ TLR 7/8 agonist activation using bioorthogonal nanozymes. The nanozymes were located inside endosomes/lysosomes following uptake by macrophages and provided on-target uncaging of an imiquimod prodrug. This uncaged imiquimod re-educated anti-inflammatory M2-like macrophages to anticancer M1-like cells. The re-educated M1-like macrophages possessed enhanced phagocytosis capabilities and resulted in diminished levels of cancer cells versus controls. This study demonstrates the polarization of macrophages for anticancer applications by harnessing the endocytosis of bioorthogonal nanozymes combined with the administration of an inert prodrug. The nanozymes act as a localized “drug factory”, enabling the innate immune system to fight cancer with minimized side effects. Translation to in vivo application will take advantage of the prodrug design, allowing pro-imiquimod to be injected intravenously due to the reduced likelihood of non-specific inflammation. The re-polarization of M2-like TAMs for cancer immunotherapy can also be coupled with in situ chemotherapy to provide immuno-chemotherapy for enhanced therapeutic effects.Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
Xianzhi Zhang: conceptualization, methodology, investigation, formal analysis, project administration, data curation, writing-original draft, visualization, writing-review & editing. Yuanchang Liu: conceptualization, methodology, investigation, formal analysis, data curation, writing-original draft. Mingdi Jiang: methodology, investigation, formal analysis, visualization, writing-review & editing. Javier A. Mas-Rosario: methodology, investigation, formal analysis. Stefano Fedeli: investigation, formal analysis, visualization. Roberto Cao-Milan: conceptualization, investigation. Liang Liu: investigation, visualization. Kyle J. Winters: Investigation. Cristina-Maria Hirschbiegel: investigation, visualization, writing-review & editing. Ahmed Nabawy: investigation. Rui Huang: investigation. Michelle E. Farkas: funding acquisition, resources, methodology, supervision, writing-review & editing. Vincent M. Rotello: conceptualization, funding acquisition, resources, methodology, supervision, project administration, visualization, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by R01 EB022641 (to V. M. R.). J. A. M.-R. was supported by a Northeast Alliance for Graduate Education and the Professoriate (NEAGEP) fellowship from the STEM Diversity Institute at UMass Amherst, and a fellowship from the Chemistry-Biology Interface (CBI) Training Program (National Research Service Award (T32 GM008515) from the National Institutes of Health (NIH)). C.-M. H. was partially supported by a fellowship from the University of Massachusetts as part of the CBI Training Program (National Research Service Award T32 GM139789). The authors thank Dr Amy S. Burnside from Flow Cytometry Core Facility at UMass Amherst, and Dr James Chambers from Light Microscopy Core Facility at UMass Amherst. The authors also thank Dr Jessie Mager from the Department of Veterinary and Animal Science and Dr Hang Xiao from the Department of Food Science, UMass Amherst for the donation of C57/B6J mice for the isolation of primary bone marrow-derived macrophages.References
- Y. Liu, J. Hardie, X. Zhang and V. M. Rotello, Semin. Immunol., 2017, 34, 25–32 CrossRef CAS.
- D. C. Luther, R. Huang, T. Jeon, X. Zhang, Y. W. Lee, H. Nagaraj and V. M. Rotello, Adv. Drug Delivery Rev., 2020, 156, 188–213 CrossRef CAS PubMed.
- P. J. Murray and T. A. Wynn, Nat. Rev. Immunol., 2011, 11, 723–737 CrossRef CAS.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J.-L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogeln and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS.
- D. M. Mosser and J. P. Edwards, Nat. Rev. Immunol., 2008, 8, 958–969 CrossRef CAS PubMed.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS.
- G. A. Duque and A. Descoteaux, Front. Immunol., 2014, 5, 491 Search PubMed.
- A. Mantovani, P. Allavena, F. Marchesi and C. Garlanda, Nat. Rev. Drug Discovery, 2022, 21, 799–820 CrossRef CAS PubMed.
- E. Y. Lin and J. W. Pollard, Cancer Res., 2007, 67, 5064–5066 CrossRef CAS.
- A. Mantovani, S. Sozzani, M. Locati, P. Allavena and A. Sica, Trends Immunol., 2002, 23, 549–555 CrossRef CAS PubMed.
- A. Mantovani, B. Bottazzi, F. Colotta, S. Sozzani and L. Ruco, Immunol. Today, 1992, 13, 265–270 CrossRef CAS.
- R. A. Franklin, W. Liao, A. Sarkar, M. V. Kim, M. R. Bivona, K. Liu, E. G. Pamer and M. O. Li, Science, 2014, 344, 921–925 CrossRef CAS PubMed.
- R. Noy and J. W. Pollard, Immunity, 2014, 41, 49–61 CrossRef CAS.
- I. Larionova, G. Tuguzbaeva, A. Ponomaryova, M. Stakheyeva, N. Cherdyntseva, V. Pavlov, E. Choinzonov and J. Kzhyshkowska, Front. Oncol., 2020, 10, 1–34 CrossRef.
- P. Pathria, T. L. Louis and J. A. Varner, Trends Immunol., 2019, 40, 310–327 CrossRef CAS.
- S. P. Arlauckas, C. S. Garris, R. H. Kohler, M. Kitaoka, M. F. Cuccarese, K. S. Yang, M. A. Miller, J. C. Carlson, G. J. Freeman, R. M. Anthony, R. Weissleder and M. J. Pittet, Sci. Transl. Med., 2017, 9, eaal3604 CrossRef PubMed.
- A. Rodriguez-Garcia, R. C. Lynn, M. Poussin, M. A. Eiva, L. C. Shaw, R. S. O'Connor, N. G. Minutolo, V. Casado-Medrano, G. Lopez, T. Matsuyama and D. J. Powell, Nat. Commun., 2021, 12, 1–17 CrossRef.
- O. C. Olson, H. Kim, D. F. Quail, E. A. Foley and J. A. Joyce, Cell Rep., 2017, 19, 101–113 CrossRef CAS PubMed.
- Q. Dai, S. Wilhelm, D. Ding, A. M. Syed, S. Sindhwani, Y. Zhang, Y. Y. Chen, P. Macmillan and W. C. W. Chan, ACS Nano, 2018, 12, 8423–8435 CrossRef CAS PubMed.
- M. De Palma and C. E. Lewis, Cancer Cell, 2013, 23, 277–286 CrossRef CAS.
- M. Xiao, J. He, L. Yin, X. Chen, X. Zu and Y. Shen, Front. Immunol., 2021, 12, 799428 CrossRef CAS.
- D. G. DeNardo and B. Ruffell, Nat. Rev. Immunol., 2019, 19, 369–382 CrossRef CAS PubMed.
- Q. Jin, Z. Liu and Q. Chen, J. Controlled Release, 2021, 329, 882–893 CrossRef CAS.
- J. Liu, Q. Chen, L. Feng and Z. Liu, Nano Today, 2018, 21, 55–73 CrossRef CAS.
- C. Zhao, X. Pang, Z. Yang, S. Wang, H. Deng and X. Chen, J. Controlled Release, 2022, 341, 272–284 CrossRef CAS PubMed.
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, 1–20 Search PubMed.
- S. Bhagchandani, J. A. Johnson and D. J. Irvine, Adv. Drug Delivery Rev., 2021, 175, 113803 CrossRef CAS PubMed.
- R. Moore, J. E. Edwards, J. Hopwood and D. Hicks, BMC Infect. Dis., 2001, 1, e3 CrossRef.
- R. I. Ceilley and J. Q. Del Rosso, Int. J. Dermatol., 2006, 45, 489–498 CrossRef.
- P. Kamath, E. Darwin, H. Arora and K. Nouri, Clin. Drug Invest., 2018, 38, 883–899 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255–270 CrossRef CAS.
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS.
- R. Walther, J. Rautio and A. N. Zelikin, Adv. Drug Delivery Rev., 2017, 118, 65–77 CrossRef CAS PubMed.
- K. A. Ryu, L. Stutts, J. K. Tom, R. J. Mancini and A. P. Esser-Kahn, J. Am. Chem. Soc., 2014, 136, 10823–10825 CrossRef CAS.
- J. D. Hantho, T. A. Strayer, A. E. Nielsen and R. J. Mancini, ChemMedChem, 2016, 11, 2496–2500 CrossRef CAS.
- H. Lei, J. H. Kim, S. Son, L. Chen, Z. Pei, Y. Yang, Z. Liu, L. Cheng and J. S. Kim, ACS Nano, 2022, 16, 10979–10993 CrossRef CAS PubMed.
- T. Liang, Z. Chen, H. Li and Z. Gu, Trends Chem., 2022, 4, 157–168 CrossRef CAS.
- X. Zhang, R. Huang, S. Gopalakrishnan, R. Cao-milán and V. M. Rotello, Trends Chem., 2019, 1, 90–98 CrossRef CAS.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- J. Wu, X. Wang, Q. Wang, Z. Lou, S. Li, Y. Zhu, L. Qin and H. Wei, Chem. Soc. Rev., 2019, 48, 1004–1076 RSC.
- M. O. N. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef.
- D. P. Nguyen, H. T. H. Nguyen and L. H. Do, ACS Catal., 2021, 11, 5148–5165 CrossRef CAS.
- M. Martínez-calvo and J. L. Mascareñas, Coord. Chem. Rev., 2018, 359, 57–79 CrossRef.
- Y. Liu and Y. Bai, ACS Appl. Bio Mater., 2020, 3, 4717–4746 CrossRef CAS PubMed.
- B. Lozhkin and T. R. Ward, Bioorg. Med. Chem., 2021, 45, 116310 CrossRef CAS.
- S. Bose, A. H. Ngo and L. H. Do, J. Am. Chem. Soc., 2017, 139, 8792–8795 CrossRef CAS.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef.
- T. Völker and E. Meggers, Chembiochem, 2017, 18, 1083–1086 CrossRef.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef.
- C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascareñas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS.
- X. Zhang, S. Fedeli, S. Gopalakrishnan, R. Huang, A. Gupta, D. C. Luther and V. M. Rotello, ChemBioChem, 2020, 21, 2759–2763 CrossRef CAS.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- X. Zhang, R. F. Landis, P. Keshri, R. Cao-Milán, D. C. Luther, S. Gopalakrishnan, Y. Liu, R. Huang, G. Li, M. Malassiné, I. Uddin, B. Rondon and V. M. Rotello, Adv. Healthcare Mater., 2020, 10, 2001627 CrossRef.
- X. Zhang, Y. Liu, S. Gopalakrishnan, L. Castellanos-Garcia, G. Li, M. Malassiné, I. Uddin, R. Huang, D. Luther, R. Vachet and V. Rotello, ACS Nano, 2020, 14, 4767–4773 CrossRef CAS PubMed.
- S. Fedeli, J. Im, S. Gopalakrishnan, J. L. Elia, A. Gupta, D. Kim and V. M. Rotello, Chem. Soc. Rev., 2021, 50, 13467–13480 RSC.
- W. Wang, X. Zhang, R. Huang, C. M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed.
- X. Zhang, S. Lin, R. Huang, A. Gupta, S. Fedeli, R. Cao-Milán, D. C. Luther, Y. Liu, M. Jiang, G. Li, B. Rondon, H. Wei and V. M. Rotello, J. Am. Chem. Soc., 2022, 144, 12893–12900 CrossRef CAS PubMed.
- M. Sancho-albero, B. Rubio-ruiz, A. M. Pérez-lópez, V. Sebastián, P. Martín-duque, M. Arruebo, J. Santamaría and A. Unciti-broceta, Nat. Catal., 2019, 2, 864–872 CrossRef CAS PubMed.
- S. Eda, I. Nasibullin, K. Vong, N. Kudo, M. Yoshida, A. Kurbangalieva and K. Tanaka, Nat. Catal., 2019, 2, 780–792 CrossRef CAS.
- C. Adam, T. L. Bray, A. M. Pérez-López, E. H. Tan, B. Rubio-Ruiz, D. J. Baillache, D. R. Houston, M. J. Salji, H. Y. Leung and A. Unciti-Broceta, J. Med. Chem., 2022, 65, 552–561 CrossRef CAS.
- R. Das, J. Hardie, B. P. Joshi, X. Zhang, A. Gupta, D. C. Luther, S. Fedeli, M. E. Farkas and V. M. Rotello, JACS Au, 2022, 2, 1679–1685 CrossRef CAS PubMed.
- Z. Chen, H. Li, Y. Bian, Z. Wang, G. Chen, X. Zhang, Y. Miao, D. Wen, J. Wang, G. Wan, Y. Zeng, P. Abdou, J. Fang, S. Li, C.-J. Sun and Z. Gu, Nat. Nanotechnol., 2021, 16, 933–941 CrossRef CAS.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, ACS Nano, 2018, 12, 12814–12826 CrossRef CAS PubMed.
- Y. You, Q. Deng, Y. Wang, Y. Sang, G. Li, F. Pu, J. Ren and X. Qu, Nat. Commun., 2022, 13, 1459 CrossRef CAS.
- Y. Wei, S. Wu, Z. Liu, J. Niu, Y. Zhou, J. Ren and X. Qu, Mater. Today, 2022, 56, 16–28 CrossRef CAS.
- Y. You, H. Liu, J. Zhu, Y. Wang, F. Pu, J. Ren and X. Qu, Chem. Sci., 2022, 13, 7829–7836 RSC.
- J. Hardie, J. M. Makabenta, A. Gupta, R. Huang, R. Cao-Milán, R. Goswami, X. Zhang, P. Abdulpurkar, M. E. Farkas and V. M. Rotello, Mater. Horiz., 2022, 9, 1489–1494 RSC.
- R. Huang, C. Li, R. Cao-milan, L. D. He, J. M. Makabenta, X. Zhang, E. Yu and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 10723–10729 CrossRef CAS PubMed.
- R. Cao-milan, S. Gopalakrishnan, L. D. He, R. Huang, L. Wang, L. Castellanos, D. C. Luther, R. F. Landis, J. M. V Makabenta, C. Li, X. Zhang, F. Scaletti, R. W. Vachet and V. M. Rotello, Chem, 2020, 6, 1–12 Search PubMed.
- A. Gupta, R. Das, J. M. Makabenta, A. Gupta, X. Zhang, T. Jeon, R. Huang, Y. Liu, S. Gopalakrishnan, R. C. Milán and V. M. Rotello, Mater. Horiz., 2021, 8, 3424–3431 RSC.
- J. Niu, L. Wang, T. Cui, Z. Wang, C. Zhao, J. Ren and X. Qu, ACS Nano, 2021, 15, 15841–15849 CrossRef CAS.
- J. Niu, C. Zhao, C. Liu, J. Ren and X. Qu, Chem. Mater., 2021, 33, 8052–8058 CrossRef CAS.
- Z. Du, C. Liu, H. Song, P. Scott, Z. Liu, J. Ren and X. Qu, Chem, 2020, 6, 2060–2072 CAS.
- S. T. Kim, K. Saha, C. Kim and V. M. Rotello, Acc. Chem. Res., 2013, 46, 681–691 CrossRef CAS PubMed.
- J. W. E. Worrall, A. Verma, H. Yan and V. M. Rotello, Chem. Commun., 2006, 2338–2340 RSC.
- X. Zhang, Y. Liu, J. Doungchawee, L. J. Castellanos-García, K. N. Sikora, T. Jeon, R. Goswami, S. Fedeli, A. Gupta, R. Huang, C.-M. Hirschbiegel, R. Cao-Milán, P. K. D. Majhi, Y. A. Cicek, L. Liu, D. J. Jerry, R. W. Vachet and V. M. Rotello, J. Controlled Release, 2023, 357, 31–39 CrossRef CAS.
- R. Huang, C. M. Hirschbiegel, X. Zhang, A. Gupta, S. Fedeli, Y. Xu and V. M. Rotello, ACS Appl. Mater. Interfaces, 2022, 14, 31594–31600 CrossRef CAS PubMed.
- M. Their, M. P. State, I. Spera, S. Ricardo, M. Favia, A. Menga, F. C. Venegas, R. Angioni, F. Munari, M. Lanza, A. Campanella, C. L. Pierri, M. Canton and A. Castegna, Cancers, 2021, 13, 5478 Search PubMed.
- M. Canel, D. Taggart, A. H. Sims, D. W. Lonergan, I. C. Waizenegger and A. Serrels, Elife, 2020, 9, e48092 CrossRef CAS.
- M. Ray, Y. W. Lee, J. Hardie, R. Mout, G. Yeşilbag Tonga, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2018, 29, 445–450 CrossRef CAS PubMed.
- L. Tian, M. Shao, Y. Gong, T. Wei, Y. Zhu, Y. Chao and Z. Liu, Bioconjugate Chem., 2022, 33, 343–352 CrossRef CAS.
- X. Zhou, X. Liu and L. Huang, Adv. Funct. Mater., 2021, 31, 2006220 CrossRef CAS PubMed.
- J. Hardie, J. A. Mas-Rosario, S. Ha, E. M. Rizzo and M. E. Farkas, Pharmacol. Res., 2019, 148, 104452 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06431j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |