
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto- and halochromism of spiropyran-based main-chain polymers†
Linh Duy
Thai
 abc,
Jochen A.
Kammerer
ab,
Hatice
Mutlu
*d and
Christopher
Barner-Kowollik
*abc
abc,
Jochen A.
Kammerer
ab,
Hatice
Mutlu
*d and
Christopher
Barner-Kowollik
*abc
aSchool of Chemistry and Physics, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
bCentre for Materials Science, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD 4000, Australia
cInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dInstitut de Science des Matériaux de Mulhouse, UMR 7361 CNRS/Université de Haute Alsace, 15 Rue Jean Starcky, Mulhouse Cedex, 68057, France. E-mail: hatice.mutlu@uha.fr
First published on 26th January 2024
Abstract
Advanced functional polymeric materials based on spiropyrans (SPs) feature multi-stimuli responsive characteristics, such as a change in color with exposure to light (photochromism) or acids (halochromism). The inclusion of stimuli-responsive molecules in general – and SPs in particular – as main-chain repeating units is a scarcely explored macromolecular architecture compared to side chain responsive polymers. Herein, we establish the effects of substitution patterns on SPs within a homopolymer main-chain synthesized via head-to-tail Acyclic Diene METathesis (ADMET) polymerization. We unambiguously demonstrate that varying the location of the ester group (–OCOR) on the chromophore, which is essential to incorporate the SPs in the polymer backbone, determines the photo- and halochromism of the resulting polymers. While one polymer shows effective photochromism and resistance towards acids, the opposite – weak photochromism and effective response to acid – is observed for an isomeric polymer, simply by changing the position of the ester-linker relative to the benzopyran oxygen on the chromene unit. Our strategy represents a simple approach to manipulate the stimuli-response of main-chain SP bearing polymers and highlights the critical importance of isomeric molecular constitution on main-chain stimuli-sensitive polymers as emerging materials.
Introduction
Spiropyrans (SPs), typically containing a benzopyran (chromene) covalently connected to a heterocyclic moiety via the spiro-carbon (Fig. 1), constitute a well-known and extensively studied class of multi-stimuli-responsive chemical structures.1–3 The ring-closed structure of SPs is typically the thermodynamically stable isomer, which can reversibly transform to the ring-opened form upon exposure to various stimuli, such as light irradiation, pH or mechanical force.1,2,4 The stimuli-responsive SP transformation is accompanied by a vivid color change (i.e., colorless to colored or vice versa) and a large polarity difference (i.e., apolar (hydrophobic) to polar/zwitterionic/merocyanine (hydrophilic) form or vice versa). Since the first report of synthetic spiropyrans at the beginning of the 20th century,5 chemists have integrated SPs into polymers, harnessing their responsive properties to generate advanced dynamic functional materials.3,6,7 Exemplary applications are smart coatings,8,9 pH/metal ion or force sensors,10–13 drug delivery,14 lithography15,16 or shape-changing materials.17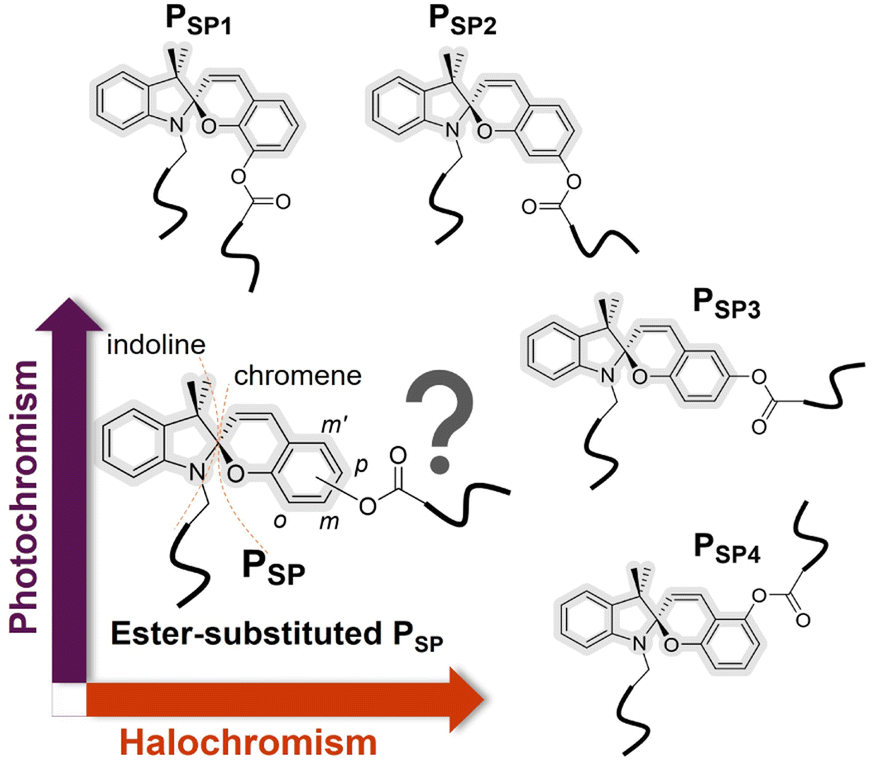 | ||
| Fig. 1 General structures of four main-chain polymers derived from four regioisomers of a spiropyran scaffold. The sole difference among these regioisomers is the relative position (with respect to the benzopyran oxygen) of the ester group (–OCOR) on the chromene moiety of the spiropyran. We demonstrate dramatic effects of these minor difference on the photo- and halochromism of the resulting polymers. Note that the covalent connection to the chromene moiety can be realized through the carbonyl carbon (–COOR), which is critically different from our design. | ||
Most stimuli-responsive moieties are tethered directly to the polymer chain as either pendant groups – or to a far lesser extent – as main-chain active groups via ester and amide bonds.18–22 In the realm of SPs, the type of the substituents decorating the SPs, e.g., electron-withdrawing or electron-donating, has a potentially significant influence on the stimuli-sensitiveness of SPs-based polymers.23–25 However, the impact of ester groups located at different sites of SPs' aromatic moieties (regioisomers) has not been investigated. In fact – to our best knowledge – only two studies explore substituent location variations of the polymerizable group on the benzopyran moiety, leading to different levels of stretching-induced ring-opening and isomerization of the covalently embedded SP units.11,26 However, the SPs-based monomers were designed such that the SP moiety and the ester group of polymerizable handles were connected via a methylene group (–CH2–) to the chromene moiety.11,26 Critically, the effect of these designs on the resulting photo- and pH-responsive properties was not explored. Interestingly, nitro-substituted SPs (NO2-SPs) have frequently been explored, most likely due to the resulting red-shifted electronic absorption and an enhanced quantum efficiency induced by the electron-withdrawing group.27,28 Remarkably, in the current literature, the electrochemical contribution of other groups, e.g., esters, is often ignored. However, tuning the properties of responsive macromolecular materials via subtle alterations of their chemical structures is undoubtedly a powerful avenue to establish varied property profiles with synthetic ease. Herein, we demonstrate such structural variations and their effect on the resulting material properties, opening a facile route to an advanced class of SP-containing responsive polymer systems.
Main-chain SP-based polymers possess – just as their well-explored side chain equivalents – photo and pH- responsiveness, yet offer additional exclusive qualities, such as mechanochromism11 and potentially larger motion change29,30 upon the ring-opening of SPs. Synthetically, however, just a limited number of methods, e.g., Ring-Opening (Metathesis) Polymerization (ROP26/ROMP31) or Suzuki polycondensation,32 were employed to install SP moieties into the polymer backbone.13,33 Here, we employ head-to-tail Acyclic Diene METathesis (ADMET) polymerization to construct SP main-chain polymers.
We thus introduce the synthesis of four main-chain polymers (PSP1, PSP2, PSP3 and PSP4) derived from four regioisomers of SPs via ADMET polymerization and investigate their photo- and acid-responsive properties. These polymers differ only in the substitution location on the SP's benzopyran (chromene) moiety (o-, m-, p- and m′- position, corresponding to PSP1, PSP2, PSP3 and PSP4, respectively, refer to Fig. 1 and 2) of the ester group (–OCOR, where R contains a polymerizable handle, inserting the chromophore into the polymer main chain). Note that the benzopyran ring is substituted with the ester oxygen (–OCOR) rather than the carbonyl carbon (–COOR) of the ester group reported elsewhere for small structures.34 To our best knowledge, we report SPs bearing an ester group (–OCOR) substituted at either the m or m′ position (refer to Fig. 1 and 2) for the first time. The synthetic accessibility of all four substitution sites allows us to systematically study the influence of regio-isomers on the stimuli response of SPs. Interestingly, our results show that such minor structural differences have dramatic effects on the solution photo- and acid-responsiveness of the SP moieties of these polymers. Particularly, while PSP1 is relatively photo-inactive yet acid sensitive, the complete opposite behavior is observed for PSP4. PSP2 responds rapidly to acids (i.e., CF3COOH and CH3SO3H), followed by PSP1 and PSP3.
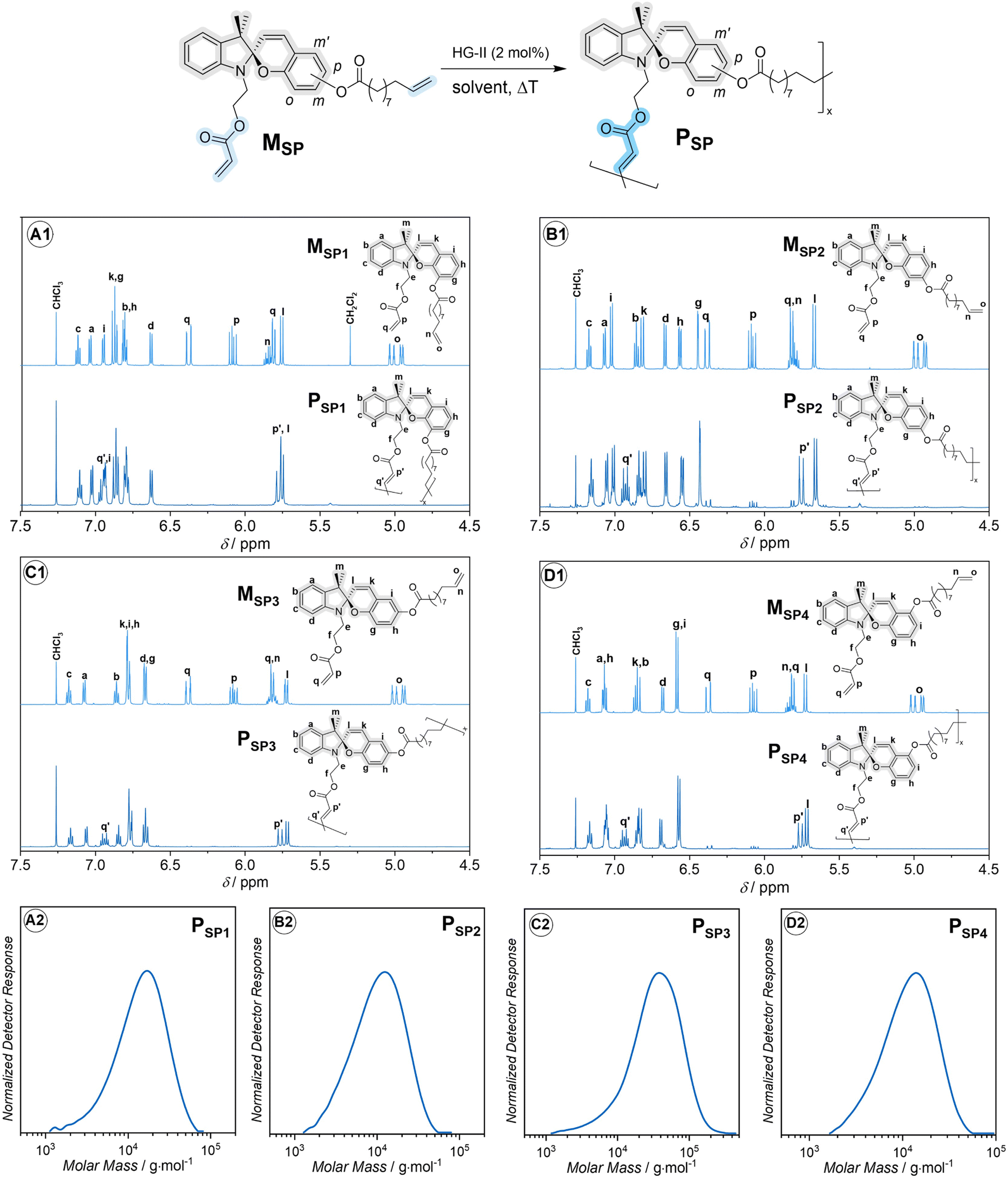 | ||
| Fig. 2 Synthesis and characterization of main-chain spiropyran-based polymers. Top: synthesis via head-to-tail ADMET polymerization and 1H NMR sectional spectra (600 MHz, 32 scans, CDCl3) of monomers and polymers: (A1) MSP1 and PSP1, (B1) MSP2 and PSP2, (C1) MSP3 and PSP3, (D1) MSP4 and PSP4. Full 1D and 2D NMR spectra are provided in the ESI, Section 9.† The assignment of the proton resonances was assisted through the analysis of 1D and 2D NMR spectra of precursors and desired products as described in ESI, Section 9.† Details of the polymerization conditions can be found in the ESI, Section 2.† Bottom: dimethylacetamide (DMAc)-size exclusion chromatography (SEC) traces of PSP1 (A2), PSP2 (B2), PSP3 (C2) and PSP4 (D2). | ||
Results and discussion
Synthesis
Initially, we designed four ADMET monomers, containing four spiropyran regioisomers as main-chain moieties. The handle containing the acrylate group is placed at the indoline nitrogen, while the phenyl ring of the chromene moiety is substituted with an ester group containing the olefin handle at one of the four sites, o-, m-, p- and m′-, yielding monomer MSP1, MSP2, MSP3 and MSP4, respectively (Fig. 2). The selective cross-metathesis using a Hoveyda–Grubbs 2nd generation catalyst (HG-II) between the acrylate and olefin functional groups affords the main-chain polymer PSP1, PSP2, PSP3 and PSP4 from the respective monomers (refer to the ESI for full synthesis protocol and characterization of monomers and polymers, Section 2 and 9†). The sectional 1H NMR spectra in Fig. 2 unambiguously evidence the formation of the internal acrylate bond (proton resonances at δ ≈ 5.75 and 6.92 ppm) from the cross-metathesis, while leaving the SP chromophores chemically intact. The apparent number-average molar mass (Mn) of these polymers falls in the range of (7500–22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) g mol−1 (Fig. 2, bottom and Table 1), in line with other stimuli-responsive polymers synthesized via ADMET polymerization.35,36 Interestingly, during the polymerization, we observed gelation of the reaction mixture of MSP2 when dichloromethane (DCM) was used as solvent. Gelation was prevented by using 1,2-dichlorobenzene (DCB) as solvent instead, successfully obtaining polymer PSP2 (Fig. 2B). Although the mechanism for the gelation of MSP2 in DCM solution is unclear, we speculate that the general structure of MSP2 with the ester group at the m-position of the benzopyran ring (Fig. 2) might allow for a ligand-exchange reaction with the Ru-based HG-II catalyst which could gradually decompose in DCM solvent.37 On the basis of the observed molar masses (Mn) of the four polymers (Table 1), the acrylate and the olefin handles in PSP3 are in an optimal orientation, enabling rapid polymerization and affording relatively high Mn compared to the other three isomeric polymers. The substitution-site dependence of Mn in ADMET polymerization is also observed in hydrazone-based main-chain polymers.38
000) g mol−1 (Fig. 2, bottom and Table 1), in line with other stimuli-responsive polymers synthesized via ADMET polymerization.35,36 Interestingly, during the polymerization, we observed gelation of the reaction mixture of MSP2 when dichloromethane (DCM) was used as solvent. Gelation was prevented by using 1,2-dichlorobenzene (DCB) as solvent instead, successfully obtaining polymer PSP2 (Fig. 2B). Although the mechanism for the gelation of MSP2 in DCM solution is unclear, we speculate that the general structure of MSP2 with the ester group at the m-position of the benzopyran ring (Fig. 2) might allow for a ligand-exchange reaction with the Ru-based HG-II catalyst which could gradually decompose in DCM solvent.37 On the basis of the observed molar masses (Mn) of the four polymers (Table 1), the acrylate and the olefin handles in PSP3 are in an optimal orientation, enabling rapid polymerization and affording relatively high Mn compared to the other three isomeric polymers. The substitution-site dependence of Mn in ADMET polymerization is also observed in hydrazone-based main-chain polymers.38
| M n /g mol−1 | M p /g mol−1 | Đ | λ max /nm | ε max /M−1 cm−1 | |
|---|---|---|---|---|---|
| a Dimethylacetamide (DMAc)-size exclusion chromatography (SEC), on the basis of poly(methyl methacrylate) (PMMA) calibration. b In dichloromethane (DCM) at 25 °C. | |||||
| PSP1 | 11000 |
17000 |
1.6 | 297 | 4130 |
| PSP2 | 8000 | 12000 |
1.6 | 299 | 4630 |
| PSP3 | 22000 |
39000 |
1.9 | 299 | 3710 |
| PSP4 | 7500 | 10500 |
1.5 | 298 | 3850 |
With regard to the optical properties of the four polymers, there is an insignificant difference in terms of the peak absorption band (λmax) in DCM, corresponding to the π–π* transition of the chromene moiety, close to the values reported for halogen- and ether-substituted SPs (λmax = (297–299) nm, Table 1).23 The molar absorptivity of the SP chromophore at λmax (i.e., εmax) for PSP1, PSP2, PSP3 and PSP4 varies between 3700 M−1 cm−1 and 4700 M−1 cm−1, respectively (Table 1).
Fundamentally, the closed- and open-conformation switching of SPs is a complex process, as there are several possible transient states, which have been subjected to careful experimental and theoretical investigation.39–44 Briefly, the photo-induced transformation of SPs first takes place via the cleavage of the labile C–O spiro bond, followed by E/Z-isomerization around specific bonds (Scheme 1) to form the merocyanine (MC) form (opened form).42 In the presence of a strong acid, the protonation of spiro oxygen and the subsequent isomerization generates thermally stable species (MCH+).42 However, probing the switching mechanism is not the main aim of the current study. Instead, we explore the effect of the molecular design on the switching efficiency of SPs main-chain polymers. Specifically, we reveal the effect of the location of the ester group on the chromene moiety on photo- and halochromism. We exploit the intense color difference between the spiropyran (closed-form, colorless) and the MC/MCH+ form (opened-form, colored), monitored by UV-vis spectroscopy, to compare the photo- and pH-responsive properties among PSP1, PSP2, PSP3 and PSP4, underpinned by careful 1H NMR spectroscopic analysis.
 | ||
| Scheme 1 Simplified mechanism of photo- and acid-induced ring-opening and isomerization of SPs. The (Z)-MC/MCH+ and (E)-MC/MCH+ can adopt other conformations via rotation around the α- and γ-bond. Details of the stimuli-responsive mechanism can be found in ref. 1, 2 and 39–44. | ||
Photochromism
Initially, we conducted the irradiation of the four polymer solutions (50–150 μM) in dichloromethane (DCM) at ambient temperature (25 °C). We selected a wavelength of 330 nm (monochromatic) to minimize unfavorable side reactions, such as photodegradation. The UV-vis spectra in Fig. 3 demonstrate the different photochromism of the investigated polymers. There is almost no vivid color change of the PSP1 polymer solution unless when irradiated with relatively large number of photons (Np) (Np = 6.0 × 1019 photons) (Fig. 3A). In contrast, the UV-vis spectra of PSP2, PSP3 and PSP4 display new and strong absorption bands in the visible region (Fig. 3B–D) upon irradiation. It is noticeable in the case of the latter polymers that beside an emerging absorption peak centered at around 458–477 nm, there is an absorption band lying at lower wavelengths close to 380–390 nm, suggesting the existence of at least two isomers of the opened form, e.g., (Z)- and (E)-isomers of MC (Scheme 1). Intriguingly, the color of the PSP1 solution in DCM gradually turned yellow in the dark after long time exposure to 330 nm irradiation (Np = 6.0 × 1019 photons) (Fig. 3A). We cannot exclude the solvent contributions, e.g., from DCM, to the photochromism of SPs, as frequently reported in the literature.45–48 Nevertheless, it is worth noting that the pristine polymer solutions in DCM did not show any change in color at 25 °C in the dark, excluding solvatochromism as the root cause of the color change. However, to our surprise, the irradiation performed in other solvents, i.e., toluene and dimethylacetamide (DMAc), did not trigger a noticeable response from all four polymers (UV-vis spectra shown in Fig. S7†). We chose toluene and DMAc alongside DCM as solvents for the irradiation study due to the good solubility of the polymers in these solvents which are of different polarity.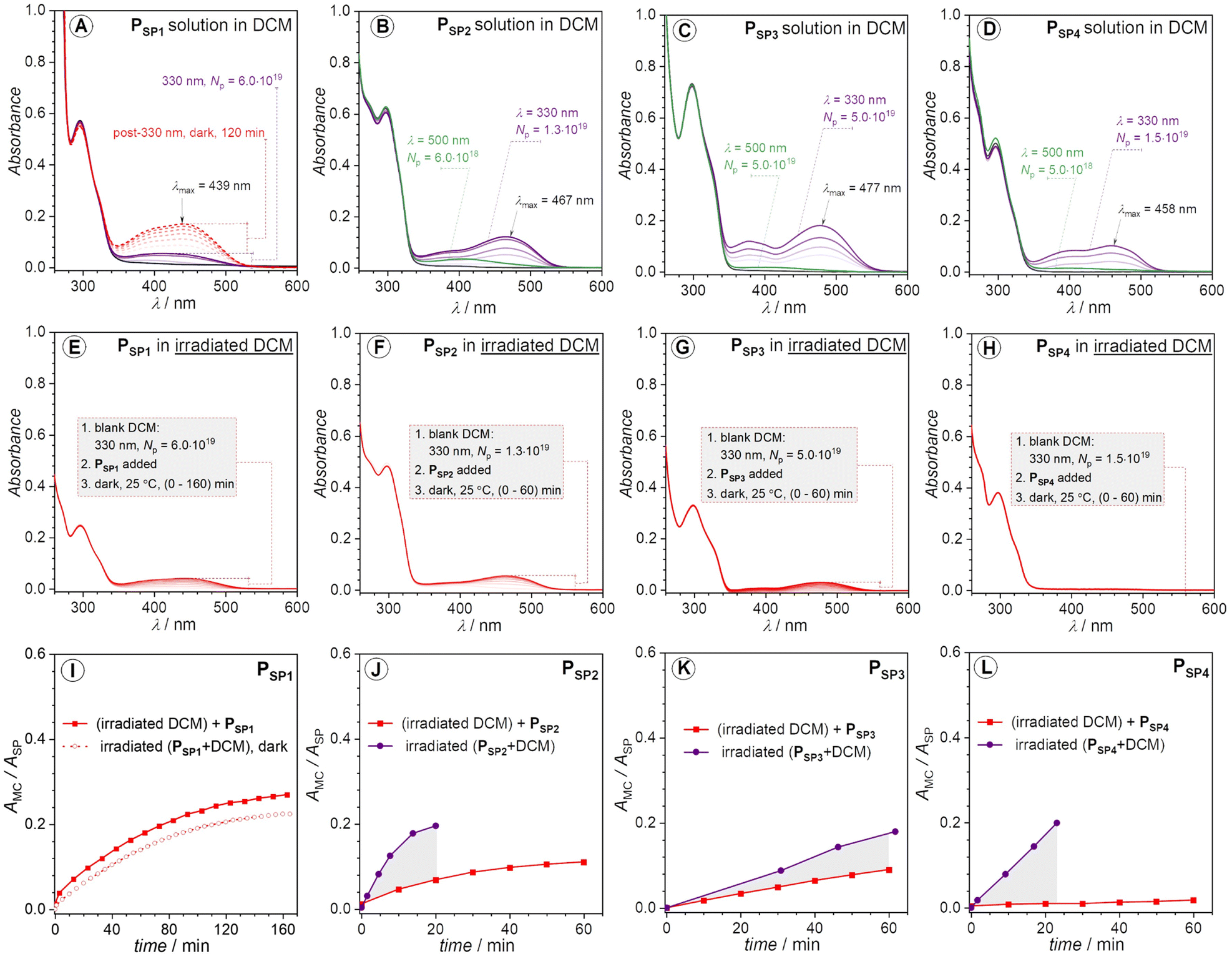 | ||
| Fig. 3 (A–D) UV-vis spectra of PSP1, PSP2, PSP3 and PSP4 solution in DCM, recorded before and after 330 nm monochromatic irradiation at 25 °C. The red dashed lines denote the spectra recorded post 330 nm irradiation; the purple solid lines represent the spectra recorded immediately after 330 nm irradiation; the green solid lines indicate the spectra recorded after 500 nm irradiation of the 330 nm irradiated solutions. (E–H) UV-vis spectra recorded upon addition of PSP1, PSP2, PSP3 and PSP4, to 330 nm irradiated DCM blank solvents. Molar concentrations were between 50 and 150 μM. Enlargements of UV-vis spectra in (A–H) are available in the ESI, Fig. S8.† The laser power at 330 and 500 nm irradiation was kept at 6.5 mW ± 5.2% and 8.5 mW ± 13%, respectively. (I–L) Plots of AMC/ASPvs. time for PSP1, PSP2, PSP3 and PSP4, respectively. Note that the protonated form of MC (MCH+) also exists but is grouped into the ‘MC’ label in AMC (i.e., absorbance of the opened form) for the sake of simplicity. The absorbance of the ‘MC’ label in this case (AMC) corresponds to the λmax of the colored species in the visible range, and the ASP refers to the absorbance at λmax of the respective spiropyran. The AMC/ASP ratio is used instead of AMC to factor in the concentration difference between experiments assuming AMC/ASP ∼ cMC/cSP with εMC/εSP = constant. The number of photons (Np) was converted into time via eqn S1.† The gray areas in (J–L) highlight the actual photochromic response of polymer solutions upon 330 nm irradiation. | ||
Protic solvents capable of forming H-bonds, such as methanol, can stabilize the merocyanine (MC) form, thus facilitating the photo-induced SP → MC conversion process.48 However, the four SP-based polymers reported in our work are hydrophobic and do not dissolve in such protic solvents. Toluene and DMAc do not possess H-bonding capabilities. In addition, DMAc can act as a Lewis base49,50 which may react with the opened form of spiropyran induced by Lewis acid addition.51 However, it is not relevant to our case where no Lewis acid was added in the irradiation study. Even though DCM is not a protic solvent, its role as a very weak H-donor was previously reported,52 which may be a contributing factor for the color change observed for PSP2, PSP3, PSP4 under 330 nm irradiation in DCM. However, we cannot discard the possibility of UV-induced degradation of the halogenated solvents, e.g., CH2Cl2, CHCl3, into halogen acids, e.g., HCl, that can cause the change in color of these polymer solutions owing to the generally acid-sensitive characteristics of SPs. For instance, Sommer and colleagues reported the in situ generation of acid from DCM-d2 and chloroform-d under sonication, which was responsible for the chromism of the reported SP-based polymers in the respective solvents.32,53
To confirm the possible in situ generation of halogen acid under UV-irradiation, we conducted control experiments in which only the blank DCM solvent was irradiated with an exact number of photons previously applied for each SP-based polymer solution, after which the stock polymer solution was rapidly added to the UV-exposed solvent. PSP1 in post-irradiated DCM shows a gradual increase in the visible absorption region (λmax = 439 nm), which is identical with the irradiated PSP1 solution mentioned above (Fig. 3A, E and I). Thus, the acid-induced ring-opening and isomerization observed for PSP1 takes place in the dark after 330 nm irradiation (6.0 × 1019 photons). In the case of PSP2 and PSP3, the HCl formed in situ also co-contributes to the color change (compare Fig. 3B, C, F, G, J and K). However, PSP4 appears to be barely affected in the irradiated blank solvent (Fig. 3D, H and L). Therefore, these results allow to conclude that PSP4 is the most photo-active polymer, while PSP1 is insensitive to solution photo-irradiation. In other words, the substitution site of the ester group on the chromene moiety does indeed critically influence the photochromic properties of the main-chain polymers. It should be noted that the comparison made here is qualitative, yet sufficiently convincing. Based on UV-vis spectra alone (Fig. 3), we cannot quantify the ratio of the opened-form induced by irradiation. However, irradiation of higher concentrated solutions (close to 0.96 mM in DCM-d2) with 330 nm laser light did not cause any change in the 1H NMR spectra, probably due to the low efficiency of the photo-induced SP → MC transformation at higher concentrations (millimolar vs. micromolar), also noted by Ballester and colleagues for a similar spiropyran compound.54
The colored state of the opened-form can be quenched via visible light irradiation (λ = 500 nm, P = 8.5 mW ± 13%) (Fig. 3B–D). However, as can be seen in Fig. 3B–D, while the absorption at around (450–480) nm completely disappears under 500 nm irradiation, the shoulder absorption band close to 390 nm persists. The incomplete visible light-induced colored to colorless reversion may be due to the stabilizing effect from either acidic protons or the solvent (DCM). In addition, we note the contribution of the polymer backbone to the thermal stability of the opened-form.55 To avoid the impact from the in situ forming acid (HCl), we performed the irradiation in thin films (solid-state) to expand the potential application scope of the polymers on the example of polymer PSP2. Under similar irradiation condition (λ = 330 nm, Np = 1.5 × 1019, P = 6.5 mW ± 4.0%), the color of the polymer film on a quartz slide turned yellow, detectable with the naked eye. Indeed, the recorded UV-vis spectrum (Fig. S2†) revealed a new broad absorption band in the visible region (up to 650 nm), suggesting SP → MC transformation in the solid state. However, the solid-state UV-vis spectrum of the MC is vastly different from the solution one, likely due to the intermolecular stacking of the formed MC species in the confined environment in the polymer film. Nevertheless, we have demonstrated the feasibility of solid-state photochromism of the SP-based polymer, i.e., PSP2.
Halochromism
The different responses of the polymers to the photogenerated acids indicate the strong effect of the molecular architecture on the halochromism of the main-chain polymers, which we systematically investigated with the addition of two acids with different pKa (trifluoroacetic acid, CF3COOH (TFA), pKa = 0.23,56 methanesulfonic acid, CH3SO3H (MsOH), pKa = −1.9)57 to the polymer solutions in DCM. To avoid the acids affecting the polymer chain, such as cleavage of ester bonds, we added only 1.0 molar equivalent of either TFA or MsOH (with respect to the SP core) to the polymer solutions. The colorimetric response was monitored by UV-vis and 1H NMR spectroscopy. As shown in Fig. 4A–C, the solution of PSP2 swiftly changes color upon TFA addition, reaching an equilibrium within 3 hours. Equilibration of the color changing process took significantly longer for PSP1 and PSP3. The observed color change is due to the formation of MCH+.40 In contrast to PSP1, PSP2, and PSP3, the PSP4 solution was hardly affected by TFA addition (Fig. 4D), which is opposite to the strong photochromism of PSP4 discussed above. | ||
| Fig. 4 Halochromism of PSP1, PSP2, PSP3, PSP4 in DCM from UV-vis spectroscopy. (A–D) TFA addition (1.0 eq.), (E–H) MsOH addition (1.0 eq.). Time until equilibration is indicated. Molar concentration of SP moiety: 100–150 μM. (I–N) Comparison of the kinetics among four polymers upon acid addition. Normalized absorbance was used in (M and N) to enable a qualitative comparison of the response rate of the polymers toward TFA and MsOH acid. The AMCH+/ASP ratio is used instead of AMCH+ to factor in the concentration difference between experiments assuming AMCH+/ASP ∼ cMCH+/cSP with εMCH+/εSP = constant. AMCH+ refers to the absorbance (A) of the MCH+ form at λmax in the visible range, and ASP refers to the absorbance of the pristine SP at λmax. | ||
Upon the addition of the stronger acid, i.e., MsOH (pKa = −1.9),57 the change in absorbance is significantly larger than in the case of TFA addition, except for PSP4 which remains largely unaffected (compare Fig. 4A–D and E–H). The absorption spectra of these MCH+ species also feature a major absorption band within the 440–465 nm range and a shoulder at around 376–393 nm (Fig. 4), indicating the existence of different stable isomers, e.g., (Z)- and (E)-MCH+ (Scheme 1). The varied sensitivity towards acids of these polymers is a strong indication of different pKa values of the respective phenolate anions upon ester substitution of the chromene ring. To compare the kinetics of the halochromic processes for PSP1, PSP2 and PSP3, we qualitatively compared the curvature of the (normalized) absorbance (at λmax of MCH+) vs. time plot (Fig. 4I–N). For both TFA (pKa = 0.23)56 and MsOH (pKa = −1.9)57 addition, PSP2 is the most responsive polymer, followed by PSP1 and PSP3 which show slightly different rates of response (Fig. 4I–N).
If one compares the acid-induced and the photo-induced UV-vis spectra of PSP2 and PSP3 in DCM (Fig. 3 and 4), no prominent shift ((3–4) nm) in the peak and shoulder absorption in the visible region can be observed. For nitrospiropyrans, a large spectral shift between the protonated MC (MCH+) and the MC form was reported.58 In the case of our ester-substituted main-chain SPs, the presence of the ester groups might bridge the electronic differences between the MC and MCH+ forms. Alternatively, the H-bonding stabilization from the solvent, i.e., DCM, might equalize the spectral characteristics of the MC and MCH+.
To quantify the ratio of the MCH+ species in each polymer, we replicated the study using more concentrated solutions (close to 0.97 mM of SP moiety in CD2Cl2) for 1H NMR measurements. Accordingly, comparison of the integral values of protons e (δ ≈ 4.25 ppm) and f (δ ≈ 3.25–3.60 ppm) in the closed-form (as shown in Fig. 2 and S3–S6†) with those in the opened-form gives the percentage of MCH+ species (Fig. S3–S6†). Approximately 50% of the spiropyran moieties in PSP2 transformed into the opened form (MCH+) upon addition of TFA (1.0 eq.), twice as high as for PSP3 (25%) under identical conditions (Fig. S4 and S5†). Similar to PSP3, the determined value for PSP1 is 29%, whereas only 9% was recorded for PSP4 (Fig. S1 and S6†). The relative amount of MCH+ increases further when MsOH is added instead: close to 75% of MCH+ for PSP1, PSP2 and PSP3 and only 10% for PSP4 (Fig. S3–S6†). Interestingly, although the former polymers show the same ratio of MCH+ species induced by MsOH addition at equilibrium, the absorbance or molar absorptivity of the MCH+ in the case of PSP2 appears to be much higher than PSP1 and PSP3. Thus, the ester-substitution pattern affects not only the acid sensitivity of SPs, but also the molar absorptivity of the resulting MCH+. In addition, the highly polar nature of MCH+ can lead to aggregation in solution at high conversion,59 as indicated by Dynamic Light Scattering (DLS) results for the MsOH-added polymer solutions (Fig. S1†). Further, addition of 2.0 eq. of triethyl amine (Et3N) fully reverted the MCH+ to the closed-form, as evidenced by 1H NMR spectroscopy (Fig. S3–S6†).
The experimental results discussed above clearly point to the significant role of the ester group substituted on the chromene ring beside the contribution from the solvent polarity. Since the chromene ring directly attaches to the oxygen of the ester (–OCOR), the ester group serves as a moderate electron-donating group (EDG), directing electrons to the ortho- and para-position (relative to that EDG) of the phenyl ring. However, the same phenyl ring is at the same time substituted with two other groups, i.e., the spiro oxygen (in the closed-form) or the phenolate oxygen (in the opened-form) and the ethene bridge (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR′′, R′′ contains the indoline moiety), which are also EDGs. Thus, there are three EDGs on the phenyl ring under discussion (Scheme 2). Depending on their relative positions on the ring, the electron density of the phenolate oxygen (O−) (forming upon ring-opening) may be different. During the photochromism of SPs, we hypothesize that the merocyanine form is more stable when electrons on the phenolate oxygen are directed to the ring or elsewhere (i.e., lower electron density on the O− group).60 In PSP4 – which is the most photoresponsive polymer – the O− and the ester groups are meta-substituted to each other, but ortho-substituted to the ethene bridge (Scheme 2), thus facilitating the synergistic pushing of electrons from these two groups to the ethene bridge, effectively reducing the electron density on the O− group. On the basis of this logic and in consideration of the steric hindrance at the o-position, the poor photochromic property observed for PSP1 can be explained (Scheme 2). Meanwhile, a less electron density in the phenolate oxygen also suggests a less susceptibility to protonation, agreeing well with the experimental result for PSP4, which is the most acid-resistant among the four polymers studied. In the same manner, PSP1 is supposed to be the most acid-sensitive. Nevertheless, PSP2 surpasses PSP1 in terms of halochromism. Close inspection of the PSP2 structure suggests possible hydrogen bonding of the phenolate O− and the carbonyl oxygen (of the ester) to the proton from acids and DCM solvent (Scheme 2), perhaps making PSP2 more sensitive towards acids than PSP1. In addition, the substitution of the ester group at the p-position with respect to the ethene bridge – where E/Z isomerization takes place upon ring-opening in PSP2 – is the most ideal among all four polymers in terms of steric hindrance, facilitating the isomerization process in PSP2. Nevertheless, an in-depth theoretical study, which is out of scope of this work, is required to fully understand the role of such ester substituents (–OCOR).
CR′′, R′′ contains the indoline moiety), which are also EDGs. Thus, there are three EDGs on the phenyl ring under discussion (Scheme 2). Depending on their relative positions on the ring, the electron density of the phenolate oxygen (O−) (forming upon ring-opening) may be different. During the photochromism of SPs, we hypothesize that the merocyanine form is more stable when electrons on the phenolate oxygen are directed to the ring or elsewhere (i.e., lower electron density on the O− group).60 In PSP4 – which is the most photoresponsive polymer – the O− and the ester groups are meta-substituted to each other, but ortho-substituted to the ethene bridge (Scheme 2), thus facilitating the synergistic pushing of electrons from these two groups to the ethene bridge, effectively reducing the electron density on the O− group. On the basis of this logic and in consideration of the steric hindrance at the o-position, the poor photochromic property observed for PSP1 can be explained (Scheme 2). Meanwhile, a less electron density in the phenolate oxygen also suggests a less susceptibility to protonation, agreeing well with the experimental result for PSP4, which is the most acid-resistant among the four polymers studied. In the same manner, PSP1 is supposed to be the most acid-sensitive. Nevertheless, PSP2 surpasses PSP1 in terms of halochromism. Close inspection of the PSP2 structure suggests possible hydrogen bonding of the phenolate O− and the carbonyl oxygen (of the ester) to the proton from acids and DCM solvent (Scheme 2), perhaps making PSP2 more sensitive towards acids than PSP1. In addition, the substitution of the ester group at the p-position with respect to the ethene bridge – where E/Z isomerization takes place upon ring-opening in PSP2 – is the most ideal among all four polymers in terms of steric hindrance, facilitating the isomerization process in PSP2. Nevertheless, an in-depth theoretical study, which is out of scope of this work, is required to fully understand the role of such ester substituents (–OCOR).
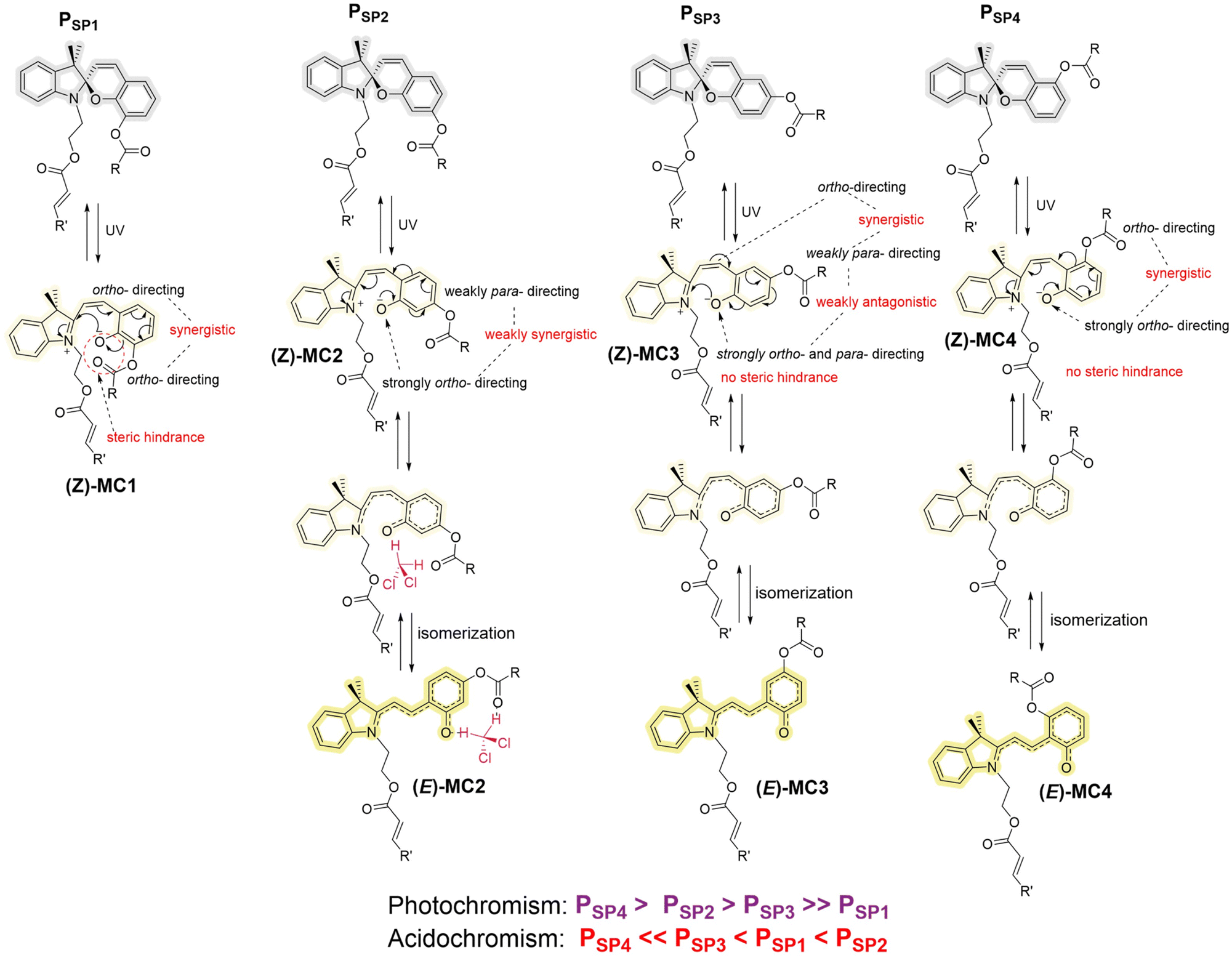 | ||
| Scheme 2 Possible impacts of the ester group substituted at one of the four possible positions on the chromene moiety (o-, m-, p- and m′-, with respect to the benzopyran oxygen) on the stability of the opened-form of spiropyran, impacting the photo- and halochromism. We hypothesize that the resulting photo- and halochromism of the four polymers in DCM solutions originate from a complex combination of electronic effects (induction and resonance), steric hindrance and hydrogen-bonding capability caused by the regio-substitution of the ester (–OCOR) group. The relative orientation of the phenolate (O−)/phenolic (OH) oxygen, the ethene bridge and the ester group (which are all electron-donating groups (EDGs)) may influence the electron densities on these groups. In the case of PSP1 and PSP3, for instance, both the ethene bridge and the ester groups are at either ortho-/ortho- or ortho-/para-positions with respect to the phenolate oxygen and vice versa. Thus, the former groups can synergistically direct electrons to the phenolate oxygen. There exists also a possibility for the phenolate oxygen (which is a much stronger EDG) to redirect electron density to the other two groups. For PSP1, the steric contribution from the ester group and the possible electronic repulsion between the phenolate oxygen (O−) and the carbonyl oxygen in the ester group may further reduce the photochromic response of the polymer compared to PSP3. However, upon protonation, a possible hydrogen bonding between these two groups can be established, co-contributing to the strong halochromism of PSP1. In the case of PSP2 and PSP4, the phenolate oxygen is meta-substituted with respect to the ester group. Thus, these two EDGs can cooperatively push electron density to the ethene bridge, leading to lower electron densities on the phenolate oxygen (which means stronger photochromism). With regard to the observed halochromism, PSP2 and PSP4 are expected to be inactive or significantly less responsive compared to PSP1 and PSP3. However, while the prediction holds true for PSP4, PSP2 responds fastest to acid-addition. Upon careful structural analysis, we submit that the possible intermolecular interaction with the solvent (DCM), i.e., hydrogen-bonding, and the geometry of the ester group (which also functions as the connecting arm) beside the electronic contribution, lead to the unexpected performance of PSP2. Within all four polymers, the ester group in PSP2 is at the para-position with respect to the ethene bridge, where E/Z-isomerization takes place upon ring-opening. In terms of steric hindrance, such a substitution site is the most ideal and thus does not interfere with the E/Z-isomerization at the ethene bridge. The term “ortho-directing” refers to the electron-donation from the substituent under consideration to its ortho-position. | ||
Furthermore – since the phenolate O− group is a much stronger EDG than the ester group – the substitution of the ester group at ortho- and para-position respective to the O− group – as seen in PSP1 and PSP3 – might make the ester group function as an electron-receiving group. As a result, the carbonyl oxygen of the ester might become more prone to protonation, leading to cleavage of the ester group. Indeed, the Size Exclusion Chromatography (SEC) traces recorded for PSP1 and PSP3 (Fig. S3 and S5†) show a drastic reduction in Mn upon addition of the strong acid, i.e., MsOH, whereas PSP2 and especially PSP4 stay relatively unaffected under the same conditions. The 1H NMR spectra (Fig. S3 and S5†) of PSP1 and PSP3 solutions upon addition of MsOH followed by quenching with Et3N also indicate some degree of degradation. Since the chromophore and the acrylate CC bonds remain intact upon quenching with Et3N, the degradation (if applicable) is highly likely due to the hydrolysis of the main-chain ester group on the chromene moiety. Polymer chains generally break in the middle of the chain owing to entropic reason, which leads to a large shift in the apparent molar mass despite only a minor degree of hydrolysis.61 The FT-IR spectrum of MsOH-added PSP3 – which showed the largest shift in the SEC – suggests only little ester hydrolysis (Fig. S9†). Thus, in addition to the inherit photo- and acidochromism of SPs, the position of the ester group (–OCOR) on the chromene moiety of SPs may complementarily affect its hydrolysis induced by acids.
Conclusions
We herein introduce the synthesis of main-chain polymers derived from four regional isomers (PSP1, PSP2, PSP3 and PSP4) of spiropyrans via head-to-tail ADMET polymerization. The photo- and halochromism of these polymers are highly dependent on the position of the ester group (–OCOR) on the phenyl ring of the chromene, which is essential to incorporate the stimuli responsive spiropyrans in the main chain. While PSP4 (ester group (–OCOR) in the m′-position) responds fastest under UV irradiation, no pronounced response is observed under acidic conditions (addition of 1.0 eq. of CF3COOH (TFA) or CH3SO3H (MsOH)). In contrast, PSP1 (ester group (–OCOR) in the o-position) reacts relatively fast to an acid stimulus, yet at the same time is inactive towards UV exposure. Both PSP2 and PSP3 (ester group (–OCOR) at m-position and p-position, respectively) are responsive toward irradiation and acids. These characteristics are clearly attributed to the change in the substitution site of the ester group on the benzopyran (chromene) moiety. The herein discussed findings thus allow to manipulate and fine-tune the properties of stimuli responsive spiropyran-based main-chain polymers for specific applications, requiring limited synthetic effort. We anticipate the influence of substitution extends towards other stimuli such as metal ions and force. Critically, our results demonstrate the significance of small isomeric molecular variations in conjunction with transitioning from side-chain decorated to main-chain based macromolecular architectures, including their potential for finely modulating the characteristics of stimuli responsive polymeric materials.Data availability
All experimental data and detailed experimental procedures are available in the main text and ESI.†Author contributions
L. D. T. conceptualized the spiropyran system, conducted all experimental work and wrote the initial draft of the manuscript. J. A. K. assisted with data interpretation and discussion and co-edited the manuscript. C. B.-K. and H. M. motivated and conceptualized the research direction, acquired funding, discussed the data and edited the manuscript.Conflicts of interest
The authors declare no conflicts of interests.Acknowledgements
C. B.-K. acknowledges the Australian Research Council (ARC) for funding in the context of a Laureate Fellowship enabling his photochemical research program. The authors acknowledge funding for the project from QUT's Centre for Materials Science. The Central Analytical Research Facility (CARF) at QUT is gratefully acknowledged. H. M. acknowledges the University of Haute-Alsace (UHA) for the financial support from the French National Research Agency (ANR) with the reference “ANR-22-CPJ1-0077-01” and from the CNRS for a junior professorship contract. C. B.-K. acknowledges additional funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy for the Excellence Cluster ‘3D Matter Made to Order’ (EXC-2082/1 – 390761711). J. A. K. acknowledges the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for his WBP fellowship – 500289223.References
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- L. Kortekaas and W. R. Browne, The evolution of spiropyran: fundamentals and progress of an extraordinarily versatile photochrome, Chem. Soc. Rev., 2019, 48, 3406–3424 RSC.
- J. K. Rad, Z. Balzade and A. R. Mahdavian, Spiropyran-based advanced photoswitchable materials: a fascinating pathway to the future stimuli-responsive devices, J. Photochem. Photobiol., C, 2022, 51, 100487 CrossRef.
- M. Li, Q. Zhang, Y.-N. Zhou and S. Zhu, Let spiropyran help polymers feel force!, Prog. Polym. Sci., 2018, 79, 26–39 CrossRef CAS.
- H. Decker and H. Felser, Über cyclische Oxoniumsalze aus Dicumarketon und über Spiropyranderivate, Ber. Dtsch. Chem. Ges., 1908, 41, 2997–3007 CrossRef.
- L. Florea, D. Diamond and F. Benito-Lopez, Photo-responsive polymeric structures based on spiropyran, Macromol. Mater. Eng., 2012, 297, 1148–1159 CrossRef CAS.
- M. Sommer, Substituent effects control spiropyran–merocyanine equilibria and mechanochromic utility, Macromol. Rapid Commun., 2021, 42, 2000597 CrossRef CAS PubMed.
- B. Di Credico, G. Griffini, M. Levi and S. Turri, Microencapsulation of a UV-responsive photochromic dye by means of novel UV-screening polyurea-based shells for smart coating applications, ACS Appl. Mater. Interfaces, 2013, 5, 6628–6634 CrossRef CAS PubMed.
- L. Kortekaas, O. Ivashenko, J. T. van Herpt and W. R. Browne, A remarkable multitasking double spiropyran: bidirectional visible-light switching of polymer-coated surfaces with dual redox and proton gating, J. Am. Chem. Soc., 2016, 138, 1301–1312 CrossRef CAS PubMed.
- F. Jiang, S. Chen, Z. Cao and G. Wang, A photo, temperature, and pH responsive spiropyran-functionalized polymer: synthesis, self-assembly and controlled release, Polymer, 2016, 83, 85–91 CrossRef CAS.
- Y. Lin, M. H. Barbee, C.-C. Chang and S. L. Craig, Regiochemical effects on mechanophore activation in bulk materials, J. Am. Chem. Soc., 2018, 140, 15969–15975 CrossRef CAS PubMed.
- F. Arjmand and Z. Mohamadnia, Fabrication of a light-responsive polymer nanocomposite containing spiropyran as a sensor for reversible recognition of metal ions, Polym. Chem., 2022, 13, 937–945 RSC.
- Y. Vidavsky, S. J. Yang, B. A. Abel, I. Agami, C. E. Diesendruck, G. W. Coates and M. N. Silberstein, Enabling room-temperature mechanochromic activation in a glassy polymer: synthesis and characterization of spiropyran polycarbonate, J. Am. Chem. Soc., 2019, 141, 10060–10067 CrossRef CAS PubMed.
- A. Fagan, M. Bartkowski and S. Giordani, Spiropyran-based drug delivery systems, Front. Chem., 2021, 9, 720087 CrossRef CAS PubMed.
- Y. Jung, H. Lee, T.-J. Park, S. Kim and S. Kwon, Programmable gradational micropatterning of functional materials using maskless lithography controlling absorption, Sci. Rep., 2015, 5, 15629 CrossRef CAS PubMed.
- H. Vijayamohanan, E. F. Palermo and C. K. Ullal, Spirothiopyran-based reversibly saturable photoresist, Chem. Mater., 2017, 29, 4754–4760 CrossRef CAS.
- G. Stoychev, A. Kirillova and L. Ionov, Light-responsive shape-changing polymers, Adv. Opt. Mater., 2019, 7, 1900067 CrossRef.
- G. D. Jaycox, Stimuli-responsive polymers. VIII. Polyesters and poly (ester amides) containing azobenzene and chiral binaphthylene segments: highly adaptive materials endowed with light-, heat-, and solvent-regulated optical rotatory power, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 207–218 CrossRef CAS.
- L. D. Thai, T. R. Guimaraes, S. Spann, A. S. Goldmann, D. Golberg, H. Mutlu and C. Barner-Kowollik, Photoswitchable block copolymers based on main chain α-bisimines, Polym. Chem., 2022, 13, 5625–5635 RSC.
- P. Min, Y. Li, L. Wang, W. Song and L. Ding, The shackling photoisomerization effect on self-assembly of azobenzene-containing side-chain homopolymers, Polymer, 2022, 258, 125301 CrossRef CAS.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Stimuli-responsive polymers and their applications, Polym. Chem., 2017, 8, 127–143 RSC.
- P. Schattling, F. D. Jochum and P. Theato, Multi-stimuli responsive polymers–the all-in-one talents, Polym. Chem., 2014, 5, 25–36 RSC.
- M. Schulz-Senft, P. J. Gates, F. D. Sönnichsen and A. Staubitz, Diversely halogenated spiropyrans-Useful synthetic building blocks for a versatile class of molecular switches, Dyes Pigm., 2017, 136, 292–301 CrossRef CAS.
- H. Görner, Photochromism of nitrospiropyrans: effects of structure, solvent and temperature, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC.
- J. H. Day, Thermochromism, Chem. Rev., 1963, 63, 65–80 CrossRef CAS.
- F. Kempe, L. Metzler, O. Brügner, H. Buchheit, M. Walter, H. Komber and M. Sommer, Substituent-controlled energetics and barriers of mechanochromic spiropyran-functionalized poly (ε-caprolactone), Macromol. Chem. Phys., 2023, 224, 2200254 CrossRef CAS.
- A. Kellmann, F. Tfibel, E. Pottier, R. Guglielmetti, A. Samat and M. Rajzmann, Effect of nitro substituents on the photochromism of some spiro [indoline-naphthopyrans] under laser excitation, J. Photochem. Photobiol., A, 1993, 76, 77–82 CrossRef CAS.
- M. Sakuragi, K. Aoki, T. Tamaki and K. Ichimura, The role of triplet state of nitrospiropyran in their photochromic reaction, Bull. Chem. Soc. Jpn., 1990, 63, 74–79 CrossRef CAS.
- S. Bhattacharyya, M. Maity, A. Chowdhury, M. L. Saha, S. K. Panja, P. J. Stang and P. S. Mukherjee, Coordination-assisted reversible photoswitching of spiropyran-based platinum macrocycles, Inorg. Chem., 2020, 59, 2083–2091 CrossRef CAS PubMed.
- A. S. Kozlenko, A. D. Pugachev, I. V. Ozhogin, I. M. El-Sewify and B. S. Lukyanov, Spiropyrans: molecules in motion, Chem. Heterocycl. Compd., 2021, 57, 984–989 CrossRef CAS.
- G. R. Gossweiler, T. B. Kouznetsova and S. L. Craig, Force-rate characterization of two spiropyran-based molecular force probes, J. Am. Chem. Soc., 2015, 137, 6148–6151 CrossRef CAS PubMed.
- M. Sommer and H. Komber, Spiropyran main-chain conjugated polymers, Macromol. Rapid Commun., 2013, 34, 57–62 CrossRef CAS PubMed.
- C. K. Lee, D. A. Davis, S. R. White, J. S. Moore, N. R. Sottos and P. V. Braun, Force-induced redistribution of a chemical equilibrium, J. Am. Chem. Soc., 2010, 132, 16107–16111 CrossRef CAS PubMed.
- V. Pantsyrnyi and M. Gal'bershtam, Preparation of spiropyrans from 3-formylsalicylic acid derivatives, Chem. Heterocycl. Compd., 1971, 7, 134–135 CrossRef.
- L. D. Thai, T. R. Guimaraes, L. C. Chambers, J. A. Kammerer, D. Golberg, H. Mutlu and C. Barner-Kowollik, Molecular photoswitching of main-chain α-bisimines in solid-state polymers, J. Am. Chem. Soc., 2023, 145, 14748–14755 CrossRef CAS PubMed.
- L. Ding, Y. Li, H. Cang, J. Li, C. Wang and W. Song, Controlled synthesis of azobenzene-containing block copolymers both in the main-and side-chain from SET-LRP polymers via ADMET polymerization, Polymer, 2020, 190, 122229 CrossRef.
- S. Mavila, C. E. Diesendruck, S. Linde, L. Amir, R. Shikler and N. G. Lemcoff, Polycyclooctadiene complexes of rhodium (I): direct access to organometallic nanoparticles, Angew. Chem., Int. Ed., 2013, 22, 5767–5770 CrossRef PubMed.
- L. D. Thai, J. Fanelli, R. Munaweera, M. O’Mara, C. Barner-Kowollik and H. Mutlu, Main-chain macromolecular hydrazone photoswitches, Angew. Chem., Int. Ed. Engl., 2023, e202315887 Search PubMed.
- V. I. Minkin, Photo-, thermo-, solvato-, and electrochromic spiroheterocyclic compounds, Chem. Rev., 2004, 104, 2751–2776 CrossRef CAS PubMed.
- J. T. Wojtyk, A. Wasey, N.-N. Xiao, P. M. Kazmaier, S. Hoz, C. Yu, R. P. Lemieux and E. Buncel, Elucidating the mechanisms of acidochromic spiropyran-merocyanine interconversion, J. Phys. Chem. A, 2007, 111, 2511–2516 CrossRef CAS PubMed.
- R. Ganesan and F. Remacle, Stabilization of merocyanine by protonation, charge, and external electric fields and effects on the isomerization of spiropyran: a computational study, Theoretical Chemistry in Belgium: A Topical Collection from Theoretical Chemistry Accounts, 2014, pp. 167–179 Search PubMed.
- L. Kortekaas, J. Chen, D. Jacquemin and W. Browne, Proton-stabilized photochemically reversible E/Z isomerization of spiropyrans, J. Phys. Chem. B, 2018, 122, 6423–6430 CrossRef CAS PubMed.
- S.-R. Keum, K.-B. Lee, P. M. Kazmaier and E. Buncel, A novel method for measurement of the merocyanine-spiropyran interconversion in non-activated 1,3,3-trimethylspiro-(2H-1-benzopyran-2,2′-indoline) derivatives, Tetrahedron Lett., 1994, 35, 1015–1018 CrossRef CAS.
- H. Shiozaki, Molecular orbital calculations for acid induced ring opening reaction of spiropyran, Dyes Pigm., 1997, 33, 229–237 CrossRef CAS.
- N. A. Murugan, S. Chakrabarti and H. Ågren, Solvent dependence of structure, charge distribution, and absorption spectrum in the photochromic merocyanine–spiropyran pair, J. Phys. Chem. B, 2011, 115, 4025–4032 CrossRef CAS PubMed.
- J. Piard, Influence of the solvent on the thermal back reaction of one spiropyran, J. Chem. Educ., 2014, 91, 2105–2111 CrossRef CAS.
- W. Tian and J. Tian, An insight into the solvent effect on photo-, solvato-chromism of spiropyran through the perspective of intermolecular interactions, Dyes Pigm., 2014, 105, 66–74 CrossRef CAS.
- S. Weber, Stabilization of the merocyanine form of photochromic compounds in fluoro alcohols is due to a hydrogen bond, Chem. Commun., 1998, 2685–2686 Search PubMed.
- J.-J. Zhang, X.-H. Duan, Y. Wu, J.-C. Yang and L.-N. Guo, Transition-metal free C–C bond cleavage/borylation of cycloketone oxime esters, Chem. Sci., 2019, 10, 161–166 RSC.
- Y. Li, K. Cheng, X. Lu and J. Sun, A Facile and Efficient Approach to N-Protected-β-Sulfinyl-enamines via C-Sulfinylation of Enamides and Enecarbamates, Adv. Synth. Catal., 2010, 352, 1876–1880 CrossRef CAS.
- K. Guo and Y. Chen, Lewis acid and base triggered molecular switch, J. Mater. Chem., 2009, 19, 5790–5793 RSC.
- A. Allerhand and P. Von Rague Schleyer, A survey of CH groups as proton donors in hydrogen bonding, J. Am. Chem. Soc., 1963, 85, 1715–1723 CrossRef CAS.
- H. Komber, S. Müllers, F. Lombeck, A. Held, M. Walter and M. Sommer, Soluble and stable alternating main-chain merocyanine copolymers through quantitative spiropyran–merocyanine conversion, Polym. Chem., 2014, 5, 443–453 RSC.
- P. Ferreira, G. Moncelsi, G. Aragay and P. Ballester, Hydrogen-bonded dimeric capsules with appended spiropyran units: towards controlled cargo release, Chem.–Eur. J., 2021, 27, 12675–12685 CrossRef CAS PubMed.
- E. Goldburt, F. Shvartsman, S. Fishman and V. Krongauz, Intramolecular interactions in photochromic spiropyran-merocyanine polymers, Macromolecules, 1984, 17, 1225–1230 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 2004 Search PubMed.
- J. P. Guthrie, Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a range of 20 p K units, Can. J. Chem., 1978, 56, 2342–2354 CrossRef CAS.
- M. E. Genovese, E. Colusso, M. Colombo, A. Martucci, A. Athanassiou and D. Fragouli, Acidochromic fibrous polymer composites for rapid gas detection, J. Mater. Chem. A, 2017, 5, 339–348 RSC.
- Y. Onai, M. Mamiya, T. Kiyokawa, K. Okuwa, M. Kobayashi, H. Shinohara and H. Sato, Colored merocyanine aggregates: long-lived crystals of large size (10-100 mu m) and deaggregation of small aggregates in solutions, J. Phys. Chem., 1993, 97, 9499–9505 CrossRef CAS.
- T. Satoh, K. Sumaru, T. Takagi, K. Takai and T. Kanamori, Isomerization of spirobenzopyrans bearing electron-donating and electron-withdrawing groups in acidic aqueous solutions, Phys. Chem. Chem. Phys., 2011, 13, 7322–7329 RSC.
- K. Pahnke, J. Brandt, G. Gryn'Ova, C. Y. Lin, O. Altintas, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Entropy-driven selectivity for chain scission: where macromolecules cleave, Angew. Chem., Int. Ed., 2016, 55, 1514–1518 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc06383f |
| This journal is © The Royal Society of Chemistry 2024 |