
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMesoionic carbene-based self-assembled monolayers on gold†
Dianne S.
Lee
 ab,
Ishwar
Singh
ab,
Alex J.
Veinot
bcd,
Mark D.
Aloisio
ab,
Justin T.
Lomax
bcd,
Paul J.
Ragogna
bc and
Cathleen M.
Crudden
*ab
ab,
Ishwar
Singh
ab,
Alex J.
Veinot
bcd,
Mark D.
Aloisio
ab,
Justin T.
Lomax
bcd,
Paul J.
Ragogna
bc and
Cathleen M.
Crudden
*ab
aDepartment of Chemistry, Queen's University, 90 Bader Lane, Kingston, Ontario K7L 3N6, Canada. E-mail: cruddenc@chem.queensu.ca
bCarbon to Metal Coating Institute, C2MCI, Queen's University, 90 Bader Lane, Kingston, Ontario K7L 4V1, Canada
cDepartment of Chemistry, Western University, London, Ontario N6A 3K7, Canada
dSurface Science Western, 999 Collip Cir, London, Ontario N6G 0J3, Canada
First published on 15th December 2023
Abstract
N-Heterocyclic carbenes (NHC) have been widely studied as ligands for surface chemistry, and have shown advantages compared to existing ligands (e.g. thiols). Herein, we introduce mesoionic carbenes (MICs) as a new type of surface ligand. MICs exhibit higher σ-donor ability compared to typical NHCs, yet they have received little attention in the area of surface chemistry. The synthesis of MICs derived from imidazo[1,2-a]pyridine was established and fully characterized by spectroscopic methods. The self-assembly of these MICs on gold was analyzed by X-ray photoelectron spectroscopy (XPS). Additionally, XPS was used to compare bonding ability in MICs compared to the typical NHCs. These results show that MIC overlayers on gold are robust, resistant to replacement by NHCs, and may be superior to NHCs for applications that require even greater levels of robustness.
Introduction
Since the first report of N-heterocyclic carbenes (NHCs) as ligands for gold surfaces by Siemeling and co-workers,1 the use of these ligands on metallic1–23 and non-metallic surfaces,2,3 has attracted considerable attention, with Au substrates being most commonly employed.1,4–23 NHC-based self-assembled monolayers (SAMs) have been integrated into molecular electronics,24–26 and biosensors,16,17,22,27 and employed in surface patterning,13,28 illustrating the importance of this emerging class of ligands for planar metal surfaces. The stronger surface binding energies of NHCs compared to the other classic ligands, such as thiolates or phosphines18,29 and their high potential for tunability30–32 positions NHCs as ideal candidates for producing the next generation of nanomaterials.Despite the high potential for structural and electronic variability with this class of ligand, most studies have focused on classical Arduengo-type NHCs,18,19,33 with select exceptions of 1,2,4-triazolylidenes,18 cyclic alkyl amino carbenes,34,35 and cyclopropenylidenes.36 Mesoionic carbenes (MICs)37,38 are superior σ-donors compared to NHCs and are powerful ligands in organometallic chemistry. However, apart from the work of the Nazemi group highlighting the use of MICs on nanoparticles,39,40 MICs are absent in the field of surface chemistry. Given the lack of experimental studies with this important class of carbene, and the possibility for enhanced properties of the resulting SAMs, we set out to explore these structural variants as ligands for self-assembled monolayers on gold.
To exclude steric issues, we focused on the imidazo[1,2-a]pyridine-type MICiPr as our target molecule, which is isostructural to the well-studied ligand NHCiPr (Fig. 1b).8,11,18 Tolman electronic parameter measurements indicate that MICs such as MICiPr are stronger σ-donors than many NHCs.41 Crystallographic analyses of isostructural Pd complexes of NHCs and MICs, reveal identical steric environments at the Pd center.42,43 Therefore, differences between the bonding of MICs and NHCs on gold should be isolated only to electronic effects. Synthetic routes to imidazo[1,2-α]pyridines with secondary aliphatic groups in the 3-position remain underdeveloped. Routes to branched 1,3-diaklylated imidazo[1,2-α]pyridinium cations have not been reported in the literature. A sulfur-promoted oxidative cyclization reaction was recently disclosed using readily available aldehydes and 2-aminopyridine.44 We examined this route, but the products of this reaction were invariably contaminated with sulfur impurities.45 To prevent surface contamination by sulfur, an alternative synthetic route was developed (Scheme 1).
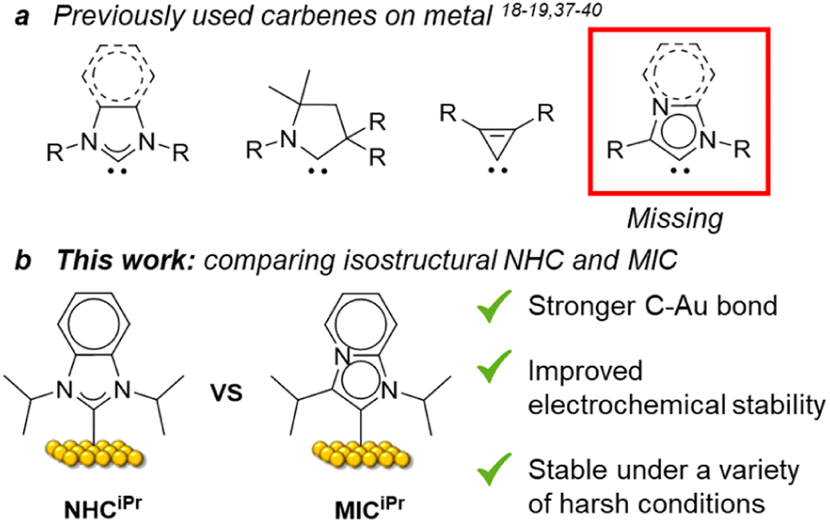 | ||
| Fig. 1 (a) Previously used carbenes on metal surfaces, (b) molecular structures and the comparison between isostructural NHC and MIC self-assembled monolayers on Au surfaces. | ||
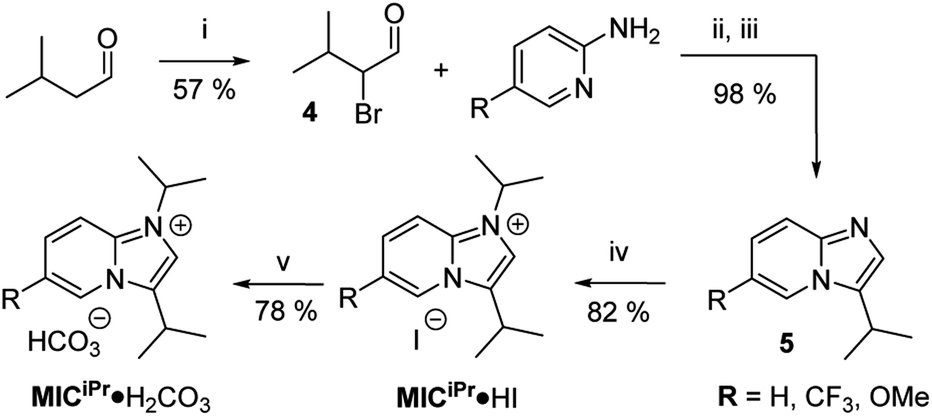 | ||
| Scheme 1 Synthesis of MICiPr·H2CO3. (i) NBS, L-proline in DCM, 0 °C → RT; (ii) anhyd. EtOH Δ; (iii) aq. NaHCO3; (iv) iPr-I, MeCN, Δ; (v) HCO3− resin, MeOH, RT. | ||
Results and discussion
The desired MIC precursor MICiPr·HI was prepared using classical α-halocarbonyl chemistry.46 Isovaleraldehyde was α-brominated using N-bromosuccinimide to afford 4 in 57% yield (Scheme 1). Reaction times longer than 2 h resulted in lower yields because of the reactive nature of α-halocarbonyl species, and thus 4 was used immediately without isolation. Reaction between an excess of 4 and 2-aminopyridine derivatives in refluxing ethanol afforded imidazo[1,2-a]pyridinium bromide salts 5·HBr in up to 98% yield. Excesses of 4 were needed to prevent contamination with unreacted 2-aminopyridine. Analytically pure 5·HBr could be isolated directly from the reaction mixture by first removing ethanol in vacuo, then suspending the resulting solids in diethyl ether and vacuum filtration. The route was amenable to the introduction of substituents on the backbone as needed (R = H, CF3, OMe).Alkylation of 5 with 2-iodopropane afforded MICiPr·HI in 82% yield. Reaction between MICiPr·HI and strong bases (NaN(SiMe3)2, LiN(iPr)2, KH) resulted in decomposition, and attempts to trap the transient carbene with CO2 to isolate a carboxylate derivative were unsuccessful. These difficulties led us to prepare the hydrogen carbonate salt MICiPr·H2CO3, which could be isolated in 78% yield as a monohydrate salt after resin exchange.17 Imidazolium hydrogen carbonate salts are valuable air-stable precursors for generating NHC SAMs in solution and ultra-high vacuum (UHV).5,6,13,17,25,27 Therefore, we expected that MICiPr·H2CO3 would be similarly effective for producing MIC SAMs, however, initial attempts to prepare films by vapour phase deposition of MICiPr·H2CO3 using techniques developed for NHCiPr proved unsuccessful.
Thermogravimetric analysis (TGA) of MICiPr·H2CO3 revealed that CO2 and H2O are liberated around 160 °C, a higher temperature than NHCiPr·H2CO3, which cleanly generates NHCiPr between 100–120 °C.47 This higher activation temperature is presumably required due to the higher pKa of the MIC precursor.48 Beyond 160 °C, volatilization of MICiPr·H2CO3 occurs, but is slower than NHCiPr·H2CO3 and has a temperature profile that suggests decomposition accompanies this process (Fig. S1†).
We next attempted to prepare SAMs of MICiPr on Au by immersing Au/Si samples in a 10 mM MeOH solution of MICiPr·H2CO3 at room temperature for 24 h.17 Although N 1s XPS analysis suggested that some deposition had occurred under these conditions (Fig. S10†), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) showed no current suppression or increase in impedance, respectively (Fig. 2a and b), suggesting minimal deposition. However, increasing the deposition temperature to 50 °C and time to 48 h yielded SAMs with significant reduction in current and notable increase in resistance, as determined by CV and EIS, respectively, consistent with the formation of densely packed SAMs on the surface (Fig. 2a–c).
 | ||
| Fig. 2 Optimization of deposition conditions for MICiPr·H2CO3 on Au using CV (a) and (b) EIS. Optimal conditions determined to be 10 mM MICiPr·H2CO3 in MeOH at 50 °C for 48 h. (c) Optimization of time required for MICiPr·H2CO3 deposition on gold by examination of CV curves. | ||
XPS analysis was employed to provide more insight into monolayer formation. We examined the XPS spectrum of the powdered MICiPr·H2CO3 starting material (Fig. S8†), which was characterized by two signals in equal intensity assigned to the pyridyl and imine groups, at 401.6 eV and 400.6 eV respectively. XP spectra of SAMs resulting from deposition of MICiPr·H2CO3 under optimized conditions also displayed two signals of equal intensity centered at 400.7 eV and 399.2 eV.49 The shift in the binding energy and increase in the FWHM for both signals suggests a change in the electronics of the ligand upon binding to gold, and data strongly support the formation of an MICiPr SAM under these conditions.
SAMs of MICiPr were subjected to a variety of conditions including pH extremes (2, 12), refluxing water and 1% hydrogen peroxide for 24 hours. During these treatments, the MICiPr SAM showed minimal changes to the overall monolayer, as illustrated when comparing XPS signals in high resolution scans of C (1s), O (1s) and N (1s) (Fig. S11–S15†). To further probe the stability of these SAMs, we exposed MICiPr SAMs and NHCiPr SAMs to pH 12 for five days. Analysis of the surface via time-of-flight secondary ion mass spectrometry (ToF-SIMS) showed virtually no loss of MICiPr on the surface even after exposure to these harsh conditions, while NHCiPr was no longer observed (Fig. 3). This illustrates the improved robustness of SAMs designed from MICiPr under these extreme conditions.
 | ||
| Fig. 3 ToF-SIMS data of MICiPr and NHCiPr before and after stability test under pH 12 for 5 days. (a) MICiPr before exposure; (b) MICiPr after exposure; (c) NHCiPr before exposure; (d) NHCiPr after exposure. | ||
To study the propensity for the two types of carbene to deposit on a bare gold surface, competition experiments were carried out at 50 °C for 48 hours with 10 mM MICiPr·H2CO3 and 10 mM CF3·NHCiPr·H2CO3 methanolic solution. To determine the surface coverage ratio between MICiPr and CF3·NHCiPr, the trifluoromethyl unit was employed as an XPS reporter along with N 1s signals (Fig. S18†). The average ratio between MICiPr and CF3·NHCiPr was calculated to be 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Table S1†), demonstrating the preferential formation of SAMs from MICiPr in comparison with CF3·NHCiPr. The inclusion of a CF3 substituent had no effect on the binding strength of CF3·NHCiPr as shown by simple replacement tests between NHCiPr and CF3·NHCiPr (Fig. S19†), since the final monolayer consists of both NHCiPr and CF3·NHCiPr in a roughly 1:1 ratio. This ratio implies that the binding strengths of NHCiPr and CF3·NHCiPr for SAM formation are comparable.
:1 (Table S1†), demonstrating the preferential formation of SAMs from MICiPr in comparison with CF3·NHCiPr. The inclusion of a CF3 substituent had no effect on the binding strength of CF3·NHCiPr as shown by simple replacement tests between NHCiPr and CF3·NHCiPr (Fig. S19†), since the final monolayer consists of both NHCiPr and CF3·NHCiPr in a roughly 1:1 ratio. This ratio implies that the binding strengths of NHCiPr and CF3·NHCiPr for SAM formation are comparable.
We then set out to compare the ability of one NHC to replace a preformed SAM of the other carbene. SAMs of MICiPr were prepared on Au/Si surfaces as previously described, and then treated with CF3·NHCiPr·H2CO3 using the trifluoromethyl unit as an XPS reporter (Fig. 4). Typically, SAMs composed of benzannulated NHCs such as NHCiPr are deposited over 24 h at room temperature, and so we employed these conditions to test the stability of MICiPrvs. replacement with NHCiPr. After exposure to these conditions, no NHC was incorporated as determined by analysis of fluorine content in the F 1s region (Fig. 4 and Table 1, entry 1). This indicates that the MICiPr SAM resisted the incorporation of CF3·NHCiPr within the error of XPS measurements.
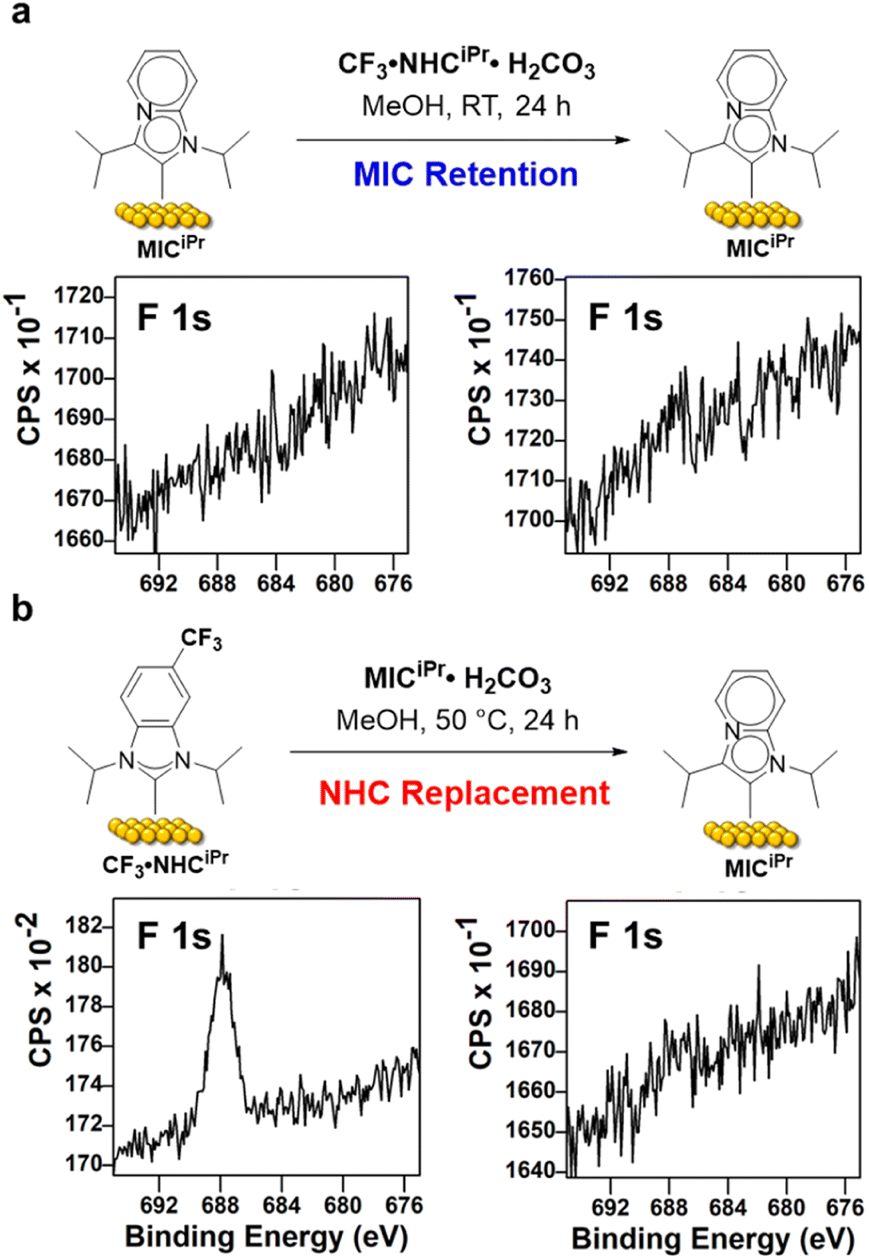 | ||
| Fig. 4 F (1s) XPS regions of the replacement experiments between MICiPr and CF3·NHCiPr on Au surfaces. (a) MICiPr replacement attempt with CF3·NHCiPr·H2CO3. (b) CF3·NHCiPr replacement with MICiPr·H2CO3. | ||
| Entry | Starting SAM | Replacement precursor | Temp. | Time | XPS (F:N) |
|---|---|---|---|---|---|
| 1 | MICiPr | CF3·NHCiPr·H2CO3 | RT | 24 h | No fluorine |
| 2 | CF3·NHCiPr | MICiPr·H2CO3 | 50 °C | 24 h | No fluorine |
| 3 | MICiPr | CF3·NHCiPr·H2CO3 | RT | 48 h | 0.7:2 |
| 4 | MICiPr | CF3·NHCiPr·H2CO3 | 50 °C | 48 h | 0.9:2 |
To test the reverse reaction, SAMs prepared from CF3·NHCiPr were treated with MICiPr·H2CO3 and the loss or retention of fluorine was assessed by XPS (Fig. 4 and Table 1, entry 2). Under these conditions, we observed the complete removal of fluorine from the surface by XPS (Fig. 4 and Table 1, entry 2).
However, the need for elevated temperatures and longer deposition times to ensure generation of MICiPr from its bicarbonate salt is problematic for a direct comparison, and so an additional set of experiments were performed in which MICiPr SAMs were treated with CF3·NHCiPr·H2CO3 at elevated temperatures and longer periods of time (Table 1, entries 3 and 4). Under these more forcing conditions, fluorine was observed by XPS analysis, but the replacement of MIC for NHC was minimal. Treatment of the MICiPr SAM with CF3·NHCiPr·H2CO3 at RT for 48 h resulted in the incorporation of one CF3·NHCiPr for every six MICiPr units (Table 1; entries 3 and 4). At 50 °C for 48 h, one CF3·NHCiPr was incorporated for every five MICiPr units on Au. These results support the conclusion that MICs form more robust SAMs than those generated from typical NHCs, presumably due to stronger carbon–metal bonds on surfaces, consistent with the well-established molecular chemistry.37,38
Contact angle measurements were also performed for Au/Si substrates functionalized by MICiPr and NHCiPr (Fig. S21 and Table S2†). Compared to bare Au (67 ± 2°), the surface hydrophobicity does not change substantially upon the surface adsorption of MICiPr (72 ± 2°) and NHCiPr (71 ± 2°).
Conclusions
A straightforward synthetic route to the MIC precursor MICiPr·H2CO3 has been developed and its deposition on Au studied. Under optimized conditions, MICiPr·H2CO3 was deposited on Au surfaces, resulting in the formation of a new MIC-based SAM. Solution deposition conditions were optimized using electrochemical methods to monitor the successful formation of a stable monolayer. Monolayer stability was confirmed by multiple CV cycles and extreme potential range, concluding that solution deposition at 50 °C for 48 h was optimal.The effect of the σ-donor ability of MICsvs.NHCs was demonstrated by a direct comparison of the two types of SAMs. This was accomplished through stability studies under extreme conditions, competitive deposition, and exchange studies by treatment of preformed monolayers with carbene precursors. These studies showed that MIC-based SAMs resist NHC incorporation, and NHC-based SAMs are replaced with MICs. When co-deposited, MICs out compete NHCs. Finally, MIC-based SAMs are more robust to long term (5 days) immersion in base. This study serves as the foundation for expanding the library of carbenes currently applied to surfaces for the next generation of carbene-based monolayers and providing more robust SAMs for use under harsher conditions. Future work in our lab will include finding appropriate conditions that allow for UHV studies of SAMs based on MICs, including spectroscopic and microscopic analyses.
Experimental
General methods
Unless otherwise stated, all solvents (including NMR solvents) and reagents were obtained from Sigma-Aldrich and used without further purification. Ethanol (anhydrous grade, Greenfield Global Commercial Alcohols) was stored over oven dried (150 °C) molecular sieves (3 Å, Alfa Aesar) prior to use. Using previously established methods,1 hydrogen carbonate exchange resin was prepared by treating Amberlyst A26 hydroxide resin treated with carbon dioxide prior to use.Author contributions
This research was conceived and supervised by C. M. C. A. J. V. and I. S. performed the original synthesis and characterization of MICs. D. S. L. performed all of the electrochemical experiments. D. S. L. prepared XPS samples and analysed the samples with I. S. D. S. L. prepared ToF-SIMS samples and J. T. L. ran and analysed the samples. M. D. A. optimized and further characterized MICs. The manuscript was written and edited by D. S. L., A. J. V., I. S. and C. M. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Queen's University, Canada Foundation for Innovation (CFI; CFI-33355), the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN/04667-2016; RGPIN/04377-2021), the New Frontiers in Research Fund – Transformation Program (NFRFT-2020-00573), and the Semiconductor Research Corporation are thanked for the financial support of this work. D. S. L. thanks Dr Gabriele Schatte for the assistance with XPS acquisition. A. J. V. thanks NSERC for the Vanier Scholarship and Banting Postdoctoral Fellowship. I. S. thanks the Ontario government for an Ontario Graduate Scholarship. J. T. L. thanks Drs Mark Biesinger and Heng-Yong Nie at Surface Science Western. C. M. C. thanks Queen's University, the Canada Foundation for Innovation, the Natural Sciences and Engineering Research Council of Canada (NSERC), and the New Frontiers in Research Fund – Transformation program.Notes and references
- T. Weidner, J. E. Baio, A. Mundstock, C. Große, S. Karthäuser, C. Bruhn and U. Siemeling, Aust. J. Chem., 2011, 64, 1177–1179 CrossRef CAS.
- M. Franz, S. Chandola, M. Koy, R. Zielinski, H. Aldahhak, M. Das, M. Freitag, U. Gerstmann, D. Liebig, A. K. Hoffmann, M. Rosin, W. G. Schmidt, C. Hogan, F. Glorius, N. Esser and M. Dähne, Nat. Chem., 2021, 13, 828 CrossRef CAS.
- A. V. Zhukhovitskiy, M. G. Mavros, K. T. Queeney, T. Wu, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 8639–8652 CrossRef CAS.
- C. R. Larrea, C. J. Baddeley, M. R. Narouz, N. J. Mosey, J. H. Horton and C. M. Crudden, ChemPhysChem, 2017, 18, 3536–3539 CrossRef CAS.
- S. Amirjalayer, A. Bakker, M. Freitag, F. Glorius and H. Fuchs, Angew. Chem., Int. Ed., 2020, 59, 21230–21235 CrossRef CAS.
- A. Bakker, A. Timmer, E. Kolodzeiski, M. Freitag, H. Y. Gao, H. Mönig, S. Amirjalayer, F. Glorius and H. Fuchs, J. Am. Chem. Soc., 2018, 140, 11889–11892 CrossRef CAS PubMed.
- G. Wang, A. Rühling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H.-J. Gao, A. Timmer, H.-Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2017, 9, 152–156 CrossRef CAS PubMed.
- G. Lovat, E. A. Doud, D. Lu, G. Kladnik, M. S. Inkpen, M. L. Steigerwald, D. Cvetko, M. S. Hybertsen, A. Morgante, X. Roy and L. Venkataraman, Chem. Sci., 2019, 10, 930–935 RSC.
- L. Jiang, B. Zhang, G. Médard, A. P. Seitsonen, F. Haag, F. Allegretti, J. Reichert, B. Kuster, J. V. Barth and A. C. Papageorgiou, Chem. Sci., 2017, 8, 8301–8308 RSC.
- E. Angove, F. Grillo, H. A. Früchtl, A. J. Veinot, I. Singh, J. H. Horton, C. M. Crudden and C. J. Baddeley, J. Phys. Chem. Lett., 2022, 13, 2051–2056 CrossRef CAS.
- A. Inayeh, R. R. K. Groome, I. Singh, A. J. Veinot, F. C. de Lima, R. H. Miwa, C. M. Crudden and A. B. McLean, Nat. Commun., 2021, 12, 4034 CrossRef CAS.
- I. Singh, D. S. Lee, S. Huang, H. Bhattacharjee, W. Xu, J. F. McLeod, C. M. Crudden and Z. She, Chem. Commun., 2021, 57, 8421 RSC.
- Z. She, M. R. Narouz, C. A. Smith, A. MacLean, H.-P. Loock, H.-B. Kraatz and C. M. Crudden, Chem. Commun., 2020, 56, 1275–1278 RSC.
- A. J. Veinot, A. Al-Rashed, J. D. Padmos, I. Singh, D. S. Lee, M. R. Narouz, P. A. Lummis, C. J. Baddeley, C. M. Crudden and J. H. Horton, Chem.–Eur. J., 2020, 26, 11431–11434 CrossRef CAS.
- Z. Li, M. R. Narouz, K. Munro, B. Hao, C. M. Crudden, J. H. Horton and H. Hao, ACS Appl. Mater. Interfaces, 2017, 9, 39223–39234 CrossRef CAS.
- Z. Li, K. Munro, I. I. Ebralize, M. R. Narouz, J. D. Padmos, H. Hao, C. M. Crudden and J. H. Horton, Langmuir, 2017, 33, 13936–13944 CrossRef CAS PubMed.
- C. M. Crudden, J. H. Horton, M. R. Narouz, Z. Li, C. A. Smith, K. Munro, C. J. Baddeley, C. R. Larrea, B. Drevniok and B. Thanabalasingam, Nat. Commun., 2016, 7, 12654 CrossRef CAS PubMed.
- C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H.-B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. G. Mavros, T. Van Voorhis and J. A. Johnson, J. Am. Chem. Soc., 2013, 135, 7418–7421 CrossRef CAS PubMed.
- S. Dery, I. Berg, S. Kim, A. Cossaro, A. Verdini, L. Floreano, F. D. Toste and E. Gross, Langmuir, 2020, 36, 697–703 CrossRef CAS PubMed.
- S. Dery, S. Kim, G. Tomaschun, D. Haddad, A. Cossaro, A. Verdini, L. Floreano, T. Klüner, F. D. Toste and E. Gross, Chem.–Eur. J., 2019, 25, 15067–15072 CrossRef CAS PubMed.
- J. F. DeJesus, M. J. Trujillo, J. P. Camden and D. M. Jenkins, J. Am. Chem. Soc., 2018, 140, 1247–1250 CrossRef CAS.
- M. J. Trujillo, S. L. Strausser, J. C. Becca, J. F. DeJesus, L. Jensen, D. M. Jenkins and J. P. Camden, J. Phys. Chem. Lett., 2018, 9, 6779–6785 CrossRef CAS PubMed.
- G. Foti and H. Vázquez, Nanotechnology, 2016, 27, 125702 CrossRef.
- G. Foti and H. Vázquez, Beilstein J. Nanotechnol., 2017, 8, 2060–2068 CrossRef CAS PubMed.
- E. A. Doud, M. S. Inkpen, G. Lovat, E. Montes, D. W. Paley, M. L. Steigerwald, H. c. Vázquez, L. Venkataraman and X. Roy, J. Am. Chem. Soc., 2018, 140, 8944–8949 CrossRef CAS.
- R. M. Mayall, C. A. Smith, A. S. Hyla, D. S. Lee, C. M. Crudden and V. I. Birss, ACS Sens., 2020, 5, 2747 CrossRef CAS.
- D. T. Nguyen, M. Freitag, M. Körsgen, S. Lamping, A. Rühling, A. H. Schaefer, M. H. Siekman, H. F. Arlinghaus, W. G. van der Wiel and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 11465–11469 CrossRef CAS PubMed.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C.-T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Häkkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS.
- C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS.
- G. Kaur, R. L. Thimes, J. P. Camden and D. M. Jenkins, Chem. Commun., 2022, 58, 13188–13197 RSC.
- L. M. Sherman, S. L. Strausser, R. K. Borsari, D. M. Jenkins and J. P. Camden, Langmuir, 2021, 37, 5864–5871 CrossRef CAS.
- A. Bakker, M. Freitag, E. Kolodzeiski, P. Bellotti, A. Timmer, J. Ren, B. Schulze Lammers, D. Moock, H. W. Roesky, H. Mönig, S. Amirjalayer, H. Fuchs and F. Glorius, Angew. Chem., Int. Ed., 2020, 59, 13643–13646 CrossRef CAS.
- J. Ren, M. Freitag, Y. Gao, P. Bellotti, M. Das, B. Schulze Lammers, H. Mönig, Y. Zhang, C. G. Daniliuc, S. Du, H. Fuchs and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202115104 CrossRef CAS.
- E. A. Doud, R. L. Starr, G. Kladnik, A. Voevodin, E. Montes, N. P. Arasu, Y. Zang, P. Zahl, A. Morgante and L. Venkataraman, J. Am. Chem. Soc., 2020, 142, 19902–19906 CrossRef CAS PubMed.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS.
- D. T. H. Nguyen, M. Bélanger-Bouliga, L. R. Shultz, A. Maity, T. Jurca and A. Nazemi, Chem. Mater., 2021, 33, 9588–9600 CrossRef CAS.
- D. T. H. Nguyen, L. R. Shultz, T. Jurca and A. Nazemi, Langmuir, 2023, 39, 3204–3215 CrossRef CAS PubMed.
- G. Song, Y. Zhang and X. Li, Organometallics, 2008, 27, 1936–1943 CrossRef CAS.
- C.-H. Ke, B.-C. Kuo, D. Nandi and H. M. Lee, Organometallics, 2013, 32, 4775–4784 CrossRef CAS.
- M. Heckenroth, A. Neels, M. G. Garnier, P. Aebi, A. W. Ehlers and M. Albrecht, Chem.—Eur. J., 2009, 15, 9375–9386 CrossRef CAS.
- J. Tan, P. Ni, H. Huang and G.-J. Deng, Org. Biomol. Chem., 2018, 16, 4227–4230 RSC.
- Determined by elemental analysis (EA). EA was not reported in the published method.
- R. Adams and J. S. Dix, J. Am. Chem. Soc., 1958, 80, 4618–4620 CrossRef CAS.
- M. Fèvre, P. Coupillaud, K. Miqueu, J.-M. Sotiropoulos, J. Vignolle and D. Taton, J. Org. Chem., 2012, 77, 10135–10144 CrossRef PubMed.
- C. Barnett, M. L. Cole and J. B. Harper, ACS Omega, 2022, 7, 34657–34664 CrossRef CAS PubMed.
- J. F. Moulder and J. Chastain, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04720b |
| This journal is © The Royal Society of Chemistry 2024 |